
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bright near-infrared emission from the Au39(SR)29 nanocluster†
Zhongyu
Liu‡
 a,
Lianshun
Luo‡
a,
Jie
Kong
b,
Ellen
Kahng
a,
Meng
Zhou
b and
Rongchao
Jin
*a
a,
Lianshun
Luo‡
a,
Jie
Kong
b,
Ellen
Kahng
a,
Meng
Zhou
b and
Rongchao
Jin
*a
aDepartment of Chemistry, Carnegie Mellon University. Pittsburgh, PA 15213, USA. E-mail: rongchao@andrew.cmu.edu
bHefei National Laboratory for Physical Sciences at the Microscale, Department of Chemical Physics, University of Science and Technology of China. Hefei, Anhui 230026, China
First published on 18th March 2024
Abstract
The synthesis of atomically precise gold nanoclusters with high photoluminescence quantum yield (PLQY) in the near-infrared (NIR) region and understanding their photoluminescence mechanism are crucial for both fundamental science and practical applications. Herein, we report a highly luminescent, molecularly pure Au39(PET)29 (PET = 2-phenylethanethiolate) nanocluster with PLQY of 19% in the NIR range (915 nm). Steady state and time-resolved PL analyses, as well as temperature-dependent PL measurements reveal the emission nature of Au39(PET)29, which consists of prompt fluorescence (weak), thermally activated delayed fluorescence (TADF), and phosphorescence (predominant). Furthermore, strong dipole–dipole interaction in the solid-state (e.g., Au39(PET)29 nanoclusters embedded in a polystyrene thin-film) is found to narrow the energy gap between the S1 and T1 states, which results in faster intersystem crossing and reverse intersystem crossing; thus, the ratio of TADF to phosphorescence varies and the total PLQY is increased to 32%. This highly luminescent nanocluster holds promise in imaging, sensing and optoelectronic applications.
Introduction
Atomically precise metal nanoclusters (NCs) have attracted much attention owing to their intriguing electronic structure and rich properties.1–6 In recent studies on the electronic and optical properties of NCs, photoluminescence has become one of the thrusts.3,4,6–25 Unlike organic dyes and quantum dots, the emission of gold nanoclusters exhibits a larger Stokes shift and falls in the near-infrared (NIR, 800–2500 nm).8,20,22,24–30 The NIR luminescence, high stability and good biocompatibility of gold nanoclusters make them ideal candidates for applications in bioimaging,22,23 medicine,31,32 and optoelectronics.6With the successful development of synthetic methods such as size focusing and transformation,33–35 molecularly pure, thiolate-protected gold NCs have been achieved.1 However, the photoluminescence quantum yields (PLQY) of most gold NCs are quite low. For instance, the PLQY of the icosahedral Au25(SR)18−, bi-icosahedral Au38(SR)24, face-centered-cubic Au28(SR)20, Au36(SR)24, Au44(SR)28 and cuboctahedral Au23(SR)16− are all found to be only ∼1%.27,30,36–39 While the Au22(SG)18 and Au10(SR)6 show very high PLQY after surface engineering,3,4 their emissions are in the visible range. In the NIR region, only few thiolate-protected gold nanoclusters with atomic precision have been found to have PLQY higher than 10%, including the body-centered-cubic Au38 NC with a 15% PLQY in the NIR reported by Li et al.,26 and the rod-shaped Au42 NC with a 12% PLQY of dual emission in the NIR reported by Luo et al.40 Therefore, strong emission is still highly desirable for thiolate-protected gold NCs and worth significant efforts.
Several strategies have been developed to improve the PLQY of gold NCs. Xie's group reported an aggregation-induced emission mechanism and found that it can significantly increase the PLQY of a series of gold NCs.41,42 This aggregation-induced enhancement was also studied by the groups of Nieh, Zang and Zhu.22,43–45 A related crystallization-induced emission enhancement was found in Au8 NCs by the Sun group.46 Lee and coworkers reported a surface rigidifying strategy for achieving highly luminescent Au22 NCs.3 Tsukuda's group found that heteroatom doping in Au25(SR)18 can tune the electron distribution and increase the stiffness of the Au13 inner core and hence largely improve the PLQY.39 They also found that the mechanism can be applied to phosphine-protected Au13 NCs.47,48 Moreover, ligand engineering was introduced by Jin's group49 to improve the PLQY of Au25(SR)18 and has been proved to be a facile and universal strategy in recent work.50–53
Understanding the PL mechanism of gold NCs is crucial for improving their PLQY. However, the excited state electron dynamics of gold NCs is much more complicated than conventional luminescent materials and requires careful analysis.30,54 In recent work, Li et al. reported a photo-induced structural distortion accompanied by electron redistribution in three bi-tetrahedral gold NCs.55 Fluorescence and phosphorescence may coexist in the NCs40 and their conversion has also been reported by Konishi.15 Wang's group identified a thermally activated delayed fluorescence (TADF) in alkynyl-protected Au22 NCs,10 and TADF also exists in Ag22 NCs.12 Therefore, the PL mechanism of gold NCs is quite complicated, and both the core structure6,7 and the ligands28,50 of NCs are involved in the PL processes.
Herein, we report an Au39(PET)29 NC with strong emission in the NIR and PLQY of 19% in solution and 32% in films at room temperature. Combining the room temperature time-resolved PL measurements and temperature-dependent PL analysis, we reveal that the room temperature PL of Au39(PET)29 consists of three components: nanosecond prompt fluorescence (weak), sub-microsecond TADF, and microsecond phosphorescence (predominant). Further analysis identifies an ultrasmall gap between the singlet and triplet states (∼50 meV), hence, an overlap of fluorescence and phosphorescence peaks in the PL spectrum. By embedding Au39(PET)29 NCs in polystyrene (PS) thin-films, much faster intersystem crossing and reverse intersystem crossing are resulted. Overall, the attainment of a highly NIR-luminescent nanocluster and mechanistic insights promotes the fundamental understanding of the PL of NCs and the development of their future applications.
Results and discussion
Synthesis and mass spectrometry characterization
The Au39(PET)29 NC was synthesized by a facile one-pot protocol that involves two primary steps: (1) the reaction between a NHC–Au–Br complex (NHC = N-heterocyclic carbene) and phenylethanethiol, which yields white insoluble Au(I)-SR complexes, and (2) a slow reduction of Au(I)-SR complexes using excess (CH3)3CNH2·BH3. The utilization of NHC–Au–Br complexes for kinetic control of Au(I)-SR polymer generation, alongside the weak reducing properties of (CH3)3CNH2BH3 compared to NaBH4, facilitated the successful synthesis of the Au39 nanocluster. Additionally, the ease of modifying the nitrogen substituents adjacent to the carbenic carbon in NHC allows for precise manipulation of electron density, thereby enabling accurate regulation of mass distribution in the generated Au(I)-SR polymers, a process being difficult to achieve with traditional thiol-Au and phosphine-Au complexes. After removing the solvent and washing with methanol, a black crude product was obtained (see ESI† for details). Pure Au39(PET)29 was isolated from the crude product by thin-layer chromatography (TLC) (Fig. S1†). Electrospray ionization (ESI) mass spectrometry was performed to determine the formula of the as-obtained gold NC. Four characteristic peaks at m/z 5830.70, 5963.63, 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 661.20 and 11793.89 are observed (Fig. 1A), which are identified as [Au39(PET)29]2+, [Au39(PET)29 + 2Cs]2+, [Au39(PET)29]+ and [Au39(PET)29 + Cs]+, respectively; note that the charges are generated by electrospray or adduction of Cs+ ions, while the native Au39(PET)29 is charge-neutral. The experimental isotope patterns match well with the calculated ones (Fig. 1B). It is worth noting that a decade ago, Meng et al. observed the Au39(PET)29 NC in ESI-MS analysis of a size-mixed product, but no pure Au39(PET)29 was isolated from the mixture56 because the TLC method for isolating NCs had not been developed until later work.57 Thus, molecularly pure Au39(PET)29 is for the first time achieved in the current work, which permits a detailed study on its PL properties (vide infra). Crystallization of Au39(PET)29 unfortunately has no success yet, thus, its structure is still unknown.
661.20 and 11793.89 are observed (Fig. 1A), which are identified as [Au39(PET)29]2+, [Au39(PET)29 + 2Cs]2+, [Au39(PET)29]+ and [Au39(PET)29 + Cs]+, respectively; note that the charges are generated by electrospray or adduction of Cs+ ions, while the native Au39(PET)29 is charge-neutral. The experimental isotope patterns match well with the calculated ones (Fig. 1B). It is worth noting that a decade ago, Meng et al. observed the Au39(PET)29 NC in ESI-MS analysis of a size-mixed product, but no pure Au39(PET)29 was isolated from the mixture56 because the TLC method for isolating NCs had not been developed until later work.57 Thus, molecularly pure Au39(PET)29 is for the first time achieved in the current work, which permits a detailed study on its PL properties (vide infra). Crystallization of Au39(PET)29 unfortunately has no success yet, thus, its structure is still unknown.
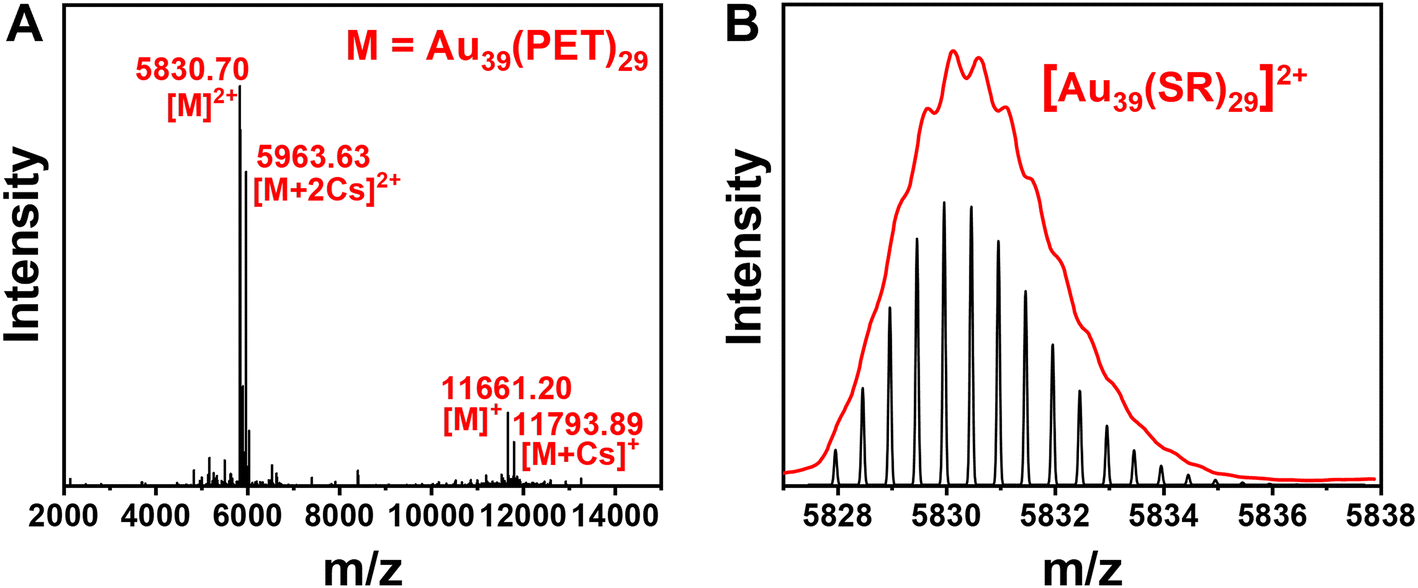 | ||
| Fig. 1 (A) ESI mass spectrum of Au39(PET)29 (CsOAc was added to facilitate ESI analysis); (B) experimental isotope pattern (red profile) of m/z at 5830.70 (corresponding to [Au39(SR)29]2+ according to the spacing of 0.5, red profile) and the theoretical pattern (black). | ||
Optical properties of Au39(PET)29
To characterize the optical properties of Au39, we use CDCl3 as the solvent because it has weaker NIR absorption (i.e., vibrational overtones) than other solvents and can thus alleviate the solvent absorption effect; the latter distorts the peaks of NCs. The dissolved Au39(PET)29 (abbrev. Au39 hereafter) in CDCl3 is a dark red solution (Fig. 2A), with the UV-vis absorption spectrum shown in Fig. 2B (solid line). Two main absorption peaks are centered at 534 and 730 nm. The optical gap of Au39 is determined to be 1.46 eV by extrapolating the lowest energy absorption peak to zero absorbance (Fig. 2B, inset).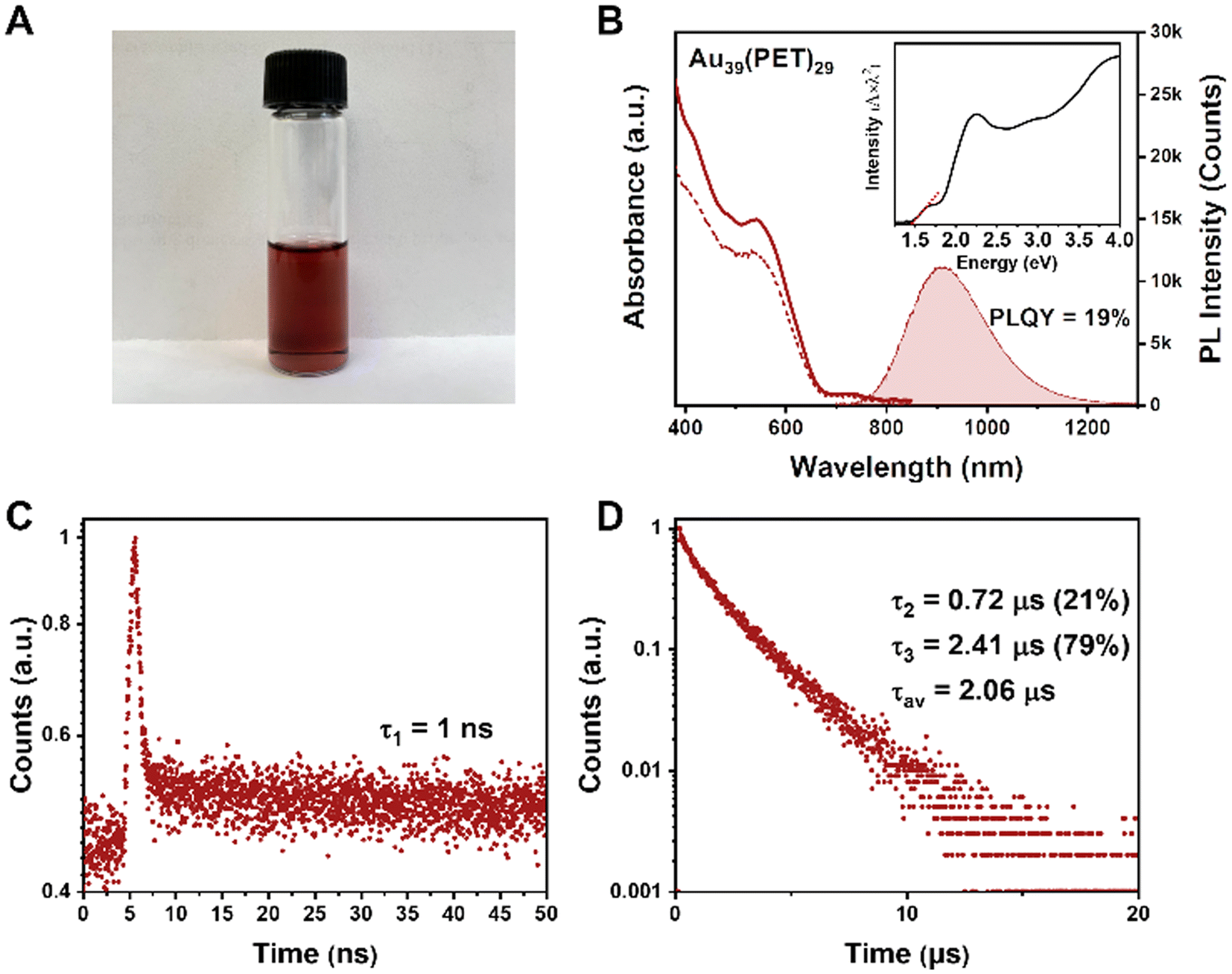 | ||
| Fig. 2 (A) A photo of Au39(PET)29 dissolved in CDCl3. (B) UV-vis absorption (solid line), PL excitation (dashed line) and PL emission (shaded) spectra of Au39. (C) PL decay profile measured on the 50 ns scale (by TCSPC mode). (D) Decay profile measured on the 20 μs scale (by MCS mode). For PL measurements: excitation at 400 nm, slit width 5 nm, and emission slit 5 nm. Deaerated CDCl3 (with N2 or helium) solutions of Au39 NCs were used in all optical measurements unless otherwise noted. | ||
The Au39 NC exhibits quite intense PL in the NIR region. Under 400, 534 or 730 nm excitation, Au39 shows an identical emission peak at 915 nm (Fig. 2B, the shaded area). The dashed line in Fig. 2B shows the photoluminescence excitation (PLE) spectrum, which is consistent with the absorption profile, indicating that the emission is from the core and its first excited state.54,58 The PLQY of Au39 in deaerated CDCl3 is measured to be 19% by the absolute method using an integrating sphere (Fig. S2†) and 17% under ambient conditions.
Interestingly, although only a single peak is observed in the emission spectrum, time-resolved PL measurements in nanosecond and microsecond regimes show multiple components (Fig. 2C and D). Of note, the microsecond decay was measured by the multi-channel scaling (MCS) single photon counting mode, whereas the nanosecond lifetime was determined by the time-correlated single-photon counting (TCSPC) mode. The resolution of MCS is 10 ns, which is why the PL components shorter than 10 ns cannot be observed in Fig. 2D. The TCSPC mode has a resolution of 0.4 ns (excitation pulses ∼100 ps), and the decay curve in the nanosecond regime suggests the existence of a 1 ns (denoted τ1) lifetime (Fig. 2C). In contrast, the decay curve in the microsecond time regime requires a biexponential function for fitting, i.e., 0.72 μs (τ2) and 2.41 μs (τ3). The fitting residuals are of 3σ or smaller (σ = standard deviation). The intensity-averaged lifetime is calculated to be 2.06 μs (Fig. 2D). A comparison of PL spectra for Au39 in different solvents is given in Fig. S3,† from which one can see that the solvent effect on the PL spectra is negligible.
Under the O2 atmosphere, the PL intensity of Au39 was found to be slightly suppressed to 87% (Fig. 3A), and the characteristic phosphorescence from singlet oxygen (1O2, an excited state of 3O2) was observed at 1273 nm (Fig. 3A, inset), suggesting that the PL of Au39 involves a contribution from phosphorescence. The lifetime measurements showed that τ3 is shortened to 1.92 μs while the other two components remain almost the same (Fig. 3B and C). It is widely acknowledged that the lifetime of fluorescence is on the nanosecond scale and the lifetime of phosphorescence is typically over one microsecond.11,15,40,47 Therefore, by combining the measured lifetimes with the observed oxygen dependence or independence, we assign τ1 as fluorescence from the first singlet state (S1) and τ3 as phosphorescence from the first triplet state (T1). The origin of τ2 is intriguing and to be discussed below. It is worth noting that the quench of phosphorescence under O2 is commonly not complete for metal NCs as reported by Mitsui et al.59 due to a combination of several factors, including low solubility and slow diffusion rate of oxygen in organic solvents, inefficient charge transfer or energy exchange between metal NCs and oxygen. The existence of a microsecond lifetime component under the O2 atmosphere supports that the phosphorescence of Au39 is not completely quenched by oxygen. The oxygen quenching rate kq is estimated to be 1.1 × 107 s−1M−1 by τ = 1/(kr + knr + [O2]kq) using the experimental results of τ at oxygen concentrations [O2] = 0 (for the N2 case) and 11.5 mM (for the O2 case).
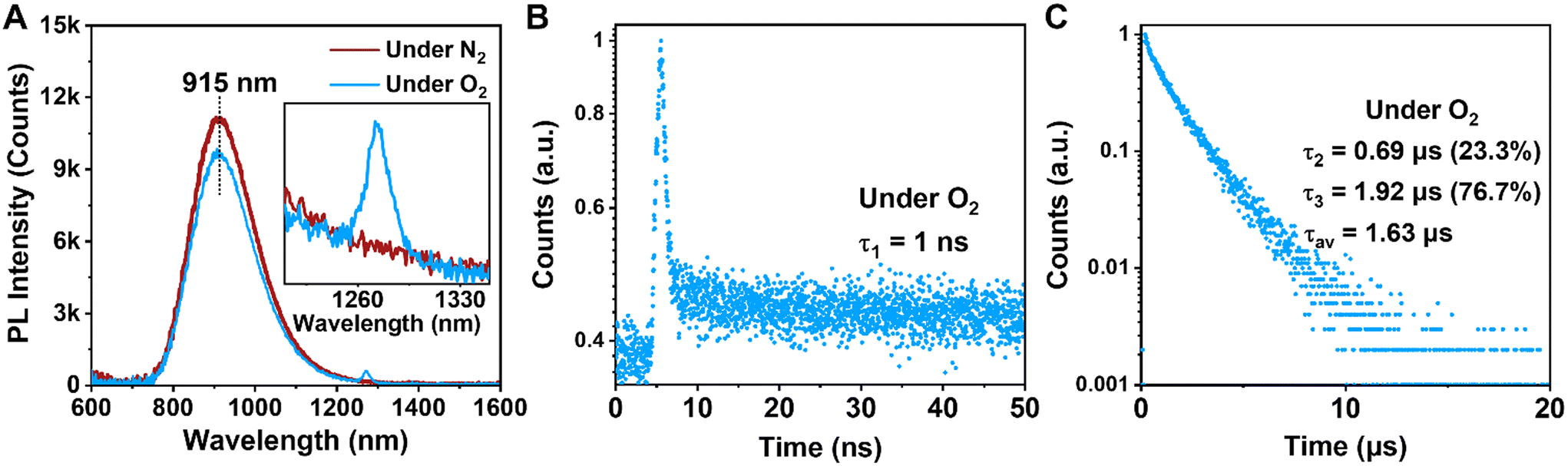 | ||
| Fig. 3 (A) PL spectra of Au39 under N2 atmosphere (dark red line) and O2 atmosphere (blue line). Inset shows the expanded region where singlet O2 emits. (B) Decay profile measured on the 50 ns scale (measured by TCSPC). (C) Decay profile measured on the 20 μs scale (measured by MCS). | ||
Temperature-dependent PL analysis
Given the fact that there is no observation of emission from higher excited states, τ2 should arise from the first excited state. To fully understand the electron dynamics, we carried out temperature-dependent PL measurements from room temperature down to 80 K using liquid nitrogen as the cryogen. The Au39 NCs were dissolved in 2-methyl-tetrahydrofuran (abbrev. 2-methyl-THF) for ‘glass’ formation at cryogenic temperatures. The sample chamber of cryostats was filled with helium gas during the measurements to avoid the interference from oxygen.With decreasing temperature from 298 K to 80 K, we observed a blue shift of the PL peak and its narrowing (from 310 to 180 meV, full width at half maximum) but no splitting of the emission peak during the entire measurements (Fig. S4A†). The PL intensity of Au39 was found to increase by 5 times from 298 K to 80 K, indicating a near unity PLQY at 80 K. Time-resolved PL measurements were performed in the same temperature range to trace the evolution of τ2 (Fig. 4A and Fig. S5, S6†). The lifetime fitting results are listed in Table 1, and the average lifetime is plotted in Fig. 4A inset. As shown in Table 1, all three lifetime components are found to increase from 298 K to 160 K. Interestingly, when the temperature was 120 K and lower, τ2 disappeared while τ1 and τ3 still remained. The disappearance of τ2 at low temperatures is attributed to a thermally activated kinetic process.10,48,60,61 We also embedded Au39 NCs in a polystyrene (PS) thin-film and measured the film's temperature-dependent PL spectra (Fig. S4B†). Similar to the solution sample, we observed a gradual blueshift of the peak position, an enhancement of PL intensity, and peak narrowing as the temperature decreases. The PL excitation spectrum for Au39 in thin films at 80 K is consistent with its 2-methyl-THF counterpart (Fig. S7†). Furthermore, temperature-dependent PL decays were measured (Fig. 4B and Fig. S8, S9†) and quantitative analysis found that all three lifetime components remained at 80 K (Table 2), suggesting that the kinetic process for τ2 can be affected by overlapping of excited state wavefunctions in the thin film,40 resulting in τ2 survival in the film state, but not in the solution state.
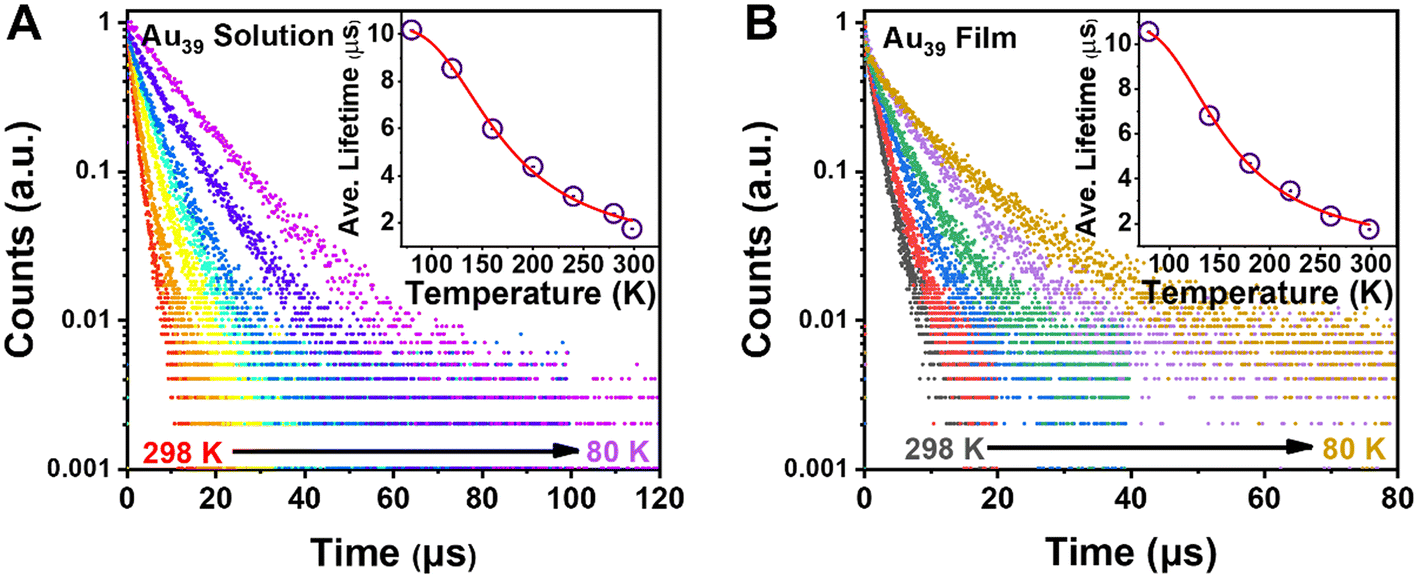 | ||
| Fig. 4 (A) Temperature-dependent PL decay profiles of Au39 in 2-methyl-THF with excitation at 400 nm. Inset: plot of average lifetime and fitting by eqn (2). (B) Temperature-dependent PL decay profiles of Au39 in films. Inset: plot of average lifetime and fitting by eqn (2) (see main text). | ||
| Temperature | τ 1 (ns) weak | τ 2 (μs) | τ 3 (μs) | τ av (μs) |
|---|---|---|---|---|
| 298 K | 0.94 | 0.87 (37.9%) | 2.29 (62.1%) | 1.75 |
| 280 K | 0.93 | 1.27 (40.7%) | 3.19 (59.3%) | 2.4 |
| 240 K | 0.95 | 1.95 (40.9%) | 4.01 (59.1%) | 3.13 |
| 200 K | 1.18 | 2.61 (28.8%) | 5.12 (71.2%) | 4.39 |
| 160 K | 1.31 | 2.72 (17.3%) | 6.68 (82.7%) | 5.99 |
| 120 K | 1.60 | None | 8.55 (100%) | 8.55 |
| 80 K | 1.68 | None | 10.18 (100%) | 10.18 |
| Temperature | τ 1 (ns) weak | τ 2 (μs) | τ 3 (μs) | τ av (μs) |
|---|---|---|---|---|
| 298 K | 1.23 | 0.55 (16.7%) | 1.99 (83.3%) | 1.75 |
| 260 K | 1.50 | 1.38 (26.2%) | 2.68 (73.8%) | 2.34 |
| 220 K | 1.18 | 1.87 (32.9%) | 4.24 (67.1%) | 3.46 |
| 180 K | 1.45 | 2.10 (24.5%) | 5.53 (75.5%) | 4.69 |
| 140 K | 1.4 | 2.53 (20.4%) | 7.91 (79.6%) | 6.81 |
| 80 K | 1.33 | 4.22 (22.1%) | 12.34 (77.9%) | 10.55 |
Mechanistic insights
Combining the results of the room temperature steady-state PL spectra and temperature-dependent PL, we assign τ2 to a TADF process. The mechanism is shown in Scheme 1A–C. Thus, the PL peak of Au39 at room temperature consists of three radiative processes (Scheme 1A): nanosecond (τ1) prompt fluorescence (PF, weak), sub-microsecond (τ2) TADF, and microsecond (τ3) phosphorescence (intense). Normally, in organic TADF materials, only PF and TADF can be observed due to weak spin–orbit coupling, but in gold nanoclusters, the heavy gold atoms provide strong spin–orbit coupling, which enables an efficient T1 to S0 transition, hence, strong phosphorescence. Among the three components, the low signal-to-noise ratio for τ1 (Fig. 2C) indicates that the contribution from the PF is much smaller than the other two emission processes. From the relative intensities (see the % values in Table 2), phosphorescence is the main radiative process that contributes to the PL spectra. We calculated the radiative rate at different temperatures. As shown in Fig. S10,† the radiative rate decreases as the temperature decreases, which is typical of TADF materials and thus supports the existence of TADF in Au39 NCs.10,12,62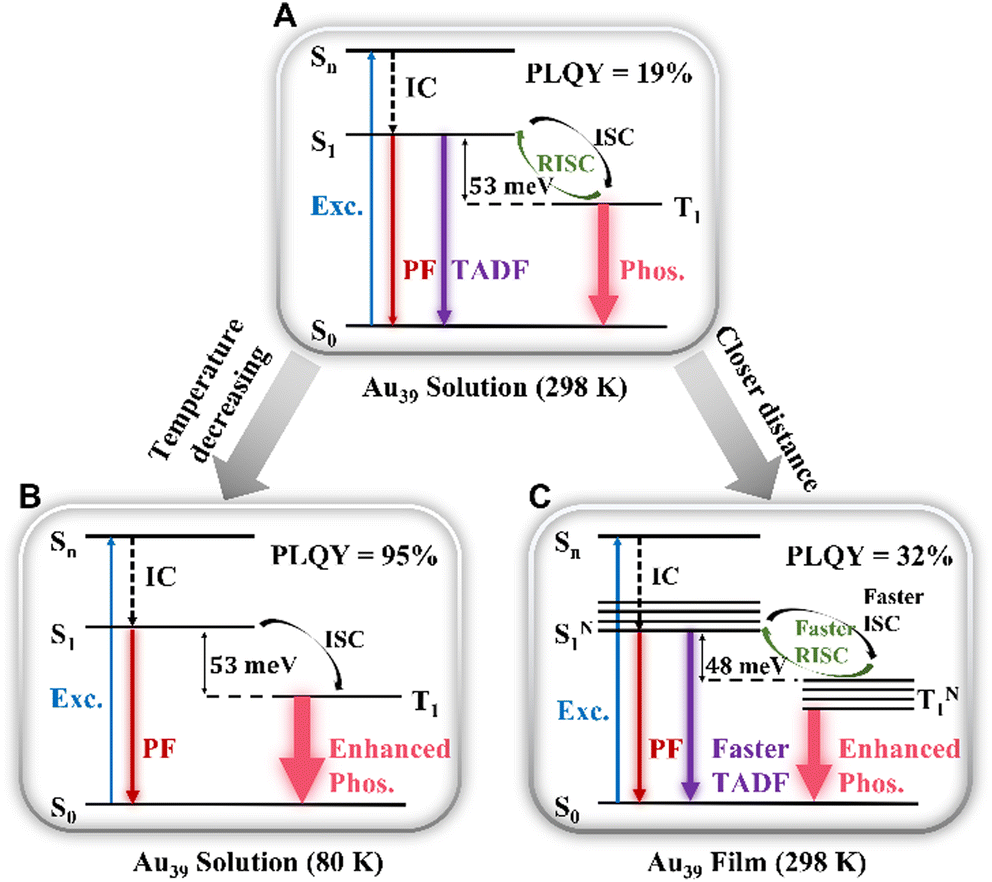 | ||
| Scheme 1 Emission mechanism of Au39 NCs in (A) solution (298 K), (B) solution (80 K), and (C) film (298 K). | ||
The temperature dependent trends of all three radiative components in Au39 solution are drawn in Scheme 1B. As the temperature decreases, the PF is only affected by the suppression of non-radiative relaxation between S1 and S0 (denoted as kSnr) which should lead to a monotonic trend. The suppression of kSnr will have an equal effect on the temperature-dependent TADF. Moreover, the temperature dependence of reverse intersystem crossing (kRISC) follows an Arrhenius formula (eqn (1)):63,64
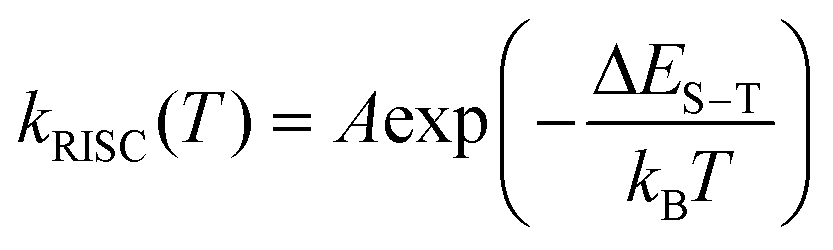 | (1) |
The observed temperature-dependent trend of Au39 NCs in 2-methyl-THF is different from the typical trend of organic TADF; for the latter, a decrease in TADF and a peak redshift would be observed. Here we further explain the Au39 results as follows. First, unlike organic TADF that exhibits no phosphorescence, phosphorescence is the main radiative process in Au39 NCs and it shows a monotonic increase as the temperature drops. The increase of phosphorescence is able to completely offset the decrease of TADF; thus, resulting in a continuous net increase of the total PL intensity (Fig. S4A†). The emission peak has a mix of three components above 140 K, but mainly the phosphorescence below 140 K. In Au39, the energy gap between S1 and T1 is extremely small (i.e., of meV, see below) as opposed to ∼0.2 eV in organic TADF, and the meV gap is overwhelmed by an enlargement (spectroscopically a blueshift) of the energy gap from the suppression of electron–phonon interaction37 when the temperature decreases, hence, a net blueshift of the peak position observed with decreasing temperature.
The kinetic process of Au39 NCs in the PS thin-film is shown in Scheme 1C. Similar to the solution phase, Au39 in PS thin-films also consists of three radiative processes but with a different ratio. As shown in Fig. S11,† the PLQY of Au39 NCs in PS thin-film is increased by 1.7 times, that is, 32%. The increase of PLQY in the Au39/PS thin-film can be ascribed to the suppression of surface staple vibrations in the solid-state.37,65 The existence of τ2 (i.e., TADF) in the Au39/PS thin-film even down to 80 K suggests that the reverse intersystem crossing is not yet sufficiently suppressed at this temperature (cf. the disappearance of τ2 at 120K in the solution system). We ascribe it to the strong dipole–dipole interaction in the solid-state because all NCs possess a dipole moment66 and they are much closer in the film than in solution. Such an interaction will lead to a narrowing of the energy gap between S1 and T1.40,67 Following the Arrhenius formula, a smaller ΔES−T will benefit both kISC and kRISC, hence, stronger TADF and phosphorescence. To completely suppress the RISC (kRISC) in the film state, a lower temperature than 80 K is needed, as opposed to the 140 K at which TADF is quenched in the solution system.
Here we estimate the singlet–triplet gap in Au39. Under the assumption of degenerate triplet states, an estimation of the ΔES−T can be given by fitting the intensity average lifetime to eqn (2):61
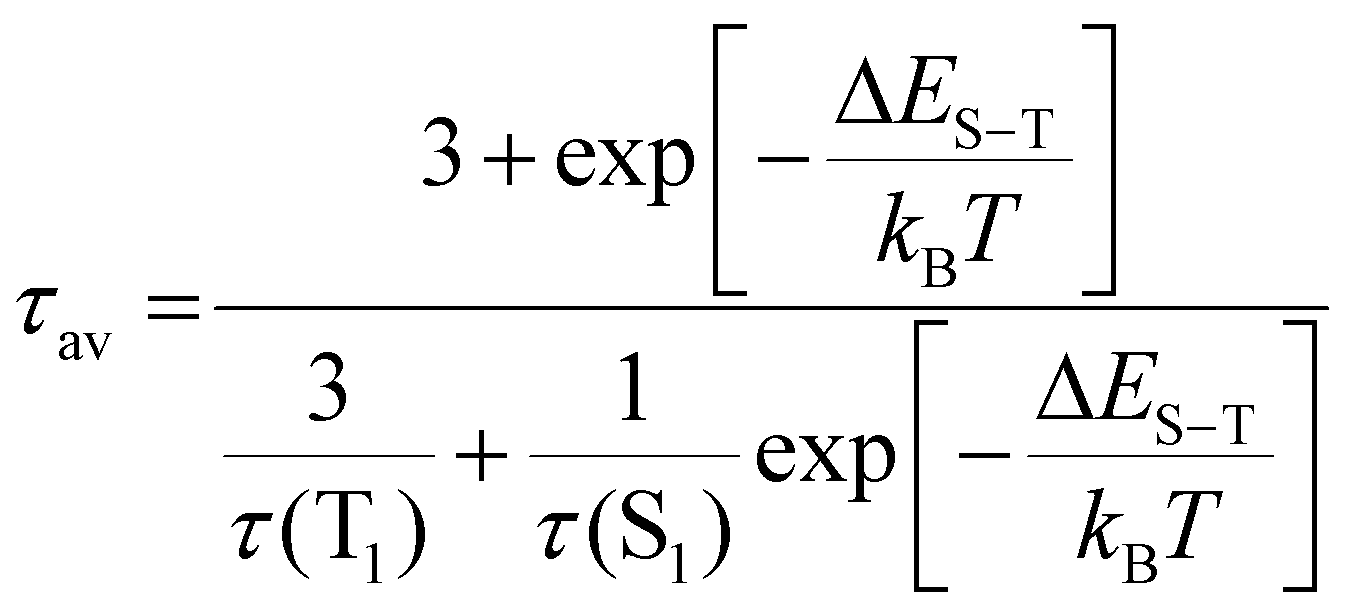 | (2) |
| Temperature | ΔES−T | τ(S1)/ns | τ(T1)/μs |
|---|---|---|---|
| Au39(PET)29 solution | 53 | 107 | 10.3 |
| Au39(PET)29/PS film | 48 | 113 | 10.8 |
Conclusion
In summary, we have synthesized a molecularly pure Au39(PET)29 nanocluster exhibiting strong near-infrared (915 nm) luminescence with QY of 19% in solution and 32% in films at room temperature, which is the highest in the NIR region among the thiolate-protected gold NCs. The steady-state and time-resolved PL measurements, as well as temperature-dependent PL measurements reveal that the single-peak emission of Au39(PET)29 consists of prompt fluorescence (weak), thermally activated delayed fluorescence, and phosphorescence (strong), and their merge into a single emission peak is due to the extremely small singlet–triplet gap (∼50 meV). Phosphorescence is identified to be the main contributor to the observed strong photoluminescence. Compared to the solution phase, Au39(PET)29 NCs in the solid-state have the suppressed surface vibrations and stronger dipolar interactions; the latter lead to a narrowing of the gap between S1 and T1 states. Both effects lead to a higher QY (32%, at room temperature) with predominant phosphorescence. The QYs of both the solution and film of Au39 reach near unity at 80 K. Overall, such NCs hold promise as a new class of luminescent (TADF/phosphorescent) materials having ultrafast ISC/RISC and short emission lifetimes (∼1 μs), which can potentially overcome the issue of roll-off of emitting efficiency in organic TADF devices and also extend to the NIR wavelengths for special applications.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
R. J. acknowledges the financial support from the Kaufman Foundation.References
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- K. Pyo, V. D. Thanthirige, K. Kwak, P. Pandurangan, G. Ramakrishna and D. Lee, J. Am. Chem. Soc., 2015, 137, 8244–8250 CrossRef CAS PubMed.
- Y. Zhong, J. Zhang, T. Li, W. Xu, Q. Yao, M. Lu, X. Bai, Z. Wu, J. Xie and Y. Zhang, Nat. Commun., 2023, 14, 658 CrossRef CAS PubMed.
- G. Deng, K. Lee, H. Deng, S. Malola, M. S. Bootharaju, H. Häkkinen, N. Zheng and T. Hyeon, Alkynyl-Protected Chiral Bimetallic Ag22Cu7 Superatom with Multiple Chirality Origins, Angew. Chem., Int. Ed., 2023, 62, e202217483 CrossRef CAS PubMed.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- J. Dong, Z. Gan, W. Gu, Q. You, Y. Zhao, J. Zha, J. Li, H. Deng, N. Yan and Z. Wu, Angew. Chem., Int. Ed., 2021, 60, 17932–17936 CrossRef CAS PubMed.
- Y. Li, M. Zhou, Y. Song, T. Higaki, H. Wang and R. Jin, Nature, 2021, 594, 380–384 CrossRef CAS PubMed.
- J. J. Li, C. Y. Liu, Z. J. Guan, Z. Lei and Q. M. Wang, Angew. Chem., 2022, 134, e202201549 CrossRef.
- X. S. Han, X. Luan, H. F. Su, J. J. Li, S. F. Yuan, Z. Lei, Y. Pei and Q. M. Wang, Angew. Chem., 2020, 132, 2329–2332 CrossRef.
- M. Mitsui, D. Arima, Y. Kobayashi, E. Lee and Y. Niihori, Adv. Opt. Mater., 2022, 10, 2200864 CrossRef CAS.
- Z.-R. Yuan, Z. Wang, B.-L. Han, C.-K. Zhang, S.-S. Zhang, Z.-Y. Zhu, J.-H. Yu, T.-D. Li, Y.-Z. Li, C.-H. Tung and D. Sun, Angew. Chem., Int. Ed., 2022, 61, e202211628 CrossRef CAS PubMed.
- J. W. Fagan, K. D. M. Weerawardene, A. Cirri, C. M. Aikens and C. J. Johnson, J. Chem. Phys., 2021, 155, 014301 CrossRef CAS PubMed.
- D. Bain, S. Maity, T. Debnath, A. K. Das and A. Patra, Mater. Res. Express, 2019, 6, 124004 CrossRef CAS.
- M. Sugiuchi, J. Maeba, N. Okubo, M. Iwamura, K. Nozaki and K. Konishi, J. Am. Chem. Soc., 2017, 139, 17731–17734 CrossRef CAS PubMed.
- E. Khatun, M. Bodiuzzaman, K. S. Sugi, P. Chakraborty, G. Paramasivam, W. A. Dar, T. Ahuja, S. Antharjanam and T. Pradeep, ACS Nano, 2019, 13, 5753–5759 CrossRef CAS PubMed.
- M. S. Bootharaju, C. P. Joshi, M. R. Parida, O. F. Mohammed and O. M. Bakr, Angew. Chem., 2016, 128, 934–938 CrossRef.
- M.-M. Zhang, X.-Y. Dong, Z.-Y. Wang, X.-M. Luo, J.-H. Huang, S.-Q. Zang and T. C. Mak, J. Am. Chem. Soc., 2021, 143, 6048–6053 CrossRef CAS PubMed.
- S.-S. Zhang, S. Havenridge, C. Zhang, Z. Wang, L. Feng, Z.-Y. Gao, C. M. Aikens, C.-H. Tung and D. Sun, J. Am. Chem. Soc., 2022, 144, 18305–18314 CrossRef CAS PubMed.
- P. J. Herbert, C. J. Ackerson and K. L. Knappenberger Jr., J. Phys. Chem. Lett., 2021, 12, 7531–7536 CrossRef CAS PubMed.
- C. Zhu, J. Xin, J. Li, H. Li, X. Kang, Y. Pei and M. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202205947 CrossRef CAS PubMed.
- A. T. Rad, Y. Bao, H. S. Jang, Y. Xia, H. Sharma, E. E. Dormidontova, J. Zhao, J. Arora, V. T. John, B. Z. Tang, T. Dainese, A. Hariri, J. V. Jokerst, F. Maran and M.-P. Nieh, Adv. Funct. Mater., 2021, 31, 2009750 CrossRef PubMed.
- Y. Huang, K. Chen, L. Liu, H. Ma, X. Zhang, K. Tan, Y. Li, Y. Liu, C. Liu, H. Wang and X.-D. Zhang, Small, 2023, 2300145 CrossRef CAS PubMed.
- H. Liu, G. Hong, Z. Luo, J. Chen, J. Chang, M. Gong, H. He, J. Yang, X. Yuan and L. Li, Adv. Mater., 2019, 31, 1901015 CrossRef CAS PubMed.
- T.-Q. Yang, B. Peng, B.-Q. Shan, Y.-X. Zong, J.-G. Jiang, P. Wu and K. Zhang, Nanomaterials, 2020, 10, 261 CrossRef CAS PubMed.
- Q. Li, C. J. Zeman IV, G. C. Schatz and X. W. Gu, ACS Nano, 2021, 15, 16095–16105 CrossRef CAS PubMed.
- L. Liao, C. Wang, S. Zhuang, N. Yan, Y. Zhao, Y. Yang, J. Li, H. Deng and Z. Wu, Angew. Chem., Int. Ed., 2020, 59, 731–734 CrossRef CAS PubMed.
- K. Konishi, M. Iwasaki and Y. Shichibu, Acc. Chem. Res., 2018, 51, 3125–3133 CrossRef CAS PubMed.
- M. Zhou and Y. Song, J. Phys. Chem. Lett., 2021, 12, 1514–1519 CrossRef CAS PubMed.
- Q. Li, M. Zhou, W. Y. So, J. Huang, M. Li, D. R. Kauffman, M. Cotlet, T. Higaki, L. A. Peteanu and Z. A. Shao, et al. , J. Am. Chem. Soc., 2019, 141, 5314–5325 CrossRef CAS PubMed.
- X. Jiang, B. Du, Y. Huang and J. Zheng, Nano Today, 2018, 21, 106–125 CrossRef CAS PubMed.
- B. Du, X. Jiang, A. Das, Q. Zhou, M. Yu, R. Jin and J. Zheng, Nat. Nanotechnol., 2017, 12, 1096–1102 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, A. Das and R. Jin, J. Phys. Chem. Lett., 2015, 6, 2976–2986 CrossRef CAS PubMed.
- S. Gratious, S. Mukherjee and S. Mandal, J. Phys. Chem. Lett., 2022, 13, 9014–9027 CrossRef CAS PubMed.
- S. Mukherjee, D. Jayakumar and S. Mandal, J. Phys. Chem. C, 2021, 125, 12149–12154 CrossRef CAS.
- T. D. Green, C. Yi, C. Zeng, R. Jin, S. McGill and K. L. Knappenberger Jr., J. Phys. Chem. A, 2014, 118, 10611–10621 CrossRef CAS PubMed.
- Z. Liu, Y. Li, E. Kahng, S. Xue, X. Du, S. Li and R. Jin, ACS Nano, 2022, 16, 18448–18458 CrossRef CAS PubMed.
- M. Zhou, C. Zeng, M. Y. Sfeir, M. Cotlet, K. Iida, K. Nobusada and R. Jin, J. Phys. Chem. Lett., 2017, 8, 4023–4030 CrossRef CAS PubMed.
- M. Suyama, S. Takano and T. Tsukuda, J. Phys. Chem. C, 2020, 124, 23923–23929 CrossRef CAS.
- L. Luo, Z. Liu, X. Du and R. Jin, J. Am. Chem. Soc., 2022, 144, 19243–19247 CrossRef CAS PubMed.
- Z. Wu, Q. Yao, S.-Q. Zang and J. Xie, Natl. Sci. Rev., 2021, 8, nwaa208 CrossRef PubMed.
- N. Goswami, Q. Yao, Z. Luo, J. Li, T. Chen and J. Xie, J. Phys. Chem. Lett., 2016, 7, 962–975 CrossRef CAS PubMed.
- Z. Han, X. Zhao, P. Peng, S. Li, C. Zhang, M. Cao, K. Li, Z.-Y. Wang and S.-Q. Zang, Nano Res., 2020, 13, 3248–3252 CrossRef CAS.
- Z. Wu, Q. Yao, O. J. H. Chai, N. Ding, W. Xu, S. Zang and J. Xie, Angew. Chem., 2020, 132, 10020–10025 CrossRef.
- S. Jin, W. Liu, D. Hu, X. Zou, X. Kang, W. Du, S. Chen, S. Wei, S. Wang and M. Zhu, Chem. – Eur. J., 2018, 24, 3712–3715 CrossRef CAS PubMed.
- W.-D. Si, K. Sheng, C. Zhang, Z. Wang, S.-S. Zhang, J.-M. Dou, L. Feng, Z.-Y. Gao, C.-H. Tung and D. Sun, Chem. Sci., 2022, 13, 10523–10531 RSC.
- S. Takano, H. Hirai, T. Nakashima, T. Iwasa, T. Taketsugu and T. Tsukuda, J. Am. Chem. Soc., 2021, 143, 10560–10564 CrossRef CAS PubMed.
- H. Hirai, S. Takano, T. Nakashima, T. Iwasa, T. Taketsugu and T. Tsukuda, Angew. Chem., Int. Ed., 2022, 61, e202207290 CrossRef CAS PubMed.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS PubMed.
- K. Sahoo and I. Chakraborty, Nanoscale, 2023, 15, 3120–3129 RSC.
- C. Yao, S. Tian, L. Liao, X. Liu, N. Xia, N. Yan, Z. Gan and Z. Wu, Nanoscale, 2015, 7, 16200–16203 RSC.
- B. Zhang, J. Chen, Y. Cao, O. J. H. Chai and J. Xie, Small, 2021, 17, 2004381 CrossRef CAS PubMed.
- M. R. Narouz, S. Takano, P. A. Lummis, T. I. Levchenko, A. Nazemi, S. Kaappa, S. Malola, G. Yousefalizadeh, L. A. Calhoun and K. G. Stamplecoskie, J. Am. Chem. Soc., 2019, 141, 14997–15002 CrossRef CAS PubMed.
- M. Zhou, T. Higaki, G. Hu, M. Y. Sfeir, Y. Chen, D.-E. Jiang and R. Jin, Science, 2019, 364, 279–282 CrossRef CAS PubMed.
- Q. Li, D. Zhou, J. Chai, W. Y. So, T. Cai, M. Li, L. A. Peteanu, O. Chen, M. Cotlet and X. W. Gu, et al. , Nat. Commun., 2020, 11, 2897 CrossRef CAS PubMed.
- X. Meng, Z. Liu, M. Zhu and R. Jin, Nanoscale Res. Lett., 2012, 7, 277 CrossRef PubMed.
- S. Tian, Y.-Z. Li, M.-B. Li, J. Yuan, J. Yang, Z. Wu and R. Jin, Nat. Commun., 2015, 6, 8667 CrossRef CAS PubMed.
- K. D. M. Weerawardene and C. M. Aikens, J. Am. Chem. Soc., 2016, 138, 11202–11210 CrossRef CAS PubMed.
- M. Mitsui, D. Arima, A. Uchida, K. Yoshida, Y. Arai, K. Kawasaki and Y. Niihori, J. Phys. Chem. Lett., 2022, 13, 9272–9278 CrossRef CAS PubMed.
- A. D. Wright, C. Verdi, R. L. Milot, G. E. Eperon, M. A. Pérez-Osorio, H. J. Snaith, F. Giustino, M. B. Johnston and L. M. Herz, Nat. Commun., 2016, 7, 11755 CrossRef PubMed.
- T. Hofbeck, U. Monkowius and H. Yersin, J. Am. Chem. Soc., 2015, 137, 399–404 CrossRef CAS PubMed.
- Y. Wang, Z. Liu, A. Mazumder, C. G. Gianopoulos, K. Kirschbaum, L. A. Peteanu and R. Jin, J. Am. Chem. Soc., 2023, 145, 26328–26338 CrossRef CAS PubMed.
- A. Niwa, T. Kobayashi, T. Nagase, K. Goushi, C. Adachi and H. Naito, Appl. Phys. Lett., 2014, 104, 213303 CrossRef.
- N. Aizawa, Y.-J. Pu, Y. Harabuchi, A. Nihonyanagi, R. Ibuka, H. Inuzuka, B. Dhara, Y. Koyama, K.-I. Nakayama and S. Maeda, Nature, 2022, 609, 502–506 CrossRef CAS PubMed.
- Z. Liu, Y. Li, W. Shin and R. Jin, J. Phys. Chem. Lett., 2021, 12, 1690–1695 CrossRef CAS PubMed.
- M. J. Cowan, T. Higaki, R. Jin and G. Mpourmpakis, J. Phys. Chem. C, 2019, 123, 20006–20012 CrossRef CAS.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the synthesis and spectroscopic characterization, supporting Fig. S1–11. See DOI: https://doi.org/10.1039/d4nr00677a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |