
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAir-stable iron phosphide catalysts for electric field-assisted low-temperature ammonia synthesis†
Ryuku
Maeda
a,
Hiroshi
Sampei
 a,
Tomohiro
Tsuda
b,
Hiromu
Akiyama
a,
Yuta
Mizutani
a,
Takuma
Higo
a,
Hideaki
Tsuneki
a,
Takato
Mitsudome
*b and
Yasushi
Sekine
*a
a,
Tomohiro
Tsuda
b,
Hiromu
Akiyama
a,
Yuta
Mizutani
a,
Takuma
Higo
a,
Hideaki
Tsuneki
a,
Takato
Mitsudome
*b and
Yasushi
Sekine
*a
aDepartment of Applied Chemistry, Waseda University, 3-4-1, Okubo, Shinjuku, Tokyo 169-8555, Japan. E-mail: ysekine@waseda.jp
bDepartment of Materials Engineering Science, Graduate School of Engineering Science, Osaka University, 1-3 Machikaneyama, Toyonaka, Osaka 560-8531, Japan. E-mail: mitsudom@cheng.es.osaka-u.ac.jp
First published on 28th March 2024
Abstract
Ammonia synthesis is crucial for fertiliser production, future hydrogen storage, and hydrogen carrier production. Herein, we present a novel ammonia synthesis system using an iron phosphide catalyst with a DC electric field. Under mild reaction conditions (i.e., 400–430 K and 0.1 MPa of H2), the air-stable iron phosphide catalyst exhibits superior performance to the conventional, unstable iron catalysts. Furthermore, this catalyst system enables precise control over ammonia production through on/off-switching of the DC electric field, demonstrating its potential for more dynamic and responsive synthesis processes. The structure–activity relationship of this catalytic system was also explored using various techniques such as TEM observation, XAFS analysis, and theoretical calculations. These findings open up promising avenues for the future development of next-generation on-site and low-temperature ammonia synthesis technologies that can be operated on demand.
1 Introduction
The production of ammonia (NH3), a chemical feedstock highly in demand for fertilisers and basic chemicals, is increasing annually with the growing world population.1 Recently, the concept of hydrogen (H2) energy has been proposed, for which renewable energy is converted into chemical energy to produce hydrogen for storage and distribution.2,3 In line with this concept, ammonia has also attracted attention as a hydrogen carrier by virtue of its carbon-free nature, high hydrogen density (17.8 wt%) and liquefaction.4 The Haber–Bosch process, established almost 110 years ago, remains the main industrial process for ammonia production. The process is operated under high-temperature and high-H2-pressure conditions (i.e., 673–773 K and 10–20 MPa H2), even though low temperature is equally favourable because it is an exothermic reaction.5,6However, to produce ammonia as a hydrogen carrier using H2 generated from renewable energy sources, ammonia synthesis using a small-scale and distributed (on-site) method that can be operated at a high rate even under mild conditions (low temperature and lower pressure) is highly desirable. It would accommodate spatial and temporal variabilities.7
Great efforts have been devoted towards catalyst development for low-temperature and low-pressure ammonia synthesis. Most of the reported low-temperature ammonia synthesis procedures to date have used precious metals such as Ru as the active metal, leaving room for improvement in terms of production cost.8–12 By contrast, Fe might be an ideal metal for use in the Haber–Bosch process as the active metal because it is earth-abundant and available at a low price.13–15 However, Fe catalysts exhibit low activity for ammonia synthesis at low temperatures. Moreover, they are difficult to handle because of their easy oxidation, leading to rapid deactivation.
Recently, we have discovered that electric field-assisted catalysis is promising for low-temperature ammonia synthesis.11 When a semi-conductor catalyst support is used for electric field-assisted catalysis by applying a weak direct current (DC), proton conduction on the catalyst support promotes surface reactions even at low temperatures. Ammonia synthesis proceeds on supported Fe catalysts even at 464 K when an electric field is applied, but this catalyst requires prereduction.16,17
For this study, we developed an on-demand-driven ammonia synthesis process with a high reaction rate at ambient pressure and low temperature (400–430 K). This synthesis was accomplished by the synergistic integration of an electric field and an air-stable iron phosphide (FeP) catalyst, thereby eliminating the need for prereduction.18 It is noteworthy that the FeP catalyst exhibits no activity, even at high temperatures, in the absence of an applied electric field, which enables precise control over ammonia production through on-/off-switching of the DC electric field. This dynamic and responsive ammonia synthesis system was not achieved with conventional gas–solid catalysis, which highlights this method's potential as a promising method for on-site and intelligent ammonia synthesis.
2 Results and discussion
2.1. Structure of unsupported FeP and supported FeP
First, iron phosphide was synthesized by a solvothermal method (Experimental section of ESI†). The iron phosphide structure was characterized (Fig. 1, Tables S1 and S2 in ESI†). ICP-AES (Inductively Coupled Plasma Atomic Emission Spectrometry) and XRF (X-ray Fluorescence) analysis (X-ray were used to determine the elemental ratios of Fe and P, respectively. The results showed that Fe/P was close to 1 in both cases, and the obtained structure was considered to be FeP. The X-ray diffraction (XRD) pattern in Fig. 1(a) shows that the synthesized iron phosphide consists of orthorhombic FeP (ICDD 03-065-2595, Table S3 in ESI†), as reported in other studies of Fe phosphide synthesis at 593 K.18–20 Nano-ordered structural images of FeP observed using transmission electron microscopy (TEM) and are presented in Fig. 1(b) and (c). The obtained FeP compounds comprise hexagonal prismatic nano-ordered rods (NRs) with 9.4 nm width and 33.2 nm length. To observe FeP NR surface details, a high-resolution TEM image and an electron diffraction (SAED) pattern were obtained, respectively, in a selected area, as presented in Fig. 1(d) and (e). These results indicate the basal plane as a (31−1) plane, whereas the (011) and (103) planes are perpendicular. Based on the above-mentioned characterization results, the synthesized Fe phosphide evidently possesses a nano-ordered rod-like structure with well-defined FeP composition. The structural information is depicted in Fig. 1(f).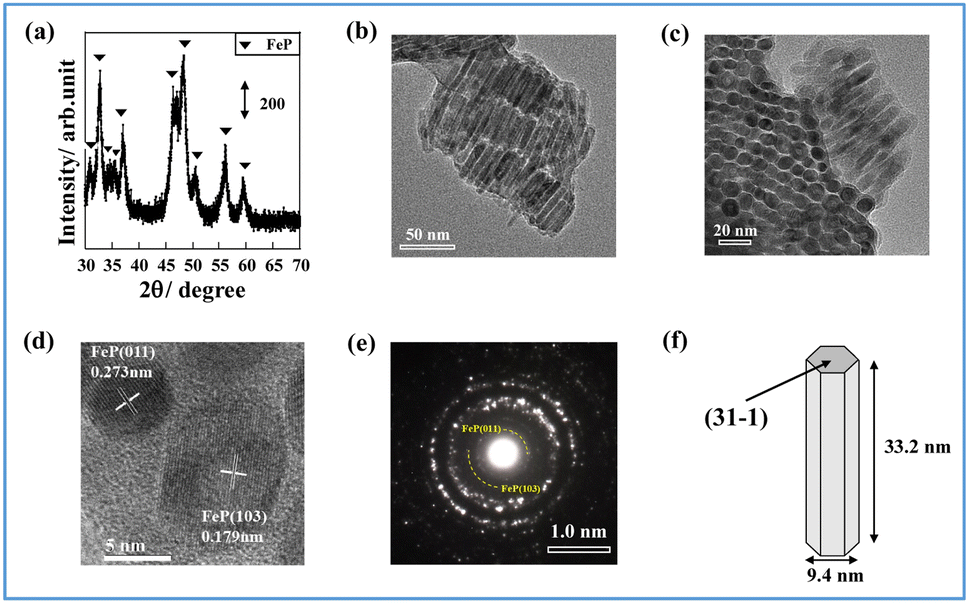 | ||
| Fig. 1 Characterization of Fe phosphide: (a) XRD measurement, (b) TEM image (side view), (c) TEM image (bottom view), (d) high-resolution TEM image, (e) electron diffraction image for selected area, and (f) illustrated structure of Fe phosphide. | ||
Catalyst supports can disperse and immobilize nanostructured metals. In fact, interaction between the support and the active metal can enhance catalytic activity. A perovskite-type oxide, Sr0.875Ba0.125ZrO3 (SBZO), was chosen as the support for FeP NRs because SBZO exhibits surface proton conduction property, which is beneficial for molecular transformation with an electric field.21 The FeP NRs were loaded on SBZO (FeP/SBZO) using the equilibrium adsorption method. Their structures were also investigated. Regarding the XRD patterns of FeP/SBZO (Fig. 2(a)), all peaks were attributed to SrZrO3 (ICDD 01-074-2231, Table S4 in ESI†). No peak of FeP or any other Fe compound was observed, indicating that FeP NRs are highly dispersed. Subsequent TEM and scanning transmission electron microscope-energy-dispersive X-ray (STEM-EDX) spectroscopy observations confirmed the high dispersion of FeP NRs, which maintained the NR shape with a uniform distribution of Fe and P (Fig. 2(b), S1 in ESI†).
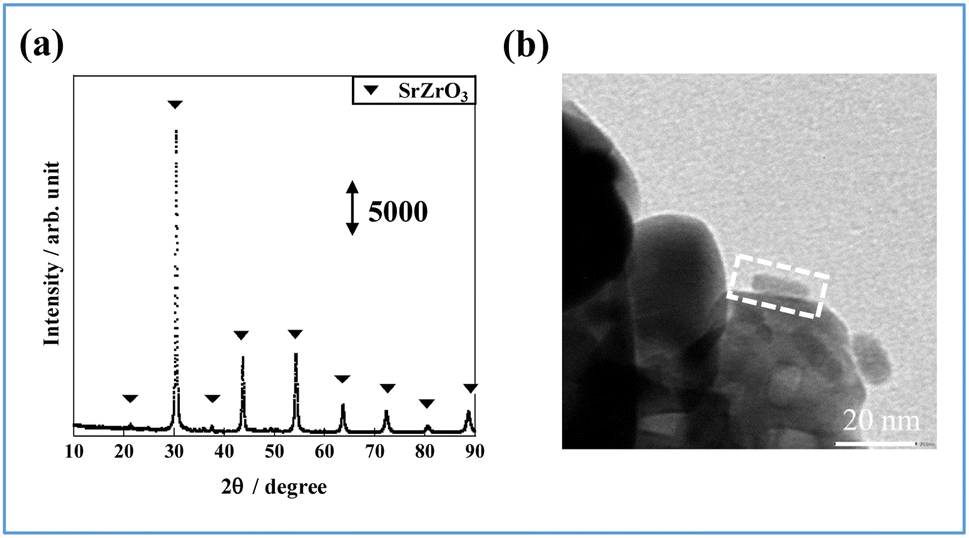 | ||
| Fig. 2 FeP/SBZO for (a) XRD patterns and (b) TEM image (the area marked by the dotted square shows the supported rod). | ||
2.2. Electronic state of FeP and FeP/SBZO
The oxidation state of Fe is known to affect ammonia synthesis activity strongly, with metallic Fe acting as the active site for ammonia synthesis, whereas oxidised Fe exhibited a considerably lower activity.16,22 To evaluate the oxidation state of the prepared catalysts, Fe K-edge XAFS (X-ray absorption fine structure) spectra of Fe foil, FeO, FeP, and FeP/SBZO were recorded. The FeP and FeP/SBZO samples were prepared after exposure to air. The X-ray absorption near-edge structure (XANES) results are depicted in Fig. 3(a). The absorption edge energy of FeP (red line) is close to that of the Fe foil (black line). The white line observed in FeO (green line) was not observed in FeP, indicating that the electronic state of Fe species in FeP after exposure to the air atmosphere is closer to metallic Fe and different from Fe oxides. Additionally, the absorption edge energy and spectral shape of FeP/SBZO (Fig. 3(a), blue) were similar to those of FeP, suggesting that the electronic state of the Fe species in FeP was not altered by loading on SBZO.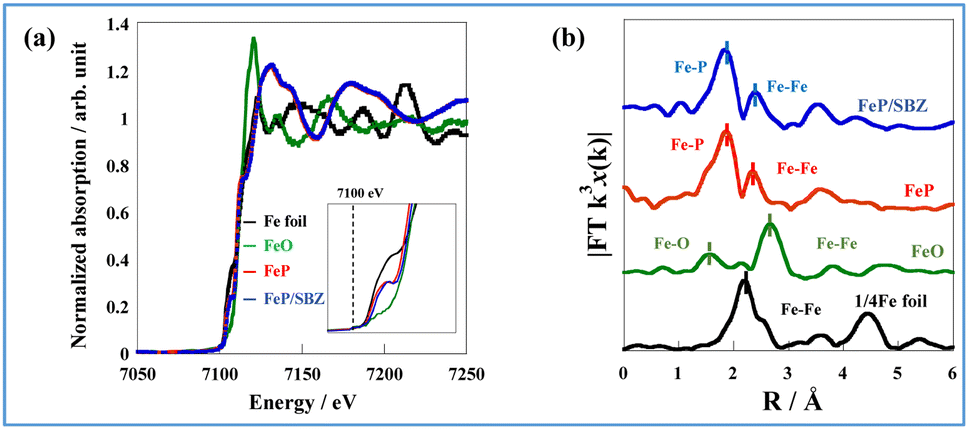 | ||
| Fig. 3 (a) Fe K-edge XANES spectra of the Fe foil, FeO, FeP, and FeP/SBZO, and (b) Fourier transformation of the k3-weighted EXAFS of the Fe foil, FeO, FeP, and FeP/SBZO. | ||
To gain insights into the atomic bonding structure of Fe species in FeP and FeP/SBZO, Fourier transform-extended X-ray absorption fine structure (FT-EXAFS) analysis was applied. The FT-EXAFS spectra of Fe foil, FeO, FeP and FeP/SBZO at the Fe-K absorption edge are depicted in Fig. 3(b) and Table S5 in ESI.† These spectra suggest that neither unsupported nor supported FeP is oxidised by exposure to air because no peak can be attributed to Fe–O bonds. These findings are consistent with the results obtained from XANES analysis. In addition, FeP and FeP/SBZO had elongated Fe–Fe bonds and a different structure from that of Fe metal. Furthermore, the coordination numbers of Fe–P and Fe–Fe bonds in FeP and FeP/SBZO indicate that the stoichiometric ratio of iron to phosphorus is 1 (Table S5 in ESI†), which is also consistent with the XRD pattern results. These XAFS results indicate that the Fe species in FeP NRs have metallic state and that Fe–P bonds have high stability in air, and these properties are maintained when FeP NRs are supported on SBZOs.
To accumulate additional information related to the electronic state near the surface of FeP, X-ray photoelectron spectroscopy (XPS) of FeP after exposure to air was performed (Fig. 4). From Fig. 4, three peaks at 707.3 eV, 709.1 eV, and 710.5 eV in Fe 2p3/2 were observed and were assigned respectively to Fe(0), Fe(II), and Fe(III).23 The existence of the major peak attributed to Fe(0) indicates that Fe on the FeP surface is in a metallic state, which is consistent with the XAFS results. The latter two peaks could be attributed to the surface oxidation of FeP or to Fe oxide formation. The XPS result of FeP/SBZO (Fig. S2 in ESI†) is similar to those of FeP. The signal-to-noise (S/N) ratio is very low because of the low loading of FeP/SBZO (5 wt%: Fe atoms).
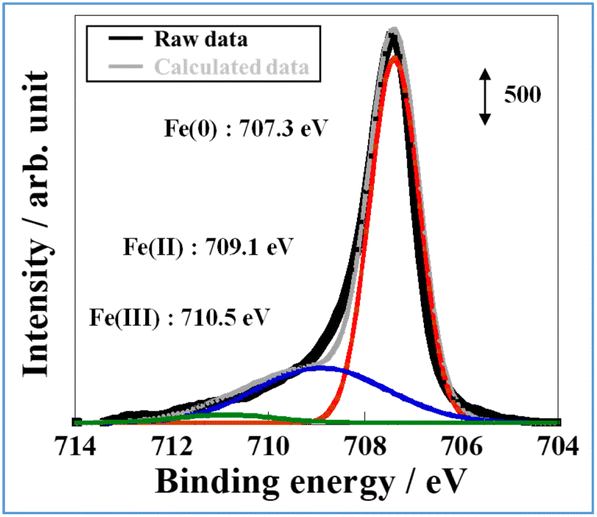 | ||
| Fig. 4 Fe 2p X-ray photoelectron spectra of FeP. | ||
Moreover, XPS analysis of P 2p and P 2s in FeP and FeP/SBZO was performed (Fig. S3(a) in ESI†). The XPS spectrum for FeP reveals three peaks attributed to P 2p3/2 (129.7 eV), P 2p1/2 (130.5 eV), PO3− (132.6 eV), and PO43− (134.0 eV).24,25 Some phosphate species might be attributed to surface oxidation of FeP and impurities during synthesis.26,27 However, the P 2p and P 2s peaks of FeP/SBZO respectively overlap with the peak of a component of the support, Sr and Zr. Therefore, they could not be assigned (Fig. S3(b) and (c) in ESI†).28–30
Theoretical calculations were carried out to elucidate the oxygen affinity of FeP NRs. The hexagonal surface of FeP was used as the FeP surface model because the active metal near the catalyst support is the main active site on the ammonia synthesis in the electric field. It has been considered that the NH3 synthesis reaction in an electric field is promoted by the collision of migrated proton and adsorbed nitrogen, and the proton migration on the catalyst surface is caused by applying an electric field.23 Therefore, in this work, the active site is set at the interface between the active metal and the catalyst support.
Hexagonal faces are present at all interfaces facing the SBZO support. Based on the FeP NR characterisation results (Fig. 1(f)), the FeP(31−1) model was used (Fig. S4 in ESI†), whereas the Fe(110) model, which represents the most stable surface of bcc Fe (Fig. S5†), was used as a reference for the Fe surface. In the FeP nanorods, the (011) and (103) facets were also exposed in the TEM images, hence two models were designed and calculated for these two surfaces, as shown in Fig. S6.† In these cases, the interfacial surfaces other than the (31−1) surface in FeP have obtuse contact angles (120°), which can be a steric hindrance for molecular adsorption.
First, the adsorption energy of oxygen atoms and di-oxygen on the slab models was calculated. The results are presented in Fig. 5. In this figure, “split” denotes dissociative adsorption. Oxygen atoms and dissociated oxygen molecules adsorb preferentially on Fe over FeP. The adsorption energy of dissociated oxygen molecules is twice as high as that of monoatomic oxygen. We also confirmed the lack of spontaneous dissociation of di-oxygen on FeP. The reaction pathway of dissociation of di-oxygen was calculated using the climbing image nudged elastic band (CI-NEB) method (Fig. S7–S9 in ESI†). Although two activation barriers and a stable intermediate state appear in the energy diagram, because of the activation barrier between the intermediate and final states, spontaneously dissociative adsorption of O2 does not occur, indicating the high antioxidant ability of FeP.
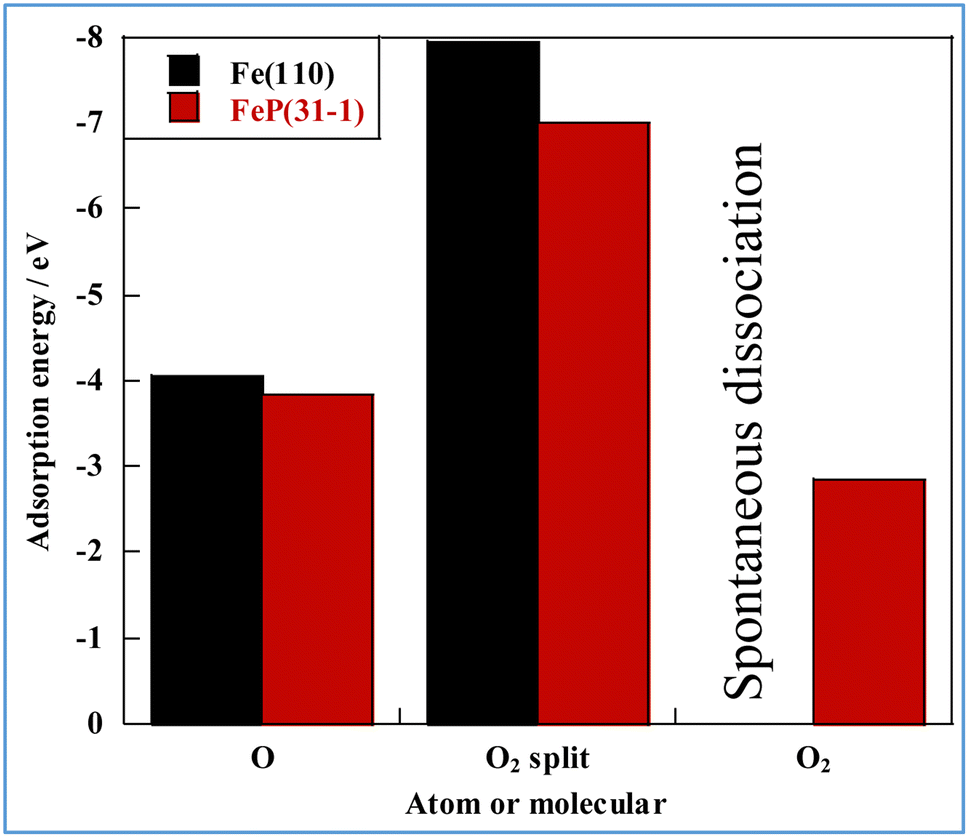 | ||
| Fig. 5 Adsorption energies of oxygen on Fe and FeP. | ||
The analysis described above indicates that the electronic state of Fe in the FeP/SBZO catalyst remains largely metallic even with existing oxygen (i.e., high air-stable nature), which is considerably different from the findings obtained for conventional Fe-based supported catalysts.
2.3. Evaluation of FeP/SBZO catalytic performance
Conventional Fe-based catalysts are susceptible to oxidation and deactivation in the presence of trace amounts of oxygen in the reaction system. They often require pre-reduction treatment at high temperatures to form metallic Fe species. In contrast, the results of the characterization of FeP revealed that the supported FeP maintains its metallic properties even when exposed to air. This finding suggests the use of supported FeP as a highly durable catalyst without the need for pre-reduction treatment. Therefore, we explored the catalytic activity of FeP/SBZO for ammonia synthesis when applying an electric field. Table 1 shows the results of catalytic activity evaluation under mild reaction conditions (i.e., 400–430 K, 0.1 MPa) with 6 mA current. It is noteworthy that the FeP/SBZO catalyst exhibited the highest ammonia production rate of 0.965 mmol g−1 h−1, surpassing the production rates for the reported iron catalysts.17,31 The results in Table 1 show that the voltage has less effects on the activity in this case. The catalysts loaded with the same amount of Fe, but not FeP, showed only a much lower activity than that of FeP in both cases, with and without reduction (shown in Table S6 in ESI†). This means that FeP is not only effective in maintaining the reduced state of Fe, but it is also necessary to consider the effect of electronic interactions between Fe and P. These will be discussed in more detail in a later section. It is intriguing that the FeP/SBZO catalyst exhibited no activity in the absence of an electric field. This pronounced electric field effect on the catalytic activity is unprecedented, strongly suggesting that a different reaction pathway than that for thermal catalysis exists on the catalyst surface.| Catalyst | Voltage/kV | NH3 synthesis rate/mmol g−1 h−1 | Reference |
|---|---|---|---|
| 5 wt% FeP/SBZO (no pre-reduction) | 0.42 | 0.965 | This work |
| 7 wt% Fe/CeO2 (pre-reduced) | 0.37 | 0.152 | 23 |
| 7 wt% Co/CeO2 (pre-reduced) | 0.39 | 0.0571 | 23 |
| 7 wt% Ni/CeO2 (pre-reduced) | 0.41 | 0.125 | 23 |
| 5.0 wt% Co/Ce0.5Zr0.5O2 (pre-reduced) | 0.47 | 0.294 | 31 |
| 5 wt% Fe/Ce0.4Al0.1Zr0.5O2-δ (pre-reduced) | 0.41 | 0.835 | 17 |
The strong effect of the electric field on ammonia synthesis when using the FeP/SBZO catalyst inspired us to explore its responsive nature. This responsiveness enables precise control over ammonia production through on/off-switching of the DC electric field. We investigated the feasibility of on-demand ammonia synthesis by toggling the electric field. The results of the cycle test, for which the heating test and the electric field application test were repeated, are depicted in Fig. 6. The catalytic activity by heating was zero at 673 K. Therefore, the temperature dependence of the FeP/SBZO catalyst was measured, and it became clear that the activity decreased at higher temperatures, which is different from the conventional Arrhenius plot (Fig. S10 in ESI†). The ammonia production rate of the FeP/SBZO catalyst was almost unchanged when only the current value was changed at the same temperature (Fig. S11 in ESI†). A more detailed study of the cause of this is required in the future. The ammonia synthesis activity appeared only when an electric field was applied, demonstrating that on-demand ammonia synthesis at low temperatures is possible by turning the electric field on and off. Furthermore, no significant decrease in activity occurred (Fig. S12 in ESI†). The FeP structure was stable during cycling (TEM images are presented in Fig. S13 in ESI,†etc.).
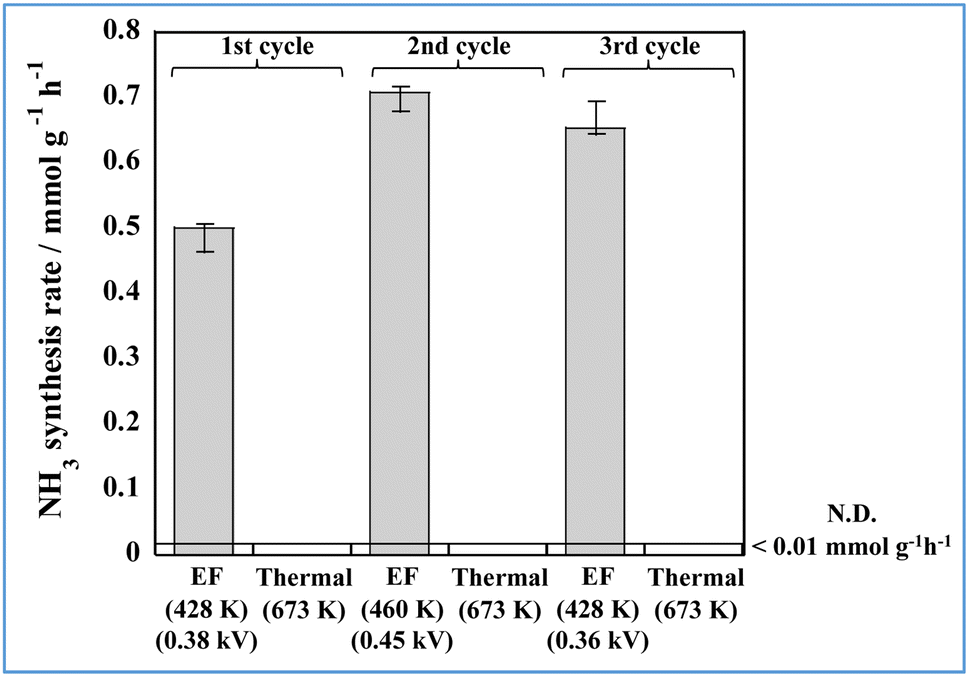 | ||
| Fig. 6 Cyclic results obtained for thermal catalysis and catalytic reactions in an electric field (denoted as EF). | ||
2.4. Investigation of the structure–activity relationship of FeP
In general, one of two mechanisms can be used for ammonia synthesis on catalyst surfaces: a dissociation mechanism, by which the N–N bond of N2 is cleaved directly, or an association mechanism, by which the N–N bond is cleaved after N2H is formed.31 According to earlier studies,31 reactions without an electric field (i.e., thermal reactions) occur through the dissociative mechanism with N2 dissociative adsorption as the rate-determining step, whereas reactions with the electric field occur though the associative mechanism with N2H formation as the rate-determining step. To elucidate the catalytic behaviour of the FeP catalyst at the rate-determining step, the adsorption energies of the main adsorbed species on the FeP surface model in comparison with the Fe(110) surface model were investigated. Fig. 7 presents the trends of N2 and N2H molecular adsorption as well as those of H2 and N2 dissociative adsorption. The results show that FeP is unfavourable for dissociative adsorption of N2 but favourable for dissociative adsorption of H2 and molecular adsorption of N2H. Bader charge analysis and density functional theory calculations have reported electron transfer from Fe to P on the FeP surface,32–34 which is regarded as a ligand effect, and which has been confirmed using XPS (Fig. 4). This ligand effect makes Fe electron-poor. As a result, the direct dissociation of N2 on FeP is suppressed because of the weak electron donation from the electron-poor Fe atom to the LUMO of the N2 molecule,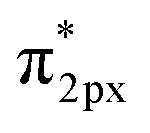 and
and 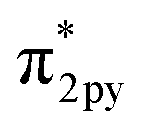 . Furthermore, because N2H is adsorbed preferentially onto positively charged metals,35 FeP was found to adsorb N2H more strongly than Fe did. The electronic state of Fe at the FeP-support interface is more positive than in other regions, which indicates that the activity of interfacial Fe is influenced more strongly by charge transfer from the catalyst support.
. Furthermore, because N2H is adsorbed preferentially onto positively charged metals,35 FeP was found to adsorb N2H more strongly than Fe did. The electronic state of Fe at the FeP-support interface is more positive than in other regions, which indicates that the activity of interfacial Fe is influenced more strongly by charge transfer from the catalyst support.
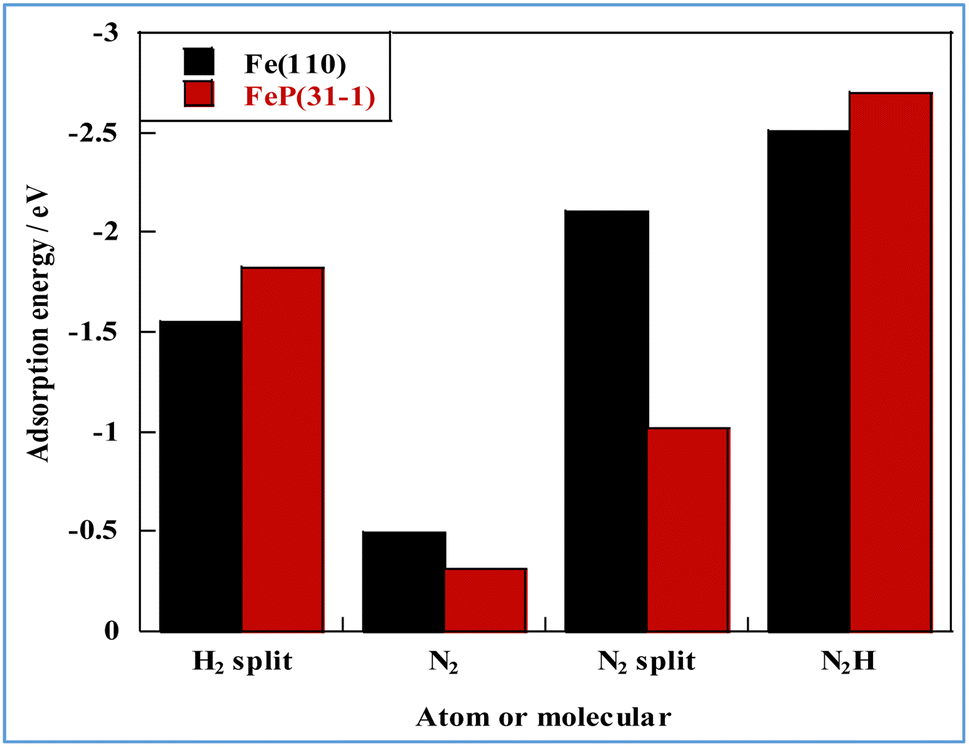 | ||
| Fig. 7 Molecular and atomic adsorption energies related to NH3 synthesis for different metal species. | ||
The states of the various stages governing the reaction rates of ammonia synthesis on Fe-catalysts and FeP catalysts, respectively, with and without an electric field, were then investigated. The energies and structures of the transition states were calculated for the Fe(110) and FeP(31−1) facets. Information related to the reaction progression in heating- and electric field-applied catalysis was organised. Fig. 8 shows the intermediate states on the respective metal surfaces in heating-applied catalysis. Similar calculations for the FeP(011) and FeP(103) surfaces as well as the FeP(31−1) surface are shown in Fig. S14–16 in ESI.† From these findings, it is apparent that P dilutes the Fe–Fe bonds and extends the Fe–Fe distance (the so-called ensemble effect). This extension prevents each N atom in N2 from getting sufficiently close to the Fe atom on the FeP surface in the dissociation of N2, which requires an Fe–Fe site. Transition states were also calculated using the CI-NEB method.
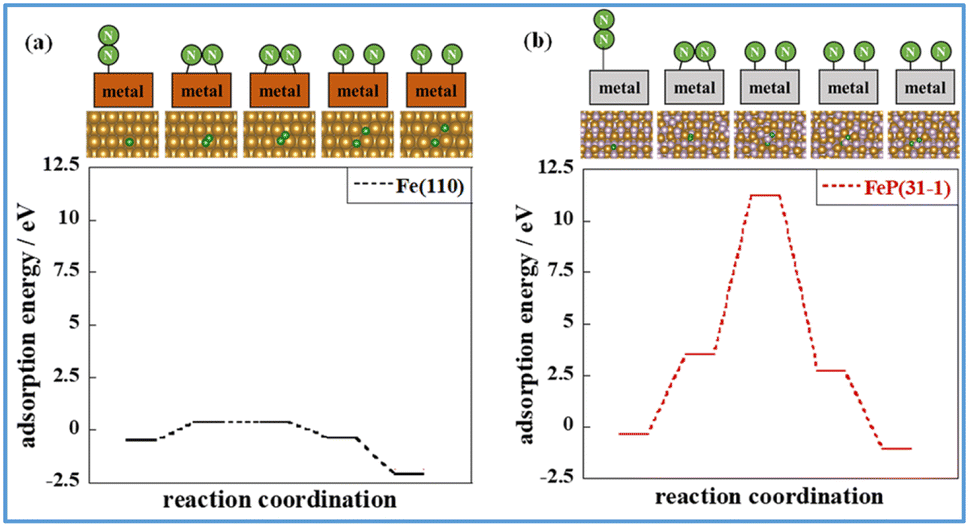 | ||
| Fig. 8 Intermediate states of N2 dissociation calculated using the NEB method. Energy diagram of N2 dissociation calculated using the NEB method: (a) Fe(110) and (b) FeP(31−1), where purple, brown, and green balls respectively represent P, Fe, and N. Because the second coordination (the first intermediate state) is the most unstable on both surface models, its energies were adopted as the energies of transition states. | ||
Comparison of these figures indicates that FeP(31−1) in Fig. 8(b) shows an extreme higher energy barrier than Fe(110) in Fig. 8(a). These results indicate that the phosphidation of Fe remarkably inhibits the dissociative mechanism in the thermal-applied catalysis in terms of both electronic state (i.e., the ligand effect) and structure (i.e., the ensemble effect). Furthermore, stronger adsorption of N2H on FeP(31−1) implies that the phosphidation of Fe facilitates the associative mechanism. Based on these results, the energy diagram of the reaction is summarised in Fig. 9 and the rate-determining energy differences on each surface are summarized in Table S7 in ESI.†Fig. 9(a) and (b) respectively present the energy diagrams involving the dissociative mechanism without electric field and the associative mechanism through the surface protonics in an external electric field. In the associative mechanism, the relative energy of N2H adsorption is calculated from the energies of N2 and each metal surface. The ammonia synthesis activity here is correlated with the N2H production energy, which is the N2H adsorption energy minus the N2 end-on adsorption energy.23,33 Subsequent cleavage of the N–N bond in N2H does not exhibit the volcano-shaped relation between the ammonia synthesis activity in an electric field and the dissociation energy of the N–N bond.23 Fe(110) exhibited a lower N2 activation energy than FeP(31−1) from comparison of the energy difference between the second state and the third state in Fig. 9(b), which seems to contradict with the trend of the N2H production energy. This contradiction arises from the difference in the activation pathway for chemisorbed N2. There are surface N2 and H as reactants when an electric field is absent; however, N2 activation is not caused by N2H-mediated N2 cleavage but by the direct dissociation of N2. This fact suggests that non-thermal equilibrium states of catalysts originate from an electric field and/or direct current may induce the characteristic N2 activation through the N2H intermediate formation. As the simulation method reported in this study cannot be applied to non-equilibrium states, the true transition state in an electric field is probably different from the third state in Fig. 9(b). Note that the N2H formation on the Fe(110) surface showed a lower activation energy than N2 direct cleavage; however, the reaction pathway of the N2H formation assumed H collision with no thermal barrier. Thus, this result is consistent with the preference for N2 direct dissociation in heat-applied catalysis, where there is a finite barrier for H migration. The energy diagram therefore suggests that FeP favours N2H-mediated N2 cleavage and not direct N2 cleavage. These simulations support that catalysis by conventional heating leads to low (almost zero) ammonia synthesis activity because of two effects: ligand effects and ensemble effects. By contrast, iron phosphide exhibits high ammonia synthesis activity via the associative mechanism in an external electric field.
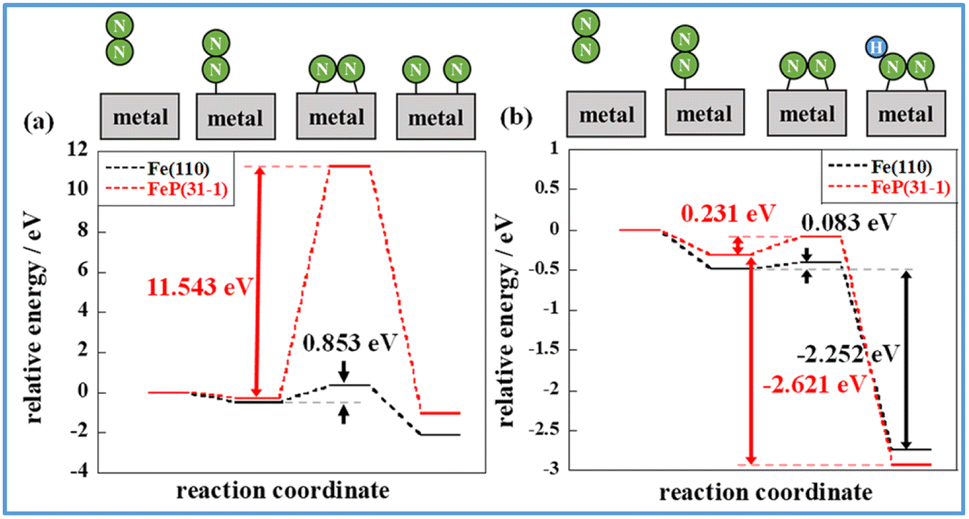 | ||
| Fig. 9 Energy diagram of N2 dissociation (a) and N2H formation (b) on Fe(110) and FeP(31−1). Activation energy for N2 dissociation in the absence of an electric field corresponds to the energy difference between the second state and the third state in (a). N2H production energy in an electric field corresponds to the energy difference between the second state and the fourth state in (b). | ||
3 Conclusions
FeP exhibits high activity in ammonia synthesis in the presence of an electric field under ambient pressure and low-temperature conditions. By virtue of its air-stable property, FeP showed high ammonia synthesis activity without pre-reduction in an electric field. FeP does not have an Fe–Fe ensemble, hence direct N2 cleavage, which is dominant in common heated iron catalysts, is unlikely to occur. This was shown both from the present experiments and from computational chemistry: on FeP/SBZO, charge transfer from Fe to P and SBZO occurs, reducing electron donation from Fe atoms to the antibonding orbital of N2 molecules, LUMO. FeP/SBZO strongly inhibits N2 direct cleavage in the absence of an electric field and promotes the associated activation of N2 with surface proton migration in an electric field. This property reflects that the activity can be switched on and off easily with and without an electric field.Author contributions
Conceptualization by T. M. and Y. S. data curation by R. M., H. S., T. T., Y. M. and Y. S. investigation by R. M. and Y. M. software by H. S. and H. A., supervision by Y. S. writing – original draft by Y. M., R. M., and H. S. writing – review & editing by T. T., T. H., H. T., T. M. and Y. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Some characterisation in this work was achieved using equipment (JEM-2100F and Versa Probe II and JSX-1000S: Material Characterization Central Laboratory in Waseda University, Agilent5100: Environmental Safety Center in Waseda University) shared with the MEXT Project for Promoting Public Utilization of Advanced Research Infrastructure (Program for supporting construction of core facilities) on grant numbers JPMXS0440500021 and JPMXS0440500022. This work was also supported by JSPS KAKENHI Grant 23H01761 and JST PRESTO Grants JPMJPR21Q9. All theoretical calculations were performed using Matlantis, provided by PFCC. XAFS measurements were conducted at SPring-8, BL14B2, Hyogo, Japan (Proposal number: 2021B1847). Some additional experiments for revision of this paper have been conducted by Ms. Reika Nakayama.References
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- M. Yue, H. Lambert, E. Pahon, R. Roche, S. Jemei and D. Hissel, Renew. Sustain. Energy Rev., 2021, 146, 111180 CrossRef.
- R. H. Lin, Y. Y. Zhao and B. D. Wu, Int. J. Hydrog. Energy, 2020, 45, 20164–20175 CrossRef CAS.
- Y. Sekine and T. Higo, Top. Catal., 2021, 64, 470–480 CrossRef CAS.
- A. J. Martın, T. Shinagawa and J. P. Ramırez, Chem, 2019, 5, 263–283 Search PubMed.
- H. Liu, Chin. J. Catal., 2014, 35, 1619–1640 CrossRef CAS.
- Y. Kobayashi, Y. Tang, T. Kageyama, H. Yamashita, N. Masuda, S. Hosokawa and H. Kageyama, J. Am. Chem. Soc., 2017, 139, 18240–18246 CrossRef CAS PubMed.
- K. Ogasawara, T. Nakao, K. Kishida, T.-N. Ye, Y. Lu, H. Abe, Y. Niwa, M. Sasase, M. Kitano and H. Hosono, ACS Catal., 2021, 11, 11005–11015 CrossRef CAS.
- Y. Ogura, K. Sato, S.-i. Miyahara, Y. Kawano, T. Toriyama, T. Yamamoto, S. Matsumura, S. Hosokawa and K. Nagaoka, Chem. Sci., 2018, 9, 2230–2237 RSC.
- K. Kishida, M. Kitano, M. Sasase, P. V. Sushko, H. Abe, Y. Niwa, K. Ogasawara, T. Yokoyama and H. Hosono, ACS Appl. Energy Mater., 2020, 3, 6573–6582 CrossRef CAS.
- R. Manabe, H. Nakatsubo, A. Gondo, K. Murakami, S. Ogo, H. Tsuneki, M. Ikeda, H. Nakai and Y. Sekine, Chem. Sci., 2017, 8, 5434–5439 RSC.
- K. Murakami, R. Manabe, H. Nakatsubo, T. Yabe, S. Ogo and Y. Sekine, Catal. Today, 2018, 303, 271–275 CrossRef CAS.
- B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 86–896 CrossRef PubMed.
- F. Bozso, G. Ertl, M. Grunze and M. Weiss, J. Catal., 1977, 49, 18–41 CrossRef CAS.
- S. Schouten, D. Pavlovic, J. S. S. Damste and J. W. D. Leeuw, Org. Geochem., 1993, 20(7), 901–909 CrossRef CAS.
- K. Murakami, Y. Mizutani, H. Sampei, A. Ishikawa, Y. Tanaka, S. Hayashi, S. Doi, T. Higo, H. Tsuneki, H. Nakai and Y. Sekine, Phys. Chem. Chem. Phys., 2021, 23, 4509–4516 RSC.
- R. Sakai, K. Murakami, Y. Mizutani, Y. Tanaka, S. Hayashi, A. Ishikawa, T. Higo, S. Ogo, H. Tsuneki, H. Nakai and Y. Sekine, ACS Omega, 2020, 5(12), 6846–6851 CrossRef CAS PubMed.
- G. Cho, H. Kim, Y. S. Park, Y.-K. Hong and D.-H. Ha, Int. J. Hydrog. Energy, 2018, 43, 11326–11334 CrossRef CAS.
- S. Yamaguchi, T. Mizugaki and T. Mitsudome, Eur. J. Inorg. Chem., 2021, 33, 3327–3331 CrossRef.
- S. Yamaguchi, S. Fujita, K. Nakajima, S. Yamazoe, J. Yamasaki, T. Mizugaki and T. Mitsudome, ACS Sustain. Chem. Eng., 2021, 9(18), 6347–6354 CrossRef CAS.
- Y. Tanaka, K. Murakami, S. Doi, K. Ito, K. Saegusa, Y. Mizutani, S. Hayashi, T. Higo, H. Tsuneki, H. Nakai and Y. Sekine, RSC Adv., 2021, 11, 7621–7626 RSC.
- L. Hu, A. Khaniya, J. Wang, G. Chen, W. E. Kaden and X. Feng, ACS Catal., 2018, 8, 9312–9319 CrossRef CAS.
- K. Murakami, Y. Tanaka, R. Sakai, K. Toko, K. Ito, A. Ishikawa, T. Higo, T. Yabe, S. Ogo, M. Ikeda, H. Tsuneki, H. Nakai and Y. Sekine, Catal. Today, 2020, 351, 119–124 CrossRef CAS.
- K. Idczak and R. Idczak, Metall. Mater. Trans. A, 2020, 51, 3076–3089 CrossRef CAS.
- A. Majjane, A. Chahine, M. Et-tabirouz, B. Echchahed, T.-O. Do and P. M. Breen, Mater. Chem. Phys., 2014, 143, 779–787 CrossRef CAS.
- X. Wu, K. Gong, G. Zhao, W. Lou, X. Wang and W. Liu, RSC Adv., 2018, 9, 4595–4603 RSC.
- S. Yamaguchi, S. Fujita, K. Nakajima, S. Yamazoe, J. Yamasaki, T. Mizugaki and T. Mitsudome, Green Chem., 2021, 23, 2010–2016 RSC.
- Y. Wang and P. M. A. Sherwood, Surf. Sci. Spectra, 2002, 9, 99–105 CrossRef CAS.
- A. Galtayries, R. Sporken, J. Riga, G. Blanchard and R. Caudano, J. Electron Spectros. Relat. Phenomena, 1998, 88, 951–956 CrossRef.
- B. M. Reddy, P. M. Sreekanth, Y. Yamada, Q. Xu and T. Kobayashi, Appl. Catal., A, 2002, 228, 269–278 CrossRef CAS.
- A. Gondo, R. Manabe, R. Sakai, K. Murakami, T. Yabe, S. Ogo, M. Ikeda, H. Tsuneki and Y. Sekine, Catal. Lett., 2018, 148, 1929–1938 CrossRef CAS.
- A. L. Garden and E. Skúlason, J. Phys. Chem. C, 2015, 119(47), 26554–26559 CrossRef CAS.
- K. Murakami, Y. Tanaka, S. Hayashi, R. Sakai, Y. Hisai, Y. Mizutani, A. Ishikawa, T. Higo, S. Ogo, J. G. Seo, H. Tsuneki, H. Nakai and Y. Sekine, J. Chem. Phys., 2019, 151, 064708 CrossRef.
- C. Ma, S. Feng, J. Zhou, R. Chen, Y. Wei, H. Liu and S. Wang, Appl. Catal., B, 2019, 259, 118015 CrossRef CAS.
- P. Liu, B. Gao, C. Wang, S. Pan, Z. Zhai, T. Wu, Y. Liu, J. Zhang and H. Lu, Nanoscale, 2021, 13, 4758–4766 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se00109e |
| This journal is © The Royal Society of Chemistry 2024 |