
Methane partial oxidation under periodic reaction conditions on Pt/Al2O3†
Surya Pratap S.
Solanki
 ab,
Zhuoran
Gan
c,
Silvia
Marino
c,
Robert J.
Davis
c,
William S.
Epling
*c and
Lars C.
Grabow
*ab
ab,
Zhuoran
Gan
c,
Silvia
Marino
c,
Robert J.
Davis
c,
William S.
Epling
*c and
Lars C.
Grabow
*ab
aWilliam A. Brookshire Department of Chemical and Biomolecular Engineering, University of Houston, Houston, Texas – 77204, USA. E-mail: grabow@uh.edu
bTexas Center for Superconductivity at the University of Houston (TcSUH), Houston, Texas – 77204, USA
cDepartment of Chemical Engineering, University of Virginia, Charlottesville, Virginia – 22903, USA. E-mail: wse2t@virginia.edu
First published on 22nd February 2024
Abstract
The increasing interest in utilizing methane, the primary component of natural gas, for chemical production has spurred research into methane partial oxidation (MPO) as an alternative to traditional steam methane reforming (SMR). MPO has lower energy requirements and potential for carbon capture, making it an attractive option for hydrogen production. Challenges remain, however, such as carbon deposition leading to degradation and achieving high hydrogen selectivity. Here, the impact of periodic reactor operation on MPO over a Pt/Al2O3 catalyst was studied, primarily via varying reactor inlet compositions. Experiments were conducted using periodic operation strategies to assess the influence of changing reactant inlet concentrations on hydrogen formation during MPO. The results suggest that cycling between mixtures with low and high oxygen content can lead to transient hydrogen formation rates that surpass those achieved at steady state. Control experiments and density functional theory (DFT) calculations show that enhanced hydrogen formation can be attributed to the reaction between CO with hydroxyl groups at the metal and alumina support interface. This work underscores the critical role of surface coverages at the metal–support interface and suggests avenues for future exploration, including alternative support materials with higher OH mobility and changes in the cycling scheme to enhance catalyst performance under periodic conditions.
1. Introduction
Given the abundance of methane, the main component of natural gas, there is growing interest in the scientific community to utilize methane in chemicals production, and not just combust it to generate heat or electricity.1,2 Even though C–H activation is feasible on metal-based catalysts, the selective conversion of methane to useful products remains a ‘Holy Grail’ for chemical engineers.1 Methane can be converted to hydrogen, which is industrially important and can be used as a clean fuel or as feedstock for a variety of reactions. Traditionally, hydrogen is produced through steam methane reforming (SMR) for industrial and energy applications.3 However SMR is energy-intensive and can generate significant greenhouse gas emissions. Methane partial oxidation (MPO) has emerged as a potential alternative for hydrogen production due to its lower energy requirements and potential for carbon capture.4,5 During MPO, methane reacts with oxygen producing H2 and CO. This reaction can be catalyzed by metals such as platinum, palladium, and rhodium supported on alumina, silica or ceria.6–10 While MPO has shown promise as a potential route for hydrogen production, there are still several challenges that must be addressed. One major challenge is the potential for carbon deposition, which can lead to catalyst deactivation and reduced efficiency.4,11 Another challenge is achieving high selectivity to H2, as other unwanted by-products such as H2O and CO2 can form via methane combustion and are thermodynamically preferred.12,13 Methane reactivity on a Pd/Pt based catalyst is a strong function of CH4/O2 ratio. Chin et al. have shown a high oxygen content poisons the Pt catalyst surface14 and can lead to Pd oxide formation.15 Thus, methane conversion to hydrogen is limited by slow kinetics and low selectivity at high oxygen partial pressures, and the tendency for coke formation increases with low oxygen partial pressure.To address this limitation, periodic reactor operation has been proposed as a promising strategy.16,17 With periodic reactor operation it is possible to modulate surface coverage and oxidation state by varying the reaction conditions, which can result in significant performance enhancement.18–20 Particularly, for redox reactions it is possible to feed pulses of reactants with different ratios of reactant gases and oxygen, often termed as rich if oxygen is lesser than the stoichiometric amount, or lean if the oxygen content is greater.20 For example, Stötzel et al. have shown that cycling methane and oxygen can improve hydrogen selectivity by inhibiting water formation on a Pd-based catalyst during methane partial oxidation to synthesis gas.21 Fathi et al. demonstrated a highly selective route for synthesis gas production from methane and oxygen in a cyclic reaction using cerium oxide.22 In this route, cerium oxide acted as an intermediate oxygen carrier. The required temperature for this process was approximately 700 °C, significantly lower than the typical temperature used in conventional synthesis gas production, which is around 900 °C. During methane oxidation on Pd-based catalysts, Franken et al. employed a short reducing pulse every 5 minutes.23 This improved the activity of the catalyst along with the catalyst lifetime by maintaining a mixed metallic and oxide phase. Karinshak et al. have shown reduction in T50 during methane combustion light-off with feed modulation over a Pt–Pd/spinel catalyst by varying the ratio of oxidants to reductants in the feed.20 Creaser et al. have demonstrated that cycling between propane and oxygen during propane dehydrogenation over a V–Mg–O catalyst resulted in higher yields than with static conditions.24 Similarly, Rambeau and Amariglio observed that switching between a feed of nitrogen and hydrogen led to higher ammonia synthesis production than that attained with steady state conditions over a ruthenium powder.25 Fu et al. also demonstrated that periodic pulsing of hydrogen on a PtWOx/C catalyst achieved alternate states that led to one order of magnitude higher activity than under constant hydrogen feed during tert-butanol dehydration.18 These studies highlight the potential advantages of periodic reactor operation, including enhanced catalytic efficiency and the ability to achieve higher yields of desired reaction products. Furthermore, periodic reactor operation offers flexible reactor dynamics by allowing for modulation of parameters such as reactor inlet composition, temperature, and pressure, among others.16
In this study, we investigated the effect of periodic reactor operation on methane partial oxidation (MPO) over Pt/Al2O3 by modulating the reactor inlet compositions. We conducted experiments that included periodic operation strategies to understand how changing the reactant inlet concentrations impact hydrogen formation during methane partial oxidation over a supported Pt catalyst. Previous research by Carlsson et al. found increased catalytic activity of Pt/Al2O3 when switching from CH4-rich to CH4-lean conditions during methane oxidation, but ultimately poor reactor utilization due to no methane conversion during the CH4-lean cycle.13,26 In our experiments, we also explored strategies that would maintain similar reactor utilization as in static inlet condition experiments. We conducted our experiments primarily under methane-rich conditions, as excess oxygen promotes the formation of combustion products. Our results show that varying the oxygen content in the feed streams can result in a transient increase in H2 formation when switching from a higher O2 content to a lower one. Through control experiments and supporting density functional theory (DFT) calculations, we determined that enhanced hydrogen formation can be attributed to the reaction of CO with hydroxyl groups on the alumina–support at the metal–support interface.
2. Methods
2.1 Experimental
The total flow rate was set to 100 sccm and the flow rates for all the gases were regulated using MKS mass flow controllers. The MKS mass flow controllers and switching valves were controlled using NI LabView software. Prior to the experiments, the catalyst was pre-treated in 5% H2 in Ar at 500 °C for 30 minutes. All the experiments were performed at 450 °C unless otherwise specified. The periodic reaction experiments were performed by cycling a specified feed and regeneration mixture at an interval of 20 s. The methane concentration was kept at 2% in all experiments and the oxygen concentration was varied between 0.1 and 3%. The outlet concentrations were monitored using a Hiden HPR-20 mass spectrometer. Masses monitored were 2, 15, 18, 28, 32, 36, 44 corresponding to H2, CH4, H2O, CO, O2, Ar, CO2 respectively, at a frequency of 33.3 Hz. Concentrations were calculated based on calibrations for each of the gases. Gases were purchased from Praxair.
Caution! Our experimental methods require the use of methane, hydrogen, carbon monoxide, carbon dioxide and oxygen gases. Methane, hydrogen and carbon monoxide are classified as GHS flammable gas, category 1. Oxygen is classified as GHS oxidizing gas, category 1. Carbon dioxide is classified as asphyxiant. The storage and use of the compressed gas cylinders were handled using UVA Environmental Health and Safety procedures.
2.2 Computational
We conducted periodic DFT calculations using the Vienna ab initio simulation package (VASP)28–33 with a plane wave basis set under the Kohn–Sham formulation. We used the Atomic Simulation Environment (ASE)34 for workflow management, visualization, and post-analysis. The electron exchange–correlation energy was determined using the Perdew–Burke–Ernzerhof (PBE)35,36 functional, which included DFT-D3 dispersion corrections.37 We used the projector augmented wave (PAW) method to describe the interaction between ion cores and electrons.38 The valence electron wavefunction was expanded into a plane wave basis set with an energy cut-off of 400 eV, and spin polarization was enabled. We used Gaussian smearing with a width of kBT = 0.1 eV and subsequently extrapolated to 0 K. We stopped the self-consistent-field (SCF) cycle when the electronic energies converged to 10−6 eV. A similar approach has been used previously for calculations for a γ-Al2O3 system.39We adopted the γ-Al2O3 bulk structure from a previous study by Raybaud and co-workers.40 For γ-Al2O3 we obtained the following unit cell parameters: a = 5.548 Å, b = 8.360 Å, c = 8.036 Å; α = γ = 90.00°, β = 90.59° which are close to the reported values.39,41 We further generated the (110) surface of γ-Al2O3 with a thickness of 8 atomic layers, similar to the setup used by Hoffman et al.41 We then grafted a Pt nanorod on top, ensuring minimal expansive strain in the Pt nanorod of only 0.1%. The open γ-Al2O3 sites were fully hydrated as the (110) surface can have close to 3 OH per nm2 at 450 °C.42 The nanorod model with full hydration is shown in Fig. 1. Ionic relaxations were performed until the atomic forces were less than 0.02 eV Å−1, and we applied dipole correction in the direction normal to the surface. The Brillouin zone of the Pt nanorod model was sampled using a 2 × 1 × 1 k-point mesh.
 | ||
| Fig. 1 Pt nanorod on alumina model. | ||
To compute gas-phase energies, we used the aforementioned parameters, except for a narrower Gaussian smearing width of 0.01 eV. We performed ionic relaxations until the atomic forces were less than 0.005 eV Å−1 and applied dipole corrections in all three Cartesian directions, keeping the gas molecule at a central position in a cell of size 15 Å × 15 Å × 15 Å.
For adsorption/desorption steps, we define the adsorption energy as
| ΔE = Emod/ads − Emod − Egas, |
3. Results and discussion
3.1 Reaction experiments under periodic inlet conditions
Fig. 2 depicts the schematic for a periodic reaction experiment, wherein a reactor inlet feed-mixture of 2% CH4 and varying oxygen concentrations (0.05–1%) were periodically cycled with a regeneration mixture containing 1% O2, balance Ar mixture. Fig. 2(b–f) illustrate the reactor outlet profile during two phases of the periodic experiment after steady cycle-to-cycle outlet concentrations were obtained. Complete oxygen conversion was observed during the phase containing CH4 (feed-phase), while the oxygen concentration rapidly attained the inlet value during the half-cycle without CH4. This rapid increase in O2 concentration to the inlet value is attributed to the saturation of oxygen atoms on the Pt surface, as platinum is known not to form bulk platinum oxide under these conditions.43,44 Methane conversion was observed to be dependent on the oxygen concentration. At lower oxygen concentrations in the feed phase containing CH4, enhanced hydrogen formation was initially observed, followed by a decrease to the steady state level of the respective mixture. A similar behavior was observed by Carlsson et al. during their periodic reaction experiments on 5% Pt/Al2O3, with cycling between 0 and 1250 ppm oxygen.13 We also performed experiments where there was a periodic switch between the CH4/O2 phase and a phase with just Ar, i.e., inert, with results shown in Fig. S2,† to verify that the peaks that we observed during the experiments are not because of any artifact from pressure changes during switching. | ||
| Fig. 2 (a) Schematic of cycling strategy 1. Reactor outlet profile at 450 °C for a periodic reactor inlet mixture containing 2% CH4 and (b) 0.05% O2, (c) 0.1% O2, (d) 0.2% O2, (e) 0.5% O2, (f) 1% O2, all balanced in Ar, with the 2nd phase containing 1% O2 in balance Ar. The shaded areas indicate excess H2 production above the equivalent steady state condition. | ||
We compared the amount of hydrogen formed during the feed-phase for all the experiments with the cycling strategy described in Fig. 2(a). The hydrogen formation during this half cycle with this scheme was generally higher than the steady state cases, except for the highest O2 concentration (Fig. 3). This latter observation is attributed to excess oxygen on the surface and feed, which is consistent with Chin et al.'s findings, where they reported methane activation to be dependent on the oxygen concentration with methane activation strongly inhibited by oxygen at O2/CH4 > 1.14 We did not observe any significant difference in CO formation during cycling as its concentration remained about 0.25% during all experiments (Fig. 2(b–f)). We can attribute the nearly constant CO concentration to favorable CO oxidation or water gas shift reaction on the Pt catalyst and lower CO selectivity as the oxygen content increases. The concentration profiles for CO2 and H2O are shown in Fig. S3.† Overall, this cycling strategy demonstrates how partial oxygen coverage can aid in achieving enhanced hydrogen formation, by avoiding oxygen poisoning which inhibits methane activation. However, this strategy leads to poor utilization of the reactor as there is no methane flow into the reactor for half of the time. Therefore, we investigated periodic experiments with cycling between a mixture containing the same methane concentration but lower oxygen (feed mixture) and higher (regeneration mixture) concentrations.
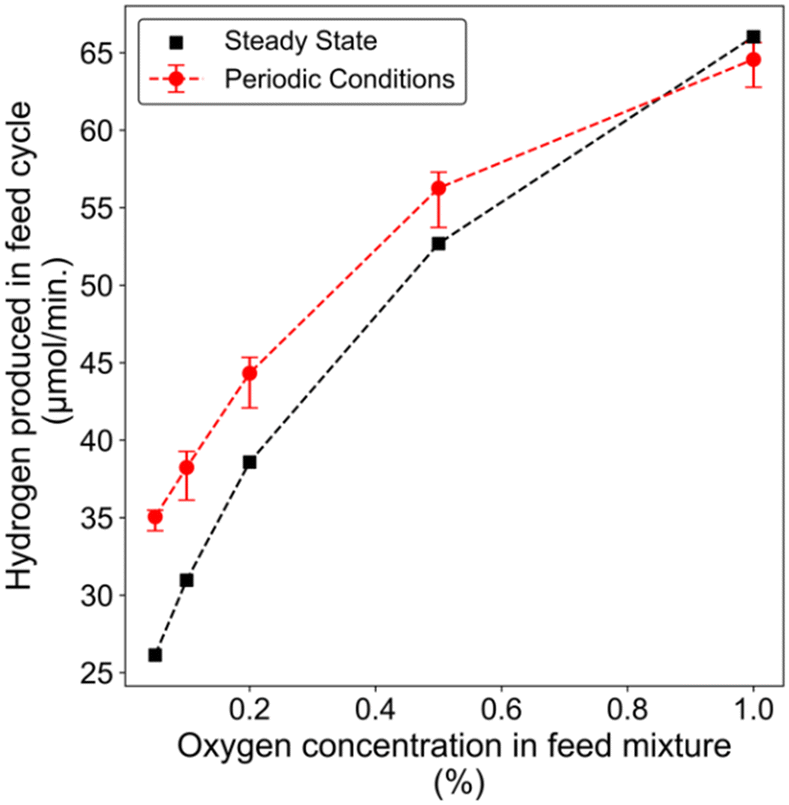 | ||
| Fig. 3 Hydrogen produced in the feed phase during the cycling conditions described in Fig. 2, and a comparison with steady state measurements. | ||
Fig. 4(a) depicts a cycling strategy where, similar to the previous experiment, the oxygen concentration in one phase was varied, but here the methane concentration was held constant at 2% through both phases. The reactor outlet profiles are shown in Fig. 4(b–f). Complete oxygen conversion was observed in both phases of the cycle, as the reaction mixture was continuously in a CH4-rich phase relative to combustion stoichiometry. Methane conversion was again dependent on the O2 concentration. Although hydrogen formation was initially high with the switch in inlet composition to the lower O2 phase (feed phase) of the cycle, it eventually decreased. Interestingly, with cycling strategy 2, hydrogen formation was higher than steady state even with the 1% O2 feed mixture, where we observed inhibition for the same mixture with cycling strategy 1. Fig. 2 shows up to 1% unreacted oxygen leaving the reactor during the regeneration cycle of strategy 1, whereas oxygen is completely consumed in strategy 2. Thus, a possible explanation for the higher hydrogen formation rate at the beginning of the feed cycle of strategy 2 is that continued methane oxidation during the regeneration phase reduces the surface coverage of oxygen. The lower oxygen surface coverage at the beginning of the feed phase of strategy 2 promotes hydrogen desorption over water formation.
 | ||
| Fig. 4 (a) Schematic of the cycling strategy 2. Reactor outlet profile at 450 °C for feed mixture containing 2% CH4 and (b) 0.05% O2, (c) 0.1% O2, (d) 0.2% O2, (e) 0.5% O2, (f) 1% O2, all balanced in Ar. The alternating phase always contained 2% CH4 and 3% O2, all balanced in Ar. The shaded areas indicate excess H2 production above the equivalent steady state condition. | ||
We calculated the amount of additional H2 formation with respect to the steady state during feed phase with variable O2 and compared this to the amount formed at steady state for each of those mixtures, with the results shown in Fig. 5. The excess H2 formation decreased with an increase in O2 concentration due to the higher side reaction to water. The profiles for water and CO2 are shown in Fig. S4,† describing the more significant water formation with a higher level of overall O2 and 3% O2 in the regeneration phase. We also experimented with different durations of the regeneration phase and discovered that we could increase feed utilization. However, this increase came at the cost of reducing the total hydrogen produced (Fig. S5†).
 | ||
| Fig. 5 Amount of additional (compared to respective steady state) hydrogen produced in the feed phase during cycling strategy 2. | ||
The excess H2 formation was up to 5 times that of the Pt active sites, which indicates that H2 originates from a catalytic process that is facilitated by the presence of O-containing surface species that have accumulated on the catalyst during the regeneration phase, or the involvement of species stored on the support. Oxygen or hydroxyl species on Pt reportedly inhibit CH4 activation14,45 and therefore cannot explain the observed increase in H2 production. The most commonly implicated support species are hydroxyl or carbonate species. For example, Becker et al. attributed the enhanced formation of CO in the rich phase during methane oxidation on Pt/Al2O3 under cycling phases of net-reducing and net-oxidizing feed to excess O-containing species (hydroxyl or carbonate species on alumina).46 Sautet's group has calculated that hydroxyls remain stable at temperatures up to 450 °C.42,47 This is consistent with Kalamaras et al.'s observation, where they exposed Pt/Al2O3 to D2O, followed by a mixture of CO and H2O, resulting in the formation of H2, HD, and D2, demonstrating the presence of OD species on the surface.48
We chose overall CH4-rich conditions for our study to mitigate oxygen inhibition on CH4 activation, but under these reducing conditions the support hydroxyl coverage may be limited under steady state operation. In contrast, the additional oxygen in the regeneration cycle of our periodic operating scheme leads to substantial water formation and presumably high hydroxyl coverages on Al2O3. We hypothesize that the transient formation of support hydroxyls favors the water-gas shift (WGS) reaction with CO at the metal–support interface (MSI) and can account for the measured increase in H2 formation. To probe the nature of surface species present on the catalyst after the regeneration phase of the cycle, we performed titration experiments where we exposed the catalyst to CO. CO was chosen as titrant, because it is a partial oxidation product that binds strongly to Pt, it has the potential to inhibit CH4 conversion,49,50 can displace other adsorbates, and it can react with hydroxyls or carbonates at the Pt/Al2O3 interface.
3.2 CO titration experiment
For the CO titration experiments we exposed the catalyst to a mixture of 2% CH4 and 3% O2 and subsequently purged the reactor system with Ar to remove residual gases. During the purging step we did not observe any H2 desorption. We then introduced 1.5% CO into the system and cycled it with Ar, as shown in Fig. S6.† The observed formation of H2 and CO2 are depicted in Fig. 6. The H2 and CO2 formation decreased over multiple cycles and ultimately stabilized. Although we used moisture and hydrocarbon traps, we suspect in the last few cycles CO2 and H2 formation is due to impurities (CO2 and H2 formation without using traps is shown in Fig. S7†). Even after removing the contribution from impurities, we observed CO2 formation of around 37 μmol and H2 formation of 2.5 μmol. Both the amount of CO2 and H2 are higher than the number of active Pt sites (1.4 μmol). A plausible source of H2 is water-gas shift chemistry involving support hydroxyls. The higher content of CO2 is also consistent with water-gas shift, or could originate from CO oxidation with residual O atoms on Pt, or from carbonate decomposition from alumina. | ||
| Fig. 6 Amount of CO2 and H2 produced during CO titration experiments at 450 °C. A schematic of the CO titration experiment and respective reactor outlet profiles are shown in Fig. S6.† | ||
Considering the large amounts of excess H2 relative to the number of Pt sites and the absence of H2 during Ar purging, hydrogen storage in the form of a Pt-surface intermediate cannot account for all excess hydrogen formed during partial oxidation. Thus, the modified catalyst surface must alter reactivity or selectivity. We can eliminate O-assisted C–H bond activation on Pt, because in our experiments we routinely see lower CH4 conversion with increasing O2 concentration. Carbonate on Al2O3 can form from CO or CO2 exposure, but it is frequently implicated as poison of metal–support interface (MSI) sites and unlikely to co-catalyze the partial oxidation of CH4 to H2 as surface carbonates have been found to inhibit methane activation as well as CO oxidation on various supported catalysts.51–54 That leaves hydroxyls on the alumina support, which can readily form during the regeneration phase where we observe significant water formation. During the feed phase, these hydroxyls may co-catalyze CH4 partial oxidation, enable steam-reforming chemistry, or react with the partial oxidation product CO to form CO2 and H2.20,55–58 The formation of CO2 and H2 from CO is confirmed in our titration experiments. Moreover, since the measured amount of H2 exceeds the total amount of Pt sites including the perimeter sites, support hydroxyls must be mobile to replenish the reactive hydroxyl groups at the interface. To provide a molecular interpretation for these observations, we have used DFT to propose a possible WGS pathway at the Pt/alumina interface.
3.3 WGS reaction mechanism at the Pt/alumina interface
The WGS reaction is a well-studied process that occurs on supported Pt catalysts.48,56,57,59–64 During this reaction, OH groups, formed by the dissociation of water, react with CO to form H2 and CO2. Several studies48,61,65–68 have been conducted to understand the H2 and CO2 formation mechanism via CO reacting with OH, and many suggest that there is some reactivity at the MSI. Interfacial activity has been supported through mechanistic DFT studies on Pt/TiO2 and Pt/ceria among others where the support is more reducible.59,60,69 Previously, our group has studied H2 activation at the Au/TiO2 interface using a nanorod model and assigned pivotal roles to support hydroxyls during CO and H2 oxidation.70,71However, despite the importance and widespread use of Pt/alumina as a catalyst, there have been no DFT studies to identify the elementary steps for the CO and OH reaction at the Pt–alumina interface. To fill this knowledge gap, we have calculated the thermodynamic potential energy diagram for WGS on extended Pt nanorods on alumina. Activation energy barrier calculations using the climbing image nudged elastic band (CI-NEB) method were attempted, but intermediate images repeatedly failed to reach electronic convergence and transition states could not be located.
To probe the reaction between CO and support hydroxyls, we considered the periodic Pt nanorod model on fully hydrated alumina support depicted in Fig. 1 and explored the mechanism illustrated in Fig. 7. The respective energy profile is shown in Fig. 8. The fully hydrated alumina surface is expected to approximate the state of the catalyst after the regeneration phase. CO adsorbs to the Pt nanorod near the MSI (Fig. S8†) with a binding energy of −2.10 eV. To initiate the interfacial reaction, we calculated the formation of a carboxyl and the reverse spillover step of H atom migration from alumina to Pt. The formation of carboxyl was strongly endothermic with 1.53 eV, which we attribute to an unstable electronic defect on alumina. Alumina is an insulator with a band gap of 8.7 eV and cannot easily accommodate the excess electron that remains after OH is removed from the support.72–74 However, the electronic surface structure of alumina can strongly deviate from its bulk properties. For example, the (100) and (110) surfaces of crystalline γ-Al2O3 are predicted to have valence bands near the Fermi level and band gaps of only ∼3 eV.75 Similarly, the band gap for ultrathin (7–10 nm) films of amorphous alumina has been measured to be 2.5 eV, which is in the semiconductor range.76
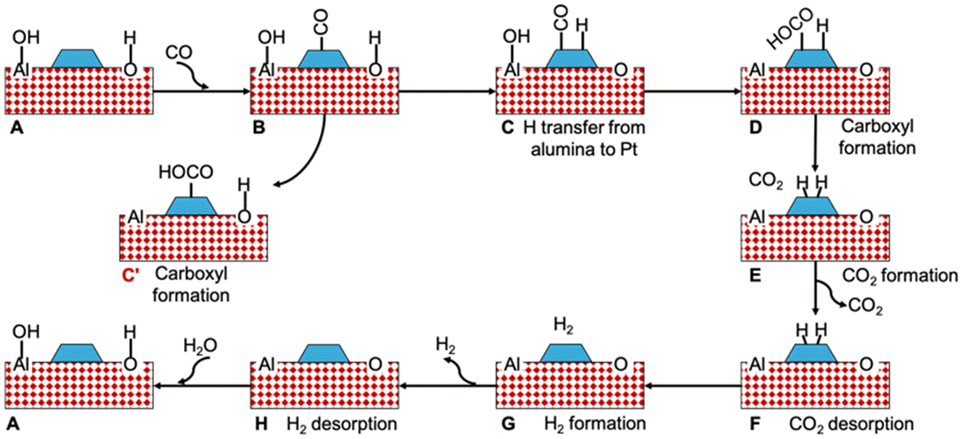 | ||
| Fig. 7 Schematic reaction pathway for CO oxidation by OH groups on Pt (nanorod)/alumina. The simplified representation shows only one water molecule in the mechanism. | ||
 | ||
| Fig. 8 Energy profile for the reaction pathway shown in Fig. 7. The labels in the profile correspond to the structures in the Fig. 7. | ||
We performed a projected density of states (PDOS) analysis of the interface Al and O atoms with the Pt nanorod model and compared this with the bare alumina surface as shown in Fig. 9. Surface hydration was not considered, to simplify the interpretation of the PDOS analysis. Interestingly we observed new O-2p and Al-2p bands, which appeared near the Fermi level and were continuous, implying alumina near the MSI becomes more metallic in character. Because of the additional electronic states near the Fermi level, the H transfer step (Fig. 7C) and carboxyl formation (Fig. 7D) were found to be only mildly endothermic. This system constitutes an example where electronic interactions between a metal particle and the support atoms near the MSI can further help in stabilizing intermediates that require electron transfer at the interface. Higher hydroxyl mobility can further increase the reactivity at the MSI.74,77
 | ||
| Fig. 9 PDOS analysis for interface Al (2p states) and O (2p states) atoms on Pt/alumina (Pt nanorod on alumina) and only alumina (bare alumina). | ||
In contrast to carboxyl formation, the transfer of H from alumina to Pt is endothermic by only 0.38 eV. Once the H atom, including its associated electron, has been transferred, carboxyl formation becomes mildly endothermic by 0.22 eV. Dehydrogenation of the carboxyl group being close to thermoneutral results in linear CO2. The recombination of H atoms to form a weakly adsorbed H2 molecule on Pt was endothermic by 0.75 eV, which is similar to calculated values on the extended Pt(111) surface.62 To close the catalytic cycle for WGS, after H2 desorption we regenerate the interfacial hydroxyl groups through water adsorption. The overall calculated WGS reaction energy is exothermic by −0.73 eV, which is in reasonable agreement with the value obtained from the NIST database (−0.43 eV).78 Overall our thermodynamic evaluation suggests that the potential energy diagram for CO oxidation by hydroxyls at the Pt/alumina interface is comparable to that of WGS on Pt(111).62 Thus, WGS involving transiently formed hydroxyls at the MSI is a plausible explanation for the excess H2 (and CO2) observed in our periodic experiments. To further improve the H2 yield of Pt/Al2O3 under periodic operating conditions it appears prudent to increase the number of accessible hydroxyls at the MSI. This may be achieved by selecting supports with high hydroxyl storage capacity and hydroxyl mobility. Alternatively, catalyst architectures that mirror the dual-layer design of CH4 oxidation catalysts integrated with CeO2 or spinel-oxides as dynamic oxygen storage materials could be explored.79,80 Possible candidates for dynamic hydroxyl storage materials are bulk metal hydroxides or oxyhydroxides.
4. Conclusions
Experimental and computational approaches were used to investigate periodic reactor operation for methane partial oxidation on a model Pt/Al2O3 catalyst. Our findings indicate that alternating between low and high oxygen content mixtures can result in transient hydrogen formation rates that exceed those obtained at steady state. While this cycling approach shows potential for attaining higher H2 levels, under the conditions used in this study, there was no overall enhancement in H2 formation when integrating the amounts formed over the entire cycle due to some portion of the cycle being less active. We attribute the higher H2 (and CO2) levels under cyclic operation to increased hydroxyl availability and WGS activity at the Pt/Al2O3 interface compared to steady state operation, with evidence for the reaction between CO and support hydroxyls provided by CO titration experiments and DFT calculations.Our study highlights the importance of surface coverages at the metal–support interface, which can play a crucial role during periodic conditions. The selective formation of hydrogen from OH groups at the interface could be exploited by supporting Pt on a different oxide with higher OH mobility and storage capacity. In this context, it is worth exploring multi-layer architectures involving stable metal hydroxides or oxyhydroxides. Additionally, changes in the cycling scheme, such as including water in the regeneration period, could further improve the catalyst's performance under periodic conditions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation, through the Emerging Frontiers in Research and Innovation award, NSF-EFMA 2029359 and U.S. Department of Energy's Office of Energy Efficiency and Renewable Energy (EERE) under the Advanced Manufacturing Office, Award Number DEEE0009410. The authors acknowledge the use of the Carya and Sabine Cluster, and the advanced support from the Research Computing Data Core at UH.References
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS.
- Z. Li, Y. Xiao, P. R. Chowdhury, Z. Wu, T. Ma, J. Z. Chen, G. Wan, T. H. Kim, D. Jing, P. He, P. J. Potdar, L. Zhou, Z. Zeng, X. Ruan, J. T. Miller, J. P. Greeley, Y. Wu and A. Varma, Nat. Catal., 2021, 4(10), 882–891 CrossRef CAS.
- HYDROGEN STRATEGY Enabling A Low-Carbon Economy, https://www.energy.gov/sites/prod/files/2020/07/f76/USDOE_FE_Hydrogen_Strategy_July2020.pdf, (accessed 30 March 2023).
- B. Christian Enger, R. Lødeng and A. Holmen, Appl. Catal., A, 2008, 346, 1–27 CrossRef.
- A. T. Ashcroft, A. K. Cheetham, M. L. H. Green and P. D. F. Vernon, Nature, 1991, 352(6332), 225–226 CrossRef CAS.
- J. H. Ryu, K. Y. Lee, H. J. Kim, J. Il Yang and H. Jung, Appl. Catal., B, 2008, 80, 306–312 CrossRef CAS.
- E. P. J. Mallens, J. H. B. J. Hoebink and G. B. Marin, Catal. Lett., 1995, 33, 291–304 CrossRef CAS.
- B. Christian Enger, R. Lødeng and A. Holmen, Appl. Catal., A, 2008, 346, 1–27 CrossRef.
- Y. Zhu, S. Zhang, J. Shan, L. Nguyen, S. Zhan, X. Gu and F. Tao, ACS Catal., 2013, 3, 2627–2639 CrossRef CAS.
- D. A. Hickman and L. D. Schmidt, Science, 1993, 259, 343–346 CrossRef CAS.
- J. B. Claridge, M. L. H. Green, S. C. Tsang, A. P. E. York, A. T. Ashcroft and P. D. Battle, Catal. Lett., 1993, 22, 299–305 CrossRef CAS.
- R. F. Hicks, H. Qi, M. L. Young and R. G. Lee, J. Catal., 1990, 122, 280–294 CrossRef CAS.
- P. A. Carlsson, E. Fridell and M. Skoglundh, Catal. Lett., 2007, 115, 1–7 CrossRef CAS.
- Y. H. Chin, C. Buda, M. Neurock and E. Iglesia, J. Am. Chem. Soc., 2011, 133, 15958–15978 CrossRef CAS PubMed.
- Y. H. Chin, C. Buda, M. Neurock and E. Iglesia, J. Am. Chem. Soc., 2013, 135, 15425–15442 CrossRef CAS PubMed.
- P. L. Silveston and R. R. Hudgins, Periodic Operation of Chemical Reactors, 2012, pp. 1–775 Search PubMed.
- M. A. Ardagh, O. A. Abdelrahman and P. J. Dauenhauer, ACS Catal., 2019, 9, 6929–6937 CrossRef CAS.
- J. Fu, S. Liu, W. Zheng, R. Huang, C. Wang, A. Lawal, K. Alexopoulos, S. Liu, Y. Wang, K. Yu, J. A. Boscoboinik, Y. Liu, X. Liu, A. I. Frenkel, O. A. Abdelrahman, R. J. Gorte, S. Caratzoulas and D. G. Vlachos, Nat. Catal., 2022, 5(2), 144–153 CrossRef CAS.
- P. J. Dauenhauer, M. A. Ardagh, M. Shetty, A. Kuznetsov, Q. Zhang, P. Christopher, D. G. Vlachos and O. A. Abdelrahman, Chem. Sci., 2020, 11, 3501–3510 RSC.
- K. Karinshak, P. W. Chen, R.-F. Liu, S. J. Golden and M. P. Harold, Appl. Catal., B, 2021, 120607 Search PubMed.
- J. Stötzel, R. Frahm, B. Kimmerle, M. Nachtegaal and J. D. Grunwaldt, J. Phys. Chem. C, 2012, 116, 599–609 CrossRef.
- M. Fathi, E. Bjorgum, T. Viig and O. A. Rokstad, Catal. Today, 2000, 63, 489–497 CrossRef CAS.
- T. Franken, M. Roger, A. W. Petrov, A. H. Clark, M. Agote-Arán, F. Krumeich, O. Kröcher and D. Ferri, ACS Catal., 2021, 11, 4870–4879 CrossRef CAS.
- D. Creaser, B. Andersson, R. R. Hudgins and P. L. Silveston, Chem. Eng. Sci., 1999, 54, 4437–4448 CrossRef CAS.
- G. Rambeau and H. Amariglio, Appl. Catal., 1981, 1, 291–302 CrossRef CAS.
- S. Fouladvand, M. Skoglundh and P. A. Carlsson, Chem. Eng. J., 2016, 292, 321–325 CrossRef CAS.
- S. Marino, L. Wei, M. Cortes-Reyes, Y. Cheng, P. Laing, G. Cavataio, C. Paolucci and W. Epling, JACS Au, 2023, 3, 459–467 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B, 1999, 59, 1758 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B, 1996, 54, 11169 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1993, 47, 558–561 CrossRef CAS PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029 CrossRef CAS.
- J. P. Perdew and K. Burke, Phys. Rev. B, 1996, 54, 16533 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- P. E. Blöchl, Phys. Rev. B, 1994, 50, 17953 CrossRef.
- H. Kubota, S. Mine, T. Toyao, Z. Maeno and K. I. Shimizu, ACS Catal., 2022, 12, 544–559 CrossRef CAS.
- A. T. F. Batista, D. Wisser, T. Pigeon, D. Gajan, F. Diehl, M. Rivallan, L. Catita, A. S. Gay, A. Lesage, C. Chizallet and P. Raybaud, J. Catal., 2019, 378, 140–143 CrossRef CAS.
- A. J. Hoffman, C. Asokan, N. Gadinas, E. Schroeder, G. Zakem, S. V. Nystrom, A. B. Getsoian, P. Christopher and D. Hibbitts, ACS Catal., 2022, 12, 11697–11715 CrossRef CAS.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, J. Catal., 2004, 226, 54–68 CrossRef CAS.
- M. Salmerón, L. Brewer and G. A. Somorjai, Surf. Sci., 1981, 112, 207–228 CrossRef.
- M. A. Van Spronsen, J. W. M. Frenken and I. M. N. Groot, Nat. Commun., 2017, 8, 429 CrossRef.
- D. Hibbitts and M. Neurock, Surf. Sci., 2016, 650, 210–220 CrossRef CAS.
- E. Becker, P. A. Carlsson, L. Kylhammar, M. A. Newton and M. Skoglundh, J. Phys. Chem. C, 2011, 115, 944–951 CrossRef CAS.
- P. Raybaud, C. Chizallet, C. Mager-Maury, M. Digne, H. Toulhoat and P. Sautet, J. Catal., 2013, 308, 328–340 CrossRef CAS.
- C. M. Kalamaras, G. G. Olympiou and A. M. Efstathiou, Catal. Today, 2008, 138, 228–234 CrossRef CAS.
- J. G. Jakobsen, M. Jakobsen, I. Chorkendorff and J. Sehested, Catal. Lett., 2010, 140, 90–97 CrossRef CAS.
- A. I. Osman, J. Meudal, F. Laffir, J. Thompson and D. Rooney, Appl. Catal., B, 2017, 212, 68–79 CrossRef CAS.
- J. Saavedra, C. J. Pursell and B. D. Chandler, J. Am. Chem. Soc., 2018, 140, 3712–3723 CrossRef CAS PubMed.
- C. Guan, Y. Yang, Y. Pang, Z. Liu, S. Li, E. I. Vovk, X. Zhou, J. P. H. Li, J. Zhang, N. Yu, L. Long, J. Hao and A. P. van Bavel, J. Catal., 2021, 396, 202–214 CrossRef CAS.
- M. J. Hazlett and W. S. Epling, Catal. Today, 2021, 360, 401–410 CrossRef CAS.
- M. J. Hazlett, M. Moses-Debusk, J. E. Parks, L. F. Allard and W. S. Epling, Appl. Catal., B, 2017, 202, 404–417 CrossRef CAS.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74–78 CrossRef CAS PubMed.
- G. Germani and Y. Schuurman, AIChE J., 2006, 52, 1806–1813 CrossRef CAS.
- T. Bunluesin, R. J. Gorte and G. W. Graham, Appl. Catal., B, 1998, 15, 107–114 CrossRef CAS.
- H.-V. Tran, H. A. Doan, B. D. Chandler and L. C. Grabow, Curr. Opin. Chem. Eng., 2016, 13, 100–108 CrossRef.
- S. C. Ammal and A. Heyden, J. Catal., 2013, 306, 78–90 CrossRef CAS.
- S. Aranifard, S. C. Ammal and A. Heyden, J. Catal., 2014, 309, 314–324 CrossRef CAS.
- D. Duprez, Catal. Today, 2006, 112, 17–22 CrossRef CAS.
- L. C. Grabow, A. A. Gokhale, S. T. Evans, J. A. Dumesic and M. Mavrikakis, J. Phys. Chem. C, 2008, 112, 4608–4617 CrossRef CAS.
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Science, 2015, 350, 189–192 CrossRef CAS PubMed.
- A. A. Phatak, N. Koryabkina, S. Rai, J. L. Ratts, W. Ruettinger, R. J. Farrauto, G. E. Blau, W. N. Delgass and F. H. Ribeiro, Catal. Today, 2007, 123, 224–234 CrossRef CAS.
- C. M. Kalamaras, P. Panagiotopoulou, D. I. Kondarides and A. M. Efstathiou, J. Catal., 2009, 264, 117–129 CrossRef CAS.
- D. C. Grenoble, M. M. Estadt and D. F. Ollis, J. Catal., 1981, 67, 90–102 CrossRef CAS.
- T. Shido and Y. Iwasawa, J. Catal., 1993, 141, 71–81 CrossRef CAS.
- G. Jacobs, L. Williams, U. Graham, G. A. Thomas, D. E. Sparks and B. H. Davis, Appl. Catal., A, 2003, 252, 107–118 CrossRef CAS.
- S. C. Ammal and A. Heyden, ACS Catal., 2019, 9, 7721–7740 CrossRef CAS.
- K. B. Sravan Kumar, T. N. Whittaker, C. Peterson, L. C. Grabow and B. D. Chandler, J. Am. Chem. Soc., 2020, 142, 5760–5772 CrossRef CAS.
- T. Whittaker, K. B. S. Kumar, C. Peterson, M. N. Pollock, L. C. Grabow and B. D. Chandler, J. Am. Chem. Soc., 2018, 140, 16469–16487 CrossRef CAS.
- A. Balzarotti and A. Bianconi, Phys. Status Solidi B, 1976, 76, 689–694 CrossRef CAS.
- B. Ealet, M. H. Elyakhloufi, E. Gillet and M. Ricci, Thin Solid Films, 1994, 250, 92–100 CrossRef CAS.
- C. Spreafico, W. Karim, Y. Ekinci, J. A. Van Bokhoven and J. Van De Vondele, J. Phys. Chem. C, 2017, 121, 17862–17872 CrossRef CAS.
- G. R. Jenness, M. A. Christiansen, S. Caratzoulas, D. G. Vlachos and R. J. Gorte, J. Phys. Chem. C, 2014, 118, 12899–12907 CrossRef CAS.
- B. Ealet, M. H. Elyakhloufi, E. Gillet and M. Ricci, Thin Solid Films, 1994, 250, 92–100 CrossRef CAS.
- D. Ciuparu, F. Bozon-Verduraz and L. Pfefferle, J. Phys. Chem. B, 2002, 106, 3434–3442 CrossRef CAS.
- R. D. Johnson III, NIST Computational Chemistry Comparison and Benchmark Database.
- Z. Zhou, M. P. Harold and D. Luss, Ind. Eng. Chem. Res., 2021, 60, 6465–6482 CrossRef CAS.
- P. W. Chen, D. Maiti, R.-F. Liu, L. C. Grabow and M. P. Harold, Catal. Sci. Technol., 2022, 12, 2618–2633 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Solanki_ESI.docx. See DOI: https://doi.org/10.1039/d3re00554b |
| This journal is © The Royal Society of Chemistry 2024 |