
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mixed polymer brushes for controlled protein adsorption: state of the art and future prospective
Muhammad
Atif
 * and
Ali
Balasini
* and
Ali
Balasini
Macromolecular Chemistry, Department of Chemistry and Biology, University of Siegen, Adolf-Reichwein-Strasse 2, Siegen 57076, Germany. E-mail: muhammad.atif@uni-siegen.de
First published on 18th January 2024
Abstract
Mixed polymer brushes (MPBs) consist of a combination of two or more disparate polymers with one end tethered to an interface by covalent bond and the other end stretched into the surroundings. Owing to the morphologies of the phase separation of these MPBs, they have been widely studied as “responsive polymers” as they belong to the category of polymer brushes having the ability to undergo chemical and conformational changes in response to external stimuli. The resulting assembly presents an exceptional opportunity to precisely control the adsorption and desorption of protein by regulating the surrounding environment, brush thickness, density, chemistry, and architecture. This precise control over adsorption and desorption of protein makes responsive polymers very useful in many applications including drug delivery, wound repairing scaffolds, antifouling surfaces, and many other biomedical and biotechnological fields. In this review, the intention is to describe the state of the art of MPBs, different synthetic techniques, morphologies, interaction with protein molecules, and responsiveness over surrounding environment. Finally, the novel applications of MPBs and their current limitations with possible solutions for upcoming studies are discussed briefly.
 Muhammad Atif | Muhammed Atif is currently pursuing his PhD in Polymer Chemistry at the University of Siegen, Germany. He has been awarded the prestigious Marie Skłodowska-Curie fellowship. He also had the opportunity to work as a visiting researcher at the University of Bath, Bath, United Kingdom, and University Medical Center (UMC), Amsterdam, Netherlands, as part of his PhD program. He has published over 15 papers in reputable scientific journals. He holds a Master's degree in Polymer Chemistry from the University of Science and Technology of China (USTC), P.R. China. Mr Atif's research interests primarily revolve around polymer chemistry, with a focus on antifouling coatings, polymer brushes, post polymerization reactions, responsive hydrogels, and drug delivery. |
 Ali Balasini | Ali Balasini, a PhD candidate affiliated with the Macro Molecular Group at the University of Siegen, Germany, has undertaken diverse research endeavors spanning various scientific disciplines. With a bachelor's degree earned in Iraq and a master's degree from the University of Düsseldorf in Germany, Ali has accumulated over two years of dedicated doctoral work. Focusing on organic synthesis, Ali's research focus includes ligands, inorganic complexes, and polymers, with the primary project aiming at the synthesis of antibacterial polymers. Ali possesses expertise in synthesizing and analyzing complexes and polymers, specializing in post-polymerization modification reactions and thermo-responsive polymers. |
1. Introduction
Proteins play a very vital role in biomaterial sciences. They interact with interfaces of many biomaterial surfaces, especially during biomedical coating,1,2 gene delivery,3 controlled drug release,4,5 and biosensors.6 When biomaterial devices engage with some kind of biological fluid, then proteins instantaneously and uncontrollably adsorb on the surface of the device.7 The adsorption of proteins is necessary for drug delivery devices, immunological tests, and the integration of prostheses.8,9 However, in the case of bioimplants, assay platforms, and protein chips, it leads to failure.10,11 The immediate adsorption of proteins also occurs on the interfaces of agricultural, biotechnological, and industrial devices, leading to the degradation of materials and eventually failure.7,12,13 Therefore, there is a demand for some simple, inexpensive assembly to modify the surfaces for the controlled adsorption of proteins, and many researchers have risen to this challenge.Over the past few decades, polymer brushes have gained much attention due to their unique structure, easy synthesis by controllable polymerization techniques, and their fabrication. In order to improve the efficiency of biomaterials, polymer brushes are one of the promising candidates to control the adsorption of proteins.14–16 Among all the polymer brushes, stimuli responsive polymer brushes are extensively studied due to their unique behavior under specific environments. At present, the most widely used stimuli responsive polymers have properties of one or more stimuli responsiveness such as pH,17,18 temperature,19,20 magnetic field,21,22 ionic strength,23 and light.24 Protein adsorption and desorption by polymer brushes is a very complicated process and is normally controlled by many factors including hydrophilicity and hydrophobicity of the polymers, surface roughness, and potential, other than the external parameters like temperature, pH, ionic strength, and magnetic field.25–27 Among all the external parameters, pH- and temperature-responsive polymer brushes are studied extensively because of their ease of regulation.
In the pH-responsive polymer brushes, the dispersion and aggregation can be controlled by changing the pH of the environment and confirmation of the protein particles. The adsorption and desorption of proteins could also be controlled by controlling the pH of the medium, which makes it possible to control the separation of proteins.28,29 Similarly, temperature-responsive polymers are investigated as potential temperature sensors owing to the alterations in size and configuration of the polymer in response to the temperature of the surrounding medium.30 These polymers play a crucial rule in regulating the adsorption and desorption of proteins as they interact with proteins through their hydrophobic properties.
The field of responsiveness increases with the design of polymer brushes composed of two different chemical species. This involves polymers containing chemically different side chains, di-block copolymers, and mixed polymer brushes (MPBs) where two or more different polymers are grafted on the side. Merging chemically different polymers having different characteristics and different responsive behavior in different environmental conditions provides an enormous combination to design specific assemblies for particular applications. A few examples contain the MPBs that are switchable between fouling and anti-fouling by changing the pH of the environment.31–34 Mixed polymer brushes are comprised of two (binary polymer brushes) or three (tertiary polymer brushes) chemically different polymers randomly tethered on the interfaces by a covalent bond (Fig. 1). This covalent bond allows phase segregation and prevents the brushes from de-wetting in contrast to the polymer blend or deposition of polymers from the solution.35,36 MPBs could be used as an effective candidate for the fabrication of smart interfaces with tunable properties for many different purposes including protein adsorption and desorption,37–39 drug release,40 and nanomotors.41
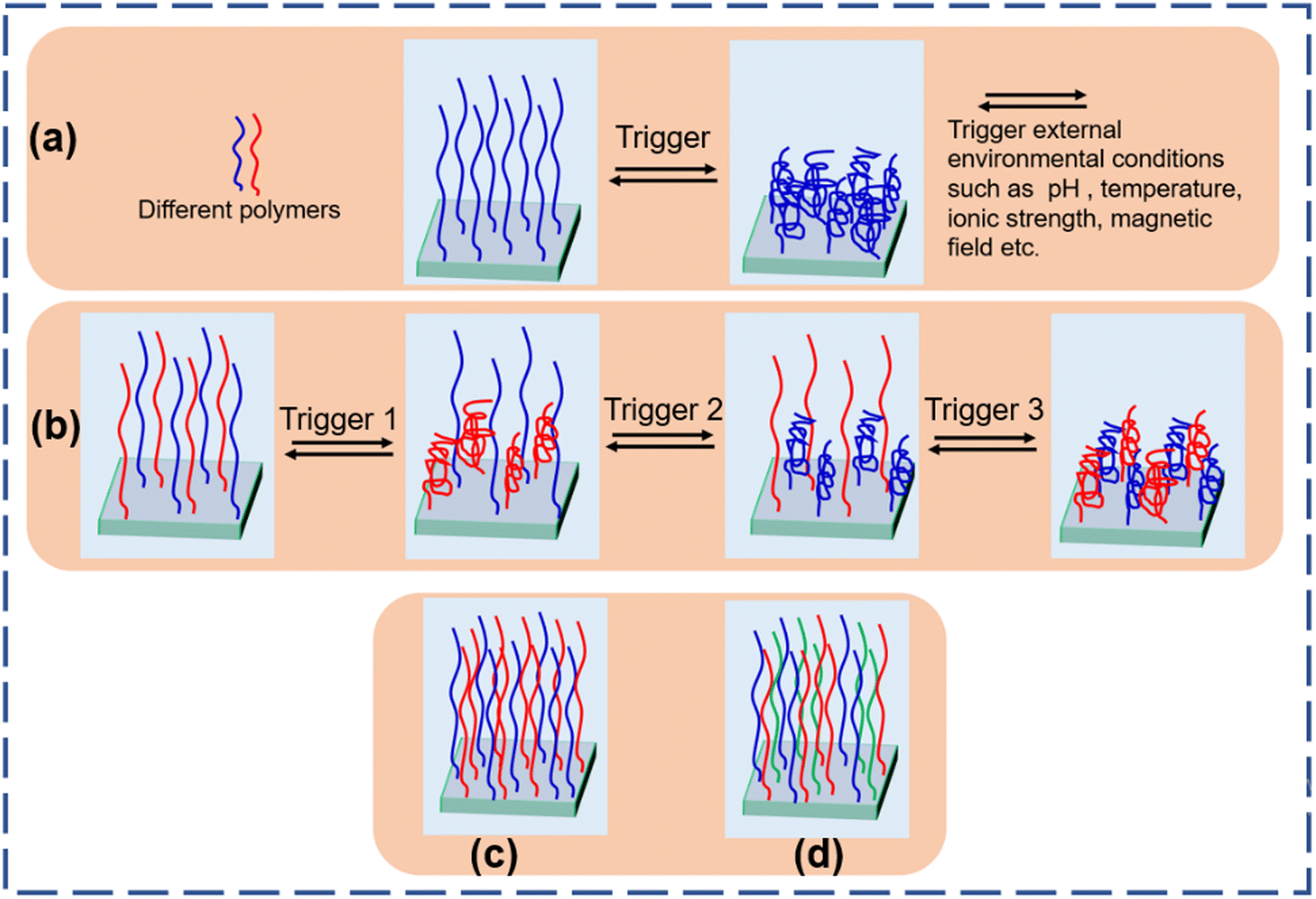 | ||
| Fig. 1 Stimuli responsive behavior of (a) homo polymer brushes and (b) binary polymer brushes. Architecture of (c) binary polymer brushes and (d) tertiary polymer brushes. | ||
This review article focuses mainly on the state of the art of responsive polymer brushes that are able to switch on/off their properties under different environments. The following sections describe the different synthetic schemes to synthesize different polymers with controlled features and current techniques to fabricate polymer brushes. Then this article will summarize the responsive properties of different polymers against different external stimuli. In this article, we will primarily focus on the protein adsorption and desorption properties of mixed polymer brushes. Finally, this review article will end with the perspective of existing challenges and provide an overview of the future paths in the field of responsive polymer brushes.
2. Design and fabrication of responsive polymer brushes
To date, there are many well-known and well-recognized techniques to prepare polymer brushes. All of them have their pros and cons and for a broader review, we suggest the reader to more exhaustive reviews.42–46 Generally, there are three techniques to prepare the polymer brushes including “polymer adsorption”, “grafting to”, and “grafting from”. The schematic illustrations of all these techniques are shown in Fig. 2. The overview of different grafting techniques used for the fabrication of different polymer brushes are shown in Table 1.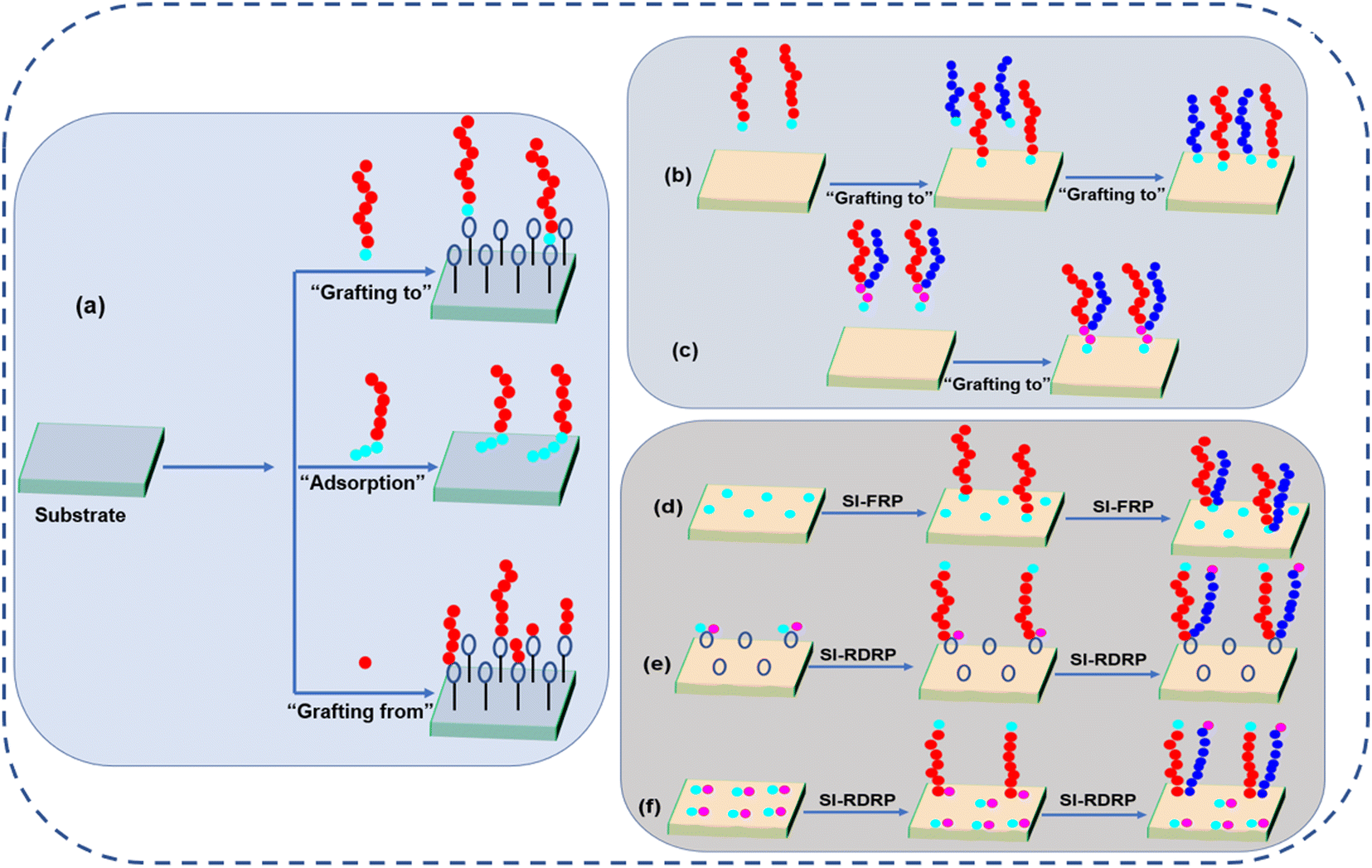 | ||
| Fig. 2 (a) Schematic illustration of different grafting techniques, (b) stepwise “grafting to” technique for homopolymer, (c) “grafting to” technique for Y-shaped di-block copolymer, (d) stepwise “grafting from” technique via surface-initiated free-radical polymerization (SI-FRP) using non-selective initiators, (e) “grafting from” technique via surface-initiated reversible-deactivation radical polymerization (SI-RDRP) using two disparate co-deposited initiators, and (f) “grafting from” technique via SI-RDRP using Y-shaped bifunctional initiator. | ||
| Polymers combination | Substrate | Grafting strategy | Ref. |
|---|---|---|---|
| PAA/P2VP | Si | “Grafting to” | 51 |
| PEO/PAA | gold | “Grafting to” | 37 |
| PDMS/P2VP | Si/ITO | “Grafting to” | 81 |
| PDMA/EPEI | Si | “Grafting to” | 82 |
| PS/P2VP | Si, Silica NP | “Grafting to” | 50, 83 and 84 |
| PMOXA/PAA | Si, glass | “Grafting to” | 32 and 33 |
| PMOXA/P4VP | Si, glass | “Grafting to” | 85 |
| PS/P4VP | silica | SI-ATRP, SI-NMP | 86 |
| PtBA/PS | Silica NP | SI-ATRP, SI-NMP | 87–90 |
| PHPMA/PREAEMA | MSN | SI-ATRP, SI-RAFT | 40 |
| PMMA/PLLA | CNT | SI-ATRP, SI-ROP | 80 |
| PS/PPhOXA | Si | SI-ROP, SI-NMP | 79 |
| PNIPAAM/PS | Si | SI-ATRP, SI-FRP | 91 |
| PS/PMMA/P4VP | Si | SI-FRP | 36 |
| PAA/PS | Silica NP | SI-ATRP, SI-NMP | 92 and 93 |
| PMMA/PS | Si, silica | SI-ATRP, SI-NMP | 94 and 95 |
2.1. Polymer adsorption technique
Polymer adsorption technique is very simple, classical, and was the first invented method for the preparation of polymer brushes. In this strategy, a di- block copolymer is required whose one side of the chain is adsorbed on the surface of the substrate while the other side of the chain is free to move and is not interacting with the surface.47,48 There are many advantages of this technique, including ease of preparation of a polymer brush using this technique simply by choosing the appropriate combination of substrate, solvent, and di-block copolymer. However, nowadays, this technique is not very convenient due to several drawbacks. One major drawback of this technique is that we need a solvent in which both blocks of copolymer should be completely dissolved; if some block or part of the block remains undissolved then micelles will be formed, due to which inhomogeneous adsorption would take place. Another serious drawback is that in this technique, polymer brushes with high grafting density cannot be prepared due to which these polymer brushes cannot be used for many specific applications including antifouling coating and resistance against protein adsorption.2.2. “Grafting to” technique
The “grafting to” technique could be performed using two strategies. The sequential co-deposition of two individual polymers and Y-shaped block copolymer in which the functional group is attached at the junction of both polymer segments. The former technique provides the opportunity to adjust the grafting ratio of both polymers, while in the latter technique, the ratio of surface tethered polymers is fixed inherently. Here, each grafting site confirms the functionalizing of both polymers as well as ensures that both polymers are in close vicinity on the molecular level.37,49In sequential co-deposition method, the order of grafting of polymers is very important. Minko and coworkers have prepared the polymer brushes of poly styrene (PS)/poly-2-vinyl pyridine (P2VP) by sequential co-deposition “grafting to” method. Glycidoxypropyltrimethoxysilane (GPS) was used to functionalize the silicon surface and its epoxy group undergoes strong reaction with the carboxy-terminated PS by annealing protocol at 150 °C. Then, in the second step, the grafting of carboxy-terminated P2VP was performed by the same mechanism in order to get the binary polymer brushes of PS/P2VP. The composition of both polymers could be adjusted by changing the annealing time.50
Investigation shows that reversing the order of grafting (grafting of P2VP first and then grafting of PS) leads to only few fractions of the composition of PS. This is due to the strong affinity between P2VP and GPC. Sequential co-deposition method is only useful if the grafting of non-polar polymer occurred at first and the polar component was grafted later. Later on, weakly charged binary polymer brushes of poly(acrylic acid) PAA/P2VP were also prepared by sequential co-deposition “grafting to” technique.51 This sequential co-deposition process was enhanced using the solution mixture of two pre-synthesized polymers. Here, the ratio of the components of both polymers is adjusted by changing the mass ratio of individual polymers in the solution. For example, previously, we have synthesized the solution mixture of poly(acrylic acid)-block-poly(glycidyl methacrylate) (PAA-b-PGMA) block copolymer and poly(2-methyl-2-oxazoline)-random-(glycidyl methacrylate) (PMOXA-r-GMA) comb copolymer. Then, the grafting of binary polymer brushes of PMOXA/PAA was done by spin coating the solution mixture, followed by an annealing protocol.32,33 Except for the glycidoxypropyltrimethoxysilane (GPS) and poly(glycidyl methacrylate) (PGMA) epoxies, there are many other epoxies to tether the polymers on the surface of the substrate.52–54
Using the “grafting to” strategy, click reaction can also be applied to prepare binary polymer brushes. For example, the mixed polymer brushes of poly2-methacryloyloxyethyl phosphorylcholine (PMPC)/poly 2-methacryloyloxy)ethyl trimethylammonium chloride (PMETA) were prepared by click reaction. Here, an adhesive layer of polydopamine (PDA) was coated on the stainless steel, which acts as the initiating anchoring layer on the surface. It reacts with the 3-azidopropyl carbonylimidazole and ethylene sulfide to produce azide and thiol on the surface, which react with the zwitterionic PMPC, followed by cationic PMETA and alkyne-azide click reaction to form the binary polymer brushes of PMPC/PMETA.54 Moreover, in our previous research group, we designed PMOXA/PAA coating using PDA as the adhesive layer. Firstly, silicon/glass substrate was modified with PDA layer over which sequential grafting of thiol-terminated PAA (PAA-SH) and amine-terminated PMOXA (PMOXA-NH2) was applied to form mixed polymer brushes. The components of the mixed polymer brushes were well modified by tuning the anchoring time of PAA-SH and PMOXA-NH2. The selection of these mentioned grafting steps prevents the polymer units from competing with similar grafting sites.15,55 The mixed polymer brushes could also be fabricated by attaching the functional group in the middle of two polymer chains. For instance, di-block copolymer of Y-shaped PS-b-poly(tert-butyl acylate)(PtBA) was prepared by tri-functional center. The hydroxyl group of this tri-functional group reacts with the carboxy terminated PtBA and PS in a stepwise protection/deprotection mechanism. The benzoic acid (third functional group) kept the di-block co-polymers chains anchored on the interface.56,57 This strategy keeps the same ratio of grafting of both polymers to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Both polymer units are chemically interlinked with each grafting site.
:1. Both polymer units are chemically interlinked with each grafting site.
2.3. “Grafting from” technique
“Grafting from” technique is also known as surface-initiated polymerization (SI-P) and can be utilized to prepare homopolymer and mixed polymer brushes.58–60 For this, firstly, a surface with physically attached or covalently linked monomer is prepared, from which initiation of polymerization occurs. By this technique, a large degree of polymerization and high grafting density can be achieved.46 Here, the degree of polydispersity strongly depends upon the polymerization reaction type. The “grafting from” approach can be used to fabricate complicated brush architectures, e.g., di-block copolymer brushes and branched polymer brushes. Here, we will discuss many SI-P approaches for the fabrication of polymer brushes. Fig. 2(d–f) illustrates the SI-P approach for fabricating polymer brushes.Although this technique is very versatile for the fabrication of polymer, still there are many disadvantages of this approach: (1) for the fabrication of binary mixed polymer brushes, both polymerization reactions use the same initiator. After the grating of the first polymer, there is no or very less initiator left over for the fabrication of the second polymer. Therefore, precise control over the ratio is not easy.62 (2) Chain length and brush architecture are difficult to control due to the termination reaction and many side reactions.63
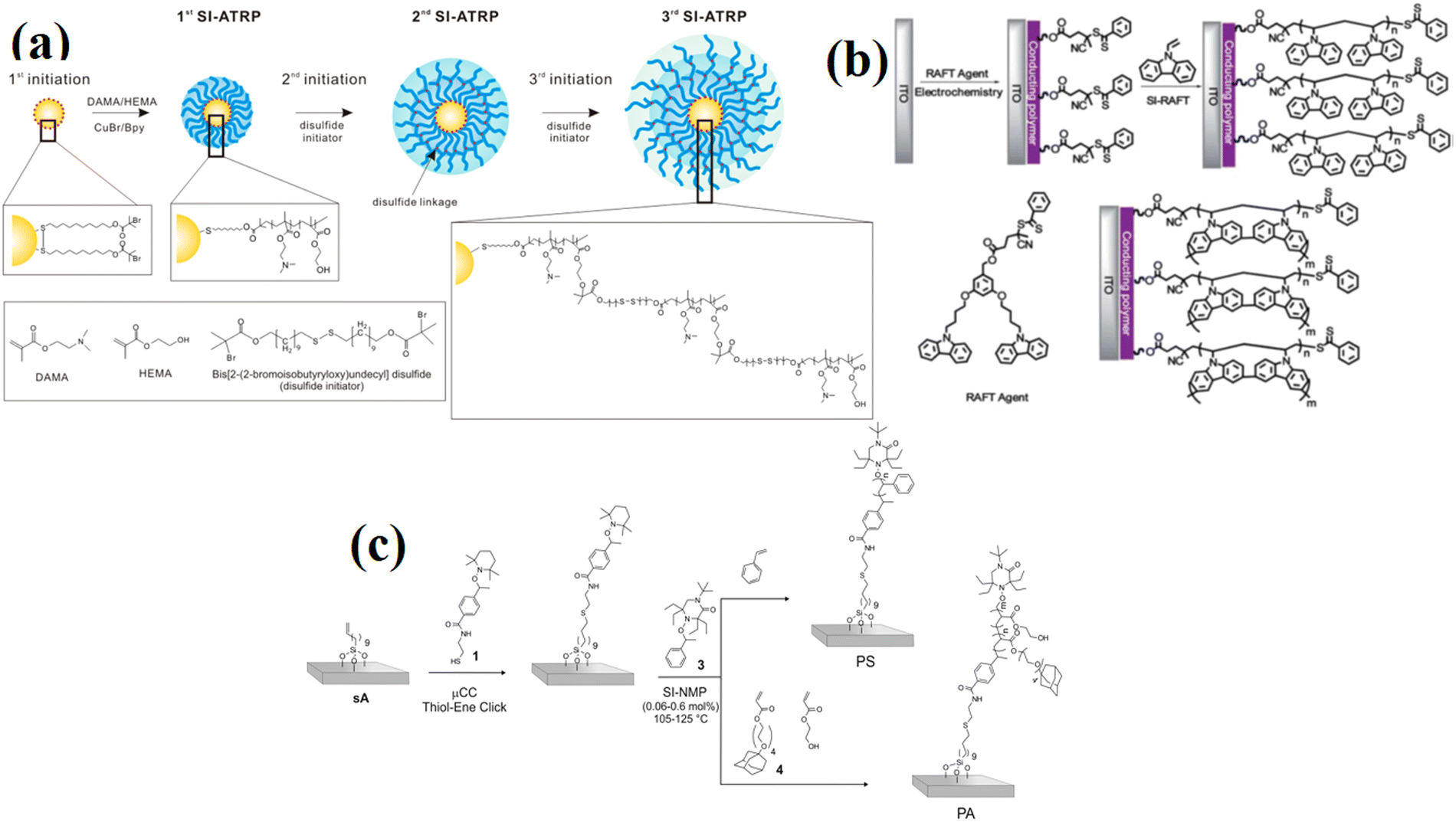 | ||
| Fig. 3 (a) Synthesis of polymer-multilayered gold nanoparticles (AuNP) via SI-ATRP. (Reproduced from ref. 72 with permission from American Chemical Society, copyright [2018]). (b) SI-RAFT polymerization for the synthesis of PVK HTL on ITO substate. (Reproduced from ref. 73 with permission from Royal Society of Chemistry, copyright [2013]). (c) Synthesis of multifunctional patterned PS and PA polymer brushes via a combination of μCC and SI-NMP. (Reproduced from ref. 74 with permission from American Chemical Society, copyright [2014]). | ||
In many laboratories, ATRP could also be accessible for commercially available ready-to-use chemicals. Sometimes, it is difficult to polymerize monomers such as pyridine or acidic monomers because, due to the usage of a metal catalyst, monomers could react or form complex with metal catalysts. Nevertheless, Matyjaszewski and coworkers65 have described a direct polymerization of methacrylic acid using a mixture of electrochemically mediated ATRP, reducing agent ATRP, and supplemental activator, which gives high yields using nontoxic and inexpensive chemicals. Many other methods such as activator (re)generated by electron transfer and initiator for continuous activator regeneration have been used to decrease the concentration of the copper catalyst that is needed for polymerization.66
Other than ATRP, RAFT polymerization uses a chain transfer agent (RAFT agent), e.g., in the form of a thiocarbonylthio compound, in order to control the polymerization process. This approach is very simple and flexible. However, only a few types of chain transfer agents are available commercially and their synthesis contains multiple steps.67
Huang and coworkers40 prepared Y-shaped bifunctional surface-initiator containing a RAFT chain transfer agent (CTA) moiety and an ATRP initiator on one grafting site. The Y-shaped initiators were anchored on aminopropyl dimethylethoxysilane (APDES)-modified mesoporous silica nanoparticles (MSNs). Poly(N-(2-hydroxypropyl)methacrylamide) (PHPMA) was polymerized using SI-RAFT, followed by washing for the removal of physiosorbed polymers. Thereafter, the second polymer poly(2-diethylaminoethyl methacrylate) (PDEAEMA) was grafted by SI-ATRP. Here, both free RAFT (CTA) and sacrificial ATRP initiators were added to the reaction solution in order to get the estimated molar mass. NMP technique uses nitroxide radicals and works based on the reversible activation and deactivation of growing polymer chains. This process does not involve any catalyst, which makes the process of purification more simple and decreases the chance of impurity involvement.68 This technique is especially useful for some applications that are very sensitive to catalysts in RAFT and ATRP. The additional synthesis of surface-reactive NMP initiator is required due to unavailability in the commercial market. In this technique, polymerization occurs at a high temperature (at which thiol-gold bond breaks); therefore, it limits the range of surface monomers.69 SI-PIMP involves the use of iniferters, which perform the role of initiators, transfer agents, and terminators. PIMP is more useful due to its synthetic ease, requiring only a few reaction components and proceeding by simple UV irradiation. It also presents a flexible route to synthesize 2D and 3D micro-structured polymer brushes without any limitations.70 However, for photosensitive monomers and surfaces, SI-PIMP is difficult to apply.
It is worthwhile to keep in mind that although both SI-ATRP and SI-NMP can be successfully accomplished without the removal of ATRP chain ends, the extension of ATRP initiators during the SI-NMP step has been observed. Therefore, chain-end deactivation (via chain-end removal) is necessary for both sequential SI-RAFT/SI-ATRP and SI-ATRP/SI-NMP to prevent possible chain extension of the first polymer brush during the second polymerization.40,71 For instance, RAFT chain ends could be removed using radical reaction with ACVA, and ATRP chain ends can be de-halogenated using tri-n-butyltin hydride (n-Bu3SnH) or via light source as an external trigger.
3. Types of stimuli responsive polymer brushes
Stimuli responsive polymers have the capability to respond under some external stimulus, e.g., ionic strength, temperature, light, pH, electric charge, and salt from the surrounding environment, and undergo an observable change in the surface properties like color, wettability, and protein adsorption/desorption properties.96–98 Among the different triggers, pH, ionic strength, temperature, and solvent have attracted a great deal of attention in the last several years due to their interesting applications. While some triggers, like light and electricity responsive polymers, attract less attention they have already been shown to have a significant degree of potential. In this section, we will examine in detail the most commonly used triggers. Examples of responsive polymers based on the trigger used are shown in Table 2.| Polymers/compounds | Trigger type | Ref. |
|---|---|---|
| Poly(acrylic acid)s | pH | 15, 32, 102 and 128–131 |
| Poly(methacrylic acid)s | pH | 101 and 132 |
| Poly(4-Vinyl pyridine) | pH | 85, 107 and 133 |
| Poly(N-isopropylacrylamide)s | Temperature | 134–139 |
| Poly(4-vinyl pyridine) | Temperature | 140 |
| Poly(sulfobetaine acrylamide) | Salt | 120 and 123 |
| Poly(sulfobetaine methacrylate) | Salt | 141–143 |
| Poly(sulfobetaine methacrylamide) | Salt | 121 and 144 |
| Poly(methyl methacrylate)–polystyrene | Solvent | 71, 127 and 145 |
| Polystyrene–poly(2-vinylpyridine) | Solvent | 127 and 146 |
| Poly(ferrocenylsilane)–poly (N-isopropylacrylamide) | Voltage | 147–150 |
| Poly(2-(methacryloyloxy)ethyl ferrocene carboxylate) | Voltage | 151 |
| Ferrocene functional polymethacrylate | Voltage | 152 |
| 6-Nitroveratryloxycarbonyl | Light | 153 |
| Poly(4,5-dimethoxy-2-nitrobenzyl methacrylate) | Light | 154 |
| Azo polymers | Light | 155–157 |
3.1. pH responsive polymer brushes
Among the responsive polymers, weak polyelectrolyte polymers especially polyacrylic acid (PAA), poly(methyl methacrylic acid) (PMAA), and polyvinyl pyridine (P4VP) polymer brushes are highly investigated due to their rapid response against pH and ionic strength of the external environment99 (Fig. 4). Because charge density on these polymer brushes strongly depends upon the solution pH, therefore, the polymer brush's thickness rapidly increases up to 4-folds by changing the environment from low pH to high pH conditions. The swelling of the polymer brushes strongly depends upon the polymer density and brush architecture. At low pH, the PMAA polymers collapse and form a rigid film, which leads to a low microbalance resonator.100 Klok and coworkers101 showed that the pKa of the PMAA depends upon the density of the grafting by comparing a thin PMAA brush having a thickness of 15 nm with low and high density covered polymer surfaces. By increasing the grafting density, the dissociation degree of PMAA brushes is reduced, which results in changes in the pKa value.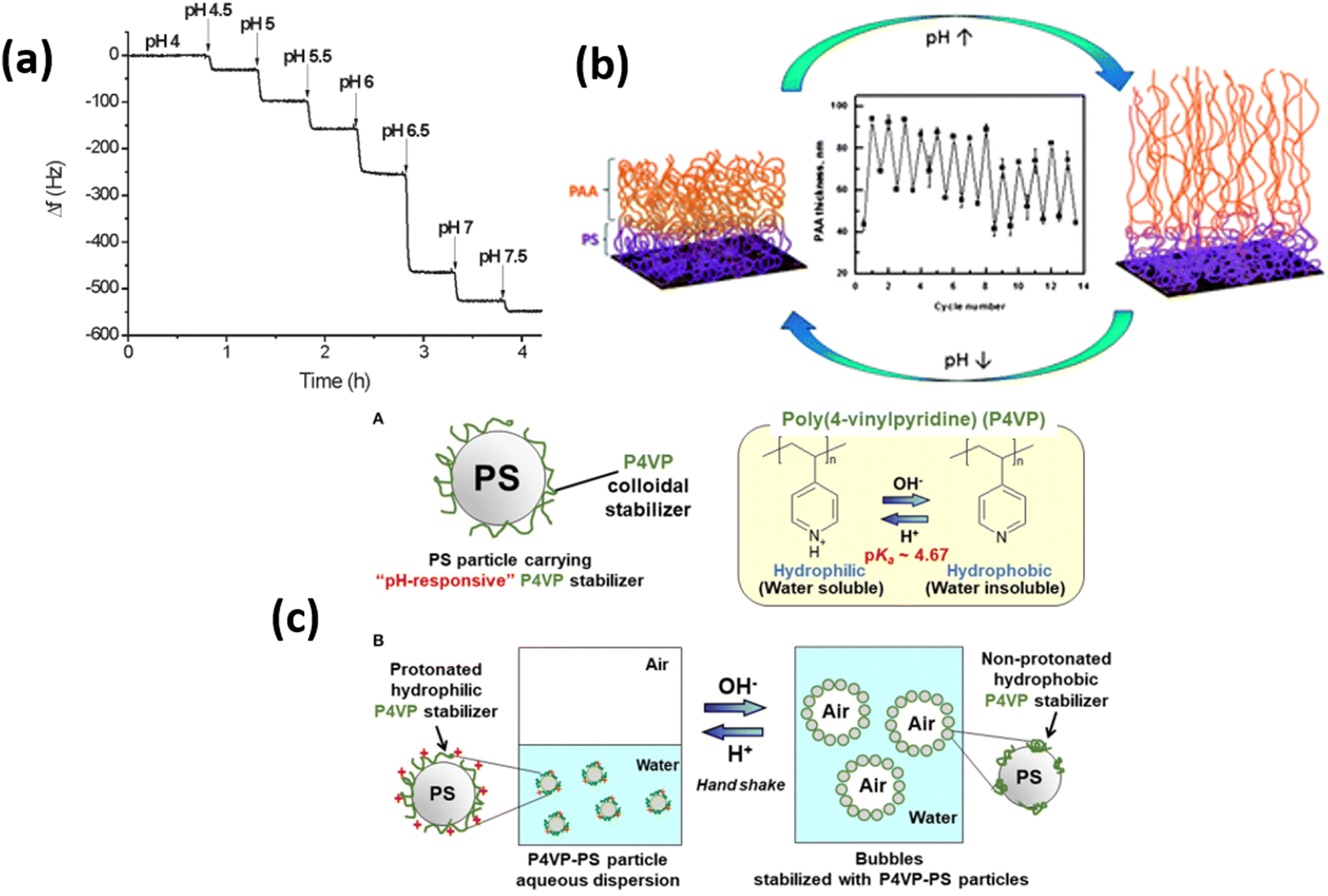 | ||
| Fig. 4 Typical response of QCM chips modified with 11 mm thick PMAA brush upon exposure to buffer solution with different pH values. (Reproduced from ref. 101 with permission from American Chemical Society, copyright [2011]). (b) Swelling and deswelling of PS-b-PAA polymer brush upon pH trigger. (Reproduced from ref. 106 with permission from Royal Society of Chemistry, copyright [2014]). (c) A-diagram of polystyrene particle carrying pH-responsive poly(4-vinylpyridine) colloidal stabilizer (P4VP-PS particle) and B-schematic illustration of pH-responsive bubbles stabilized with the P4VP-PS particles. (Reproduced from ref. 107 with permission from Frontiers Media S.A., copyright [2018]). | ||
Similarly, for PAA polymer brushes, in response to different pH conditions, the characteristics of PAA chains, e.g., thickness, hydrophilicity, and wettability, can be determined by knowing the ionization degree of carboxyl groups (–COOH). PAA chains start to shrink when pH values remain below the pKa of PAA, which is estimated to be about 5. On the other hand, by increasing the pH (above pKa), carboxyl groups dissociate, which induces strong electrostatic repulsions between charged repeating units that help to swell the PAA units.39,102 P4VP polymer, which is considered a weak polybase having pKa values between 4.5–4.7, also attains great attention. By changing the pH values, P4VP units show stimuli responsive characteristics due to the protonation of pyridine. By decreasing the pH values below pKa, the protonation of P4VP chains occur due to which chains tend to swell and ultimately become hydrophilic. When pH values are greater than pKa, P4VP chains becomes collapsed and illustrate hydrophobic behavior due to deprotonating effect.103–105
3.2. Temperature-responsive polymer brushes
In this category, attention is given to polymers where solvent quality depends strongly on surrounding environmental temperature. One of the best-studied temperature responsive polymers is poly-N-isopropyl acrylamide (PNIPAAM)108,109 (Fig. 5). It swells easily in an aqueous solution at 20 °C and undergoes a sharp transition above its lower critical solution temperature (LCST) at 32 °C. Grafted onto solid substrates, PNIPAAM caused temperature dependent changes in the physicochemical surface properties toward higher hydrophobicity above the LCST. At the LCST, a phase transition could be observed. The structure of the temperature responsive polymer depends upon the grafting density.110 For instance, at low molecular weight and low grafting densities, an octopus-like micelle formation of end-grafted PNIPAAM was seen through atomic force microscopy (AFM).111 In contrast, PNIPAAM chains collapse, which leads to the formation of continuous thin films resulting conformational changes of very thin (dry thickness of 3–24 nm) PNIPPAM brushes at low grafting densities and molecular weight.111,112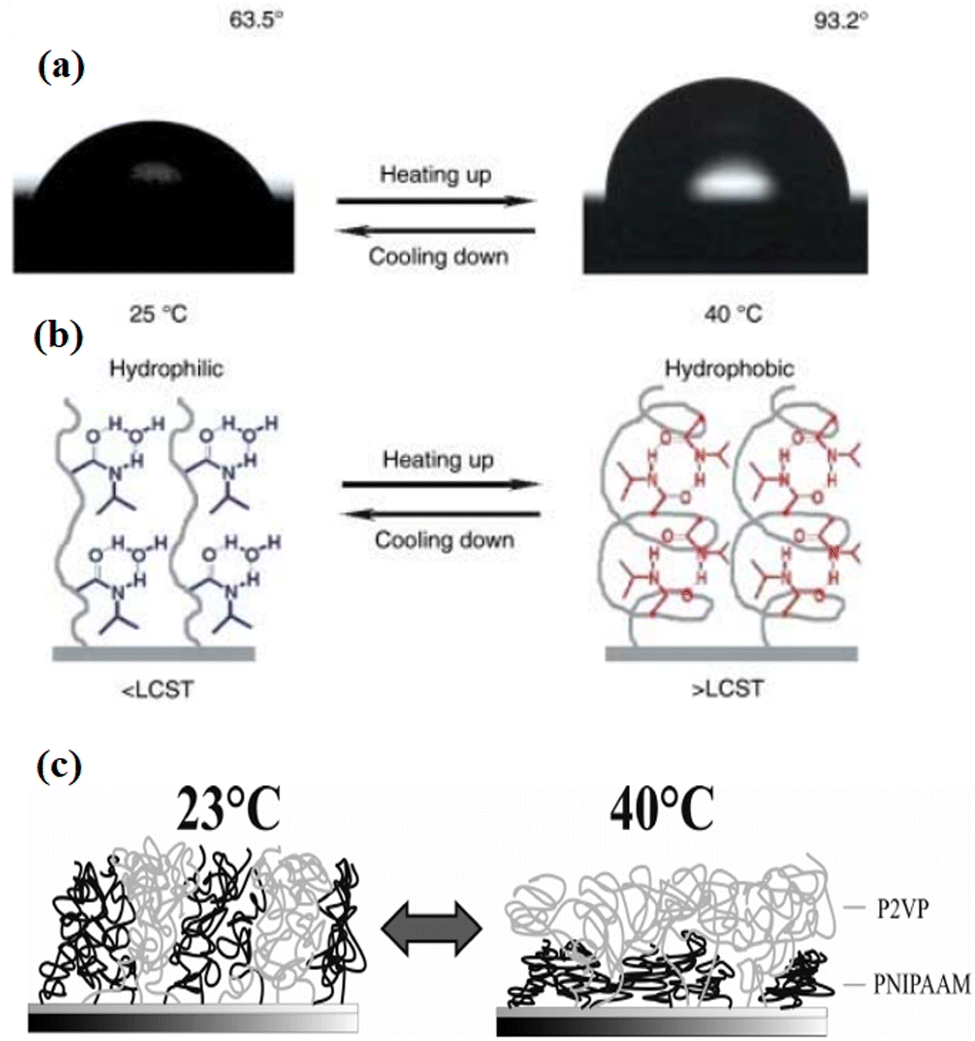 | ||
Fig. 5 (a) Change of water drop profile when the temperature was elevated from 25 °C (left) to 40 °C (right) with water CAs of 63.5° 93.2°, respectively. (b) Diagram of reversible formation of intermolecular hydrogen bonding between PNIPAAm chains and water molecules (left) and intramolecular hydrogen bonding between C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H groups in PNIPAAm chains (right) below and above the LCST, which is considered to be the molecular mechanism of the thermally responsive wettability of a PNIPAAm thin film.113 (c) Schematic swelling effects in a mixed brush consisting of P2VP and PNIPAAm. (Reproduced from ref. 114 with permission from Willey Online Library, copyright [2010]). O and N–H groups in PNIPAAm chains (right) below and above the LCST, which is considered to be the molecular mechanism of the thermally responsive wettability of a PNIPAAm thin film.113 (c) Schematic swelling effects in a mixed brush consisting of P2VP and PNIPAAm. (Reproduced from ref. 114 with permission from Willey Online Library, copyright [2010]). | ||
3.3. Salt-responsive polymer brushes
All polyelectrolyte polymers (both strong and weakly charged) show responsive behavior upon change in ionic strength.115,116 At the osmotic brush regime (low ionic strength), polyelectrolyte polymers swell to their maximum extend due to maximum repulsive force between the polymer chains (Fig. 6). Their degree of swelling depends upon the degree of dissociation of their functional group. However, in the salted regime (high ionic strength), ionic interactions are screened, and therefore polyelectrolyte polymers are de-swelled. Here, the degree of deswelling depends upon the grafting density rather than the dissociation degree of the functional group. It is also known that particle swelling depends on the nature of the anions present in the solution. Anions like Cl− cause particle deswelling because they strongly compete for the water molecules hydrating the polymer.117,118 Delcroix with coworkers119 explained the effect of ionic strength of PAA polymer brushes. At low ionic strength of 10−5 M, PAA chains swell to their maximum extend due to the dissociation of carboxylic group, while PAA chains de-swell by increasing the ionic strength to 10−1 M. There are many other examples of salt-responsive polymer brushes. The zwitterionic polymer brushes also show responsive nature upon ionic strength trigger.120,121 Zwitterionic polymer brushes contain both positive and negative moiety. These oppositely charged agents/moieties interact with each other at low salt contents and cause the polymer chains to collapse, while at high salt contents, these moieties interact with the more salted ions in the aqueous solution, leading to a higher degree of swelling of polymer chains. The swelling of the zwitterionic polymer at high salt contents and deswelling at low ionic strength is also due to the anti-polyelectrolyte effect.122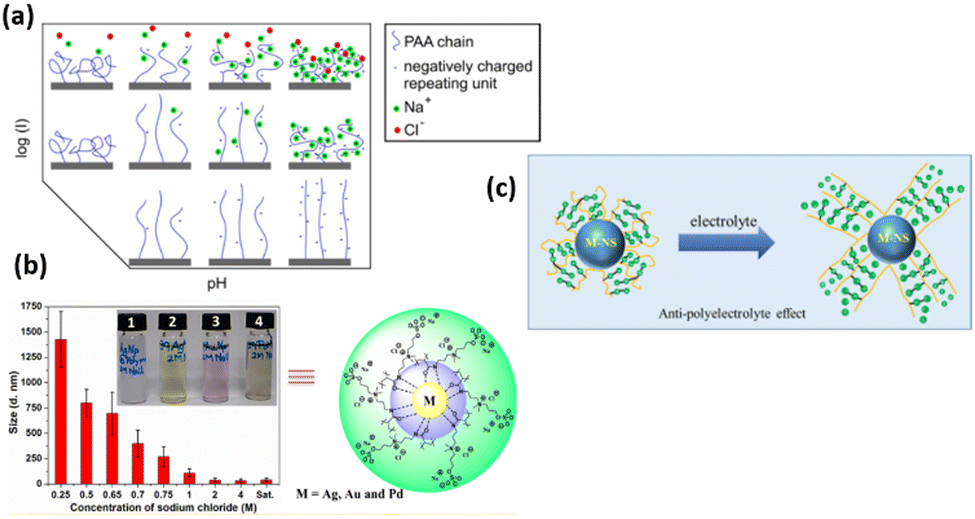 | ||
| Fig. 6 (a) Scheme of PAA conformation upon pH and ionic strength trigger. (Reproduced from ref. 119 with permission from American Chemical Society, copyright [2013]). (b) Size of AgNPs end-capped using polysulfabetaines methacrylamide (PSBMAm) in NaCl solutions of various concentrations. (Reproduced with the permission from ref. 123). (c) Schematic illustration of anti-polyelectrolyte effect. (Reproduced from ref. 124 with permission from American Chemical Society, copyright [2020]). | ||
3.4. Solvent-responsive polymer brushes
Some polymer brushes undergo responsive nature under the explosion of different solvents (Fig. 7). For example, Gupta and coworkers125 have designed an immobilization of gold nanoparticles on end-functionalized and solvent-responsive polystyrene brushes, grafted on an underlying substrate. The resulting polystyrene-Au nano assemblies underlying the mechanism of solvent-induced reversible swelling–deswelling of polystyrene chains. A dramatic blue shift of 32 nm in the surface resonance band was observed as the surrounding media of Au immobilized polystyrene brushes (Au-PS) was changed from air to the toluene. Similarly, P2VP polymer brushes also show swelling and deswelling behavior by changing the solvent from ethanol to toluene.126 Another interesting example of solvent-responsive polymer brush is the fabrication of a phase separation method to move the CdS nanoparticles reversibly in the perpendicular direction. The CdS nanoparticles were chemically bonded to polystyrene-b-(poly(methyl methacrylate)-co-poly(methacrylic acid) (CdS)) (PS-b-(PMMA-co-PMAA(CdS))) brushes and could be vertically lowered into the brush by exposure to toluene, while they were lifted out of the brush by exposure to ethanol (or acetone). Therefore, the authors illustrate the possible extension of movement by controlling the thickness of the two blocks of polymer brushes.127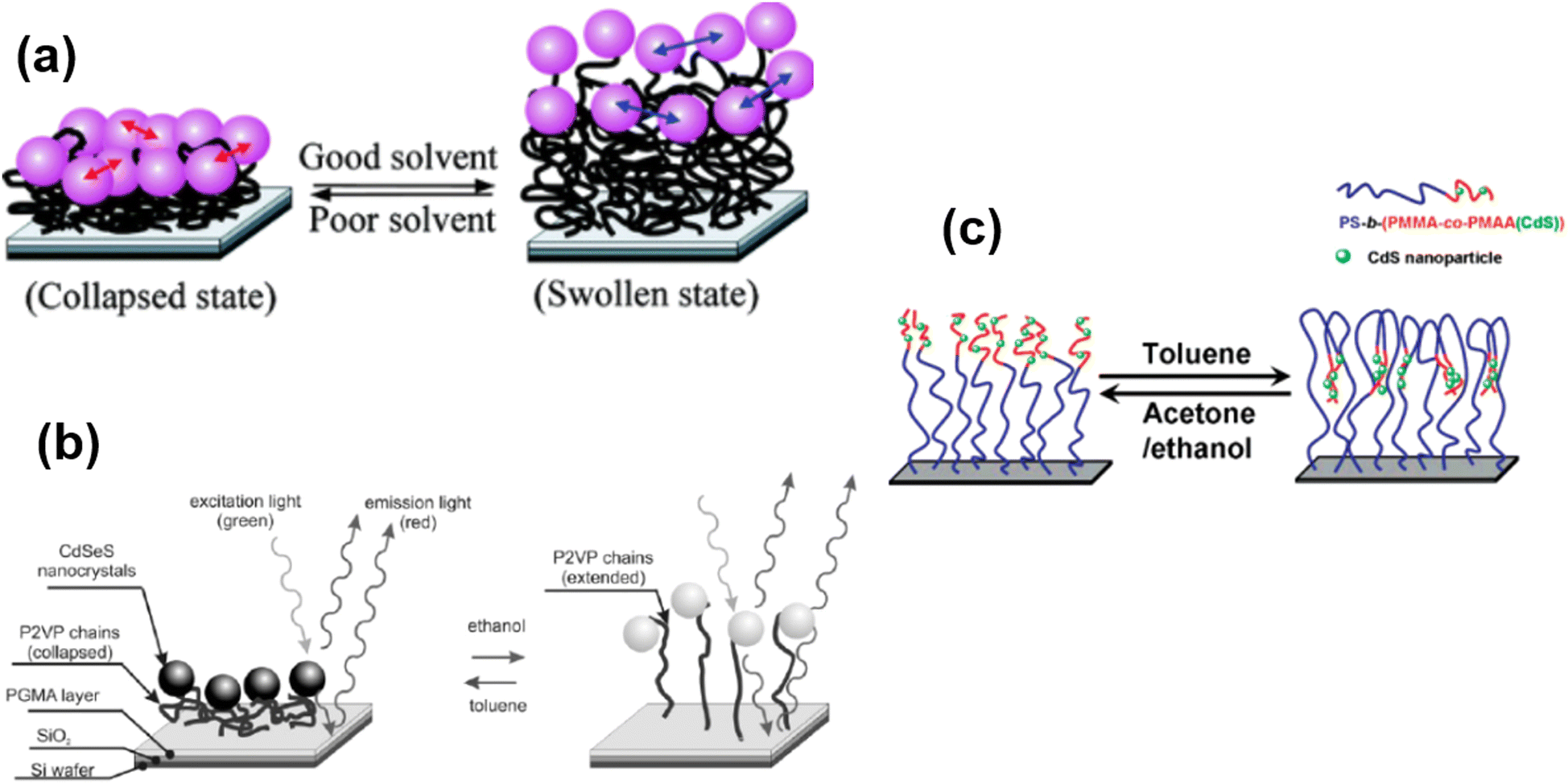 | ||
| Fig. 7 (a) Schematic representation of solvent-induced reversible swelling of Au-PS brushes. (Reproduced from ref. 125 with permission from American Chemical Society, copyright [2008]). (b) Schematic illustration of swelling and deswelling of P2VP chains upon solvent trigger. (Reproduced from ref. 126 with permission from Willey Online Library, copyright [2006]). (c) Proposed Mechanism of motion of integrated CdS nanoparticles by perpendicular phase Separation of polymer brushes. (Reproduced from ref. 127 with permission from American Chemical Society [2007]). | ||
4. Protein interaction
Polymer brushes have attained great attention of researchers for the manipulation and immobilization of protein due to the reason that they have enough available volume to bind the protein molecules on the surface. When a protein molecule comes in contact with the polymer brush interfaces, the protein molecule experiences adsorption/desorption due to various forces including electrostatic interaction (ion exchange), covalent binding, and binding to metal ion complexes.158–160In case of charged polymers like polyelectrolyte polymers, the major driving forces considered behind the adsorption of protein are the electrostatic interaction with protein molecules as well as change in entropy of the system. In order to control the adsorption and desorption of protein, many factors including isoelectric points (IEP) of equal and opposite charged protein molecules and grafted polymer chains, and applied environmental conditions could be adjusted precisely.31 A recent investigation on the adsorption of model protein molecules on the spherical polyelectrolyte polymer brushes shows that the amount of counter ions present inside the mono polymer brush has a tremendous influence on the entropic driving force behind the protein adsorption mechanism.161 Here, the counter ions consist of potassium or sodium ions in an aqueous solution, which is the main reason for local charge neutrality in the polyelectrolyte polymer brushes.
Non-charged polymers like poly(N-isopropyl acrylamide) (PNIPAAM) or poly(ethylene glycol) (PEG), which are soluble in water, are promising candidates, especially in brush confirmation because in these polymer brushes, the molecular weight and grafting density can be adjusted easily to stop the primary and secondary protein adsorption due to steric repulsion.162 Moreover, these polymer chains interact with water molecules, leading to the growth of stable water layers on the brush interfaces, which are also responsible for resistance against nonspecific protein adsorption.112 These polymer coating surfaces with strong resistance against protein adsorption are very important for application in the field of blood-containing devices, contact lenses, bioprocessing, etc.
4.1. Protein structure and its adsorption mechanism for polyelectrolyte polymers
Proteins are considered as complex biopolymers. Protein molecules consist of twenty natural amino acids, and even some extra chains like lipids, phosphates, and oligosaccharides are added to them after they undergo the translation process.163 This exceptional combination of chains, complex structure, and functional intricacy makes it difficult for researchers to define the adsorption mechanism of protein molecules.163 There are four main types of structure of protein molecules based on their structure, namely, primary, secondary, tertiary, and quaternary structure. The initial structure consists of a specific series of 20 L-amino acids that are encoded by DNA. Here, every unit of amino acids consists of a backbone structure, where R represents the specific side group structure that describes its certain functional properties. On this basis, the amino acid can be further divided into three classes including polar, nonpolar, and ± charged, in which each type has its own particular physicochemical properties.164Proteins can be regarded as polyampholytes because they contain positive and negative charges on their interfaces. Their behavior in the electrolyte solution as the function of pH was investigated quite early.165,166 Based on the pH of the solution, protein molecules may either have a negative or positive charge and are balanced at the isoelectric point. Due to this obvious fact, a protein molecule interacts very strongly with the linear polyelectrolyte polymer chains having opposite charges in the solution. The uptake and release of protein from spherical polyelectrolyte brush (SPB) containing chains of poly(acrylic acid) (PAA) interestingly adsorbs protein by maintaining low salt contents irrespective of the presence of net charge on the protein units.167 The amount of adsorbed protein units on polyelectrolyte brushes changes linearly with the concentration of protein molecules in the solution. Also, it was observed that by increasing the salt contents, the brush surface becomes hydrophilic and shows strong resistance against protein adsorption.161,167,168 In this system, there is steric repulsion between the brush layer of the SPB and dissolved proteins; therefore, only a small amount of protein adsorption would take place. Consequently, it is very easy to release the adsorbed protein just by increasing the ionic strength to 100 mM. However, when the protein adsorbs on the surface of polyelectrolyte brush surfaces by opposite charge, then there is strong Coulomb force behind the protein adsorption mechanism. It has been observed that when protein molecules and polyelectrolyte brushes have the same charges, then the protein could still adsorb on the brush interfaces, which is called as “wrong side adsorption” of the isoelectric point.169
Dubin and coworkers170 illustrated that soluble complexes are formed at the pH values above the isoelectric point and with the function of the concentration of salt contents, these pH values show a distinct maximum. Dubin and coworkers describe these unexpended results in the presence of positive charge patches on the interfaces of the protein molecules beyond the isoelectric point. When the linear polyelectrolyte polymer chains interact with the protein molecules having the same charge, then polyelectrolyte polymer chains are rationalized into the balance of attraction by the patches of positive charge and repulsion by the overall negative charge of the protein. Therefore, another mechanism was developed that is based mainly on the entropy gain in the release of counterions.171,172 The major requirement in this mechanism is the presence of positively charged patches on the surface of protein molecules.169 De Vries and coworkers173 explain another theoretical mechanism of the interaction of protein molecules with the polyelectrolyte brushes. They explained the mechanism of complex formation based on polyelectrolyte adsorption on randomly charged surfaces. By altering the pH and ionic strength of the surrounding environment, the strength of interaction changes linearly. Furthermore, soluble complexes are expected to form on the “wrong side” by maintaining low salt contents.
4.2. Influence of polymer surface engineering on protein adsorption
Interactions of protein and biomaterials, which potentially impact the cell response, are directly affected by the individual properties of biomaterial surface and protein.174,175 In this section, we will explain the influence of various physical and chemical properties of biomaterial surfaces on protein adsorption and, subsequently, the cell behaviors. Moreover, we will also describe some recent findings in this field.4.2.1.1. The influence of roughness and curvature of polymers on protein adsorption. Previous studies show that the topography of polymer surfaces might affect the behavior of cell reactions in two ways; either by a direct effect on the cytoskeleton or indirectly by protein orientation and unfolding.177,178 Investigations also show that the hydrophobicity of the polymer surfaces increases with increasing surface roughness.179,180 Moreover, it has been seen that when a smooth surface adsorbs protein, then its roughness values increase, even though the adsorption of protein on the rough surface blocks valleys between lumps and causes a decrease in its roughness. Moreover, studies show that for some specific types of cells like corneal cells, increment in the roughness values of the surfaces causes irregular spreading of the protein as well as increase in the migration of cells on the surface.181 Furthermore, Capsular contracture, the most common delay in a patient, has been observed to be more likely to form on soft surfaces.182 From previous observation, on smooth surfaces, collagen and fibroblasts (key players for capsular contracture formation) begin to build up around the surface. Nevertheless, raising the surface roughness values causes the cells to arrange around the surface indiscriminately, which leads to a decrease in the growth of fibroblasts and then the random formation of the collagen matrix.182 Studies give evidence that the amount of protein adsorption changes by changing the surface roughness values.183 Roach and coworkers184 have examined the effect of the degree of curvature and type of protein on the adsorption amount. The results illustrated that by increasing the curvature size, Fg starts the denaturation, while reducing the curvature size denatures bovine serum albumin (BSA). Recently, Akkas and coworkers185 have studied the influence of surface roughness, hydrophilicity, and swelling behavior of polyurethane (PU) films (or layers) on the adsorption mechanism of BSA. In this experiment, they used plasma treatment, and the surfaces were modified with PAA polymer and prepared coatings having the same hydrophilicity but different roughness values. The results illustrated that surface roughness, swelling degree, and hydrophilicity play a major role in the adsorption of proteins. Furthermore, Zhang and coworkers186 have examined the effect of roughness values of pyrrole-like (PPpy) film on the adsorption properties of BSA. They have characterized the surface roughness using atomic force microscopy (AFM), and they observed the behavior of the adsorption of BSA using electrochemical impedance spectroscopy (EIS) and surface plasmon resonance spectroscopy (SPR). The results illustrated that a greater amount of BSA adsorbed on the PPpy film surface due to their higher surface roughness and hydrophobic properties. Moreover, the adsorption amount of BSA decreases with increasing pH values of the solution. It is due to the fact that higher pH values impart negative charges on BSA molecules and also convert the nitrogen atom, present in the PPpy film, from protonated to deprotonated form, as shown in Fig. 8.
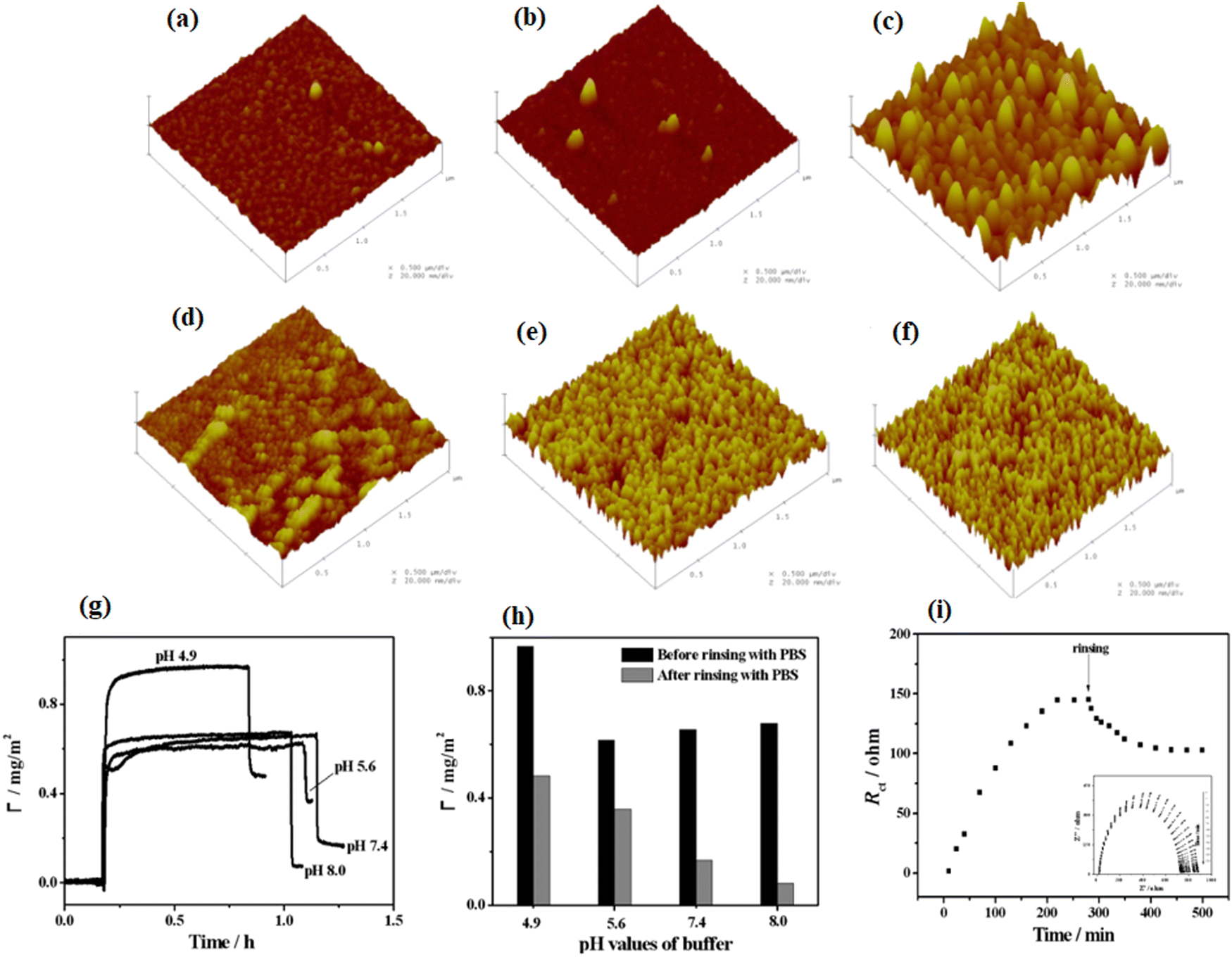 | ||
| Fig. 8 Tapping mode AFM 3-D photos of PPy substrates having thickness about 128 nm placed under (1 × 1 μM) (a) 5, (b) 50, and (c) 100 W with average roughness values of 0.57, 0.72, and 2.9 nm, respectively. Also placed under 100 W for (d) 5, (e) 15, and (f) 20 min with a normal roughness of 1.06, 1.98, and 2.13 nm, respectively. (g) SPR binding curvatures of 1% BSA adsorption on PPpy substrates set below 100 W and for 1 min in PBS solution having the pH value of 4.9, 5.6, 7.4, and 8.0. (h) Level of BSA adsorption before and after cleaning with PBS. (i) Nyquist plots of PPpy set under 100 W and for 1 min in 0.10 M PBS (pH = 7.4) enclosing 10 mM K3Fe(CN)6-10 mM K4Fe(CN)6 and 1%BSA for times ranging from 0 to 250 min. (Reproduced from ref. 186 with permission from De Gruyter, copyright [2012]). | ||
4.2.1.2. The influence of the size of polymers on the adsorption of protein. Even though there is no sufficient literature on the influence of the size of the polymers on protein adsorption, still, the polymer size could have a strong effect on the adsorption mechanism of protein by affecting the surface curvature of polymers. Few researchers have used nanoscale polymers having different values of hydrophobicity and observed the quality of the adsorption of protein on the surfaces.187 Studies have shown that by changing the size of the polymers, the adsorption of protein changes, and the maximum adsorption of protein takes place where the size of the polymers is larger. Recently, Satzer and coworkers188 have studied the influence of the size of the polymers on the adsorption of protein by determining the conformational changes of BSA and myoglobin throughout their adsorption process on silica-based polymer surfaces. The results illustrated that conformational changes in the protein depend upon the size of the silica-based polymers. They have prepared the size of silica-based polymers in the range of 30–1000 nm, upon which conformational changes in the protein only occur when the size of the polymer was greater than 200 nm. Wang and coworkers189 prepared patterned polymer brushes of poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) with the size of sub-100 nm structures across major parts. The prepared surface undergoes swollen state at pH 4 and neutralizes and collapses at pH 9. They have investigated the adsorption of BSA using AFM, QCM-D, and laser scanning confocal microscopy, and their results are shown in Fig. 9. The results depict that BSA could be adsorbed outside and inside the polymer brushes. Moreover, using the AFM technique, the protein adsorption mechanism in nanoscale could be observed on sub-100 nm topographies of the patterned polymer brushes of PDMAEMA. The suggested system might be used as a prototype system for the observation of protein adsorption and desorption mechanism at the nanoscale.
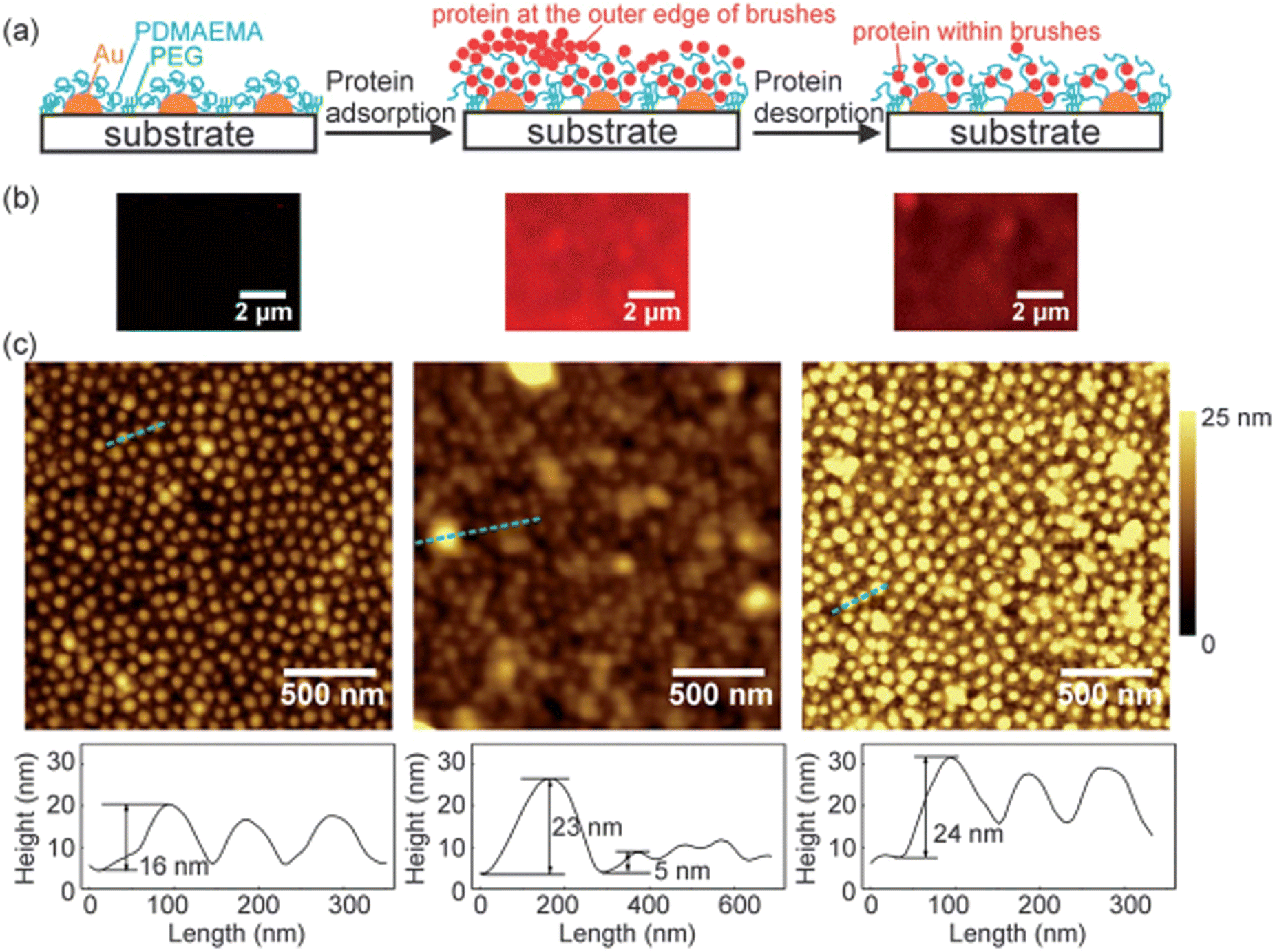 | ||
| Fig. 9 Schematic illustrations (a), laser scanning confocal microscopy samples (b), and AFM topography and corresponding cross-sectional profiles (c) of patterned PDMAEMA brush prior to protein adsorption (left), following protein adsorption (middle), and after protein desorption (right). In the protein adsorption process, the brushes were dipped in BSA solution (0.1 mg mL−1, pH 5.8), washed with NaCl solution (1 mM, pH 5.8), and dried under nitrogen. In the protein desorption process, the brushes were successively rinsed in NaCl solution (1 M, pH 4), NaCl solution (pH 9, 1 M), and Milli-Q Water, and dried under nitrogen. (Reproduced from ref. 189 with permission from Royal Society of Chemistry, copyright [2014]. | ||
4.2.1.3. The impact of density, charges, and wettability of polymers on protein adsorption. Polymer chains undergo molecular and cellular response and in response, electrostatic interactions have a major role, because all of the boundaries in the solution are charged, in addition to the cell membranes, which are negatively charged.179,190 Literature review shows that when a cell interacts with any surface, then the membrane of the cell interacts only with the positively charged surface. However, if the surface is negatively charged, then interaction takes place only in certain areas.191 Therefore, it is recommended that positively charged polymer chains interact with the cells more potentially. Thus, if any resorbable polymer has negative charge, then surface alteration of the polymer is required before cell seeding. Some studies have proposed that these described variations are due to electrostatic attraction and repulsion, which leads to a distinction in the arrangement of protein adsorption and desorption.192 Several studies have been undertaken to observe the adsorption of certain proteins having different chemical structures on the interfaces of positive and negatively charged polymers, and various results have been obtained on protein adsorption based on the structure of the protein. For example, it has been examined that FN could be adsorbed only on a positively charged surface, while an unidentified 30-kDa protein was adhered on the surface containing high negative charge. The FN retains its functionality on the hydrophilic interfaces and it is continuously evaluated, which represents a great advantage in enhancing the cellular reactions. On the contrary, vitronectin protein could be adsorbed on the polymers having variably charged interfaces, and its functional properties do not change with the wettability of the surface. Other studies also show that other than the charge present on the surface, hydrophobicity could also play a major role in the adsorption of protein. Some researchers have examined the adsorption of fibronectin (FN) on the surface of tissue culture PS and Primaria tissue culture. The results illustrated that polarized and positive charged Primaria surface could adsorb more FN and subsequently show the adhesion of monocyte, as compared to the tissue culture PS surface.193 However, researchers194 have revealed the adsorption of FN on a negatively charged surface. The results show that on a neutral polar surface, only a few molecules of FN could be adsorbed, while no adsorption was detected on a hydrophobic nonpolar surface. Moreover, the adsorption of fibrinogen (Fg) decreases while that of albumin increases by increasing the hydrophilicity of the polymer. Many studies also examined that hydrophilic surfaces could be used to stop the adhesion of leukocyte and macrophage fusion, and therefore the secretion of inflammatory factors.195 Recently, Recek and coworkers196 analyzed the rate of adsorption of protein on plasma-treated PET surfaces through QCM-D technique. The PET surface was prepared under hydrophobic and also hydrophilic environments. The results indicated that when the incubation time is low, then, as compared to the untreated surface, almost same amount of protein adsorption took place on plasma-treated hydrophobic and hydrophilic surfaces. This variation of protein adsorption on treated and untreated surfaces decreases by increasing the incubation time. Moreover, the adsorption of protein on hydrophilic interfaces gives the maximum difference in both dissipation and occurrence, which is a green signal for the maximum adsorption of protein. Although, the amount of adsorption of protein was very low for untreated surfaces, it is somewhat in between for the hydrophobically-treated surfaces. Additionally, it was investigated that the presence of various functional groups on the surface of polymers creates interesting cellular responses.
In this section, by reviewing the current research in the last few years, we will see the potential role of various chemical properties of polymers on the protein adsorption mechanism.
4.2.2.1. Effect of functional groups and the chemical structure of polymers on protein adsorption. Many studies have revealed the influence of chemical functionalities of surfaces on the adsorption of protein and cellular responses to the polymer interfaces.197,198 Many researchers chemically modified the surfaces to study and manipulate the cellular response to polymers. In fact, polymer surfaces having different charges and carrying functional groups with variant hydrophobicity have a great influence on protein adsorption behavior and cell responses. Some studies suggested that a functional group having protein resistance properties could help polymers regulate protein adsorption at their interfaces.199 Recent studies have also shown that other than hydrophilicity and hydrophobicity, the surface functional groups could also play an important role in protein adsorption and cell responses. Commonly examined functional groups that are related to the interaction of polymers with the protein molecules are hydroxyl (–OH), carboxyl (–COOH), amino (–NH2), and methyl (–CH3) groups.200 Other studies have examined the effect of these functional groups on the adsorptive properties of protein and cell response. It was illustrated that on a surface containing hydroxyl group, water molecules resist the adsorption of Fg, conforming to the strong interaction of Fg with the –OH group.201 More studies have demonstrated that using an ideal amount of –OH functional group, the affinity of albumin binding over Fg could be improved. Moreover, it has been examined that Fg might make a hydrophobic link with the surfaces containing (–CH3) functional group. In addition, Fg could also have a strong affinity with the amine group. However, Fg has less interaction with the negatively charged carboxyl group. Therefore, it is suggested that surfaces containing both amine and hydroxyl groups lead to an increase in hydrophilicity. Besides, the functional groups can be covalently bonded with complementary proteins to protect them against directing proteins.202 It has been proved that the surfaces containing (–OH) group could adsorb IgG from serum selectively, which results in the deposition of C3 on biomaterials interfaces. On the other hand, surfaces containing carboxyl or amine group had less C3 activation. Roach and coworkers203 have investigated the effect of hydrophilic (OH) and hydrophobic (CH3) groups on the adsorptive behavior of BSA and Fg with the help of QCM and grazing angle infrared spectroscopy. The results depicted that BSA could be adsorbed easily through a single step, while the adsorption of Fg is a very complicated process. Moreover, BSA has more affinity toward CH3 as compared to OH-terminated surfaces. Results also show that Fg adsorbed more rapidly on both surfaces with slightly more affinity toward methyl0terminated surfaces. After the incubation time of one hour, few time-dependent alterations were observed. Both Fg and BSA showed a less ordered secondary structure when adsorbed on a hydrophobic surface compared to a hydrophilic surface, especially for BSA.203
4.2.2.2. Influence of conformational flexibility of biomaterials on protein adsorption. A conformational change in the field of biochemistry can be defined as a change in the formation of macromolecules, which is normally promoted by the surrounding environmental elements. The main conformational properties of polymers are rigidity and flexibility, and they have much influence on the protein adsorption process. For example, Jin and coworkers204 have recently determined the adsorption behavior of lysozyme (LYZ) and BSA on the PEG layer using PEG 5000 and PEG 2000, which show flexible and rigid mushroom conformation, respectively. The results demonstrated that an insignificant quantity of LYZ could form hydrogen bond with the interfacial water near the rigid mushroom PEG2000, resulting in the adsorption of protein. However, PEG 5000 mushroom having more flexibility shows resistance against BSA and LYZ due to its greater elastic repulsion energy. Moreover, when the grafting density of PEG is higher than the critical value (brush conformation), the interaction between the PEG chains and LYZ is a key factor. However, if the grafting density of PEG is below the critical value (mushroom conformation), an elastic repulsion was observed between protein and PEG via the conformation entropy of PEG. Therefore, due to high elastic repulsion energy, BSA could not be adsorbed on the surface, while the adsorption of LYZ could be well adjusted due to the interaction of entropy elasticity repulsion and hydrogen bonding.204
4.3. Controlled adsorption/desorption of protein
In the development of advanced biomaterials, an important direction is to create bioresponsive surfaces that show responsiveness upon signaling molecules or the external environment. The first step in this advancement is the fabrication of surfaces that undergo protein adsorption and desorption and tune their properties between inertness and active in a controlled manner. Designing and understanding the behavior of mixed polymer brushes is more complex, but they offer promising tunable properties due to the combination of different stimuli-responsive functionalities. Brushes containing carboxylic acid and epoxide groups are particularly common because they can be readily derivatized.205,206 Poly(acrylic acid) (PAA) brushes are especially attractive for protein immobilization because in an aqueous solution, these films swell up to four times their initial thickness to facilitate the binding of large biomolecules.206Delcroix and coworkers39 designed mixed brushes of poly(acrylic acid) (PAA) and poly(ethylene oxide) (PEO) via the “grafting-to” approach for tuning protein adsorption onto smart materials. Proteins were shown to adsorb effectively into pure PAA brushes but only partially desorbed from the brushes by changes in the pH and ionic strength. The properties of the mixed polymer brushes were, however, adjusted by different ratios of PEO and PAA, allowing them to achieve a highly switchable behavior toward protein adsorption. High amounts of albumin were indeed adsorbed on PEO/PAA brushes, and 86% of these could then be desorbed upon pH and ionic strength change. Further cycles of adsorption/desorption could also be achieved.
In another research, Delcroix and coworkers119 studied the influence of conformations of polymer brushes toward the adsorption of HSA protein. For this purpose, homo-layers of PEO or PAA were subjected in the aqueous solutions having pH values ranging from 3 to 9 and ionic strength varying from 10−5 M to 10−1 M. The conformation of polymer units and protein adsorption behavior was then investigated using QCM-D. Results show that at low ionic strength values, swollen PAA chains allow HSA protein to enter into the mixed polymer brush with a maximum adsorption at pH 5 and ionic strength = 10−5 M. However, with increasing pH and ionic strength, i.e., pH 9 and 10−1 M, PAA chains tend to collapse, which decreases the adsorption of HSA, and PEO/PAA mixed brushes also expose more PEO chains; thus, the HSA was released from the mixed PEO/PAA brushes at high pH and high ionic strength conditions.
Later on, the author extended their research207 to observe the tunable behavior of mixed polymer brushes of PEO/PAA against lysozyme (Lyso), HSA, collagen (Col), and immunoglobulin G (lgG) adsorption/desorption reversibly and repeatedly. They observed that by choosing the appropriate condition of pH and ionic strength, the mixed polymer brushes provide a good combination of the characteristics of PAA and PEO. For homopolymer brushes, PEO brush exhibited negligible and irreversible adsorption of proteins, whereas PAA brush presented more adsorption of protein as compared to PEO. The PEO/PAA mixed polymer brush also exhibited substantial adsorption and desorption for all types of proteins for 3 to 4 continuous successive cycles. They observed that Lyso adsorption occurred because of swollen PAA chains and desorption was due to the collapse state of chains of PAA, which reduces the electrostatic interactions between the PAA chains and Lyso. When PAA chains collapse, PEO chains are dominant over the surface, which also causes the desorption of Lyso and HSA. In addition to this, electrostatic repulsive interactions between PAA and HSA also cause the desorption of HSA protein at high pH and high ionic strength value. The protein desorption behavior of collagen in the mixed polymer brush was due to the protein-repellent characteristics of PEO chains. Finally, IgG adsorption and desorption were switched by changing the ionic strength value, triggering PAA chains to swell or shrink (Table 1 (a, b)).207
Moreover, Belegriou and coworkers208 designed mixed polymer brushes of PEG and PAA and observed the tunable adsorption and desorption of bovine serum albumin (BSA) protein at different pH values. They concluded that the maximum adsorption of the protein took place in the range of 4.8–5.5, which is close to the isoelectric point of BSA (4.8). They also observed that hydrophobic interaction took place due to a reduction in electrostatic interaction between BSA and PAA at the lower pH range of 4–7. On the other hand, at pH values 8.5 (above pI), strong electrostatic repulsion took place due to negatively charged BSA and PAA, which cause hindrance in the adsorption of protein. However, some attractive attractions, e.g., hydrophobic, still allowing protein adsorption at a higher pH.
Hoy and coworkers34 have determined the protein adsorption and desorption at pH 7.4 on the homopolymer brush of poly(acrylic acid)-block-polystyrene (PAA-b-PS) and mixed polymer brushes of PEG and PAA-b-PS in the presence of CaCl2. They concluded that in the presence of CaCl2, the homopolymer brush does not show any significant tunability of protein adsorption and desorption. On the other hand, mixed polymer brushes of PEG and PAA-b-PS undergo very fine tuning of protein adsorption and desorption having adsorption thickness between 0.3 nm and 4 nm by changing the calcium ion contents.
Psarra and coworkers209 have fabricated binary mixed polymer brushes based on poly(N-isopropylacrylamide) (PNIPAAm) with thermal reactivity and poly(acrylic acid) (PAA) with pH responsive capability. It was examined that at pH 7.4, the ratio of the components forming the brushes is significant for protein adsorption on those brushes: when PAA contents are higher than that of PNIPAAm, protein adsorption decreases while the temperature is greater than the low critical solution temperature (LCST) of PNIPAAm. On the other hand, when PNIPAAm contents are higher than PAA, the sensitive behavior of PNIPAAm causes to increase in the protein adsorption by keeping the temperature higher than LCST. Likewise, Burket and coworkers210 also determined the protein adsorption behavior of mixed polymer brushes of PNIPAAm and poly (2-vinylpyridine) (P2VP) (weight ratio of PNIPAAm and P2VP is 50/50). It was seen that PNIPAAm chains have greater impact on the protein adsorption and desorption properties. PNIPAAm chains swell and show good resistance against human serum albumin (HSA) adsorption when the temperature is below the LCST of PNIPAAm. On the other hand, by increasing the temperature above the LCST, PNIPAAm chains collapse and P2VP chains become more prominent but still HSA doesn’t show any considerable amount of adsorption, because P2VP chains could not completely cover the collapsed chains of PNIPAAm.
Wang and coworkers15,32,33,55 fabricated mixed binary brushes of PMOXA/PAA using spin coating the mixture of poly[(2-methyl-2-oxazoline)-random-glycidyl methacrylate] (PMOXA-r-GMA) comb copolymer and poly(acrylic acid)-block-poly(glycidyl methacrylate) (PAA-b-PGMA) block copolymer solutions onto the substrates materials of glass/silicon and examined the protein adsorption and desorption behavior. The results illustrated that at low pH (pH 5, I = 10−5 M), PAA chains swell and cover the interfaces and adsorb BSA and lysozyme protein. By increasing the pH (pH 9, I = 10−1 M), PAA chains collapse and go to the bottom leaving the hydrophilic PMOXA chains on the surface. Therefore >90% of the adsorption amount of protein could be desorbed (Fig. 10 c). The results also verified PMOXA/PAA based coatings represent outstanding repeatability of adsorption and desorption of protein in multiple cycles.
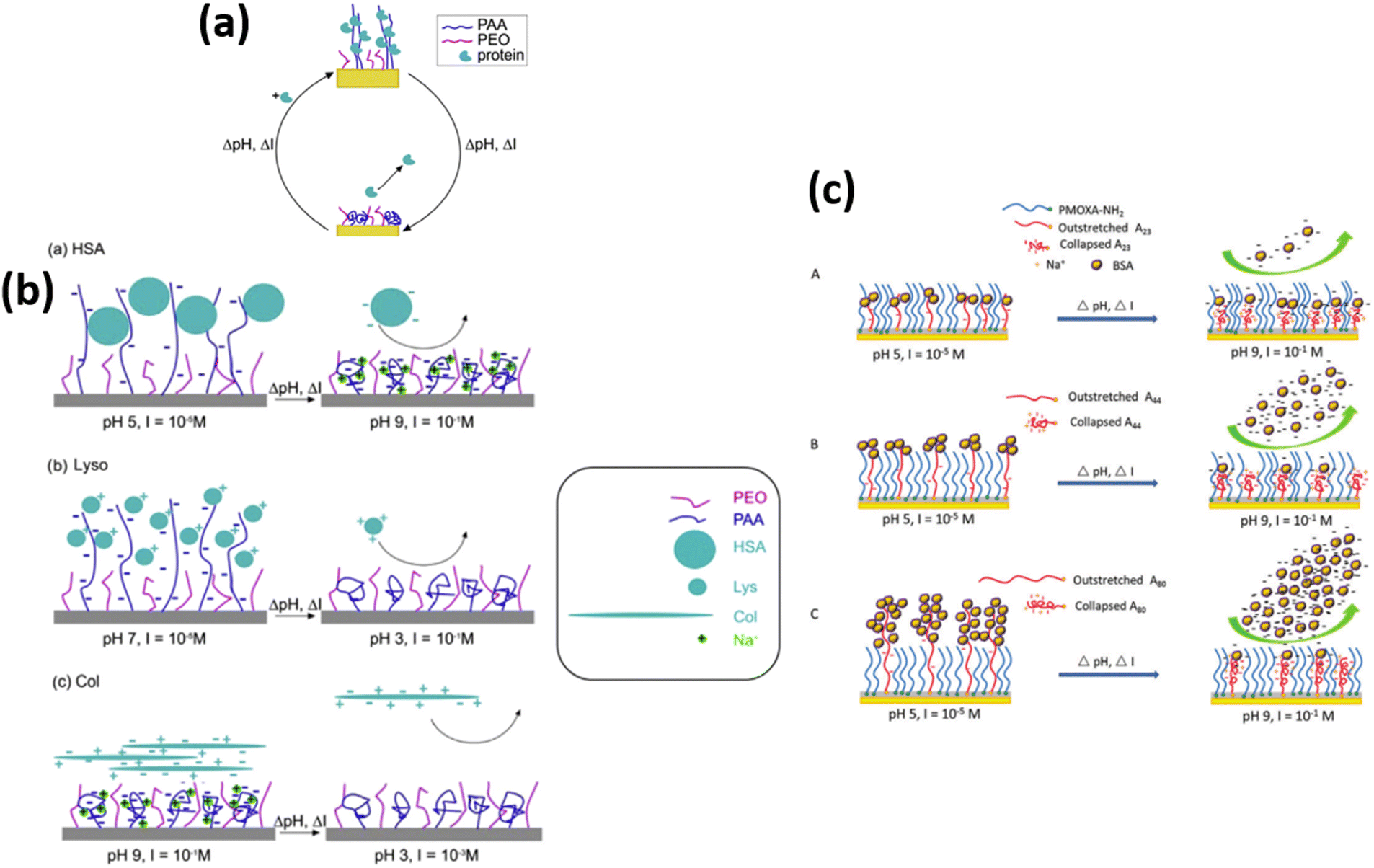 | ||
| Fig. 10 (a) Representation of the mixed PEO/PAA polymer brushes that can reversibly adsorb large amounts of proteins. Adsorption and desorption are triggered by changing the salt or acid contents of the medium. (Reproduced from permission from the ref. 207). (b) Representation of HSA (a), Lyso (b), and Col (c) adsorption and desorption on mixed PEO/PAA brushes. The conditions used for adsorption and desorption are indicated (pH, I). Note that the scale used to represent polymer chains and proteins is not realistic, for the sake of clarity (c) Schematic representation of the process of BSA adsorption and desorption on mixed brushes of (A) PDA/M9h/A2324h, (B) PDA/M9h/A4424h, and (C) PDA/M9h/A8024h. (Reproduced from ref. 15 with permission from Royal Society of Chemistry, copyright [2018]. | ||
Uhlmann and coworkers31 designed the binary mixed polymer brushes of the combination of two oppositely charged polyelectrolyte polymers of PAA and P2VP and observed the influence of pH, buffer salt concentration, and isoelectric point of the bush and proteins on the protein adsorption amount. They demonstrated that at higher ionic strength, behavior of protein adsorption is almost similar to that at hydrophobic surfaces. In the osmotic system, however, the balance between electrostatic repulsion and a strong entropic driving force, the “counterion release”, proved to be the main influence on protein adsorption. The adsorption amount of chymotrypsin increases significantly by increasing the pH values from 4 to 9 keeping the low ionic strength. While at high ionic strength, increment of adsorption of chymotrypsin with the rising of pH values is almost negligible. Whereas almost reverse behavior of adsorption of lactalbumin was observed as compared to chymotrypsin and its adsorption amount decreases by increasing the pH values from 4 to 9.
Lei and coworkers211 fabricate the poly(2-(dimethylamino ethyl) methacrylate)-block-poly(methacrylic acid) (PDMAEMA-b-PMAA) di-block copolymer brushes and observed good control of lysozyme adsorption by pH in the range of 4–10. They showed that the thickness of the outer PMAA block (lPMAA) is crucial for adsorption. When lPMAA was less than 10 nm, adsorption increased with increasing pH, and the difference in adsorption between high and low pH increased with lPMAA. The adsorption ratio at pH 10 and pH 4 reached values up to 16.4. When lPMAA was more than 10 nm, the adsorption tendency on the PDMAEMA-b-PMAA di-block copolymer brushes was similar to that on PMAA homopolymer brushes (Fig. 11). These results indicate that the combination of PDMAEMA and PMAA gives adsorption behavior reflecting the properties of both polymers. However, if the outer PMAA block is thicker than a critical value, then the protein-resistant effect of the inner PDMAEMA block is screened.
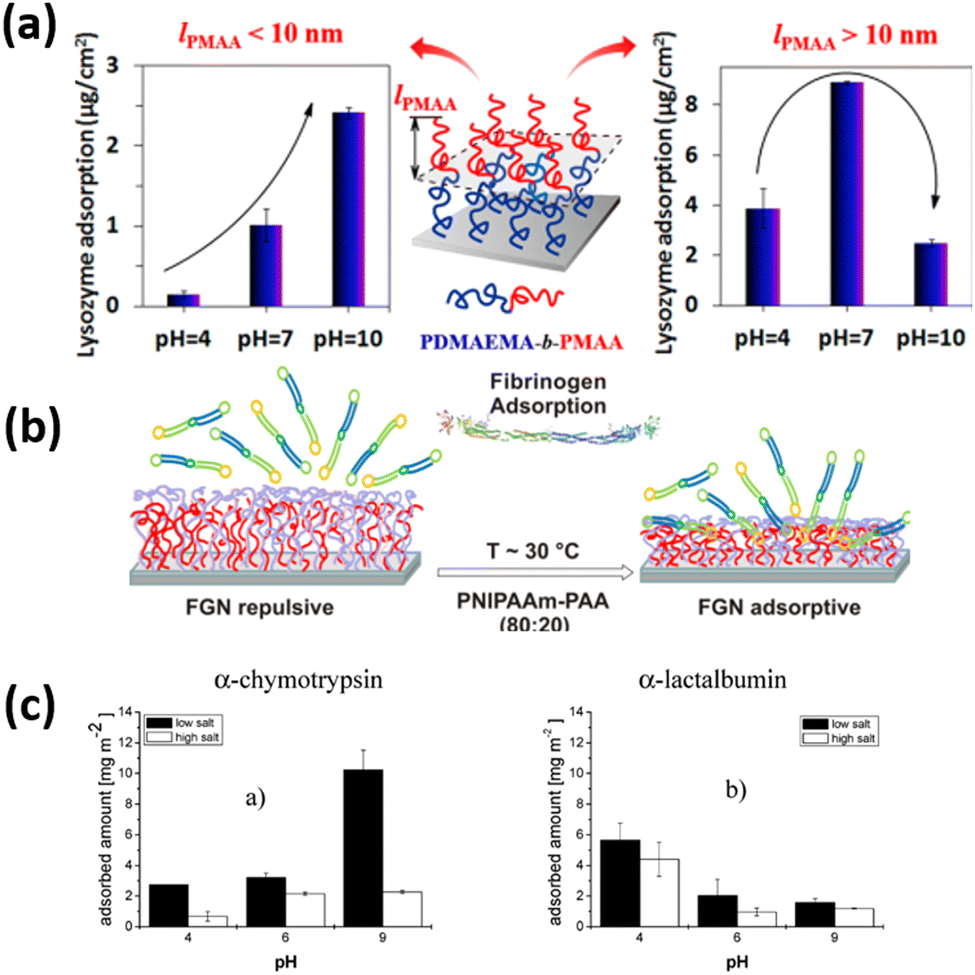 | ||
| Fig. 11 (a) Schematic representation of the adsorption/desorption of Lysozyme on PDMAEMA-b-PMAA polymer brush. (Reproduced from ref. 211 with permission from American Chemical Society, copyright [2014]) (b) Schematic figure of the adsorption/desorption of fibrinogen on PNIPAAm- PAA coatings. (Reproduced from ref. 209 with permission from American Chemical Society, copyright [2015]) (c) quantities of a-chymotrypsin and a-lactalbumin (plateau values) adsorbed from different buffer solutions on P2VP-PAA polymer brushes. (Reproduced from ref. 31 with permission from American Chemical Society, copyright [2007]). | ||
Conclusions
This paper is a comprehensive review of mixed polymer brushes (MPBs) for controlled protein adsorption. It provides a detailed analysis of the synthetic techniques used to create MPBs, including the different types of polymers used and the methods for tethering them to an interface. The review also covers the interaction of polymer brushes with protein molecules, including the factors that influence protein adsorption and desorption, and how MPBs could be used to precisely control these processes. Furthermore, the review discussed the potential applications of MPBs in biomedical and biotechnological fields. It highlights the advantages of using MPBs over traditional biomaterials, such as their ability to provide a high degree of control over protein adsorption and desorption, and their potential for use in drug delivery and tissue engineering. Overall, the review aims to provide insights into the state of the art and future prospects of MPBs. It concludes that MPBs are a promising platform for developing advanced biomaterials, and that further research is needed to fully understand their potential and limitations.Future prospective
Future research should be focused on:(1) The viable protein adsorption over the well-defined surfaces should be investigated systematically. Such a study must include the time factor to gain the much-needed quantitative results of the long-term protein resistance of surfaces in the complex biological environment.
(2) During protein adsorption, the conformational changes as well as the influence of these changes over biocompatibility should be examined systematically.
(3) Based on the developing theoretical understanding, new bio-inert coatings should be developed that effectively prevent the adsorption of proteins from highly complex protein mixtures (e.g. blood plasma).
(4) Since most of the coatings over implanted devices are poorly anchored or enzymatically degraded, therefore, the cells may integrate with those implanted surfaces and replace the coatings with the extracellular matrix (ECM) components. Vivo viability for the long term has not yet been fabricated by any protein-resistant coating. This should be examined on this subject.
(5) Although many biomedical applications have already investigated but still the combination of bio-specific surfaces and bioinert remains a major challenge for future studies.
Author contributions
Muhammad Atif: conceptualization, methodology, data curation, investigation, validation, formal analysis, writing, and review & editing. Ali Balasini: review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Muhammad Atif greatly acknowledges the financial support from the European Union's Horizon 2020 research and innovation programme under the Marie SKŁODOWSKA-CURIE grant agreement No 955664. The authors are also thankful to Prof. Dr Ulrich Jonas at Macromolecular Chemistry, Department of Chemistry and Biology, University of Siegen, Germany, for consistently providing invaluable guidance and introducing the scientific writing tools that significantly contributed to the development of this review paper.References
- T. J. Webster, L. S. Schadler, R. W. Siegel and R. Bizios, Tissue Eng., 2001, 7, 291–301 CrossRef CAS PubMed
.
- D. S. Wilson and S. Nock, Angew. Chem., Int. Ed., 2003, 42, 494–500 CrossRef CAS PubMed
- P. Zhang, J. Yang, W. Li, W. Wang, C. Liu, M. Griffith and W. Liu, J. Mater. Chem., 2011, 21, 7755–7764 RSC
- I. Brigger, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2012, 64, 24–36 CrossRef
- X. Zhang, P. Yang, Y. Dai, P. A. Ma, X. Li, Z. Cheng, Z. Hou, X. Kang, C. Li and J. Lin, Adv. Funct. Mater., 2013, 23, 4067–4078 CrossRef CAS
- J.-M. Nam, C. S. Thaxton and C. A. Mirkin, Science, 2003, 301, 1884–1886 CrossRef CAS PubMed
- A. Brzeska, C. Dupont and S. Demoustier-Champagne.
- P. Zhuang, A. Dirani, K. Glinel and A. M. Jonas, Langmuir, 2016, 32, 3433–3444 CrossRef CAS PubMed
- M. Badoux, M. Billing and H.-A. Klok, Polym. Chem., 2019, 10, 2925–2951 RSC
- H. Zhang, H. Xu, M. Wu, Y. Zhong, D. Wang and Z. Jiao, J. Mater. Chem. B, 2015, 3, 6480–6489 RSC
- L. Ionov, J. Mater. Chem., 2010, 20, 3382–3390 RSC
- Z. Wang, K. Chen, C. Hua and X. Guo, Polymers, 2020, 12, 566 CrossRef CAS PubMed
- Y. Xu, Y. Engel, Y. Yan, K. Chen, D. F. Moyano, P. L. Dubin and V. M. Rotello, J. Mater. Chem. B, 2013, 1, 5230–5234 RSC
-
H. F. Mark, Encyclopedia of polymer science and technology, 15 volume set, Wiley, New York, NY, USA, 2014 Search PubMed
- C. Pan, X. Liu, K. Gong, F. Mumtaz and Y. Wang, J. Mater. Chem. B, 2018, 6, 556–567 RSC
- H.-X. Wu, X.-H. Zhang, L. Huang, L.-F. Ma and C.-J. Liu, Langmuir, 2018, 34, 11101–11109 CrossRef CAS PubMed
- R. Lupitskyy, M. Motornov and S. Minko, Langmuir, 2008, 24, 8976–8980 CrossRef CAS PubMed
- J. Xie, K. Nakai, S. Ohno, H.-J. Butt, K. Koynov and S.-I. Yusa, Macromolecules, 2015, 48, 7237–7244 CrossRef CAS
- T. Gillich, C. Acikgöz, L. Isa, A. D. Schlüter, N. D. Spencer and M. Textor, ACS Nano, 2013, 7, 316–329 CrossRef CAS PubMed
- Y. Gotoh, H. Suzuki, N. Kumano, T. Seki, K. Katagiri and Y. Takeoka, New J. Chem., 2012, 36, 2171–2175 RSC
-
V. Parmar, G. Patel and N. Y. Abu-Thabit, Stimuli Responsive Polymeric Nanocarriers for Drug Delivery Applications, Elsevier, 2018, vol. 1, pp. 555–580 Search PubMed
- P. Du, T. Wang and P. Liu, Colloids Surf., B, 2013, 102, 1–8 CrossRef CAS PubMed
- N. Su, H. Li, Y. Huang and X. Zhang, J. Nanomater., 2015, 956819 Search PubMed
- Y. Shi, V. Selin, Y. Wang and S. A. Sukhishvili, Part. Part. Syst. Charact., 2013, 30, 950–957 CrossRef CAS
- A. Hasan, S. K. Pattanayek and L. M. Pandey, ACS Biomater. Sci. Eng., 2018, 4, 3224–3233 CrossRef CAS PubMed
- A. Hasan, G. Waibhaw and L. M. Pandey, Langmuir, 2018, 34, 8178–8194 CrossRef CAS PubMed
- Y. Arima and H. Iwata, J. Mater. Chem., 2007, 17, 4079–4087 RSC
- W. Wang, L. Li, K. Henzler, Y. Lu, J. Wang, H. Han, Y. Tian, Y. Wang, Z. Zhou and G. Lotze, Biomacromolecules, 2017, 18, 1574–1581 CrossRef CAS PubMed
- X. Yan, J. Kong, C. Yang and G. Fu, J. Colloid Interface Sci., 2015, 445, 9–15 CrossRef CAS PubMed
- M. Ballauff and Y. Lu, Polymer, 2007, 48, 1815–1823 CrossRef CAS
- P. Uhlmann, N. Houbenov, N. Brenner, K. Grundke, S. Burkert and M. Stamm, Langmuir, 2007, 23, 57–64 CrossRef CAS PubMed
- F. Mumtaz, C. Chen, H. Zhu, C. Pan and Y. Wang, Appl. Surf. Sci., 2018, 439, 148–159 CrossRef CAS
- F. Mumtaz, C.-S. Chen, H.-K. Zhu, M. Atif and Y.-M. Wang, Chin. J. Polym. Sci., 2018, 36, 1328–1341 CrossRef CAS
- O. Hoy, B. Zdyrko, R. Lupitskyy, R. Sheparovych, D. Aulich, J. Wang, E. Bittrich, K. J. Eichhorn, P. Uhlmann and K. Hinrichs, Adv. Funct. Mater., 2010, 20, 2240–2247 CrossRef CAS
- B. M. Okrugin, R. P. Richter, F. A. Leermakers, I. M. Neelov, O. V. Borisov and E. B. Zhulina, Soft Matter, 2018, 14, 6230–6242 RSC
- C. K. Simocko, A. L. Frischknecht and D. L. Huber, ACS Macro Lett., 2016, 5, 149–153 CrossRef CAS PubMed
- A. Bratek-Skicki, P. Eloy, M. Morga and C. Dupont-Gillain, Langmuir, 2018, 34, 3037–3048 CrossRef CAS PubMed
- A. Bratek-Skicki, V. Cristaudo, J. R. M. Savocco, S. Nootens, P. Morsomme, A. Delcorte and C. Dupont-Gillain, Biomacromolecules, 2019, 20, 778–789 CrossRef CAS PubMed
- M. Delcroix, G. Huet, T. Conard, S. Demoustier-Champagne, F. E. Du Prez, J. Landoulsi and C. C. Dupont-Gillain, Biomacromolecules, 2012, 14, 215–225 CrossRef PubMed
- X. Huang, N. Hauptmann, D. Appelhans, P. Formanek, S. Frank, S. Kaskel, A. Temme and B. Voit, Small, 2012, 8, 3579–3583 CrossRef CAS PubMed
- S. Santer, A. Kopyshev, J. Donges, H. K. Yang and J. Rühe, Adv. Mater., 2006, 18, 2359–2362 CrossRef CAS
- R. Advincula, W. Brittain, K. Caster and J. Rühe.
- E. Currie, W. Norde and M. C. Stuart, Adv. Colloid Interface Sci., 2003, 100, 205–265 CrossRef PubMed
- S. Edmondson, V. L. Osborne and W. T. Huck, Chem. Soc. Rev., 2004, 33, 14–22 RSC
-
J. Rühe, M. Ballauff, M. Biesalski, P. Dziezok, F. Gröhn, D. Johannsmann, N. Houbenov, N. Hugenberg, R. Konradi and S. Minko, Polyelectrolytes with Defined Molecular Architecture I, Springer, 2004, pp. 79–150 Search PubMed
-
Y. Tsujii, K. Ohno, S. Yamamoto, A. Goto and T. Fukuda, Surface-initiated polymerization I, Springer, 2006, pp. 1–45 Search PubMed
- J. Marra and M. L. Hair, Colloids Surf., 1988, 34, 215–226 CrossRef CAS
- H. Watanabe and M. Tirrell, Macromolecules, 1993, 26, 6455–6466 CrossRef CAS
- F. Leonforte and M. Muller, ACS Appl. Mater. Interfaces, 2015, 7, 12450–12462 CrossRef CAS PubMed
- S. Minko, S. Patil, V. Datsyuk, F. Simon, K.-J. Eichhorn, M. Motornov, D. Usov, I. Tokarev and M. Stamm, Langmuir, 2002, 18, 289–296 CrossRef CAS
- N. Houbenov, S. Minko and M. Stamm, Macromolecules, 2003, 36, 5897–5901 CrossRef CAS
- B. Zhao and W. J. Brittain, Prog. Polym. Sci., 2000, 25, 677–710 CrossRef CAS
- J. Escorihuela, A. T. Marcelis and H. Zuilhof, Adv. Mater. Interfaces, 2015, 2, 1500135 CrossRef
- G. Xu, P. Liu, D. Pranantyo, L. Xu, K.-G. Neoh and E.-T. Kang, Ind. Eng. Chem. Res., 2017, 56, 14479–14488 CrossRef CAS
- K. Gong, C. Pan, K. He, H. Zhu, L. Chen, M. Hou and Y. Wang, J. Appl. Polym. Sci., 2019, 136, 48135 CrossRef
- D. Julthongpiput, Y.-H. Lin, J. Teng, E. R. Zubarev and V. V. Tsukruk, J. Am. Chem. Soc., 2003, 125, 15912–15921 CrossRef CAS PubMed
- D. Julthongpiput, Y.-H. Lin, J. Teng, E. R. Zubarev and V. V. Tsukruk, Langmuir, 2003, 19, 7832–7836 CrossRef CAS
- T. Chen, I. Amin and R. Jordan, Chem. Soc. Rev., 2012, 41, 3280–3296 RSC
- L. Wu, U. Glebe and A. Böker, Polym. Chem., 2015, 6, 5143–5184 RSC
- A. Olivier, F. Meyer, J.-M. Raquez, P. Damman and P. Dubois, Prog. Polym. Sci., 2012, 37, 157–181 CrossRef CAS
- O. Prucker and J. Rühe, Macromolecules, 1998, 31, 592–601 CrossRef CAS
- A. Sidorenko, S. Minko, K. Schenk-Meuser, H. Duschner and M. Stamm, Langmuir, 1999, 15, 8349–8355 CrossRef CAS
-
P. C. Hiemenz and T. P. Lodge, Polymer chemistry, CRC press, 2007 Search PubMed
- R. Barbey, L. Lavanant, D. Paripovic, N. Schuwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS PubMed
- M. Fantin, A. A. Isse, A. Venzo, A. Gennaro and K. Matyjaszewski, J. Am. Chem. Soc., 2016, 138, 7216–7219 CrossRef CAS PubMed
- K. Min, H. Gao and K. Matyjaszewski, Macromolecules, 2007, 40, 1789–1791 CrossRef CAS
- S. b Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS
-
S. Kobayashi and K. Müllen, Encyclopedia of polymeric nanomaterials, Springer Berlin Heidelberg, 2015 Search PubMed
- V. Sciannamea, R. Jérôme and C. Detrembleur, Chem. Rev., 2008, 108, 1104–1126 CrossRef CAS PubMed
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed
- B. Zhao, R. T. Haasch and S. MacLaren, J. Am. Chem. Soc., 2004, 126, 6124–6134 CrossRef CAS PubMed
- H. S. Kim, Y. J. Son, W. Mao, K. W. Leong and H. S. Yoo, Nano Lett., 2018, 18, 314–325 CrossRef CAS PubMed
- M. C. Tria, K.-S. Liao, N. Alley, S. Curran and R. Advincula, J. Mater. Chem., 2011, 21, 10261–10264 RSC
- O. Roling, A. Mardyukov, J. A. Krings, A. Studer and B. J. Ravoo, Macromolecules, 2014, 47, 2411–2419 CrossRef CAS
- C.-H. Wong and S. C. Zimmerman, Chem. Commun., 2013, 49, 1679–1695 RSC
- M. B. Runge, S. Dutta and N. B. Bowden, Macromolecules, 2006, 39, 498–508 CrossRef CAS
- C. Fu, J. Xu, M. Kokotovic and C. Boyer, ACS Macro Lett., 2016, 5, 444–449 CrossRef CAS PubMed
- C. Fu, J. Xu and C. Boyer, Chem. Commun., 2016, 52, 7126–7129 RSC
- Y. Wang and W. J. Brittain, Macromol. Rapid Commun., 2007, 28, 811–815 CrossRef CAS
- D. Priftis, G. Sakellariou, D. Baskaran, J. W. Mays and N. Hadjichristidis, Soft Matter, 2009, 5, 4272–4278 RSC
- M. Motornov, R. Sheparovych, E. Katz and S. Minko, ACS Nano, 2008, 2, 41–52 CrossRef CAS PubMed
- M. Motornov, R. Sheparovych, I. Tokarev, Y. Roiter and S. Minko, Langmuir, 2007, 23, 13–19 CrossRef CAS PubMed
- J. Draper, I. Luzinov, S. Minko, I. Tokarev and M. Stamm, Langmuir, 2004, 20, 4064–4075 CrossRef CAS PubMed
- M. K. Vyas, K. Schneider, B. Nandan and M. Stamm, Soft Matter, 2008, 4, 1024–1032 RSC
- M. Atif, C. Chen, M. Irfan, F. Mumtaz, K. He, M. Zhang, L. Chen and Y. Wang, Eur. Polym. J., 2019, 120, 109199 CrossRef CAS
- W. Thiessen and T. Wolff, Des. Monomers Polym., 2011, 14, 287–302 CrossRef CAS
- X. Jiang, G. Zhong, J. M. Horton, N. Jin, L. Zhu and B. Zhao, Macromolecules, 2010, 43, 5387–5395 CrossRef CAS
- X. Jiang, B. Zhao, G. Zhong, N. Jin, J. M. Horton, L. Zhu, R. S. Hafner and T. P. Lodge, Macromolecules, 2010, 43, 8209–8217 CrossRef CAS
- J. M. Horton, S. Tang, C. Bao, P. Tang, F. Qiu, L. Zhu and B. Zhao, ACS Macro Lett., 2012, 1, 1061–1065 CrossRef CAS PubMed
- S. Tang, T.-Y. Lo, J. M. Horton, C. Bao, P. Tang, F. Qiu, R.-M. Ho, B. Zhao and L. Zhu, Macromolecules, 2013, 46, 6575–6584 CrossRef CAS
- N. C. Estillore and R. C. Advincula, Langmuir, 2011, 27, 5997–6008 CrossRef CAS PubMed
- T. L. Fox, S. Tang, J. M. Horton, H. A. Holdaway, B. Zhao, L. Zhu and P. L. Stewart, Langmuir, 2015, 31, 8680–8688 CrossRef CAS PubMed
- D. Li, X. Sheng and B. Zhao, J. Am. Chem. Soc., 2005, 127, 6248–6256 CrossRef CAS PubMed
- B. Zhao, Polymer, 2003, 44, 4079–4083 CrossRef CAS
- S. Santer, A. Kopyshev, J. Donges, J. Rühe, X. Jiang, B. Zhao and M. Müller, Langmuir, 2007, 23, 279–285 CrossRef CAS PubMed
- J. Guo, L. Peng and J. Yuan, Eur. Polym. J., 2015, 69, 449–459 CrossRef CAS
- A. Kroning, A. Furchner, D. Aulich, E. Bittrich, S. Rauch, P. Uhlmann, K.-J. Eichhorn, M. Seeber, I. Luzinov and S. M. Kilbey, ACS Appl. Mater. Interfaces, 2015, 7, 12430–12439 CrossRef CAS PubMed
- Y. Ren, X. Jiang and J. Yin, Eur. Polym. J., 2008, 44, 4108–4114 CrossRef CAS
- E. Currie, A. Sieval, G. Fleer and M. C. Stuart, Langmuir, 2000, 16, 8324–8333 CrossRef CAS
- A. J. Parnell, S. J. Martin, C. C. Dang, M. Geoghegan, R. A. Jones, C. J. Crook, J. R. Howse and A. J. Ryan, Polymer, 2009, 50, 1005–1014 CrossRef CAS
- N. Schüwer and H.-A. Klok, Langmuir, 2011, 27, 4789–4796 CrossRef PubMed
- D. Aulich, O. Hoy, I. Luzinov, M. Brücher, R. Hergenröder, E. Bittrich, K.-J. Eichhorn, P. Uhlmann, M. Stamm and N. Esser, Langmuir, 2010, 26, 12926–12932 CrossRef CAS PubMed
- R. Burtovyy and I. Luzinov, Langmuir, 2008, 24, 5903–5910 CrossRef CAS PubMed
- Y. Sun, K. R. Shull, D. A. Walko and J. Wang, Langmuir, 2010, 27, 201–208 CrossRef PubMed
- N. Li, B. He, S. Xu, J. Yuan, J. Miao, L. Niu and J. Song, Mater. Chem. Phys., 2012, 133, 726–734 CrossRef CAS
- O. Borozenko, C. Ou, W. Skene and S. Giasson, Polym. Chem., 2014, 5, 2242–2252 RSC
- M. Ito, K. Takano, H. Hanochi, Y. Asaumi, S.-I. Yusa, Y. Nakamura and S. Fujii, Front. Chem., 2018, 6, 269 CrossRef PubMed
- Y. G. Takei, T. Aoki, K. Sanui, N. Ogata, Y. Sakurai and T. Okano, Macromolecules, 1994, 27, 6163–6166 CrossRef CAS
- T. Russell, Science, 2002, 297, 964–967 CrossRef CAS PubMed
- K. N. Plunkett, X. Zhu, J. S. Moore and D. E. Leckband, Langmuir, 2006, 22, 4259–4266 CrossRef CAS PubMed
- B.-C. Choi, S. Choi and D. E. Leckband, Langmuir, 2013, 29, 5841–5850 CrossRef CAS PubMed
- E. Bittrich, S. Burkert, M. Müller, K.-J. Eichhorn, M. Stamm and P. Uhlmann, Langmuir, 2012, 28, 3439–3448 CrossRef CAS PubMed
- T. Sun, G. Wang, L. Feng, B. Liu, Y. Ma, L. Jiang and D. Zhu, Angew. Chem., Int. Ed., 2004, 43, 357–360 CrossRef CAS PubMed
- S. Burkert, E. Bittrich, M. Kuntzsch, M. Müller, K.-J. Eichhorn, C. Bellmann, P. Uhlmann and M. Stamm, Langmuir, 2010, 26, 1786–1795 CrossRef CAS PubMed
- M. Balastre, F. Li, P. Schorr, J. Yang, J. W. Mays and M. V. Tirrell, Macromolecules, 2002, 35, 9480–9486 CrossRef CAS
- O. Borisov, E. Zhulina and T. Birshtein, Macromolecules, 1994, 27, 4795–4803 CrossRef CAS
- A. F. Mak and S. Sun, Intelligent chitosan-based hydrogels as multi-functional materials, Royal Society of Chemistry, 2008 Search PubMed
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS PubMed
- M. Delcroix, S. Demoustier-Champagne and C. C. Dupont-Gillain, Langmuir, 2013, 30, 268–277 CrossRef PubMed
- A. T. Nguyen, J. Baggerman, J. M. Paulusse, C. J. van Rijn and H. Zuilhof, Langmuir, 2011, 27, 2587–2594 CrossRef CAS PubMed
- R. Quintana, M. Gosa, D. Janczewski, E. Kutnyanszky and G. J. Vancso, Langmuir, 2013, 29, 10859–10867 CrossRef CAS PubMed
- D. Schulz, D. Peiffer, P. Agarwal, J. Larabee, J. Kaladas, L. Soni, B. Handwerker and R. Garner, Polymer, 1986, 27, 1734–1742 CrossRef CAS
- V. Arjunan Vasantha, C. Junhui, T. B. Ying and A. Parthiban, Langmuir, 2015, 31, 11124–11134 CrossRef CAS PubMed
- J. Sun, X. Chang, F. Zhang, Y. Bai, K. Lv, J. Wang, X. Zhou and B. Wang, Energy Fuels, 2020, 34, 1669–1679 CrossRef CAS
- S. Gupta, M. Agrawal, P. Uhlmann, F. Simon, U. Oertel and M. Stamm, Macromolecules, 2008, 41, 8152–8158 CrossRef CAS
- L. Ionov, S. Sapra, A. Synytska, A. L. Rogach, M. Stamm and S. Diez, Adv. Mater., 2006, 18, 1453–1457 CrossRef CAS
- K. Yu, H. Wang and Y. Han, Langmuir, 2007, 23, 8957–8964 CrossRef CAS PubMed
- W. Van Camp, F. E. Du Prez, H. Alem, S. Demoustier-Champagne, N. Willet, G. Grancharov and A.-S. Duwez, Eur. Polym. J., 2010, 46, 195–201 CrossRef CAS
- E. Bittrich, K. B. Rodenhausen, K.-J. Eichhorn, T. Hofmann, M. Schubert, M. Stamm and P. Uhlmann, Biointerphases, 2010, 5, 159–167 CrossRef PubMed
- P. Akkahat and V. P. Hoven, Colloids Surf., B, 2011, 86, 198–205 CrossRef CAS PubMed
- T. Wu, P. Gong, I. Szleifer, P. Vlcek, V. Subr and J. Genzer, Macromolecules, 2007, 40, 8756–8764 CrossRef CAS
- M. G. Santonicola, G. W. de Groot, M. Memesa, A. Meszynska and G. J. Vancso, Langmuir, 2010, 26, 17513–17519 CrossRef CAS PubMed
- K. H. Kan, J. Li, K. Wijesekera and E. D. Cranston, Biomacromolecules, 2013, 14, 3130–3139 CrossRef CAS PubMed
- P. Heinz, F. Bretagnol, I. Mannelli, L. Sirghi, A. Valsesia, G. Ceccone, D. Gilliland, K. Landfester, H. Rauscher and F. Rossi, Langmuir, 2008, 24, 6166–6175 CrossRef CAS PubMed
- M. Klein Gunnewiek, A. Di Luca, X. Sui, C. A. van Blitterswijk, L. Moroni and G. J. Vancso, Isr. J. Chem., 2012, 52, 339–346 CrossRef CAS
- R. Elashnikov, P. Slepička, S. Rimpelova, P. Ulbrich, V. Švorčík and O. Lyutakov, Mater. Sci. Eng., C, 2017, 72, 293–300 CrossRef CAS PubMed
- T. Peng and Y. L. Cheng, J. Appl. Polym. Sci., 1998, 70, 2133–2142 CrossRef CAS
- T. H. Lee and J. Y. Jho, Macromol. Res., 2018, 26, 659–664 CrossRef CAS
- C. Xue, B. C. Choi, S. Choi, P. V. Braun and D. E. Leckband, Adv. Funct. Mater., 2012, 22, 2394–2401 CrossRef CAS
- J. Raczkowska, Y. Stetsyshyn, K. Awsiuk, J. Zemła, A. Kostruba, K. Harhay, M. Marzec, A. Bernasik, O. Lishchynskyi and H. Ohar, RSC Adv., 2016, 6, 87469–87477 RSC
- A. T. Nguyen, J. Baggerman, J. M. Paulusse, H. Zuilhof and C. J. van Rijn, Langmuir, 2012, 28, 604–610 CrossRef CAS PubMed
- F. Çalılı, P. Kaner, G. Aro, A. Asatekin and P. Z. Çulfaz-Emecen, React. Funct. Polym., 2020, 156, 104738 CrossRef
- J. Yang, H. Chen, S. Xiao, M. Shen, F. Chen, P. Fan, M. Zhong and J. Zheng, Langmuir, 2015, 31, 9125–9133 CrossRef CAS PubMed
- K. M. Johnson, M. J. Fevola and C. L. McCormick, J. Appl. Polym. Sci., 2004, 92, 647–657 CrossRef CAS
- M. Han, J.-S. Ryu and J.-W. Park, React. Funct. Polym., 2013, 73, 66–72 CrossRef CAS
- A. Jung, S. Rangou, C. Abetz, V. Filiz and V. Abetz, Macromol. Mater. Eng., 2012, 297, 790–798 CrossRef CAS
- B. M. Okrugin, R. P. Richter, F. A. Leermakers, I. M. Neelov, E. B. Zhulina and O. V. Borisov, Polymers, 2020, 12, 898 CrossRef CAS PubMed
- E. Kutnyánszky, M. A. Hempenius and G. J. Vancso, Polym. Chem., 2014, 5, 771–783 RSC
- X. Sui, X. Feng, A. Di Luca, C. A. van Blitterswijk, L. Moroni, M. A. Hempenius and G. J. Vancso, Polym. Chem., 2013, 4, 337–342 RSC
- X. Sui, X. Feng, J. Song, M. A. Hempenius and G. J. Vancso, J. Mater. Chem., 2012, 22, 11261–11267 RSC
- M. Mazurowski, M. Gallei, J. Li, H. Didzoleit, B. Stühn and M. Rehahn, Macromolecules, 2012, 45, 8970–8981 CrossRef CAS
- B. Y. Kim, E. L. Ratcliff, N. R. Armstrong, T. Kowalewski and J. Pyun, Langmuir, 2010, 26, 2083–2092 CrossRef CAS PubMed
- J. Erath, J. Cui, J. Schmid, M. Kappl, A. N. D. Campo and A. Fery, Langmuir, 2013, 29, 12138–12144 CrossRef CAS PubMed
- A. A. Brown, O. Azzaroni and W. T. Huck, Langmuir, 2009, 25, 1744–1749 CrossRef CAS PubMed
- D. Wang, G. Ye, X. Wang and X. Wang, Adv. Mater., 2011, 23, 1122–1125 CrossRef CAS PubMed
- J. Cui, T.-H. Nguyen, M. Ceolín, R. D. Berger, O. Azzaroni and A. N. Del Campo, Macromolecules, 2012, 45, 3213–3220 CrossRef CAS
- M. J. Salierno, A. J. García and A. del Campo, Adv. Funct. Mater., 2013, 23, 5974–5980 CrossRef CAS
- L. Sun, J. Dai, G. L. Baker and M. L. Bruening, Chem. Mater., 2006, 18, 4033–4039 CrossRef CAS
- J. Dai, G. L. Baker and M. L. Bruening, Anal. Chem., 2006, 78, 135–140 CrossRef CAS PubMed
- L. Sage, 2004.
- A. Wittemann and M. Ballauff, Phys. Chem. Chem. Phys., 2006, 8, 5269–5275 RSC
- A. Halperin, Langmuir, 1999, 15, 2525–2533 CrossRef CAS
-
G. E. Schulz and R. H. Schirmer, Principles of protein structure, Springer Science & Business Media, 2013 Search PubMed
- R. A. Latour, Encycl. Biomater. Biomed. Eng., 2005, 1, 270–278 Search PubMed
-
E. J. Cohn, Proteins, amino acids and peptides as ions and dipolar ions/by E.J.Cohn J.T.Edsall and others, Reinhold, New York, 1943 Search PubMed
-
C. Tanford, Phys. Chem. Macromol., Wiley, 1961 Search PubMed
- A. Wittemann, B. Haupt and M. Ballauff, Phys. Chem. Chem. Phys., 2003, 5, 1671–1677 RSC
- O. Hollmann and C. Czeslik, Langmuir, 2006, 22, 3300–3305 CrossRef CAS PubMed
- C. Czeslik and A. Wittemann, Colloid Polym. Sci., 2020, 1–15 Search PubMed
- C. Cooper, Curr. Opin. Colloid Interface Sci., 2005, 10, 52–78 CrossRef CAS
- K. R. Grymonpré, B. A. Staggemeier, P. L. Dubin and K. W. Mattison, Biomacromolecules, 2001, 2, 422–429 CrossRef PubMed
- E. Seyrek, P. L. Dubin, C. Tribet and E. A. Gamble, Biomacromolecules, 2003, 4, 273–282 CrossRef CAS PubMed
- R. De Vries, F. Weinbreck and C. De Kruif, J. Chem. Phys., 2003, 118, 4649–4659 CrossRef CAS
- L. Feller, Y. Jadwat, R. A. Khammissa, R. Meyerov, I. Schechter and J. Lemmer, BioMed Res. Int., 2015, 171945 Search PubMed
- B. Young, W. Pitt and S. Cooper, J. Colloid Interface Sci., 1988, 125, 246–260 CrossRef CAS
- S. Mitragotri and J. Lahann, Nat. Mater., 2009, 8, 15–23 CrossRef CAS PubMed
- A. P. Liu, O. Chaudhuri and S. H. Parekh, Integr. Biol., 2017, 9, 383–405 CrossRef PubMed
- E. Kaivosoja, G. Barreto, K. Levon, S. Virtanen, M. Ainola and Y. T. Konttinen, Ann. Med., 2012, 44, 635–650 CrossRef CAS PubMed
- Z. Yoshimitsu, A. Nakajima, T. Watanabe and K. Hashimoto, Langmuir, 2002, 18, 5818–5822 CrossRef CAS
- I. Yoda, H. Koseki, M. Tomita, T. Shida, H. Horiuchi, H. Sakoda and M. Osaki, BMC Microbiol., 2014, 14, 1–7 CrossRef PubMed
- J. Carpenter, D. Khang and T. J. Webster, Nanotechnology, 2008, 19, 505103 CrossRef PubMed
- G. P. Barnsley, L. J. Sigurdson and S. E. Barnsley, Plast. Reconstr. Surg., 2006, 117, 2182–2190 CrossRef CAS PubMed
- B. D. Boyan, E. M. Lotz and Z. Schwartz, Tissue Eng., Part A, 2017, 23, 1479–1489 CrossRef CAS PubMed
- P. Roach, D. Farrar and C. C. Perry, J. Am. Chem. Soc., 2006, 128, 3939–3945 CrossRef CAS PubMed
- T. Akkas, C. Citak, A. Sirkecioglu and F. S. Güner, Polym. Int., 2013, 62, 1202–1209 CrossRef CAS
- Z. Zhang, G. Li, F. Yan, X. Zheng and X. Li, Open Chem., 2012, 10, 1157–1164 CrossRef CAS
- O. Gamucci, A. Bertero, M. Gagliardi and G. Bardi, Coatings, 2014, 4, 139–159 CrossRef
- P. Satzer, F. Svec, G. Sekot and A. Jungbauer, Eng. Life Sci., 2016, 16, 238–246 CrossRef CAS PubMed
- X. Wang, R. Berger, J. I. Ramos, T. Wang, K. Koynov, G. Liu, H.-J. Butt and S. Wu, RSC Adv., 2014, 4, 45059–45064 RSC
- M. P. Calatayud, B. Sanz, V. Raffa, C. Riggio, M. R. Ibarra and G. F. Goya, Biomaterials, 2014, 35, 6389–6399 CrossRef CAS PubMed
- R. Shelton, A. Rasmussen and J. Davies, Biomaterials, 1988, 9, 24–29 CrossRef CAS PubMed
- Y. Wu, F. I. Simonovsky, B. D. Ratner and T. A. Horbett, J. Biomed. Mater. Res., Part A, 2005, 74, 722–738 CrossRef PubMed
- D. MacDonald, N. Deo, B. Markovic, M. Stranick and P. Somasundaran, Biomaterials, 2002, 23, 1269–1279 CrossRef CAS PubMed
- C. A. Scotchford, C. P. Gilmore, E. Cooper, G. J. Leggett and S. Downes, J. Biomed. Mater. Res., Part A, 2002, 59, 84–99 CrossRef CAS PubMed
- W. G. Brodbeck, G. Voskerician, N. P. Ziats, Y. Nakayama, T. Matsuda and J. M. Anderson, J. Biomed. Mater. Res., Part A, 2003, 64, 320–329 CrossRef PubMed
- N. Recek, M. Jaganjac, M. Kolar, L. Milkovic, M. Mozetič, K. Stana-Kleinschek and A. Vesel, Molecules, 2013, 18, 12441–12463 CrossRef CAS PubMed
- Y. Arima and H. Iwata, Biomaterials, 2007, 28, 3074–3082 CrossRef CAS PubMed
- Z. Zhang, S. Chen and S. Jiang, Biomacromolecules, 2006, 7, 3311–3315 CrossRef CAS PubMed
- E. A. Dubiel, Y. Martin and P. Vermette, Chem. Rev., 2011, 111, 2900–2936 CrossRef CAS PubMed
- L. Tang, P. Thevenot and W. Hu, Curr. Top. Med. Chem., 2008, 8, 270–280 CrossRef PubMed
- J. Benesch, S. Svedhem, S. C. Svensson, R. N. Valiokas, B. Liedberg and P. Tengvall, J. Biomater. Sci., Polym. Ed., 2001, 12, 581–597 CrossRef CAS PubMed
- L. Liu and H. Elwing, J. Biomed. Mater. Res., 1994, 28, 767–773 CrossRef CAS PubMed
- P. Roach, D. Farrar and C. C. Perry, J. Am. Chem. Soc., 2005, 127, 8168–8173 CrossRef CAS PubMed
- J. Jin, Y. Han, C. Zhang, J. Liu, W. Jiang, J. Yin and H. Liang, Colloids Surf., B, 2015, 136, 838–844 CrossRef CAS PubMed
- F. Xu, Q. Cai, Y. Li, E. Kang and K. Neoh, Biomacromolecules, 2005, 6, 1012–1020 CrossRef CAS PubMed
- S. P. Cullen, X. Liu, I. C. Mandel, F. J. Himpsel and P. Gopalan, Langmuir, 2008, 24, 913–920 CrossRef CAS PubMed
- M. Delcroix, S. Laurent, G. Huet and C. C. Dupont-Gillain, Acta Biomater., 2015, 11, 68–79 CrossRef CAS PubMed
- S. Belegrinou, I. Mannelli, P. Lisboa, F. Bretagnol, A. Valsesia, G. Ceccone, P. Colpo, H. Rauscher and F. Rossi, Langmuir, 2008, 24, 7251–7261 CrossRef CAS PubMed
- E. Psarra, U. König, Y. Ueda, C. Bellmann, A. Janke, E. Bittrich, K.-J. Eichhorn and P. Uhlmann, ACS Appl. Mater. Interfaces, 2015, 7, 12516–12529 CrossRef CAS PubMed
- S. Burkert, E. Bittrich, M. Kuntzsch, M. Müller, K.-J. Eichhorn, C. Bellmann, P. Uhlmann and M. Stamm, Langmuir, 2009, 26, 1786–1795 CrossRef PubMed
- H. Lei, M. Wang, Z. Tang, Y. Luan, W. Liu, B. Song and H. Chen, Langmuir, 2014, 30, 501–508 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2024 |