
Microfluidic-assisted single-cell RNA sequencing facilitates the development of neutralizing monoclonal antibodies against SARS-CoV-2
Ziwei
Wang†
 a,
Amelia Siqi
Huang†
b,
Lingfang
Tang
b,
Jianbin
Wang
a and
Guanbo
Wang
*c
a,
Amelia Siqi
Huang†
b,
Lingfang
Tang
b,
Jianbin
Wang
a and
Guanbo
Wang
*c
aSchool of Life Sciences, Tsinghua University, Beijing, 100084, China
bDalton Academy, The Affiliated High School of Peking University, Beijing, 100190, China
cInstitute for Cell Analysis, Shenzhen Bay Laboratory, Shenzhen, 518132, China. E-mail: guanbo.wang@pku.edu.cn
First published on 2nd January 2024
Abstract
As a class of antibodies that specifically bind to a virus and block its entry, neutralizing monoclonal antibodies (neutralizing mAbs) have been recognized as a top choice for combating COVID-19 due to their high specificity and efficacy in treating serious infections. Although conventional approaches for neutralizing mAb development have been optimized for decades, there is an urgent need for workflows with higher efficiency due to time-sensitive concerns, including the high mutation rate of SARS-CoV-2. One promising approach is the identification of neutralizing mAb candidates via single-cell RNA sequencing (RNA-seq), as each B cell has a unique transcript sequence corresponding to its secreted antibody. The state-of-the-art high-throughput single-cell sequencing technologies, which have been greatly facilitated by advances in microfluidics, have greatly accelerated the process of neutralizing mAb development. Here, we provide an overview of the general procedures for high-throughput single-cell RNA-seq enabled by breakthroughs in droplet microfluidics, introduce revolutionary approaches that combine single-cell RNA-seq to facilitate the development of neutralizing mAbs against SARS-CoV-2, and outline future steps that need to be taken to further improve development strategies for effective treatments against infectious diseases.
Introduction
The COVID-19 pandemic, caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that first emerged at the end of 2019, has since caused significant global disruption. While non-pharmaceutical interventions such as mask usage, regional lockdowns, and social distancing have been actively implemented, pharmaceutical interventions remain crucial to ending the pandemic. Effective pharmaceutical interventions, both preventative and therapeutic, are important for controlling disease spread and relieving symptoms.1 Although vaccination can effectively prevent both symptomatic and severe COVID-19,2 it requires several weeks for antibody levels to attain a protective concentration in the serum.3 Its relatively slow immune response and diminished effectiveness in certain populations, including immunocompromised patients,4 the elderly,5 and organ transplant recipients,6 underscore the necessity for therapeutics that directly target pathogens without relying on the stimulation of the body's immune response. Small-molecule drugs and neutralizing monoclonal antibodies are the two major types of medical therapeutics.7 In the early stages of the disease outbreak, the potential preventive benefits of vitamin D8 and hydroxychloroquine9 were investigated. The RNA-dependent RNA polymerase (RdRp) and 3C-like (3CL) proteases of viruses have served as key targets for a range of antiviral small-molecule drugs.10 Owing to their structural similarity, numerous small molecules initially designed to combat other viruses have been repurposed for SARS-CoV-2 treatment prior to introducing newly developed drugs. Remdesivir, initially developed for Ebola prevention,11 is one such example. While various small-molecule drugs have been developed and some, such as nirmatrelvir/ritonavir (Paxlovid), have been authorized to treat COVID-19 patients, these treatments have proven effective only in the initial stages of the disease.12 Despite challenges in efficient screening, production cost, scalability, and logistic requirements, neutralizing mAbs remain a preferred choice for combating COVID-19 due to their high specificity and efficacy in treating serious infections.13Since the first approval in 1986,14 monoclonal antibodies (mAbs) have been used as therapeutics to treat a wide range of diseases, including cancers,15 inflammatory diseases,16 and infectious diseases,13 among others. mAbs developed for cancer treatment, such as cetuximab, trastuzumab and pembrolizumab, have achieved both therapeutic and commercial successes. For the treatment of infectious diseases, the US FDA has approved several mAbs or their combinations, such as ibalizumab for HIV and ansuvimab for Ebola.17
One of the primary advantages of using mAbs in clinical applications is their high specificity, derived from the host's natural immune response to stimuli18 and optimized through a meticulous selection or re-engineering process when identifying antibody candidates. In the case of infections, various antibodies are produced by B-lymphocytes, with each mAb representing a specific type of antibody molecule with an identical peptide sequence. Each antibody within a clone binds to the same epitope, a specific part of the target, such as a component of the pathogen. Some clones of antibodies may prevent effective binding between the pathogen and its receptor on the host cell and are therefore referred to as neutralizing antibodies (NAbs).19 NAbs function through either direct competitive binding at the viral-receptor interfaces or binding to regions distant from these interfaces. In the context of SARS-CoV-2, while most NAbs isolated from COVID-19 convalescent patients target the RBD, some NAbs recognize the N-terminal domain (NTD) of the S protein20 and may potentially interfere with the conformational changes necessary for fusion or disrupt proposed interactions with attachment receptors, such as the transmembrane lectins DC-SIGN, L-SIGN, and SIGLEC1.21,22
Another inherent advantage of mAbs is their lower immunogenicity, particularly when the antibodies are human-derived or humanized. High immunogenicity can lead to the formation of anti-drug antibodies, eventually causing a loss of efficacy.23 Although some humanized and fully human antibodies may still carry immunological risk, these molecules have highly similar or identical constant regions to those produced in the human body, resulting in significantly reduced immunogenicity.24 With the successful application of antibodies in treating infectious pathogens, such as raxibacumab and obiltoxaximab for inhaled anthrax and palivizumab for respiratory syncytial virus,25 neutralizing mAbs have been a major focus of drug development since the beginning of the COVID-19 pandemic.13
The mammalian immune system is an efficient factory for producing antibodies, with B cells, a type of white blood cell of the lymphocyte subtype, working in conjunction with other immune cells to create these antibodies upon encountering antigens. Human-derived mAbs can be obtained by sequencing B cells from convalescent patients, a common practice in the treatment of infectious diseases. While neutralizing antibodies in convalescent plasma from patients have induced clinical improvement in mild and severe COVID-19 patients, the therapeutic use of such polyclonal antibodies is limited due to insufficient scalability and neutralization efficiency. In contrast, neutralizing mAbs, which are screened from the polyclonal population for higher neutralizing capability followed by structural and functional characterization, are expected to exhibit superior performance through better determined molecular mechanisms and feature higher yields, higher optimization potential, and lower batch effects. MAbs can be used for both therapeutic and prophylactic purposes, offering both prevention and treatment functions. In contrast to vaccines, neutralizing mAbs offer instant protection when applied to patients, making them especially vital for emergency uses and for those who cannot be effectively immunized.6
The development of highly potent neutralizing mAbs against COVID-19 presents significant challenges. One of the primary obstacles is the high mutation rate of SARS-CoV-2,26 with multiple replacement events leading to the emergence of new variants that become the dominant strains in global circulation (Fig. 1). The efficacy of existing antibodies is greatly reduced when newly emerged mutants can escape their binding. For instance, bebtelovimab was recently withdrawn by the US FDA27 due to its inability to neutralize the new omicron BQ1.1 and XBB strains.28 The combinations of tixagevimab plus cilgavimab (EvuSheld) and bamlanivimab plus etesevimab, which were issued emergency use authorization (EUA) by the US FDA for preexposure prophylaxis and treatment or post-exposure prophylaxis of COVID-19, are not currently authorized for use in the U.S. due to the high frequency of SARS-CoV-2 variants. In fact, almost all neutralizing mAbs developed and proven effective during the early phase of the COVID-19 pandemic are now ineffective against prevalent variants.29
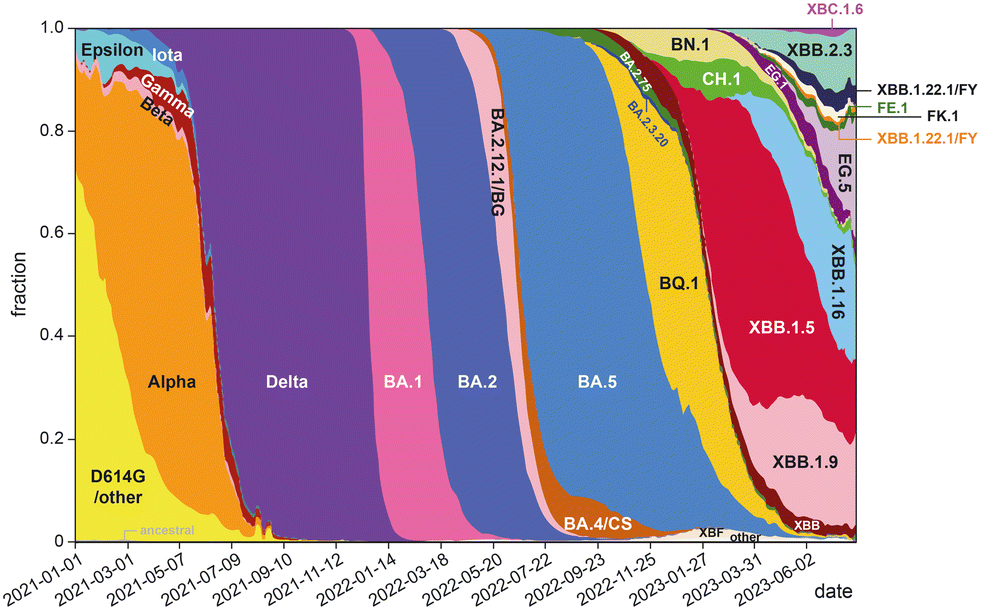 | ||
| Fig. 1 Change of global frequencies of grouped Pango lineages of SARS-CoV-2 over time (graph prepared based on data from ref. 30). | ||
In this ‘catch-me-if-you-can’ battle to combat infectious diseases, developing effective neutralizing mAbs is a time-sensitive task. Conventional approaches involve immunization of animals, hybridoma culture, functional screenings, candidate identification, and sophisticated production and validation procedures.31 Many of these steps have been developed and optimized over decades, and large-scale production, animal tests, and clinical trials are conducted following regulations to ensure both efficacy and safety.32 However, certain steps in this process can be significantly accelerated using state-of-the-art single-cell sequencing technologies. Since each B cell has a unique transcript sequence pair that can be translated into a B cell receptor (BCR) or secreted immunoglobulin, the identification of a mAb candidate is essentially a typical problem for single-cell biology. Since Tang et al. reported the first single-cell RNA sequencing (RNA-seq) method in 2009,33 the throughput of single-cell transcriptomic analysis has increased from a few cells per experimental batch to several thousand or more per batch,34–36 thanks to advances in droplet microfluidics. The application of such high throughput single-cell RNA-seq technologies has led to a dramatic increase in screening effectiveness, expanded screening scale, shortened turnaround time, and accelerated process of drug development.37 High throughput single-cell RNA-seq technologies eliminate the need for time-consuming steps such as cell fusion and culture in hybridoma technology and multiple rounds of biopanning in phage display. In the case of identifying human-derived neutralizing mAbs, the humanization process is also circumvented.
From this perspective, we focus on the development of anti-SARS-Cov-2 neutralizing monoclonal antibodies through the aspect of single-cell transcriptomics enabled by advanced microfluidic techniques. The first section provides a brief overview of the general procedure for high-throughput single-cell RNA-seq, with a particular emphasis on how droplet microfluidics has enabled breakthroughs in this field. The second section introduces immunology related to B cells and the molecular mechanism in sequencing immunoglobulin transcripts. The third section provides an overview of various strategies for single-cell immunoglobulin gene sequencing (Ig-seq) and discusses the selection of strategies under different circumstances. The fourth section highlights revolutionary approaches that combine single-cell RNA-seq to facilitate the development of neutralizing mAbs against SARS-CoV-2. The fifth section emphasizes novel and significant findings gleaned from the rapid development of a single-cell sequencing-based antibody development workflow and outlines future steps that need to be taken.
Development of microfluidic-assisted single-cell RNA-seq technologies
Cells, the building blocks of life, exhibit a high degree of heterogeneity in complex processes such as embryonic development, neural system organization, cancer initiation, and progression.38 The invention of ‘next-generation’ sequencing (NGS) technology39 has rapidly propelled the study of such complex systems into a new paradigm: data-driven science. RNA-seq, a prominent application of NGS, involves converting RNA into cDNA via reverse transcription, followed by constructing sequencing libraries that can be accepted by the sequencers. This process was first successfully implemented at the single-cell level in 2009 (ref. 33) and has since become a routine approach to reveal heterogeneity at the finest level of biology.40However, experimental operations at the single-cell level are invariably challenging. Over the past decade, hundreds of specific experimental protocols have been creatively developed.41 A particular requirement in single-cell RNA-seq is the tagging of mRNA with information regarding its cell of origin. This can be achieved by introducing a short and unique DNA oligo during reverse transcription, which serves as an ideal label for this purpose.42 However, traditional tube-based strategies are limited in terms of throughput and scalability. This bottleneck was overcome by microfluidics, which specializes in handling liquid at nanoliter to picoliter scales.43 Microfluidics has been demonstrated in many labs to perform small-volume liquid-phase experiments in parallel with high flexibility and scalability,44 making it ideal for integration with various single-cell-based protocols. Practically, the primary objective is to segregate cells into numerous tiny reactors (microreactors) and to minimize the possibility of multiple cells being sealed in a single reactor.
Microreactors can be classified as either micro-wells (Fig. 2A) or micro-droplets (Fig. 2B).44 Both schemes have proven effective in single-cell RNA-seq, with the throughput extendable to thousands of cells per experiment. Despite minor methodological differences, the primary objective of all droplet-based single-cell RNA-seq approaches remains consistent – allowing most cells to be distributed in a one-cell-per-droplet fashion, with each cell co-captured with a microbead containing barcoded primers. To address the challenge of double Poisson-distribution difficulty50 that limits the isolation of single cells, and to maximize the use of the precious cells in the sample, the current leading approach involves employing gel-based microbeads to ensure a high occupation rate of single beads within droplets.36 With proper dilution of cells, one-to-one matching of beads and cells in the droplets can now be achieved. Typically, each droplet-based single-cell RNA-seq experiment generates transcriptomes for approximately 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells. To improve quantification and minimize PCR amplification bias between different RNA transcripts, many applications employ a unique molecule index (UMI; Fig. 2C).51
000 cells. To improve quantification and minimize PCR amplification bias between different RNA transcripts, many applications employ a unique molecule index (UMI; Fig. 2C).51
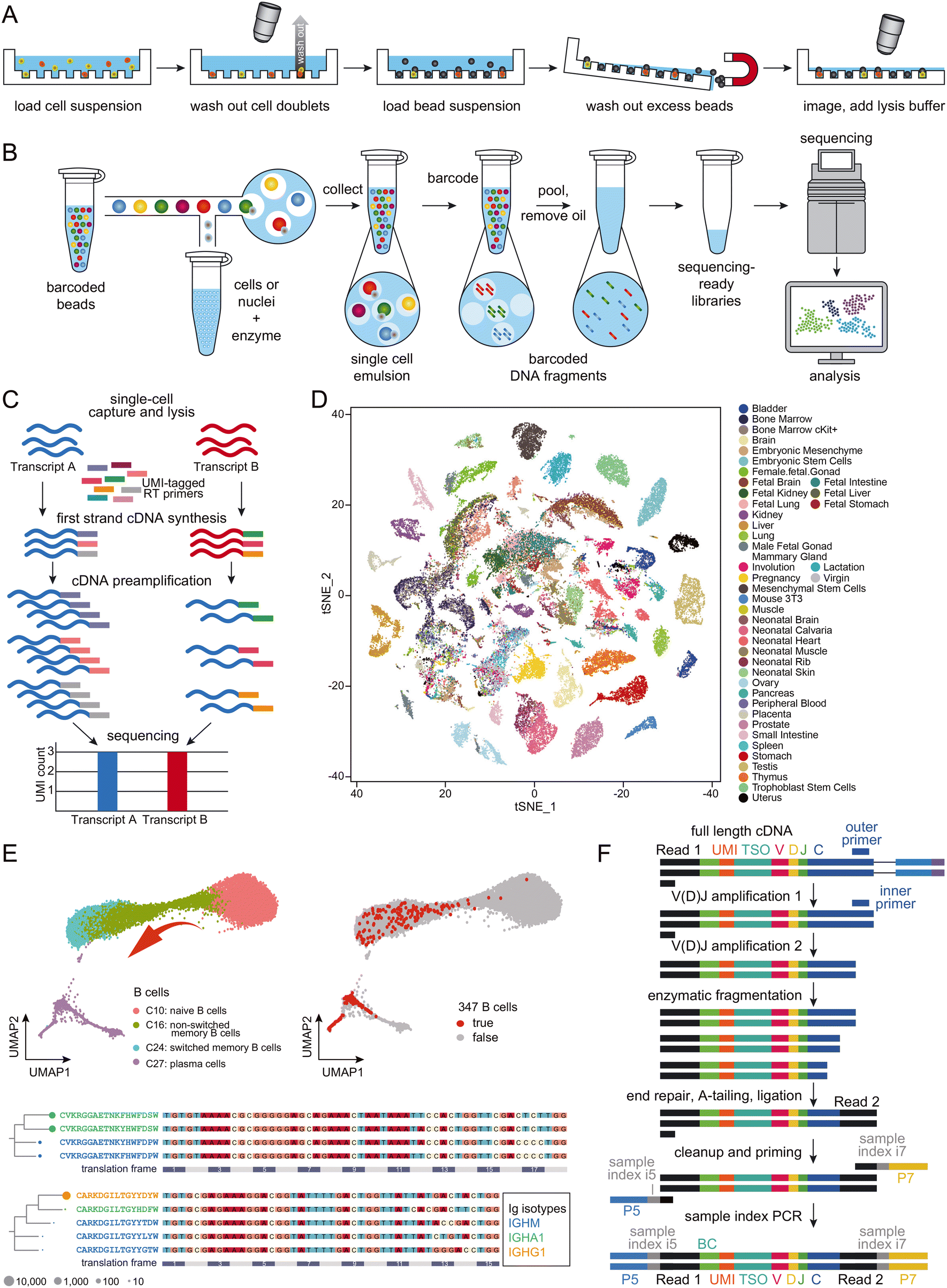 | ||
| Fig. 2 Schematic presentation of the workflows of (A) micro-well-based and (B) micro-droplet-based microfluidic-assisted single-cell RNA-seq technologies. Cartoons reproduced from ref. 45 with permission from Elsevier, copyright 2018 and from ref. 46 with 10x Genomics' permission respectively. (C) Schematic presentation of the workflow of UMI. Adapted from ref. 47 with permission from Oxford University Press, copyright 2017. (D) A representative set of mouse cell atlas data, showing t-SNE analysis of 60000 single cells. Adapted from ref. 45 with permission from Elsevier, copyright 2018. (E) UMAP map of B cells from 12 COVID-19 patients and 8 healthy controls, which formed a gradient of transcriptional states from naïve B cells to activated memory B cells and then to plasma cells (top left); 347 potential antigen-specific BCRs were enriched in activated B cells (C24) and plasma cells (C27; top right). The selected BCR heavy chain groups are presented in lineages trees (bottom), with aligned DNA sequences as reference. Each node represents a BCR clone, with Ig isotypes color-coded. Adapted from ref. 48 with permission from Oxford University Press, copyright 2020. (F) Schematic presentation of BCR enrichment. Reproduced with 10x Genomics' permission from ref. 49. | ||
While it may be feasible for some laboratories to set up microfluidics instrumentation from scratch, commercialized products offer consistent reagents and highly robust operation, making them increasingly popular among researchers. Droplet-based microfluidics, characterized by its compact design and user-friendly operation, has significantly boosted the appeal of single-cell RNA-seq in biological laboratories. This technology enables high-throughput scaling, allowing for analyzing several thousand single cells in a single batch. This has facilitated the development of cell atlases, which aim to dissect organisms into single cells and identify each one, providing the highest resolution analysis of organs (Fig. 2D). Notable achievements include the mouse cell atlas,45,52,53 the human cell atlas,54,55 and the fly cell atlas,56 among others. The generation of such an atlas provides valuable resources to support both fundamental studies in biology and medicine.
While droplet-based single-cell RNA-seq offers numerous advantages, it does have certain limitations. One such limitation is its lower detection sensitivity when compared to many non-microfluidic approaches.64 This necessitates a trade-off between throughput and sensitivity. However, for most B-cell-related works discussed in this article, the sensitivity provided by droplet reactions is sufficient, and a higher throughput better serves the experiment. Another dynamic and related research field is the data computation and analyses of high-dimensional large-scale datasets.65 Developments in this computational aspect, including recent advances in machine learning for data interpretation,66,67 have greatly improved the understanding of the complex immune system.
Utilizing single-cell RNA-seq for infectious disease research
Large-scale single-cell RNA-seq has generated considerable interest among researchers in not only cancer but also infectious disease fields. A primary focus of these studies is the profiling of immune cells within tissues.57 For example, scientists have characterized the landscapes of tumor-infiltrating myeloid (TIM) cells58 and tumor-infiltrating T cells59 in various types of cancers, unveiling heterogeneities in cell composition and distinct functions associated with tumor types, disease progress, and treatment outcomes. While cancer studies may dominate the field of single-cell immune landscaping, and most work has focused on T cells, the single-cell sequencing protocol is identical for studies on B cells.55 In malaria-related research, both antigen-specific and total B cells were sequenced at the single-cell level, revealing the presence of an alternative B cell lineage that contributes to immune responses in humans following vaccination and infection.60 T-cell receptors (TCRs) and BCRs are crucial for understanding cellular development, evolution, activation, maturation, and clone expansion in immune cell sequencing. With the availability of protocols for sequencing the 5′-ends of transcripts, single-cell BCR sequencing data can be easily obtained and integrated with whole transcriptome data. Large-scale single-cell sequencing experiments have significantly advanced our understanding of B-cell development.61 For instance, Wilson and colleagues studied the pathogenic and clonally expanded B cell transcriptome in active multiple sclerosis.62 Such strategies have been widely applied to study immune responses to SARS-CoV-2 infection (Fig. 2E; BCR enrichment is illustrated in Fig. 2F).48,63 These results, along with others not mentioned in this review, provide valuable resources for the development of antibody drugs, which will be discussed in more detail later.B cell immunology and molecular mechanism in sequencing immunoglobulin transcripts
Consequently, obtaining new antibodies from the B cells of immunized or infected animals is a logical and straightforward process. Although the traditional hybridoma screening approach has proven effective in developing first-generation monoclonal antibodies, it is undoubtedly a laborious and expensive process.68,69 Therefore, sequencing immunoglobulin transcripts from B cells followed by antibody synthesis has become increasingly prevalent. This strategy bypasses the limitations of the traditional approach while still utilizing the natural antibody maturation and optimization process of the mammalian immune system.Acquiring immunoglobulin gene sequences in B cells requires careful consideration of their developmental stage and subgroup characteristics. From a developmental perspective, immature B cells develop from common lymphoid precursor cells in the bone marrow and mature further after migrating to secondary lymphoid organs, such as the spleen.70 Upon antigen stimulation and with the help of other immune cells, antigen-specific B cells differentiate into various types of effector cells, such as antibody-secreting cells and memory cells.71 Some of these cells enter germinal centers and undergo hypermutation and affinity selection, resulting in the maturation of high-affinity immunoglobulin sequences.18 When sequencing B cells from an immunized animal's bone marrow or spleen, it is necessary to carefully sort B cell sub-types to ensure the selection of cells with high affinity. Some of these cells, such as memory cells, have a BCR,72 which has the same variable region sequence as secretory antibodies and can recognize the antigen. From a spatial perspective, B cells can either be tissue-resident or circulating between secondary lymphoid organs.73 Tissue-resident B cells have been reported in the lung and gut74,75 and may play a crucial role in developing antibodies against pathogens in these areas. Thus, additional attention should be paid to this type of B cell when targeting pathogens in the respiratory and digestive systems.
Understanding the characteristics of immunoglobulin composition is also essential for immunoglobulin transcript sequencing. Most antibodies consist of four subunits: two identical heavy chains and two identical light chains. Antibodies function by specifically binding to an antigen, and the variable region of the heavy and light chains determines the epitope specificity. Therefore, sequencing variable regions should be the focus of antibody development projects. The variable regions of heavy and light chains are primarily determined by V(D)J rearrangements,76 and mutation makes the sequences more diverse. In brief, the variable region of the heavy chain is a fusion gene product that randomly assembles three different groups of fragments, while the light chain only fuses two groups of gene fragments in the corresponding region. The resulting gene products have highly mutated regions that play critical roles in antigen binding, known as CDR1, CDR2, and CDR3, while the parts in between are called framework regions.77
Sequencing immunoglobulin transcripts has been made possible by utilizing the characteristics of immunoglobulin gene sequences. The limited number of germline fragments makes it feasible to design primers to amplify the variable region.78,79 Initially, Sanger sequencing was used,80,81 but due to its inefficiency, it was quickly replaced by NGS. Bulk Ig-seq has been used to investigate the immune repertoires of different animals in various immune response situations.82–84 However, the linkage information between heavy and light chains needs to be recovered. The method of exhaustion, where light and heavy chains are randomly paired one by one and subsequently tested, can be employed to search for pairing information during antibody production.85,86 However, it is a costly and time-consuming process. Hence, maintaining the pairing information became the next obstacle to overcome.
Strategies involving microfluidics to physically link the nucleotide sequences of the variable regions of the heavy and light chains (VH and VL) were developed. Georgiou and colleagues employed a two-step strategy to obtain linked VH–VL sequences. Single B cells were captured and lysed within droplets generated by a microfluidic system, and the mRNAs were captured by oligo-dT beads within these droplets. After breaking the emulsion, the mRNA-captured beads were emulsified again with reagents for reverse transcription and overlap-extension PCR, resulting in the formation of the linked VH–VL sequences.87 This VH–VL sequence can either be directly sequenced87 or cloned into yeast for rounds of display selection.88 However, it's important to note that these strategies are complex and can only be effectively managed in a limited number of labs.
Single-cell RNA-seq offers an alternative method for preserving the pairing information of heavy and light chains. By barcoding the transcripts from a single cell, it becomes straightforward to retain the pairing information of heavy and light chain transcripts.89 In addition, factual antibody clone type information is acquired, which helps to distinguish antigen-specific antibodies.90 Moreover, the transcriptome information from the same cell is obtained alongside immunoglobulin sequence information. This information has been used to reduce false positives in the search for antigen-specific antibodies. After infection or vaccination, the activated B cell types and developmental stages are diverse at different time points and in different organisms. Transcriptome combined with BCR sequence information has been used to help researchers focus on specific B cell subgroups and exclude others.85 In finding the neutral antibodies from COVID-19 patients, Jiang and colleagues used the transcriptome to mark activated B cells and plasma cells, and used this information along with BCR information to find potential neutralizing antibodies.48
Phenotypic B cell screening for discovery of antigen-specific mAbs
Since the advent of the first single-cell RNA-seq study, various strategies and workflows have been adopted in antibody development projects. The selection of a specific strategy is determined by the characteristics of the cells, antigens, and sampling time after infection or vaccination. These factors must be carefully considered to ensure that the most effective and efficient approach is chosen for each individual project.Phenotypic B cell screening involves characterizing the antigen specificity of the B cells. By doing this, one can select the cells of interest for downstream sequencing and generate antigen-binding mAbs in a more efficient manner. Different types of cells are involved in different phenotypic B cell screening strategies.
Memory B cells are a popular subject of study in antibody development projects. Since they have cell membrane-anchored antibodies, antigens modified with fluorophores can easily bind to them.91 This allows antigen-specific cells to be easily identified and isolated for sequencing in a microfluidic system. However, antigen-specific memory B cells might be rare in the peripheral blood mononuclear cell (PBMC) population,92 particularly when sampling occurs long after infection, depending on the disease. In such situations, fluorescence-activated cell sorting (FACS) followed by Smart-seq2 has been widely used.93 This workflow does not require the enrichment of BCR sequences and is a more economical choice than higher-throughput commercial kits. When antigen-specific B cells are present at higher frequencies, microfluidic-assisted strategies come into the spotlight. Fluorophore labeling can be performed before entering the microfluidic systems, making it compatible with both micro-wells and micro-droplet systems. One such strategy is LIBRA-seq,94 which involves dual-labeling multiple types of antigens with fluorophores and different DNA oligos. Antigen-specific B cells are enriched by FACS/MACS, and DNA oligo can mark B cells that bind to different antigens. The advantage of this technology is obvious: multiple antigens can be screened in a single run, and due to the characteristics of the droplet-based strategy, the throughput is high.
In the acute phase of infection or shortly after vaccination, disease-related plasma cells increase and should be investigated.95 However, determining antigen-specificity becomes more challenging without the presence of membrane-anchored antibodies. Intracellular staining of antigens followed by FACS is a straightforward approach that has been used in some research.96,97 However, cell membrane permeabilization and standard fixation with formaldehyde substantially affect RNA quality and increase subsequent sequencing difficulties.98 Alternatively, visualization of antigen-binding capacity can be accomplished by microfluidic systems. Single cells can be separated in microwells, and after culture, secreted antibodies can be captured on a surface by antigen or non-specific antibody binding protein.99 Following a procedure similar to the immunofluorescence technique, a fluorescence-labeled secondary antibody or antigen can be detected, and the antigen specificity of the cells can be identified.100,101 In droplet-based systems, fluorescence co-localization information of the antigen and antibody in droplets has been used to identify cells of interest.102 Single antibody-secreting cells can be separated into droplets with VHH anti-mouse κ light chain coated magnetic beads, antigen, and fluorescent secondary antibody. After a period of culture, the secreted antibody can be captured on magnetic beads and labeled with fluorescence. If the antigen fluorescently co-localizes with the secondary antibody instead of being evenly distributed throughout the droplet, the cell will be recognized as antigen-specific and sorted for downstream sequencing. Additionally, physically linking secreting type IgG to the membrane has been proposed in flow cytometry research.103
Certain antigens are known to non-specifically bind to other proteins. This can make sorting antigen-specific B cells using fluorescently labeled antigens challenging. To solve this problem, microfluidic-assisted single-cell sequencing can be combined with other techniques to identify antigen-specific cells. Based on the high throughput characteristics of microfluidic-assisted single-cell sequencing, a large antibody sequence library can be generated from animal B cells. Antigen-specificity identification can be accomplished by performing antigen-specific antibody pull-down from serum followed by mass spectrometry analysis.86,104,105 This approach can tolerate harsher washing conditions and may solve the problem of non-specific binding. Additionally, the antibody serum level can be determined, helping researchers assess which antibodies should be considered as candidates.106 Moreover, antibody binding affinity can be roughly measured by gradient acid washing the antibody binding to beads.107 However, access to a mass spectrometer and the need for proteomic experts must be considered. Fig. 3 provides a visual summary of the various strategies employed to access sequence information of specific antibodies from blood samples.
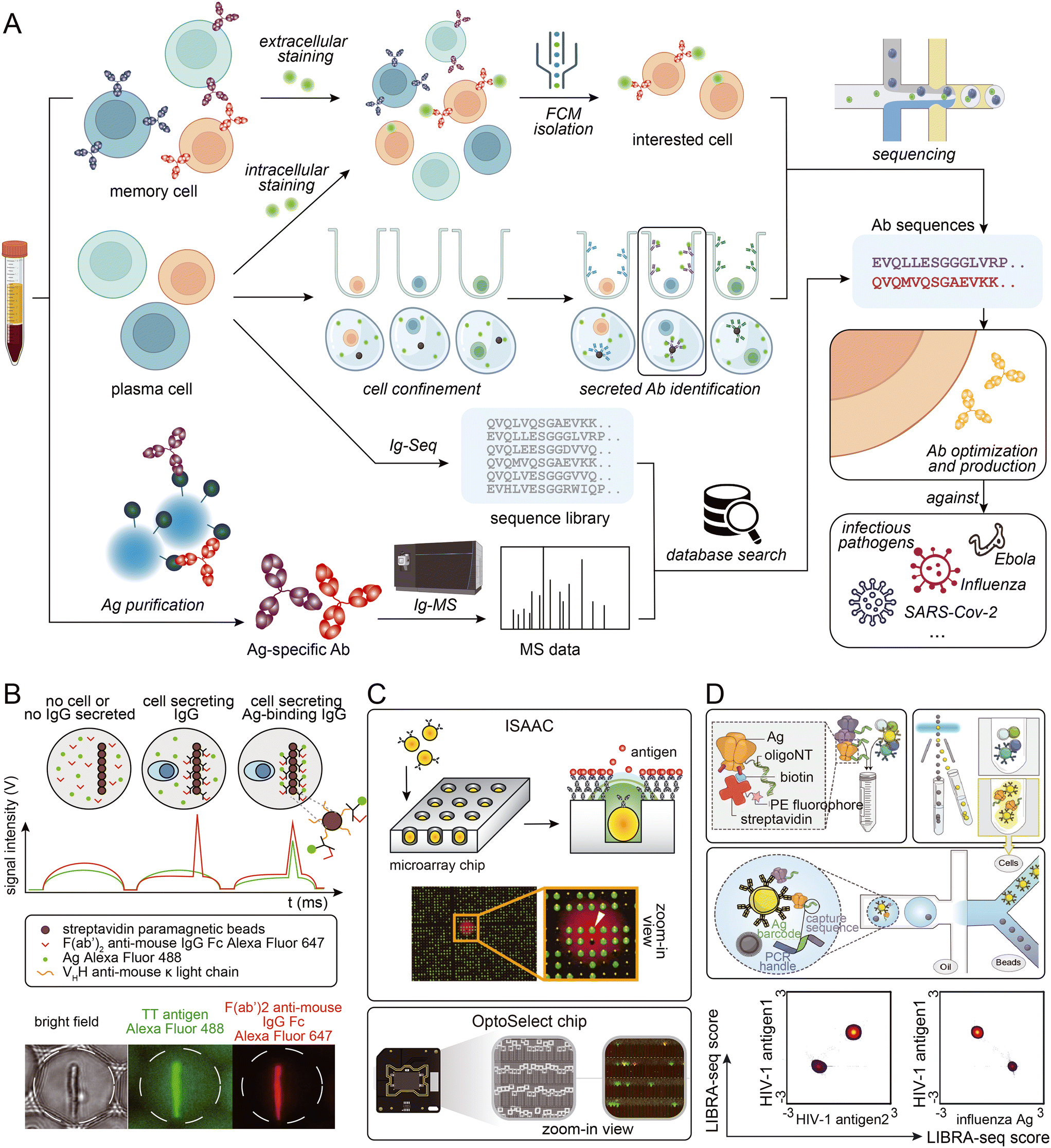 | ||
| Fig. 3 (A) Schematic presentation of strategies employed to access Ab sequence information from biological samples. (B) Phenotypic screening and sorting of IgG-secreting cells: binding assay in droplets, which were scanned one-by-one as they pass the laser line (top), and bright field and epifluorescence micrographs of a sorted droplet containing a splenocyte secreting a TT-binding IgG (bottom). Adapted from ref. 102 with permission from Springer Nature, copyright 2020. (C) Detection and retrieval of single antigen-specific antibody-secreting cells (ASCs) on an “immunospot array assay on a chip” (ISAAC; top; reproduced from ref. 100 with permission from Springer Nature, copyright 2009), and clone of single B cells and function characterization using multiple assays on an OptoSelect Chip (bottom; adapted from ref. 111 with permission from Berkeley Lights). (D) Schematic of the LIBRA-seq assay: fluorescently labeled, DNA-barcoded antigens (top left) are used to sort antigen-positive B cells (top right) before co-encapsulation of single B cells with bead-delivered oligos using droplet microfluidics (middle). For each B cell, the LIBRA-seq scores for each pair of antigens were plotted. Adapted from ref. 94 with permission from Elsevier, copyright 2019. | ||
Selecting candidates for further neutralizing tests is the next step. This selection process is not trivial and requires human experience, as the abundance of specific immunoglobulin gene sequences does not necessarily correlate with neutralizing efficacy. It is often the case that the strongest neutralizing antibody comes from small clones, as clonal size is determined by the stimulation and proliferation of B cells, which is linked to the binding capacity of the BCR with the antigen, rather than the neutralizing capability. Besides, the length of the CDR3 region, the frequency of somatic hypermutations, and other factors also affect the choice of candidates for further validation.
Once the candidates are identified, the antibodies can be expressed from synthesized DNA fragments that contain light chains and matched heavy chains. These synthesized antibodies will then undergo a set of stringent assays before clinical trials. Firstly, an affinity test to assess the binding capacity of the antibody is crucial, as phenotypic B cell screening and Ig-MS mentioned earlier may produce some false-positive signals. This process is typically qualitatively or semi-quantitatively evaluated using enzyme-linked immunosorbent assay (ELISA) and quantitatively assessed using surface plasmon resonance (SPR).108 Since binding affinity is not inherently linked to neutralizing capacity, it is essential to conduct neutralization assays using pseudo-viruses or actual viruses at the cellular level and further in vivo tests in animals.109 Once a candidate is selected, further optimization is pursued to enhance the affinity, neutralizing capacity, and durability110 of the mAbs.
Single-cell RNA-seq facilitates the discovery of neutralizing mAbs against COVID-19
With the integration of single-cell sequencing in the development of new mAbs, many mAbs have been developed using this approach. Due to its technical simplicity, FACS-based antigen-specific memory B cell sequencing, as shown at the top of Fig. 3A, has become popular. mAbs against diseases such as Ebola,112 HIV,113 and influenza114 have been successfully developed using this workflow. Additionally, droplet-based sequencing methods have been used to generate mAbs against infectious diseases such as influenza90 and dengue virus.115Typically, a patient who has recently recovered from COVID-19 will possess a sufficient concentration of effective antibodies in his blood.116 Anti-COVID-19 treatment using plasma from convalescent patients is considered effective, but plasma batch effects are severe, and sources are limited. Among the diversified antibodies in convalescent patients' serum, neutralizing antibodies are responsible for blocking the viruses from invading cells. Fishing for high-efficacy neutralizing antibodies from COVID-19 convalescents has been ongoing since the beginning of the pandemic.117 This is a rational approach that takes advantage of the natural immune response, as recovery from the disease somewhat proves the efficacy of the possible targeting antibodies. Although traditional large-volume cell culture systems can produce kg-scale monoclonal antibodies at a time, the throughput of functional screening is difficult to scale up.118 This is further complicated when the pathogen undergoes continuous mutation, potentially compromising the ability of antibodies to suppress it and necessitating renewed antibody development and an increase in the amount of time required to combat the pathogen.
The sudden outbreak of COVID-19 has provided a battleground for the single-cell RNA-seq approach to revolutionize the conventional process of mAb screening and selection. This novel approach, which eliminates the need for harvesting immunized spleen cells or generating hybridoma cells, saves both time and labor. As mentioned in the previous section, identifying antigen-specific plasma cells, which lack BCRs that can bind the RBD of S protein, is more challenging than identifying memory cells. Hence, most applied research focuses on memory B-cells. Using RBD-tethered magnetic beads or fluorescence-labeled RBD, memory B-cells with strong binding affinity to the RBD can be separated from other cells. Subsequently, single B-cells enriched from convalescents can be sequenced to analyze their immunoglobulin gene sequences, which are the binding antibodies. After the neutralizing test and optimization steps mentioned in the previous section, neutralizing mAbs can be produced and then proceed to animal-level safety and pharmacokinetic experiments.
The development of novel neutralizing antibodies has seen numerous successful attempts that were facilitated by single-cell RNA-seq. The most prevalent and popular methods currently in use are workflow strategies that focus on the selection of antigen-specific B cells through the interaction of antigens with the BCR, as illustrated in the upper portion of Fig. 3A. For example, during the initial stages of the COVID-19 pandemic, Xie and his collaborators employed high-throughput single-cell sequencing to sequence B cells from 60 convalescents and discovered the most potent antibody, BD-368-2, which demonstrated both highly therapeutic and prophylactic efficacy in SARS-CoV-2 infected mice.119 In this research, a biotinylated RBD and S protein were employed to isolate antigen-specific B cells through magnetic bead separation. This was followed by single-cell RNA-seq to obtain BCR sequences, a method that closely resembles the upper pathway illustrated in Fig. 3A, albeit with the use of biotinylated antigens instead of fluorescently labeled ones. Subsequently, Qin and collaborators discovered that SARS-CoV-2 neutralizing antibodies could bind to different effective epitopes without interfering with each other's function. This led to the development of a cocktail design.120 However, as anticipated, these antibodies were all escaped by currently prevalent variants. Despite the identification of an increasing number of antibodies with strong neutralizing functions, most become ineffective within months. Recently, Xie and collaborators published an article that provides a comprehensive overview of the selection of epitopes, mutation prediction and convergence, and predictable escapes of the existing SARS-CoV-2 neutralizing antibodies (Fig. 4).26 This extensive dataset offers valuable insights for the design or targeted search of a broad-spectrum neutralizing antibody capable of binding to most, if not all, variants of SARS-CoV-2. Corti and colleagues successfully employed biotinylated SARS-CoV-2 S ectodomain trimers in combination with fluorescent streptavidin to identify the mAb known as S2X35.121 The LIBRA-seq technique, which involves the dual-labeling of multiple types of antigens with fluorophores and distinct DNA oligonucleotides as previously mentioned, was utilized by Chen and her collaborators based on a droplet microfluidic system in antibody discovery. This led to the successful isolation of two antibodies against the RBD of SARS-CoV-2.117
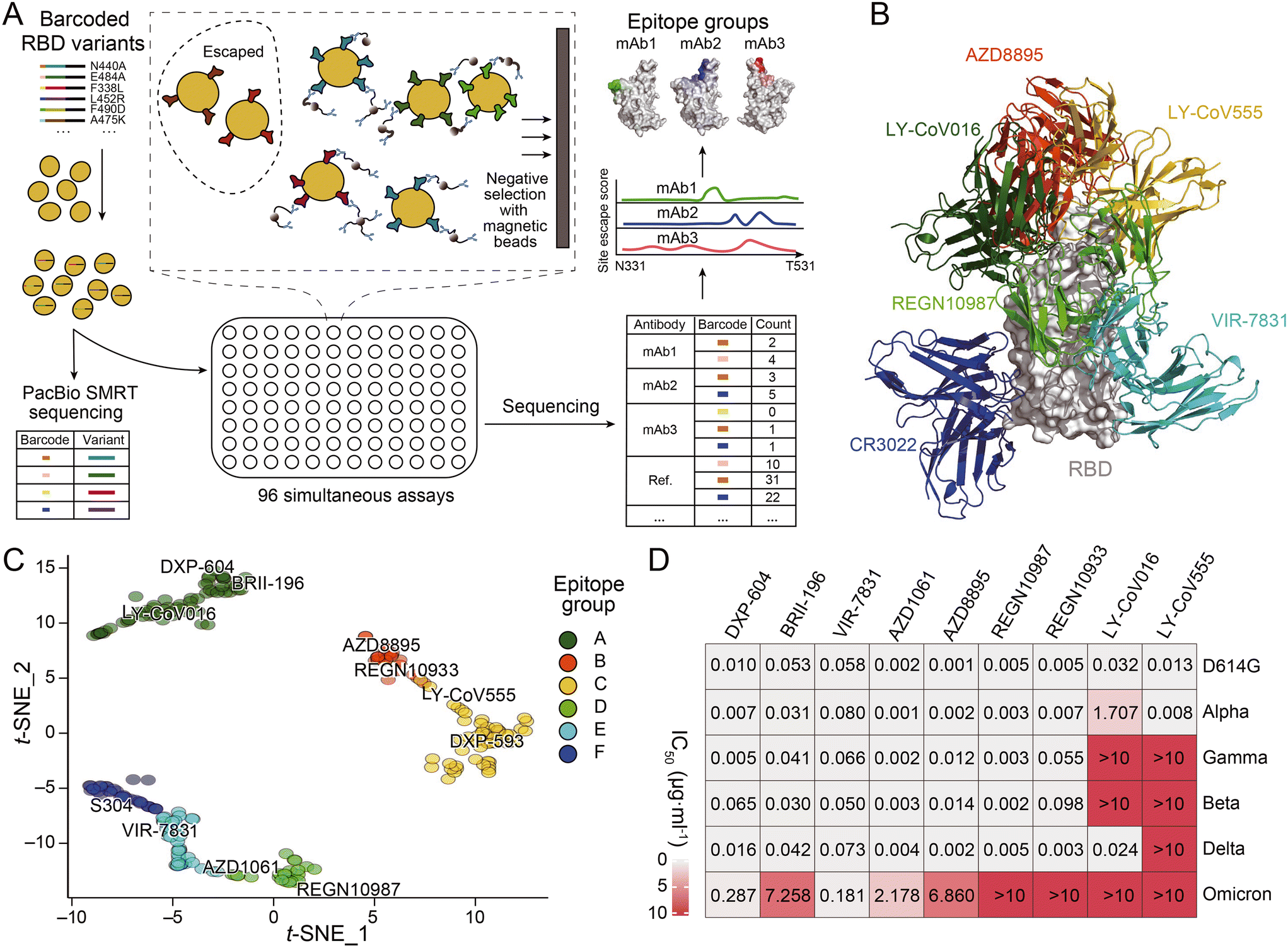 | ||
| Fig. 4 (A) Schematic of MACS-based high-throughput yeast display mutation screening. (B) Representative antibody structures of each epitope group. (C) t-distributed stochastic neighbor embedding (t-SNE) and unsupervised clustering of SARS-CoV-2 human neutralizing antibodies on the basis of each antibody escaping mutation profile. A total of six epitope groups (groups A–F) could be defined. (D) Neutralization of SARS-CoV-2 variants of concern (pseudotyped VSV) by nine neutralizing-antibody-based drugs. Adapted from ref. 26 with permission from Springer Nature, copyright 2021. | ||
Several research groups have focused on the isolation of SARS-CoV-2 antibodies from plasma cells or plasmablasts, as depicted in the central section of Fig. 3A. One particular study employed single-cell sequencing of plasma cells in combination with mammalian display techniques to generate SARS-CoV-2 antibodies. The identification of expanded plasma cell lineages served as a crucial criterion for selecting candidates for mammalian display and subsequent functional characterization.122 Furthermore, strategies involving the cultivation of individual plasma cells and the assessment of their secreted antibodies123 have been applied in the development of SARS-CoV-2 mAbs. Notably, the discovery of mAbs S2H13 and S2H14 is a prime example of this approach.121
Ig-MS, a workflow represented at the bottom in Fig. 3A, as also played a role in the development of SARS-CoV-2 antibodies. Qiu and colleagues used affinity chromatography followed by LC-MS/MS to identify sequences of SARS-CoV-2 antibodies.124 They used four B cell epitope peptides for affinity purification of serum antibodies, and epitope-specific mAbs were obtained from the sera of 15 patients.
Despite the accelerated process facilitated by microfluidic-assisted single-cell RNA-seq, numerous challenges persist. A significant challenge lies in the selection of COVID-19 convalescents. Two primary considerations exist: tracking viral mutations and identifying or designing a broad-spectrum antibody that remains effective despite variant differences. It is crucial for researchers to continue working with convalescents of different variants, as it has been found that an effective neutralizing antibody can rapidly lose its potency as a therapeutic due to escape.125 Many of the previously developed and now obsolete antibodies have been reviewed,126,127 leading to a more profound understanding of epitopes, especially those on the RBD, than at the onset of the pandemic. This knowledge is invaluable in the ongoing pursuit of effective treatments for COVID-19.
What have we learned from these results, and what can we do next?
The development of single-cell sequencing methods has been critical in the discovery of new monoclonal antibodies. Single-cell sequencing enables the preservation of paired sequence information, which is essential for sequencing-and-synthesis workflows. In addition, transcriptome information from the same cell can assist in reducing false positives. Microfluidic-assisted single-cell RNA-seq has made it possible to sequence a large number of B cells and acquire a paired antibody library. With advances in microfluidic technology, memory B cells for multiple antigens can be screened in a single run, and antigen-specific antibody-secreting cells can be distinguished and sequenced on chips. Microfluidic-assisted single-cell RNA-seq has indeed accelerated the development cycle of antibodies. However, the rapid mutation rate of SARS-CoV-2 results in continuous antibody escape, necessitating quick tracking of mutations and further shortening the antibody development cycle.The cost of microfluidic-assisted single-cell RNA-seq can limit the throughput of acquiring paired Ig sequences. Antigen-specific B cells are rare in the B cell population,92 and even antibodies bound to the same antigen can exhibit different epitopes and non-specific binding levels.128 Increasing the throughput can improve the chances of obtaining antibodies with satisfactory performance. While designing and constructing microfluidic systems may not pose a significant challenge for certain labs, most antibody development groups rely on highly integrated commercial kits. These one-stop solutions, such as the microwell-based Beacon system from Berkeley Lights, the droplet-based system CelliGO from HiFiBiO Therapeutics, or the Chromium Single Cell Immune Profiling Solution from 10x Genomics, employed different strategies but share one common characteristic: their high cost.
After the sequencing experiment, candidates should be selected from the sequencing results for downstream synthesis. Even using an ingenious microfluidic system to select antigen-specific B cells, this process still requires a significant amount of expert guidance and human experience.90 As previously mentioned, certain characteristics of the immunoglobulin sequence, such as V-gene frequency, hypermutation rates, CDR3 length, and lineage tracing information combined with transcriptome clustering results, are still used to assess the potential of the immunoglobulin sequence. However, clear criteria and workflows are still needed to streamline this process.
In order to optimize the antibody development workflow, it is crucial to focus not only on the sequencing process itself, but also on the level of integration of sequencing with upstream and downstream steps. The development of a more integrated microfluidic system for antibody discovery is a worthwhile endeavor. By integrating antibody characterization units into a microfluidic-assisted single-cell sequencing system, cells with higher potential for downstream sequencing can be retrieved, thereby improving the efficiency and effectiveness of antibody development. Antibody characterization encompasses many aspects. Antibodies can perform functions through neutralization, opsonization, and complement activation, with binding to antigens being the essential requirement of these pathways. The affinity level of antibodies can be estimated by continuously observing the speed at which fluorescently labeled secreted antibodies and antigens move toward antibody-capturing beads.129,130 Competitive assays have also been used by monitoring the competitiveness of newly added fluorescently labeled antigens compared to pre-bound antigens.131 Cross-reactivity testing can be easily achieved by adding antigens with different fluorescence. Functional assays, such as testing an antibody's capability to inhibit bacterial growth, have also been developed.132 However, in many situations, including COVID-19, the neutralization function of the antibody is the most useful. Only antibodies that bind to certain regions of the virus can provide protection,128 so it is crucial to integrate an epitope testing unit into the workflow in the future. Moreover, a multi-dimension functional screen might further expedite the antibody development process by more precisely measuring a cell's potential.
An antibody expression microfluidic system can accelerate the antibody discovery cycle in rapid verification of candidates' binding capabilities. A significant challenge in the antibody development cycle is the decoupling between sequencing and rapid antibody synthesis, which hampers the verification steps. Traditionally, gene synthesis and bacterial cloning are required to construct plasmids for antibody expression. However, this workload becomes substantial when the number of candidates is significant. Berkeley Lights has launched a new Opto BCR Rapid Re-expression kit that enables the enrichment of BCR sequences from reverse transcription cDNA and the construction of expression vectors without prior knowledge of the candidate sequences. This strategy allows for parallel NGS and rapid candidate re-expression. However, this kit is off-chip, and further effort may be needed to integrate it into its microfluidic system. Within academic circles, efforts have been made to explore the use of cell-free protein synthesis microfluidic systems for the rapid verification of mAb binding capacity. Jacková and colleagues developed a strategy that employs a droplet system containing VHH-coding DNA, fluorescent antigen, cell-free expression components, and a VHH capture scaffold to assess the antigen binding capacity of VHH. Following a 3-hour incubation, the aggregation status of the fluorescence signal can be used as an indicator to evaluate the binding capacity of mAbs.133 In the future, efforts should be made to reduce the setup and operating complexity of microfluidic systems to more conveniently obtain a large number of cells of interest for single-cell sequencing library construction. Reducing the cost of sequencing is also important to expand sequencing throughput. As a result, the antibody sequence pool can be larger, increasing the likelihood of obtaining potential candidates. Moreover, microfluidic systems have high integration potential, allowing for the integration of multi-functional characterization units to select cells with high potential and alleviate the throughput pressure of sequencing and antibody synthesis. With advances in microfluidic systems, the time required to identify cells of interest, perform sequencing, and construct expression vectors can be further compressed. However, antibody expression via cell culture takes several days due to cellular metabolism. Therefore, cell-free antibody expression systems may warrant further exploration.
The battle against infectious diseases is an ongoing arms race, with monoclonal antibodies serving as a crucial weapon for humanity in treating acute infections and critical illnesses. As viruses mutate at an increasingly rapid pace, the iteration of antibody drugs is necessary to keep up. Microfluidic-assisted single-cell sequencing technology has already played a significant role in developing antibodies against viruses and will undoubtedly continue to do so in the future. This technique offers a powerful tool for accelerating the development of effective treatments and staying ahead in the fight against infectious diseases.
Concluding remarks
The COVID-19 pandemic has completely reshaped our lives and presented challenges for researchers in the field of antibody development. The time frame for developing novel and effective neutralizing antibodies is narrow, as mutated virus variants can quickly escape the antibody's effects. Single-cell sequencing has introduced a new possibility for accelerating the speed of antibody development, with the introduction of the microfluidic-assisted droplet isolation and barcoding process providing a ready tool. The effectiveness of such an approach has been demonstrated over the past three years, generating various neutralizing monoclonal antibodies against different variants of the coronavirus. This approach is a prime example of how the integration of engineering, chemistry, biology, and bioinformatics can shift paradigms in medical research. Data collected and expanded rapidly through high-throughput experiments has become a valuable resource for improving our understanding of the evolutionary characteristics of viruses and the possible design strategies of wide-spectrum neutralizing monoclonal antibodies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (NSFC T2225005, 22050004, 21927802, 21974069), the Ministry of Science and Technology of the People's Republic of China (2018YFA0800200) and the Open Fund Programs of Shenzhen Bay Laboratory (SZBL2020090501001). The authors would like to express their gratitude to Dr. Lu Liu and Dr. Lin Di for demonstrating certain experimental details involved in techniques discussed in this review and for providing valuable comments on the manuscript.References
- W. R. Strohl, Z. Ku, Z. An, S. F. Carroll, B. A. Keyt and L. M. Strohl, BioDrugs, 2022, 36, 231–323 CrossRef CAS PubMed
.
- E. Stadler, M. T. Burgess, T. E. Schlub, S. R. Khan, K. L. Chai, Z. K. McQuilten, E. M. Wood, M. N. Polizzotto, S. J. Kent, D. Cromer, M. P. Davenport and D. S. Khoury, Nat. Commun., 2023, 14, 4545 CrossRef CAS PubMed
- M. Sadarangani, A. Marchant and T. R. Kollmann, Nat. Rev. Immunol., 2021, 21, 475–484 CrossRef CAS PubMed
- J. S. K. Teh, J. Coussement, Z. C. F. Neoh, T. Spelman, S. Lazarakis, M. A. Slavin and B. W. Teh, Blood Adv., 2022, 6, 2014–2034 CrossRef CAS PubMed
- D. A. Collier, I. A. T. M. Ferreira, P. Kotagiri, R. P. Datir, E. Y. Lim, E. Touizer, B. Meng, A. Abdullahi, S. Baker, G. Dougan, C. Hess, N. Kingston, P. J. Lehner, P. A. Lyons, N. J. Matheson, W. H. Owehand, C. Saunders, C. Summers, J. E. D. Thaventhiran, M. Toshner, M. P. Weekes, P. Maxwell, A. Shaw, A. Bucke, J. Calder, L. Canna, J. Domingo, A. Elmer, S. Fuller, J. Harris, S. Hewitt, J. Kennet, S. Jose, J. Kourampa, A. Meadows, C. O’Brien, J. Price, C. Publico, R. Rastall, C. Ribeiro, J. Rowlands, V. Ruffolo, H. Tordesillas, B. Bullman, B. J. Dunmore, S. Fawke, S. Gräf, J. Hodgson, C. Huang, K. Hunter, E. Jones, E. Legchenko, C. Matara, J. Martin, F. Mescia, C. O’Donnell, L. Pointon, N. Pond, J. Shih, R. Sutcliffe, T. Tilly, C. Treacy, Z. Tong, J. Wood, M. Wylot, L. Bergamaschi, A. Betancourt, G. Bower, C. Cossetti, A. De Sa, M. Epping, S. Fawke, N. Gleadall, R. Grenfell, A. Hinch, O. Huhn, S. Jackson, I. Jarvis, B. Krishna, D. Lewis, J. Marsden, F. Nice, G. Okecha, O. Omarjee, M. Perera, M. Potts, N. Richoz, V. Romashova, N. S. Yarkoni, R. Sharma, L. Stefanucci, J. Stephens, M. Strezlecki, L. Turner, E. M. D. D. De Bie, K. Bunclark, M. Josipovic, M. Mackay, A. Michael, S. Rossi, M. Selvan, S. Spencer, C. Yong, A. Ansaripour, A. Michael, L. Mwaura, C. Patterson, G. Polwarth, P. Polgarova, G. di Stefano, C. Fahey, R. Michel, S.-H. Bong, J. D. Coudert, E. Holmes, J. Allison, H. Butcher, D. Caputo, D. Clapham-Riley, E. Dewhurst, A. Furlong, B. Graves, J. Gray, T. Ivers, M. Kasanicki, E. Le Gresley, R. Linger, S. Meloy, F. Muldoon, N. Ovington, S. Papadia, I. Phelan, H. Stark, K. E. Stirrups, P. Townsend, N. Walker, J. Webster, A. Elmer, N. Kingston, B. Graves, E. Le Gresley, D. Caputo, L. Bergamaschi, K. G. C. Smith, J. R. Bradley, L. Ceron-Gutierrez, P. Cortes-Acevedo, G. Barcenas-Morales, M. A. Linterman, L. E. McCoy, C. Davis, E. Thomson, P. A. Lyons, E. McKinney, R. Doffinger, M. Wills and R. K. Gupta, Nature, 2021, 596, 417–422 CrossRef CAS PubMed
- C. Milito, F. Cinetto, A. Palladino, G. Garzi, A. Punziano, G. Lagnese, R. Scarpa, M. Rattazzi, A. M. Pesce, F. Pulvirenti, G. Di Napoli, G. Spadaro, R. Carsetti and I. Quinti, Biomedicines, 2022, 10, 1026 CrossRef CAS PubMed
- P. C. Robinson, D. F. L. Liew, H. L. Tanner, J. R. Grainger, R. A. Dwek, R. B. Reisler, L. Steinman, M. Feldmann, L.-P. Ho, T. Hussell, P. Moss, D. Richards and N. Zitzmann, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2119893119 CrossRef PubMed
- M. A. Villasis-Keever, M. G. López-Alarcón, G. Miranda-Novales, J. N. Zurita-Cruz, A. S. Barrada-Vázquez, J. González-Ibarra, M. Martínez-Reyes, C. Grajales-Muñiz, C. E. Santacruz-Tinoco, B. Martínez-Miguel, J. Maldonado-Hernández, Y. Cifuentes-González, M. Klünder-Klünder, J. Garduño-Espinosa, B. López-Martínez and I. Parra-Ortega, Arch. Med. Res., 2022, 53, 423–430 CrossRef CAS PubMed
- B. S. Abella, E. L. Jolkovsky, B. T. Biney, J. E. Uspal, M. C. Hyman, I. Frank, S. E. Hensley, S. Gill, D. T. Vogl, I. Maillard, D. V. Babushok, A. C. Huang, S. D. Nasta, J. C. Walsh, E. P. Wiletyo, P. A. Gimotty, M. C. Milone, R. K. Amaravadi and Prevention and Treatment of COVID-19 With Hydroxychloroquine Investigators, JAMA Intern. Med., 2021, 181, 195–202 CrossRef CAS PubMed
- S. Lei, X. Chen, J. Wu, X. Duan and K. Men, Signal Transduction Targeted Ther., 2022, 7, 387 CrossRef CAS PubMed
- M. G. Santoro and E. Carafoli, Biochem. Biophys. Res. Commun., 2021, 538, 145–150 CrossRef CAS PubMed
- D. E. Malden, V. Hong, B. J. Lewin, B. K. Ackerson, M. Lipsitch, J. A. Lewnard and S. Y. Tartof, Morb. Mortal. Wkly. Rep., 2022, 71, 830–833 CrossRef CAS PubMed
- H. Ledford, Nature, 2021, 591, 513–514 CrossRef CAS PubMed
- D. M. Ecker, S. D. Jones and H. L. Levine, mAbs, 2015, 7, 9–14 CrossRef CAS PubMed
- A. M. Scott, J. D. Wolchok and L. J. Old, Nat. Rev. Cancer, 2012, 12, 278–287 CrossRef CAS PubMed
-
S. Kotsovilis and E. Andreakos, Human Monoclonal Antibodies: Methods Protocols, 2014, pp. 37–59 Search PubMed
- O. Tshiani Mbaya, P. Mukumbayi and S. Mulangu, Front. Immunol., 2021, 12, 721328 CrossRef PubMed
- G. D. Victora and M. C. Nussenzweig, Annu. Rev. Immunol., 2012, 30, 429–457 CrossRef CAS PubMed
- D. C. Ekiert, G. Bhabha, M.-A. Elsliger, R. H. Friesen, M. Jongeneelen, M. Throsby, J. Goudsmit and I. A. Wilson, Science, 2009, 324, 246–251 CrossRef CAS PubMed
- M. Guo, M. Xiong, J. Peng, T. Guan, H. Su, Y. Huang, C. G. Yang, Y. Li, D. Boraschi, T. Pillaiyar, G. Wang, C. Yi, Y. Xu and C. Chen, Natl. Sci. Rev., 2023, 10, nwad161 CrossRef PubMed
- F. A. Lempp, L. B. Soriaga, M. Montiel-Ruiz, F. Benigni, J. Noack, Y.-J. Park, S. Bianchi, A. C. Walls, J. E. Bowen, J. Zhou, H. Kaiser, A. Joshi, M. Agostini, M. Meury, E. Dellota, S. Jaconi, E. Cameroni, J. Martinez-Picado, J. Vergara-Alert, N. Izquierdo-Useros, H. W. Virgin, A. Lanzavecchia, D. Veesler, L. A. Purcell, A. Telenti and D. Corti, Nature, 2021, 598, 342–347 CrossRef CAS PubMed
- M. McCallum, A. De Marco, F. A. Lempp, M. A. Tortorici, D. Pinto, A. C. Walls, M. Beltramello, A. Chen, Z. Liu, F. Zatta, S. Zepeda, J. di Iulio, J. E. Bowen, M. Montiel-Ruiz, J. Zhou, L. E. Rosen, S. Bianchi, B. Guarino, C. S. Fregni, R. Abdelnabi, S.-Y. C. Foo, P. W. Rothlauf, L.-M. Bloyet, F. Benigni, E. Cameroni, J. Neyts, A. Riva, G. Snell, A. Telenti, S. P. J. Whelan, H. W. Virgin, D. Corti, M. S. Pizzuto and D. Veesler, Cell, 2021, 184, 2332–2347.e16 CrossRef CAS PubMed
- A. Vaisman-Mentesh, M. Gutierrez-Gonzalez, B. J. DeKosky and Y. Wine, Front. Immunol., 2020, 11, 1951 CrossRef CAS PubMed
- F. A. Harding, M. M. Stickler, J. Razo and R. B. Dubridge, mAbs, 2010, 2, 256–265 CrossRef PubMed
- E. Sparrow, M. Friede, M. Sheikh and S. Torvaldsen, Bull. W. H. O., 2017, 95, 235 CrossRef PubMed
- Y. Cao, J. Wang, F. Jian, T. Xiao, W. Song, A. Yisimayi, W. Huang, Q. Li, P. Wang, R. An, J. Wang, Y. Wang, X. Niu, S. Yang, H. Liang, H. Sun, T. Li, Y. Yu, Q. Cui, S. Liu, X. Yang, S. Du, Z. Zhang, X. Hao, F. Shao, R. Jin, X. Wang, J. Xiao, Y. Wang and X. S. Xie, Nature, 2021, 602, 657–663 CrossRef PubMed
- K. E. Kip, E. K. McCreary, K. Collins, T. E. Minnier, G. M. Snyder, W. Garrard, J. C. McKibben, D. M. Yealy, C. W. Seymour, D. T. Huang, J. R. Bariola, M. Schmidhofer, R. J. Wadas, D. C. Angus, P. L. Kip and O. C. Marroquin, Ann. Intern. Med., 2023, 176, 496–504 CrossRef PubMed
- M. Imai, M. Ito, M. Kiso, S. Yamayoshi, R. Uraki, S. Fukushi, S. Watanabe, T. Suzuki, K. Maeda, Y. Sakai-Tagawa, K. Iwatsuki-Horimoto, P. J. Halfmann and Y. Kawaoka, N. Engl. J. Med., 2023, 388, 89–91 CrossRef PubMed
- K. Sariahmed, M. LaRochelle and B. P. Linas, Health Affairs Forefront, 2023.
- Triad National Security, LLC for the U.S. Department of Energy's National Nuclear Security Administration. COVID-19 Viral Genome Analysis Pipeline, https://cov.lanl.gov.
- J. W. Goding, J. Immunol. Methods, 1980, 39, 285–308 CrossRef CAS PubMed
- B. Kelley, Nat. Biotechnol., 2020, 38, 540–545 CrossRef CAS PubMed
- F. Tang, C. Barbacioru, Y. Wang, E. Nordman, C. Lee, N. Xu, X. Wang, J. Bodeau, B. B. Tuch, A. Siddiqui, K. Lao and M. A. Surani, Nat. Methods, 2009, 6, 377–382 CrossRef CAS PubMed
- A. M. Klein, L. Mazutis, I. Akartuna, N. Tallapragada, A. Veres, V. Li, L. Peshkin, D. A. Weitz and M. W. Kirschner, Cell, 2015, 161, 1187–1201 CrossRef CAS PubMed
- E. Z. Macosko, A. Basu, R. Satija, J. Nemesh, K. Shekhar, M. Goldman, I. Tirosh, A. R. Bialas, N. Kamitaki, E. M. Martersteck, J. J. Trombetta, D. A. Weitz, J. R. Sanes, A. K. Shalek, A. Regev and S. A. McCarroll, Cell, 2015, 161, 1202–1214 CrossRef CAS PubMed
- G. X. Y. Zheng, J. M. Terry, P. Belgrader, P. Ryvkin, Z. W. Bent, R. Wilson, S. B. Ziraldo, T. D. Wheeler, G. P. McDermott, J. Zhu, M. T. Gregory, J. Shuga, L. Montesclaros, J. G. Underwood, D. A. Masquelier, S. Y. Nishimura, M. Schnall-Levin, P. W. Wyatt, C. M. Hindson, R. Bharadwaj, A. Wong, K. D. Ness, L. W. Beppu, H. J. Deeg, C. McFarland, K. R. Loeb, W. J. Valente, N. G. Ericson, E. A. Stevens, J. P. Radich, T. S. Mikkelsen, B. J. Hindson and J. H. Bielas, Nat. Commun., 2017, 8, 14049 CrossRef CAS PubMed
- X. Zhang, T. Li, F. Liu, Y. Chen, J. Yao, Z. Li, Y. Huang and J. Wang, Mol. Cell, 2019, 73, 130–142.e5 CrossRef PubMed
- T. Graf and M. Stadtfeld, Cell Stem Cell, 2008, 3, 480–483 CrossRef CAS PubMed
- L. Liu, Y. Li, S. Li, N. Hu, Y. He, R. Pong, D. Lin, L. Lu and M. Law, J. Biomed. Biotechnol., 2012, 2012, 251364 Search PubMed
- F. Buettner, K. N. Natarajan, F. P. Casale, V. Proserpio, A. Scialdone, F. J. Theis, S. A. Teichmann, J. C. Marioni and O. Stegle, Nat. Biotechnol., 2015, 33, 155–160 CrossRef CAS PubMed
- V. Svensson, R. Vento-Tormo and S. A. Teichmann, Nat. Protoc., 2018, 13, 599–604 CrossRef CAS PubMed
- R. Zilionis, J. Nainys, A. Veres, V. Savova, D. Zemmour, A. M. Klein and L. Mazutis, Nat. Protoc., 2017, 12, 44–73 CrossRef CAS PubMed
- K. Hosokawa, T. Fujii and I. Endo, Anal. Chem., 1999, 71, 4781–4785 CrossRef CAS
- T. A. Duncombe, A. M. Tentori and A. E. Herr, Nat. Rev. Mol. Cell Biol., 2015, 16, 554–567 CrossRef CAS PubMed
- X. Han, R. Wang, Y. Zhou, L. Fei, H. Sun, S. Lai, A. Saadatpour, Z. Zhou, H. Chen, F. Ye, D. Huang, Y. Xu, W. Huang, M. Jiang, X. Jiang, J. Mao, Y. Chen, C. Lu, J. Xie, Q. Fang, Y. Wang, R. Yue, T. Li, H. Huang, S. H. Orkin, G. C. Yuan, M. Chen and G. Guo, Cell, 2018, 172, 1091–1107.e17 CrossRef CAS PubMed
- 10xGenomics, https://www.10xgenomics.com.
- A. A. Kolodziejczyk and T. Lönnberg, Briefings Funct. Genomics, 2017, 17, 209–219 CrossRef PubMed
- F. Li, M. Luo, W. Zhou, J. Li, X. Jin, Z. Xu, L. Juan, Z. Zhang, Y. Li, R. Liu, Y. Li, C. Xu, K. Ma, H. Cao, J. Wang, P. Wang, Z. Bu and Q. Jiang, Protein Cell, 2020, 12, 751–755 CrossRef PubMed
- 10xGenomics, Chromium Single Cell V(D)J Reagent Kits User Guide, CG000086 Rev M.
- D. J. Collins, A. Neild, A. DeMello, A.-Q. Liu and Y. Ai, Lab Chip, 2015, 15, 3439–3459 RSC
- T. Kivioja, A. Vähärautio, K. Karlsson, M. Bonke, M. Enge, S. Linnarsson and J. Taipale, Nat. Methods, 2012, 9, 72–74 CrossRef CAS PubMed
- N. M. Negretti, E. J. Plosa, J. T. Benjamin, B. A. Schuler, A. C. Habermann, C. S. Jetter, P. Gulleman, C. Bunn, A. N. Hackett and M. Ransom, Development, 2021, 148, dev199512 CrossRef CAS PubMed
- H. Van Hove, L. Martens, I. Scheyltjens, K. De Vlaminck, A. R. Pombo Antunes, S. De Prijck, N. Vandamme, S. De Schepper, G. Van Isterdael, C. L. Scott, J. Aerts, G. Berx, G. E. Boeckxstaens, R. E. Vandenbroucke, L. Vereecke, D. Moechars, M. Guilliams, J. A. Van Ginderachter, Y. Saeys and K. Movahedi, Nat. Neurosci., 2019, 22, 1021–1035 CrossRef CAS PubMed
- K. J. Travaglini, A. N. Nabhan, L. Penland, R. Sinha, A. Gillich, R. V. Sit, S. Chang, S. D. Conley, Y. Mori, J. Seita, G. J. Berry, J. B. Shrager, R. J. Metzger, C. S. Kuo, N. Neff, I. L. Weissman, S. R. Quake and M. A. Krasnow, Nature, 2020, 587, 619–625 CrossRef CAS PubMed
- S. He, L.-H. Wang, Y. Liu, Y.-Q. Li, H.-T. Chen, J.-H. Xu, W. Peng, G.-W. Lin, P.-P. Wei, B. Li, X. Xia, D. Wang, J.-X. Bei, X. He and Z. Guo, Genome Biol., 2020, 21, 1–34 CrossRef PubMed
- H. Li, J. Janssens, M. De Waegeneer, S. S. Kolluru, K. Davie, V. Gardeux, W. Saelens, F. P. A. David, M. Brbić, K. Spanier, J. Leskovec, C. N. McLaughlin, Q. Xie, R. C. Jones, K. Brueckner, J. Shim, S. G. Tattikota, F. Schnorrer, K. Rust, T. G. Nystul, Z. Carvalho-Santos, C. Ribeiro, S. Pal, S. Mahadevaraju, T. M. Przytycka, A. M. Allen, S. F. Goodwin, C. W. Berry, M. T. Fuller, H. White-Cooper, E. L. Matunis, S. DiNardo, A. Galenza, L. E. O’Brien, J. A. T. Dow, F. C. A. Consortium, H. Jasper, B. Oliver, N. Perrimon, B. Deplancke, S. R. Quake, L. Luo and S. Aerts, Science, 2022, 375, eabk2432 CrossRef CAS PubMed
- S. Chevrier, J. H. Levine, V. R. T. Zanotelli, K. Silina, D. Schulz, M. Bacac, C. H. Ries, L. Ailles, M. A. S. Jewett, H. Moch, M. van den Broek, C. Beisel, M. B. Stadler, C. Gedye, B. Reis, D. Pe’er and B. Bodenmiller, Cell, 2017, 169, 736–749.e18 CrossRef CAS PubMed
- S. Cheng, Z. Li, R. Gao, B. Xing, Y. Gao, Y. Yang, S. Qin, L. Zhang, H. Ouyang, P. Du, L. Jiang, B. Zhang, Y. Yang, X. Wang, X. Ren, J.-X. Bei, X. Hu, Z. Bu, J. Ji and Z. Zhang, Cell, 2021, 184, 792–809.e23 CrossRef CAS PubMed
- L. Zheng, S. Qin, W. Si, A. Wang, B. Xing, R. Gao, X. Ren, L. Wang, X. Wu, J. Zhang, N. Wu, N. Zhang, H. Zheng, H. Ouyang, K. Chen, Z. Bu, X. Hu, J. Ji and Z. Zhang, Science, 2021, 374, abe6474 CrossRef PubMed
- H. J. Sutton, R. Aye, A. H. Idris, R. Vistein, E. Nduati, O. Kai, J. Mwacharo, X. Li, X. Gao, T. D. Andrews, M. Koutsakos, T. H. O. Nguyen, M. Nekrasov, P. Milburn, A. Eltahla, A. A. Berry, N. Kc, S. Chakravarty, B. K. L. Sim, A. K. Wheatley, S. J. Kent, S. L. Hoffman, K. E. Lyke, P. Bejon, F. Luciani, K. Kedzierska, R. A. Seder, F. M. Ndungu and I. A. Cockburn, Cell Rep., 2021, 34, 108684 CrossRef CAS PubMed
- H. J. Sutton, R. Aye, A. H. Idris, R. Vistein, E. Nduati, O. Kai, J. Mwacharo, X. Li, X. Gao, T. D. Andrews, M. Koutsakos, T. H. O. Nguyen, M. Nekrasov, P. Milburn, A. Eltahla, A. A. Berry, N. Kc, S. Chakravarty, B. K. L. Sim, A. K. Wheatley, S. J. Kent, S. L. Hoffman, K. E. Lyke, P. Bejon, F. Luciani, K. Kedzierska, R. A. Seder, F. M. Ndungu and I. A. Cockburn, Cell Rep., 2021, 35, 109286 CrossRef PubMed
- A. Ramesh, R. D. Schubert, A. L. Greenfield, R. Dandekar, R. P. Loudermilk, J. J. Sabatino, M. T. Koelzer, E. B. Tran, K. Koshal, K. Kim, A.-K. Pröbstel, D. Banerji, C.-Y. Guo, A. J. Green, R. M. Bove, J. L. Derisi, J. M. Gelfand, B. A. C. Cree, S. S. Zamvil, S. E. Baranzini, S. L. Hauser and M. R. Wilson, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 22932–22943 CrossRef CAS PubMed
- X. Ren, W. Wen, X. Fan, W. Hou, B. Su, P. Cai, J. Li, Y. Liu, F. Tang, F. Zhang, Y. Yang, J. He, W. Ma, J. He, P. Wang, Q. Cao, F. Chen, Y. Chen, X. Cheng, G. Deng, X. Deng, W. Ding, Y. Feng, R. Gan, C. Guo, W. Guo, S. He, C. Jiang, J. Liang, Y.-m. Li, J. Lin, Y. Ling, H. Liu, J. Liu, N. Liu, S.-Q. Liu, M. Luo, Q. Ma, Q. Song, W. Sun, G. Wang, F. Wang, Y. Wang, X. Wen, Q. Wu, G. Xu, X. Xie, X. Xiong, X. Xing, H. Xu, C. Yin, D. Yu, K. Yu, J. Yuan, B. Zhang, P. Zhang, T. Zhang, J. Zhao, P. Zhao, J. Zhou, W. Zhou, S. Zhong, X. Zhong, S. Zhang, L. Zhu, P. Zhu, B. Zou, J. Zou, Z. Zuo, F. Bai, X. Huang, P. Zhou, Q. Jiang, Z. Huang, J.-X. Bei, L. Wei, X.-W. Bian, X. Liu, T. Cheng, X. Li, P. Zhao, F.-S. Wang, H. Wang, B. Su, Z. Zhang, K. Qu, X. Wang, J. Chen, R. Jin and Z. Zhang, Cell, 2021, 184, 1895–1913.e19 CrossRef CAS PubMed
- C. Ziegenhain, B. Vieth, S. Parekh, B. Reinius, A. Guillaumet-Adkins, M. Smets, H. Leonhardt, H. Heyn, I. Hellmann and W. Enard, Mol. Cell, 2017, 65, 631–643.e4 CrossRef CAS PubMed
- O. B. Poirion, X. Zhu, T. Ching and L. Garmire, Front. Genet., 2016, 7, 163 Search PubMed
- V. Greiff, G. Yaari and L. G. Cowell, Curr. Opin. Syst. Biol., 2020, 24, 109–119 CrossRef
- E. Miho, A. Yermanos, C. R. Weber, C. T. Berger, S. T. Reddy and V. Greiff, Front. Immunol., 2018, 9, 224 CrossRef PubMed
- C. Milstein and G. Kohler, Nature, 1975, 256, 495–497 CrossRef PubMed
- J. K. Liu, Ann. Med. Surg., 2014, 3, 113–116 CrossRef PubMed
- K. Pieper, B. Grimbacher and H. Eibel, J. Allergy Clin. Immunol., 2013, 131, 959–971 CrossRef CAS PubMed
- M. McHeyzer-Williams, S. Okitsu, N. Wang and L. McHeyzer-Williams, Nat. Rev. Immunol., 2012, 12, 24–34 CrossRef CAS PubMed
-
R. J. Brezski and J. G. Monroe, Multichain Immune Recognition Receptor Signaling: From Spatiotemporal Organization to Human Disease, 2008, pp. 12–21 Search PubMed
- S. R. Allie and T. D. Randall, Viral Immunol., 2020, 33, 282–293 CrossRef CAS PubMed
- S. R. Allie, J. E. Bradley, U. Mudunuru, M. D. Schultz, B. A. Graf, F. E. Lund and T. D. Randall, Nat. Immunol., 2019, 20, 97–108 CrossRef CAS PubMed
- N. M. Weisel, F. J. Weisel, D. L. Farber, L. A. Borghesi, Y. Shen, W. Ma, E. T. Luning Prak and M. J. Shlomchik, Blood, 2020, 136, 2774–2785 CrossRef CAS PubMed
- D. Jung and F. W. Alt, Cell, 2004, 116, 299–311 CrossRef CAS PubMed
- J. L. Xu and M. M. Davis, Immunity, 2000, 13, 37–45 CrossRef CAS PubMed
- C. M. Johnston, A. L. Wood, D. J. Bolland and A. E. Corcoran, J. Immunol., 2006, 176, 4221–4234 CrossRef CAS PubMed
- Z. Wang, M. Raifu, M. Howard, L. Smith, D. Hansen, R. Goldsby and D. Ratner, J. Immunol. Methods, 2000, 233, 167–177 CrossRef CAS PubMed
- J. Wrammert, K. Smith, J. Miller, W. A. Langley, K. Kokko, C. Larsen, N.-Y. Zheng, I. Mays, L. Garman, C. Helms, J. James, G. M. Air, J. D. Capra, R. Ahmed and P. C. Wilson, Nature, 2008, 453, 667–671 CrossRef CAS PubMed
- K. Smith, L. Garman, J. Wrammert, N.-Y. Zheng, J. D. Capra, R. Ahmed and P. C. Wilson, Nat. Protoc., 2009, 4, 372–384 CrossRef CAS PubMed
- B. Briney, A. Inderbitzin, C. Joyce and D. R. Burton, Nature, 2019, 566, 393–397 CrossRef PubMed
- K. B. Hoehn, A. Gall, R. Bashford-Rogers, S. Fidler, S. Kaye, J. Weber, M. McClure, S. T. Investigators, P. Kellam and O. G. Pybus, Philos. Trans. R. Soc., B, 2015, 370, 20140241 CrossRef PubMed
- J. D. Galson, E. A. Clutterbuck, J. Trück, M. N. Ramasamy, M. Münz, A. Fowler, V. Cerundolo, A. J. Pollard, G. Lunter and D. F. Kelly, Immunol. Cell Biol., 2015, 93, 885–895 CrossRef CAS PubMed
- S. T. Reddy, X. Ge, A. E. Miklos, R. A. Hughes, S. H. Kang, K. H. Hoi, C. Chrysostomou, S. P. Hunicke-Smith, B. L. Iverson, P. W. Tucker, A. D. Ellington and G. Georgiou, Nat. Biotechnol., 2010, 28, 965–969 CrossRef CAS PubMed
- J. J. Lavinder, Y. Wine, C. Giesecke, G. C. Ippolito, A. P. Horton, O. I. Lungu, K. H. Hoi, B. J. Dekosky, E. M. Murrin, M. Wirth, A. D. Ellington, T. Dörner, E. M. Marcotte, D. R. Boutz and G. Georgiou, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2259–2264 CrossRef CAS PubMed
- B. J. DeKosky, T. Kojima, A. Rodin, W. Charab, G. C. Ippolito, A. D. Ellington and G. Georgiou, Nat. Med., 2015, 21, 86–91 CrossRef CAS PubMed
- B. Wang, B. J. DeKosky, M. R. Timm, J. Lee, E. Normandin, J. Misasi, R. Kong, J. R. McDaniel, G. Delidakis, K. E. Leigh, T. Niezold, C. W. Choi, E. G. Viox, A. Fahad, A. Cagigi, A. Ploquin, K. Leung, E. S. Yang, W.-P. Kong, W. N. Voss, A. G. Schmidt, M. A. Moody, D. R. Ambrozak, A. R. Henry, F. Laboune, J. E. Ledgerwood, B. S. Graham, M. Connors, D. C. Douek, N. J. Sullivan, A. D. Ellington, J. R. Mascola and G. Georgiou, Nat. Biotechnol., 2018, 36, 152–155 CrossRef CAS PubMed
- C. E. Busse, I. Czogiel, P. Braun, P. F. Arndt and H. Wardemann, Eur. J. Immunol., 2014, 44, 597–603 CrossRef CAS PubMed
- W. Wen, W. Su, H. Tang, W. Le, X. Zhang, Y. Zheng, X. Liu, L. Xie, J. Li, J. Ye, L. Dong, X. Cui, Y. Miao, D. Wang, J. Dong, C. Xiao, W. Chen and H. Wang, Cell Discovery, 2020, 6, 31 CrossRef CAS PubMed
- E. Roose, G. Vidarsson, K. Kangro, O. J. H. M. Verhagen, I. Mancini, L. Desender, I. Pareyn, N. Vandeputte, A. Vandenbulcke, C. Vendramin, A.-S. Schelpe, J. Voorberg, M.-A. Azerad, L. Gilardin, M. A. Scully, D. Dierickx, H. Deckmyn, S. F. De Meyer, F. Peyvandi and K. Vanhoorelbeke, Thromb. Haemostasis, 2018, 118, 1729–1742 CrossRef PubMed
- K. S. Cox, A. Tang, Z. Chen, M. Horton, H. Yan, X.-M. Wang, S. Dubey, D. J. Distefano, A. Ettenger, R. H. Fong, B. J. Doranz, D. R. Casimiro and K. A. Vora, mAbs, 2015, 8, 129–140 CrossRef PubMed
- E. Waltari, A. McGeever, N. Friedland, P. S. Kim and K. M. McCutcheon, Front. Immunol., 2019, 10, 1452 CrossRef CAS PubMed
- I. Setliff, A. R. Shiakolas, K. A. Pilewski, A. A. Murji, R. E. Mapengo, K. Janowska, S. Richardson, C. Oosthuysen, N. Raju, L. Ronsard, M. Kanekiyo, J. S. Qin, K. J. Kramer, A. R. Greenplate, W. J. McDonnell, B. S. Graham, M. Connors, D. Lingwood, P. Acharya, L. Morris and I. S. Georgiev, Cell, 2019, 179, 1636–1646.e15 CrossRef CAS PubMed
- R. Stephens, F. Ndungu and J. Langhorne, Parasite Immunol., 2009, 31, 20–31 CrossRef CAS PubMed
- M. J. Price, S. L. Hicks, J. E. Bradley, T. D. Randall, J. M. Boss and C. D. Scharer, J. Immunol., 2019, 203, 2121–2129 CrossRef CAS PubMed
- Z. Lin, N. Y. Chiang, N. Chai, D. Seshasayee, W. P. Lee, M. Balazs, G. Nakamura and L. R. Swem, Nat. Protoc., 2014, 9, 1563–1577 CrossRef CAS PubMed
- D. Guez-Barber, S. Fanous, B. K. Harvey, Y. Zhang, E. Lehrmann, K. G. Becker, M. R. Picciotto and B. T. Hope, J. Neurosci. Methods, 2012, 203, 10–18 CrossRef CAS PubMed
- J. C. Love, J. L. Ronan, G. M. Grotenbreg, A. G. van der Veen and H. L. Ploegh, Nat. Biotechnol., 2006, 24, 703–707 CrossRef CAS PubMed
- A. Jin, T. Ozawa, K. Tajiri, T. Obata, S. Kondo, K. Kinoshita, S. Kadowaki, K. Takahashi, T. Sugiyama, H. Kishi and A. Muraguchi, Nat. Med., 2009, 15, 1088–1092 CrossRef CAS PubMed
- A. Winters, K. McFadden, J. Bergen, J. Landas, K. A. Berry, A. Gonzalez, H. Salimi-Moosavi, C. M. Murawsky, P. Tagari and C. T. King, mAbs, 2019, 11, 1025–1035 CrossRef PubMed
- A. Gérard, A. Woolfe, G. Mottet, M. Reichen, C. Castrillon, V. Menrath, S. Ellouze, A. Poitou, R. Doineau, L. Briseno-Roa, P. Canales-Herrerias, P. Mary, G. Rose, C. Ortega, M. Delincé, S. Essono, B. Jia, B. Iannascoli, O. Richard-Le Goff, R. Kumar, S. N. Stewart, Y. Pousse, B. Shen, K. Grosselin, B. Saudemont, A. Sautel-Caillé, A. Godina, S. McNamara, K. Eyer, G. A. Millot, J. Baudry, P. England, C. Nizak, A. Jensen, A. D. Griffiths, P. Bruhns and C. Brenan, Nat. Biotechnol., 2020, 38, 715–721 CrossRef PubMed
- C. L. Pinder, S. Kratochvil, D. Cizmeci, L. Muir, Y. Guo, R. J. Shattock and P. F. McKay, J. Immunol., 2017, 199, 4180–4188 CrossRef CAS PubMed
- Y. Wine, D. R. Boutz, J. J. Lavinder, A. E. Miklos, R. A. Hughes, K. H. Hoi, S. T. Jung, A. P. Horton, E. M. Murrin, A. D. Ellington, E. M. Marcotte and G. Georgiou, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2993–2998 CrossRef CAS PubMed
- J. Lee, P. Paparoditis, A. P. Horton, A. Frühwirth, J. R. McDaniel, J. Jung, D. R. Boutz, D. A. Hussein, Y. Tanno, L. Pappas, G. C. Ippolito, D. Corti, A. Lanzavecchia and G. Georgiou, Cell Host Microbe, 2019, 25, 367–376.e5 CrossRef CAS PubMed
- A. Bondt, M. Hoek, S. Tamara, B. de Graaf, W. Peng, D. Schulte, D. M. H. van Rijswijck, M. A. den Boer, J.-F. Greisch, M. R. J. Varkila, J. Snijder, O. L. Cremer, M. J. M. Bonten and A. J. R. Heck, Cell Syst., 2021, 12, 1131–1143.e5 CrossRef CAS PubMed
- W. C. Cheung, S. A. Beausoleil, X. Zhang, S. Sato, S. M. Schieferl, J. S. Wieler, J. G. Beaudet, R. K. Ramenani, L. Popova, M. J. Comb, J. Rush and R. D. Polakiewicz, Nat. Biotechnol., 2012, 30, 447–452 CrossRef CAS PubMed
- L. Heinrich, N. Tissot, D. J. Hartmann and R. Cohen, J. Immunol. Methods, 2010, 352, 13–22 CrossRef CAS PubMed
- A. E. Muruato, C. R. Fontes-Garfias, P. Ren, M. A. Garcia-Blanco, V. D. Menachery, X. Xie and P.-Y. Shi, Nat. Commun., 2020, 11, 4059 CrossRef CAS PubMed
- S. Ko, M. Jo and S. T. Jung, BioDrugs, 2021, 35, 147–157 CrossRef CAS PubMed
- BerkeleyLights, Antibody Discovery, https://www.berkeleylights.com/workflows/antibody-discovery.
- Z. A. Bornholdt, H. L. Turner, C. D. Murin, W. Li, D. Sok, C. A. Souders, A. E. Piper, A. Goff, J. D. Shamblin, S. E. Wollen, T. R. Sprague, M. L. Fusco, K. B. J. Pommert, L. A. Cavacini, H. L. Smith, M. Klempner, K. A. Reimann, E. Krauland, T. U. Gerngross, K. D. Wittrup, E. O. Saphire, D. R. Burton, P. J. Glass, A. B. Ward and L. M. Walker, Science, 2016, 351, 1078–1083 CrossRef CAS PubMed
- J. Huang, B. H. Kang, E. Ishida, T. Zhou, T. Griesman, Z. Sheng, F. Wu, N. A. Doria-Rose, B. Zhang, K. McKee, S. O'Dell, G.-Y. Chuang, A. Druz, I. S. Georgiev, C. A. Schramm, A. Zheng, M. G. Joyce, M. Asokan, A. Ransier, S. Darko, S. A. Migueles, R. T. Bailer, M. K. Louder, S. M. Alam, R. Parks, G. Kelsoe, T. Von Holle, B. F. Haynes, D. C. Douek, V. Hirsch, M. S. Seaman, L. Shapiro, J. R. Mascola, P. D. Kwong and M. Connors, Immunity, 2016, 45, 1108–1121 CrossRef CAS PubMed
- Y. Fu, Z. Zhang, J. Sheehan, Y. Avnir, C. Ridenour, T. Sachnik, J. Sun, M. J. Hossain, L.-M. Chen, Q. Zhu, R. O. Donis and W. A. Marasco, Nat. Commun., 2016, 7, 12780 CrossRef CAS PubMed
- N. D. Durham, A. Agrawal, E. Waltari, D. Croote, F. Zanini, M. Fouch, E. Davidson, O. Smith, E. Carabajal, J. E. Pak, B. J. Doranz, M. Robinson, A. M. Sanz, L. L. Albornoz, F. Rosso, S. Einav, S. R. Quake, K. M. McCutcheon and L. Goo, eLife, 2019, 8, e52384 CrossRef CAS PubMed
- S. Saadat, Z. R. Tehrani, J. Logue, M. Newman, M. B. Frieman, A. D. Harris and M. M. Sajadi, JAMA, J. Am. Med. Assoc., 2021, 325, 1467–1469 CrossRef CAS PubMed
- B. He, S. Liu, Y. Wang, M. Xu, W. Cai, J. Liu, W. Bai, S. Ye, Y. Ma, H. Hu, H. Meng, T. Sun, Y. Li, H. Luo, M. Shi, X. Du, W. Zhao, S. Chen, J. Yang, H. Zhu, Y. Jie, Y. Yang, D. Guo, Q. Wang, Y. Liu, H. Yan, M. Wang and Y.-Q. Chen, Signal Transduction Targeted Ther., 2021, 6, 195 CrossRef CAS PubMed
-
C. Zhang, Antibody methods and protocols, 2012, pp. 117–135 Search PubMed
- Y. Cao, B. Su, X. Guo, W. Sun, Y. Deng, L. Bao, Q. Zhu, X. Zhang, Y. Zheng, C. Geng, X. Chai, R. He, X. Li, Q. Lv, H. Zhu, W. Deng, Y. Xu, Y. Wang, L. Qiao, Y. Tan, L. Song, G. Wang, X. Du, N. Gao, J. Liu, J. Xiao, X.-d. Su, Z. Du, Y. Feng, C. Qin, C. Qin, R. Jin and X. S. Xie, Cell, 2020, 182, 73–84.e16 CrossRef CAS PubMed
- S. Du, Y. Cao, Q. Zhu, P. Yu, F. Qi, G. Wang, X. Du, L. Bao, W. Deng, H. Zhu, J. Liu, J. Nie, Y. Zheng, H. Liang, R. Liu, S. Gong, H. Xu, A. Yisimayi, Q. Lv, B. Wang, R. He, Y. Han, W. Zhao, Y. Bai, Y. Qu, X. Gao, C. Ji, Q. Wang, N. Gao, W. Huang, Y. Wang, X. S. Xie, X.-d. Su, J. Xiao and C. Qin, Cell, 2020, 183, 1013–1023.e13 CrossRef CAS PubMed
- L. Piccoli, Y.-J. Park, M. A. Tortorici, N. Czudnochowski, A. C. Walls, M. Beltramello, C. Silacci-Fregni, D. Pinto, L. E. Rosen, J. E. Bowen, O. J. Acton, S. Jaconi, B. Guarino, A. Minola, F. Zatta, N. Sprugasci, J. Bassi, A. Peter, A. De Marco, J. C. Nix, F. Mele, S. Jovic, B. F. Rodriguez, S. V. Gupta, F. Jin, G. Piumatti, G. Lo Presti, A. F. Pellanda, M. Biggiogero, M. Tarkowski, M. S. Pizzuto, E. Cameroni, C. Havenar-Daughton, M. Smithey, D. Hong, V. Lepori, E. Albanese, A. Ceschi, E. Bernasconi, L. Elzi, P. Ferrari, C. Garzoni, A. Riva, G. Snell, F. Sallusto, K. Fink, H. W. Virgin, A. Lanzavecchia, D. Corti and D. Veesler, Cell, 2020, 183, 1024–1042.e21 CrossRef CAS PubMed
- R. A. Ehling, C. R. Weber, D. M. Mason, S. Friedensohn, B. Wagner, F. Bieberich, E. Kapetanovic, R. Vazquez-Lombardi, R. B. Di Roberto and K.-L. Hong, Cell Rep., 2022, 38, 110242 CrossRef CAS PubMed
- D. Corti, J. Voss, S. J. Gamblin, G. Codoni, A. Macagno, D. Jarrossay, S. G. Vachieri, D. Pinna, A. Minola, F. Vanzetta, C. Silacci, B. M. Fernandez-Rodriguez, G. Agatic, S. Bianchi, I. Giacchetto-Sasselli, L. Calder, F. Sallusto, P. Collins, L. F. Haire, N. Temperton, J. P. M. Langedijk, J. J. Skehel and A. Lanzavecchia, Science, 2011, 333, 850–856 CrossRef CAS PubMed
- M. Yu, Z. Zhu, Y. Wang, P. Wang, X.-Y. Jia, J. Wang, L. Liu, W. Liu, Y. Zheng, G. Kou, W. Xu, J. Huang, F. Lu, X. Zou, S. Zheng, Y. Lu, J. Zhao, H. Dai and X. Qiu, Mol. Biomed., 2022, 3, 20 CrossRef CAS PubMed
- D. Planas, N. Saunders, P. Maes, F. Guivel-Benhassine, C. Planchais, J. Buchrieser, W.-H. Bolland, F. Porrot, I. Staropoli, F. Lemoine, H. Péré, D. Veyer, J. Puech, J. Rodary, G. Baele, S. Dellicour, J. Raymenants, S. Gorissen, C. Geenen, B. Vanmechelen, T. Wawina-Bokalanga, J. Martí-Carreras, L. Cuypers, A. Sève, L. Hocqueloux, T. Prazuck, F. A. Rey, E. Simon-Loriere, T. Bruel, H. Mouquet, E. André and O. Schwartz, Nature, 2022, 602, 671–675 CrossRef CAS PubMed
- A. C. Hurt and A. K. Wheatley, Viruses, 2021, 13, 628 CrossRef CAS PubMed
- M. Guo, M. Xiong, J. Peng, T. Guan, H. Su, Y. Huang, C.-G. Yang, Y. Li, D. Boraschi, T. Pillaiyar, G. Wang, C. Yi, Y. Xu and C. Chen, Natl. Sci. Rev., 2023, 10, nwad161 CrossRef PubMed
- H. Qi, B. Liu, X. Wang and L. Zhang, Nat. Immunol., 2022, 23, 1008–1020 CrossRef CAS PubMed
- K. Eyer, R. C. L. Doineau, C. E. Castrillon, L. Briseño-Roa, V. Menrath, G. Mottet, P. England, A. Godina, E. Brient-Litzler, C. Nizak, A. Jensen, A. D. Griffiths, J. Bibette, P. Bruhns and J. Baudry, Nat. Biotechnol., 2017, 35, 977–982 CrossRef CAS PubMed
- K. Eyer, C. Castrillon, G. Chenon, J. Bibette, P. Bruhns, A. D. Griffiths and J. Baudry, J. Immunol., 2020, 205, 1176–1184 CrossRef CAS PubMed
- A. Singhal, C. A. Haynes and C. L. Hansen, Anal. Chem., 2010, 82, 8671–8679 CrossRef CAS PubMed
- B. E. Debs, R. Utharala, I. V. Balyasnikova, A. D. Griffiths and C. A. Merten, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11570–11575 CrossRef PubMed
- B. Jacková, G. Mottet, S. Rudiuk, M. Morel and D. Baigl, Adv. Biol., 2023, 7, 2200266 CrossRef PubMed
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |