
Immobilization of enzymes on polymers with upper critical solution temperature: promising engineering of enzymes for biocatalysis
Lin
Huang
 *,
Xirui
Li
and
Zhi
Li
*,
Xirui
Li
and
Zhi
Li
Department of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, 117585, Singapore. E-mail: huanglin@awfa.com.sg
First published on 29th April 2024
Abstract
Polymers with upper critical solution temperature (UCST) can find promising applications in biocatalysis using immobilized enzymes, in that UCST-type polymer-immobilized enzymes use lower temperatures for biocatalyst separation than homogeneous biocatalytic reactions to ensure the prevention of the immobilized enzymes from deactivation as well as producing high biocatalytic performance. Herein, we have provided an overview of the developments of UCST-type polymer-immobilized enzymes for biocatalysis by reviewing the fundamentals of thermoresponsive polymers and the application of UCST-type polymers in the fabrication and biocatalysis of immobilized enzymes present in the literature. Furthermore, published studies on UCST-type polymer-immobilized enzymes involving various categories of polymers in this research area are reviewed. The characteristics of various UCST-type polymer-immobilized enzymes and their advantages and disadvantages in terms of biocatalytic performance, separation and reusability are summarized with an outlook on the engineering of enzymes with UCST-type polymers for biocatalysis.
 Lin Huang | Dr. Lin Huang graduated with a bachelor's degree in chemistry from Nanjing University, China. He obtained his DEA and doctorate in physical chemistry from Université Lyon 1, France. He successively joined the Dalian Institute of Chemical Physics, China; Institute of Chemical and Engineering Sciences, Singapore; and National University of Singapore. His research interests include surface organometallic chemistry, catalysis towards Fischer–Tropsch, CO2 hydrogenation to methanol, hydroformylation, carbon–carbon coupling, ethanol steam reforming, and dehydration of lactic acid. |
 Xirui Li | Dr. Xirui Li graduated with a bachelor's degree in chemistry from Zhengzhou University, China. She obtained her PhD in chemistry from the Nanyang Technological University, Singapore. She joined the Department of Chemical and Biomolecular Engineering, National University of Singapore, as a postdoc. Her research interests include catalysis and green and sustainable pharmaceutical synthesis via biocatalysis. |
 Zhi Li | Prof. Zhi Li received his bachelor's degree in chemistry in 1982 from Nanjing University, China, his master's degree in chemical engineering in 1985 from the Chinese Academy of Forestry, China. In 1991, he graduated with a PhD in organic chemistry from the University of Vienna, Austria. He joined the Department of Chemical and Biomolecular Engineering, National University of Singapore, to continue his research on advanced biocatalysis for the green and sustainable manufacturing of chemicals and pharmaceuticals, production of bio-based fuels and chemicals, and development of functional and smart materials. |
1. Introduction
Because of their outstanding characteristics such as high selectivity, activity and specificity, enzymes are promising biocatalysts in many fields such as food chemistry, fine chemistry, medical chemistry, fuel industry and environmental protection.1,2 However, free enzymes are sensitive to harsh environments, and the production of these enzymes is expensive. Many enzymes are water-soluble and therefore are not collectible or reusable in most circumstances. Immobilization of enzymes improves enzyme strength, turning enzymes into heterogeneous biocatalysts and allowing their recycling and operation under a flow process in various bioreactors without losing the outstanding characteristics of free enzymes.3–11 Many immobilized enzymes are known to even outperform free enzymes in activity and stability in chemical and biochemical processes.10,11Immobilization of enzymes on polymers has become a research hotspot to endow enzymes with extraordinary properties and broader usage. It is generally admitted that inorganic supports are materials of choice because of their advantages of thermal, mechanical and microbial resistance and low cost,12,13 whereas organic supports, especially polymers, are attractive as they possess some unique features. Compared with free enzymes, polymer-immobilized enzymes exhibit improved operational and thermal stabilities in harsh environments such as extreme pH, concentration and temperature. Compared with other supports, polymers are simple to produce and can be modified to meet the requirements for the interaction between polymers and enzymes.14,15 Particularly, polymers possess underlying stimulus-responsive features, which allow them to undergo a conformational change or coil-to-globule transition upon changes in external stimuli such as temperature, counterions, pH, electricity and light.16–20
Amid stimulus-responsive polymers, thermoresponsive polymers have been extensively studied in academic and applied polymer sciences over the last decades.10,17–22 A thermoresponsive polymer undergoes a change in solubility in a polymer–solvent system with varying temperatures.23,24 As the temperature increases, the polymer dissolves in the solvent through a UCST process and/or precipitates from the solution through a lower critical solution temperature (LCST) process. Scheme 1 shows the representative phase diagram patterns of UCST- and LCST-type polymer–solvent systems, which depict the solution behaviour of the polymers as a function of temperature versus polymer concentration. In such phase diagrams, the maxima and the minima of the solubility curves are the respective UCST and LCST values.17,18,24 Any point along the respective solubility curves is defined as a cloud point (Tcp) or demixing point (Tdem). Typical examples of UCST- and LCST-type polymers are polystyrene in cyclohexane24 and poly(N-isopropyl acrylamide), i.e., PNIPAM in water,23 respectively. Water-soluble thermoresponsive polymers are of special interest because water is the cheapest and safest solvent as well as the solvent of living systems, which allow practical applications in the fields of biochemistry and medicine.
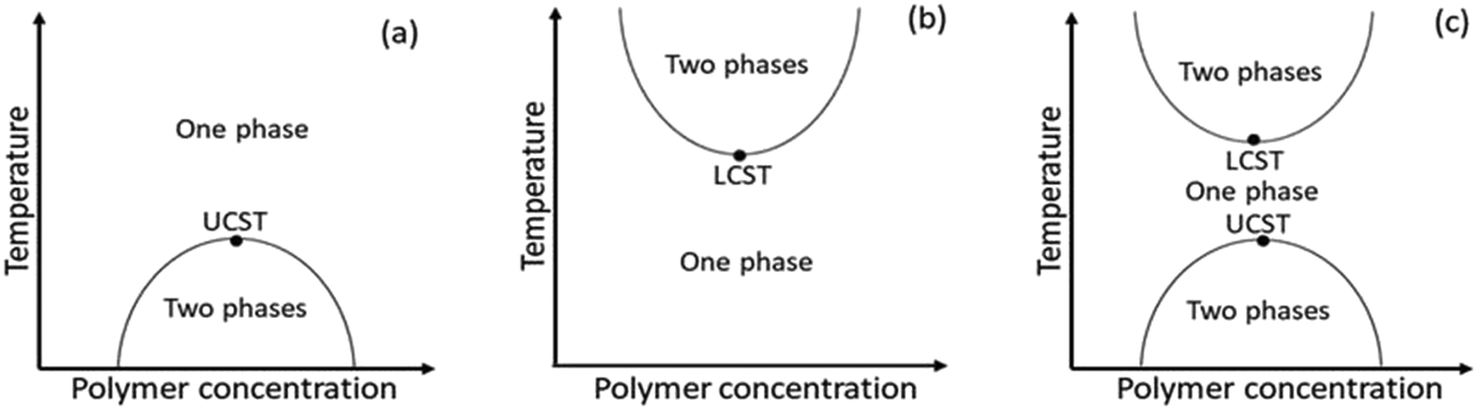 | ||
| Scheme 1 Phase diagram patterns of (a) UCST-type polymer–solvent, (b) LCST-type polymer–solvent and (c) UCST- and LCST-type polymer–solvent systems. | ||
Thermoresponsive polymers are expected to find “smart” applications that exploit temperature responsivity. The applications of latent materials of this kind may involve switchable hydrophilic–hydrophobic surfaces,25–27 chromatography,28 targeted drug delivery,17,18,29 gene therapy,30,31 thermally switchable optical devices,32 bioseparation33–35 and supported biocatalysis.10,36–52 As far as the application of thermoresponsive polymers in immobilized biocatalysis is concerned, the “smartness” consists of integrating the advantage of homogeneous catalysis with the convenience of biocatalyst separation from two-phase systems via a thermoresponsive polymer phase transition, so as to meet the goal of high biocatalytic efficiency of enzymes. When a thermoresponsive polymer-immobilized enzyme is used to biocatalyze a chemical or biochemical reaction, the immobilized enzyme can be shifted from the solid to liquid phase by varying the temperature so that a homogeneously biocatalytic reaction can be carried out with high efficiency. After the reaction, the liquid-phase system can be shifted back to the two-phase system by changing the temperature for the purpose of separation and reuse of the immobilized enzyme.
In the area of thermoresponsive polymer-immobilized enzyme biocatalysis, only 1.4 dozen publications have appeared involving UCST-type polymers,36–52 while several dozens of publications are available relating to LCST-type polymers. Up to date, only a couple of reviews on LCST-type polymer-immobilized enzyme biocatalysis have been published,53,54 while reviews or minireviews on UCST-type polymer-immobilized enzyme biocatalysis have yet to be published. It is worth pointing out that UCST-type polymer-immobilized enzyme biocatalysis is of greater significance. This is because UCST-type polymer-immobilized enzymes use a lower temperature for biocatalyst separation than for homogeneously biocatalytic reactions, which ensures the prevention of the immobilized enzymes from deactivation. By contrast, LCST-type polymer-immobilized enzymes adopt a higher temperature for biocatalyst separation than for homogeneous biocatalytic reactions, which may cause deactivation of the immobilized enzymes. Note that many enzymes can hardly withstand a higher temperature environment that causes thermal denaturation.55 This contribution presents an explicit overview of the branch of biocatalysis by UCST-type polymer-immobilized enzymes. We focus on the unique characteristics of UCST-type polymers and the roles they play during biocatalytic processes. We review the published studies in this branch with our critiques. We deal with the advantages and disadvantages of various UCST-type polymer-immobilized enzymes in terms of biocatalytic performance, separation and reusability. An outlook is made on the new engineering of enzymes for biocatalysis by highlighting the available literature on the developments of UCST-type polymer-immobilized enzymes for biocatalysis. In the available literature, the reported effects of enzyme immobilization on UCST-type polymers on biocatalytic performance are limited to the aspects of activity and stability. Thus, the effects of enzyme immobilization on UCST-type polymers on selectivity and specificity are not included in this review, although immobilization of enzymes generally affects selectivity and specificity to a certain extent while improving stability and changing activity.
2. Phase transition principles of UCST- and LCST-type polymer solutions
Let us begin with the fundamentals of phase transitions of UCST- and LCST-type polymer solutions. A polymer dissolves in a solvent to a different extent as long as the Gibbs energy change (ΔG) of dissolution (eqn (1)) becomes negative. Thermoresponsive polymers display drastic and discontinuous changes in their solubility in a given solvent with temperature, in contrast to temperature-sensitive materials, which change their solubility continuously with environmental conditions. In a strict sense, thermoresponsive polymers exhibit a miscibility gap in their temperature–concentration diagrams.| ΔG = ΔH − TΔS | (1) |
Most typical of the phase transitionMost typical of the phase transition properties of UCST- and LCST-type polymer solutions for comparison are the thermoresponsive behaviour of poly(N-acryloyl glycinamide), i.e., PNAGA in water and PNIPAM in water, respectively. The structures of PNAGA and PNIPAM are illustrated in Scheme 2. In aqueous solutions, the dissolution of polymer proceeds from the globule to the coil state via the demolition of polymer–polymer interaction and the formation of polymer–water hydrogen bonds. PNAGA is a nonionic polymer that exhibits UCST behaviour in water with a thermoreversible Tcp of 7.6–33 °C.56 The thermoresponsiveness of PNAGA consists of the hydrogen bonding between the polymer side groups.56–58 Upon the dissolution of PNAGA in water, the hydrogen bonds of polymer–polymer (between the carbonyl and amine groups) and water–water as well as the van der Waals forces of polymer–polymer are broken in an endothermic process, and they are replaced by the hydrogen bonds of polymer–water in an exothermic process.57 The phase transition of PNAGA is fully thermoreversible via hydrogen bonding,56,57 as indicated by turbidity measurements in Fig. 1 and 2.56 Because of the particular dependence on hydrogen bonding, the net enthalpy change (ΔH) of the dissolution process of PNAGA is positive and low, being determined to be 90 J (mol monomer)−1.57 Now that PNAGA dissolves in water, the ΔG of dissolution must be negative. Thus, the entropy change (ΔS) of mixing must be positive with TΔS > ΔH above the Tcp. Because the ΔS of mixing is much lower for polymers than for small molecules and moreover decreases with increasing polymer molecular weight, it was observed that a low PNAGA molecular weight favoured the dissolution of PNAGA and thus even caused the disappearance of UCST behaviour,59–61 and that the Tcp increased with increasing molecular weight in 1% PNAGA solution.62 Although the phase transition properties of PNAGA can be tuned by copolymerization, the ΔH of dissolution would be affected by comonomers to a limited extent. It is easily understood that hydrophilic comonomers affect little the ΔH of dissolution and enhance the ΔS of mixing, thus lowering the Tcp of PNAGA. In the case of hydrophobic comonomers, it is deemed that the hydrogen bonds of polymer–polymer and polymer–water are little affected, so the contribution to the ΔH of dissolution from hydrophobes is unimportant.63 However, the ΔS of mixing is notably lowered due to the hydrophobic effect.64 In fact, the hydration shell of hydrophobic segments of the polymer forms in water because the water molecules are unable to hydrogen bond with the hydrophobic segments. The decreased ΔS of mixing results in an increased Tcp. Apart from the homopolymer of NAGA, some derivatives of PNAGA such as poly(N-acryloylasparagineamide), poly(N-acryloylglutamineamide) and poly(N-methacryloylasparagineamide), and some copolymers of NAGA with N-acetylacrylamide, biotin-functionalized methacrylamide, butyl acrylate, styrene and vinyl phenylboronic acid (VBA) likewise show UCST behaviour in water.52,65–67
 | ||
| Scheme 2 Structural formulas of PNAGA and PNIPAM. | ||
 | ||
| Fig. 1 Turbidity curves of a 1% PNAGA aqueous solution at a heating or cooling rate of 1 °C min−1. Reprinted with permission from ref. 56, copyright 2010, John Wiley and Sons. | ||
 | ||
| Fig. 2 UCST and gel melting temperature of PNAGA as a function of PNAGA concentration in water with a heating rate of 1 °C min−1. Reprinted with permission from ref. 56, copyright 2010, John Wiley and Sons. | ||
PNIPAM is a rich hydrophobe-containing nonionic polymer that displays LCST behaviour in water with a thermoreversible Tcp of 31–36 °C.68–72 It undergoes a thermoreversible coil-to-globule transition on heating.73 Below the Tcp, the formation of hydrogen bonds between PNIPAM and water leads to a negative ΔG of dissolution with negative ΔH and ΔS of dissolution, which enables PNIPAM to dissolve in water. The ΔH of dissolution was determined to be (−4)–(−15) kJ (mol monomer)−1.68,70,74–76 The negative ΔS of dissolution is attributed to the highly ordered hydration shell of the isopropyl groups induced by the hydrophobic effect. Under such a circumstance, PNIPAM orders itself in water to hydrogen bond with the already arranged water molecules on the one hand, and the water molecules must reorient around the hydrophobic segments of PNIPAM on the other hand. Below the Tcp, the negative ΔH term from the hydrogen bonding effect dominates the ΔG (which is negative) so as to facilitate the globule-to-coil transition or dissolution of PNIPAM, which gives one phase in water. When the temperature is raised to the Tcp or above, the negative TΔS term from the hydrophobic effect dominates the ΔG (which is positive) so as to promote the coil-to-globule transition or precipitation of PNIPAM, which produces the two phases in water. The phase transition of PNIPAM is fully thermoreversible and very abrupt on turbidity curves irrespective of the molecular weight over a wide range,73,76,77 as illustrated by light scattering measurements in Fig. 3.73 Apart from the homopolymer of NIPAM, many copolymers of NIPAM such as poly(NIPAM-co-acrylic acid (AAc)), poly(NIPAM-co-acrylamide (AAm)), poly(NIPAM-co-butyl methacrylate-co-X) with X = butyl methacrylate, AAm, AAc, (diethyl amino)ethyl methacrylate, and poly(NIPAM-co-N-ethylacryl amide), etc. show LCST behaviour in water.74,78–81
 | ||
| Fig. 3 Temperature dependences of the average radius of gyration <Rg> and the average hydrodynamic radius <Rh> in the coil-to-globule (heating) and the globule-to-coil (cooling) transitions of PNIPAM, respectively, with the inset showing the temperature dependences of <Rg>/<Rh> in the heating and the cooling processes. Reprinted with permission from ref. 73, copyright 1998, American Chemical Society. | ||
From the comparative phase transition properties of PNAGA in water and PNIPAM in water, the UCST of PNAGA susceptibly fluctuates with factors such as polymer molecular weight, polymer concentration, hydrophilic fraction, counterions, electricity, light and pH, whereas the LCST of PNIPAM is little affected by these factors. This may be explained in terms of the huge difference in their absolute magnitudes of ΔH of dissolution from the hydrogen bonding effect, leading to different sensitivities of Tcp to these factors. The ΔH of dissolution of PNIPAM is deemed to be 44–167 times that of PNAGA. According to ΔG = 0 at the Tcp values, eqn (1) simplifies to Tcp = ΔH/ΔS. The change in the Tcp caused by the variation of ΔH and/or ΔS due to any of these factors is much stronger for PNAGA than for PNIPAM. For example, Liu et al. used Tepper turbidity photometry to show that the Tcp of PNAGA in phosphate buffer on heating decreases from 38 to 26 °C with increasing molecular weight from Mn = 8270 to 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 at a PNAGA concentration of 0.2% in water.59 By contrast, Fujishige et al. found by light scattering that the Tcp of PNIPAM (ca. 30 °C) either on heating or cooling is independent of molecular weight over a range of Mn = 50000–8400000 with a PNIPAM concentration of 1% in water.76 According to Afroze et al., the temperature-concentration demixing curves of PNIPAM in water on heating with three different molecular weights at Mn = 2200, 36000 and 83300 almost coincide over the whole range, as studied by differential scanning calorimetry.77 At a very low PNIPAM concentration in water, the Tdem values of PNIPAM on heating are 34, 31 and 31 °C with Mn = 2200, 36000 and 83300, respectively.77
700 at a PNAGA concentration of 0.2% in water.59 By contrast, Fujishige et al. found by light scattering that the Tcp of PNIPAM (ca. 30 °C) either on heating or cooling is independent of molecular weight over a range of Mn = 50000–8400000 with a PNIPAM concentration of 1% in water.76 According to Afroze et al., the temperature-concentration demixing curves of PNIPAM in water on heating with three different molecular weights at Mn = 2200, 36000 and 83300 almost coincide over the whole range, as studied by differential scanning calorimetry.77 At a very low PNIPAM concentration in water, the Tdem values of PNIPAM on heating are 34, 31 and 31 °C with Mn = 2200, 36000 and 83300, respectively.77
According to Seuring et al., 0.2 mol% of AAc in an aqueous solution of PNAGA (fed prior to polymerization) causes the Tcp of PNAGA to disappear, as measured by Tepper turbidity photometry,57 whereas according to Qiu et al., 0.8 mol% of AAc in an aqueous solution of PNIPAM (fed prior to polymerization) increases the Tcp of PNIPAM by just 1 °C (from 32 to 33 °C), as determined by light scattering.79 It is just the drawback of UCST-type polymers in this aspect, in combination with the advantage of UCST-type polymers in biocatalytic processes, that has been attracting researchers to seek to surmount the challenge worldwide.
3. Application of UCST-type polymers in the immobilization of enzymes for biocatalysis
In light of the characteristics and advantages, UCST-type polymers have long been expected to be smartly applied in the fabrication and the biocatalysis of immobilized enzymes. Although the number of published studies is not enormous,36–52 these studies have a profound impact on the perspective of the developing branch of UCST-type polymer-immobilized biocatalysis. The UCST-type polymers involved in the literature can be categorized into sulfobetaine homopolymers,36,37,47 nonionic homopolymers45,46 and copolymers.38–44,48–52Sulfobetaine homopolymers
As early as 2006, Liu's group reported on biocatalysis of trypsin, a protease enzyme, conjugated with monodispersed carboxyl-terminated poly(3-dimethyl(methacryloyloxyethyl) ammonium propane sulfonate) (PDMAPS) for hydrolysis of biochemical substrates such as N-R-benzoyl-D,L-arginine p-nitroanilide hydrochloride (BAPNA) and a casein.36 This report marked, to our knowledge, the first application of UCST-type polymers in the immobilization of enzymes for biocatalysis. The enzyme–polymer conjugation adopted a “grafting to” approach, where presynthesized or end-functionalized polymers are coupled to accessible amino acid side chains or end termini on the enzyme surface.82,83 The conjugated trypsin was prepared via covalent linking of trypsin to PDMAPS at 4 °C with a PDMAPS:trypsin molar ratio of 2.4. The PDMAPS of 10 mg mL−1 and the conjugate of 10 mg mL−1 in 0.02 M phosphate buffers with pH 7.8 exhibited their Tcp values of 16 and 14 °C, respectively, in UV-vis transmittance at 600 nm with temperature, as shown in Fig. 4. The slight fall in the Tcp may result from more hydrophiles in the conjugate sample than in the PDMAPS sample. This is not as expected. In the expected case of the chemical interaction between trypsin and PDMAPS, it is supposed that the lysine groups of trypsin are able to soundly react with the carboxyl groups of PDMAPS with the concomitant decrease in the fraction of hydrophiles (i.e., increase in the fraction of hydrophobes) in the conjugate, which leads to an increase in the Tcp. The converse observation confirms the drawback of “grafting to” in excess of polymer to specific amino acid residues on the enzyme surface for the dichroic spectroscopic synthesis of enzyme–polymer conjugates due to steric limitations. The fluorescence emission and circular studies testified that the conjugated trypsin retained its native conformation. Moreover, the conjugation was demonstrated to strongly improve the thermal enzymatic stability of trypsin, i.e., strongly decrease both the thermal denaturation and the autolysis of trypsin, especially at higher temperatures, through the dispersion of trypsin on PDMAPS. The half-life of 0.5 mg mL−1 trypsin at 50 °C was enhanced from 0.42 to 9.0 h (by 20-fold) after the conjugation. In the enzymatic hydrolysis of BAPNA, the activities of trypsin and the conjugate almost coincided at 25 °C and different pH values, confirming the structural stability of trypsin after the conjugation. Such a highly retained activity of the conjugate suggests the existence of no mass transfer limitations between the conjugate and the substrate. The conjugate had incomparable thermal stability than trypsin, as shown by deactivation kinetic curves at 50 °C in Fig. 5. By means of the Lineweaver–Burk plot,84 similar curves of the limiting reaction rate (Vm) variation with reaction temperature at pH 7.8 over trypsin and over the conjugate were obtained as in Fig. 6, which indicated an increased Vm with increasing temperature at 10–40 °C over both of them. The similar Vm values imply similar activities for the reaction over them. Meanwhile, it was found that the Michaelis constant (Km) over the conjugate was lower than that over trypsin while the Km values over both of them decreased with increasing reaction temperature; the difference increased with increasing reaction temperature. The decrease in Km implies an increased BAPNA's affinity with the biocatalyst. The relative decrease in Km over the conjugate to that over trypsin indicates a stronger BAPNA's affinity with the conjugate than with trypsin. However, the stronger affinity was misunderstood to arise from the interaction between the hydrophobic conjugated trypsin and the hydrophobic BAPNA.36 Furthermore, the reaction data above 40 °C were lacking so the trends of Vm and Km variations could not be estimated at higher temperatures for comparison. Actually, the BAPNA is highly hydrophilic,85 and the conjugate is much more hydrophilic than trypsin, taking into account the BAPNA's affinity with the entire conjugate, including the enzyme and the polymer. In this case, the stronger affinity by conjugation should be assumed to be primarily from the higher BAPNA's hydrophilicity with the conjugate. In light of reported comparative results concerning free trypsin- and immobilized trypsin-biocatalyzed hydrolysis of BAPNA at room temperature,36,86–96 the immobilization of trypsin leads to an increased Km in most cases, corresponding to decreased BAPNA's affinity and decreased Vm.87,90,92,95,96 This appears to support the assumption of a decreased hydrophilicity on the conjugate. In the cases with about the same or a decreased Km, the corresponding Vm remains unchanged or increases, depending on other factors.36,86 When the reaction is carried out at 45–85 °C, the activity of the conjugate is reportedly superior to that of free trypsin in all cases.86–88,90,92,93,95,96 From the comparative reaction data and thermal stability in this case, the conjugation is able to create the stabilization of trypsin without changing the activity of trypsin towards the hydrolysis of BAPNA at 10–40 °C. Indeed, the conjugate could be recycled 10 times with losing only 15% of the initial activity in the hydrolysis of BAPNA at 40 °C and pH 7.8, as shown in Fig. 7. In the conjugate recycling operation, the soluble conjugate precipitated from the reaction solution upon cooling from 40 to 4 °C after each run. After the solid conjugate was washed at 4 °C, it was subjected to the next reaction cycle at 40 °C. By temperature swinging this way, the recovery and the recycling of the UCST-type conjugate were eased. The high recyclability and the convenient operation embody the advantages of the UCST-type conjugate in practical applications. In the enzymatic hydrolysis of the casein as a high molecular weight substrate, the activities of trypsin and the conjugate were quite different with varying temperatures at pH 7.8, as shown in Fig. 8. The optimal reaction temperatures were 40 and 60 °C for trypsin and the conjugate, respectively. The corresponding optimal activity of the conjugate reached 2.6 times that of trypsin, indicative of an enhancement in the activity of trypsin by the conjugation. This enhancement was unprecedented towards conjugated trypsin biocatalysis for hydrolysis of caseins.88,92,97–99 Generally, immobilized enzymes by conventional methods have higher stability but lower activity than free enzymes, mainly due to the loss of activity during immobilization and the mass transfer limitations between the enzymes and substrates,10,11 whereas immobilized enzymes by nanotechnology may have a higher activity than free enzymes because of higher specific surface area and less mass transfer limitations in heterogeneous biocatalysis.10,100,101 As for thermoresponsive polymer-immobilized enzymes, increased activity compared to that of free enzymes is expected to result from homogeneous biocatalysis with the concurrent promoting effect via the enzyme–polymer interaction. In the case of the PDMAPS-conjugated trypsin for the hydrolysis of casein, the tremendously increased activity may be attributed to an enhanced casein's hydrophilicity with the conjugate. Although caseins are relatively hydrophobic, they possess discrete hydrophilic and hydrophobic segments in their peptide sequences. From the other reported studies, the increase in the faction of hydrophobic segments after the covalent linking of trypsin to the polymers leads to either about the same or a decreased activity at the optimal temperatures compared to that of free trypsin.88,92,97–99 Moreover, the immobilization of trypsin on the polymers does not affect the native conformation of trypsin.97,99 The effect of the casein's hydrophobicity with the conjugate on the increased activity is thus considered unimportant.
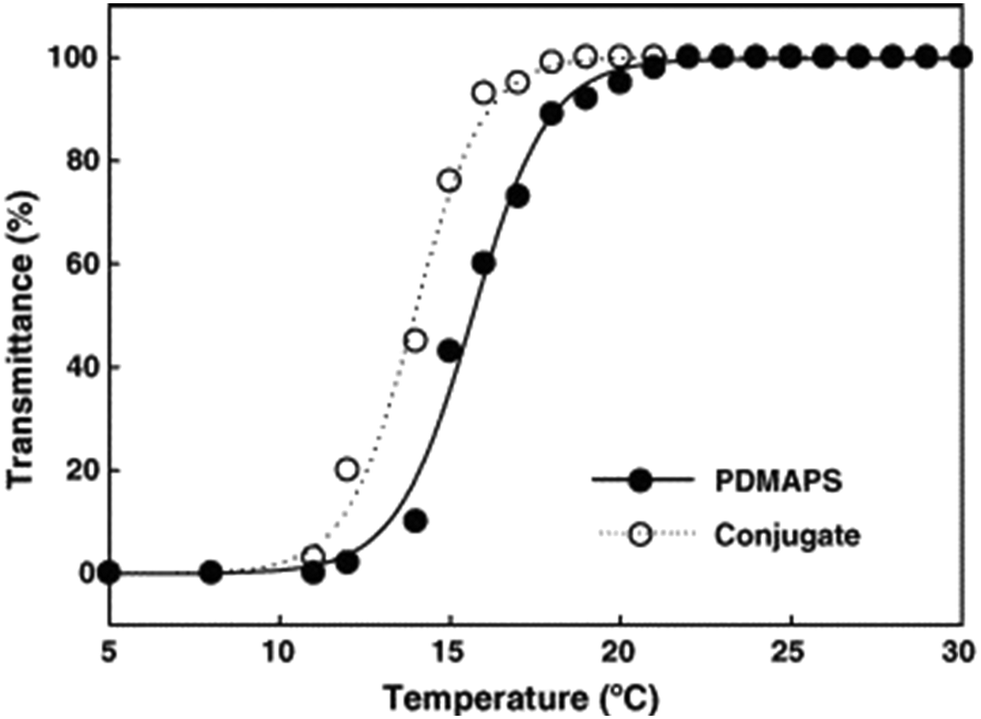 | ||
| Fig. 4 UV-vis transmittance transitions of PDMAPS of 10 mg mL−1 and the PDMAPS-conjugated trypsin of 10 mg mL−1 in 0.02 M phosphate buffer with pH 7.8. Reprinted with permission from ref. 36, copyright 2006, Elsevier Inc. | ||
 | ||
| Fig. 5 Deactivation behaviour of trypsin and the PDMAPS-conjugated trypsin at 0.50 mg mL−1 in the hydrolysis of BAPNA at 50 °C and pH 7.8. | ||
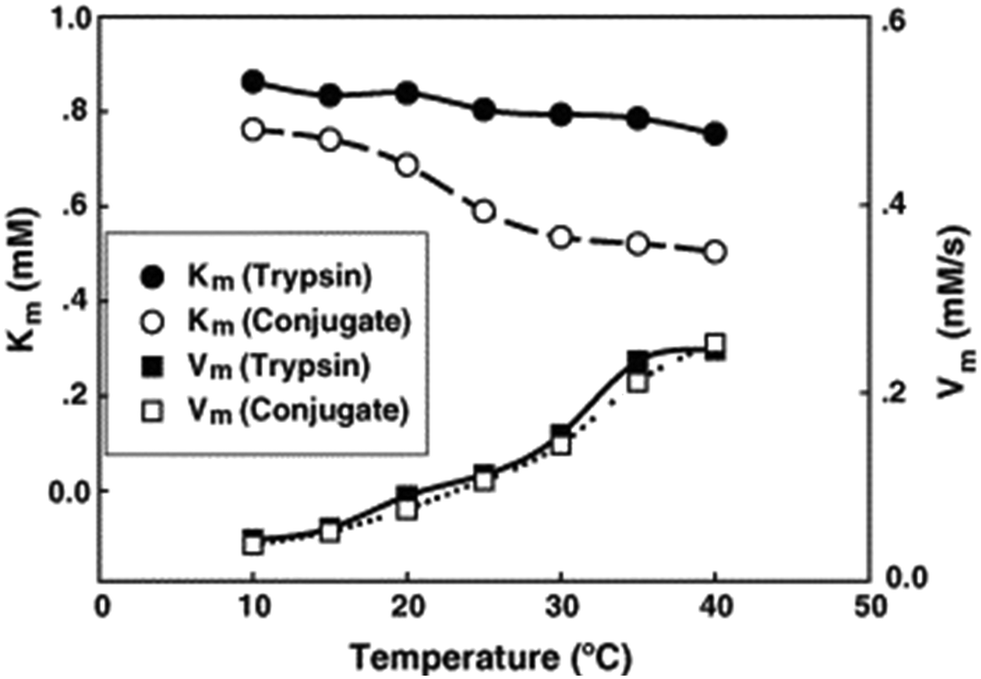 | ||
| Fig. 6 Kinetic parameters of trypsin and the PDMAPS-conjugated trypsin at different temperatures and pH 7.8 in the hydrolysis of BAPNA. Reprinted with permission from ref. 36, copyright 2006 Elsevier Inc. | ||
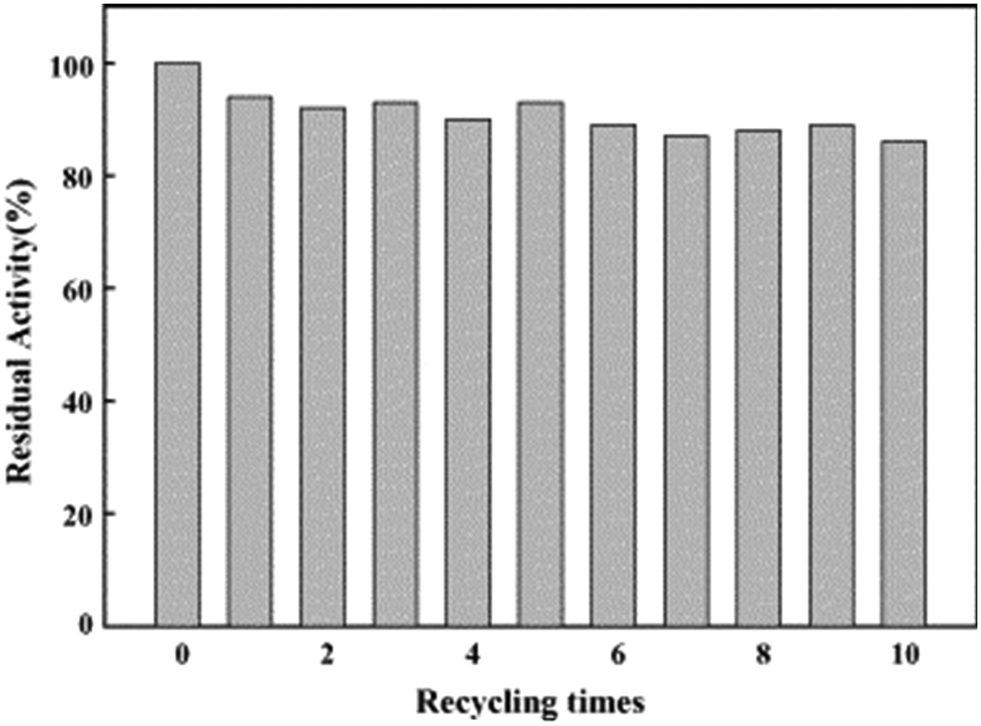 | ||
| Fig. 7 Recycling of the PDMAPS-conjugated trypsin in the hydrolysis of BAPNA at 40 °C and pH 7.8. Reprinted with permission from ref. 36, copyright 2006, Elsevier Inc. | ||
 | ||
| Fig. 8 Relative activities of trypsin and the PDMAPS conjugated trypsin at different temperatures and pH 7.8 in the hydrolysis of the casein using that of trypsin at 40 °C and pH 7.8 as the reference. Reprinted with permission from ref. 36, copyright 2006, Elsevier Inc. | ||
In 2013, Cummings et al. published a paper on biocatalysis of nano-sized chymotrypsin (CT)-PDMAPS conjugates for hydrolysis of N-succinyl-Ala-Ala-Pro-Phe p-nitroanilide (Suc-AAPF-pNA).37
Thus far, most of the UCST-type polymers reported for application in immobilized enzyme biocatalysis belong to micro-sized polymers.36,41–44,46–49,51 Nano-sized polymers are attractive supports for achieving higher specific enzyme loading, higher specific activity and less mass transfer limitations than their micro-sized counterparts in heterogeneous biocatalysis. The enzyme–polymer conjugation used a “grafting from” approach, where enzyme molecules themselves serve as the initiating sites for controlled radical polymerization.82,83 The “grafting from” approach led to high yields (or high polymer density) and facile purification of the resultant conjugates.82,83 The conjugated CT was synthesized via the covalent attachment of the atom transfer radical polymerization (ATRP) initiator to accessible primary amines either on lysine residues or N-terminus of CT (forming a macroinitiator) at 4 °C, followed by the growth of PDMAPS from the initiator-modified CT at 4 °C, as depicted in Scheme 3.37 In UV-vis absorbance at 490 nm, the PDMAPS and the CT-PDMAPS conjugate in phosphate buffers responded to changes in temperature in a similar manner, giving their Tcp values of 12 and 13 °C, respectively. The slight rise in Tcp is deemed to arise from more hydrophobes on the conjugate than on the PDMAPS. This is as expected and reflects the advantage of “grafting from” in the achievement of high polymer density for the synthesis of enzyme–polymer conjugates owing to the avoidance of steric limitations. By dynamic light scattering, the CT-PDMAPS conjugate was found to have a maximal temperature-dependent hydrodynamic particle radius of 6.5 nm at 19 °C, corresponding to the one-phase state above the Tcp. Towards the enzymatic hydrolysis of Suc-AAPF-pNA, the conjugate and free CT showed close activities both at 25 and 40 °C in terms of Vm/Km. Such a highly retained activity of the conjugate can be attributed to the fact that there are no mass transfer limitations between the conjugate and the substrate. Although free CT had a higher Vm, the conjugate gained a lower Km because of an enhanced Suc-AAPF-pNA's affinity with the conjugate, mainly through the hydrophobic interaction between Suc-AAPF-pNA and PDMAPS. The lower Vm over the conjugate than over free CT implies that the conjugation causes a conformational change of part of CT. The conjugate exhibited a higher enzyme thermal stability than free CT both at 25 and 40 °C, especially at 40 °C, as in Fig. 9. The conjugation led to a 3.1-fold enhancement in the half-life of CT (at 1 mg mL−1) at 40 °C (from 2.4 to 9.8 h). However, the conjugate had not only markedly decreased thermal stability with time but also markedly decreased activity during biocatalyst cycling above and below the Tcp. These results show that the CT stability is enhanced by the conjugation to a limited extent only.
 | ||
| Scheme 3 Thermoresponsive polymer-conjugated CT engineering: (1) initiator immobilization onto the CT surface and (2) “grafting from” ATRP reaction to produce CT-PDMAPS and CT-PNIPAM conjugates. Reprinted with permission from ref. 37, copyright 2013, Elsevier Inc. | ||
 | ||
| Fig. 9 Deactivation behaviour of free CT and the CT-PDMAPS conjugate at 1 mg mL−1 in the hydrolysis of Suc-AAPF-pNA at 40 °C. | ||
Until now, the vast majority of the applications of UCST-type polymers in biocatalysis involve transformations of chemical substrates.38,40–52 In order to efficiently recycle cellulases for lignocellulosic enzymatic hydrolysis, Li et al. recently investigated the recovery and recycling of cellulase Cellic CTec2 (CTec2) for biocatalytic cellulolysis using PDMAPS as the adsorbent by regulating the temperature.47 The additions of CTec2 to 0.2 g L−1 PDMAPS in acetate buffer resulted in continuously increased Tcp values from 38 to 49 °C, which was an indication that the resultant CTec2–PDMAPS systems are more hydrophobic than PDMAPS. The higher hydrophobicity probably arises from the formation of covalent bonds between CTec2 and PDMAPS, which reduces the fraction of hydrophiles. CTec2 efficiently adsorbed on PDMAPS via the covalent bonding and precipitated in acetate buffer at 25 °C, its recovery rate being up to 63%. In the biocatalytic hydrolysis of Avicel, corncob residues (CCRs) and eucalyptus chips treated with dilute acid (Eu-DA), the presence of PDMAPS hardly affected the hydrolysis efficiencies of free CTec2 at 60, 70 and 50%, respectively, at 50 °C. After the reaction, PDMAPS precipitated with the concomitant adsorption of CTec2 on PDMAPS upon cooling to 25 °C. The recovered PDMAPS-held CTec2 led to hydrolysis efficiencies of 28, 32 and 22%, respectively, at 50 °C. The experimental data demonstrated that adding PDMAPS can save more than 50% CTec2 for the hydrolyses of Avicel, CCR and Eu-DA. This work is of practical significance in reducing the cost of lignocellulosic enzymatic hydrolysis. However, the adsorption of CTec2 on PDMAPS was inappropriately explained by the authors for the recovery of CTec2. The hydrophobic interaction between CTec2 and PDMAPS was suggested to be the driving force for the adsorption of CTec2 on PDMAPS, in contradiction with the observations of the increased Tcp values upon the addition of CTec2 to PDMAPS buffer solution. The increase in Tcp implies that the more hydrophobic CTec2–PDMAPS systems form at the expense of the hydrophilic interaction between CTec2 and PDMAPS. The hydrophilic interaction probably involves covalent bonding. In the meantime, although the study of the effect of urea, usually as a hydrogen bond breaker with biomaterials,102,103 on the adsorption of CTec2 on PDMAPS showed that after adding urea, the adsorption capacity of CTec2 increased from 187 to 258 ng cm−2 at 25 °C; the authors could not rule out the possibility that urea acts as a hydrogen bond producer between CTec2 and PDMAPS while speculating the possibility of the hydrophobic interaction between CTec2 and PDMAPS. Lastly, the preparation of hydrogen-bonded poly(vinyl alcohol) (PVA)-urea hydrogels by drying PVA-urea solutions was reported by Wu et al., which revealed for the first time that urea can play a hydrogen bond producer role other than a hydrogen bond breaker role.104
Nonionic homopolymers
Despite that, the UCST behaviour of nonionic polymers leads to broader phase transition temperature intervals with larger hysteresis upon heating and cooling due to the lower strength of hydrogen bonds, PNAGA is the most studied UCST-type polymer as a platform for biocatalysis.17,19,45,46In 2013 and 2020, Hietala and coworkers published their work on chemo- and biocatalytic applications of nano-sized chemically crosslinked PNAGA hydrogels.45,105 The chemically crosslinked PNAGA hydrogels and the β-D-glucosidase (β-D-GS)–PNAGA hydrogels were synthesized using N,N′-methylenebis(acrylamide) (BIS) as the crosslinker by free radical polymerization in water at 0–22 °C.45,105 The silver nanoparticle (AgNP)-containing hybrid hydrogels were prepared by reduction of AgNO3 to AgNPs using NaBH4 inside the PNAGA hydrogels and the BG-PNAGA hydrogels, respectively.45,105 The dynamic light scattering study indicated that both the chemically crosslinked PNAGA hydrogels and the hybrid chemically crosslinked PNAGA hydrogels continuously swelled upon heating up to 70 °C in water.45,105 From Fig. 10, the particle size of the PNAGA-3.5% BIS hydrogels in phosphate buffer increased by 1.9 times in volume upon heating from 10 to 52 °C.45 It approached the maximum above 52 °C. The broad temperature range from 10 to 52 °C was deemed to correspond to the phase transition temperature interval, analogous to the cases of other UCST-type polymers.37,52,106,107 The changes in the concentrations of BIS and β-D-GS added in the chemically crosslinked PNAGA hydrogels apparently did not change the phase transition of the PNAGA hydrogels, which followed the same trend of particle size variation with temperature,45,105 differing from what happens to linear homologous PNAGA hydrogels.57,59–61,108 In the latter cases, the phase transition is sensitive to the polymer molecular weight, the polymer structure and the environmental factors, so the Tcp values are susceptible to vary.57,59–61,108 Based on the comparative observation that the particle size of free β-D-GS dramatically rose at around 50 °C due to aggregation, it was assumed that the aggregation of β-D-GS encapsulated inside the PNAGA hydrogels is suppressed at elevated temperatures.45 The suppression of β-D-GS aggregation may result from the hydrogen bonding between β-D-GS and the PNAGA hydrogels, which renders β-D-GS dispersed on the PNAGA hydrogels. Likewise, the hybrid Ag-PNAGA-BIS hydrogels retained the phase transition of the PNAGA hydrogels, displaying an increased dispersion or swelling with increasing temperatures up to at least 70 °C in water.105 Towards the enzymatic cleavage of p-nitrophenyl-β-D-glucopyranoside (pNGP), the β-D-GS–PNAGA hydrogels generally displayed similar activity to that of free β-D-GS at pH 3–11 and 40 °C, implying the occurrence of no mass transfer limitations between the conjugate and the substrate.45 Their optimal activities were found to be 5.0 × 10−3 s−1 mmol−1 at pH 7.0 and 3.0 × 10−3 s−1 mmol−1 at pH 6.1, respectively. Attempts to recycle the BG-PNAGA hydrogels after recovery by cooling down were unsuccessful, with the activity dropping at each recycling round. The problem was ascribed mainly to the inefficient sedimentation of the chemically crosslinked β-D-GS–PNAGA hydrogels upon cooling. While chemically crosslinked PNAGA hydrogels possess the advantage of stable UCST behaviour with low stimulus responsivity, they have trouble with a very broad phase transition temperature interval where the hydrogels gradually swell or dissolve in water from 0 to at least 70 °C in water.105,109 Not only is a preferred higher temperature difficult to adapt for the reaction efficiency, but a lower temperature is also not necessarily ideal for the hydrogel recovery after the reaction. Significantly, the cascade reaction of pNPG cleavage and subsequent 4-nitrophenol reduction with NaBH4 to 4-aminophenol was effectively biocatalyzed by Ag-β-D-GS–PNAGA hydrogels.45
 | ||
| Fig. 10 (A) Particle size distributions of BG, PNAGA and BG-PNAGA at 40 °C. (B) Particle size variations with temperature upon heating; the inset showing the aggregation of BG. Reprinted with permission from ref. 45, copyright 2020, Elsevier Inc. | ||
Later on, Kappauf et al. reported a biocatalytic application of a physically crosslinked PNAGA hydrogel.46 The physically crosslinked PNAGA hydrogel and the transaminase from Bacillus megaterium (BmTA)–PNAGA hydrogels were synthesized by photo-initiated free radical polymerization and physical in situ gel formation without chemical crosslinker in water at 30 °C. In the case of the BmTA–PNAGA hydrogels, BmTA was added to the pre-gel solution prior to irradiation. The swelling experiments showed a gradual swelling of the PNAGA hydrogel with increasing temperature and reversible temperature-dependent swelling behaviour of the PNAGA hydrogel both in water and 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer between 20 and 35 °C. With respect to the swelling degree in water, the PNAGA hydrogel shrank in the HEPES buffer severely (by 60% at 35 °C) and in a HEPES-containing reaction medium even more pronouncedly (by 73% at 35 °C). Moreover, the swelling of the PNAGA hydrogel heavily lost its thermoresponsiveness in these two media. These facts uncover the disadvantages of physically crosslinked PNAGA, suggesting that not only does the broad phase transition temperature interval of crosslinked PNAGA persists45,105,109 but also the pronounced effect of electrolytes on the phase transition of linear PNAGA remains.19,57,59 There were no significant changes in the swelling of the PNAGA hydrogel in the HEPES buffer at 20–35 °C after the immobilization of BmTA, implying that the immobilization of BmTA does not alter the thermoresponsiveness of the PNAGA hydrogel. In the enzymatic conversion from 1-hydroxy-1-(3-hydroxyphenyl)propan-2-one and α-methylbenzylamine to (1S,2S)-3-(2-amino-1-hydroxypropyl)phenol and acetophenone at 20–35 °C, the PNAGA-immobilized BmTA exhibited a lower activity for the product formation than free BmTA. The rates of product formation were 20 and 32 μmol min−1 mg−1 at 20 and 35 °C, respectively, over free BmTA, whereas the rates of product formation were 12 and 28 μmol min−1 mg−1at 20 and 35 °C, respectively, over the PNAGA-immobilized BmTA. It is noteworthy that the swelling degree of crosslinked PNAGA is supposedly far from reaching the maximum at 35 °C, referring to the reported cases with chemically crosslinked PNAGA, where the swelling degree gradually increased with increasing temperature from 0 to at least 52–60 °C in acid buffer.45,109 This implies that the BmTA–PNAGA hydrogel is likely far from being ideally dispersed in water at 35 °C so that it fails to produce an activity similar or superior to that of free BmTA even if the native conformation of BmTA is intactly retained after the immobilization. Due to incomplete precipitation at room temperature, the BmTA–PNAGA hydrogel could not be satisfactorily recovered by filtration on cooling down after the reaction at 35 °C, which obstructed its recycling. Meanwhile, the BmTA–PNAGA hydrogel more or less underwent BmTA leaching during the reaction despite the hydrogen bonding between BmTA and PNAGA. These two issues led to the unsatisfactory reusability of the PNAGA-immobilized BmTA in the reaction at 35 °C and the rate dropping of product formation by 56 and 80% in the second and third cycles, respectively.
Copolymers
The vast majority of the UCST-type polymers reported for application in biocatalysis are copolymers.38–44,48–52 Generally, the phase transition properties of thermoresponsive polymers can be tuned by taking advantage of copolymerization. By using different comonomers and adjusting related synthesis parameters adapted to the variation of hydrogen bonding with temperature, the properties of UCST-type copolymers can be expectedly optimized. Meanwhile, the use of copolymers can improve the enzyme–polymer interaction to meet the needs of biocatalytic applications.Most of the UCST-type copolymers reported for application in biocatalysis belong to diblock copolymers.38,39,41–44,51,52 In 2013, Liu's group communicated the conjugation of different enzymes to Pluronic F-127 and biocatalysis of the resultant enzyme–Pluronic nanoconjugates.38 This communication marked the first case of the nano-sized UCST-type polymers for engineering enzyme biocatalysts. Pluronic F-127 is an amphiphilic nonionic diblock nano-sized copolymer consisting of a central hydrophobic block of poly(propylene glycol) flanked by two hydrophilic blocks of poly(ethylene glycol), i.e., EG100PG65EG100.110 It is the most studied UCST-type copolymer for application in drug delivery owing to its high stability, thermoresponsiveness at room temperature, bioadhesive characteristics and nontoxic properties.111 Its phase transition in water is abrupt on turbidity curves, showing a Tcp of 23 °C with 20% Pluronic F-127.112,113 The nanoconjugates were prepared by grafting aldehyde-functionalized Pluronic F-127 onto the enzymes via covalent linking. According to the electrophoretic and fluorometric analyses, high conjugation yields were achieved on all the enzymes, including bovine serum albumin (BSA), Candida rugosa lipase (CRL), Candida antarctic lipase B (CALB) and cytochrome c (Cyt C). In the enzymatic hydrolysis of p-nitrophenyl butyrate in aqueous solution at room temperature, the relative activities of the CRL-, CALB- and Cyt C-Pluronic conjugates to their respective free enzymes were found to be 58, 98 and 568%, respectively. Such results can rule out the possibility of mass transfer limitations between the nano-sized conjugates and the substrate in water. However, the higher relative activity of the conjugated Cyt C to free Cyt C was explained by the stronger substrate's affinity with the conjugated Cyt C with the assistance of the hydrophobic segments of Pluronic. This argument is unconvincing because p-nitrophenyl butyrate is highly hydrophilic, while Pluronic F-127 contains more hydrophilic segments than hydrophobic segments. Moreover, the relative activities in the cases of the CRL- and CALB-Pluronic conjugates are incompatible with that in the case of the Cyt C-Pluronic conjugate under the identical conjugation and reaction circumstances, assuming no conformational change of the enzymes upon conjugation.
In this work, the enzymatic activity investigation was focused on enzymatic reactions in organic solvents. Biocatalysis in organic solvents has been vitally pursued in that organic media are advantageous in dissolving substrates, inhibiting side reactions and preventing adverse effects of water. Usually, free enzymes are little active in organic solvents in that they have an extremely low solubility. The integration of enzymes with nanomaterials such as thermoresponsive polymers has offered a new opportunity to improve enzymatic performance in organic media. This communication included the earliest examples of thermoresponsive enzyme–polymer conjugates that exhibit higher activity than their native counterparts in organic solvents. At 40 °C, all the enzyme–Pluronic conjugates readily dissolved in most commonly used organic solvents, including toluene, tetrahydrofuran, methanol, dichloromethane and chloroform, in which the free enzymes remained as precipitates. Zhu et al. reported for the first time the thermoresponsiveness of Pluronic F-127 in organic solvents. According to the UV-vis absorbance measurements at 500 nm with temperature, Tcp appeared at around 12 °C with 10% Pluronic F-127 in toluene, while no UCST behaviour came about with 1% Pluronic F-127 in toluene. Nevertheless, the BSA-Pluronic conjugate as a representative nanoconjugate displayed a Tcp with a broad phase transition temperature interval of 23–43 °C with 1% conjugate in toluene. The dramatic appearance of Tcp is definitely attributed to the positive effect of BSA conjugation on the conjugate–conjugate interaction in toluene, indicating that the conjugate–conjugate interaction rather than the conjugate–toluene interaction dominates the ΔG of dissolution (which is positive) below the Tcp. Chemically grafting aldehyde-functionalized Pluronic onto BSA decreases the fraction of hydrophilic segments by 30%, i.e., increases the fraction of hydrophobic segments by 30%, unlike stated by the authors that the coupling of Pluronic to a protein introduced more hydrophilic parts into the conjugate. Hence, the enhanced conjugate–conjugate interaction is assumed to be between the hydrophobic segments of Pluronic in toluene. According to the dynamic light scattering measurements, the average particle size of the conjugates dissolved fell in the range of 27–42 nm at room temperature with 0.01% conjugate in toluene, which is in agreement with the TEM observations. Scheme 4 and Fig. 11 and 12 illustrate the on–off biocatalytic cycling and the biocatalytic performance of the enzyme–Pluronic conjugates in organic solvents, respectively. In the enzymatic esterification of palmitic acid and n-octanol in toluene at 40 °C, the CRL-Pluronic conjugate displayed an increased activity by 58-fold compared to free CRL. In the enzymatic esterification of hexanoic acid and n-butyl alcohol in toluene at 40 °C, the CALB-Pluronic conjugate gave rise to an increased activity by 68-fold compared to free CALB. In the enzymatic oxidation of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt by hydrogen peroxide in methanol at 40 °C, the peroxidase activity of Cyt C was 670-fold that of free Cyt C. The excellent activities can rule out the possibility of mass transfer limitations between the nano-sized conjugates and the substrates in organic solvents. By cycling the temperature between 4 and 40 °C with 1% conjugate in toluene, the CALB-Pluronic conjugate was successfully reused 9 times for the esterification of hexanoic acid and n-butyl alcohol, giving 98% of the initial activity after 10 cycles. The reusability test is essential evidence that the recovery of the CALB-Pluronic conjugate proceeds smoothly at 4 °C and that the Pluronic-immobilized CALB via covalent linking is quite stable under the reaction conditions.
 | ||
| Scheme 4 Dissolution of the enzyme–Pluronic conjugates in organic solvents for homogeneous biocatalysis at 40 °C, and precipitation of the enzyme–Pluronic conjugates in organic solvents at 4 °C for recycling. | ||
 | ||
| Fig. 11 Comparative activities of the enzyme–Pluronic conjugates and the free enzymes in the esterification of palmitic acid and n-octanol in organic solvents at 40 °C. | ||
 | ||
| Fig. 12 Recycling of the CALB-Pluronic conjugate via temperature switching in the esterification of palmitic acid and n-octanol in toluene at 40 °C. The relative activity was expressed using the initial one as the reference. | ||
The development of UCST-type diblock copolymers in immobilized enzyme biocatalysis was also explored by several other groups.39,41–44,51,52 Wang's group developed reversibly soluble–insoluble biocatalysts for hydrolysis of celluloses by immobilizing cellulases onto a diblock copolymer of methacrylamide (MAA) and AAc, i.e., PMAAc.43 The PMAAc was synthesized by copolymerization of MAA and AAc using ammonium persulfate as the initiator at 60 °C (Scheme 5). The cellulase-PMAAc conjugate was made by grafting aldehyde-functionalized PMAAc onto a mixture of endo-β-1,4-glucanase, cellobiohydrolase and β-GS via covalent binding (Scheme 5). The PMAAc and the cellulase-PMAAc conjugate in phosphate buffers exhibited their Tcp values of 16 and 19 °C, respectively. As expected, the conjugation led to a rise in the Tcp, likely because of a higher fraction of hydrophobes produced in the conjugate. In the enzymatic hydrolysis of carboxymethylcellulose sodium, the cellulase-PMAAc conjugate retained 93% of the activity of the free cellulase. The slightly lower activity suggested the existence of less mass transfer limitations caused by the binding copolymer. The cellulase-PMAAc conjugate obviously improved the enzymatic stabilities against pH and temperature: the relative activity of 86–100% was retained at pH 4.0–8.0 and 50 °C, and the relative activity of 86–100% was maintained at pH 5.0 and 30–80 °C, as shown in Fig. 13. The results seem to uncover the stabilization of the PMAAc on the biocatalytic properties against pH and temperature. The conjugate meanwhile displayed a much higher enzyme thermal stability than the free cellulase at 70 °C, as shown in Fig. 14(b) and (c). All these improved stabilities may be attributed to the strong interaction of the cellulase with PMAAc by covalent binding. In biocatalyst reusability, the conjugate was recycled 9 times in the reaction at 50 °C, retaining 82% of the initial activity after 10 cycles, as illustrated in Fig. 14(d). The biocatalyst recovery proceeded smoothly by cooling to 5 °C, followed by centrifugation at 5 °C after each run. In the enzymatic hydrolysis of water-insoluble microcrystalline cellulose at 50 °C, the enzyme-PMAAc conjugate was slightly more active than the free enzyme. Under the optimized reaction conditions, the β-GS-supplemented cellulase-PMAAc (cellu&β-GS-PMAAc) conjugate was 1.9 times more active than the cellulase-PMAAc conjugate, the respective yields of glucose being 89 and 31% after 24 h of reaction. The cellu&β-GS-PMAAc conjugate was reused 7 times, retaining 61% of the initial activity after 8 cycles, as shown in Fig. 15. The biocatalyst recovery was operated by cooling to 5 °C followed by centrifugation at 5 °C after each run.
 | ||
| Scheme 5 Synthesis strategy for UCST-type PMAAc and immobilization of cellulase on PMAAc. Reprinted with permission from ref. 43, copyright 2018, American Chemical Society. | ||
 | ||
| Fig. 13 Relative activities in the hydrolysis of carboxymethylcellulose sodium versus (a) pH and (b) temperature. Reprinted with permission from ref. 43, copyright 2018, American Chemical Society. | ||
 | ||
| Fig. 14 Relative activities in the hydrolysis of carboxymethylcellulose sodium versus (a) incubation pH at 50 °C, (b) incubation time at 70 °C, (c) incubation days at 70 °C and (d) cycles at 50 °C (for PMAAc-cellulase). Reprinted with permission from ref. 43, copyright 2018, American Chemical Society. | ||
 | ||
| Fig. 15 Recycling of PMAAc-cellu&β-G in the hydrolysis of microcrystalline cellulose at 50 °C. Reprinted with permission from ref. 43, copyright 2018, American Chemical Society. | ||
This group lastly utilized VBA as an affinity comonomer with horseradish peroxidase (HRP) to make nano-sized P(NAGA-co-VBA)-immobilized HRP for use in phenol degradation by hydrogen peroxide.52 The diblock P(NAGA-co-VBA) was synthesized by RAFT polymerization. HRP was immobilized onto P(NAGA-co-VBA) by the “grafting to” approach without a grafting linker. In terms of the UV-vis transmittance measurements, the influence of stimulating factors such as polymerization degree, anion and urea on the phase transition of P(NAGA-co-VBA) implied that the presence of PVBA in the copolymer increased the Tcp of PNAGA, the presence of kosmotrope anions increased the Tcp of P(NAGA-co-VBA), and the presence of chaotrope anions or urea decreased the Tcp of P(NAGA-co-VBA). The increase in the Tcp of PNAGA by the presence of the PVBA block may give rise to higher precipitation of P(NAGA-co-VBA) below the Tcp. The representative P(NAGA-co-VBA) with an NAGA: VBA molar ratio of 200:5 at 1% concentration had a Tcp of 44 °C. Its particle size varied from 29 nm below the Tcp to 46 nm above the Tcp in terms of the TEM analysis. The corresponding HRP-P(NAGA-co-VBA) conjugate had an increased Tcp to 60 °C, as expected. The recovery rate of the conjugate was 92% after one dissolution–precipitation cycle at pH 8.0, implying that the precipitation of the conjugate is far from being complete below the Tcp. In the enzymatic phenol degradation by hydrogen peroxide, the conjugate displayed 108% of the activity of free HRP in terms of Vm/Km, which assumed the existence of no mass transfer limitations between the conjugate and the substrate. The conjugate slightly increased the enzymatic stabilities against the pH and temperature compared to free HRP: the relative activity of 82–100% was retained at pH 6.0–8.0 and 60 °C, and the relative activity of 87–100% was maintained at pH 7.0 and 30–70 °C. The conjugate also slightly enhanced the enzyme's thermal stability relative to free HRP. All these improved stabilities may be attributed to the strong interaction of HRP with P(NAGA-co-VBA) by covalent binding. Concerning the biocatalyst recyclability test, the reaction was run in phosphate buffer at pH 7.0, phenol: hydrogen peroxide molar ratio of 1:1 and 50 °C for 10 min. After each run, the reaction solution was cooled to 4 °C and kept at 4 °C for 20 min. Then, the conjugate was separated via centrifugation at 4 °C. Based on the biocatalyst recycling experiments illustrated in Fig. 16, the activity of the conjugate declined with cycles and with 78% of the initial activity after 5 cycles, coinciding with the recovery rates of the conjugate after both each and the 5 cycles. The coincidence of the activity with the recovery rate of the conjugate strongly suggests that the conjugate undergoes no HRP leaching during the reaction process as a consequence of the strong VBA affinity with HRP. The notable decline in the activity after each cycle most likely results from the incomplete precipitation of the conjugate at 4 °C and loss of the conjugate nanoparticles due to the trouble in separation and recovery. The presence of a second block in a UCST-type copolymer may tune the phase transition properties and modify the interaction with enzymes on the one hand but may reduce the precipitation of the copolymer and the conjugates below the Tcp values on the other hand.39 The reduction in the precipitation unfavours the recovery of the conjugates for recycling. Without comparisons between HRP-PNAGA and HRP-P(NAGA-co-VBA) in biocatalytic performance and biocatalyst reusability, the advantage of P(NAGA-co-VBA) as a copolymer in the engineering of HRP for biocatalysis remains questionable.
 | ||
| Fig. 16 Recycling and recovery rate of HRP-P(NAGA-co-VBA) for phenol degradation by hydrogen peroxide at 50 °C and pH 7.0. Reprinted with permission from ref. 52, copyright 2023, Elsevier Inc. | ||
Zhu et al. applied a diblock copolymer of methacrylic acid (MA) and AAc, i.e., PMAc, to a cascade biocatalytic process of hydrolysis of insoluble starch wastes.51 The PMAc was first synthesized by copolymerization of MA and AAc using ammonium persulfate as the initiator at 60 °C and was subsequently mixed with gelatin under acidic conditions to become a UCST-type copolymer system, as depicted in Scheme 6. The enzyme-PMAc conjugate was prepared by grafting glycidyl methacrylate (GMA)-modified PMAc onto α-amylase or/and glucoamylase via covalent binding. In UV transmittance at 300 nm with temperature, the PMAc in acetic acid buffer exhibited a broad phase transition temperature interval from 8 to 40 °C. The conjugation of the enzymes slightly lowered the Tcp, which is indicative of the production of slightly more hydrophiles after the immobilization of the enzymes containing rich hydroxyl groups on the PMAc. In the enzymatic cascade hydrolysis of insoluble starch wastes to glucose, the PMAc-conjugated coenzymes mixed with α-amylase and glucoamylase with a weight ratio of 1:3 as the best biocatalyst displayed much higher activity than the PMAc-conjugated α-amylase and the PMAc-conjugated glucoamylase, as well as slightly higher activity than the free coenzymes. Such a slightly higher retained activity can be ascribed to the fact that there are no mass transfer limitations between the PMAc-conjugated coenzymes and the substrate. After 24 h of reaction at 50 °C, a glucose yield of 88% was obtained over the PMAc-conjugated coenzymes, whereas glucose yields of 26 and 36% were produced over the PMAc-conjugated α-amylase and the PMAc-conjugated glucoamylase, respectively. The PMAc-conjugated coenzymes showed larger activity movements against pH and temperature: the relative activity changed from 62 to 100% at pH 4.0–6.5 and 50 °C, and the relative activity changed from 34 to 100% at pH 5.0 and 35–60 °C. The results hardly account for the stabilization of PMAc on the biocatalytic properties against pH and temperature. In biocatalyst reusability, the PMAc-conjugated coenzymes were recycled 9 times with 71% of the initial activity after 10 cycles in the reaction at 50 °C. The efficiency of the PMAc-conjugated coenzymes in enzymatic hydrolysis is of economic significance for the utilization of insoluble starch wastes.
 | ||
| Scheme 6 Synthesis strategy of a UCST-type PMAA-co-PAA (i.e., PMAc) system. Reprinted with permission from ref. 51, copyright 2023 Elsevier Inc. | ||
Cummings et al. engineered nano-sized UCST- and LCST-type bifunctional diblock copolymer-conjugated CT for hydrolysis of Suc-AAPF-pNA.39 The conjugated CT was fabricated by the “grafting from” approach. Following the synthesis of the CT-Cl macroinitiator, poly(sulfobetaine methacrylamide), i.e., PSBAM, was first grown from the initiator-modified CT. Then, PNIPAM was grown from CT-PSBAM using chain extension to yield CT-PSBAM-block-PNIPAM conjugates. The hydrodynamic diameters of the resultant conjugate nanoparticles fell in the range of 46 to 64 nm. The conjugates displayed both UCST and LCST behaviour on turbidity curves, as in Fig. 17. As expected, the UCST-type phase transition was sensitively dependent on the polymer chain length while the LCST-type phase transition was almost independent of the polymer chain length. The presence of PSBAM in the copolymer resulted in a slightly broadened LCST-type phase transition temperature interval and almost unchanged Tcp relative to the phase transition of CT-PNIPAM,37 whereas the presence of PNIPAM in the copolymer caused severely broadened UCST-type phase transition temperature interval and decreased Tcp by 9–17 °C compared to the phase transition of CT-PSBAM.37 In the meantime, the turbidities of the CT-PSBAM-block-PNIPAM solutions were much lower than that of a CT-PSBAM or CT-PNIPAM solution.37 It follows that CT-PSBAM below its Tcp in solution and CT-PNIPAM above its Tcp in solution considerably precipitate, whereas CT-PSBAM-block-PNIPAM in solution finitely precipitates. This may be explained by the assumption that the PNIPAM block located on the outside of the CT conjugates sterically hinders PSBAM association among the CT-PSBAM-block-PNIPAM molecules. In the enzymatic hydrolysis of Suc-AAPF-pNA, the CT-PSBAM-block-PNIPAM conjugates showed significantly increased thermal stability at pH 8.0 and 37 °C and notably increased acid stability at pH 1.0 and 37 °C compared to free CT. The increased stabilities may be attributed to the strong interaction of CT with PSBAM-block-PNIPAM by covalent bonding. The CT-PSBAM-block-PNIPAM conjugates exhibited Km similar to that of free CT and a lower reaction rate constant (kcat) than that of free CT at 25 °C. Overall, the former had lower activity than the latter to a different extent in terms of kcat/Km at 25 °C, likely due to steric hindrance from the copolymer towards the substrate. While the CT-PSBAM-block-PNIPAM conjugates with both UCST and LCST behaviour provide lower Tcp of PSBAM, largely dissolved PNIPAM amount above the Tcp of PNIPAM and good thermal and acid stabilities, they produce finite precipitation of PSBAM below the Tcp of PSBAM. The finite precipitation of PSBAM undoubtedly impedes the biocatalyst recovery upon cooling for recycling. To increase the biocatalyst performance and reusability, the solubility of CT-PSBAM-block-PNIPAM should be decreased below the Tcp of PSBAM and should be increased above the Tcp of PNIPAM. To this end, the relative amount of PSBAM to PNIPAM is recommended to be increased to an optimum. To evaluate the value and advantage of CT-PSBAM-block-PNIPAM, it is recommended to compare the biocatalyst performance and reusabilities of CT-PSBAM, CT-PNIPAM and CT-PSBAM-block-PNIPAM.
 | ||
| Fig. 17 Turbidity curves of CT-PSBAM-block-PNIPAM with temperature variations (CT-35/39-open diamond, CT-50/67-closed square, CT90/100-closed circle). Each conjugate of 3 mg mL−1 was incubated in 0.1 M sodium phosphate buffer at pH 8.0 and heated or cooled at 0.5 °C min−1. Reprinted with permission from ref. 39, copyright 2014, American Chemical Society. | ||
Chado et al. investigated the modification of Bacillus subtilis lipase A (LipA) with poly(acryloylmorpholine (AcMO)-co-NIPAM), i.e., PAN, to tune the molecular interaction of the enzyme in diverse solvent environments for the purpose of effective biocatalysis.41 The diblock PAN was synthesized by copolymerization of AcMO and NIPAAM via the reversible addition-fragmentation chain-transfer (RAFT) approach. The LipA-PAN conjugates were prepared by reacting the terminal N-hydroxysuccinimide ester on the copolymer with free amines on the surface of LipA in the buffer. The incorporation of NIPAAM repeat units into a PAcMO polymer imparts UCST behaviour to the lipase-polymer conjugates. The measurements of optical transmittance at 658 nm with temperature indicated that the UCST-type LipA-PAN conjugates had Tcp values of 10–70 °C in [BMIM][PF6] and of 16–76 °C in [BMIM][PF6] containing 1 M ethanol with the molar fractions of NIPAAM in PAN ranging from 0.58 to 0.86. The Tcp was linearly dependent on the molar fraction of NIPAAM in the LipA-PAN conjugates over this molar fraction range. As such, the Tcp is sensitively dependent on the composition and molecular weight of PAN. At the same time that the transmittance values at 658 nm of the conjugates both in [BMIM][PF6] and [BMIM][PF6] containing 1 M ethanol above the Tcp values were 85% and above, those below the Tcp values were superior to 0. This implies that the conjugates almost completely dissolve above the Tcp values, whereas they incompletely precipitate below the Tcp values. High dissolution will favour the activity of the conjugates, whereas incomplete precipitation will affect the recovery and recycling of the conjugates when the conjugates serve as biocatalysts. In the enzymatic transesterification of nitrophenyl butyrate with ethanol in [BMIM][PF6], the conjugates were cycled between 40 and 4 °C. Typically, in the case of LipA-PAN61 with a molar fraction of NIPAAM in PAN at 0.61, the conjugate precipitated on cooling at 4 °C after the reaction at 40 °C. The dissolution and precipitation process was reversible for at least 10 cycles. At 40 °C, the activity of LipA-PAN61 was 21-fold greater than that of free LipA, being nearly identical to that of LipA-poly(AcMO). Such a high activity can rule out the possibility of mass transfer limitations between LipA-PAN61 and the substrate. After precipitation at 4 °C, the residual soluble conjugate exhibited 7% of the activity of the whole conjugate, indicative of a noticeable loss of the conjugate upon recovery due to the incomplete precipitation. The result is consistent with the transmittance values superior to 0 at 658 nm at 4 °C during the 10 cycles, which poses a noticeable issue of activity loss for biocatalyst recycling.
Lou et al. published preliminary research on pseudomonas cepacia lipase (PSL) immobilized onto poly(AAm-co-acrylonitrile (AN)), i.e., PAA, for biocatalysis.42 This work presented the first application of UCST-type polymers in the immobilization of enzymes for asymmetric biocatalysis. The diblock PAA was synthesized via copolymerization of AAm and AN with a molar ratio of 82:18 using 2,2-azobisisobutyronitrile as the initiator. PSL was covalently immobilized onto PAA by the “grafting to” approach using glutaraldehyde (GA) as the grafting linker. The turbidity observations of 0.5% PAA and 0.5% PAA-PSL in phosphate buffers gave their Tcp values of 26 and 22 °C, respectively. The decreased Tcp is accounted for by an increased fraction of hydrophiles on the PAA-PSL conjugate, as expected. In the enzymatic hydrolysis of tributyrin, the PAA-PSL conjugate obviously improved the enzymatic stabilities against pH and temperature: the relative activity of 89–100% was retained at pH 6.0–8.0 and 55 °C, and the relative activity of 88–100% was maintained at pH 7.0 and 40–65 °C. The results further show the stabilization of UCST-type copolymers on the biocatalytic properties against pH and temperature. In the meantime, the PAA-PSL conjugate notably enhanced the enzyme thermal stability towards the reaction at 45 °C, following incubation in the phosphate buffer at 60 °C as a result of the covalent immobilization of PSL, as shown in Fig. 18. In the enzymatic kinetic resolution of (R,S)-α-methylbenzyl butyrate to (R)-1-phenylethanol in phosphate buffer, the PAA-PSL conjugate generally produced a slightly higher substrate conversion than free PSL with about the same enantiomeric excess of the target product. With 0.25 M substrate at pH 7.0 and 55 °C, 15 h of reaction gave conversions of 22 and 17% over the PAA-PSL conjugate and over free PSL, respectively, with the same enantiomeric excess of the target product at 99%. Such a highly retained activity can be attributed to the fact that there are no mass transfer limitations between the PAA-PSL conjugate and the substrate. Regarding biocatalyst reusability, the reaction was carried out with 0.05 M substrate at pH 7.0 and 55 °C for 12 h. After each run, the biocatalyst was easily recovered upon cooling to 0 °C. The Bradford assay indicated that there was no detectable PSL leaching out of the PAA-PSL conjugate. According to the biocatalyst recycling experiments depicted in Fig. 19, the PAA-PSL conjugate could maintain 81% of the initial activity after 6 cycles. This good biocatalyst recyclability is attributed to the good enzymatic stability of the covalently immobilized PSL on PAA. PAA can be viewed as a promising UCST-type polymer for use in enzyme immobilization for biocatalysis.
 | ||
| Fig. 18 Thermal stabilities of free PSL and the PAA-PSL conjugate in the hydrolysis of tributyrin at 45 °C, following the incubation at 60 °C. | ||
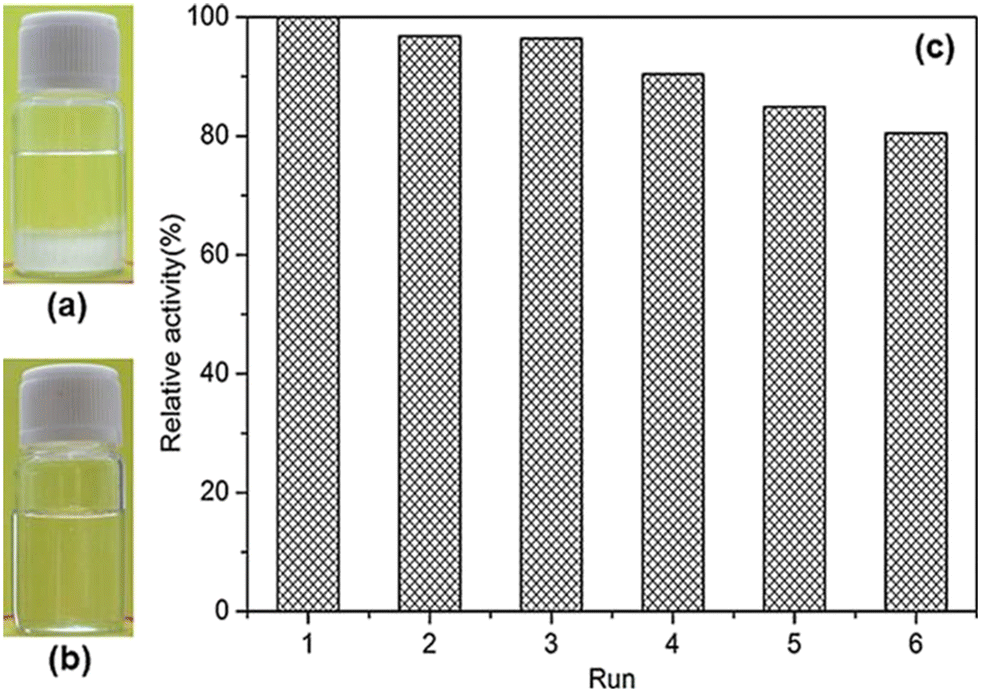 | ||
| Fig. 19 Photographs of the solutions of the kinetic resolution of (R,S)-α-methylbenzyl butyrate to (R)-1-phenylethanol over PAA-PSL at pH 7.0 (a) after cooling to 0 °C and (b) under a new cycle at 55 °C (b). (c) Recycling PAA-PSL in the kinetic resolution of (R,S)-α-methylbenzyl butyrate to (R)-1-phenylethanol at 55 °C and pH 7.0. Reprinted with permission from ref. 42, copyright 2018, John Wiley and Sons. | ||
Besides, the application of UCST-type polymers was integrated with the optical control technique of enzymes.44 Zhang et al. developed an off–on manipulation of the activity of enzyme–polymer conjugates for hydrolysis of biochemical and chemical substrates by near-infrared light.44 They fabricated Pt nanoparticle-embedded enzymes (E/Pt) immobilized onto diblock PAA by the “grafting from” approach. In the preparation of such an immobilized enzyme PAA-E/Pt, E/Pt was first synthesized inside the enzyme via reduction of H2PtCl4 with NaBH4 using the enzyme as a template, followed by modification with N-acryloxysuccinimide (NAS). Then, PAA-E/Pt was obtained on NAS-E/Pt via copolymerization of AAm and AN. It is supposed that below the Tcp, PAA-E/Pt is insoluble as micro aggregates in an aqueous solution, which prevents a soluble macromolecular substrate from approaching the insoluble immobilized enzyme; and upon near-infrared irradiation, the Pt nanoparticles generate heat through a photothermal effect and thus increase the local temperature to above the Tcp, so that the copolymer becomes soluble and the enzyme–copolymer aggregates disassemble to activate the immobilized enzyme. The enzymes included glucoamylase (GluA), proteinase K (ProK) and deoxyribonuclease 1(DNase 1). The UV-vis transmittance measurements at 600 nm with temperature showed a Tcp of PAA-E/Pt at 16–47 °C, consistent with the TEM study. The fluctuation of Tcp of PAA-E/Pt relies on the type of enzyme, content of the enzyme, molar ratio of AN:AAm and molecular weight of PAA. The sensitivity increase in the Tcp of PAA-E/Pt with increasing molar ratio of AN:AAm and with increasing molecular weight of PAA is in accordance with the Tcp variation (from 5.5 to 57 °C) in the case of PAA.67,106 The increased Tcp of PAA with increasing molar ratio of AN:AAm may be ascribed to the weakening of hydrogen bonding between PAA and water due to the presence of more hydroneutral AN groups in the PAA chain. The increased Tcp of PAA leads to higher precipitation of PAA.106 In the enzymatic hydrolyses of starch at 45 °C over the GluA systems, casein over the ProK systems and plasmid over the (DNase 1) systems, E/Pt and NAS-E/Pt produced slightly decreased activity while PAA-E/Pt gave rise to tremendously decreased activity (by 60–70%) compared to that of E. The slight decrease in the retained activity probably arises from less mass transfer limitations between E/Pt or NAS-E/Pt and the substrate, while the large decrease in the retained activity is probably due to more mass transfer limitations between PAA-E/Pt and the substrate. Despite the severe fall in the activity with the uncertainty of whether the conjugated enzymes changed their native conformations, PAA-GluA/Pt and PAA-(DNase 1)/Pt could retain their 90% initial activities after 48 h of hydrolysis of papain. In contrast, GluA and DNase 1 almost lost their initial activities under the equivalent reaction conditions. The comparative data showed enhanced stability of the enzymes after the conjugation. It was verified that the off–on switching of the enzyme activities of PAA-GluA/Pt, PAA-ProK/Pt and PAA-(DNase 1)/Pt is able to proceed reversibly between 25 and 45 °C through the photothermal effect to cause the phase transitions of these PAA-immobilized enzyme biocatalysts by near-infrared irradiation. The enzyme activity of PAA-E/Pt when on was increased by 50–61 fold compared to that of PAA-E/Pt when off. In biocatalyst reusability, the dissolution and precipitation process of the conjugate was reversible for at least 5 cycles. After 6 cycles, 68% of the initial activity was maintained. The loss of activity may be due to the incomplete precipitation of the conjugate at 4 °C and GluA leaching out of the conjugate.
Apart from the above UCST-type diblock copolymers, 3 UCST-type triblock copolymers were reportedly applied in the engineering of enzymes for biocatalysis.48–50 Wang's group synthesized triblock PEG-b-PAA and utilized this series of copolymers to immobilize crude β-GS for use in hydrolysis of a cellobiose.48 PEG-b-PAA was prepared by RAFT polymerization. The PEG113-based macroRAFT agent was first synthesized from the esterification of 3-(benzylthiocarbonothioyl-thio) propanoic acid and PEG113-OH. Then, AAm and AN were copolymerized in the presence of the PEG113-based macroRAFT agent. The PEG-b-PAA-conjugated crude β-GS, (LytA-Glu)-(PEG-b-PAA) was prepared by the “grafting to” approach using GA as the grafting linker via covalent binding. The Tcp of PEG-b-PAA sensitively changed with AN fraction in the copolymer, increasing from 14 to 48 °C with increasing AN fraction from 0.21 to 0.28. This behaviour is analogous to that of PAA.67,106 Meanwhile, the Tcp of PEG-b-PAA notably increased in the presence of SO42− and notably decreased in the presence of SCN− or urea, indicative of the stimulating influences of the kosmotrope and chaotrope anions and urea on the phase transition of PEG-b-PAA. As expected, the immobilization of LytA-Glu increased the Tcp of PEG-b-PAA as a consequence of an increased fraction of hydrophobes. In the enzymatic hydrolysis of the cellobiose, (LytA-Glu)-(PEG-b-PAA) generally improved the enzymatic stabilities against pH and temperature compared to free LytA-Glu, above all at higher temperatures. Meanwhile, the conjugate strongly enhanced the thermal stability of the enzyme relative to free LytA-Glu at higher temperatures. However, the conjugate retained only ca. 63% of the activity of free LytA-Glu, consistent with slightly higher Km, and much lower Vm and kcat/Km. The slightly higher Km is as expected because of a decreased cellobiose affinity with the conjugate as a result of the decreased fraction of hydrophiles on the conjugate after the cellobiase immobilization. The much lower Vm and kcat/Km are presumably due mostly to the reduction of the enzyme's conformational flexibility after the immobilization and/or to steric hindrance from the copolymer towards the substrate. With regards to biocatalyst recyclability, (LytA-Glu)-(PEG-b-PAA) with an AN fraction of 0.29 was investigated as the selected conjugate in the hydrolysis of the cellobiose in phosphate buffer at 50 °C. After 10 min of reaction, the reaction solution was cooled down to 4 °C followed by centrifugation at 4 °C. The conjugate was separated from the solution for recycling. The activity of the conjugate declined with each cycle and with 51% of the initial activity after 10 cycles. The marked decline in the activity after each cycle probably arises from incomplete precipitation of the conjugate at 4 °C. The presence of more blocks in a UCST-type copolymer may readily reduce the precipitation of the copolymer and the conjugates below the Tcp values.
On the basis of PEG-b-PAA, Wang's group synthesized triblock PVBA-b-PAA, i.e., PVAA, and applied this series of copolymers to immobilize glucose isomerase (GI) for biocatalysis of isomerization of glucose to fructose.49 To attempt to make up a decrease in the conjugate's affinity with the substrate, VBA was introduced as an affinity comonomer in the synthesis of the triblock copolymer. PVAA was synthesized by RAFT copolymerization of VBA, AAm and AN. GI was immobilized onto PVAA by the “grafting to” approach using GA as the grafting linker. The turbidity measurements of 1.0% PVAA solutions gave phase transition temperature intervals of 24 °C with Tcp values, which sensitively increased from 19 to 23 °C, with increasing molecular weight from 9312 to 10128. The immobilization of GI led to an increased Tcp of PVAA, as expected. In the enzymatic isomerization of glucose to fructose for the production of high-fructose syrup, GI-PVAA slightly increased the enzymatic stability against pH at pH 5–10 and the enzyme thermal stability at 50–100 °C relative to free GI. The slight increase in the enzyme thermal stability was consistent with the thermal denaturation kinetic constants, half-life values and ΔH values of GI-PVAA and free GI at the higher temperatures. Although the presence of PVBA was expected to enhance the conjugate's affinity with glucose and thus the biocatalytic performance, the conjugate gained slightly higher Km, and significantly lower Vm and kcat/Km than free GI, which was consistent with a ca. 72% retained activity. The lower retained activity is probably due to more mass transfer limitations between GI-PVAA and the substrate. With the lack of a comparative study on GI-PVAA and GI-PAA, the claim that the conjugate's affinity with glucose and biocatalytic efficiency could be improved by introducing PVBA is unconvincing. Regarding biocatalyst reusability, GI-PVAA with a Tcp of 46 °C was investigated as the selected conjugate in the isomerization of glucose to fructose at pH 7.0 and ca. 75 °C. After each run, the conjugate was separated via low-temperature centrifugation before recycling. The activity of the conjugate declined with each cycle and with 65% of the initial activity after 10 cycles. The marked decline in the activity after each cycle probably arises from incomplete precipitation of the conjugate at low temperatures. Like in the case of PEG-b-PAA,47 the presence of more blocks in a UCST-type copolymer may readily reduce the precipitation of the copolymer and the conjugates below the Tcp values.
Sun et al. designed nano-sized triblock copolymers using AAm and NAS as comonomers that enabled both covalent binding and hydrogen bonding with enzymes such as BSA and ovalbumin (OVA) in favour of the facile formation of enzyme–copolymer hybrids for applications in biocatalysis and biomedicine.50 P(AAm-co-NAS-co-AAc) was synthesized by RAFT copolymerization from AAm and NAS, taking advantage of in situ partial hydrolysis of NAS to AAc. The enzyme-P(AAm-co-NAS-co-AAc) nanoparticles were obtained by simply mixing a P(AAm-co-NAS-co-AAc) solution with BSA or OVA and incubating at 25–50 °C. From the turbidity measurements of 0.1% P(AAm-co-NAS-co-AAc) solutions, the Tcp increased with increasing NAS fraction, being tunable from 12 to 81 °C over an NAS fraction range from 1.5 to 7.3 mol%. The Förster resonance energy transfer study suggested that both the covalent bonds and the hydrogen bonds contribute to the enzyme–copolymer hybridization in the enzyme-P(AAm-co-NAS-co-AAc): the covalent bonding makes a strong chemical linkage between the enzyme and the copolymer, while the hydrogen bonding produces a loose enzyme–copolymer complex. The circular dichroism study demonstrated that such a BSA-copolymer coassembly process does not alter the thermal stability of BSA. Towards the enzymatic hydrolysis of p-nitrophenyl acetate, BSA-P(AAm-co-NAS-co-AAc) could retain 95% or above of the activity of free BSA, assuming the occurrence of less mass transfer limitations between BSA-P(AAm-co-NAS-co-AAc) and the substrate. However, no information on the enzyme leaching, phase transition, recovery and recyclability of the enzyme-P(AAm-co-NAS-co-AAc) was reported. Thus, the biocatalytic efficiency and stability of the enzyme-P(AAm-co-NAS-co-AAc) obtained by coassembly remains unclear.
Li′s group successfully synthesized nano-sized P(AAm-co-AAc) and developed good performance P(AAm-co-AAc)-immobilized cellulase and cellobiase nanoparticle biocatalysts for hydrolysis of insoluble celluloses such as filter paper and pretreated oil palm empty fruit bunch to glucose.40 The nano-sized P(AAm-co-AAc), which was categorized as an alternating copolymer,40 was synthesized by controlling the copolymerization between AAm and AAc using lower comonomer concentration, shorter reaction time, lower reaction temperature, alternative initiator and no crosslinker. It was produced in 90% yield after 1 h of the reaction of AAm of 0.01 g mL−1 and AAc of 0.02 g mL−1 using ammonium persulfate of 0.6 mg mL−1 as the initiator in water at 60 °C. The nanoparticles had a mean size of 107 nm and hydrodynamic size of 120 nm. The enzymes, including a cellulase (consisting of cellobiohydrolase, endoglucanase and cellobiase at a ratio of 20:80:1) and cellobiase (i.e., β-GS), were covalently immobilized onto the P(AAm-co-AAc) by the “grafting to” approach using GMA as the grafting linker, as illustrated in Scheme 7. Functionalizing the P(AAm-co-AAc) nanoparticles with GMA gave an interpenetrating polymer network of PGMA-P(AAm-co-AAc) (i.e., IPN PGA) nanoparticles with a yield of 71% (Scheme 7). The nanoparticles had a mean size of 109 nm and hydrodynamic size of 122 nm. Reacting the IPN PGA nanoparticles with the cellulase or cellobiase resulted in IPN PGA-immobilized cellulase (IPN-Cellu) or cellobiase (IPN-Cello) nanoparticles with a yield of 100% (Scheme 7). Either the IPN-Cellu or IPN-Cello nanoparticles had a mean size of 120 nm. In UV-vis transmittance at 500 nm with varying temperature, 0.5% (w/v), the P(AAm-co-AAc), the IPN PGA, the IPN-Cellu and the IPN-Cello in citric acid buffers with pH 3.0 exhibited steep phase transition temperature intervals with their Tcp values of 9, 14, 13 and 14 °C, respectively, as in Fig. 20(c) and 21(c). The phase transition behaviour and Tcp of the 0.5% (w/v) nano-sized P(AAm-co-AAc) solution are similar to those of a recently reported 4.0% micro-sized P(AAm-co-AAc) solution with an AAm:AAc molar ratio of 0.0091.114 The rise in Tcp with the IPN PGA relative to that of the P(AAm-co-AAc) implies the rise in the fraction of hydrophobes after the functionalization of P(AAm-co-AAc) with GMA. The slight fall in the Tcp with the IPN-Cellu relative to that of the IPN PGA likely results from the slight rise in the fraction of hydrophiles after the immobilization of cellulase. No change in Tcp with the IPN-Cello relative to that of the IPN PGA may be explained by the fact that there was no change in the fraction of hydrophiles after the immobilization of cellobiase.
 | ||
| Scheme 7 Synthesis of P(AAm-co-AAc), IPN PGA and IPN-enzyme nanoparticles. | ||
 | ||
| Fig. 20 (a) TEM image of P(AAm-co-AAc) nanoparticles. (b) Photographs of P(AAm-co-AAc) nanoparticles in water (29 mg mL−1) at (1) 25 °C and (2) 4 °C for 5 min. (c) UV transmittance transitions of the P(AAm-co-AAc) nanoparticles (0.5% (w/v)) and IPN PGA nanoparticles (0.5% (w/v)) in citric acid buffers (50 mM, pH 3) with temperature. (d) TEM image of the IPN PGA nanoparticles. | ||
 | ||
| Fig. 21 (a) TEM image of IPN-Cellu nanoparticles. (b) TEM image of IPN-Cello nanoparticles. (c) UV transmittance transitions of IPN-Cellu nanoparticles (0.5% (w/v)) and IPN-Cello nanoparticles (0.5% (w/v)) in citric acid buffers (50 mM, pH 3) with temperature. (d) Photographs of the mixture of IPN-Cellu nanoparticles (5.3 mg mL−1) and IPN-Cello nanoparticles (11 mg mL−1) in citric acid buffer (1) at 25 °C for 5 min, (2) at 4 °C for 5 min and (3) after centrifugation at 4 °C. | ||
In the enzymatic hydrolyses of carboxymethylcellullose as water-soluble substrate and filter paper as water-insoluble substrate at 50 °C, the IPN-Cellu could retain 90 and 86% activities of the free cellulase, respectively. In the enzymatic hydrolysis of cellobiose as a slightly water-soluble substrate at 50 °C, the IPN-Cello could retain a 78% activity of free cellobiase. The retained activities of 78–90% over the soluble nano-sized IPN-Cellu or IPN-Cello implies that there more or less exist mass transfer limitations between the conjugate and the substrate during the hydrolyses.
Li's group focused their work on the recyclability of the nano-sized IPN-Cellu and the nano-sized IPN-Cello towards the hydrolyses of the soluble and insoluble celluloses.40 The reactions were carried out at 50 °C. After each run, the insoluble substrate was removed by centrifugation at room temperature, and the supernatant was cooled down to 4 °C and was subjected to centrifugation at 4 °C to recover the conjugate for recycling. The supernatant was also used for product analysis. No enzyme leaching from the conjugates in each cycle was detected by the Bradford assay, demonstrating a strong attachment of the enzymes to the nanocarrier. In the hydrolyses of carboxymethylcellullose, filter paper and pretreated oil palm empty fruit bunch, the IPN-Cellu with 1% cellulase relative to the substrate was able to produce glucose yields of 56, 42 and 31%, respectively. For the hydrolysis of carboxymethylcellullose, 72% of the initial activity remained after 10 cycles, which represents one of the prominent recyclability results of immobilized cellulases.43,115–118 For the hydrolysis of pretreated oil palm empty fruit bunch, 70% of the initial activity remained after 10 cycles, which shows a strong biocatalyst recyclability. Due to the very low fraction of cellobiase in the cellulase used restricting the conversion efficiency of cellobiose (as the intermediate sugar) to glucose, a mixture of the IPN-Cellu and the IPN-Cello was investigated as a mixed biocatalyst for increasing the glucose productivity from the hydrolysis of celluloses. As a result, the addition of IPN-Cello obviously increased the glucose yields in the hydrolyses of filter paper and pretreated oil palm empty fruit bunch. The mixture of IPN-Cellu and the IPN-Cello with a weight ratio of 1:2 was able to produce glucose yields of 97 and 93% in the hydrolyses of filter paper and pretreated oil palm empty fruit bunch, respectively. This work presented unprecedented high glucose yields from the hydrolysis of lignocelluloses with immobilized enzymes. For the former reaction, 71% of the initial activity was retained after 8 cycles. For the latter reaction, 73% of the initial activity was retained after 6 cycles. Such biocatalyst recyclabilities are good enough compared to the reported strong recyclabilities of immobilized cellulases and cellobiase for hydrolysis of lignocelluloses.43,119,120 The decline in the activities of the conjugates with each cycle probably arises from incomplete precipitation of the conjugates at 4 °C and loss of the conjugate nanoparticles due to the trouble in separation and recovery. Although the nano-sized P(AAm-co-AAc) enables high specific enzyme loading and thus provides for the high specific activity of the nano-size immobilized enzymes, the advantage of the nano-sized IPN-Cellu and the nano-sized IPN-Cello in biocatalysis requires to be further assessed through a comparative study of micro-sized IPN-Cellu and micro-sized IPN-Cello prepared from well-known micro-sized P(AAm-co-AAc).114,121–123
4. Conclusions and outlook
All the enzymes engineered on UCST-type polymers for application in biocatalysis possess the common characteristics in response to the thermal stimulation:16–20 they effectively biocatalyze the chemical and biochemical reactions in the soluble state above the Tcp values and are recovered by precipitation below the Tcp values. In the reversible dissolution–precipitation processes, the conjugates can be recycled between the higher temperatures (above the Tcp values) and the lower temperatures (below the Tcp values) so as to enable the prevention of the immobilized enzymes from thermal deactivation, contrary to the case of enzymes engineered on LCST-type polymers. Such “smart” recycling, which the enzymes engineered on UCST-type polymers undergo, can provide for high biocatalytic performance and convenient operation towards chemical and biochemical reactions.From the available literature, the UCST-type polymer-conjugated enzymes inevitably have a sensitive Tcp fluctuation with the stimulating factors, including polymer molecular weight, polymer concentration, hydrophile fraction, counterions and pH, differing from LCST-type polymer-conjugated enzymes whose Tcp values are little affected by these stimulating factors. Even if an LCST-type block is introduced to a UCST-type homopolymer, the sensitive Tcp fluctuation behaviour remains unchanged.39 In order to prevent an obvious change or the disappearance of Tcp in UCST-type homopolymer and conjugate solutions, it is recommended that either the polymer molecular weight be changed or a second proper comonomer would be chosen to make UCST-type diblock copolymers and conjugates. The expected Tcp can be obtained in a narrow range either by adjusting the amount of the monomer in the synthesis of the homopolymer47,52 or by tuning the molar ratio of the two comonomers in the synthesis of copolymers.52,67
Compared to their free counterparts, the UCST-type polymer-conjugated enzymes improve the thermal enzymatic stability in a systematic way and fluctuate to a small extent with varying pH and temperature, depending on the nature of the interaction between the enzymes and the polymers. The strong interaction via covalent bonding favours the stability of all the UCST-type polymer-immobilized enzymes. The observed high retained activities are deemed to result from the formation of multiple covalent bonds between the enzymes and the polymers, which makes the enzymes dispersed and stabilized on the polymers against the denaturation and autolysis of the enzymes. In order to achieve a stable biocatalytic performance of a UCST-type polymer-conjugated enzyme against pH and temperature, it is advisable that an affinity comonomer with the enzyme would be used to make a UCST-type diblock copolymer in cases where the interaction between the enzyme and the UCST-type homopolymer is insufficiently strong.
Among all the categories of UCST-type polymers documented up to date, the UCST-type homopolymers can be regarded as the ideal category applied in the fabrication and biocatalysis of engineered enzymes. The UCST-type homopolymers and related conjugates are not only simple to prepare,36,37,45,46,105 but also completely precipitate to gain a high recovery rate because of the efficient polymer–polymer interaction with lower steric hindrance upon cooling down to below the Tcp values after heating above the Tcp values.36,37 In the meantime, the related conjugates are able to deliver a strong retained activity owing to less mass transfer limitations between the conjugates and the substrates.36,37,45 These definitely enable the related conjugates to exhibit a strong activity or recyclability in biocatalysis.36,37,45 In contrast, the UCST-type copolymers are encountering some challenges, although viewed as a promising category in the application. The UCST-type copolymers and related conjugates are more or less complicated to produce.38–44,48–52 Moreover, they hardly produce a high recovery rate when precipitating upon cooling down to below the Tcp values after heating above the Tcp values due to higher steric hindrance from more blocks towards copolymer molecule association.39–41,44,48,49,52 Meanwhile, they are unable to deliver a strong retained activity due to more mass transfer limitations between the conjugates and the substrates caused by the presence of more blocks.39,40,44,48,49 These certainly affect the related conjugates to display a strong activity or recyclability in biocatalysis.39–41,44,48,49,52
From the available literature, most of the micro-sized UCST-type polymer-conjugated enzymes exhibit superior or close activity to that of their free counterparts as well as superior stability to that of their free counterparts.36,41–44,51 Such high biocatalytic performance shows the advantage of UCST-type polymers over non-thermoresponsive polymers in the engineering of enzymes for biocatalysis. The rest of the micro-sized UCST-type polymer-conjugated enzymes present significantly lower activity to that of their free counterparts as well as superior stability to that of their free counterparts,44,46,48,49 mainly as a result of more mass transfer limitations between the conjugates and the substrates.44,48,49 It follows that the high biocatalytic performance of the UCST-type polymer-conjugated enzymes is attained via homogeneous biocatalysis, whereas the biocatalytic properties of non-thermoresponsive polymer-conjugated enzymes generally trade-off activity for stability.10,11 In the meantime, almost all of the nano-sized UCST-type polymer-conjugated enzymes display superior or close activity to that of their free counterparts as well as superior stability to that of their free counterparts.37,38,40,45,50,52 Some of them even produce a wonderful activity, which is (59–670)-fold that of their free counterparts.38 The incomparable advantage of nano-sized UCST-type polymers in terms of activity is due to the higher specific surface area, which enables high dispersion of enzymes and less mass transfer limitations between the conjugates and the substrates. However, the disadvantage of nano-sized UCST-type polymers in terms of separation and recovery ought not to be ignored. In designing and developing nano-sized UCST-type polymer-engineered enzyme biocatalysts, the biocatalytic performance should be preferably compared to that of their micro-sized counterparts. In making an optimal choice between nano-sized and micro-sized UCST-type polymer counterparts, a trade-off is required between activity and separation and recovery.
The limited available literature shows that the UCST-type polymer-immobilized enzymes definitely affect the kinetics of the chemical or biochemical reactions relative to the free enzymes.36,37,39,48 The immobilization of the enzymes on the UCST-type polymers changes the Vm, kcat and Km values of the free enzymes to a different extent.36,37,39,48 As the activity is assessed in terms of Vm/Km or kcat/Km, the increase in the Vm or kcat and the decrease in the Km are favourable for the enhancement in the activity. Km values can be expectedly increased by enhancing substrates' affinity with conjugates (i.e., by enhancing substrates' hydrophilicity or hydrophobicity with conjugates). However, Vm and kcat values are strongly dependent on the category of UCST-type polymers used. When UCST-type homopolymers are involved, higher Vm or kcat values can be achieved owing to less mass transfer limitations between conjugates and substrates. When UCST-type copolymers are involved, lower Vm or kcat values are gained due to more mass transfer limitations between conjugates and substrates caused by the presence of more blocks. The limited number of cases show that the UCST-type homopolymer-immobilized enzymes result in the Vm/Km or kcat/Km values not lower than those over their free enzymes, corresponding to the high retained activities,36,37 whereas the UCST-type copolymer-immobilized enzymes give rise to the Vm/Km or kcat/Km values inferior to those over their free enzymes, corresponding to the inferior retained activities.39,48
The fascination with UCST-type polymers lies in reversible temperature-dependent phase transitions in solution, which are related to polymer–polymer and polymer–solvent interactions. The phase transitions of UCST-type polymers reflect an equilibrium shift between these two kinds of interactions. Such phase transition processes are associated with reversible swelling of the polymers or reversible increase in polymer particle size with increasing temperature in solution, corresponding to reversible dissociation of the polymer molecules and reversible association of polymer and solvent molecules with increasing temperature. UCST-type polymers can be homo- or copolymers. UCST-type homopolymers must possess UCST behaviour. The phase transition properties of the homopolymers are solely determined by the physical properties of the homopolymers. UCST-type copolymers can be composed of both UCST- and non-UCST type blocks39,43,48,52,65–67 (e.g., P(NAGA-co-VBA)52) or just non-UCST-type blocks40–42,44,49–51,122–125 (e.g., P(AAm-co-NAS-co-AAc)50). In the former case, non-UCST-type blocks can play a promotional role in the modification of phase transition properties, immobilization of enzymes, and enhancement of conjugates' affinity with substrates.39,43,48,52,67 In the latter case, the copolymers still can display UCST behaviour as long as the interaction between the different blocks and the interaction between the blocks and solvents meet the conditions of reversible temperature-dependent polymer association and dissociation, as observed in the case of the IPN series.40–42,44,49,50,114,122–126 Even if the interaction between the different blocks themselves fails to fit the conditions of reversible temperature-dependent polymer association, the modification of the copolymers (e.g., with gelatin in water) enables fulfilling the task, thus making the copolymers UCST-type systems.51 As such, there is bigger developing room for UCST-type copolymer systems applied in the engineering of enzymes for biocatalysis. It can be envisioned that various types of copolymers would be developed to meet the needs of rationally designing and fabricating versatile immobilized enzyme biocatalysts.
Other than the wide use of water as the reaction medium, the use of organic solvents, such as toluene and methanol, as the reaction media has brought about the exciting biocatalytic performance of the nano-sized UCST-type polymer-conjugated enzymes, including CRL-Pluronic, CALB-Pluronic and Cyt C.38 In toluene, CALB-Pluronic has also presented the excellent reusability for the esterification of hexanoic acid and n-butyl alcohol at 40 °C, giving more than 95% of the initial activity after 10 cycles.38 In organic solvents, most free enzymes are little active for chemical and biochemical reactions just due to their extremely low solubility. The immobilization of enzymes on UCST-type polymers has made the dissolution of the conjugates in organic solvents above Tcp values possible and thus paved a potential pathway for the achievement of high efficiency in enzymatic reactions. In order to develop biocatalysis by UCST-type polymer-engineered enzymes in organic solvents, it can be expected that systematic research should be conducted on the chemistry of the polymers with organic solvents, the UCST phase transition behaviour of the polymers and the enzyme–polymer conjugates in organic solvents, and the biocatalytic properties of the enzyme–polymer conjugates in organic solvents. In contrast to the case in water, where reversible temperature-dependent phase transitions are associated with the polymer–polymer interaction (including hydrogen bonding and hydrophobic interaction) and the polymer–water hydrogen bonding, it is supposed that reversible temperature-dependent phase transitions in organic solvents would be related to the polymer–polymer hydrophobic interaction and the polymer–organic solvent hydrophobic interaction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by GlaxoSmithKline (GSK) and the Economic Development Board (EDB) through a Green and Sustainable Manufacturing (GSM) grant (project no. 279-000-506-592/A-0005295-01-00) as well as the Agency for Science, Technology and Research (A*STAR) through an IAF-PP grant (R-279-000-585-305/A-0005342-01-00) from the Pharma Innovation Program Singapore (PIPS). The assistance of Dr. Willy See and Jieran Yi for this contribution is acknowledged.References
- J. M. Guisan, Immobilization of Enzymes and Cells, Springer, New York Heidelberg Dordrecht London, 3rd edn, 2013 Search PubMed.
- X. Lyu, R. Gonzalez, A. Horton and T. Li, Catalysts, 2021, 11, 1211 CrossRef CAS.
- A. Dwevedi, in Enzyme Immobilization: Advances in Industry, Agriculture, Medicine, and the Environment, Springer, Switzerland, 2016, ch. 2, pp. 21–44 Search PubMed.
- J. Zdarta, A. S. Meyer, T. Jesionowski and M. Pinelo, Adv. Colloid Interface Sci., 2018, 258, 1–20 CrossRef CAS PubMed.
- F. Zhao, H. Li, X. Wang, L. Wu, T. Hou, J. Guan, Y. Jiang, H. Xu and X. Mu, J. Mater. Chem. B, 2015, 3, 9315–9322 RSC.
- F. Zhao, Q. Wang, J. Dong, M. Xian, J. Yu, H. Yin, Z. Chang, X. Mu, T. Hou and J. Wang, Process Biochem., 2017, 57, 87–94 CrossRef CAS.
- W. Tischer and F. Wedekind, Top. Curr. Chem., 1999, 200, 95–126 CrossRef CAS.
- A. Rodriguez-Abetxuko, D. Sánchez-deAlcázar, P. Muñumer and A. Beloqui, Front. bioeng. biotechnol., 2020, 8, 830 CrossRef PubMed.
- S. Liu, M. Bilal, K. Rizwan, I. Gul, T. Rasheed and H. M. N. Iqbal, Int. J. Biol. Macromol., 2021, 190, 396–408 CrossRef CAS PubMed.
- K. K. Bansal1, P. K. Upadhyay, G. K. Saraogi, A. Rosling and J. M. Rosenholm, eXPRESS Polym. Lett., 2019, 13, 974–992 CrossRef.
- K. T. sriwong and T. Matsuda, Org. Process Res. Dev., 2022, 26, 1857–1877 CrossRef.
- P. Zucca and E. Sanjust, Molecules, 2014, 19, 14139–14194 CrossRef PubMed.
- E. Magner, Chem. Soc. Rev., 2013, 42, 6213–6222 RSC.
- M. C. C. Pinto, N. L. de Souza e Castro, E. P. Cipolatti, R. Fernandez-Lafuente, E. A. Manoel, D. M. G. Freire and J. C. Pinto, Macromol. React. Eng., 2019, 13, 1800055 CrossRef.
- T. Jesionowski, J. Zdarta and B. Krajewska, Adsorption, 2014, 20, 801–821 CrossRef CAS.
- Y. Ito, J. Biomater. Sci., Polym. Ed., 1999, 10, 1237–1249 CrossRef CAS PubMed.
- J. Seuring and S. Agarwal, Macromol. Rapid Commun., 2012, 33, 1898–1920 CrossRef CAS PubMed.
- Q. Zhang and R. Hoogenboom, Prog. Polym. Sci., 2015, 48, 122–142 CrossRef CAS.
- J. Niskanen and H. Tenhu, Polym. Chem., 2017, 8, 220–232 RSC.
- Y. Pei, A. B. Lowe and P. J. Roth, Macromol. Rapid Commun., 2017, 38, 1600528 CrossRef PubMed.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- V. Aseyev, H. Tenhu and F. M. Winnik, Adv. Polym. Sci., 2011, 242, 29–89 CrossRef CAS.
- Y. C. Bae, S. M. Lambert, D. S. Soane and J. M. Prausnitz, Macromolecules, 1991, 24, 4403–4407 CrossRef CAS.
- V. Aseyev, H. Tenhu and F. M. Winnik, in Self Organized Nanostructures of Amphiphilic Block Copolymers II, ed. A. H. E. Müller and O. Borisov, Springer, Berlin Heidelberg, 2011, pp. 29–89 Search PubMed.
- N. Yamada, T. Okano, H. Sakai, F. Karikusa, Y. Sawasaki and Y. Sakurai, Macromol. Rapid Commun., 1990, 11, 571–576 CrossRef CAS.
- N. Mori, H. Horikawa, H. Furukawa and T. Watanabe, Macromol. Mater. Eng., 2007, 292, 917–922 CrossRef CAS.
- P. Muthiah, S. M. Hoppe, T. J. Boyle and W. Sigmund, Macromol. Rapid Commun., 2011, 32, 1716–1721 CrossRef CAS PubMed.
- H. Kanazawa, K. Yamamoto, Y. Matsushima, N. Takai, A. Kikuchi, Y. Sakurai and T. Okano, Anal. Chem., 1996, 68, 100–105 CrossRef CAS PubMed.
- V. Bulmus, S. Patir, S. A. Tuncel and E. Piskin, J. Controlled Release, 2001, 76, 265–274 CrossRef CAS PubMed.
- W. L. J. Hinrichs, N. M. E. Schuurnmans-Nieuwenbroek, P. van de Wetering and W. E. Hennink, J. Controlled Release, 1999, 60, 249–259 CrossRef CAS PubMed.
- S. Dincer, A. Tuncel and E. Piskin, Macromol. Chem. Phys., 2002, 203, 1460–1465 CrossRef CAS.
- J. M. Weissman, H. B. Sunkura, A. S. Tse and S. A. Asher, Science, 1996, 274, 959–963 CrossRef CAS PubMed.
- J. P. Chen and A. S. Hoffman, Biomaterials, 1990, 11, 631–634 CrossRef CAS PubMed.
- A. Kondo, T. Kaneko and K. Higashitani, Biotechnol. Bioeng., 1994, 44, 1–6 CrossRef CAS PubMed.
- S. Anastase-Ravion, Z. Ding, A. Pelle, A. S. Hoffman and D. Letourneur, J. Chromatogr., B, 2001, 761, 247–254 CrossRef CAS PubMed.
- M. Yan, J. Ge, W. Dong, Z. Liu and P. Ouyang, Biochem. Eng. J., 2006, 30, 48–54 CrossRef CAS.
- C. Cummings, H. Murata, R. Koepsel and A. J. Russell, Biomaterials, 2013, 34, 7437–7443 CrossRef CAS PubMed.
- J. Zhu, Y. Zhang, D. Lu, R. N. Zare, J. Ge and Z. Liu, Chem. Commun., 2013, 49, 6090–6092 RSC.
- C. Cummings, H. Murata, R. Koepsel and A. J. Russell, Biomacromolecules, 2014, 15, 763–771 CrossRef CAS PubMed.
- P. A. Limadinata, A. Li and Z. Li, Green Chem., 2015, 17, 1194–1203 RSC.
- G. R. Chado, E. N. Holland, A. K. Tice, M. P. Stoykovich and J. L. Kaar, ACS Catal., 2018, 8, 11579–11588 CrossRef CAS.
- L.-L. Lou, H. Qu, W. Yu, B. Wang, L. Ouyang, S. Liu and W. Zhou, ChemCatChem, 2018, 10, 1166–1172 CrossRef CAS.
- J. Han, J. Wan, Y. Wang, L. Wang, C. Li, Y. Mao and L. Ni, ACS Sustainable Chem. Eng., 2018, 6, 7779–7788 CrossRef CAS.
- S. Zhang, C. Wang, H. Chang, Q. Zhang and Y. Cheng, Sci. Adv., 2019, 5, 4252 CrossRef PubMed.
- D. Yang, H. Tenhu and S. Hietala, Eur. Polym. J., 2020, 133, 109760 CrossRef CAS.
- K. Kappauf, N. Majstorovic, S. Agarwal, D. Rother and C. Claaßen, ChemBioChem, 2021, 22, 3452–3461 CrossRef CAS PubMed.
- F. Li, F. Qin, Y. Pang, H. Lou, C. Cai, W. Liu, Y. Qiana and X. Qiu, Green Chem., 2021, 23, 2738–2746 RSC.
- W. Huang, W. Zheng, J. Han, J. Wu, Y. Li, Y. Mao, L. Wang and Y. Wang, Colloids Surf., B, 2022, 217, 112694 CrossRef CAS PubMed.
- X. Ma, W. Huang, Y. Song, J. Han, J. Wu, L. Wang and Y. Wang, J. Agric. Food Chem., 2022, 70, 13959–13968 CrossRef CAS PubMed.
- J. Sun, J. Lu, C. Li, Y. Tian, K. Liu, L. Liu, C. Zhao and M. Zhang, Biomacromolecules, 2022, 23, 1291–1301 CrossRef CAS PubMed.
- X. Zhu, Y. Tian and B. He, Environ. Res., 2023, 222, 115414 CrossRef CAS PubMed.
- Z. Chen, J. Wu, W. Huang, Y. Li, Y. Mao, J. Han, Y. Wang and L. Ni, J. Environ. Chem. Eng., 2023, 11, 109072 CrossRef CAS.
- M. Ballauff and Y. Lu, Polymer, 2007, 48, 1815–1823 CrossRef CAS.
- J. Zhang, M. Zhang, K. Tang, F. Verpoort and T. Sun, Small, 2014, 10, 32–46 CrossRef CAS PubMed.
- The Open Oregon Online, https://openoregon.pressbooks.pub/mhccbiology112/chapter/changes-in-enzyme-activity, (accessed April 2024).
- J. Seuring and S. Agarwal, Macromol. Chem. Phys., 2010, 211, 2109–2117 CrossRef CAS.
- J. Seuring, F. M. Bayer, K. Huber and S. Agarwal, Macromolecules, 2012, 45, 374–384 CrossRef CAS.
- J. Seuring and S. Agarwal, ACS Macro Lett., 2013, 2, 597–600 CrossRef CAS PubMed.
- F. Liu, J. Seuring and S. Agarwal, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4920–4928 CrossRef CAS.
- F. Liu, J. Seuring and S. Agarwal, Polym. Chem., 2013, 4, 3123–3131 RSC.
- M. Boustta, P.-E. Colombo, S. Lenglet, S. Poujol and M. Vert, J. Controlled Release, 2014, 174, 1–6 CrossRef CAS PubMed.
- S. Glatzël, A. Laschewsky and J.-F. Lutz, Macromolecules, 2011, 44, 413–415 CrossRef.
- T. P. Silverstein, J. Chem. Educ., 1998, 75, 116–118 CrossRef CAS.
- C. Tanford, The Hydrophobic Esect, John Wiley and Sons, New York, 2nd edn, 1973 Search PubMed.
- H. Nagaoka, N. Ohnishi and M. Eguchi, US Pat., 20070203313A1, 2007 Search PubMed.
- N. Ohnishi, H. Furukawa, K. Kataoka and K. Ueno, US Pat., 20077195925B2, 2007 Search PubMed.
- J. Seuring and S. Agarwal, Macromolecules, 2012, 45, 3910–3918 CrossRef CAS.
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Chem., 1968, 2, 1441–1455 CAS.
- F. M. Winnik, Macromolecules, 1990, 23, 233–242 CrossRef CAS.
- H. G. Schild and D. A. Tirrell, J. Phys. Chem., 1990, 94, 4352–4356 CrossRef CAS.
- F. M. Winnik, Polymer, 1990, 31, 2125–2134 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- C. Wu and X. Wang, Phys. Rev. Lett., 1998, 80, 4092–4094 CrossRef CAS.
- P. S. Mumick and C. L. McCormick, Polym. Eng. Sci., 1994, 34, 1419–1428 CrossRef CAS.
- Y. Ding, X. Ye and G. Zhang, Macromolecules, 2005, 38, 904–908 CrossRef CAS.
- S. Fujishige, K. Kubota and I. Ando, J. Phys. Chem., 1989, 93, 3311–3313 CrossRef CAS.
- F. Afroze, E. Nies and H. Berghmans, J. Mol. Struct., 2000, 554, 55–68 CrossRef CAS.
- L. D. Taylor and L. D. Cerankowski, J. Polym. Sci., Polym. Chem. Ed., 1975, 13, 2551–2570 CrossRef CAS.
- X. Qiu, C. M. S. Kwan and C. Wu, Macromolecules, 1997, 30, 6090–6094 CrossRef CAS.
- H. Feil, Y. H. Bae, J. Feijen and S. W. Kim, Macromolecules, 1993, 26, 2496–2500 CrossRef CAS.
- R. Liu, M. Fraylich and B. R. Saunders, Colloid Polym. Sci., 2009, 287, 627–643 CrossRef CAS.
- R. M. Broyer, G. N. Grover and H. D. Maynard, Chem. Commun., 2011, 47, 2212–2226 RSC.
- V. Bulmus, C. Boyer, X. Huang, M. R. Whittaker and T. P. Davis, Soft Matter, 2011, 7, 1599–1614 RSC.
- The Wikipedia Online, https://en.wikipedia.org/wiki/Lineweaver%E2%80%93Burk_plot, (accessed April 2024).
- The Santa Cruz Biotechnology Online, https://www.scbt.com/p/n-alpha-benzoyl-dl-arginine-4-nitroanilide-monohydrochloride-911-77-3#:~:text=Solubility%20%3A%2Fml)%2C%20and%20water, (accessed April 2024).
- I. P. G. Amaral, M. G. Carneiro-da-Cunha, L. B. Carvalho Jr. and R. S. Bezerra, Process Biochem., 2006, 41, 1213–1216 CrossRef CAS.
- G. Bayramoğlu, M. Yılmaz, A. Ü. Şenel and M. Y. Arıca, Biochem. Eng. J., 2008, 40, 262–274 CrossRef.
- S. S. Caramori, F. N. de Faria, M. P. Viana, K. F. Fernandes and L. B. Carvalho Jr., Mater. Sci. Eng., C, 2011, 31, 252–257 CrossRef CAS.
- H. Hinterwirth, W. Lindner and M. Lämmerhofer, Anal. Chim. Acta, 2012, 733, 90–97 CrossRef CAS PubMed.
- M. Kamburov and I. Lalov, Biotechnol. Biotechnol. Equip., 2012, 26, 156–163 CrossRef.
- S. Li, Z. Wu, M. Lu, Z. Wang and Z. Li, Molecules, 2013, 18, 1138–1149 CrossRef CAS PubMed.
- G. Bayramoğlu, V. C. Ozalp and M. Y. Arica, Ind. Eng. Chem. Res., 2014, 53, 132–140 CrossRef.
- K. Atacana and M. Özacar, Colloids Surf., B, 2015, 128, 227–236 CrossRef PubMed.
- J. Stolarow, M. Heinzelmann, W. Yeremchuk, C. Syldatk and R. Hausmann, BMC Biotechnol., 2015, 15, 77 CrossRef PubMed.
- K. Atacan, B. Çakıroğlu and M. Özacar, Food Chem., 2016, 212, 460–468 CrossRef CAS PubMed.
- K. Atacan, B. Çakıroğlu and M. Özacar, Int. J. Biol. Macromol., 2017, 97, 148–155 CrossRef CAS PubMed.
- D.-F. Li, H.-C. Ding and T. Zhou, J. Agric. Food Chem., 2013, 61, 10447–10453 CrossRef CAS PubMed.
- D. Agyei, S. Tambimuttu, B. Kasargod, Y. Gao and L. He, J. Biotechnol., 2014, 188, 53–60 CrossRef CAS PubMed.
- E. C. S. Júnior, M. P. F. Santos, V. S. Sampaio, S. P. B. Ferrão, R. C. I. Fontan, R. C. F. Bonomo and C. M. Veloso, J. Chromatogr., B, 2020, 1146, 122124 CrossRef PubMed.
- J. Cui and S. Jia, Coord. Chem. Rev., 2017, 352, 249–263 CrossRef CAS.
- J. Cui, S. Ren, B. Sun and S. Jia, Coord. Chem. Rev., 2018, 370, 22–41 CrossRef CAS.
- B. K. Patel, O. H. Campanella and S. Janaswamy, Carbohydr. Polym., 2013, 92, 1873–1879 CrossRef CAS PubMed.
- H. Lou, M. Lin, M. Zeng, C. Cai, Y. Pang, D. Yang and X. Qiu, BioEnergy Res., 2018, 11, 456–465 CrossRef CAS.
- Y. Wu, Y. Shi and H. Wang, Macromolecules, 2023, 56, 4491–4502 CrossRef CAS.
- D. Yang, M. Viitasuo, F. Pooch, H. Tenhu and S. Hietala, Polym. Chem., 2013, 9, 517–524 RSC.
- J. Yin, J. Hu, G. Zhang and S. Liu, Langmuir, 2014, 30, 2551–2558 CrossRef CAS PubMed.
- A. Asadujjaman, B. Kent and A. Bertin, Soft Matter, 2017, 13, 658–669 RSC.
- S. Glatzël, N. Badi, M. Päch, A. Laschewsky and J. Lutz, Chem. Commun., 2010, 46, 4517–4519 RSC.
- F. Liu, J. Seuring and S. Agarwal, Macromol. Chem. Phys., 2014, 215, 1466–1472 CrossRef CAS.
- The Wikipedia Online, https://en.wikipedia.org/wiki/Poloxamer_407, (accessed April 2024).
- R. Jaquilin P J, O. S. Oluwafemi, S. Thomas and A. O. Oyedeji, J. Drug Delivery Sci. Technol., 2022, 72, 103390 CrossRef CAS.
- G. Bonacucina, M. Spina, M. Misici-Falzi, M. Cespi, S. Pucciarelli, M. Angeletti and G. F. Palmieri, Eur. J. Pharm. Sci., 2007, 32, 115–122 CrossRef CAS PubMed.
- A. M. Pragatheeswaran and S. B. Chen, Langmuir, 2013, 29, 9694–9701 CrossRef CAS PubMed.
- G. Beaudoin, A. Lasri, C. Zhao, B. Liberelle, G. De Crescenzo and X.-X. Zhu, Macromolecules, 2021, 54, 7963–7969 CrossRef CAS.
- J. Sánchez-Ramírez, J. L. Martínez-Hernández, P. Segura-Ceniceros, G. López, H. Saade, M. A. Medina-Morales, R. Ramos-González, C. N. Aguilar and A. Ilyina, Bioprocess Biosyst. Eng., 2017, 40, 9–22 CrossRef PubMed.
- Y. Wang, D. Chen, G. Wang, C. Zhao, Y. Ma and W. Yang, Chem. Eng. J., 2018, 336, 152–159 CrossRef CAS.
- I. N. Ahmed, X.-L. Yang, A. A. Dubale, R.-F. Li, Y.-M. Ma, L.-M. Wang, G.-H. Hou, R.-F. Guan and M.-H. Xie, Bioresour. Technol., 2018, 270, 377–382 CrossRef CAS PubMed.
- S. S. Rashid, A. H. Mustafa, M. H. Ab Rahim and B. Gunes, Int. J. Biol. Macromol., 2022, 209, 1048–1053 CrossRef CAS PubMed.
- P. Zheng, J. Wang, C. Lu, Y. Xu and Z. Sun, Process Biochem., 2013, 48, 683–687 CrossRef CAS.
- C.-T. Tsai and A. S. Meyer, Molecules, 2014, 19, 19390–19406 CrossRef PubMed.
- O. V. Klenina, E. G. Fain, V. I. Klenin, L. V. Mineyev, V. A. Maslennikov and V. P. Gorodnov, Kolloidn. Zh., 1980, 42, 558 CAS.
- O. V. Klenina and E. G. Fain, Polym. Sci., 1981, 23, 1439–1446 Search PubMed.
- M. Yang, C. Liu, Z. Li, G. Gao and F. Liu, Macromolecules, 2010, 43, 10645–10651 CrossRef CAS.
- H. Katono, A. Maruyama, K. Sanui, N. Ogata, T. Okano and Y. Sakurai, J. Controlled Release, 1991, 16, 215–228 CrossRef CAS.
- H. Sasase, T. Aoki, H. Katono, K. Sanui and N. Ogata, Makromol. Chem., Rapid Commun., 1992, 13, 577–581 CrossRef CAS.
- F. Ilmain, T. Tanaka and E. Kokufuta, Nature, 1991, 349, 400–401 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |