
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA metal and a metalloid Lewis acid bridged by a μ2-phosphinidene†
David
Biskup
 a,
Gregor
Schnakenburg
a,
Arturo
Espinosa Ferao
*b and
Rainer
Streubel
*a
a,
Gregor
Schnakenburg
a,
Arturo
Espinosa Ferao
*b and
Rainer
Streubel
*a
aInstitut für Anorganische Chemie, Rheinische Friedrich-Wilhelms-Universität Bonn, Gerhard-Domagk-Str.1, 53121 Bonn, Germany. E-mail: r.streubel@uni-bonn.de
bDepartamento de Química Orgánica, Facultad de Química, Campus de Espinardo, Universidad de Murcia, 30100 Murcia, Spain. E-mail: artuesp@um.es
First published on 24th May 2024
Abstract
Dinuclear phosphinidene complexes bridging two transition metal centres are now well established. However, a phosphinidene bridging a metal centre and a main group Lewis acid has not yet been reported. Herein, we describe the generation of a highly reactive phosphinidene complex bridging a tungsten and a boron centre. Furthermore, the synthesis and dyotropic rearrangement of a P-borane adduct of an N-methylimidazole-stabilized neutral, electrophilic terminal phosphinidene complex is reported.
In 1975, Huttner reported on the synthesis of the first μ2-phosphinidene-bridged dinuclear complex1 and subsequently, μ3- and μ4-phosphinidene complexes.2,3 Dinuclear complexes are commonly prepared via salt metathesis starting from dihalophosphanes,4 metallophosphanes5,6 or anionic phosphinidene complexes.7 Additionally, syntheses via deprotonation of phosphanyl ligands,6,8 dehydrohalogenation9 or elimination reactions10 are possible.
Complexes have either a trigonal pyramidal geometry at phosphorus, which can be regarded as doubly metallated phosphanes I (Fig. 1), e.g. [Pt2(dippe)2(μ2-PPh)]11 or [Fe2Cp2(μ2-PMes*)(μ2-CO)-(CO)2],12 or a trigonal planar geometry (II) representing a μ2-bridged phosphinidene complex, e.g., [W2(μ2-PCp*)(CO)10]13 or [W2Cp2(μ2-PMes*)(CO)4].7 However, complexes II could be better described by a 3c,4e π-bond as confirmed by short metal–phosphorus distances.14,15 Examples with slightly longer bond distances13,16 can be explained by steric repulsion of the metal fragments and the P-substituent, however, they are still shorter than a typical single dative bond as in [W(CO)5(PMe3)].17
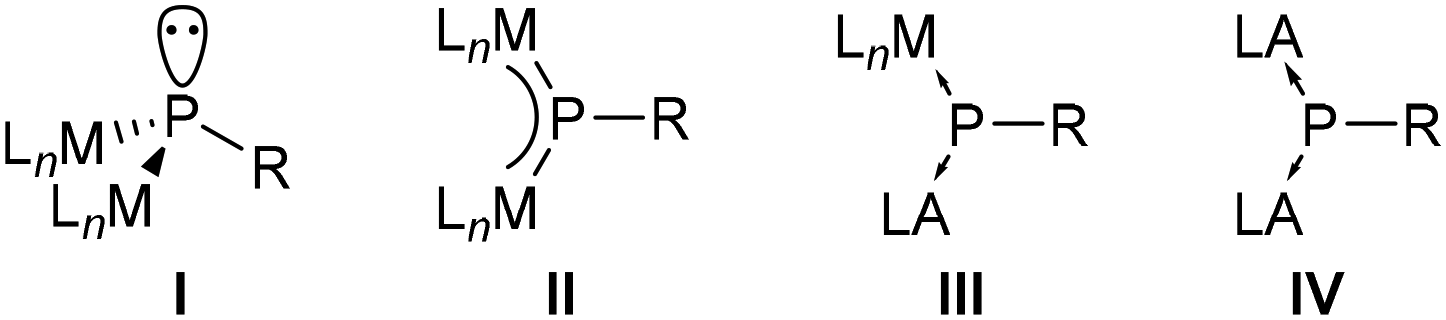 | ||
| Fig. 1 General structures of a dimetallophosphane (I) and μ2-phosphinidene bridging two metal fragments (MLn) (II), a metal and a main group Lewis acid (LA) (III), and two main group Lewis acids (IV). | ||
Trigonal planar complexes II with an M–P–M π-bonding have small HOMO–LUMO gaps, thus allowing for an admixture of excited states to the electronic ground state configuration (paramagnetic contribution). Therefore, phosphorus nuclei reveal 31P NMR chemical shifts at a very low field (593–1077 ppm).2,18,19 A broad range of synthetic applications are known for symmetric (M = M) dinuclear phosphinidene complexes II, including reactions with nucleophiles, electrophiles, E–H bonds (E = B and P), unsaturated organic molecules and p-block elements, as well as addition of metal fragments, thermolysis and photolysis reactions.2 While mixed metal μ-phosphinidene complexes have been reported,15,20 phosphinidenes bridging a metal and a main group Lewis acid are not known.
We recently reported on the synthesis of the donor-stabilised, neutral, electrophilic, terminal phosphinidene complex adduct 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 and we showed that the phosphorus centre in N-donor adducts can be protonated by Brønsted–Lowry acids, but only in the case of superacids were N-donor adducts of P–H phosphenium complexes formed.22
21 and we showed that the phosphorus centre in N-donor adducts can be protonated by Brønsted–Lowry acids, but only in the case of superacids were N-donor adducts of P–H phosphenium complexes formed.22
Herein, we describe the reactions of an N-MeIm donor adduct 1 of a terminal phosphinidene complex with a transition metal and a main group Lewis acid leading to homo- and hetero-dinuclear-bridged phosphinidene complexes, and report on a unique case of a dyotropic rearrangement.
To probe the Lewis basicity and kinetic availability of the P-centre of complex 1, it was first treated with the labile tungsten(0) complexes [W(CO)5(NCMe)] and [W(CO)5(thf)] (Scheme 1). Addition of [W(CO)5(MeCN)] to 1 in benzene-d6 led to a deep violet solution within a few minutes but the 31P{1H} NMR spectrum revealed only a marginal conversion of 1 under ambient conditions (4% via31P{1H} NMR integration). However, reaction of 1 with [W(CO)5(thf)] revealed an improved outcome. The main product 3 showed a signal at 791.4 ppm (1JW,P = 185.2 Hz) but the maximum content was only about 46%. Besides the homo-dinuclear μ2-phosphinidene complex 2, other products were observed in the 31P{1H} NMR spectrum at 247.0 ppm (2%), 182.2 ppm (1JW,P = 193.9 Hz) (6%), 150.4 ppm (1JW,P = 192.3 Hz) (6%) and 69.9 ppm (1JP,H = 371.9 Hz, 1JW,P = 274.7 Hz) (5%). We assumed that the broad signal at 150.4 ppm (1JW,P = 191.6 Hz) can be assigned to the primarily formed compound 2 (Scheme 1) which converts into 3via loss of the N-donor. Unfortunately, no 31P NMR data of N-donor adducts of homo-dinuclear μ2-phosphinidene complexes were reported by Huttner, but the colour change from blue to yellow was described to be indicative.19,23
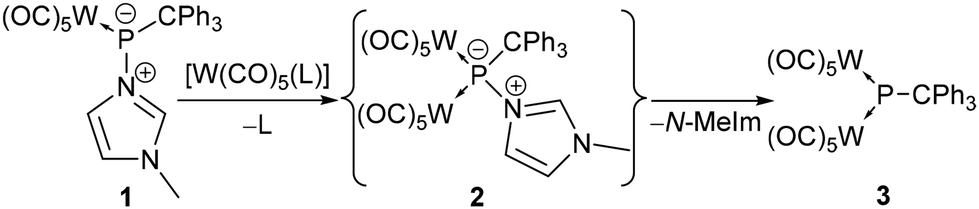 | ||
| Scheme 1 Proposed formation of the N-donor adduct of dinuclear phosphinidene complex 2 and its conversion into 3. | ||
An in situ UV/vis spectrum of the reaction mixture of complex 1 with [W(CO)5(MeCN)] in THF containing 3 shows a characteristic absorption at 540 nm which is assigned to a transition in the W–P–W π-system. Similar homo-dinuclear μ2-phosphinidene complexes were described earlier by Huttner, Jutzi and Kuchen; the 31P NMR parameters and UV/vis data are compiled in Table 1.19,24–26
Going from a transition metal to a main group Lewis acid, namely tris(pentafluorophenyl)borane (B(C6F5)3), addition of a slight excess (1.1 eq.) of B(C6F5)3 to complex 1 in benzene-d6 under ambient conditions resulted in a colour change from yellow to deep turquoise-blue within 20 minutes and the formation of hetero-dinuclear complex 4 (Scheme 2). Formation of the diradical ion pair via a single electron transfer (SET) from the HOMO of complex 1 (−6.07 eV) to the LUMO of B(C6F5)3 (−2.31 eV) seems to be unlikely, as demonstrated by the high computed value of ΔGSET = +47.8 kcal mol−1 for the separated ion pair. Indeed, an in situ UV/vis spectrum of the reaction mixture reveals an absorption at 578 nm which is in accordance with the observed colour, and significantly differs from that of the reported tris(pentafluorophenyl)borane radical anion (λmax = 603 nm).27
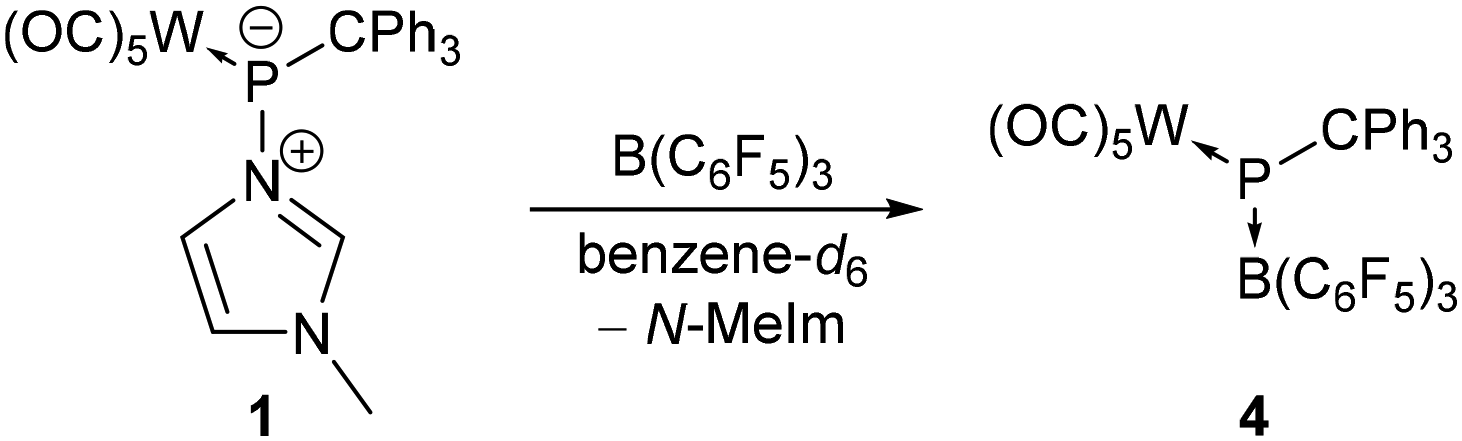 | ||
| Scheme 2 Formation of complex 4 as a μ2-phosphinidene bridging a tungsten and a boron centre. | ||
The 31P{1H} NMR spectrum of the reaction mixture (after 1.5 h) showed a strongly deshielded 31P resonance at 1040.3 ppm (1JW,P = 180.1 Hz) and hence, it is assigned to the hetero-dinuclear complex 4. The 31P chemical shift of this molecular species is in rather good agreement with the computed value of 974.3 ppm (see the ESI†). To improve the formation of complex 4, various changes were examined but the best outcome was obtained when 1 was fully consumed (3.4 eq. of B(C6F5)3, stirring for 6.5 hours at rt) and only a 24% content of 4 was determined (via integration). Unfortunately, complex 4 converted rather rapidly into a main product (δ(31P{1H}) = −132.8 ppm, 1JW,P = 260.3 Hz) which could not be isolated.
The limited success of achieving the formation of a stable borane adduct led us to conclude that the steric demand of the system and the borane used were the origin of the instability. Therefore, complex 1 was treated with the borane adduct H3B·SMe2 which underwent the reaction smoothly and selectively formed the hetero-dinuclear phosphinidene complex 5 having N-methylimidazole still bound to phosphorus (Scheme 3). This product was also thermally unstable but could be isolated via precipitation at −20 °C, and a low solubility in common organic solvents was beneficial. 31P NMR spectra (dichloromethane-d2) showed a broad resonance signal of 5 at 163.5 ppm which was corroborated by a calculated value of 133.0 ppm (see the ESI†) for the model complex 5′ (prime numbering indicates P-tBu substitution).
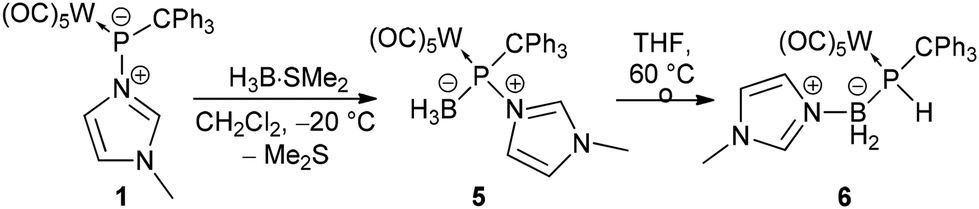 | ||
| Scheme 3 Synthesis of the N-methylimidazole-to-phosphinidene complex borane P-adduct 5 and its thermal rearrangement to 6. | ||
Due to the adduct formation, rotation about the P–C bond is hindered and the ortho-CH protons of all phenyl groups differ significantly. The BH3 protons were determined as a very broad doublet between 1.24 ppm and 1.52 ppm. The 11B NMR spectrum showed a resonance at −24.9 ppm as a very broad singlet. The ATR FTIR spectrum of complex 5 showed an absorption at 2402 cm−1 attributed to the B–H valence vibration. HRMS experiments (APCI method) confirmed the elemental composition of the molecular ion (m/z 693.0944). The final confirmation of the molecular structure of the borane P-adduct 5 came from X-ray diffraction analysis. The molecular structure (Fig. 2, left) discloses a slightly elongated P–CPh3 bond of 1.9540(19) Å and an angular sum at phosphorus of 313.76(3)°, being slightly smaller than expected for a tetrahedral geometry.
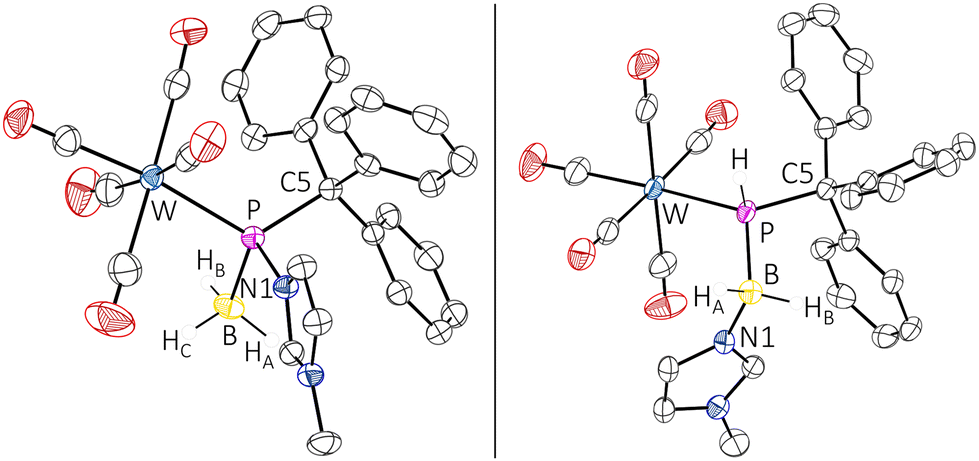 | ||
| Fig. 2 Molecular structures of 5 (left) and 6 (right). Thermal ellipsoids are set at 50% probability, and solvent molecules and hydrogen atoms are omitted for clarity, except for those bound to boron and phosphorus atoms. The selected bond lengths/Å and bond angles/°: 5: W–P 2.5688(5), P–C5 1.9540(19), P–N1 1.8138(16), P–B 1.959(2), N1–P–W 107.17(5), N1–P–C5 101.73(8), N1–P–B 100.26(9), C5–P–W 121.14(16), C5–P–B 111.77(9), B–P–W 111.89(7); 6: W–P 2.5650(4), P–C5 1.9179(14), P–B 1.9904(16), N1–B 1.5586(19), C5–P–W 123.10(4), C5–P–B 107.48(6), B–P–W 118.20(5), N1–B–P 111.31(9). | ||
When a solution of complex 5 in THF was heated to 60 °C, formation of a P–H containing compound was observed while the N-MeIm donor had shifted to the boron centre (Scheme 4).
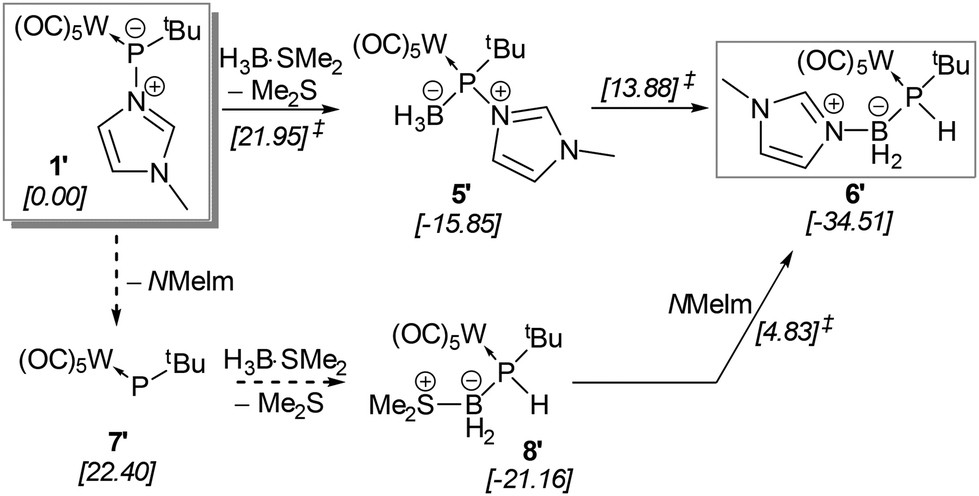 | ||
| Scheme 4 Proposed mechanism for the formation of the rearranged model product 6′ from phosphinidene adduct complex 1′. The computed [CPCMTHF/PWPB95-D3/def2-QZVPP//CPCMTHF/PBEh-3c] relative Gibbs free energies (kcal mol−1) are given in square brackets for ground and transition states (the latter indicated by the ‡ superscript). The dashed arrows indicate barrierless processes. | ||
The conversion of 5 into 6 came with a strong highfield-shifted signal in the 31P NMR spectrum at −42.6 ppm (1JP,H = 289.3 Hz, 1JP,B = 49.2 Hz), which is in good agreement with a computed value of −30.9 ppm for the P-tBu model complex (6′), revealing also the presence of the N-methylimidazole unit. It is noteworthy that the diminished steric congestion allowed for free rotation about the P–C bonds at ambient temperature. Complex 6 was further confirmed by APCI-HRMS and single-crystal X-ray diffraction analysis (Fig. 2, right). Similar base-stabilised phosphanylborane complexes have been reported by Scheer, e.g. [W(CO)5P(H)2B(H)2NMe3] (δ31P = −184.2 ppm, 1JP,B = 63 Hz), although the route is different.28
Quantum chemical calculations (see computational details) reveal the expected trigonal planar geometry at P for both 3 (∑<P = 359.6°) and 4 (∑<P = 359.0°) confirming the sp2 hybridisation. As expected, the vacant p atomic orbital at P constitutes the LUMO (Fig. 3), showing very low energy values for 3 and 4 (−2.69 and −3.32 eV, respectively), in line with a high positive natural charge at P (1.15 and 0.59 e, for 3 and 4, respectively), thus indicating the highly electrophilic character of these bridging phosphinidene species compared to adduct complex 1 (the LUMO is mostly located at the metal fragment, with εLUMO = 1.33 eV, qn(P) = 0.42 e). Carbonyl ligands intertwining in 3 are favoured by a non-covalent interaction between the different metal fragments (Fig. S113†). A significant shortening of the P–W bond distance in 3 (2.48 and 2.46 Å) and 4 (2.42 Å), in the same range as similar complexes,13,15,16 is due to the partial double bond character (WBI = 0.735/0.774 and 1.263, respectively) compared to 1 (d = 2.63 Å; WBI 0.457). The P–CPh3 distances in 3 (1.95 Å) and 4 (1.96 Å) fall in the normal range, but steric crowding at P remarkably elongates the P–B(C6H5)3 distance in 4 (2.07 Å; compared to 1.93 Å for H3P–BH3 at the same level).
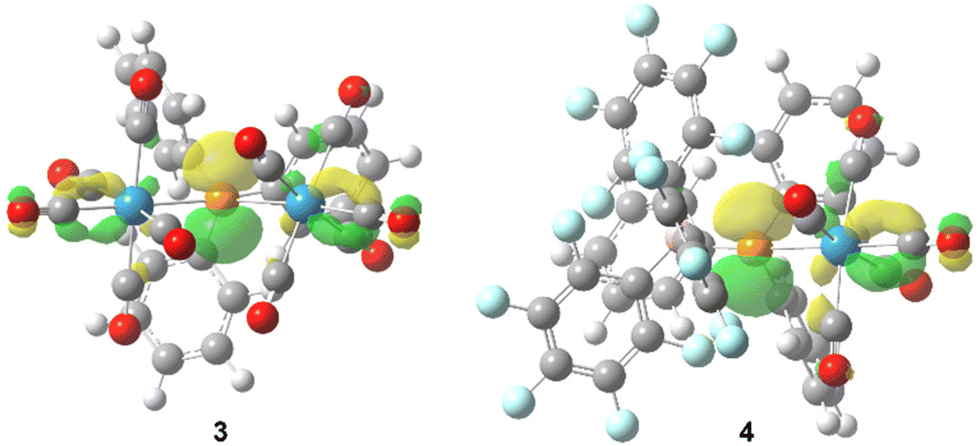 | ||
| Fig. 3 Computed (B3LYP-D3/def2-TZVP(ecp)) Kohn–Sham LUMO isosurfaces (0.05 au) for 3 and 4 viewed from the P–CPh3 axis. | ||
The mechanism of the formation of the P-borane adduct 5 and its rearrangement to 6 was investigated starting from the tert-butyl (instead of trityl)-substituted model compound 1′ (Scheme 4). A thorough analysis of the potential energy surface (see Fig. S114† for a full description) reveals that the lowest energy path proceeds over an exergonic low barrier addition of borane to phosphorus (5′), followed by the moderate barrier (ΔΔG‡ = 29.73 kcal mol−1) of the rate-determining formal type I dyotropic migration29 of hydride and N-MeIm between B and P giving (model) complex 6′, in sum, a remarkable case.
Author contributions
DB, AEF and RS wrote the manuscript and the ESI.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the Deutsche Forschungsgemeinschaft (STR 411/45-1) and the University of Bonn. We are grateful to Prof. Dr A. C. Filippou and Prof. Dr S. Höger for the use of X-ray facilities. A. E. F. is thankful for the computational resources used at the computation centre at Servicio de Cálculo Científico (SCC – University of Murcia).References
- G. Huttner, H.-D. Müller, A. Frank and H. Lorenz, Angew. Chem., Int. Ed. Engl., 1975, 14, 705–706 CrossRef.
- M. E. García, D. García-Vivó, A. Ramos and M. A. Ruiz, Coord. Chem. Rev., 2017, 330, 1–36 CrossRef.
- G. Huttner and K. Evertz, Acc. Chem. Res., 1986, 19, 406–413 CrossRef CAS.
- (a) J. Sánchez-Nieves, B. T. Sterenberg, K. A. Udachin and A. J. Carty, Can. J. Chem., 2004, 82, 1507–1516 CrossRef; (b) J. Sánchez-Nieves, B. T. Sterenberg, K. A. Udachin and A. J. Carty, J. Cluster Sci., 2004, 15, 151–162 CrossRef; (c) A. H. Cowley, D. M. Giolando, C. M. Nunn, M. Pakulski, D. Westmoreland and N. C. Norman, J. Chem. Soc., Dalton Trans., 1988, 2127–2134 RSC; (d) A. M. Arif, A. H. Cowley, N. C. Norman, A. G. Orpen and M. Pakulski, Organometallics, 1988, 7, 309–318 CrossRef CAS; (e) A. M. Arif, A. H. Cowley, M. Pakulski, M.-A. Pearsall, W. Clegg, N. C. Norman and A. G. Orpen, J. Chem. Soc., Dalton Trans., 1988, 2713–2721 RSC; (f) A. M. Arif, A. H. Cowley, M. Pakulski, N. C. Norman and A. G. Orpen, Organometallics, 1987, 6, 189–191 CrossRef CAS.
- (a) T. A. Bazhenova, A. V. Kulikov, A. F. Shestakov, A. E. Shilov, M. Y. Antipin, K. A. Lyssenko, Y. T. Struchkov and v. d. Makhaev, J. Am. Chem. Soc., 1995, 117, 12176–12180 CrossRef CAS; (b) J. Ho, R. J. Drake and D. W. Stephan, J. Am. Chem. Soc., 1993, 115, 3792–3793 CrossRef CAS.
- J. Ho, Z. Hou, R. J. Drake and D. W. Stephan, Organometallics, 1993, 12, 3145–3157 CrossRef CAS.
- R. Schmitt, PhD thesis, Julius-Maximilian-Universität Würzburg, 2005.
- (a) T. Pugh, F. Tuna, L. Ungur, D. Collison, E. J. L. McInnes, L. F. Chibotaru and R. A. Layfield, Nat. Commun., 2015, 6, 7492 CrossRef PubMed; (b) A. C. Colson and K. H. Whitmire, Organometallics, 2010, 29, 4611–4618 CrossRef CAS; (c) U. Vogel, P. Sekar, R. Ahlrichs, U. Huniar and M. Scheer, Eur. J. Inorg. Chem., 2003, 2003, 1518–1522 CrossRef; (d) J. E. Davies, M. J. Mays, E. J. Pook, P. R. Raithby and P. K. Tompkin, J. Chem. Soc., Dalton Trans., 1997, 3283–3286 RSC; (e) S. V. Maslennikov, D. S. Glueck, G. P. A. Yap and A. L. Rheingold, Organometallics, 1996, 15, 2483–2488 CrossRef CAS; (f) H.-J. Haupt, M. Schwefer and U. Flrke, Z. Anorg. Allg. Chem., 1995, 621, 1098–1105 CrossRef CAS; (g) H.-J. Haupt, M. Schwefer and U. Floerke, Inorg. Chem., 1995, 34, 292–297 CrossRef CAS; (h) H.-J. Haupt, M. Schwefer, H. Egold and U. Floerke, Inorg. Chem., 1995, 34, 5461–5467 CrossRef CAS; (i) U. Flörke and H. J. Haupt, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 573–575 CrossRef; (j) I.-P. Lorenz, W. Pohl, H. Nöth and M. Schmidt, J. Organomet. Chem., 1994, 475, 211–221 CrossRef CAS; (k) C. Klasen, G. Effinger, S. Schmid and I.-P. Lorenz, Z. Naturforsch., B: Chem. Sci., 1993, 48, 705–712 CrossRef CAS; (l) U.-A. Hirth and W. Malisch, J. Organomet. Chem., 1992, 439, C16–C19 CrossRef CAS; (m) A. J. Deeming, S. Doherty, M. W. Day, K. I. Hardcastle and H. Minassian, J. Chem. Soc., Dalton Trans., 1991, 1273–1279 RSC; (n) S. B. Colbran, B. F. Johnson, J. Lewis and R. M. Sorrell, J. Organomet. Chem., 1985, 296, C1–C5 CrossRef CAS.
- (a) H. Aktaş, J. C. Slootweg and K. Lammertsma, Angew. Chem., Int. Ed., 2010, 49, 2102–2113 CrossRef PubMed; (b) A. T. Termaten, T. Nijbacker, A. W. Ehlers, M. Schakel, M. Lutz, A. L. Spek, M. L. McKee and K. Lammertsma, Chem. – Eur. J., 2004, 10, 4063–4072 CrossRef CAS PubMed; (c) G. A. Abdul Hadi, K. Fromm, S. Blaurock, S. Jelonek and E. Hey-Hwakins, Polyhedron, 1997, 16, 721–731 CrossRef.
- (a) A. C. Behrle, L. Castro, L. Maron and J. R. Walensky, J. Am. Chem. Soc., 2015, 137, 14846–14849 CrossRef CAS PubMed; (b) J. Zhou, T. Li, L. Maron, X. Leng and Y. Chen, Organometallics, 2015, 34, 470–476 CrossRef CAS; (c) J. D. Masuda, K. C. Jantunen, O. V. Ozerov, K. J. T. Noonan, D. P. Gates, B. L. Scott and J. L. Kiplinger, J. Am. Chem. Soc., 2008, 130, 2408–2409 CrossRef CAS PubMed; (d) M. P. Shaver and M. D. Fryzuk, Organometallics, 2005, 24, 1419–1427 CrossRef CAS; (e) T. L. Breen and D. W. Stephan, Organometallics, 1996, 15, 4509–4514 CrossRef CAS; (f) J. Ho, R. Rousseau and D. W. Stephan, Organometallics, 1994, 13, 1918–1926 CrossRef CAS.
- R. Waterman and T. D. Tilley, Chem. Sci., 2011, 2, 1320 RSC.
- (a) M. A. Alvarez, M. E. García, R. González, A. Ramos and M. A. Ruiz, Organometallics, 2011, 30, 1102–1115 CrossRef CAS; (b) M. A. Alvarez, M. E. García, R. González, A. Ramos and M. A. Ruiz, Organometallics, 2010, 29, 1875–1878 CrossRef CAS.
- M. Scheer, E. Leiner, P. Kramkowski, M. Schiffer and G. Baum, Chem. – Eur. J., 1998, 4, 1917–1923 CrossRef CAS.
- (a) M. A. Alvarez, I. Amor, M. E. García, D. García-Vivó, M. A. Ruiz, D. Sáez, H. Hamidov and J. C. Jeffery, Inorg. Chim. Acta, 2015, 424, 103–115 CrossRef CAS; (b) I. Amor, M. E. García, M. A. Ruiz, D. Sáez, H. Hamidov and J. C. Jeffery, Organometallics, 2006, 25, 4857–4869 CrossRef CAS; (c) T. W. Graham, K. A. Udachin and A. J. Carty, Inorg. Chim. Acta, 2007, 360, 1376–1379 CrossRef CAS.
- J. Sánchez-Nieves, B. T. Sterenberg, K. A. Udachin and A. J. Carty, Inorg. Chim. Acta, 2003, 350, 486–494 CrossRef.
- (a) M. Seidl, R. Weinzierl, A. Y. Timoshkin and M. Scheer, Chem. – Eur. J., 2016, 22, 5484–5488 CrossRef CAS PubMed; (b) J. Borm, G. Huttner, L. Zsolnai, K. Evertz and H. Berke, J. Organomet. Chem., 1987, 327, 223–235 CrossRef CAS.
- F. A. Cotton, D. J. Darensbourg and B. W. S. Kolthammer, Inorg. Chem., 1981, 20, 4440–4442 CrossRef CAS.
- G. Huttner, J. Organomet. Chem., 1986, 308, C11–C13 CrossRef CAS.
- G. Huttner, J. Borm and L. Zsolnai, J. Organomet. Chem., 1984, 263, C33–C36 CrossRef CAS.
- M. A. Alvarez, M. E. García, D. García-Vivó, M. A. Ruiz and P. Vega, J. Organomet. Chem., 2022, 977, 122460 CrossRef CAS.
- D. Biskup, G. Schnakenburg, R. T. Boeré, A. Espinosa Ferao and R. K. Streubel, Nat. Commun., 2023, 14, 6456 CrossRef CAS PubMed.
- D. Biskup, G. Schnakenburg, A. Espinosa Ferao and R. Streubel, Dalton Trans., 2024, 53, 2517–2525 RSC.
- (a) H. Lang, G. Mohr, O. Scheidsteger and G. Huttner, Chem. Ber., 1985, 118, 574–596 CrossRef CAS; (b) H. Lang, L. Zsolnai and G. Huttner, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1985, 40, 500–506 CrossRef.
- H. Lang, O. Orama and G. Huttner, J. Organomet. Chem., 1985, 291, 293–309 CrossRef CAS.
- P. Jutzi and R. Kroos, J. Organomet. Chem., 1990, 390, 317–322 CrossRef CAS.
- A.-M. Hinke, A. Hinke, W. Kuchen and W. Hönle, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1986, 41, 629–639 CrossRef.
- R. J. Kwaan, C. J. Harlan and J. R. Norton, Organometallics, 2001, 20, 3818–3820 CrossRef CAS.
- U. Vogel, P. Hoemensch, K.-C. Schwan, A. Y. Timoshkin and M. Scheer, Chem. – Eur. J., 2003, 9, 515–519 CrossRef CAS PubMed.
- (a) M. T. Reetz, Adv. Organomet. Chem., 1977, 16, 33–65 CrossRef CAS; (b) M. T. Reetz, Angew. Chem., Int. Ed. Engl., 1972, 11, 129–130 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols and methods, NMR, MS and X-ray data, theoretical results and methods. CCDC 2320467 and 2320468. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01276c |
| This journal is © The Royal Society of Chemistry 2024 |