
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIonic liquid-coated lipid nanoparticles increase siRNA uptake into CNS targets†
Purva
Khare
a,
Sara X.
Edgecomb
b,
Christine M.
Hamadani
 b,
James F.
Conway
c,
Eden E. L.
Tanner
b and
Devika
S Manickam
*a
b,
James F.
Conway
c,
Eden E. L.
Tanner
b and
Devika
S Manickam
*a
aGraduate School of Pharmaceutical Sciences, Duquesne University, 600 Forbes Avenue, 453 Mellon Hall, Pittsburgh, PA 15282, USA. E-mail: soundaramanickd@duq.edu; Tel: +1 (412) 396-4722
bDepartment of Chemistry and Biochemistry, The University of Mississippi, MS, USA
cDepartment of Structural Biology, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA
First published on 5th December 2023
Abstract
Lipidoid nanoparticles (LNPs) have transformed the field of drug delivery and are clinically used for the delivery of nucleic acids to liver and muscle targets. Post-intravenous administration, LNPs are naturally directed to the liver due to the adsorption of plasma proteins like apolipoprotein E. In the present work, we have re-engineered LNPs with ionic liquids (ILs) to reduce plasma protein adsorption and potentially increase the accumulation of LNPs in hard-to-deliver central nervous system (CNS) targets such as brain endothelial cells (BECs) and neurons. We have developed two approaches to re-engineer LNPs using a choline trans-2-hexenoate IL: first, we have optimized an IL-coating process using the standard LNP formulation and in the second approach, we have incorporated ILs into the LNPs by replacing the PEG-lipid component in the standard formulation using ILs. IL-coated as well as IL-incorporated LNPs were colloidally stable with morphologies similar to the standard LNPs. IL-coated LNPs showed superior uptake into mouse BECs and neurons and demonstrated reduced mouse plasma protein adsorption compared to the standard LNPs. Overall, our results (1) demonstrate the feasibility of re-engineering the clinically approved LNP platform with highly tunable biomaterials like ILs for the delivery of therapeutics to CNS targets like BECs and neurons and (2) suggest that the surface properties of LNPs play a critical role in altering their affinity to and uptake into hard-to-deliver cell types.
Introduction
Lipid nanoparticles (LNPs) are clinically used for the delivery of siRNA to the liver (Onpattro) and are mRNA carriers in the Pfizer and Moderna COVID-19 vaccines.1,2 The superior drug (siRNA/mRNA) loading efficiencies of ∼70–80% in LNPs make them an attractive choice for drug delivery to challenging targets such as the brain. The high drug loading in LNPs may allow the delivered nucleic acids to exert a therapeutic response, even in the event of lower accumulation in the brain endothelial cells (BECs) lining the blood–brain barrier (BBB) followed by subsequent entry into the brain tissue.3,4 While LNPs have been studied extensively for the delivery of macromolecules to liver hepatocytes and muscle targets,5–13 only a handful of reports have studied LNPs as drug carriers for extra-hepatic targets such as the brain.14–19 We wanted to leverage the high encapsulation efficiency and the established clinical safety profile of LNPs for drug delivery to challenging CNS targets such as BECs and neurons.7,20,21Apolipoprotein E adsorbs onto the LNP surfaces post intravenous administration that serve as ligands for the lipoprotein receptors on hepatocytes, facilitating their uptake by endocytosis and accumulation in the liver.6,21 Thus, LNPs are inherently directed to the liver post-intravenous administration. Though there are methods to temporarily disrupt the BBB to increase the passage of drugs to the brain tissue,22,23 we wanted to re-engineer LNPs using ionic liquids (ILs) to reroute their biodistribution to the BECs without disrupting the BBB. BECs line the BBB and we propose that increasing accumulation of LNPs in the BECs will allow subsequent entry into the brain tissue.4
ILs are a class of materials that have the potential to advance the delivery of conventionally barrier-hindered drugs through various administration routes, such as intranasal,24 transdermal,25–28 oral,29,30 and buccal.31 ILs exist as viscous liquids below 100 °C and are composed of bulky, asymmetric cations and anions.32–34 Coordinating interactions between the cation and anion direct the biophysical and physicochemical properties within the ILs and the resulting interactions with the external microenvironment. Thus, structural alterations to the cation or anion, (e.g., alkyl chain length, functionalization with chemical moieties, branching, or steric organization)35–42 can cause radical changes in bulk properties of ILs and their interplay with other biomolecules. When comprised of moieties with high evidenced biosafety (i.e., derived from biological sources), they create a versatile biomaterial. Their tunability and promise for applications in biological drug delivery systems stems from their highly correlated structure–function dynamics.43,44 ILs have advanced the field of drug delivery by improving the pharmacokinetic and pharmacodynamic profiles of small molecules24,26,27 and increasing their biocompatibility, and reducing the immunogenicity of drug delivery systems.43 Thus, we propose to leverage and extend the physicochemical attributes of ILs to the clinically-validated LNP platform to control and regulate the interactions of LNPs in the bloodstream. This approach can be a potential strategy to overcome traditionally-hindered barriers for intravenously administered therapeutics aimed at brain drug delivery. In this study, we have used the choline trans-2-hexenoate IL, which consists of a choline cation and a trans-2-hexenoic acid anion to re-engineer LNPs. When coated on polymeric nanoparticles such as poly(lactide-co-glycolide), the choline trans-2-hexenoate IL has been previously established to act as both a protein-avoidant coating on polymeric nanoparticles, as well as mediate “red blood cell (RBC) hitchhiking” in whole blood, to lower off-target effects and overcome clearance of drug delivery systems post intravenous injection.45,46
RBC hitchhiking is a mechanism wherein the IL-associated nanoparticles attach onto RBC membranes, which mediates shuttling of membrane-adsorbed nanoparticles to selective target organs.45,46 As trans-2-hexenoic acid constitutes the outermost layer of the IL-associated nanoparticle coating, RBC hitchhiking may be derived from the anion's interaction with certain RBC membrane components (potentially surface-lipids like sphingomyelin to carboxylate transporters like MCT-1 or anion exchange transporters like AE-1).47,48 Subsequently, target organ accumulation results from RBCs exiting circulation as they shear through blood vessel capillaries, depositing nanoparticles onto endothelial cells.47,49 As an extension of this concept, IL-modified LNPs may also hitch a ride on the RBCs post-intravenous administration.43,50,51 Also, since ∼15–20% of the body's blood supply is directed to the brain,52 a comparatively higher dose of IL-LNPs might potentially reach the BBB along with the RBCs. In addition, the protein-avoidant properties of the IL coating may also translate to the reduction of adsorbed serum proteins on LNPs post-intravenous injection.47 Thus, this approach may considerably redirect the biodistribution of LNPs from the liver to the BBB. Additionally, choline trans-2-hexenoate's choline cation may potentially mediate receptor-mediated transcytosis of LNPs across the BBB to reach the brain tissue. The receptor-mediated transcytosis may be facilitated by choline transporter-like proteins 1 and 2 that are expressed on the luminal side of BBB that can aid in the transport of LNPs from the luminal to the abluminal side of the BBB.53,54 A second mechanism for the transport of LNPs across the BBB may be facilitated by the trans-2-hexenoic acid anion of the IL that may interact with the anion transporters on the BBB to enable the transport of LNPs across the BBB.55,56 Collectively, we propose that re-engineering of the standard LNP formulation with ILs may improve delivery to the BBB and potentially, into the brain tissue. Herein, we propose two methods of re-engineering LNPs with IL: coating of the choline trans-2-hexenoate IL on LNPs pre-loaded with siRNA or its integration into siRNA-LNPs (Fig. 1).
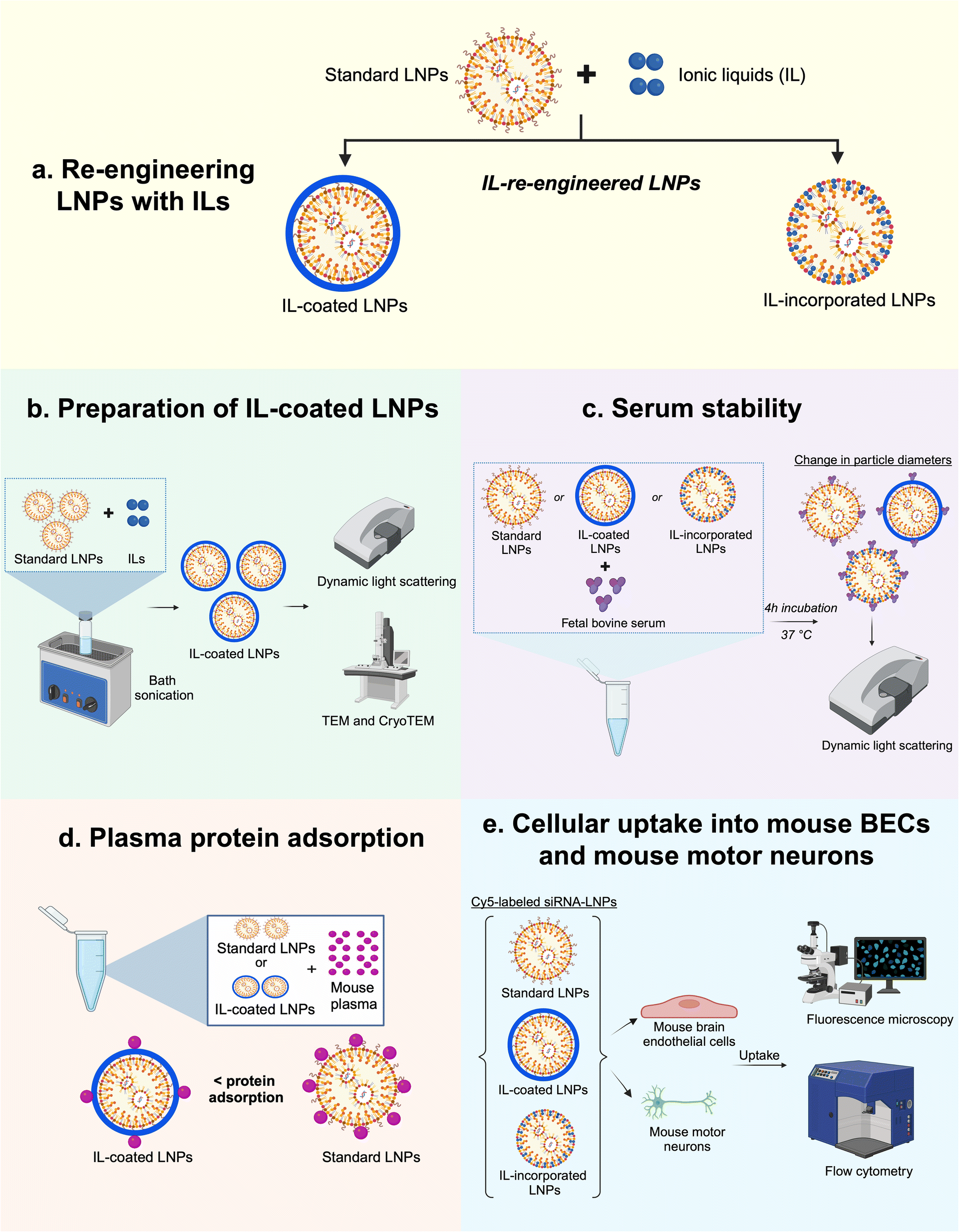 | ||
| Fig. 1 Schematic depicting the study outline. (a) Two approaches were used to re-engineer LNPs with ILs: IL-coating on LNP surfaces and IL incorporation into the LNPs; (b) bath sonication was used to prepare IL-coated LNPs whereas a direct incorporation method was used to prepare IL-incorporated LNPs followed by characterization using dynamic light scattering, TEM, and cryoTEM; (c) the colloidal stability of standard and IL-re-engineered LNPs were tested post-four-hour incubation in FBS; (d) total plasma protein adsorption onto LNP surfaces was studied in mouse plasma and (e) cellular uptake of standard and IL-re-engineered LNPs in mouse BECs and mouse motor neurons were determined using fluorescence microscopy and flow cytometry. | ||
In the first approach to re-engineer LNPs, LNPs loaded with siRNA were subsequently coated with ILs. Coating LNPs with ILs may modify the surface properties of LNPs causing lower protein adsorption onto LNP surfaces. The second approach to re-engineering LNPs was to incorporate ILs to replace the PEG-lipid (1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000: PEG-DMG) component of LNPs with IL. PEG-DMG is a widely used PEG-lipid that provides steric stability to siRNA-loaded LNPs and limits the particle–particle aggregation.7,57 PEG-DMG is also responsible for maintaining the thermodynamic stability and providing a stealth effect to LNPs thus increasing their circulation times in vivo. Additionally, the PEG-DMG component is key to regulating the structural self-assembly of LNPs. Briefly, the positively charged ionizable cationic lipid (at pH 4) interacts with anionic siRNA upon rapid mixing of the ethanolic and aqueous phases, generating hydrophobic inverted micellar structures.11,21 As mixing continues, the hydrophobic micellar structures coalesce to form the LNP core which is surrounded by a monolayer of more polar lipids (i.e., PEG-DMG and DSPC). Thus, PEG-DMG plays a crucial role in governing the structural self-assembly of LNPs by forming a monolayer around the pre-formed inverted micellar structures.
Seminal studies by Langer, Anderson, and colleagues developed libraries of lipidoids comprising several ionizable cationic lipids. Among the library of lipids, C12-200 demonstrated the highest uptake and gene knockdown in vitro and in vivo.58–61 The novelty of our work lies in re-engineering LNPs using ILs to amalgamate and leverage their key features for brain drug delivery. In this pilot study, we prepared siRNA-loaded LNPs using C12-200 as the ionizable cationic lipid and coated them using two different cation: anion (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2) ratios of the choline trans-2-hexenoate IL (IL-coated LNPs). Additionally, we also incorporated IL into the lipid monolayer of the LNPs (IL-incorporated LNPs). We optimized the coating and formulation processes and characterized the colloidal properties and colloidal stability of IL-coated- and IL-incorporated LNPs using dynamic light scattering, and studied their morphologies using negative-stain TEM and cryogenic TEM (see experimental scheme in Fig. 1). We also demonstrated the cytocompatibility of IL-coated LNPs and IL-incorporated LNPs with mouse BECs (bEnd.3) and mouse motor neuron cells (NSC-34). The qualitative and quantitative uptake of a fluorescently-labeled Cy5 siRNA into bEnd.3 and differentiated NSC-34 neurons were studied using fluorescence microscopy and flow cytometry, respectively (see experimental scheme in Fig. 1).
:1 and 1:2) ratios of the choline trans-2-hexenoate IL (IL-coated LNPs). Additionally, we also incorporated IL into the lipid monolayer of the LNPs (IL-incorporated LNPs). We optimized the coating and formulation processes and characterized the colloidal properties and colloidal stability of IL-coated- and IL-incorporated LNPs using dynamic light scattering, and studied their morphologies using negative-stain TEM and cryogenic TEM (see experimental scheme in Fig. 1). We also demonstrated the cytocompatibility of IL-coated LNPs and IL-incorporated LNPs with mouse BECs (bEnd.3) and mouse motor neuron cells (NSC-34). The qualitative and quantitative uptake of a fluorescently-labeled Cy5 siRNA into bEnd.3 and differentiated NSC-34 neurons were studied using fluorescence microscopy and flow cytometry, respectively (see experimental scheme in Fig. 1).
Experimental section
Materials
1,2-Distearoyl-sn-glycero-3-phosphocholine (DSPC) (850365P) and 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (PEG-DMG) (8801518) were purchased from Avanti Polar Lipids (Alabaster, AL). Cholesterol (8667) and cyanine 5 (Cy5)-labeled siRNA were procured from Sigma-Aldrich (St. Louis, MO). Choline bicarbonate (80% in water) and trans-2-hexenoic acid (99%) were procured from Sigma-Aldrich (St. Louis, MO). An ionizable cationic lipid, 1,1′-((2-(4-(2-((2-(bis(2-hydroxy dodecyl)amino)ethyl)(2-hydroxydodecyl)amino)ethyl)piperazin-1-yl)ethyl)azanediyl) bis(dodecan-2-ol) (C12-200) was generously provided by Dr Muthiah Manoharan, Alnylam Pharmaceuticals (Cambridge, MA). Phosphate-buffered saline (PBS) and heat-inactivated fetal bovine serum (FBS) were purchased from Hyclone Laboratories (Logan, UT). Penicillin streptomycin (Pen-Strep) was purchased from Gibco (Grand Island, NY). TrypLE Express, DMEM high glucose, and Neurobasal medium were obtained from Life Technologies Corporation (Grand Island, NY). Mouse plasma was obtained from Millipore Sigma (St. Louis, MO). Mouse brain endothelial bEnd.3 cells were procured from American Type Culture Collection (Manassas, VA) and mouse motor neuron-neuroblastoma NSC-34 cells were obtained from CELLutions biosystems (Burlington, Canada). siGFP (AM4626) was purchased from Thermo Fisher Scientific (Austin, TX). All reagents were used as received unless stated otherwise.Ionic liquid synthesis and characterization
Choline and trans-2-hexenoate IL was synthesized via a salt metathesis reaction at a 1:1 and 1:2 molar ionic ratio as previously reported.62 Briefly, choline bicarbonate (Sigma Aldrich, # C7519) was combined dropwise at 40 °C in a 500 mL round bottom flask with 98% pure trans-2-hexenoic acid (Sigma Aldrich, #W316903) at a 1:1 or 1:2 cation: anion molar ionic ratio. Once reacted (evidenced by the evolution of bicarbonate), the solution underwent magnetic stirring overnight and the next day was subsequently rotary evaporated (chiller set to −10 °C) at 60 °C and 15 mbar for 2 hours. Afterward, choline trans-2-hexenoate 1:1 and 1:2 IL were dried in a vacuum oven for 48 hours at −760 mmHg and 60 °C. The viscous IL product was a honey-gold color and was both chemically characterized by 1H-NMR (ESI Fig. 1†) in DMSO-D6 as well as water content by Karl Fischer titration, below:
1:1 Choline trans-2-hexenoate IL (1:1 IL) (MW: 218.31 g mol−1, water content: 2.9% wt wt−1, yield: 94.9%, density: 1.56 g cm−3): 1H NMR (400 MHz, DMSO) δ 6.27 (dt, J = 15.4, 7.0 Hz, 1H), 5.62 (dd, J = 15.4, 1.6 Hz, 1H), 3.85 (dq, J = 8.0, 2.7 Hz, 2H), 3.49–3.42 (m, 2H), 3.15 (s, 9H), 1.99 (qd, J = 7.1, 1.5 Hz, 2H), 1.36 (p, J = 7.4 Hz, 2H), 0.86 (t, J = 7.4 Hz, 3H).62
1:2 Choline trans-2-hexenoate IL (1:2 IL) (MW: 332.5 g mol−1, water content: 1.68% wt wt−1, yield: yield: 82.6%, density 1.66 g cm−3) 1H NMR (300 MHz, DMSO) δ 6.41–6.29 (m, 2H), 5.50 (dt, J = 15.6, 1.5 Hz, 2H), 3.62 (dq, J = 5.0, 2.6 Hz, 2H), 3.20 (dd, J = 6.3, 3.7 Hz, 2H), 2.89 (s, 9H), 1.83–1.72 (m, 4H), 1.10 (p, J = 7.3 Hz, 4H), 0.58 (t, J = 7.3 Hz, 6H).62,63
Preparation of standard and IL-coated siRNA-LNPs
Standard/uncoated LNPs were formulated using previously described methods.61 A one mg mL−1 siRNA solution was prepared in 10 mM citrate buffer pH 4.0. The lipid components: ionizable cationic lipidoid C12-200, cholesterol, DSPC, and PEG-DMG were dissolved at a molar ratio of 50/10/38.5/1.5 in ethanol. The standard formulation scheme used for preparing LNPs is shown in Table 1. The lipid phase was added in a dropwise manner to the aqueous siRNA under continuous vortexing for 30 seconds using a Fisher benchtop vortexer at knob position ‘7’. A precalculated volume of 1× PBS pH 7.4 was added to the LNPs to adjust the final siRNA concentration to 400 nM. For all LNP formulations, the cationic ionizable lipidoid/siRNA w/w ratio was maintained at 5:1 and the final siRNA concentration was 400 nM unless stated otherwise. For coated LNPs, the prepared siRNA-LNPs were further coated using 12.5, 25, and 50 μL of the 1:1 and 1:2 cation:anion choline trans-2-hexenoate IL. First, LNPs were diluted with nuclease-free water instead of 1× PBS and transferred from the Eppendorf tube to a glass scintillation vial and sonicated for one minute using a bath sonicator. After one minute, either 12.5 μL, 25 μL, or 50 μL of the 1:1 or 1:2 (cation:anion) IL were added to the LNPs. The sonication was performed for 60 minutes, and an ice pack was placed in the bath sonicator to prevent over-heating the samples.
| Component | Molar ratio in final LNP mixture | Stock concentration (mg mL−1) | Volume of the stock solution required (μL) |
|---|---|---|---|
| Ethanolic phase | |||
| C12-200 | 50 | 1 | 12.5 |
| Cholesterol | 38.5 | 1 | 3.1 |
| PEG-DMG | 1.5 | 0.1 | 13.6 |
| DSPC | 10 | 0.1 | 16.9 |
| Ethanol | — | — | 78.9 |
| Total ethanolic phase volume | 125 | ||
|
|||
| Aqueous phase | |||
| siRNA | 400 nM | 1 | 2.5 |
| 10 mM citrate buffer | — | — | 122.5 |
| PBS | — | — | 250 |
| Total aqueous phase volume | 375 | ||
| Final volume of LNPs prepared (μL) | 500 | ||
Preparation of IL-incorporated siRNA-LNPs
IL-incorporated LNPs were prepared as described in Table 2 wherein the PEG-DMG component of standard LNPs was replaced by ILs. The ethanolic phase of LNPs was prepared with C12-200, cholesterol, and DSPC whereas the aqueous phase was prepared by adding 12.5, 25, or 50 μL of 1:1 or 1:2 (cation:anion) choline trans-2-hexenoate IL to the citrate buffer containing siRNA. Due to the hydrophilic nature of IL, they were incorporated into the aqueous phase instead of the organic phase. The last step of LNP preparation was consistent with standard LNPs wherein the LNPs were diluted with 250 μL nuclease-free water.
| Component | Molar ratio in final LNP mixture | Stock concentration (mg mL−1) | Volume of the stock solution required (μL) |
|---|---|---|---|
| Ethanolic phase | |||
| C12-200 | 50 | 1 | 12.5 |
| Cholesterol | 38.5 | 1 | 3.1 |
| DSPC | 10 | 0.1 | 16.9 |
| Ethanol | — | — | 92.5 |
| Total ethanolic phase volume | 125 | ||
|
|||
| Aqueous phase | |||
| siRNA | 400 nM | 1 | 2.5 |
| 1:1 or 1:2 IL |
— | — | 12.5/25/50 |
| 10 mM citrate buffer | — | — | q.s. to 125 μL |
| Nuclease-free water | 250 | ||
| Final volume of LNPs prepared (μL) | 500 | ||
Dynamic light scattering
To determine the physical stability of LNPs, the average particle diameters and zeta potentials of the standard, IL-coated, and IL-incorporated LNPs were measured by dynamic light scattering (DLS) using a Malvern Zetasizer Pro (Malvern Panalytical Inc., Westborough, PA) over a period of seven days with intermittent storage at 2–8 °C. Formulation parameters were screened by measuring the average particle diameters, polydispersity indices, and zeta potentials of LNPs at 15–20 minutes post-preparation. The particle diameters of standard, IL-coated, and IL-incorporated LNPs were measured at 0 and 4 h post-preparation in samples supplemented with fetal bovine serum (FBS) (10%) in nuclease-free water to determine their serum stability. The standard, IL-coated, and IL-incorporated LNPs were diluted to a final siRNA concentration of 40 nM using 10 mM 1× PBS pH 7.4 or nuclease-free water for particle size measurements and with deionized water for zeta potential measurements. All DLS measurements were carried out in triplicates and the data are presented as mean ± standard deviation (SD).Transmission electron microscopy
LNPs were prepared for negative stain electron microscopy by applying 3 μL to a freshly glow-discharged copper grid coated with a continuous layer of graphitized carbon. Excess sample was blotted off, and the remaining sample was stained with a 2% uranyl acetate solution, blotted and air-dried. Grids were inserted into a Thermofisher TF20 electron microscope (Thermofisher Scientific, Waltham, Massachusetts, USA) equipped with a field emission gun and imaged on a TVIPS XF416 CMOS camera (TVIPS GmbH, Gauting, Germany) to visualize nanoparticles. The microscope was operated at 200 kV and contrast was enhanced with a 40 μm objective aperture. Electron micrographs were collected at a nominal 19000× magnification with a post-column magnification of 1.3× corresponding to a calibrated pixel size of 5.6 Ångstroms in the sample. Images were acquired with TVIPS Emplified software using movie-mode for drift correction.
Plasma protein adsorption to LNPs
Standard LNPs, IL-coated LNPs, and IL-incorporated LNPs were prepared as described above. A 150 μL aliquot of mouse plasma was incubated with 50 μL of the LNPs for 30 min at 37 °C. Then, the mixtures were centrifuged at 16000×g for 40 min to separate plasma protein-bound LNPs from unbound plasma proteins. Unbound and loosely-bound plasma proteins were removed by two cycles of washing with deionized water at 16000×g for 40 min. Plasma protein-bound LNPs were reconstituted in 1× RIPA buffer to determine the total protein content using MicroBCA assay.Agarose gel electrophoresis
To determine whether siRNA remains encapsulated in the re-engineered LNPs, we qualitatively confirmed the encapsulation of siRNA using agarose gel electrophoresis. Briefly, a 2% agarose gel was prepared in 1× Tris–borate buffer. The 10× Tris–borate buffer was composed of 108 g Tris, 55 g boric acid and 7.4 g Na2EDTA dissolved in 900 mL distilled water. Ethidium bromide (0.05%) was added to the gel for visualization of siRNA bands. Samples containing 1 μg siRNA were pre-mixed with 1× RNA loading buffer and were electrophoresed in an Owl EasyCast B2 Mini Gel Electrophoresis System (Thermo Fisher Scientific, St. Louis, MO) for 90 min. The gel was imaged under UV light using a Gel Doc system (ProteinSimple, San Jose, CA).Cell culture
NSC-34 is a hybrid cell line, produced by the fusion of mouse neuroblastoma cells with motor neuron-enriched, embryonic mouse spinal cord cells. NSC-34 cultures contain two subpopulations of cells: small, undifferentiated cells that have the capacity to undergo cell division and larger, multi-nucleate cells. The larger cells exhibit key characteristics of motor neurons, including the expression of choline acetyltransferase, neurofilament triplet proteins and they synthesize, store and release acetylcholine.64 NSC-34 were cultured in DMEM high glucose medium supplemented with 10% FBS and 1% Pen-Strep. NSC-34 cells were cultured until ∼70% confluency before differentiating them into a motor neuron phenotype by switching the culture medium to Neurobasal medium (supplemented with 4 mM glutamine and 1% Pen-Strep).65 Non-differentiated cells were continuously maintained in the differentiation medium and passaged as required. The differentiated cells were cultured until cells showed neurite-like structures observed under an EVOS microscope (EVOS Cell Imaging Systems, Thermo Fisher Scientific). bEnd.3 cells were maintained in DMEM high glucose medium supplemented with 10% FBS and 1% Pen-Strep. The growth medium was changed every 48 hours until the cells reached 90% confluency. All cultures were maintained in a humidified incubator at 37 °C and 5% CO2.Cytocompatibility of LNPs
bEnd.3 and NSC-34 cells were seeded at a density of 16500 cells per well in a clear, flat-bottomed poly-D-lysine coated 96-well plate (Azer Scientific, Morgantown, PA). Untreated cells and cells treated with 10–100 μg per mL PEI were used as controls in all studies. Cells were transfected for 4 h using standard, IL-coated, and IL-incorporated LNPs containing 50 nM of siRNA in complete growth medium in a 100 μL per well volume. Post-4 h transfection, the treatment mixture was replaced with 200 μL of fresh complete growth medium per well, which was followed by a 24 hour incubation at 37 °C and 5% CO2. A Cell Titer-Glo luminescence assay (ATP assay) was used to measure the relative intracellular ATP levels 24 h-post transfection using previously described methods.66–68 The intracellular ATP levels linearly correlate with the cell numbers, which is used as a measure of cell viability. The ATP levels of cells treated with the LNP samples were measured against those of the control, untreated control cells. Sixty μL of the complete growth medium and 60 μL of Cell Titer Glo 2.0 reagent were added to each well of the 96-well plate to lyse the cells. The 96-well plate was incubated for 15 minutes at room temperature in the dark in an incubator-shaker (Thermo Fisher Scientific, Waltham, MA). Post 15 minute incubation, 60 μL of the mixture from each well was aliquoted into a flat-bottom, white opaque 96-well plate (Azer Scientific, Morgantown, PA). The luminescence of the samples was measured using a SYNERGY HTX multi-mode reader (BioTek Instruments, Winooski, VT). The relative ATP levels (%) were calculated by normalizing the luminescence of the treatment groups to the luminescence of the untreated cells as shown in eqn (1).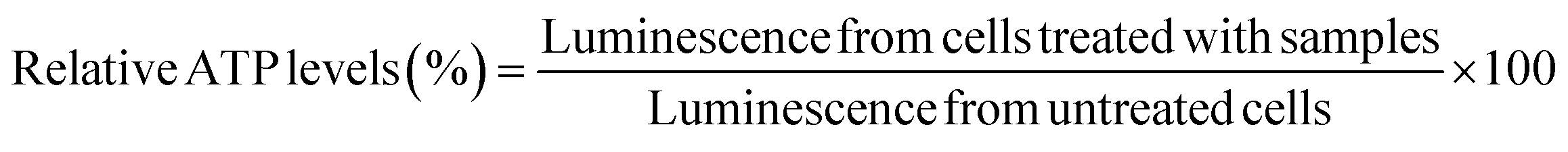 | (1) |
Cy5 siRNA uptake into b.End3 BECs and NSC-34 motor neurons using flow cytometry
b.End3 BECs and NSC-34 motor neurons were plated at a density of 100000 cells per well in complete growth medium in a clear, flat-bottom poly-D-lysine coated 24-well plate (Genesee Scientific, San Diego, CA). Cells were allowed to acclimatize in a humidified incubator at 37 °C and 5% CO2 for 2–3 days. Standard, IL-coated, and IL-incorporated LNPs loaded with Cy5-labeled siRNA were prepared as described earlier and diluted with the respective growth medium to achieve a final siRNA concentration of 50 nM per well. Cells were incubated with 350 μL of the treatment mixture for 4 h. The transfection mixture consisted of either Cy5 siRNA containing standard LNPs or 12.5 and 50 μL IL-coated LNPs or 50 μL IL-incorporated LNPs or Cy5 siRNA-Lipofectamine RNAiMAX complexes. Post-transfection, the cells were washed with 200 μL of 1× PBS, gently detached with 100 μL trypsin–EDTA solution from the wells and collected in microcentrifuge tubes. The cell suspension was centrifuged at 900×g for 10 minutes and the resulting pellet was resuspended in 500 μL flow cytometry running buffer (5% FBS and 2 mM EDTA in 1× PBS) for bEnd.3 cells and 5% FBS in 1× PBS for NSC-34 motor neuron cells. The cell suspension was analyzed using the Attune NxT Acoustic Focusing Cytometer (Singapore) equipped with Attune NxT software. The Cy5 siRNA fluorescence intensity was detected at excitation and emission wavelengths of 638 nm and 720/30 nm, respectively. A total of 30000 events were recorded for each sample. Forward vs. side scatter plots were used to gate the cells to exclude dead cells and cell debris. Untreated cells were used as negative-staining controls to account for the autofluorescence of cells and to set a gate for Cy5-negative and Cy5-positive cell populations. Data are presented as a fold increase in the percentage of Cy5-positive (Cy5 (+)) events normalized to untreated controls for each treatment group.
Cy5 siRNA uptake into bEnd.3 BECs using fluorescence microscopy
bEnd.3 cells were plated at a density of 100000 cells per well in complete growth medium in a clear, flat-bottom poly-D-lysine coated 24-well plate (Genesee Scientific, San Diego, CA). Cells were allowed to acclimatize in a humidified incubator at 37 °C and 5% CO2 for 2–3 days. Cy5 siRNA containing standard, IL-coated, and IL-incorporated LNPs were prepared as described earlier and diluted with complete growth medium to a final Cy5 siRNA concentration of 50 nM per well. Cells were incubated with 350 μL of the treatment mixture for 4 h. The transfection mixture consisted of either Cy5 siRNA containing standard LNPs or 12.5 and 50 μL IL-coated LNPs or 50 μL IL-incorporated LNPs or Cy5 siRNA-lipofectamine RNAiMAX complexes. Four hours post-transfection, the cells were washed with pre-warmed PBS followed by the addition of 350 μL of complete, phenol red-free growth medium per well. Cell nuclei were stained using Hoechst dye (10 μg mL−1) for five minutes followed by washing with 350 μL of 1× PBS. Untreated cells and cells treated with Cy5 siRNA-Lipofectamine RNAiMAX complexes were used as controls. Olympus IX 73 epifluorescent inverted microscope (Olympus, Center Valley, PA) was used to image the cells, and Cy5 signals were detected at excitation and emission wavelengths of 650 nm and 665 nm, respectively. Hoechst signals were detected at excitation and emission wavelengths of 350 nm and 461 nm, respectively. Cy5 signal intensities were normalized to untreated cells using the CellSens Software.
Statistical analysis
The data are represented as mean ± standard deviation (SD) with the numbers of replicates as stated for each experiment. Statistical analyses were performed with comparative tests using either one-way or two-way ANOVA using GraphPad Prism 9 (GraphPad Software, San Diego, CA). Bonferroni's multiple comparisons test was performed for comparative analyses using one-way ANOVA. Šídák's and Tukey's multiple comparisons test was performed using two-way ANOVA for statistical comparisons, as applicable. Alpha was set at 0.05.Results and discussion
Coating LNPs with choline trans-2-hexenoate IL resulted in significant changes in the sizes and zeta potentials of nanoparticles
LNPs were originally designed for drug delivery to the liver.5,7,21,57,60,69–71 Post intravenous administration, apolipoprotein E adsorption onto LNP surfaces plays a major role in their uptake into liver cells.72 Apolipoprotein E-LNP complexes function as ligands for the lipoprotein receptors on liver hepatocytes, thus enabling their uptake and accumulation in the liver.6 ILs are a class of materials that have considerably advanced the delivery of drugs through traditionally barrier-hindered sites of action (e.g., intranasal, transdermal, etc.).24–28 Re-engineering LNPs via surface modification with biocompatible ILs may considerably impact their protein binding properties and as a consequence, may allow re-routing the LNPs to distant sites such as the BBB. Hence, we hypothesized that surface modification of LNPs by coating them with ILs can potentially increase their ‘stealthiness’ as compared to standard LNPs and allow increased accumulation in the BECs lining the BBB.We used two different approaches to re-engineer LNPs. First, we used a coating approach where the LNPs were coated with ILs post-preparation, and in the second approach, ILs were incorporated into LNPs during preparation instead of the PEG-DMG component used in the standard LNPs. Choline trans-2-hexenoate IL at 1:1 and 1:2 cation:anion ratios (1:1 IL or 1:2 IL) were chosen to coat standard LNPs using a bath sonication protocol. ILs were synthesized and characterized as previously described in the Experimental section.73 Standard LNPs were bath sonicated for one minute before the addition of ILs. 12.5, 25, or 50 μL of the 1:1 or 1:2 IL were added to the 0.5 mL batch of standard LNPs followed by bath sonication for 4, 60, and 180 min. Significant changes in the particle diameters and/or zeta potential in comparison to uncoated LNPs were used as determinants of IL coating. Non-significant changes were observed in the particle diameters of LNPs post-four mins of bath sonication upon the addition of either 12.5, 25, or 50 μL of 1:1 IL (Fig. 2a). However, significant changes were observed in the particle diameters of LNPs post-60 and 180 min of bath sonication upon the addition of 12.5 (p < 0.0001), 25 (p < 0.05), and 50 (p < 0.0001) μL of 1:1 IL (Fig. 2a). The dispersity indices of uncoated as well as IL-coated LNPs were <0.15 with non-significant changes upon IL addition (Fig. 2a). An interesting observation was that a significant decrease in particle diameters was observed upon the addition of 12.5 and 25 μL IL whereas a significant increase was observed upon the addition of 50 μL IL at sonication times >60 minutes. ILs are known to stabilize nanoparticles by forming ion shells around them, which can cause an increase in the diameters of the resulting IL-coated LNPs.47 However, bath sonication may simultaneously cause particle deagglomeration and particle size reduction. An additional confounding factor can be the formation of single or multiple ion shells around the LNPs that can influence the particle diameters of the resulting IL-coated LNPs.
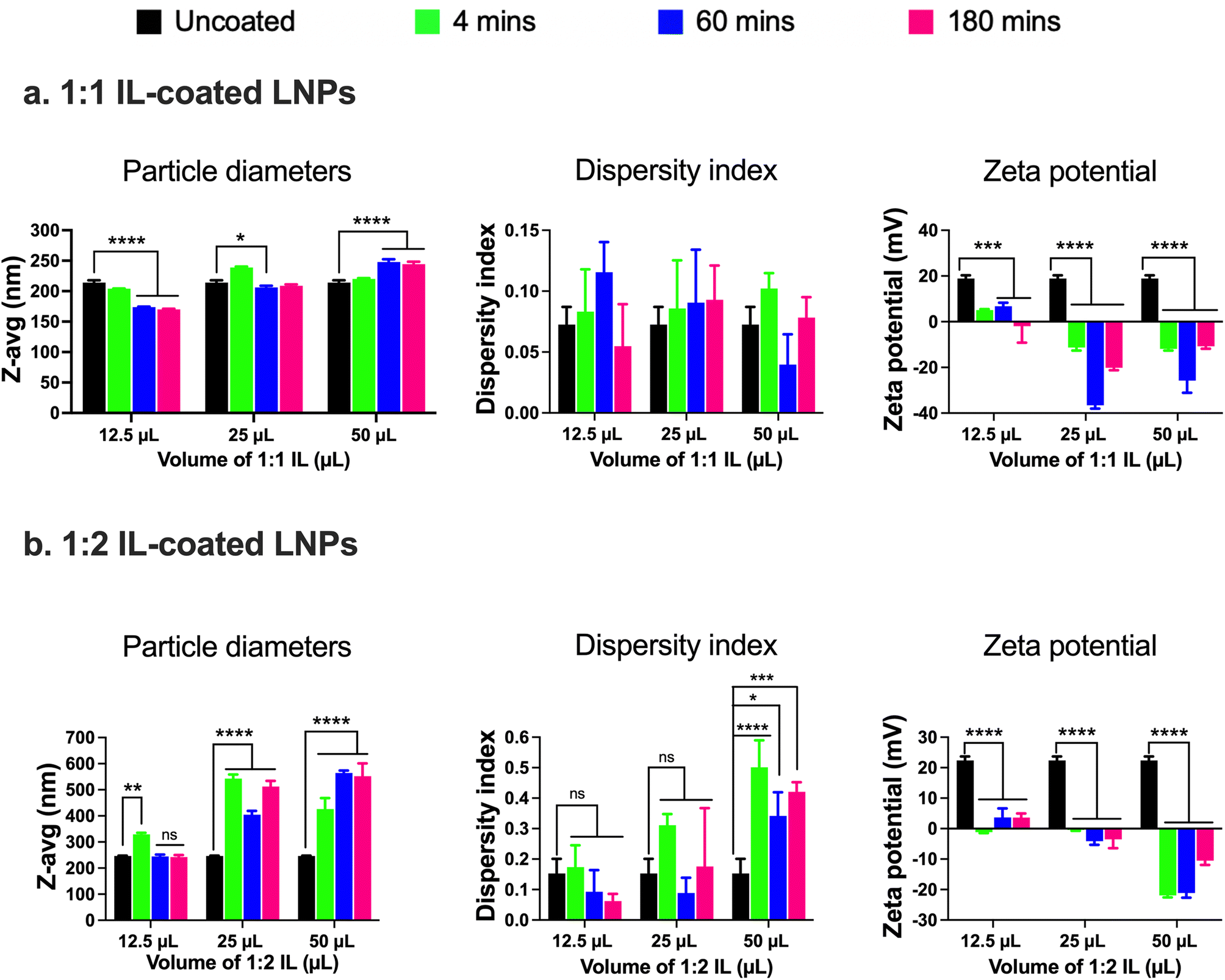 | ||
| Fig. 2 Particle diameters, dispersity indices, and zeta potential of 1:1 (a) and 1:2 (b) IL-coated siRNA-LNPs measured using dynamic light scattering. siRNA-LNPs (400 nM) were initially prepared in DI water and further diluted 10× using DI water. 12.5, 25, and 50 μL of 1:1 or 1:2 choline trans-2-hexenoate IL were added to the siRNA-LNPs followed by bath sonication for 4, 60, or 180 minutes. Z-Average particle diameters, dispersity indices, and zeta potential were measured after each time point using a Malvern Zetasizer Pro. Statistical analysis was done using two-way ANOVA with Dunnett's multiple comparisons test. Data are presented as mean ± SD of n = 3 measurements. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 and ns: non-significant. | ||
We studied changes in zeta potential as an orthogonal measure to determine the IL coating of LNPs. Post-60 min of bath sonication, the zeta potential of LNPs upon IL coating shifted from +18.9 mV to +6.8 mV (12.5 μL), −36.6 mV (25 μL), and −25.8 mV (50 μL) (Fig. 2a). The zeta potentials of IL-coated LNPs after 180 min of bath sonication for different volumes of 1:1 IL were −1.9 mV (12.5 μL), −20.1 mV (25 μL), and −10.7 mV (50 μL) (Fig. 2a). The significant changes observed in the surface charge of LNPs are one of the determinants of IL coating. The cationic charge on uncoated LNPs acts as a driving force for the anion and cation clusters in the ILs to form coatings on LNP surfaces. Furthermore, IL coating can alter the surface charge of LNPs resulting in significant changes in the zeta potential of IL-coated LNPs.
Next, we used 1:2 IL to coat LNPs and studied the changes (if any) in the particle diameters, dispersity indices, or zeta potential in comparison to the uncoated LNPs. The 1:2 IL has an excess of the 2-hexenoic acid anion in comparison to the choline cation. We noted a significant increase in the particle diameters of LNPs post-four minutes of sonication with 12.5 μL IL (329 nm) in comparison to uncoated LNPs (246.2 nm) (Fig. 2b). However, there was a non-significant increase in the particle diameters of LNPs post-60 (244.8 nm) and 180 minutes sonication (242.2 nm) (Fig. 2b). A significant increase in the particle diameters of LNPs was observed upon sonication with 12.5 and 50 μL IL for 4, 60, and 180 min, and the sizes were <400 nm (Fig. 2b). Non-significant increases in the dispersity indices of LNPs were observed upon sonication with 12.5 and 25 μL IL, nevertheless, we noted significant increases with 50 μL IL (Fig. 2b). However, the dispersity index values for LNPs sonicated with 50 μL IL were >0.3, indicative of a polydisperse sample (Fig. 2b). Lastly, we measured the zeta potential to determine changes (if any) in the surface charge of LNPs after sonication with IL. Post-60 minutes of bath sonication the zeta potential of LNPs upon IL coating shifted from +22.4 mV to +3.7 mV (12.5 μL), −4.1 mV (25 μL), and −21.1 mV (50 μL) (Fig. 2b). The zeta potentials of IL-coated LNPs after 180 min of bath sonication for different volumes of 1:2 IL were 3.6 mV (12.5 μL), −3.5 mV (25 μL), and −10.5 mV (50 μL) (Fig. 2b). While the significant changes in the zeta potential of LNPs are suggestive of coating, the sizes of LNPs sonicated with 25 and 50 μL of IL and dispersity indices of LNPs sonicated with 50 μL of IL were >400 nm and >0.3, respectively. A direct comparison of LNPs coated with 1:1 and 1:2 ILs is also shown in ESI Fig. 2.†
LNPs coated with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 choline-2-hexenoate IL are colloidally stable
:1 choline-2-hexenoate IL are colloidally stable
In addition to examining the colloidal properties of IL-coated LNPs, we also studied their colloidal stability upon storage. Self-assembled polyelectrolyte complexes like LNPs are prone to particle aggregation and instability over time which can affect their therapeutic efficacy. We also predicted that hard-to-transfect cell models such as BECs or neurons can potentially show a slower rate of uptake of LNPs in comparison to other non-CNS cell types due to their limited pinocytic capabilities. Moreover, introducing charged species like ILs can potentially cause particle–particle aggregation due to electrostatic attractions among the IL-coated LNPs. To study this, we tested LNPs coated with 12.5, 25, and 50 μL 1:1 and 1:2 IL for changes (if any) in their particle diameters and zeta potential over a period of seven days upon storage at 2–8 °C (Fig. 3). We did not note significant changes in the particle diameters of uncoated LNPs and LNPs coated with 12.5 and 50 μL 1:1 IL over the seven-day storage regime (Fig. 3a). However, the particle sizes of LNPs coated with 25 μL IL increased significantly post-seven days i.e., from 206.1 nm on day 0 to 425.7 nm on day 7 (Fig. 3a). Lower sizes of LNPs coated with 50 μL 1:1 IL compared to 25 μL 1:1 IL-coated LNPs can be attributed to the excess ILs (two-fold higher volume compared to 25 μL) in the LNP dispersion that can limit particle–particle contact and therefore, decrease agglomeration. While excess ILs are unable to efficiently coat the LNPs, they can still remain in the LNP dispersion/loosely bound to LNPs providing a “stabilizing” effect to the LNPs over time. The dispersity indices of uncoated and 12.5 μL IL-coated LNPs showed a non-significant change over seven days (Fig. 3a). However, LNPs coated with 25 and 50 μL IL showed a significant increase in dispersity indices over the seven-day storage period (Fig. 3a). We noted a similar trend with the zeta potential of LNPs wherein uncoated and 12.5 μL IL-coated LNPs showed a non-significant change in zeta potential over seven days, whereas LNPs coated with 25 and 50 μL IL showed a significant decrease over the seven-day storage period (Fig. 3a). Overall, we noted significant changes in the particle diameters, dispersity indices, and zeta potential of LNPs coated with relatively higher volumes of IL i.e., LNPs coated with 12.5 μL IL maintained their colloidal attributes over seven days. The above observations may potentially be due to excess, loosely-bound ILs forming non-specific interactions (electrostatic or hydrogen bonding) with uncoated, partially-coated, or IL-coated LNPs.
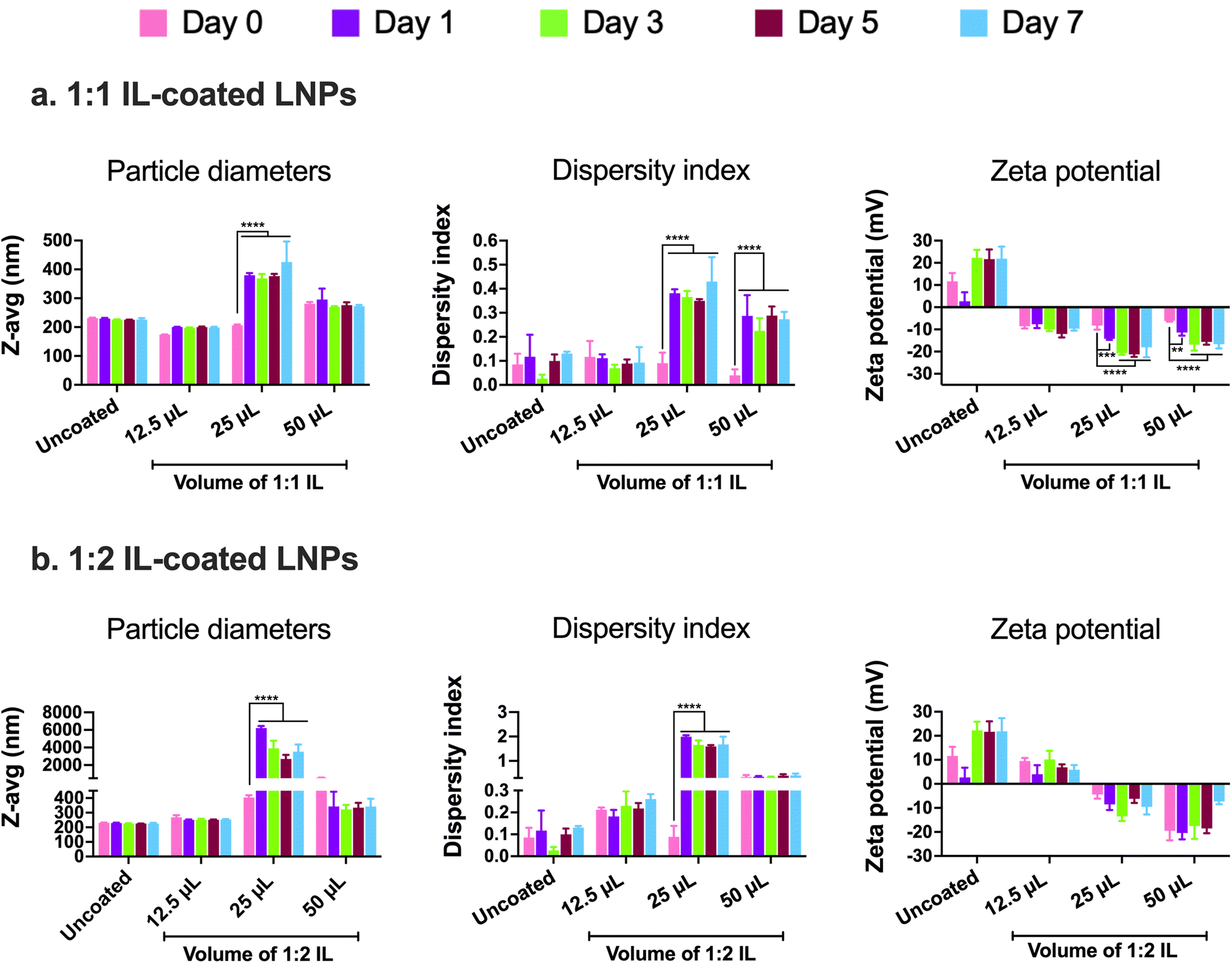 | ||
| Fig. 3 Particle diameters, dispersity indices, and zeta potential of 1:1 (a) and 1:2 (b) IL-coated siRNA-LNPs over seven days measured using dynamic light scattering. siRNA-LNPs (400 nM) were initially prepared in DI water and further diluted 10× using DI water. 12.5, 25, and 50 μL of 1:1 or 1:2 choline trans-2-hexenoate IL were added to the siRNA-LNPs followed by bath sonication for 60 minutes. Z-Average particle diameters, dispersity indices, and zeta potential were measured on days 0 (the day of preparation), 1, 3, 5, and 7 upon storage at 2–8 °C using a Malvern Zetasizer Pro. Statistical analysis was done using two-way ANOVA with Dunnett's multiple comparisons test. Data are presented as mean ± SD of n = 3 measurements. **p < 0.01, ***p < 0.001, and ****p < 0.0001. | ||
Similar to 1:1 IL-coated LNPs, we tested the colloidal stability of LNPs coated with 1:2 IL. The presence of excess anions in comparison to cations may prove both beneficial and detrimental to the colloidal stability of LNPs. As IL-coated LNPs have an overall negative surface charge, the presence of excess anions could cause increased surface repulsion between particles, thus increasing their colloidal stability. Conversely, the excess anions can form non-specific electrostatic and hydrogen bonding interactions with uncoated, partially-coated, or IL-coated LNPs. We tested 12.5, 25, and 50 μL 1:2 IL-coated LNPs for changes in their particle diameters, dispersity indices, and zeta potential over a seven-day storage regime at 2–8 °C (Fig. 3b). We observed non-significant changes in the particle diameters of uncoated LNPs and LNPs coated with 12.5 and 50 μL 1:1 IL post-the-seven-day storage period (Fig. 3b). However, the particle sizes of LNPs coated with 25 μL IL increased significantly after seven days i.e., from 404.5 nm on day 0 to ∼3 μm on day 7 (Fig. 3b). This trend was similar to that of LNPs coated with 1:1 IL wherein, non-significant changes were noted with LNPs coated with 12.5 and 50 μL IL whereas the sizes increased significantly for 25 μL IL-coated LNPs (Fig. 3b). The dispersity indices followed an analogous trend wherein the dispersity indices of uncoated, 12.5, and 50 μL IL-coated LNPs showed a non-significant change over seven days (Fig. 3b). Nonetheless, LNPs coated with 25 μL IL depicted a significant increase in dispersity indices over the seven-day storage period (Fig. 3b). Unlike the significant changes observed in the zeta potential of 1:1 IL-coated LNPs, we did not note any significant changes in the zeta potential values of 1:2 IL-coated LNPs over a period of seven days (Fig. 3b). While the presence of excess anions in 1:2 IL do not impact the sizes and dispersity indices of LNPs considerably over seven days in comparison to 1:1 IL, they do affect the zeta potential of LNPs.
ILs can replace the PEG-DMG component of standard LNPs
As presented in the introduction, PEG-DMG has an important role in the structural self-assembly of LNPs and limits particle–particle aggregation through steric effects. Although PEG-DMG plays an important role in LNP formation, it also has a few notable limitations on the drug delivery potential of LNPs. Firstly, the uptake of LNPs by cells is limited by the desorption rates of PEG-DMG from LNP surfaces which can hamper the gene silencing efficacy of siRNA.74 While the PEG chain length, which plays a dominant role in determining the desorption rate can be optimized, it still affects the pharmacokinetics and biodistribution of LNPs. A higher molar concentration of PEG-lipids is used to achieve smaller LNPs, however, the concentration of PEG-DMG in the final LNP formulation also limits LNP uptake by the target cells.57 Studies have shown that 1.5 mol% PEG-lipid is an optimized concentration wherein the chain length plays a minor role in affecting the gene-silencing ability of LNP.74 However, it still has the potential to modify the pharmacokinetics and biodistribution of LNPs. Furthermore, increasing the concentration of the PEG-lipid from 1.5 to 3.5 mol% drastically reduces the gene-silencing ability of LNPs.74 Mui et al. have shown that the in vivo desorption rates of C-14, C-16, and C-18 PEG-lipids from LNPs are >45%/hr, >1.3%/hr, and >0.2%/hr, respectively.74 Although shorter chain lengths of PEG-lipids show faster desorption rates from LNPs, they also have shorter circulation half-lives. Thus, it is important to maintain the balance of the use of PEG-lipids in LNPs since the mol% and chain length of PEG-lipids affect the colloidal stability, uptake, and the resulting pharmacokinetics of LNPs. The second major challenge with using PEG-lipids in nanoformulations is the development of anti-PEG immunity due to the generation of anti-PEG antibodies. Numerous patients have pre-existing antibodies (IgM and IgG) that bind to PEG whereas PEGylated nanoparticles can induce the secretion of additional antibodies against PEG thus impacting the safety and efficacy of therapeutics.75–77 The production of anti-PEG antibodies in response to PEGylated nanoparticles also induces their accelerated blood clearance from the system.78 This phenomenon negates one of the key beneficial effects of PEGylated nanoparticles i.e., increasing the circulation time of nanoparticles by inducing a stealth effect.To address the “PEG dilemma”, we formulated IL-incorporated LNPs wherein we replaced the PEG-DMG component of LNPs with ILs. For IL-incorporated LNPs, the ethanolic phase of LNPs was prepared with C12-200, cholesterol, and DSPC whereas the aqueous phase was prepared by adding either 12.5, 25, or 50 μL of 1:1 or 1:2 (cation: anion) choline trans-2-hexenoate IL to the citrate buffer containing siRNA (see Table 2). Owing to the hydrophilic nature of the IL, they were incorporated into the aqueous phase instead of the organic phase. We first tested the feasibility of replacing PEG-DMG with IL since PEG-DMG is key to the self-assembly and thermodynamic stability of LNPs.
The particle size, dispersity index, and zeta potential of IL-incorporated LNPs prepared with 12.5, 25, or 50 μL of 1:1 or 1:2 (cation:anion) choline trans-2-hexenoate IL were measured using DLS over a period of seven days (Fig. 4). Standard LNPs and LNPs prepared without adding PEG or IL (LNPs without PEG-DMG) were used as controls. IL-incorporated LNPs prepared by adding 25 and 50 μL of 1:2 IL were physically unstable wherein precipitation of the citrate buffer salts was seen immediately after the addition of IL to the aqueous phase (data not shown). There was a non-significant change in the sizes of standard LNPs over seven days (295.8 nm on day 0 and 270.4 nm on day 7) (Fig. 4a). The sizes of LNPs (without PEG-DMG) increased significantly from 793.3 nm on day 0 to ∼3 μm on day 7 (p < 0.0001) (Fig. 4a). It should be noted that the sizes of LNPs (without PEG-DMG) are significantly high due to the absence of a stabilizing agent in the formulation. The sizes of IL-incorporated LNPs prepared with 12.5 and 25 μL of 1:1 IL on day 0 were 248 nm and 133.3 nm, however, these values increased significantly to ∼2 μm and ∼3 μm, respectively (p < 0.0001) (Fig. 4a). This is an interesting observation to note as the sizes of IL-incorporated LNPs prepared with 12.5 and 25 μL of 1:1 IL were similar to or smaller than that of standard LNPs on day 0, however, they increased significantly post-seven-day storage. This suggests that although IL-incorporated LNPs form immediately after mixing the ethanolic and aqueous phases, they are not able to retain their sizes as seen from the significant increase in their sizes on days 3 and 7. A slightly different effect was seen when IL-incorporated LNPs were prepared with 12.5 μL of 1:2 IL wherein the size of LNPs on day 0 was 980.5 nm that increased significantly to ∼1.5 μm after seven days (p < 0.01) (Fig. 4a). This suggests that IL-incorporated LNPs prepared with 1:2 IL are not able to form or maintain their sizes over the storage regime. Upon increasing the volume of 1:1 IL to 50 μL for preparing IL-incorporated LNPs, we noted sizes of 172.3 μm on day 0 that increased to 209.9 nm on day 7 (non-significant change) (Fig. 4a). This implies that IL-incorporated LNPs prepared with 50 μL of 1:1 IL were smaller than standard LNPs (ca. 250–300 nm) and maintained their sizes over the seven-day storage regime.
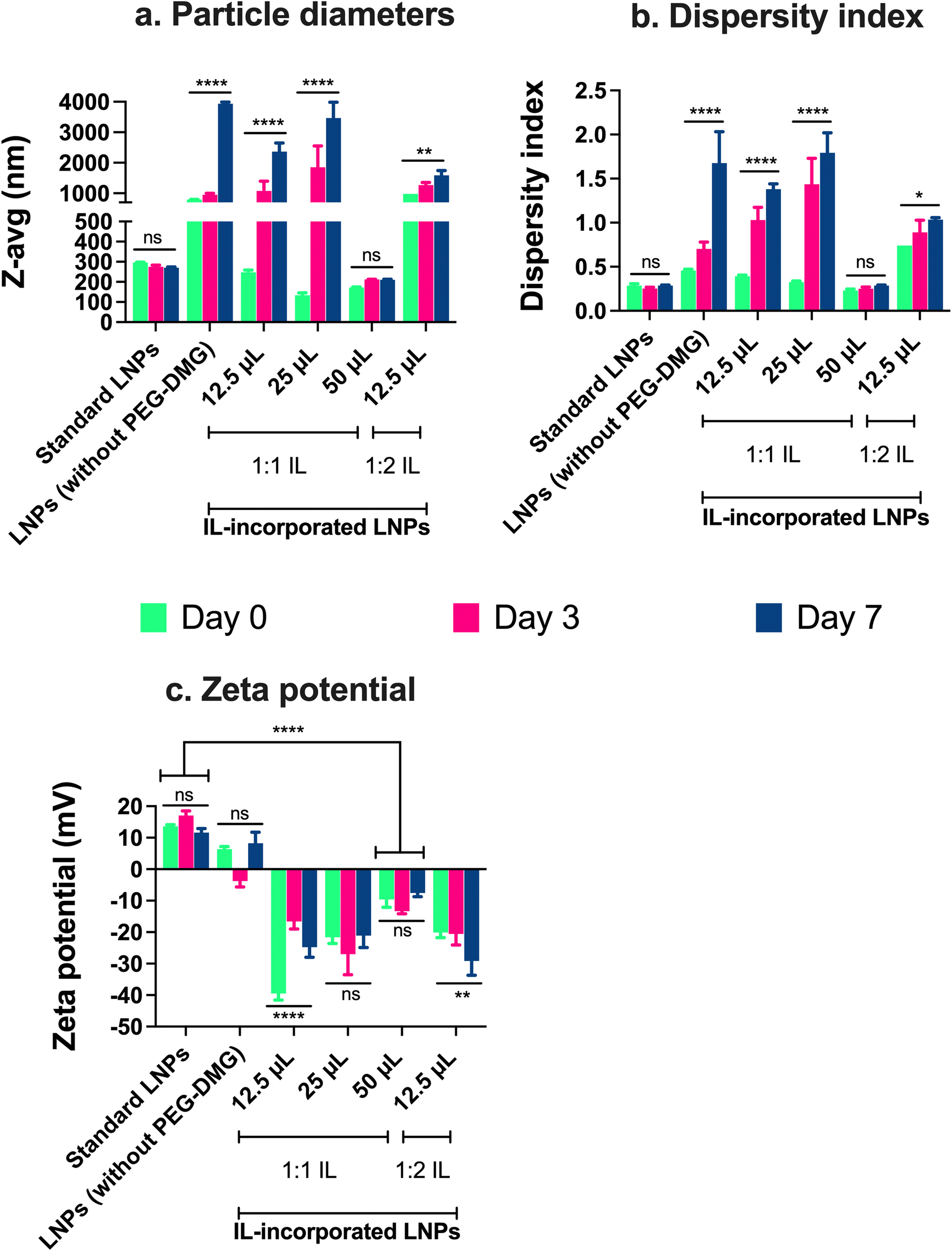 | ||
| Fig. 4 Particle diameters (a), dispersity indices (b), and zeta potential (c) of IL-incorporated siRNA-LNPs measured using dynamic light scattering. Standard LNPs or LNPs (without PEG-DMG) or IL-incorporated siRNA-LNPs (400 nM) were prepared in DI water and further diluted 10× using DI water. 12.5, 25, and 50 μL of 1:1 and 12.5 μL of 1:2 choline trans-2-hexenoate IL were incorporated in the LNPs. Z-average particle diameters and dispersity indices were measured post-preparation and on days 3 and 7, whereas zeta potential was measured post-preparation using a Malvern Zetasizer Pro. Statistical analysis was done using two-way ANOVA with Dunnett's multiple comparisons test. Data are presented as mean ± SD of n = 3 measurements. *p < 0.05, **p < 0.01, ****p < 0.0001 and ns: non-significant. | ||
We noted a non-significant change in the dispersity indices of standard LNPs over seven days (∼0.28 on days 0 and 7) (Fig. 4b). The dispersity indices of LNPs (without PEG-DMG) increased significantly from 0.45 on day 0 to 1.67 on day 7 (p < 0.0001) (Fig. 4b). It is important to note that LNPs (without PEG-DMG) are highly polydisperse due to the absence of a stabilizing agent in the formulation. The dispersity indices of IL-incorporated LNPs prepared with 12.5 and 25 μL of 1:1 IL on day 0 were 0.39 and 0.32, however, these values increased significantly to 1.38 and 1.79, respectively (p < 0.0001) (Fig. 4b). This trend was consistent with the sizes of IL-incorporated LNPs prepared with 12.5 and 25 μL of 1:1 IL wherein their dispersity indices were similar to that of standard LNPs on day 0 but increased significantly post-seven-day storage. This observation suggests that although IL-incorporated LNPs have a lower dispersity index immediately after mixing the ethanolic and aqueous phases, they are not able to retain their dispersity indices over seven days. A slightly different trend was observed when IL-incorporated LNPs were prepared with 12.5 μL of 1:2 IL wherein the dispersity index of LNPs on day 0 was 0.74 that increased significantly to 1.03 after seven days (p < 0.05) (Fig. 4b). This suggests that IL-incorporated LNPs prepared with 1:2 IL form highly polydisperse particles and are not able to maintain their dispersity indices over the storage regime. When we increased the volume of 1:1 IL to 50 μL, we noted that the dispersity indices of IL-incorporated LNPs were 0.23 on day 0 which increased to 0.28 on day 7 (non-significant) (Fig. 4b). This suggests that IL-incorporated LNPs prepared with 50 μL of 1:1 IL show dispersity index values similar to standard LNPs and maintain these values over the seven-day storage regime.
We also measured the zeta potential of IL-incorporated LNPs prepared with 12.5, 25, or 50 μL of 1:1 IL and 12.5 μL of 1:2 IL. The polar, hydrophilic nature of PEG-DMG in comparison to the other lipids i.e., C12-200, cholesterol, and DSPC, drives the self-assembly of LNPs such that the PEG moiety faces the outer aqueous phase.7,21,57 Since ILs are hydrophilic, similar to PEG-DMG, we hypothesized that ILs too would be incorporated in the monolayer of LNPs. The bulky cations and anions in ILs would be a stabilizer (electrostatic and steric) by interacting with the lipids in the LNPs and the outer aqueous phase.4,79,80 This would consecutively cause a change in the surface charge (zeta potential) of IL-incorporated LNPs in comparison to standard LNPs. Thus, we studied the difference in the zeta potential of IL-incorporated LNPs in comparison with standard LNPs. We also tested for changes (if any) in the zeta potential of LNPs over the seven-day storage regime.
We noted a non-significant change in the zeta potential of standard LNPs over seven days (13.6 mV on day 0 and 11.6 mV on day 7) (Fig. 4c). The zeta potential of LNPs (without PEG-DMG) was 6.3 mV on day 0 and 8.2 mV on day 7 (non-significant) (Fig. 4c). The ionizable cationic lipid, C12-200, accounts for the slightly positive surface charge on LNPs. IL-incorporated LNPs showed a significant change in the surface charge wherein they showed negative zeta potential values as compared to standard LNPs. The zeta potential of IL-incorporated LNPs prepared with 12.5 μL of 1:1 IL on days 0 and 7 were −39.5 mV and −24.8 mV, respectively (p < 0.0001) (Fig. 4c). IL-incorporated LNPs prepared with 25 μL of 1:1 IL showed zeta potential values of −21.6 mV on day 0 and −21.1 mV on day 7 (non-significant difference) (Fig. 4c). The zeta potential values of IL-incorporated LNPs prepared using 50 μL of 1:1 IL were −9.6 mV on day 0 and −7.5 mV on day 7 (non-significant difference) (Fig. 4c). We noted a trend between the volume of 1:1 IL incorporated and the zeta potential of the resulting IL-incorporated LNPs. The zeta potential of IL-incorporated LNPs became less anionic upon increasing the volume of 1:1 IL. The more anionic zeta potential indicates the presence of free IL as compared to the less anionic zeta potential that suggests IL incorporation in the LNP monolayer. Thus, a higher volume of IL i.e., 50 μL, was required to be incorporated into the complete LNP monolayer as compared to the lower volumes (12.5 and 25 μL). This observation also supports the instability seen with IL-incorporated LNPs prepared with 12.5 and 25 μL IL. A lower volume of IL is likely not sufficiently incorporated in the LNP monolayer thus causing aggregation and a consequent increase in sizes and dispersity indices. On the other hand, IL-incorporated LNPs prepared with 50 μL IL maintain their colloidal properties over the storage regimen. For IL-incorporated LNPs prepared with 12.5 μL of 1:2 IL, the zeta potential on day 0 was −20.1 mV that decreased significantly to −29.1 mV after seven days (p < 0.01) (Fig. 4c). Thus, we chose a 50 μL 1:1 IL volume for all future studies based on the colloidal properties of IL-incorporated LNPs prepared with different volumes of ILs.
We imaged IL-incorporated LNPs prepared with 50 μL 1:1 IL using transmission electron microscopy (TEM) and cryo-TEM. Standard LNPs and LNPs coated with 12.5 μL and 50 μL 1:1 IL were used as controls (Fig. 5 and 6). The LNPs appeared to have a spherical morphology with sizes similar to the particle diameters obtained by DLS. LNPs coated with 12.5 μL IL appeared significantly smaller than standard LNPs (Fig. 5a and b). LNPs coated with 50 μL of 1:1 IL are unstable and aggregate into larger particles (data not shown). We also noted heterogeneously-sized particles in this sample which were consistent with the DLS data. IL-incorporated LNPs were spherical with sizes similar to standard LNPs (Fig. 5c). The shape and morphology of IL-incorporated LNPs were similar to that of standard LNPs. The cryo-TEM image of standard LNPs showed multilamellar structures (Fig. 6a). A “halo” surrounding the LNP core was observed for 12.5 μL 1:1 IL-coated LNPs which may be indicative of the formation of a shell/coat of IL around the LNPs (Fig. 6b). LNPs coated with 50 μL of 1:1 IL did not appear to have a lipid/IL envelope. Heterogeneously-sized particles were also observed in the sample (data not shown). IL-incorporated LNPs showed sizes and morphology similar to standard LNPs (Fig. 6c). A direct comparison table for the particle sizes, dispersity indices, and zeta potentials of IL-coated vs. IL-incorporated LNPs is provided in ESI Table 1.†
 | ||
| Fig. 5 TEM images of standard LNPs (a), IL-coated LNPs (b), and IL-incorporated LNPs (c) acquired using a Thermofisher T12 electron microscope. siRNA-LNPs (400 nM) were prepared in nuclease-free water. IL-coated LNPs were prepared by adding 12.5 μL of 1:1 choline trans-2-hexenoate IL to the siRNA-LNPs followed by bath sonication for 60 minutes. IL-incorporated LNPs were prepared by adding 50 μL of 1:1 choline trans-2-hexenoate IL to the aqueous phase during LNP formulation. Negative-stain electron micrographs were acquired using a Thermofisher TF12 electron microscope. | ||
 | ||
| Fig. 6 Cryo-TEM images of standard LNPs (a), IL-coated LNPs (b), and IL-incorporated LNPs (c) acquired using a Thermofisher T12 electron microscope. siRNA-LNPs (400 nM) were prepared in nuclease-free water. IL-coated LNPs were prepared by adding 12.5 μL of 1:1 choline trans-2-hexenoate IL to the siRNA-LNPs followed by bath sonication for 60 minutes. IL-incorporated LNPs were prepared by adding 50 μL of 1:1 choline trans-2-hexenoate IL to the aqueous phase during LNP formulation. Cryo-TEM micrographs were acquired using a Thermofisher TF12 electron microscope. Scale bar = 50 nm. | ||
To test whether siRNA remains encapsulated in the IL-coated and IL-incorporated LNPs, we qualitatively confirmed the encapsulation of siRNA by agarose gel electrophoresis. Ethidium bromide was added to the gel which intercalates between the base pairs of naked/free siRNA and emits fluorescence under UV light. The lack of ethidium bromide fluorescence would suggest that siRNA remains encapsulated in the LNPs without leakage. This also qualitatively confirms the integrity of siRNA-loaded LNPs post-sonication. As seen in ESI Fig. 3,† naked siRNA showed an intense band (lane 2) whereas, siRNA-LNP groups (lanes 3–6) showed no signal suggesting the encapsulation of siRNA in LNPs and the absence of any siRNA leakage from the formulations.
The serum/cell culture stability of standard LNPs, IL-coated LNPs, and IL-incorporated LNPs was studied by incubating them in 10% fetal bovine serum (FBS). In this experiment, we measured the particle diameters of LNPs in the presence of serum proteins to study the stability of LNPs in cell culture conditions. We incubated standard LNPs, IL-coated LNPs, and IL-incorporated LNPs in 10% FBS for 4 h at 37 °C. We then examined their intensity-size distribution plots to study the effect of FBS proteins on the particle diameters of LNPs. Serum proteins can potentially adsorb onto LNP surfaces causing changes to LNP particle sizes.81,82 We compared the intensity-size distribution graphs of standard LNPs, IL-coated LNPs, and IL-incorporated LNPs in nuclease-free water (ESI Fig. 4,† pink) or in 10% FBS (ESI Fig. 4,† blue) immediately post-preparation (0 h) and upon storage for 4 h at 37 °C. We used the intensity-size distribution plots of a control sample containing 10% FBS in nuclease-free water to study the Z-average particle diameter and size distribution of serum proteins (ESI Fig. 4,† green). A bimodal peak of serum proteins was seen with a Z-average diameter of ∼20 nm (ESI Fig. 4e,† green).
Standard LNPs (without FBS) showed a Z-average diameter of 229.8 nm at 0 and 4 h (ESI Fig. 4a,† pink). Upon the addition of 10% FBS, we noted a peak of Z-average (∼197 nm) although with a reduced % intensity at 0 and 4 h (ESI Fig. 4a,† blue). A similar trend was noted for IL-coated LNPs wherein the Z-average of FBS-supplemented samples was similar to the non-FBS-supplemented samples with a reduced %intensity (ESI Fig. 4b and c†). However, for IL-incorporated LNPs, we did not note a sharp peak corresponding to the Z-average of LNPs when supplemented with 10% FBS (ESI Fig. 4d†). We speculate the aggregation or instability of IL-incorporated LNPs in the presence of serum proteins may be caused due to the ionic interactions between the serum proteins and IL. So, this suggests that while IL-incorporated LNPs maintain colloidal stability, the PEG-DMG component is critical for the serum stability of LNPs.
The IL-addition step affects the colloidal properties of the formed IL-incorporated LNPs
The IL-addition step during LNP preparation plays a critical role in ensuring the formation and colloidal stability of LNPs. We wanted to incorporate IL in the LNP monolayer rather than forming a “shell” or a coat of IL on the surface of pre-formed LNPs. As PEG-DMG is omitted from the IL-LNP formulation, we speculated that an additional “stabilizer” should be able to promote particle–particle repulsion during LNP formation. The stabilizer should also be able to maintain the colloidal stability of LNPs during the storage regime. To determine the effect of the IL-addition step during LNP formulation and to ascertain whether the IL is incorporated into the LNPs, we added IL during one of the three different steps of LNP formation (Table 3).| Step 1 | Step 2 | Step 3 |
|---|---|---|
| IL added in the aqueous phase consisting of siRNA and citrate buffer | IL added immediately after mixing of the aqueous and organic phases | IL added during the final volume makeup step with nuclease-free water |
LNPs are formed during and after the mixing of the aqueous and organic phases. Thus, IL addition during steps 1 and 2 would suggest IL incorporation in the LNPs whereas IL addition during dilution with nuclease-free water would potentially form an IL shell/coat around the pre-formed LNPs. The particle diameters, dispersity indices, and zeta potential of IL-incorporated LNPs prepared upon IL-addition during steps 1, 2, or 3 were measured using DLS on days 0 and 7. The particle diameters of IL-incorporated LNPs when IL was added in the aqueous phase on days 0 and 7 were 218 and 224.4 nm (Fig. 7a), their dispersity indices were 0.06 and 0.27 (Fig. 7b) and zeta potential values were −2.5 and −1.9 mV (Fig. 7c). It is important to note that there was only a non-significant change in the particle diameters, dispersity indices, and zeta potential of IL-incorporated LNPs prepared upon IL-addition during step 1 i.e., when ILs were added in the aqueous phase. The particle diameters of IL-incorporated LNPs when IL were added immediately post-mixing of aqueous and organic phases on days 0 and 7 were 225.8 and 2425 nm (p < 0.0001) (Fig. 7a), dispersity indices were 0.3 and 1.51 (p < 0.0001) (Fig. 7b) and zeta potential values were −8.3 and −5.5 mV (non-significant) (Fig. 7c). The significant increases (day 0 vs. day 7) in the sizes and dispersity indices of IL-incorporated LNPs upon IL-addition immediately post mixing of aqueous and organic phases suggests the colloidal instability of these systems. When IL was added immediately after mixing aqueous and organic phases, although LNPs with sizes ∼225 nm form, they did not retain their sizes after a seven-day storage period. This could be a result of the formation of an IL shell around pre-formed LNPs or simply the presence of IL in the LNP dispersion. The particle diameters of IL-incorporated LNPs when IL was added during volume makeup with nuclease-free water on days 0 and 7 were 760 and 1547 nm (p < 0.01) (Fig. 7a), dispersity indices were 0.42 and 0.87 (p < 0.01) (Fig. 7b) and zeta potential values were −35.6 and −16.5 mV (p < 0.0001) (Fig. 7c). Upon IL addition during volume makeup with nuclease-free water, larger LNPs with sizes ∼760 nm were formed. Additionally, their zeta potential values were significantly negative in comparison to IL-incorporated LNPs when IL was added during steps 1 and 2. This is indicative of the presence of IL in the colloidal dispersion of pre-formed LNPs. An interesting trend was noted in the surface charge of LNPs wherein the zeta potential shifted to more anionic values upon IL addition during steps 1, 2, and 3. The zeta potential values of IL-incorporated LNPs upon IL-addition during steps 1, 2, and 3 were −2.5 mV, −8.3 mV, and −35.6 mV, respectively (Fig. 7c). This shift of zeta potential to more anionic values indicates the presence of free IL in IL-incorporated LNPs formed upon IL addition during volume makeup with nuclease-free water. When IL was added during volume makeup with nuclease-free water, it can either form a shell/coat around the pre-formed LNPs or exist freely in the colloidal LNP dispersion, resulting in more anionic zeta potential values. However, IL was incorporated into the LNP monolayer when IL was added in the aqueous phase before mixing the two phases or added immediately after mixing the aqueous and organic phases. This results in near-neutral zeta potential values of IL-LNP due to the absence of free IL in the LNP dispersion. Thus, we conclude that adding IL to the aqueous phase before mixing the aqueous and organic phases is critical to allow IL incorporation in the LNPs.
 | ||
| Fig. 7 Effect of IL-addition during different steps of LNP formation on the particle diameters, dispersity indices, and zeta potential of LNPs. siRNA-LNPs (400 nM) were initially prepared in nuclease-free water with IL addition during different steps of LNP preparation. LNPs were further diluted 10× using nuclease-free water for DLS measurements. 50 μL of 1:1 choline trans-2-hexenoate IL were added to the siRNA-LNPs either in the aqueous phase or post-mixing of the phases or during volume makeup with nuclease-free water. Z-Average particle diameters (a), dispersity indices (b), and zeta potential (c) were measured on days 0 and 7 upon storage at 2–8 °C using a Malvern Zetasizer Pro. Statistical analysis was done using two-way ANOVA with Sidak's multiple comparisons test. Data are presented as mean ± SD of n = 3 measurements. **p < 0.01, ****p < 0.0001 and ns: non-significant. | ||
IL-coated LNPs adsorb significantly lower plasma proteins compared to standard LNPs
Nanoparticles are subject to plasma protein adsorption upon intravenous administration which can result in immunogenicity and rapid clearance.83–85 PEGylated nanoparticles exhibiting stealth properties have also shown reports of plasma protein adsorption and induction of anti-PEG antibodies.57,74–78 Plasma protein adsorption on nanoparticles triggers their opsonization causing rapid elimination of nanoparticles by phagocytes, thus reducing their therapeutic efficacy.86–89 To address this issue, we modified the surfaces of standard LNPs using ILs to form IL-coated LNPs or IL-incorporated LNPs. Choline trans-2-hexenoate IL is known to have ‘protein avoidant’ properties that can potentially reduce opsonization and increase the circulation time of IL-associated nanoparticles.47 We hypothesized that surface modification with ILs can significantly reduce protein adsorption on LNPs as compared to standard LNPs.90 To test this hypothesis, we incubated standard LNPs, IL-coated LNPs, and IL-incorporated LNPs with mouse plasma at 37 °C for 30 min to induce plasma protein adsorption on LNPs.91–93 Protein-bound LNPs were separated from unbound proteins through multiple cycles of washing. Total protein content adsorbed/bound to LNPs were quantified using a MicroBCA assay. We noted a significant reduction (p < 0.01) in the plasma protein binding of 12.5 μL IL-coated LNPs (∼18% reduction) and 50 μL IL-coated LNPs (∼15% reduction) compared to standard LNPs (Fig. 8). A non-significant difference was observed in the plasma protein adsorption of standard LNPs vs. 50 μL IL-incorporated LNPs (Fig. 8). However, we noted a significant difference between the protein adsorption by 12.5 μL IL-coated LNPs vs. 50 μL IL-incorporated LNPs (p = 0.0042). This trend is also reflected in the serum stability study wherein 50 μL IL-incorporated LNPs were unable to maintain their sizes in the presence of 10% FBS (ESI Fig. 4†). However, 12.5 μL and 50 μL IL-coated LNPs retained their intensity-size distribution peaks upon incubation with 10% FBS (ESI Fig. 4†). This phenomenon can be attributed to the absence of the stabilizing PEG-DMG component in IL-incorporated LNPs. PEG-DMG serves as a stealth component of nanoparticles and helps maintain their colloidal properties in the presence of serum proteins.7,21,74 PEG-DMG sterically decreases protein corona formation on nanoparticles and preserves colloidal properties of the formed nanoparticles. When the PEG-DMG component of LNPs were replaced with ILs in IL-incorporated LNPs, it may have caused higher protein adsorption on their surface compared to IL-coated LNPs that have both ILs and PEG-DMG (Fig. 8 and ESI 4†) in the formulation. Thus, the lower protein binding to IL-coated LNPs in comparison to standard LNPs could result in reduced immunogenicity and clearance upon systemic administration. Our data suggests that ILs can be used to surface-modify LNPs to improve their pharmacokinetics and enhance their stealth properties. | ||
| Fig. 8 Total protein adsorption on standard LNPs, IL-coated LNPs, and IL-incorporated LNPs determined using MicroBCA assay. Standard LNPs, IL-coated LNPs, and IL-incorporated LNPs were incubated with mouse plasma at a volume:volume ratio of 1:3, and protein-bound LNPs were separated from unbound proteins using centrifugation. The total protein adsorbed on LNPs was determined quantitatively using the MicroBCA assay. Data are presented as mean ± SD. Statistical analysis was done using one-way ANOVA with Dunnett's multiple comparisons test. **p < 0.001, and ns: non-significant. | ||
Mouse BECs and mouse motor neurons internalize IL-coated LNPs and IL-incorporated LNPs to a greater extent compared to standard LNPs
Our next goal was to determine the uptake of standard LNPs, IL-coated LNPs, and IL-incorporated LNPs in BECs and a neuronal cell model. Before that, we tested the cytocompatibility of LNPs with a mouse BEC line, bEnd.3 and NSC-34, a motor neuron-enriched, embryonic mouse spinal cord cell line fused with mouse neuroblastoma cells, (ESI Fig. 5†). The cell viability for bEnd.3 cells treated with standard LNPs, 12.5 and 50 μL IL-coated and IL-incorporated LNPs were 107.5%, 97.5%, 96%, and 87.6% respectively (ESI Fig. 5a†). The cell viability for NSC-34 cells treated with standard LNPs was 112.5% whereas the cell viability for cells treated with 12.5 μL and 50 μL IL-coated LNPs was 112.2% and 86.9%, respectively (ESI Fig. 5b†). The cell viability for cells treated with IL-incorporated LNPs was 74.3% (ESI Fig. 5b†). We observed a significant difference in the cell viability for cells treated with 50 μL IL-coated LNPs and IL-incorporated LNPs when compared to standard LNPs (p < 0.0001) (ESI Fig. 5b†). The lower cellular viability can be due to the 4-fold higher IL volume (50 μL) compared to 12.5 μL IL-coated LNPs. In general, we observed that cells are comparatively more sensitive to 50 μL ILs than 12.5 μL ILs. However, we did not note changes in the cell viability of b.End3 cells upon treatment with 50 μL IL-coated and IL-incorporated LNPs compared to standard LNPs (ESI Fig. 5a†). Overall, we observed that b.End3 cells are resilient and NSC-34 cells are comparatively more sensitive to LNP treatments. This cell-line dependent behavior of nanoparticle tolerance has been previously reported with other nanoparticle systems as well.94–96After determining the cytocompatibility of LNPs, we used flow cytometry to study the uptake of LNPs loaded with Cy5 siRNA in bEnd.3 mouse BECs and differentiated NSC-34 mouse motor neurons. Flow cytometry is a quantitative technique for single-cell analysis that measures the scattered light and the fluorescence emitted from a single-cell.97 Complexes of Cy5 siRNA with Lipofectamine RNAiMAX, a cationic lipid, were used as a positive control. We analyzed untreated/unstained cells to gate out their autofluorescence. The flow gating strategy and raw histogram plots of bEnd.3 BECs and NSC-34 motor neurons are presented in ESI Fig. 6 and 7.†
bEnd.3 cells treated with standard LNPs showed a 3.9-fold increase in the uptake of Cy5 siRNA (Fig. 9a). We noted a significant increase in the uptake of Cy5 siRNA by cells treated with 12.5 μL (14.2-fold; p < 0.0001) and 50 μL (6.5-fold, p < 0.05) 1:1 IL-coated LNPs in comparison to standard LNPs (Fig. 9a). We also noted a significant increase in the uptake of Cy5 siRNA by cells treated with 50 μL IL-incorporated LNPs (7.3-fold; p < 0.01) (Fig. 9a).
 | ||
| Fig. 9 Cellular uptake of standard, IL-coated, and IL-incorporated LNPs containing Cy5 siRNA into bEnd.3 BECs (a) and NSC-34 motor neurons (b) using flow cytometry. bEnd.3 BECs and NSC-34 motor neurons were treated for 4 h with standard LNPs, IL-coated LNPs, IL-incorporated LNPs, and siRNA-Lipofectamine RNAiMAX complexes loaded with 50 nM Cy5 siRNA in complete growth medium or Neurobasal medium in a humidified incubator at 37 °C and 5% CO2. Lipofectamine-siRNA complexes and untreated cells were used as positive and negative controls, respectively. Data are presented as a fold increase in Cy5 siRNA uptake (% Cy5+ cells) relative to untreated cells (n = 4). Statistical analysis was done using one-way ANOVA with Dunnett's multiple comparisons test. *p < 0.05, **p < 0.001, ****p < 0.0001. | ||
NSC-34 motor neurons treated with standard LNPs showed a 21-fold increase in Cy5 siRNA uptake (Fig. 9b). We noted a significant increase in Cy5 siRNA uptake by cells treated with 12.5 μL (26-fold; p < 0.05) and 50 μL (37.7-fold, p < 0.0001) 1:1 IL-coated LNPs in comparison to standard LNPs (Fig. 9b). We also noted a significant increase in the uptake of Cy5 siRNA by cells treated with 50 μL IL-incorporated LNPs (42.4-fold; p < 0.0001) (Fig. 9b).
Overall, the significantly higher uptake of Cy5 siRNA in bEnd.3 BECs and NSC-34 motor neurons treated with 12.5 μL IL-coated LNPs compared to standard LNPs can be attributed to the smaller sizes of IL-coated LNPs compared to standard LNPs (Fig. 2). Cells may preferentially internalize smaller particles as compared to larger particles.98–101 Additionally, the ability of ILs to permeate BEC membranes may have potentially resulted in higher uptake of IL-coated LNPs compared to standard LNPs.102–104 Thus, in the case of BECs, the surface characteristics of the particles, i.e., IL-coating but not IL-incorporation seemed to drive differences in the extent of uptake. However, in the case of motor neurons, both IL-coated as well as incorporated LNPs showed greater uptake compared to the standard LNPs. In fact, IL-incorporated LNPs showed similar levels of uptake as the positive control, Lipofectamine RNAiMAX. The overall differences in the magnitude of uptake (relative to untreated cells) of Cy5 siRNA by mouse BECs and mouse motor neurons may be attributed to the differences in membrane characteristics of these cell types. BECs have extremely low pinocytic activity that limits their capability to internalize nanoparticles.105–108 Conversely, we have previously shown the ability of LNPs to internalize into rat primary dorsal root ganglion neurons to a considerable extent (at levels similar to the positive control, Lipofectamine RNAiMAX).15 It appears that neurons are amenable to delivery as long as the particle surfaces promote affinity to cell membranes, at least in vitro. To summarize, IL-modified LNPs promote affinity and internalization of LNPs into hard-to-deliver cell types such as BECs and neurons.
In addition to studying the uptake of IL-coated LNPs and IL-incorporated LNPs, we also determined their cytoplasmic localization using fluorescence microscopy (ESI Fig. 8†). While the overall Cy5 signal intensities are low, we observed cytosolic uptake of Cy5 siRNA via colocalization of Hoechst + Cy5 channels in ESI Fig. 8b and c† (white arrows show colocalization). This observation suggests the cytoplasmic presence of Cy5 siRNA which is essential for the therapeutic activity of siRNA.
Conclusions
For the first time, we have re-engineered LNPs using IL to formulate IL-coated LNPs and IL-incorporated LNPs for the delivery of siRNA to BECs and neurons. IL-coated LNPs and IL-incorporated LNPs showed similar morphologies to standard LNPs and showed significant changes in size and surface charge compared to standard LNPs. IL-coated LNPs and IL-incorporated LNPs were colloidally stable over seven days of storage. IL-coated LNPs were stable for 4 h in the presence of serum proteins. IL-coated LNPs and IL-incorporated LNPs showed superior uptake into mouse BECs and neurons compared to standard LNPs. Additionally, IL-coated LNPs showed a significant reduction in plasma protein adsorption compared to standard LNPs. Overall, our results demonstrate the potential of re-engineering LNPs as a novel carrier for the delivery of siRNA to BECs and neurons—extra-hepatic, CNS targets.Abbreviations
| BBB | Blood–brain barrier |
| BECs | Brain endothelial cells |
| CNS | Central nervous system |
| DI water | Deionized water |
| DLS | Dynamic light scattering |
| ILs | Ionic liquids |
| LNPs | Lipid nanoparticles |
| PBS | Phosphate-buffered saline |
| PEG-DMG | Polyethylene glycol-dimyristoyl glycerol |
| *IL-coated LNPs | standard LNPs coated with ILs |
| *IL-incorporated LNPs | PEG-DMG component in standard LNPs replaced with ILs |
Conflicts of interest
E. E. L. T. is an inventor on patents covering ionic liquids, P. K., E. E. L. T. and D. S. are named inventors in a patent application covering IL-incorporated LNPs.Acknowledgements
This work was supported via start-up funds for the Manickam laboratory from Duquesne University (DU) and the Charles Henry Leach II Fund to the PI. The authors are thankful to Drs Lauren O'Donnell, Manisha Chandwani and Yashika Kamte (DU) for their kind support with the flow cytometry studies. EELT acknowledges the PhRMA Foundation, and the College of Liberal Arts at the University of Mississippi for financial support.References
- R. Cross, Without these lipid shells, there would be no mRNA vaccines for COVID-19, https://cen.acs.org/pharmaceuticals/drug-delivery/Without-lipid-shells-mRNA-vaccines/99/i8 Search PubMed.
- F. N. RELEASE, FDA Approves First COVID-19 Vaccine, https://www.fda.gov/news-events/press-announcements/fda-approves-first-covid-19-vaccine Search PubMed.
- K. B. Johnsen, A. Burkhart, F. Melander, P. J. Kempen, J. B. Vejlebo, P. Siupka, M. S. Nielsen, T. L. Andresen and T. Moos, Sci. Rep., 2017, 7, 1–13 CrossRef CAS PubMed.
- P. Khare, S. X. Edgecomb, C. M. Hamadani, E. E. L. Tanner and D. S. Manickam, Adv. Drug Delivery Rev., 2023, 197, 114861 CrossRef CAS PubMed.
- A. Akinc, M. Maier, M. Manoharan, K. Fitzgerald, M. Jayaraman, S. Barros, S. Ansell, X. Du, M. Hope, T. Madden, B. Mui, S. Semple, Y. Tam, M. Ciufolini, D. Witzigmann, J. Kulkarni, R. van der Meel and P. Cullis, Nat. Nanotechnol., 2019, 14, 1084–1087 CrossRef CAS PubMed.
- A. Akinc, W. Querbes, S. De, J. Qin, M. Frank-Kamenetsky, K. Jayaprakash, M. Jayaraman, K. Rajeev, W. Cantley, R. Dorkin, J. Butler, L. Qin, T. Racie, A. Sprague, F. Eugenio, A. Zeigerer, M. Hope, M. Zerial, D. Sah and M. Maier, Mol. Ther., 2010, 18, 1357–1364 CrossRef CAS PubMed.
- N. Belliveau, J. Huft, P. Lin, S. Chen, A. Leung, T. Leaver, A. Wild, J. Lee, R. Taylor, Y. Tam, C. Hansen and P. Cullis, Mol. Ther.–Nucleic Acids, 2012, 1, e37 CrossRef PubMed.
- A. K. Blakney, P. F. McKay, B. I. Yus, Y. Aldon and R. J. Shattock, Gene Ther., 2019, 26, 363–372 CrossRef CAS PubMed.
- M. Evers, J. Kulkarni, R. van der Meel, P. Cullis, P. Vader and R. Schiffelers, Small Methods, 2017, 2, 1700375 CrossRef.
- J. Kulkarni, M. Darjuan, J. Mercer, S. Chen, R. van der Meel, J. Thewalt, Y. Tam and P. Cullis, ACS Nano, 2018, 12, 4787–4795 CrossRef CAS PubMed.
- J. A. Kulkarni, P. R. Cullis and R. van der Meel, Nucleic Acid Ther., 2018, 28, 146–157 CrossRef CAS PubMed.
- R. Van der Meel, S. Chen, J. Zaifman, J. Kulkarni, X. Zhang, Y. Tam, M. Bally, R. Schiffelers, M. Ciufolini, P. Cullis and Y. Tam, Small, 2021, 17, 2103025 CrossRef CAS PubMed.
- K. Whitehead, R. Dorkin, A. Vegas, P. Chang, O. Veiseh, J. Matthews, O. Fenton, Y. Zhang, K. Olejnik, V. Yesilyurt, D. Chen, S. Barros, B. Klebanov, T. Novobrantseva, R. Langer and D. Anderson, Nat. Commun., 2014, 5, 4277 CrossRef CAS PubMed.
- P. Khare, J. F. Conway and D. S. Manickam, Eur. J. Pharm. Biopharm., 2022, 180, 238–250 CrossRef CAS PubMed.
- P. Khare, K. M. Dave, Y. S. Kamte, M. A. Manoharan, L. A. O'Donnell and D. S. Manickam, AAPS J., 2021, 24, 8 CrossRef PubMed.
- J. F. Nabhan, K. M. Wood, V. P. Rao, J. Morin, S. Bhamidipaty, T. P. LaBranche, R. L. Gooch, F. Bozal, C. E. Bulawa and B. C. Guild, Sci. Rep., 2016, 6, 20019 CrossRef CAS PubMed.
- R. L. Rungta, H. B. Choi, P. J. Lin, R. W. Ko, D. Ashby, J. Nair, M. Manoharan, P. R. Cullis and B. A. MacVicar, Mol. Ther.–Nucleic Acids, 2013, 2, e136 CrossRef CAS PubMed.
- M. Tamaru, H. Akita, T. Nakatani, K. Kajimoto, Y. Sato, H. Hatakeyama and H. Harashima, Int. J. Nanomed., 2014, 4267–4276 Search PubMed.
- T. Wei, Q. Cheng, Y.-L. Min, E. N. Olson and D. J. Siegwart, Nat. Commun., 2020, 11, 3232 CrossRef CAS.
- X. Hou, T. Zaks, R. Langer and Y. Dong, Nat. Rev. Mater., 2021, 6, 1078–1094 CrossRef CAS PubMed.
- A. Leung, I. Hafez, S. Baoukina, N. Belliveau, I. Zhigaltsev, E. Afshinmanesh, D. Tieleman, C. Hansen, M. Hope and P. Cullis, J. Phys. Chem. C, 2012, 116, 18440–18450 CrossRef CAS.
- S. V. Morse, A. Mishra, T. G. Chan, R. T. de Rosales and J. J. Choi, J. Controlled Release, 2022, 341, 605–615 CrossRef CAS PubMed.
- L. H. Treat, N. McDannold, Y. Zhang, N. Vykhodtseva and K. Hynynen, Ultrasound Med. Biol., 2012, 38, 1716–1725 CrossRef PubMed.
- H. Tanigawa, N. Suzuki and T. Suzuki, Eur. J. Pharm. Sci., 2022, 178, 106290 CrossRef CAS PubMed.
- A. Banerjee, K. Ibsen, Y. Iwao, M. Zakrewsky and S. Mitragotri, Adv. Healthcare Mater., 2017, 6, 1601411 CrossRef PubMed.
- T. Hattori, H. Tagawa, M. Inai, T. Kan, S.-i. Kimura, S. Itai, S. Mitragotri and Y. Iwao, Sci. Rep., 2019, 9, 1–11 CrossRef.
- E. E. Tanner, K. N. Ibsen and S. Mitragotri, J. Controlled Release, 2018, 286, 137–144 CrossRef CAS PubMed.
- M. Zakrewsky, K. S. Lovejoy, T. L. Kern, T. E. Miller, V. Le, A. Nagy, A. M. Goumas, R. S. Iyer, R. E. Del Sesto and A. T. Koppisch, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13313–13318 CrossRef CAS.
- A. Banerjee, K. Ibsen, T. Brown, R. Chen, C. Agatemor and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7296–7301 CrossRef CAS PubMed.
- M. Nurunnabi, K. N. Ibsen, E. E. Tanner and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 25042–25047 CrossRef CAS PubMed.
- A. Vaidya and S. Mitragotri, J. Controlled Release, 2020, 327, 26–34 CrossRef CAS PubMed.
- M. A. Gebbie, A. M. Smith, H. A. Dobbs, G. G. Warr, X. Banquy, M. Valtiner, M. W. Rutland, J. N. Israelachvili, S. Perkin and R. Atkin, Chem. Commun., 2017, 53, 1214–1224 RSC.
- J. F. Rudzinski, S. Kloth, S. Wörner, T. Pal, K. Kremer, T. Bereau and M. Vogel, J. Phys.: Condens. Matter, 2021, 33, 224001 CrossRef CAS PubMed.
- Y.-L. Wang, B. Li, S. Sarman, F. Mocci, Z.-Y. Lu, J. Yuan, A. Laaksonen and M. D. Fayer, Chem. Rev., 2020, 120, 5798–5877 CrossRef CAS PubMed.
- A. Banerjee and J. K. Shah, J. Chem. Phys., 2020, 153, 034306 CrossRef CAS PubMed.
- K. N. Ibsen, H. Ma, A. Banerjee, E. E. Tanner, S. Nangia and S. Mitragotri, ACS Biomater. Sci. Eng., 2018, 4, 2370–2379 CrossRef CAS PubMed.
- M. M. Islam, S. Barik and M. Sarkar, J. Phys. Chem. B, 2019, 123, 1512–1526 CrossRef CAS PubMed.
- J. Y. Lee, K. M. Selfridge, E. M. Kohn, T. D. Vaden and G. A. Caputo, Biomolecules, 2019, 9, 264 CrossRef CAS PubMed.
- R. Markiewicz, A. Klimaszyk, M. Jarek, M. Taube, P. Florczak, M. Kempka, Z. Fojud and S. Jurga, Int. J. Mol. Sci., 2021, 22, 5935 CrossRef CAS PubMed.
- I. R. Nichols, C. M. Hamadani, C. M. Chism, A. N. Hunter, H. Ahmad, K. Wontor, A. E. Williams, P. Vashisth, N. I. Hammer and S. Kundu, Adv. Ther., 2022, 2200096 Search PubMed.
- Q. M. Qi and S. Mitragotri, J. Controlled Release, 2019, 311, 162–169 CrossRef PubMed.
- T. Singh, P. Bharmoria, M.-a. Morikawa, N. Kimizuka and A. Kumar, J. Phys. Chem. B, 2012, 116, 11924–11935 CrossRef CAS PubMed.
- A. M. Curreri, S. Mitragotri and E. E. Tanner, Advanced Science, 2021, 8, 2004819 CrossRef CAS PubMed.
- K. S. Egorova, E. G. Gordeev and V. P. Ananikov, Chem. Rev., 2017, 117, 7132–7189 CrossRef CAS.
- J. Brenner, S. Mitragotri and V. Muzykantov, Annu. Rev. Biomed. Eng., 2021, 23, 225–248 CrossRef CAS PubMed.
- Z. Zhao, J. Kim, V. V. Chandran Suja, N. Kapate, Y. Gao, J. Guo, V. Muzykantov and S. Mitragotri, Advanced Science, 2022, 9, 2201293 CrossRef CAS PubMed.
- C. Hamadani, M. Goetz, S. Mitragotri and E. Tanner, Sci. Adv., 2020, 6, eabd7563 CrossRef CAS PubMed.
- I. I. Slowing, C. W. Wu, J. L. Vivero-Escoto and V. S. Y. Lin, Small, 2009, 5, 57–62 CrossRef CAS.
- J. S. Brenner, D. C. Pan, J. W. Myerson, O. A. Marcos-Contreras, C. H. Villa, P. Patel, H. Hekierski, S. Chatterjee, J.-Q. Tao and H. Parhiz, Nat. Commun., 2018, 9, 2684 CrossRef.
- C. M. Hamadani, M. J. Goetz, S. Mitragotri and E. E. Tanner, Sci. Adv., 2020, 6, eabd7563 CrossRef CAS PubMed.
- E. E. Tanner, Nat. Chem., 2022, 14, 842 CrossRef CAS.
- N. A. Lassen, D. H. Ingvar and E. Skinhøj, Sci. Am., 1978, 239, 62–71 CrossRef CAS.
- D. Allen and P. Lockman, Life Sci., 2003, 73, 1609–1615 CrossRef CAS PubMed.
- M. Inazu, Nutrients, 2019, 11, 2265 CrossRef CAS PubMed.
- K.-i. Hosoya and M. Tachikawa, Clin. Exp. Nephrol., 2011, 15, 478–485 CrossRef CAS.
- T. Taogoshi, A. Nomura, T. Murakami, J. Nagai and M. Takano, J. Pharm. Pharmacol., 2010, 57, 61–66 CrossRef.
- J. Kulkarni, P. Cullis and R. van der Meel, Nucleic Acid Ther., 2018, 28, 146–157 CrossRef CAS.
- Y. Dong, K. T. Love, J. R. Dorkin, S. Sirirungruang, Y. Zhang, D. Chen, R. L. Bogorad, H. Yin, Y. Chen and A. J. Vegas, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 3955–3960 CrossRef CAS.
- X. Hou, T. Zaks, R. Langer and Y. Dong, Nat. Rev. Mater., 2021, 6, 1078–1094 CrossRef CAS PubMed.
- K. T. Love, K. P. Mahon, C. G. Levins, K. A. Whitehead, W. Querbes, J. R. Dorkin, J. Qin, W. Cantley, L. L. Qin and T. Racie, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1864–1869 CrossRef CAS.
- K. A. Whitehead, J. R. Dorkin, A. J. Vegas, P. H. Chang, O. Veiseh, J. Matthews, O. S. Fenton, Y. Zhang, K. T. Olejnik, V. Yesilyurt, D. Chen, S. Barros, B. Klebanov, T. Novobrantseva, R. Langer and D. G. Anderson, Nat. Commun., 2014, 5, 4277 CrossRef CAS.
- C. M. Hamadani, I. Chandrasiri, M. L. Yaddehige, G. S. Dasanayake, I. Owolabi, A. Flynt, M. Hossain, L. Liberman, T. P. Lodge and T. A. Werfel, Nanoscale, 2022, 14, 6021–6036 RSC.
- C. M. Hamadani, F. Mahdi, A. Merrell, J. Flanders, R. Cao, P. Vashisth, M. C. Pride, A. N. Hunter, G. Singh and G. Roman, Res. Sq., 2023, rs.3.rs–2574352 Search PubMed.
- C. Biosystems, Cedarlane Laboratories, Catalog Search PubMed.
- R. D. Madison, C. McGee, R. Rawson and G. A. Robinson, J. Extracell. Vesicles, 2014, 3, 22865 CrossRef PubMed.
- K. M. Dave, L. Ali and D. S. Manickam, PLoS One, 2020, 15, 1–28 CrossRef PubMed.
- K. M. Dave, L. Han, M. A. Jackson, L. Kadlecik, C. L. Duvall and D. S. Manickam, Pharm. Res., 2020, 37, 176 CrossRef CAS.
- K. M. Dave, W. Zhao, C. Hoover, A. D'Souza and D. S. Manickam, AAPS PharmSciTech, 2021, 22, 18 CrossRef CAS PubMed.
- M. Jayaraman, S. M. Ansell, B. L. Mui, Y. K. Tam, J. Chen, X. Du, D. Butler, L. Eltepu, S. Matsuda and J. K. Narayanannair, Angew. Chem., 2012, 124, 8657–8661 CrossRef.
- A. Pharmaceuticals, Onpattro (Patisiran) Lipid Complex Injection, https://www.onpattro.com Search PubMed.
- S. C. Semple, A. Akinc, J. Chen, A. P. Sandhu, B. L. Mui, C. K. Cho, D. W. Sah, D. Stebbing, E. J. Crosley and E. Yaworski, Nat. Biotechnol., 2010, 28, 172–176 CrossRef CAS.
- R. W. Mahley, Science, 1988, 240, 622–630 CrossRef CAS PubMed.
- E. E. Tanner, A. M. Curreri, J. P. Balkaran, N. C. Selig-Wober, A. B. Yang, C. Kendig, M. P. Fluhr, N. Kim and S. Mitragotri, Adv. Mater., 2019, 31, 1901103 CrossRef.
- B. L. Mui, Y. K. Tam, M. Jayaraman, S. M. Ansell, X. Du, Y. Y. C. Tam, P. J. C. Lin, S. Chen, J. K. Narayanannair, K. G. Rajeev, M. Manoharan, A. Akinc, M. A. Maier, P. Cullis, T. D. Madden and M. J. Hope, Mol. Ther.–Nucleic Acids, 2013, 2, e139 CrossRef CAS PubMed.
- B.-M. Chen, T.-L. Cheng and S. R. Roffler, ACS Nano, 2021, 15, 14022–14048 CrossRef CAS.
- Q. Yang and S. Lai, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 655–677 CAS.
- P. Zhang, F. Sun, S. Liu and S. Jiang, J. Controlled Release, 2016, 244, 184–193 CrossRef CAS.
- T.-C. Chang, B.-M. Chen, J.-Y. Wu, T.-L. Cheng and S. Roffler, Biomed. Pharmacother., 2022, 146, 112502 CrossRef CAS PubMed.
- Z. He and P. Alexandridis, Adv. Colloid Interface Sci., 2017, 244, 54–70 CrossRef CAS.
- J. Obliosca, I. H. Arellano, M. Huang and S. Arco, Mater. Lett., 2010, 64, 1109–1112 CrossRef CAS.
- P. Aggarwal, J. B. Hall, C. B. McLeland, M. A. Dobrovolskaia and S. E. McNeil, Adv. Drug Delivery Rev., 2009, 61, 428–437 CrossRef CAS PubMed.
- F. Sebastiani, M. Yanez Arteta, L. Lindfors and M. Cardenas, J. Colloid Interface Sci., 2022, 610, 766–774 CrossRef CAS.
- N. E. Elsadek, E. Hondo, T. Shimizu, H. Takata, A. S. Abu Lila, S. E. Emam, H. Ando, Y. Ishima and T. Ishida, Mol. Pharm., 2020, 17, 2964–2970 CrossRef CAS PubMed.
- V. Kumar, J. Qin, Y. Jiang, R. G. Duncan, B. Brigham, S. Fishman, J. K. Nair, A. Akinc, S. A. Barros and P. V. Kasperkovitz, Mol. Ther.–Nucleic Acids, 2014, 3, e210 CrossRef PubMed.
- T. Suzuki, Y. Suzuki, T. Hihara, K. Kubara, K. Kondo, K. Hyodo, K. Yamazaki, T. Ishida and H. Ishihara, Int. J. Pharm., 2020, 588, 119792 CrossRef CAS PubMed.
- N. Bertrand, P. Grenier, M. Mahmoudi, E. M. Lima, E. A. Appel, F. Dormont, J.-M. Lim, R. Karnik, R. Langer and O. C. Farokhzad, Nat. Commun., 2017, 8, 777 CrossRef.
- S. A. Dilliard and D. J. Siegwart, Nat. Rev. Mater., 2023, 8, 282–300 CrossRef CAS.
- A. S. A. Lila, H. Kiwada and T. Ishida, J. Controlled Release, 2013, 172, 38–47 CrossRef.
- D. E. Owens III and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef.
- C. M. Hamadani, M.S. Thesis, Harvard University, 2020 Search PubMed.
- T. Blunk, D. F. Hochstrasser, J. C. Sanchez, B. W. Muller and R. H. Muller, Electrophoresis, 1993, 14, 1382–1387 CrossRef CAS PubMed.
- R. Gref, M. Lück, P. Quellec, M. Marchand, E. Dellacherie, S. Harnisch, T. M. Blunk and R. H. Müller, Colloids Surf., B, 2000, 18, 301–313 CrossRef CAS PubMed.
- Z. He, L. Miao, R. Jordan, S. Manickam, R. Luxenhofer and A. V. Kabanov, Macromol. Biosci., 2015, 15, 1004–1020 CrossRef CAS PubMed.
- A. Kroll, C. Dierker, C. Rommel, D. Hahn, W. Wohlleben, C. Schulze-Isfort, C. Göbbert, M. Voetz, F. Hardinghaus and J. Schnekenburger, Part. Fibre Toxicol., 2011, 8, 1–19 CrossRef PubMed.
- B. Pradines, V. Lievin-Le Moal, C. Vauthier, G. Ponchel, P. M. Loiseau and K. Bouchemal, Int. J. Pharm., 2015, 491, 17–20 CrossRef CAS PubMed.
- E. Sulheim, T.-G. Iversen, V. To Nakstad, G. Klinkenberg, H. Sletta, R. Schmid, A. R. Hatletveit, A. M. Wågbø, A. Sundan and T. Skotland, Int. J. Mol. Sci., 2017, 18, 2454 CrossRef PubMed.
- H. M. Davey and D. B. Kell, Microbiol. Rev., 1996, 60, 641–696 CrossRef CAS PubMed.
- E. C. Cho, L. Au, Q. Zhang and Y. Xia, Small, 2010, 6, 517–522 CrossRef CAS PubMed.
- C. Freese, M. I. Gibson, H.-A. Klok, R. E. Unger and C. J. Kirkpatrick, Biomacromolecules, 2012, 13, 1533–1543 CrossRef CAS PubMed.
- C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Biomaterials, 2010, 31, 3657–3666 CrossRef CAS PubMed.
- Q. He, Z. Zhang, Y. Gao, J. Shi and Y. Li, Small, 2009, 5, 2722–2729 CrossRef CAS.
- K. Cook, K. Tarnawsky, A. J. Swinton, D. D. Yang, A. S. Senetra, G. A. Caputo, B. R. Carone and T. D. Vaden, Biomolecules, 2019, 9, 251 CrossRef CAS.
- N. Gal, D. Malferarri, S. Kolusheva, P. Galletti, E. Tagliavini and R. Jelinek, Biochim. Biophys. Acta, Biomembr., 2012, 1818, 2967–2974 CrossRef CAS PubMed.
- N. Kaur, S. Kumar, Shiksha, G. K. Gahlay and V. S. Mithu, J. Phys. Chem. B, 2021, 125, 3613–3621 CrossRef CAS PubMed.
- B. Engelhardt and L. Sorokin, Semin. Immunopathol., 2009, 31, 497–511 CrossRef.
- D. Furtado, M. Björnmalm, S. Ayton, A. I. Bush, K. Kempe and F. Caruso, Adv. Mater., 2018, 30, 1801362 CrossRef.
- S. Nakagawa, M. A. Deli, H. Kawaguchi, T. Shimizudani, T. Shimono, A. Kittel, K. Tanaka and M. Niwa, Neurochem. Int., 2009, 54, 253–263 CrossRef CAS PubMed.
- S. Yazdani, J. R. Jaldin-Fincati, R. V. Pereira and A. Klip, Traffic, 2019, 20, 390–403 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00699a |
| This journal is © The Royal Society of Chemistry 2024 |