
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigation on the coordination between methylpyridine additives and the [Cu(dmp)2]2+/+ redox couple and its improvement towards the stability of the dye-sensitized solar cells†
Vinh Son
Nguyen
a,
Kala
Kannankutty
a,
Yu-Hsuan
Chen
 b,
Ding-Cheng
Wang
b,
Chen-Yu
Yeh
*b and
Tzu-Chien
Wei
*a
b,
Ding-Cheng
Wang
b,
Chen-Yu
Yeh
*b and
Tzu-Chien
Wei
*a
aDepartment of Chemical Engineering, National Tsing-Hua University, No. 101, Sec. 2, Guangfu Rd., East Dist., Hsinchu City 300, Taiwan. E-mail: tcwei@mx.nthu.edu.tw
bDepartment of Chemistry, i-Center for Advanced Science and Technology (i-CAST), Innovation and Development Center of Sustainable Agriculture (IDCSA), National Chung-Hsing University, No. 145, Xingda Rd., South Dist., Taichung 402, Taiwan. E-mail: cyyeh@dragon.nchu.edu.tw
First published on 12th April 2024
Abstract
In dye-sensitized solar cells (DSSCs), the use of copper(II/I) complex redox couples enables a high open-circuit voltage of greater than 1.0 V. However, the widely used electrolyte additive, 4-tert-butylpyridine (TBP), poisons the copper(II) complex, resulting in poor device stability. In this study, a series of pyridine derivatives 2-methylpyridine (2MP), 3-methylpyridine (3MP), 4-methylpyridine (4MP), and 3,5-dimethylpyridine (35DMP) were studied as alternatives to TBP for solving or mitigating the TBP-induced performance degradation. The coordination between the additives and copper(II) complex was extensively studied using ultraviolet-visible spectrophotometric titration, cyclic voltammetry, and 1H-nuclear magnetic resonance spectroscopy. The moiety position on the pyridine ring was found to greatly affect the electrochemical properties of the redox couple. The 35DMP penta-coordinated copper(II) was found to least affect the electrochemical activity at the counter electrode and ionic diffusion in the bulk electrolyte. A DSSC with a 35DMP additive outperforms its TBP-based counterparts in not only power conversion efficiency but also long-term stability.
Introduction
Dye-sensitized solar cells (DSSCs), invented by Grätzel and co-workers, are a practical technology for harvesting the abundant energy of photons and converting this energy into electricity. DSSCs have achieved power conversion efficiencies (PCEs) of greater than 15% for simulated sunlight and 37% for artificial fluorescent light.1–5 Conventional liquid-type DSSCs comprise three components: a dye-adsorbed mesoporous wide-band-gap semiconductor film, a counter electrode containing a catalytic material, and an electrolyte containing a redox couple that maintains the charge neutrality of the device. Of these components, the redox couple plays a key role in determining the overall performance of the DSSC because it affects not only the open-circuit voltage VOC but also the charge collection efficiency of the device. Two-electron redox couples, such as the iodide/triiodide (I−/I3−) system, have been widely used for their slow electron recombination kinetics and high stability; however, they have a large potential loss that results in considerable VOC loss.6 By contrast, one-electron redox systems such as cobalt(III/II)7,8 and copper(II/I)9–11 have tunable redox potentials, enabling mitigation of the potential loss and achieving a device VOC approaching or exceeding 1.0 V. Cobalt(III/II) redox mediators are octahedral, bulky complexes that cause large mass transport resistance in the DSSCs.12,13 By contrast, copper(II/I) complex mediators are preferably tetrahedral with a highest occupied molecular orbital position that leads to high VOC and efficient dye regeneration.9 Fukuzumi et al. synthesized a series of copper(II/I) complexes with various ligands to create distinct geometries. Because of the mismatch between the ground state energy of N719 and the redox potential of bis(2,9-dimethyl-1,10-phenanthroline) copper(II/I) (abbreviated as [Cu(dmp)2]2+/+), a PCE of only 1.4% was achieved in their study.14 Wang et al. subsequently developed a [Cu(dmp)2]2+/+-based device with a PCE of 7% by using an energetically suitable dye in the same redox couple system.9 A breakthrough in copper-complex-mediated DSSCs occurred in 2016 when Freitag et al. reported a PCE of over 10% with a VOC exceeding 1.0 V for a DSSC comprising [Cu(dmp)2]2+/+ and bis(4,4′,6,6′-tetramethyl-2,2′-bipyridine) copper(II/I), known as [Cu(tmby)2]2+/+.15 Very recently, a DSSC with a PCE of 13.5% and a record-high VOC of 1.24 V was achieved through co-sensitization of the dyes MS5 and XY1b with a [Cu(tmby)2]2+/+ redox couple.16Electrolyte additives are essential for enhancing the photovoltaic performance of a DSSC.17,18 For example, the addition of a Lewis acid, such as Li+ and H+, to the electrolyte was found to increase JSC,19–21 whereas addition of a Lewis base, such as 4-tert-butylpyridine (TBP), was reported to enhance VOC.22,23 TBP is also reported to adsorb on the defect sites of TiO2 and thus reduce the frequency of electron recombination between injected electrons and oxidized species in the electrolyte.24 Unfortunately, many studies have reported that TBP has a poisoning effect on copper(II) complexes.25–27 Grätzel et al. reported that the charge transfer kinetics and diffusion of [Cu(tmby)2]2+/+ is sluggish in the presence of TBP.28 We also confirmed that new TBP-coordinated [Cu(dmp)2]2+ complexes form when TBP is added to an electrolyte solution.29 These newly formed compounds are bulky and electrochemically inactive on the counter electrode, negatively affecting the photovoltaic performance of the DSSC.29 Very recently, Bach et al. confirmed the presence of [Cu(dmp)2TBP]2+ in an electrolyte containing TBP.30 Aiming to overcome TBP-induced degradation, many studies have replaced TBP with other pyridine-derived additives. Kavan et al. investigated the device performance of 2,6-bis-tert-butylpyridine, 4-methoxypyridine, and 4-(5-nonyl)pyridine.31 They found that coordination ability and basicity of a Lewis base strongly affects the performance of the DSSC. Batch et al. compared 1-methyl-benzimidazole (NMBI) and TBP in a [Cu(dmp)2]2+/+ redox system.30 They reported that the NMBI-based devices achieved a PCE of 8.89%, higher than a device using standard TBP (7.26%). However, they did not report any improvement in stability.
In this work, we aimed to solve the problem of TBP-induced degradation in the [Cu(dmp)2]2+/+ system. The effects of Lewis bases on the coordination behavior of the [Cu(dmp)2]2+/+ redox system were investigated, and a series of Lewis bases based on pyridine derivatives 2-methylpyridine (2MP), 3-methylpyridine (3MP), 4-methylpyridine (4MP), and 3,5-dimethylpyridine (35DMP; Fig. 1) were employed as novel replacements for TBP as the additive in the [Cu(dmp)2]2+/+ redox system. The 35DMP-based device was found to outperform the benchmark TBP-based device in terms of cell efficiency and stability. This is explained in terms of the coordination chemistry and the electrochemical properties between the moiety position of pyridine derivatives and [Cu(dmp)2]2+.
 | ||
| Fig. 1 Molecular structure of Lewis base additives used in this study. | ||
Results and discussion
To study the coordination of Lewis bases with [Cu(dmp)2]2+, the ultraviolet–visible (UV-vis) absorption spectra of [Cu(dmp)2]2+ with added Lewis bases were obtained; the results are presented in Fig. 2. The absorption spectrum of pure [Cu(dmp)2]2+ indicates a metal-to-ligand charge transfer (MLCT) and a d–d band at approximately 464 and 749 nm, respectively. The addition of 15 equivalents of a Lewis base (the same dosing as in the actual electrolyte in a DSSC device) resulted in a considerably different absorption spectrum; the MLCT bands of all of the samples except that containing 2MP were blue-shifted, and the d–d band was blue-shifted to 742, 720, 714, and 720 nm when the additive was 2MP, 3MP, 4MP, and 35DMP, respectively. Notably, the largest shift in the d–d band, by 40 nm to 709 nm, was that for the standard TBP addition. This suggests that all of the Lewis bases coordinate with the CuII metal center. The band shift indicated that the most reactive additive was TBP (shifted 40 nm), whereas the least reactive was 2MP (shifted 7 nm). A sharp peak at approximately 450 nm was present in the MLCT region only in the spectrum of the sample containing 2MP; this is the characteristic MLCT band for the CuI species and implies that 2MP may be a reductant for CuII. To prove this, absorption titrations were performed by adding various concentrations of 2MP to [Cu(dmp)2]2+ separately; the results are shown in Fig. S1a.† The absorption of the d–d band of [Cu(dmp)2]2+ changed little even after titration with 21.4 equivalents of 2MP was performed, indicating that the coordinating interaction of 2MP with the CuII center was negligible. A sharp peak in the MLCT region implied the formation of monovalent [Cu(dmp)2]+. Further addition of 2MP up to 50 equivalents resulted in an increase in the MLCT peak at approximately 450 nm, but the d–d transition remained unchanged, confirming that the strong Lewis base 2MP reduced the CuII center to CuI. For the 3MP samples, three isosbestic points were observed at 450, 570, and 975 nm, respectively (Fig. S1b†). Similarly, two isosbestic points were discovered at approximately 560 and 960 nm for the samples containing 4MP (Fig. S1c†) or 35DMP (Fig. S1d†). At higher concentrations of 4MP (up to 15 equivalents), the MLCT band (∼450 nm) region increased, and the absorption intensity of the d–d band decreased and shifted slightly to a shorter wavelength; the precipitation of unknown compounds also occurred. The presence of more than two isosbestic points in Fig. S1b and c† indicated that Lewis base ligand coordination processes occurred on [Cu(dmp)2(MeCN)x]2+ to [Cu(dmp)2(Lewis base)(MeCN)x]2+. Here and elsewhere, x can be 0 or 1. The binding stoichiometry and stability constant values of Lewis bases coordinated with [Cu(dmp)2(MeCN)]2+ were determined using the host ([Cu(dmp)2]2+) and guest (Lewis base) analysis method, conducted with the Bindfit software using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–guest binding model (Fig. S2†).32 A larger stability constant indicates that a reaction is more thermodynamically favorable. The reactions and their stability constants are presented in Scheme 1. The stability constant of [Cu(dmp)2(2MP)(MeCN)x]2+ was small (9.0 M−1) because the substituted position of the methyl group was unfavorable for the coordination. Because the dose of additive in the actual electrolyte is 15-fold relative to the CuII concentration, only a negligible amount of 2MP (pKa = 5.96) reacted with the CuII center; the remainder remained in the solution. This increased the basicity and was responsible for the reduction of CuII into CuI. After 24 h, [Cu(dmp)2(MeCN)]2+ had been completely converted into [Cu(dmp)2]+, as evident from the absorption spectra displayed in Fig. S3.† Meanwhile, the methyl groups located at the 3- and 4-substituted positions (3MP and 4MP, respectively) reduced the steric hindrance, increasing the stability constants to 342.7 and 631.8 M−1, respectively. The stability constant of 35DMP was the highest at 700.4 M−1 because this base had the highest basicity (pKa = 6.15). Overall, the stability constants of 3MP, 4MP, and 35DMP were all larger than that of TBP (239 M−1, from the previous report). This result was attributable to the smaller molecular size of the investigated Lewis bases, where smaller size results in better coordination with CuII ions. The results of the UV-vis titration and stability constant tests suggested that the true form of [Cu(dmp)2(MeCN)]2+ after the addition of a Lewis base was [Cu(dmp)2(Lewis bases)(MeCN)x]2+. For 4MP, the Bindfit calculation indicated that a small fraction of [Cu(dmp)2(4MP)(MeCN)x]2+ formed [Cu(dmp)2(4MP)2]2+; this may have been the aforementioned precipitated compound.
:1 host–guest binding model (Fig. S2†).32 A larger stability constant indicates that a reaction is more thermodynamically favorable. The reactions and their stability constants are presented in Scheme 1. The stability constant of [Cu(dmp)2(2MP)(MeCN)x]2+ was small (9.0 M−1) because the substituted position of the methyl group was unfavorable for the coordination. Because the dose of additive in the actual electrolyte is 15-fold relative to the CuII concentration, only a negligible amount of 2MP (pKa = 5.96) reacted with the CuII center; the remainder remained in the solution. This increased the basicity and was responsible for the reduction of CuII into CuI. After 24 h, [Cu(dmp)2(MeCN)]2+ had been completely converted into [Cu(dmp)2]+, as evident from the absorption spectra displayed in Fig. S3.† Meanwhile, the methyl groups located at the 3- and 4-substituted positions (3MP and 4MP, respectively) reduced the steric hindrance, increasing the stability constants to 342.7 and 631.8 M−1, respectively. The stability constant of 35DMP was the highest at 700.4 M−1 because this base had the highest basicity (pKa = 6.15). Overall, the stability constants of 3MP, 4MP, and 35DMP were all larger than that of TBP (239 M−1, from the previous report). This result was attributable to the smaller molecular size of the investigated Lewis bases, where smaller size results in better coordination with CuII ions. The results of the UV-vis titration and stability constant tests suggested that the true form of [Cu(dmp)2(MeCN)]2+ after the addition of a Lewis base was [Cu(dmp)2(Lewis bases)(MeCN)x]2+. For 4MP, the Bindfit calculation indicated that a small fraction of [Cu(dmp)2(4MP)(MeCN)x]2+ formed [Cu(dmp)2(4MP)2]2+; this may have been the aforementioned precipitated compound.
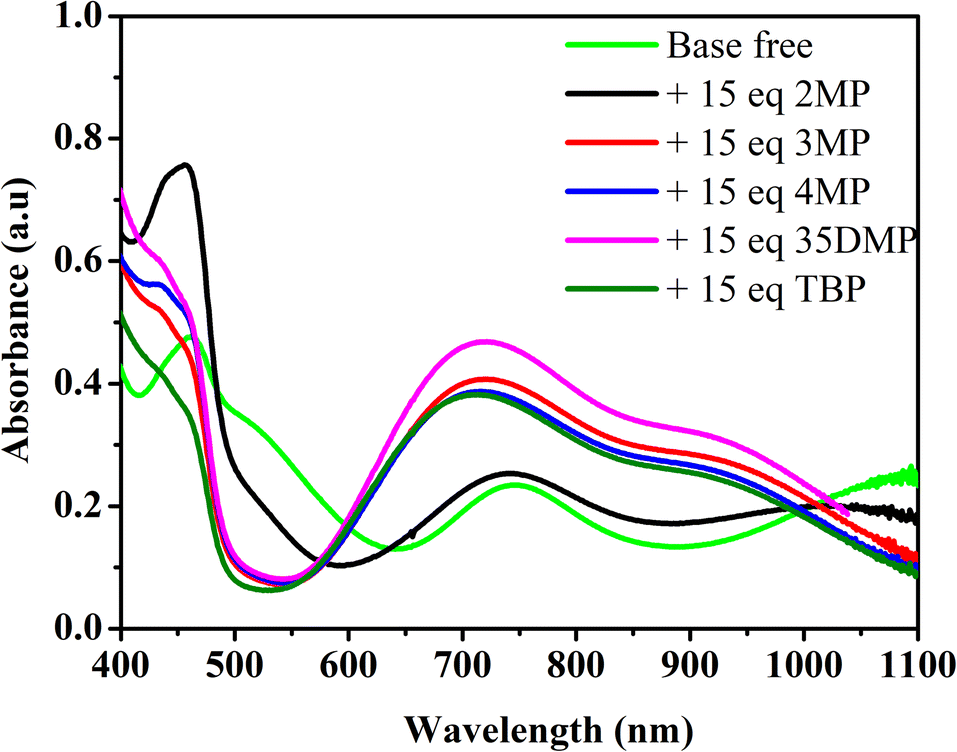 | ||
| Fig. 2 Absorption spectra of [Cu(dmp)2]2+ with addition of different Lewis bases in non-coordinating solvent dichloromethane. | ||
 | ||
| Scheme 1 Reactions of [Cu(dmp)2(MeCN)]2+ with Lewis base additives. | ||
To further understand the coordination of Lewis bases with [Cu(dmp)2]2+, 1H nuclear magnetic resonance (NMR) was performed with d-MeCN as the solvent. The 1H-NMR spectra (Fig. 3) revealed that the pure [Cu(dmp)2]2+ complex had paramagnetic characteristic peaks at 23.88, 16.37, and −8.08 ppm. MeCN is known to coordinate with [Cu(dmp)2]2+ to form [Cu(dmp)2(MeCN)]2+.21 The addition of a Lewis base did not remove these paramagnetic peaks but did reduce their intensity, and new peaks formed at approximately 7 to 10 ppm; these both indicated that the dmp ligand still coordinated with CuII. Notably, the smallest intensity reduction of the paramagnetic peaks was observed for the addition of 15 equivalents of 2MP (Fig. 3), strongly implying that 2MP has the least effective coordination of the investigated Lewis bases. This was also consistent with the result of the UV-vis titration study. To observe the details of the ligand exchange process, titration experiments were performed by adding d2-DCM internal standard to evaluate whether the dmp ligand would be displaced (Fig. S4–S7†). The results of the titrations of [Cu(dmp)2(MeCN)]2+ with 2MP (Fig. S4†) and 4MP (Fig. S6†) revealed that the reactions were irreversible and that a nonequilibrium between the reactants and products occurred. During the titration experiment, some crystals were deposited on the NMR tube of the sample containing 4MP. The color of [Cu(dmp)2(MeCN)]2+ with added 2MP also changed to red-orange, revealing the presence of [Cu(dmp)2]+. The paramagnetic [Cu(dmp)2]2+ was entirely changed into diamagnetic [Cu(dmp)2]+ after 24 h, as clearly evidenced by the 1H-NMR spectra presented in Fig. S8.† However, the interactions of the [Cu(dmp)2(MeCN)]2+ complex with 3MP (Fig. S5†) and 35DMP (Fig. S7†) were reversible, with the same behavior as the interaction with the standard TBP additive.
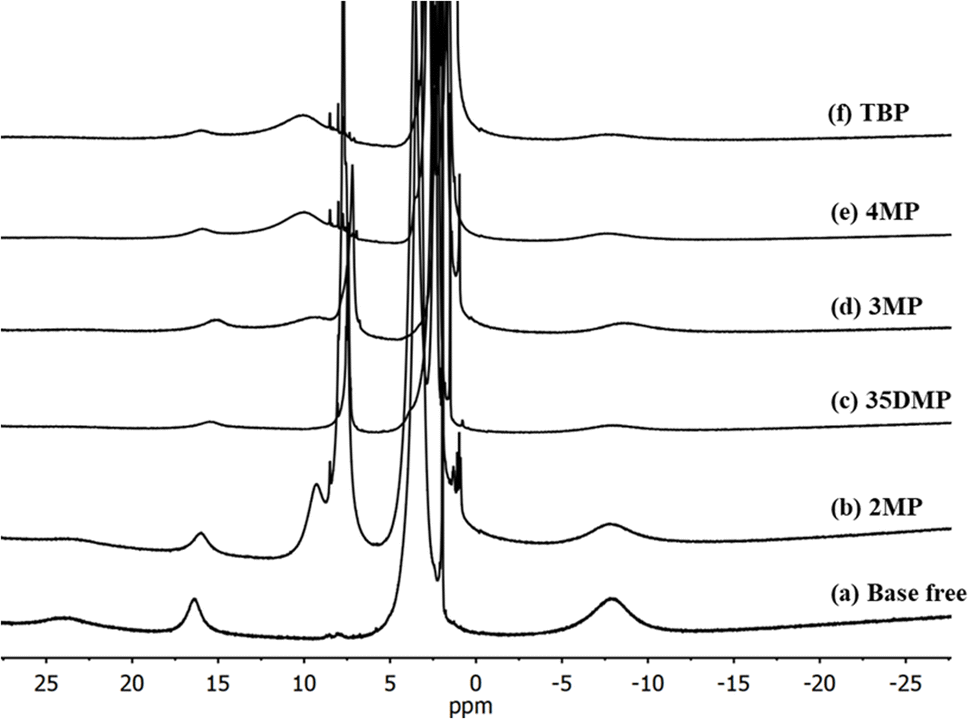 | ||
| Fig. 3 1H NMR spectra of [Cu(dmp)2]2+ (a) without the addition of Lewis bases, with the addition of 15 equivalents of (b) 2MP, (c) 35DMP, (d) 3MP (e) 4MP and (f) TBP in d3-MeCN. | ||
A summary of the reactions involved inside the electrolyte system for the addition of various Lewis bases is presented in Scheme 2; the reactions were reversible for TBP, 3MP, and 35DMP but irreversible for 2MP and 4MP. Addition of 2MP reduced [Cu(dmp)2]2+ to [Cu(dmp)2]+; this is expected to result in inferior device performance due to an imbalance of the redox couple ratio. However, for 4MP, the intermediate ([Cu(dmp)2(4MP)(MeCN)x]2+) was reversible with [Cu(dmp)2]2+. At higher 4MP concentrations, the sixth valency of Cu2+ was satisfied, resulting in precipitation of a solid product. This precipitation is irreversible, and further exchange of the ligand with [Cu(dmp)2(4MP)2]2+ in solution is difficult because of its poor solubility in the solvent.
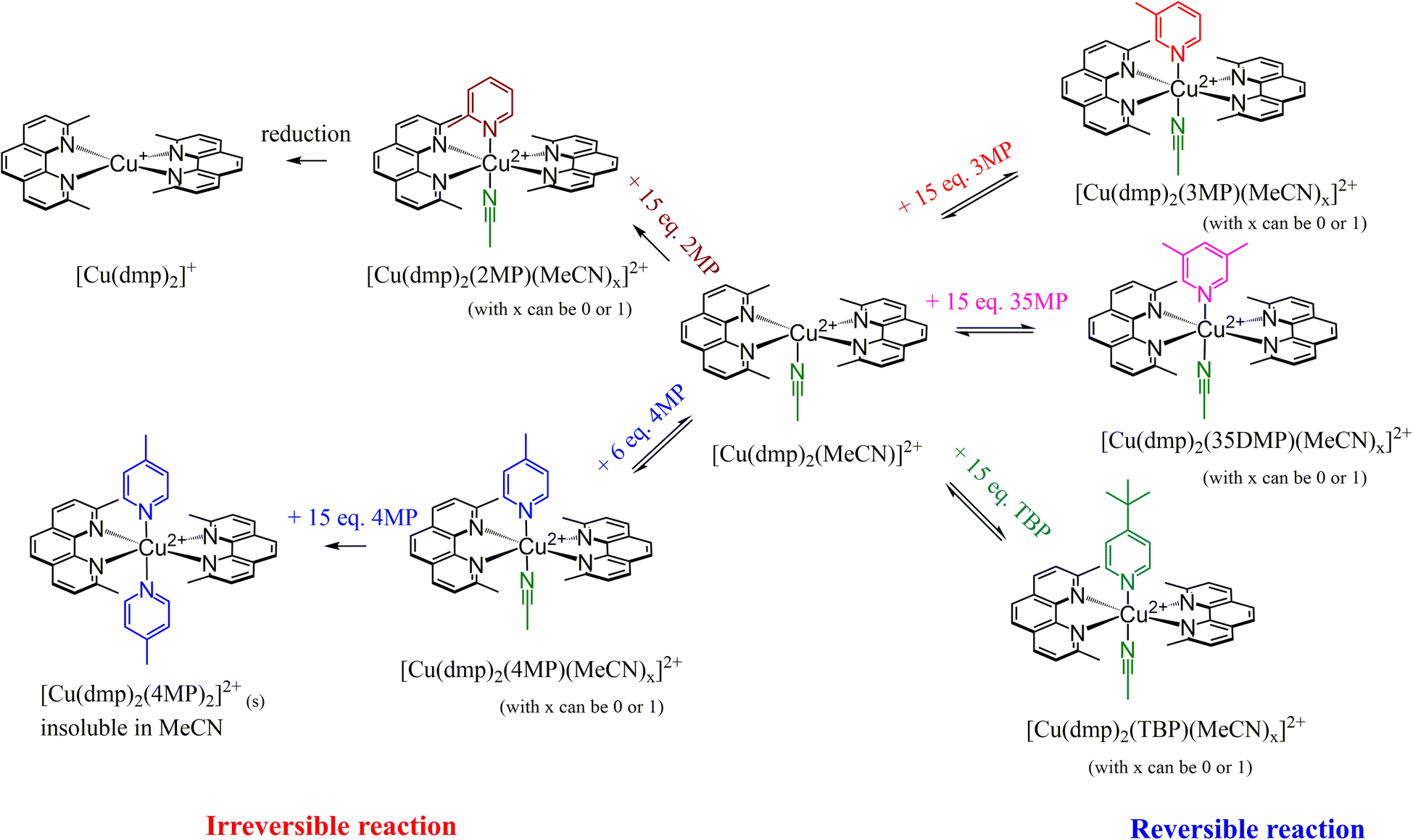 | ||
| Scheme 2 The reactions occurring inside the electrolyte system in the presence of different Lewis bases. | ||
The electrochemical properties of Lewis-base-coordinated [Cu(dmp)2]2+ were studied using cyclic voltammetry (CV) in a three-electrode cell. The cyclic voltammograms are presented in Fig. 4. The cyclic voltammogram of pure [Cu(dmp)2]2+ contains a characteristic reduction peak at 0.71 V. The addition of 15 equivalents of all Lewis bases except for 2MP resulted in a considerable negative shift of this reduction potential. Specifically, the redox potential shifted the most for TBP and 4MP coordination by approximately 100 mV, and new reduction peaks at 0.43 V appeared (Fig. S9†); this result implied that TBP and 4MP had similar coordination behavior toward [Cu(dmp)2(MeCN)]2+. Consistent with the UV-vis and 1H-NMR results, the 2MP-coordinated complex was found to have the smallest change in redox potential because of the steric hindrance of the methyl group on 2MP, which is unfavorable for the coordination.23,24 The samples containing 3MP and 35DMP had slightly smaller shifts (∼50 mV) than that for the sample containing 4MP. The current densities in the CV waves for all coordinated complexes except for the 2MP-coordinated complex were smaller than the current densities for pristine [Cu(dmp)2]2+, implying that charge transfer was more sluggish after the coordination, especially in the case of TBP and 4MP. One week after the Lewis bases were added, CV of the aged solutions was performed again. As illustrated in Fig. 4, the redox potentials of [Cu(dmp)2(Lewis)(MeCN)x]2+ slightly shifted to more negative potentials. Reduction peaks at 0.43 V in the fresh scans of TBP and 4MP samples shifted to 0.4 V and 0.35 V, respectively. Noticeably, new reduction peaks appeared at 0.4 V for 35DMP and 0.44 V for 3MP. This result indicates that new complexes formed in the solution, and the substitution of methyl groups for 3MP and 35DMP had a weaker effect on shifting the redox potential as compared to those of 4MP and TBP. No considerable potential shift of [Cu(dmp)2(MeCN)]2+ was recorded for the 2MP solution after 1 week of storage; this was attributable to most of the [Cu(dmp)2]2+ being reduced to [Cu(dmp)2]+. However, a small redox signal was found at 0.17 V that had not been detected in the initial scan, indicating that a new redox species had formed. This behavior was unique for 2MP among the Lewis bases. The CV study further confirmed that no pristine [Cu(dmp)2]2+ or individual Lewis base additives existed in the MeCN-based solution; the compounds were [Cu(dmp)2 (Lewis base)]2+ and newly formed complexes.
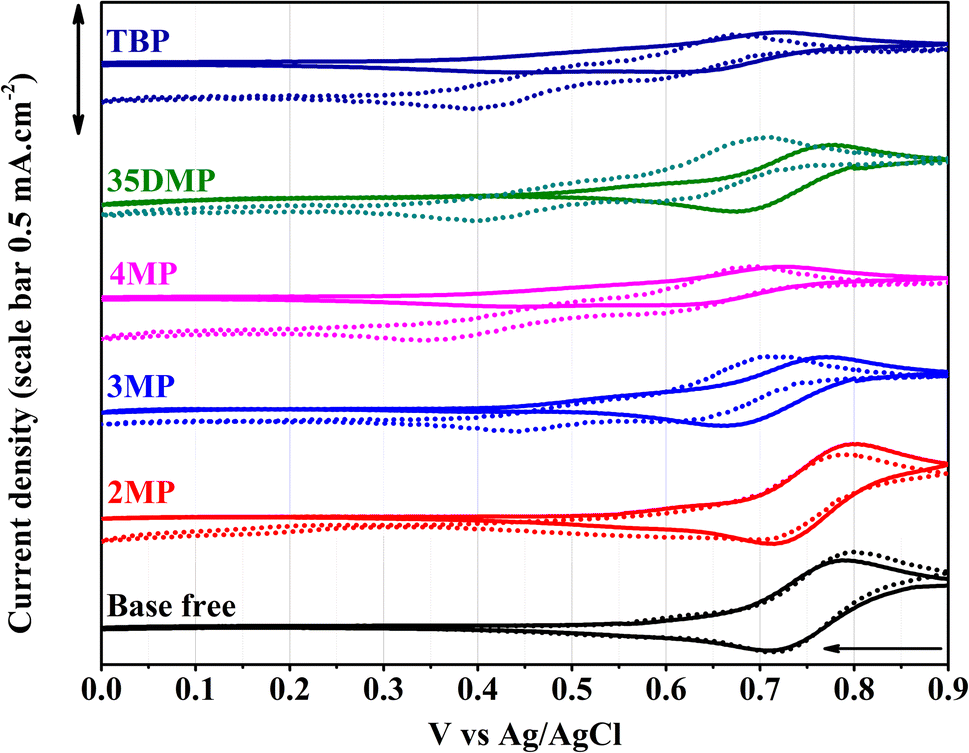 | ||
| Fig. 4 Cyclic voltammograms of fresh (solid line) and one-week aged (dash line) 3 mM solutions of [Cu(dmp)2]2+ in acetonitrile without and with the addition of 15 equivalents of Lewis bases. The solution contains 0.1 M LiTFSI as a supporting electrolyte. The reference electrode is Ag/AgCl (saturated LiCl in ethanol). The scan rate is 10 mV s−1. The arrow indicates the scan direction. | ||
Electrochemical impedance spectroscopy (EIS) was performed with a symmetric cell configuration in which two pieces of poly-N-vinyl-2-pyrrolidone (PVP)-capped Pt nanocluster (PVP-Pt) counter electrodes were used to sandwich the electrolyte. The composition of the electrolyte in the EIS cell was identical to that in an actual device. A typical Nyquist plot of a symmetric cell (Fig. S10†) has three elements, namely an intercept at high frequency (>104 Hz), which represents the series resistance RS of the cell; a semicircle at approximately 104 to 102 Hz, which represents the charge transfer resistance (RCT) of CuII to CuI at the counter electrode surface; and a Warburg impedance RD of finite diffusion length in the low-frequency region (<102 Hz), which accounts for the diffusion of ions. In general, RCT and RD are part of the total internal resistance of the DSSC and should be minimized to achieve high efficiency. Nyquist plots of the investigated samples and the fit results are presented in Fig. 5 and Table 1. Addition of TBP led to a large increase in RCT from 0.03 to 5.17 Ω cm2. This increase in RCT was attributable to the coordination of TBP with [Cu(dmp)2]2+ and the steric hindrance of tert-butyl moieties, which prevented adsorption of [Cu(dmp)2(TBP)(MeCN)x]2+ on catalytic sites of the Pt surface, increasing the charge transfer barrier. The bulky structure of [Cu(dmp)2(TBP)(MeCN)x]2+ also caused RD to increase to 17.33 Ω cm2 s−0.5. For 2MP, the substituted position of the methyl group hindered coordination; hence, RCT was almost unchanged (0.02 Ω cm2). The slight increase in RD may have been due to the higher viscosity of the electrolyte after 2MP addition. Addition of 3MP to the electrolyte, resulting in the formation of [Cu(dmp)2(3MP)(MeCN)x]2+, restrained regeneration at the catalyst interface, resulting in RCT and RD increasing to 0.83 Ω cm2 and 8.28 Ω cm2 s−0.5, respectively. Similarly, the RCT and RD for the cell with the 35DMP additive increased to 1.31 Ω cm2 and 9.14 Ω cm2 s−0.5, respectively. The RCT of the 35DMP-based device also increased because 35DMP is more basic (pKa = 6.15) than 3MP (pKa = 5.63), which resulted in interactions between the Lewis base and [Cu(dmp)2]2+ and hampered charge transfer. A similar finding was reported by Ferdowsi et al.; RCT and RD are strongly dependent on the basicity of the Lewis base additive.31 The irreversible reaction between [Cu(dmp)2]2+ and 4MP may have led to the formation of a solid product [Cu(dmp)2(4MP)2]2+, which could have reduced the concentration of the CuII complex in the electrolyte, suppressing charge transfer kinetics at the counter electrode surface and thereby inducing a large RCT of 2.92 Ω cm2 and RD of 15.62 Ω cm2 s−0.5.
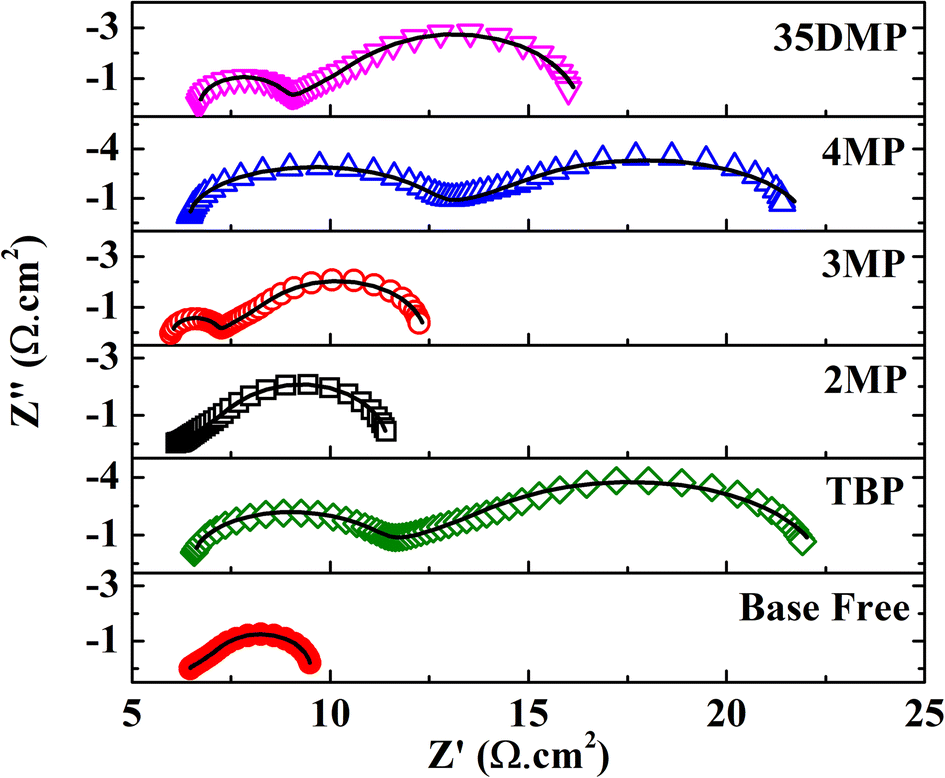 | ||
| Fig. 5 Nyquist plot of symmetric cells with and without the addition of Lewis bases obtained at 0 V in the dark. Scatters are measured data; solid lines are fit data. | ||
| R CT (Ω cm2) | R D (Ω cm2 s−0.5) | J SC (mA cm−2) | V OC (V) | FFb | PCEb (%) | |
|---|---|---|---|---|---|---|
| a Electrochemical properties of [Cu(dmp)2]2+/+ redox with and without the addition of Lewis bases obtained from EIS measurements of symmetric cells. b Photovoltaic performance of the [Cu(dmp)2]2+/+-mediated DSSC on adding different Lewis bases. Data are averaged from 5 separate devices. The bold data indicate the best performing cells. | ||||||
| Base free | 0.03 | 4.10 | — | — | — | — |
| 2MP | 0.02 | 5.06 | 5.4 ± 0.3 | 0.962 ± 0.012 | 0.58 ± 0.05 | 3.0 ± 0.2 |
| 5.6 | 0.971 | 0.60 | 3.2 | |||
| 3MP | 0.83 | 8.28 | 11.7 ± 0.5 | 1.051 ± 0.002 | 0.76 ± 0.01 | 9.3 ± 0.2 |
| 12.4 | 1.048 | 0.74 | 9.7 | |||
| 4MP | 2.92 | 15.62 | 10.2 ± 0.3 | 1.062 ± 0.006 | 0.73 ± 0.03 | 7.9 ± 0.2 |
| 10.3 | 1.063 | 0.75 | 8.2 | |||
| 35DMP | 1.31 | 9.14 | 12.0 ± 0.4 | 1.073 ± 0.003 | 0.74 ± 0.03 | 9.6 ± 0.2 |
| 11.9 | 1.074 | 0.76 | 9.7 | |||
| TBP | 5.17 | 17.33 | 11.7 ± 0.1 | 1.066 ± 0.012 | 0.73 ± 0.02 | 9.2 ± 0.4 |
| 11.8 | 1.072 | 0.76 | 9.7 | |||
DSSCs based on [Cu(dmp)2]2+/+ mediators with a Lewis base additive were fabricated. Their JV curves, their incident photon-to-electron conversion efficiency (IPCE) spectra, and details of the photovoltaic parameters are presented in Fig. 6 and Table 1. The benchmark TBP-based device had an average and maximum PCE of 9.2% and 9.7%, respectively. The 2MP-based devices had an average efficiency of 3.0%. The JV parameters suggested that this low PCE was attributable to the halving of JSC to 5.4 mA cm−2, a low VOC of 0.96 V, and a low fill factor (FF) of 0.58. The low JSC could be further confirmed from the IPCE spectra, which indicated that the 2MP-based device suffered from poor photon-to-electron conversion, which was a result of the imbalance of CuI and CuII, as revealed in the preceding discussion. The 4MP-based device had lower PCE (7.9%; 8.2% for the best-performing cell) than the TBP-based device did. This result is attributable to the formation of irreversible coordination deposition that also caused an imbalance of CuI and CuII. Although the efficiencies of the devices with 3MP, 35DMP, and TBP were all comparable within experimental errors, the 3MP- and 35DMP-based devices had slightly higher average PCEs of 9.3% and 9.6%, respectively, compared with the average PCE for the TBP-based device (9.2%); this indicated that the function of TBP on TiO2 could instead be achieved by substituting it with 3MP or 35DMP.
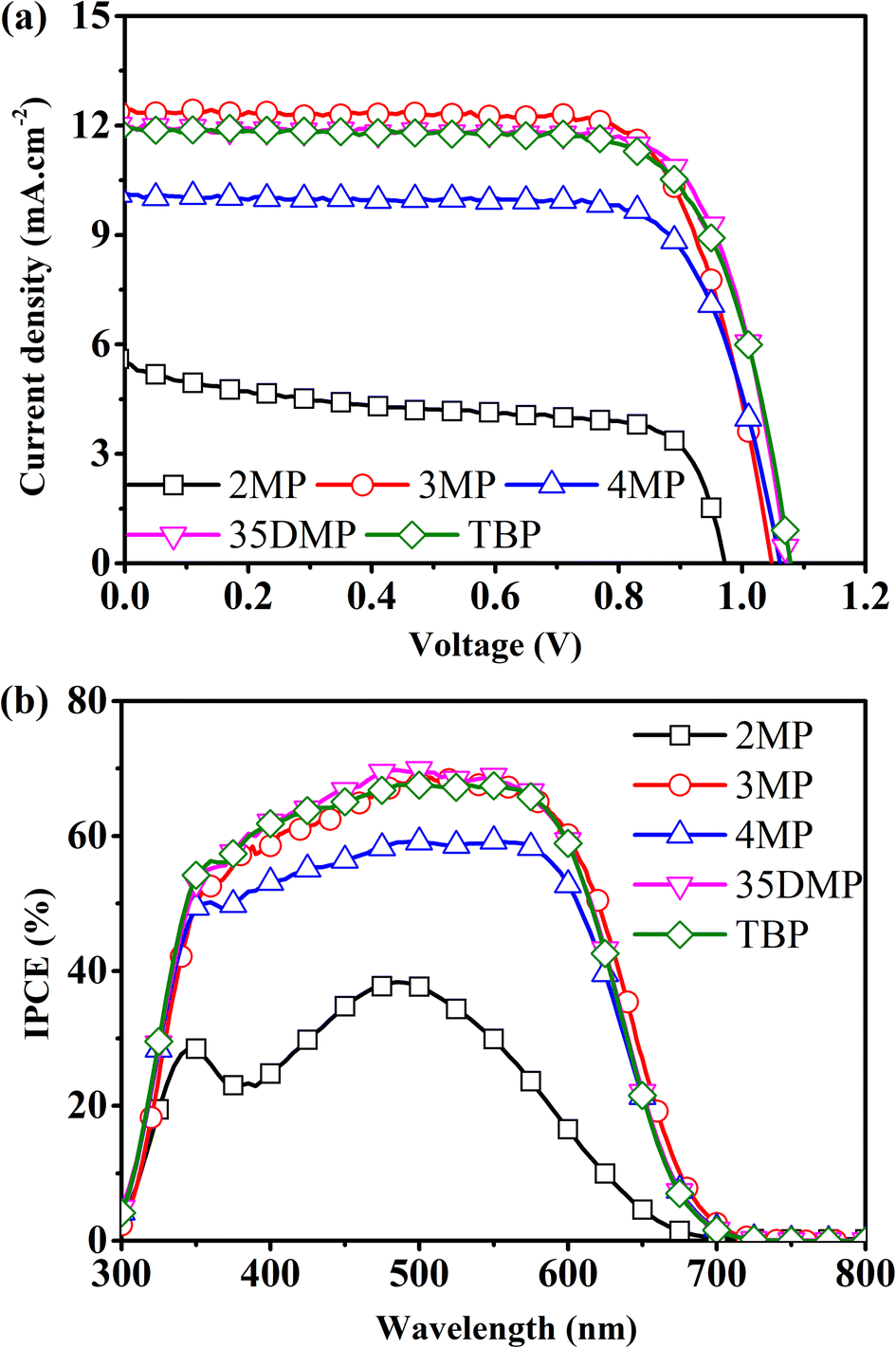 | ||
| Fig. 6 (a) J–V curves of a [Cu(dmp)2]2+/+-mediated DSSC on adding different Lewis bases and (b) their corresponding IPCE spectra. | ||
The purpose of adding a Lewis base to the electrolyte is to mitigate recombination between photoelectrons and [Cu(dmp)2]2+ at the TiO2 surface. To examine this effect for the investigated Lewis bases, EIS spectra of the DSSCs were recorded in the dark and analyzed using a transmission line model (Fig. S11a†).33,34 A typical Nyquist plot obtained with a forward bias of 0.75 V is shown in Fig. S11b.† A transmission line that represents the electron transport resistance RT in the mesoporous structure in the high-frequency region can be identified, and an arc follows in the middle-frequency region that represents the parallel connection of the recombination resistance RREC and chemical capacitance Cμ. The electron lifetime τ = RREC × Cμ, and the effective diffusion length Ln = L × (RREC/RT)1/2 can then be calculated. Generally, Ln/L greater than 1 indicates efficient charge collection for a DSSC. Fig. S12a† reveals that the RREC values for DSSCs with 4MP, 35DMP, and TBP additives were all approximately two orders of magnitude greater than that for the 2MP-based DSSC, indicating that 2MP addition failed to sufficiently inhibit recombination.
Because 2MP acted as a reductant for [Cu(dmp)2]2+, in 2MP-added electrolyte, the redox species gradually transformed into [Cu(dmp)2]+. In addition, the hindrance of the methyl group at the 2-position may have contributed to its poor ability to adsorb on TiO2, thus failing to provide the desired recombination inhibition. Severe electron recombination at the TiO2/electrolyte interface of the 2MP-based device induced poor electron density in TiO2, as shown in Fig. S12b;† the RT values were two to three orders of magnitude higher for 2MP than for 4MP, 35DMP, and TBP. Therefore, the Ln/L ratio (Fig. S12c†) of the 2MP-based device was far below 1, and its τ was the shortest among the investigated devices (Fig. S12d†). The methyl group at the 3-position (3MP) reduced the degree of steric hindrance, enhancing adsorption on the TiO2 surface and leading to negative shifts of the TiO2 conduction band and suppressing interfacial electron recombination. This caused the RREC of the 3MP-based DSSC to be much higher than that of the 2MP-based DSSC; this could be observed as improvements in VOC and JSC. An increase in the electron density at the Fermi level of TiO2 leads to a small RT. The additives 4MP, 35DMP, and TBP have much higher basicity than 3MP; thus, 3MP had stronger adsorption, which caused the TiO2 conduction bands to shift to a more negative region, providing a better driving force for recombination inhibition. The fitting of the RREC and RT results suggested that the τ of the 3MP-added device was smaller than those of the devices containing 4MP, 35DMP, and TBP. However, the 3MP-based device achieved an Ln/L ratio greater than 1 and thus had sufficiently high charge collection efficiency.
The long-term stability of [Cu(dmp)2]2+/+-mediated DSSCs containing TBP, 3MP, or 35DMP additives was evaluated. Stability tests were conducted with the devices in the dark and under ambient conditions for 46 days. The results are presented in Fig. 7. The PCE of the device with the TBP additive decayed sharply from 8.95% to 2.04%, corresponding to poor 22.8% retention. However, the final PCE retention of the 3MP and 35DMP devices was 68.8% and 77.3%, respectively. The photovoltaic parameters illustrated in Fig. 7b–d suggest that FF decay was mainly responsible for the PCE decrease, whereas the values of VOC and JSC did not generally change. The decay of FF could be explained by the slowed charge transfer process at the counter electrode's surface.29 The evolution of the RCT and RD of symmetrical cells over 1 week for the 3MP-, 35DMP-, and TBP-based devices is illustrated in Fig. S13.† The kinetics and mass transportation of the counter electrode were significantly slowed when TBP was added; both RCT and RD increased by a factor of almost 50. This was due to the accumulation of bulky [Cu(dmp)2(TBP)(MeCN)x]2+ in the electrolyte. The tert-butyl group on TBP prevented its adsorption on catalytic active sites of the counter electrode, leading to an increase in RCT. For the 3MP and 35DMP additives, the bulkiness of their coordinated complexes was mitigated because of their smaller methyl moieties. The RCT of the 3MP-based and 35DMP-based devices remained small. These results further confirmed that the size of the alkyl moiety on the pyridine ring has a major effect on the reduction of coordinated-[Cu(dmp)2]2+ compounds. Because the increases in the RCT and RD for the counter electrode were mitigated, FF retention in the stability trial was better for the 3MP and 35DMP-added devices than the other device. Compared with the 3MP-based device, the 35DMP-based device had better retention because interfacial recombination was more strongly inhibited. Hence, 35DMP is a viable candidate for replacing TBP in electrolytes.
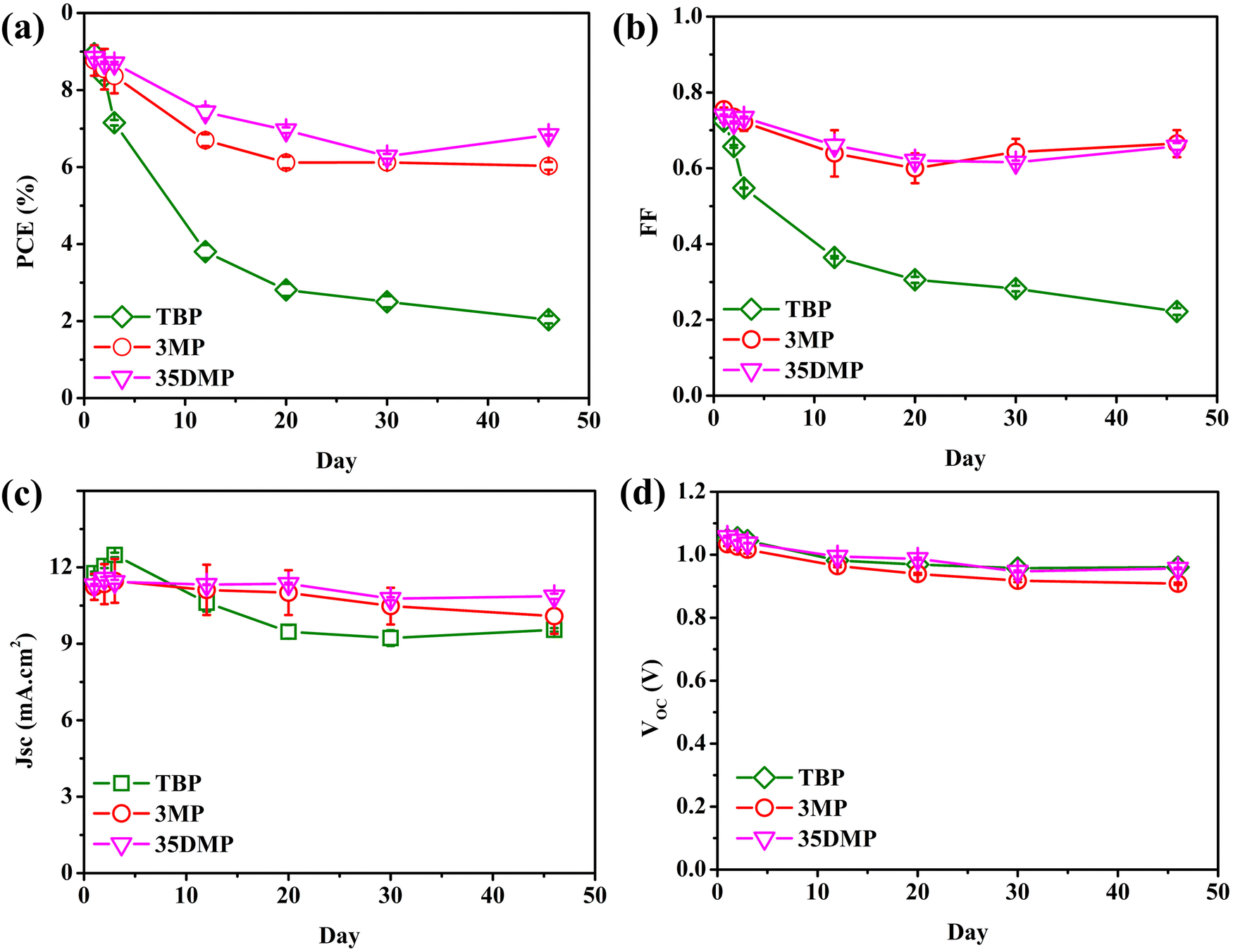 | ||
| Fig. 7 The evolution of (a) PCE, (b) FF, (c) JSC, and (d) VOC of the [Cu(dmp)2]2+/+-mediated DSSC employing TBP, 3MP and 35DMP as a Lewis base within 46 days. | ||
Conclusions
The coordination of methylpyridine-based Lewis bases with [Cu(dmp)2]2+ in MeCN was extensively studied. The additive 2MP reduced [Cu(dmp)2]2+ to a monovalent species, and adding 4MP resulted in a solid precipitate of [Cu(dmp)2(4MP)2]2+. Hence, neither effectively shifted the TiO2 conduction band. The additives 3MP and 35DMP were found to form [Cu(dmp)2(3MP) (MeCN)x]2+ and [Cu(dmp)2(35DMP)(MeCN)x]2+, respectively, through a reaction mechanism similar to that of standard TBP. Absorption titrations, 1H-NMR spectra, and electrochemical studies confirmed that both [Cu(dmp)2(3MP)(MeCN)x]2+ and [Cu(dmp)2(35DMP)(MeCN)x]2+ can alleviate degradation of the performance of devices. The improvement of the poisoning effect for 3MP and 35DMP is attributable to the position of the methyl group in the pyridyl ring and the lower steric hindrance of this methyl group compared with that of the tert-butyl group in TBP. The efficiencies of the 3MP- and 35DMP-based devices were 9.3% and 9.6%, slightly better than that of the TBP device (9.2%). Moreover, the 3MP- and 35DMP-based DSSCs exhibited superior stability with 68.8% and 77.3% PCE retention, respectively, after a 46-day period.Experimental section
Chemicals and reagents
[Cu(dmp)2]2+/+ redox couples were purchased from Everlight (Taiwan) or synthesized as per procedures reported in the literature.15 All other reagents and solvents were obtained from Sigma-Aldrich or Alfa Aesar and used without further purification unless otherwise noted.Device fabrication
The DSSC fabrication procedure was mentioned previously.29 Briefly, a piece of fluorine doped tin oxide (FTO) glass (2.2 mm thick, 8 Ω per square, Dyesol) was sequentially cleaned by using detergent, DI water and ethanol. Cleaned FTO was then treated with UV/ozone for 15 minutes before electrodepositing a thin layer of TiOx in a 0.2 M TiCl3 aqueous solution for 200 seconds.35 Then the substrate was rinsed with DI water and pure ethanol sequentially. The TiO2 electrode was prepared by screen-printing a 2 μm-thick TiO2 film using a commercial TiO2 paste (Ti-2105, Eternal Materials, Taiwan), followed by screen printing an additional 2 μm-thick TiO2 film using modified commercial TiO2 paste. The modification was performed by uniformly dispersing 5 wt% of ethyl cellulose in commercial Ti-2105 paste (Ti-2105M). After a Ti-2105 + Ti2105M bilayer was prepared, a 4 μm-thick scattering layer was overcoated onto the substrate using commercial paste (400 nm, PST-400, JGC C&C, Japan). The TiO2 trilayer was subsequently sintered at 500 °C for 30 minutes under air flow. The screen-printed area is 0.25 cm2. Before soaking in dye solution, the TiO2 electrode was treated again with 40 mM TiCl4 and annealed at 450 °C for 30 minutes. TiO2 electrodes were immersed in a dye solution containing 0.1 mM Y123 dye (Dyenamo) and 5.0 mM chenodeoxycholic acid (CDCA) in an acetonitrile (MeCN)/tert-butanol mixture (v/v, 1:1) for 16 hours and rinsed with pure MeCN. A home-made Pt counter electrode was prepared by a two-step dip-coating process, which involves immersion of FTO glass in a surfactant agent (ML371, MacDiarmid, USA) and then dipping into an aqueous solution containing poly-N-vinyl-2-pyrrolidone (PVP)-capped Pt nanoclusters (PVP-Pt) for 5 minutes to adsorb a thin layer of PVP-Pt.36–38 After rinsing with deionized water, the PVP-Pt counter electrode was sintered at 325 °C for 30 minutes The DSSCs were assembled by sandwiching a dye-sensitized TiO2 electrode and a PVP-Pt counter electrode with a piece of thermoplastic film (25 μm, Surlyn, Dupont, Japan) at 125 °C for a few seconds. Then 5 μL of electrolyte containing 0.2 M [Cu(dmp)2] (TFSI), 0.04 M [Cu(dmp)2](TFSI)2, 0.1 M LiTFSI and 0.6 M of the corresponding Lewis bases in MeCN was injected into a hole on the counter electrode. Finally, the injection hole was sealed by a Surlyn sheet and a thin glass to avoid electrolyte leakage.
Characterization methods
To control the illumination area of the device during photovoltaic performance measurement, a photomask with an opening area of 0.16 cm2 was attached on the photoanode before measurement. A solar simulator (PEC-L15, PECCELL, Japan) with an AM 1.5G spectrum was calibrated to an intensity of 100 mW cm−2 by using a reference cell (KG3, ORIEL, USA). The current–voltage (IV) curves were measured by using a computer-controlled digital source meter (Keithley 2400C). The IPCE was performed using commercial equipment (PEC-S20, PECCELL, Japan). For stability trials, the devices were stored in the dark at 25 °C. UV-vis spectra of [Cu(dmp)2](TFSI)2 in acetonitrile (MeCN) and dichloromethane (DCM) were measured by using a UV-vis spectrophotometer (Hewlett Packard 8453). Electrochemical impedance spectroscopy (EIS) measurements were performed by using an Autolab PGSTAT302N. The impedance data of symmetric cells were recorded with the frequency ranged from 100 kHz to 0.1 Hz along with an AC amplitude of 10 mV. A DC potential equivalent to the open-circuit voltage of the device was applied. The data were then fit with a relevant equivalent circuit by using Z-View software. Cyclic voltammetry was performed based on a three-electrode system using an Autolab PGSTAT204. The working electrode was a platinum plate, while the reference and auxiliary electrodes were homemade Ag/AgCl (saturated LiCl in ethanol) and coiled platinum wire, respectively. The solvent used in CV was acetonitrile with the addition of 0.1 M LiTFSI. The scanning rate is 10 mV s−1. The 1H-NMR spectra were recorded using a Varian Mercury 400 FT-NMR spectrometer using d3-acetonitrile as the solvent and d2-DCM as the internal standard.Author contributions
Tzu-Chien Wei & Chen-Yu Yeh supervised the study and finalized the manuscript. Vinh Son Nguyen conceived the idea, designed the experiments, and wrote the first manuscript draft. Kala Kannankutty helped in data analysis and manuscript modification. Yu-Hsuan Chen synthesized the copper(II) complex. All authors contributed to the writing and revision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the Ministry of Science and Technology, Taiwan (108-2221-E-007-102-MY3), and the Ministry of Economic Affairs, Taiwan (E111-EC-17-A-08-S6-013).References
- M. Grätzel, J. Photochem. Photobiol., C, 2003, 4, 145–153 CrossRef.
- Y. Ren, D. Zhang, J. Suo, Y. Cao, F. T. Eickemeyer, N. Vlachopoulos, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Nature, 2023, 613, 60–65 CrossRef CAS.
- H. Michaels, M. Rinderle, I. Benesperi, R. Freitag, A. Gagliardi and M. Freitag, Chem. Sci., 2023, 14, 5350–5360 RSC.
- K. Kakiage, Y. Aoyama, T. Yano, K. Oya, J.-i. Fujisawa and M. Hanaya, Chem. Commun., 2015, 51, 15894–15897 RSC.
- C.-C. Chen, V. S. Nguyen, H.-C. Chiu, Y.-D. Chen, T.-C. Wei and C.-Y. Yeh, Adv. Energy Mater., 2022, 12, 2104051 CrossRef CAS.
- H. J. Snaith, Adv. Funct. Mater., 2010, 20, 13–19 CrossRef CAS.
- T. Stergiopoulos and P. Falaras, Adv. Energy Mater., 2012, 2, 616–627 CrossRef CAS.
- J.-H. Yum, E. Baranoff, F. Kessler, T. Moehl, S. Ahmad, T. Bessho, A. Marchioro, E. Ghadiri, J.-E. Moser, C. Yi, M. K. Nazeeruddin and M. Grätzel, Nat. Commun., 2012, 3, 631 CrossRef PubMed.
- Y. Bai, Q. Yu, N. Cai, Y. Wang, M. Zhang and P. Wang, Chem. Commun., 2011, 47, 4376–4378 RSC.
- J. Cong, D. Kinschel, Q. Daniel, M. Safdari, E. Gabrielsson, H. Chen, P. H. Svensson, L. Sun and L. Kloo, J. Mater. Chem. A, 2016, 4, 14550–14554 RSC.
- H. Michaels, I. Benesperi, T. Edvinsson, A. B. Muñoz-Garcia, M. Pavone, G. Boschloo and M. Freitag, Inorganics, 2018, 6, 53 CrossRef.
- E. Mosconi, J.-H. Yum, F. Kessler, C. J. Gómez García, C. Zuccaccia, A. Cinti, M. K. Nazeeruddin, M. Grätzel and F. De Angelis, J. Am. Chem. Soc., 2012, 134, 19438–19453 CrossRef CAS.
- J. J. Nelson, T. J. Amick and C. M. Elliott, J. Phys. Chem. C, 2008, 112, 18255–18263 CrossRef CAS.
- S. Hattori, Y. Wada, S. Yanagida and S. Fukuzumi, J. Am. Chem. Soc., 2005, 127, 9648–9654 CrossRef CAS PubMed.
- Y. Saygili, M. Söderberg, N. Pellet, F. Giordano, Y. Cao, A. B. Muñoz-García, S. M. Zakeeruddin, N. Vlachopoulos, M. Pavone, G. Boschloo, L. Kavan, J.-E. Moser, M. Grätzel, A. Hagfeldt and M. Freitag, J. Am. Chem. Soc., 2016, 138, 15087–15096 CrossRef CAS PubMed.
- D. Zhang, M. Stojanovic, Y. Ren, Y. Cao, F. T. Eickemeyer, E. Socie, N. Vlachopoulos, J.-E. Moser, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Nat. Commun., 2021, 12, 1777 CrossRef CAS PubMed.
- T. Stergiopoulos, E. Rozi, C.-S. Karagianni and P. Falaras, Nanoscale Res. Lett., 2011, 6, 307 CrossRef PubMed.
- G. Boschloo, L. Häggman and A. Hagfeldt, J. Phys. Chem. B, 2006, 110, 13144–13150 CrossRef CAS PubMed.
- J. R. Jennings and Q. Wang, J. Phys. Chem. C, 2010, 114, 1715–1724 CrossRef CAS.
- Y.-C. Liu, H.-H. Chou, F.-Y. Ho, H.-J. Wei, T.-C. Wei and C.-Y. Yeh, J. Mater. Chem. A, 2016, 4, 11878–11887 RSC.
- Z.-S. Wang, T. Yamaguchi, H. Sugihara and H. Arakawa, Langmuir, 2005, 21, 4272–4276 CrossRef CAS PubMed.
- M. Dürr, A. Yasuda and G. Nelles, Appl. Phys. Lett., 2006, 89, 061110 CrossRef.
- J.-Y. Kim, J. Y. Kim, D.-K. Lee, B. Kim, H. Kim and M. J. Ko, J. Phys. Chem. C, 2012, 116, 22759–22766 CrossRef CAS.
- S. Yu, S. Ahmadi, C. Sun, P. Palmgren, F. Hennies, M. Zuleta and M. Göthelid, J. Phys. Chem. C, 2010, 114, 2315–2320 CrossRef CAS.
- W. L. Hoffeditz, M. J. Katz, P. Deria, G. E. Cutsail Iii, M. J. Pellin, O. K. Farha and J. T. Hupp, J. Phys. Chem. C, 2016, 120, 3731–3740 CrossRef CAS.
- Y. Saygili, M. Stojanovic, H. Michaels, J. Tiepelt, J. Teuscher, A. Massaro, M. Pavone, F. Giordano, S. M. Zakeeruddin, G. Boschloo, J.-E. Moser, M. Grätzel, A. B. Muñoz-García, A. Hagfeldt and M. Freitag, ACS Appl. Energy Mater., 2018, 1, 4950–4962 CrossRef CAS.
- Y. Wang and T. W. Hamann, Chem. Commun., 2018, 54, 12361–12364 RSC.
- L. Kavan, Y. Saygili, M. Freitag, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Electrochim. Acta, 2017, 227, 194–202 CrossRef CAS.
- K. Kannankutty, C.-C. Chen, V. S. Nguyen, Y.-C. Lin, H.-H. Chou, C.-Y. Yeh and T.-C. Wei, ACS Appl. Mater. Interfaces, 2020, 12, 5812–5819 CrossRef CAS PubMed.
- S. O. Fürer, R. A. Milhuisen, M. K. Kashif, S. R. Raga, S. S. Acharya, C. Forsyth, M. Liu, L. Frazer, N. W. Duffy, C. A. Ohlin, A. M. Funston, Y. Tachibana and U. Bach, Adv. Energy Mater., 2020, 10, 2002067 CrossRef.
- P. Ferdowsi, Y. Saygili, S. M. Zakeeruddin, J. Mokhtari, M. Grätzel, A. Hagfeldt and L. Kavan, Electrochim. Acta, 2018, 265, 194–201 CrossRef CAS.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- J. Bisquert, Phys. Chem. Chem. Phys., 2000, 2, 4185–4192 RSC.
- F. Fabregat-Santiago, J. Bisquert, G. Garcia-Belmonte, G. Boschloo and A. Hagfeldt, Sol. Energy Mater. Sol. Cells, 2005, 87, 117–131 CrossRef CAS.
- T.-S. Su, T.-Y. Hsieh, C.-Y. Hong and T.-C. Wei, Sci. Rep., 2015, 5, 16098 CrossRef CAS PubMed.
- T.-C. Wei, C.-C. Wan, Y.-Y. Wang, C.-m. Chen and H.-s. Shiu, J. Phys. Chem. C, 2007, 111, 4847–4853 CrossRef CAS.
- J.-L. Lan, Y.-Y. Wang, C.-C. Wan, T.-C. Wei, H.-P. Feng, C. Peng, H.-P. Cheng, Y.-H. Chang and W.-C. Hsu, Curr. Appl. Phys., 2010, 10, S168–S171 CrossRef.
- T. C. Wei, C. C. Wan and Y. Y. Wang, Appl. Phys. Lett., 2006, 88, 103122 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3se00983a |
| This journal is © The Royal Society of Chemistry 2024 |