
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Monitoring structure and coordination chemistry of Co4O4-based oxygen evolution catalysts by nitrogen-14/-15 and cobalt-59 NMR spectroscopy†‡
Felix
Uhlig§
a,
Michael B.
Stammler¶
a,
Florian
Meurer
ab,
Ilya G.
Shenderovich
 c,
Jan
Blahut
*d and
Florian M.
Wisser
*ae
c,
Jan
Blahut
*d and
Florian M.
Wisser
*ae
aUniversity of Regensburg, Institute of Inorganic Chemistry, Universitätsstraße 31, 93053 Regensburg, Germany. E-mail: florian.wisser@fau.de
bRossendorf Beamline, Helmholtz-Zentrum Dresden-Rossendorf, Dresden, Germany
cUniversity of Regensburg, Institute of Organic Chemistry, Universitätsstraße 31, 93040 Regensburg, Germany
dInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nám. 2, 166 10, Prague 6, Czech Republic. E-mail: jan.blahut@uochb.cas.cz
eErlangen Center for Interface Research and Catalysis, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstraße 3, 91058 Erlangen, Germany
First published on 1st May 2024
Abstract
The structural features of cobalt-based oxygen evolution catalysts are elucidated by combining high-field MAS NMR spectroscopy and DFT calculations. The superior photocatalytic activity of the heterogeneous system over its homogeneous counterpart is rationalised by the structural features. The higher activity is caused by a more favourable electron-withdrawing character of the framework.
Transition metal (TM) cubanes are a class of organometallic compounds that contain an M4O4 cube as a core, typically surrounded by nitrogen and oxygen donor ligands.1–3 Most complexes have pyridine- or bipyridine-based axial ligands while the equatorial facets of the cube are bridged by carboxylates. TM cubanes have been studied extensively in the last decade mainly for three reasons. First, their structure is similar to the active site in photo system II (PS II), which consists of a {CaMn4O5} oxo cluster, making them ideal model systems to study the oxygen evolution reaction (OER). Second, the large number of TM and TM combinations opens up the possibility to vary the metal core by design. Third, the rich coordination chemistry of TM allows the design of an almost infinite number of cubanes by exchanging axial pyridines and/or equatorial carboxylates.1,3 Although cubanes have been widely studied as OER catalysts or in selective oxidation reactions, surprisingly their characterisation by (solid-state) nuclear magnetic resonance spectroscopy remains scarce, except for the standard 1H and 13C liquid NMR characterisation. As with other metal–organic complexes, 15N NMR and NMR studies of the metal are expected to provide important insights into the local structure.4,5 To this end tetrakis[(μ-acetato-κO′κO′)-μ3-oxo-(pyridine)cobalt(III)]6 (Co4O4(OAc)4Py4, Fig. 1) is an ideal model, not only because of its high catalytic activity, but also because the central Co is NMR active (100% natural abundance) with the largest chemical shift range.7 Furthermore, 59Co is a quadrupolar nucleus with a spin of 7/2, so the linewidth of the 59Co NMR signal is therefore highly sensitive to the symmetry of the ligand atom combination.
 | ||
| Fig. 1 (A) Molecular structure of Co4O4(OAc)4Py4 along the crystallographic c direction. Colour code: C: black, O: red, N: blue, Co: cyan and H white. (B) Solid state NMR spectra of Co4O4(OAc)4{15N}Py4 with and without MAS as well as a deconvoluted spectra. (C) Comparison of 15N NMR spectra of Co4O4(OAc)4{15N}Py4 dissolved in D2O (red), D2O (pH > 9, blue), ACN-d3 (cyan), CD2Cl2 (orange) with the CP MAS spectrum (black) as well as the spectrum of pyridine in D2O. # labels 15N signal from ACN-d3. (D) Comparison of 59Co NMR spectra of Co4O4(OAc)4{15N}Py4 dissolved in D2O (red), D2O (pH > 9, blue), ACN-d3 (cyan) and CD2Cl2 (orange). | ||
Here we showcase the detailed insight into the local structure of Co4O4(OAc)4Py4 available from both liquid- and solid-state NMR spectroscopy. In addition to Co4O4(OAc)4Py4, we also studied its 15N-labelled analogue Co4O4(OAc)4{15N}Py4 and a Co4O4(OAc)4Py4 derived porous coordination polymer (PCP).8 Solid-state 15N NMR spectroscopic studies, either under magic angle spinning (MAS) or static, provide additional information not accessible from liquid-state NMR spectroscopy. By combining 15N NMR with DFT calculations, we highlight the effect of Co coordination on the chemical shift tensors of 15N and on the resulting isotropic chemical shift. In addition, liquid-state 15N and 59Co NMR spectroscopy reveal the stable coordination of axial pyridine ligands, the (partial) exchange of equatorial acetate ligands, and the intact structure of the Co4O4 core, even after photocatalysis. In order to obtain useful nitrogen-15 NMR spectra in a reasonable time, labelling schemes are essential.9 While this is feasible for molecular complexes with simple ligands such as pyridine, solid materials require more complex ligands whose labelling leads to multi-step synthesis schemes. Although the natural abundance of 14N is >99.6%, 14N NMR spectroscopy remains scarce.10 Therefore, we have finally extended the investigation to the 3D solid material using proton-detected nitrogen-14 NMR spectroscopy at fast MAS and high field,11,12 adding another proton-detected technique to the toolbox for PCP characterisation.13
The 15N chemical shift of pyridine is very sensitive to the coordination or protonation of the N atom.5 It is therefore an ideal descriptor to monitor changes in e.g. the structure of a pyridine-based complex. The solid state 15N NMR spectrum of Co4O4(OAc)4{15N}Py4 measured under static conditions shows a typical powder pattern from which the 15N chemical shift tensor and the symmetry of the tensor have been extracted to be δt = 465(5) ppm (δ11), δr = 405(5) ppm (δ22) and δ⊥ = −54(1) ppm (δ33, δref(15NH3 liq.) = 0 ppm). To obtain a deeper insight, DFT calculations of the chemical shifts were performed using the Polarisable Continuum Model approximation assuming that the effects of non-covalent interactions and the crystal field on the chemical shielding are small, and that a change in the dielectric constant has a negligible effect (see ESI‡ for details).14,15 The calculated tensors vary between 480 ppm (δt), 440 ppm (δr) and −91 ppm (δ⊥), in good agreement with the observed values.
The solid-state 15N NMR spectrum of Co4O4(OAc)4{15N}Py4 recorded under MAS conditions, gives an isotropic chemical shift of 246.6 ppm, with a linewidth of 730 Hz. This rather large linewidth is most likely caused by the distribution of the N⋯Co distances, between 1.954(3) and 1.965(2) Å (Table S13‡).|| At first sight, it can be assumed that there are three overlapping signals (239(3), 248(2), 259(2) ppm, Fig. 1b), with the central signal being twice as intense as the side ones. Indeed, we would expect such a pattern from the single crystal structure, as we see three sets of N⋯Co distances (Tables S5 and S7‡).
The effect of Co coordination on the 15N chemical shift of pyridine is very different from that caused by hydrogen bonding. Hydrogen bonding leads to a strong change in the tangential component of the tensor.16,17 Coordination to CoIII, however, leads to changes in both the tangential and the perpendicular components of the tensor (Tables S5 and S7‡). These changes are unidirectional and result in a large change in the isotropic value, compared to the chemical shift of free pyridine in the solid state (314 ppm (ref. 18)). Also compared to the impact of hydrogen bonding in the solid state, the change upon coordination in chemical shift (Δδ15N) of pyridine is large, with −68 ppm compared to −20 ppm in case of hydrogen bonding to water.18 The Δδ15N values observed both in the solid state and in solution (vide infra) for pyridine coordinated to CoIII in the cubane are in line with the reported Δδ15N value for transition metal complexes containing pyridine-moieties as ligands.5
Next, we studied the cubane dissolved in different solvents. In aprotic solvents i.e. acetonitrile (244.8 ppm) and dichloromethane (243.3 ppm), the 15N chemical shifts are in excellent agreement with the isotropic chemical shift observed by MAS NMR (246.6 ppm, Fig. 1c). When dissolved in neutral water or basic media (pH of catalysis, see below), the signal shifts to 233.3 ppm independent of the pH value. Thus, the N atom is more shielded in aqueous environments compared to the solid state or dissolved in aprotic solvents. Importantly, no traces of free or hydrogen-bonded pyridine are observed.18 Thus, the small changes in chemical shift observed between aqueous and aprotic conditions as well as the solid state cannot be explained by de-coordination of a pyridine even in a very fast equilibrium. In the case of a fast equilibrium, a low field shift would have been expected, as only the time average of the chemical shifts would have been observed.18 An increase in the Co⋯N bond length should result in a reduction of Δδ15N (Tables S5 and S7‡) and thus in a low field shift of the isotopic chemical shift, which we did not observe. Although, the isotropic 15N chemical shifts of the cubane are different in aqueous solution and in the polycrystalline state, the structure of the complex is preserved, i.e. the Co⋯N bond not only does not break in aqueous solutions but, apparently, becomes somewhat shorter.
We further studied the catalytically active Co4O4 core by 59Co NMR spectroscopy. The linewidth of the 59Co NMR signal is highly sensitive to the symmetry of the ligand atom combination.19 With decreasing symmetry of the ligand atom combination, the NMR signal becomes broader or vanishes completely. The linewidth of the 59Co chemical shift increases from 3.7 kHz in acetonitrile, 4.4 kHz in dichloromethane to more than 22.5 kHz in aqueous solution, again independent of the pH value (Fig. 1d). Similarly, for a series of Co complexes, a change in linewidth by a factor of approx. 6 has been reported when acetonitrile was replaced by water, the increase in linewidth has been attributed to additional hydrogen bonding interactions.20 To allow for significant hydrogen bonding interaction with water, some of the labile acetate ligands are (partially) replaced by a water molecule and a hydroxyl group.8 This ligand exchange not only allows for a better hydrogen bonding interaction, but also decreases the ligand group symmetry which may also contribute to the observed increase in linewidth.21 Other effects that may affect the linewidth such as scalar coupling can be neglected as the 59Co NMR spectra for complexes with 15N-labelled and natural abundance pyridine gave the same linewidth (Fig. S1, and section S4‡ for further discussion). The chemical shift is also affected by the solvent and thus partial ligand exchange: from about 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 770 ppm for the pristine Co4O4(OAc)4Py4 to 12070 ppm for the cubane in aqueous solution. Similarly, 59Co NMR spectra can be acquired under MAS conditions (with spinning speed exceeding the second order quadrupolar shift). By recording spectra at two different magnetic fields, an isotropic 59Co shift of 12030 ppm was extracted for Co4O4(OAc)4Py4 in the solid state. Partial exchange of acetate by H2O/OH is further evidenced by 1H and 1H–13C double-CP MAS NMR spectra showing a signal at approx. −1 ppm, characteristic for isolated OH groups such as in MgO, γ-Al2O3 or zinc-based metal–organic frameworks.22–24
770 ppm for the pristine Co4O4(OAc)4Py4 to 12070 ppm for the cubane in aqueous solution. Similarly, 59Co NMR spectra can be acquired under MAS conditions (with spinning speed exceeding the second order quadrupolar shift). By recording spectra at two different magnetic fields, an isotropic 59Co shift of 12030 ppm was extracted for Co4O4(OAc)4Py4 in the solid state. Partial exchange of acetate by H2O/OH is further evidenced by 1H and 1H–13C double-CP MAS NMR spectra showing a signal at approx. −1 ppm, characteristic for isolated OH groups such as in MgO, γ-Al2O3 or zinc-based metal–organic frameworks.22–24
Advantageously, the NMR experiments discussed offer sufficient sensitivity to study Co4O4(OAc)4Py4 under catalytic conditions. In order to verify the catalytic activity, Co4O4(OAc)4Py4 was dissolved in an aqueous borate buffer at pH 8 containing 6 mM Na2S2O8 as a sacrificial electron acceptor and 1 mM Ru(bpy)3Cl2 as photosensitiser. Irradiation with a 100 W lamp yields turnover frequencies of up to 1.3 × 10−3 s−1 (Fig. S3‡), in line with reports from Dismukes and co-workers (10−2 s−1, pH 7, carbonate buffer, 250 W lamp)25 or Sartorel and co-workers (0.2 × 10−3 s−1, pH 8, borate buffer, 450 nm LEDs, Table S9‡).26 To shed light on the molecular structure of the cubane after catalysis, the photocatalytic experiments were also performed in an NMR tube and in deuterated solvents under otherwise similar conditions. The 15N NMR and 59Co NMR spectra of the cubane dissolved in the reaction mixture and recorded before and after the reaction are the same as those observed in aqueous solutions (Fig. S4‡). No change in the 59Co NMR linewidth is observed, confirming that no change in the molecular structure has occurred. Thus, a coordination of borate to the cubane core can be ruled out, which has been discussed in stabilizing the cubane core.27 However, such a coordination would change the coordination sphere of the Co centre, affecting its linewidth (see above). Similarly, 1H NMR spectra did not show any changes in the cubane signals, in particular no free pyridine is observed, nor are there any changes in the acetate region of the spectrum, in contrast to bipyridine-based analogues.27
To showcase the potential of NMR in establishing the local structure, e.g. Co's ligand group symmetry and N coordination, we also studied an amorphous PCP in which the cubane core acts as a node. In this PCP, the cubane nodes are interconnected with each other by replacing monodentate pyridine with tridentate 2,4,6-tri(4-pyridyl)-1,3,5-triazine (TPT) to form Co4TPT (Fig. 2a).8 First we demonstrate the activity of Co4TPT as an OER photocatalyst in the presence of Ru(bpy)3Cl2 as a photosensitiser. Under otherwise identical conditions to Co4O4(OAc)4Py4, Co4TPT gives a TOF of 3.0 × 10−3 s−1, which is stable over several cycles (Fig. S5‡). Compared to the molecular Co4O4(OAc)4Py4, the TOF increases by a factor of 3 when using the heterogeneous catalyst (Fig. S3‡). The higher activity of Co4TPT is not due to decomposition of the cubane core after heterogenization. First, only traces of leached Co species are detected in the supernatant after each cycle (Table S10‡). Note that traces of decomposed cubane, the presence of which depends on the reaction conditions, have been reported to be catalytically inactive.26 Second, the IR spectrum of the spent catalyst confirms the stable nature of the framework, i.e. the Co⋯TPT coordination bonds, with the labile acetate ligands most likely replaced by water and OH groups (Fig. S6 & ESI‡ for further discussion).8,28 Finally, the presence of free pyridine moieties,8 which could be responsible for the change in activity, was ruled out by 14N solid-state NMR spectroscopy (see below). Thus, the higher activity of Co4TPT compared Co4O4(OAc)4Py4 is most likely due to the change of the electron density on the active cubane core. The replacement of a proton (Hammett constant of 0) in the 4-position of the pyridine by a triazine core (Hammett constant of 0.2929) in the framework TPT-linker causes a lower electron density on the pyridine and subsequently on the Co core. For a series of molecular cubanes, Wang et al. correlated a lower electron density on the Co core with a higher reaction rate.30 The PCP framework controls the catalytic activity in the same way as a molecular ligand in homogeneous catalysis and thus behaves as a macroligand in oxidative reactions. Similar behaviour of different PCPs has already been demonstrated for various reduction reactions.31–33
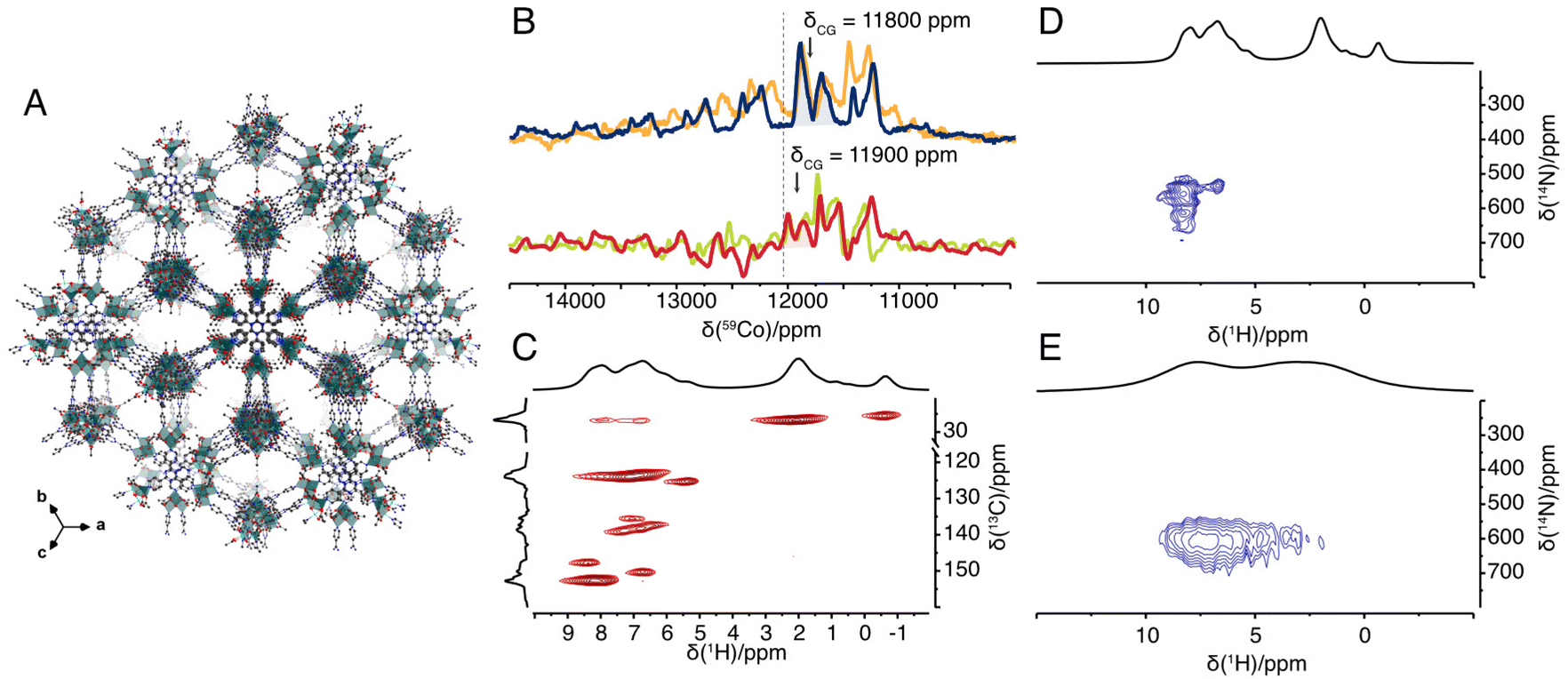 | ||
| Fig. 2 (A) Idealised structural model of Co4TPT.8 (B) 59Co solid-state NMR spectrum of Co4O4(OAc)4Py4 acquired under 70 kHz (blue), 62.5 kHz (orange) MAS rate at 14.09 T B0 field (600 MHz) and under 55 kHz (red), 50 kHz (green) MAS rate at 18.79 T B0 field (800 MHz) with indicated centre of gravity of the isotropic signal and pure isotropic chemical shift of 12030 ppm (dashed line). (C) 2D 1H–13C double-CP spectrum of Co4O4(OAc)4Py4. (D and E) 2D 1H–14N TRAPDOR-HMQC spectrum of Co4O4(OAc)4Py4 (D) and of Co4TPT (E). All spectra in (C–E) were recorded at a MAS frequency of 62.5 kHz MAS rate at 14.09T. | ||
To rule out that free pyridine moieties8 are responsible for the change in activity, we studied Co4TPT by solid-state NMR spectroscopy. Selective labelling of the linker TPT with 15N on its pyridyl moiety would require multi-step synthesis schemes. To avoid such costly schemes, we have instead studied the amorphous PCP using proton-detected nitrogen-14 NMR spectroscopy (TRAPDOR-HMQC).12 For comparison, the molecular cubane was also studied using the same techniques. In contrast to the molecular complex, the 1H NMR spectrum of the amorphous PCP shows two broad peaks at around 7 and 3 ppm of aromatic and aliphatic protons, respectively. Likewise, the 13C NMR spectrum shows broader peaks and thus loses some of the fine information obtained for the molecular complex (Fig. S2‡). However, the proton-detected 14N NMR experiments yield very similar 14N chemical shifts for the pyridine moiety centred at around 600 ppm (Fig. 2D and E). This confirms the very similar coordination geometry of the pyridine N atoms in Co4O4(OAc)4Py4 and Co4TPT. In addition, observed difference between 15N and 14N shift allows estimation of 14N quadrupolar coupling constant to CQ = 2.9 MHz (assuming η = 0 see ESI 5.3‡). Note that similar differences between the observed nitrogen-14 and nitrogen-15 shifts have previously been described and are caused by the contribution of the isotropic second order quadrupolar shift to the nitrogen-14 shift.12,34 All together with the knowledge obtained from pair distribution function analysis and X-ray absorption spectroscopy that the Co4O4 core remains intact after integration into the amorphous PCP,8 we now have a complete picture of the molecular structure of the active site.
Conclusions
We have demonstrated a step forward in the analysis of Co4O4-based model catalyst of nature's PS II, made possible by combining 14/15N and 59Co liquid-state NMR with solid-state NMR under high MAS rates and carefully tuned pulse sequences, and DFT calculations. This allows to understand changes in N⋯Co coordination distance on 15N chemical shift, to assign all nitrogen resonances resolved in the spectrum of the molecular complex Co4O4(OAc)4Py4 and to disclose its behaviour under catalytic conditions. For the first time the coordination chemistry of pyridine moieties in an amorphous PCP has been determined, which is crucial for the understanding of their catalytic performance. The reported proton-detected 14N NMR experiments are an important addition to the conventional repertoire of solid-state NMR. Notably, they allow to avoid costly and challenging labelling schemes to introduce 15N atoms at selected positions on the molecules, and allow a reduction in the amount of sample required, which in turn allows to use of fast MAS to achieve high spectral resolution, thus increasing the amount of information.Author contributions
F.U.: investigation (synthesis, catalysis), writing – review. M.S.: investigation (synthesis, characterization), writing – review. F.M.: investigation (SC XRD), formal analysis, writing – review. I.G.S.: investigation (solid state NMR & calculations), writing – original draft, review & editing. J.B. investigation (solid state NMR), conception, writing – original draft, review & editing. F.M.W.: formal analysis, conception, writing – original draft, review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. B. acknowledge Prof. Eike Brunner (TU Dresden) for experimental time for high-field 59Co and Czech Science Foundation (24-10843S) for financial support. F. M. is grateful to the Studienstiftung des Deutschen Volkes for a PhD fellowship.References
- J. Li, C. A. Triana, W. Wan, D. P. Adiyeri Saseendran, Y. Zhao, S. E. Balaghi, S. Heidari and G. R. Patzke, Chem. Soc. Rev., 2021, 50, 2444–2485 RSC.
- J. Amtawong, A. I. Nguyen and T. D. Tilley, J. Am. Chem. Soc., 2022, 144, 1475–1492 CrossRef CAS PubMed.
- Y. Zhao, D. P. Adiyeri Saseendran, C. Huang, C. A. Triana, W. R. Marks, H. Chen, H. Zhao and G. R. Patzke, Chem. Rev., 2023, 123, 6257–6358 CrossRef CAS PubMed.
- R. W. Schurko and R. E. Wasylishen, J. Phys. Chem. A, 2000, 104, 3410–3420 CrossRef CAS.
- L. Pazderski, in Annual Reports on NMR Spectroscopy, Elsevier Ltd, 1st edn, 2020, vol. 101, pp. 151–284 Search PubMed.
- R. Chakrabarty, S. J. Bora and B. K. Das, Inorg. Chem., 2007, 46, 9450–9462 CrossRef CAS PubMed.
- P. Granger, in Encyclopedia of Magnetic Resonance, John Wiley & Sons, Ltd, Chichester, UK, 2007 Search PubMed.
- A. I. Nguyen, K. M. Van Allsburg, M. W. Terban, M. Bajdich, J. Oktawiec, J. Amtawong, M. S. Ziegler, J. P. Dombrowski, K. V. Lakshmi, W. S. Drisdell, J. Yano, S. J. L. Billinge and T. D. Tilley, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 201815013 Search PubMed.
- R. Jabbour, C. W. Ashling, T. C. Robinson, A. H. Khan, D. Wisser, P. Berruyer, A. C. Ghosh, A. Ranscht, D. A. Keen, E. Brunner, J. Canivet, T. D. Bennett, C. Mellot-Draznieks, A. Lesage and F. M. Wisser, Angew. Chem., Int. Ed., 2023, 62, e202310878 CrossRef CAS.
- H.-L. Deng, X.-S. Luo, Z. Lin, J. Niu and M.-H. Huang, J. Org. Chem., 2021, 86, 16699–16706 CrossRef CAS PubMed.
- Y. Nishiyama and N. T. Duong, J. Magn. Reson. Open, 2022, 10–11, 100062 CrossRef.
- B. P. Tatman, H. Modha and S. P. Brown, J. Magn. Reson., 2023, 352, 107459 CrossRef CAS.
- J. Blahut, A. L. Lejeune, S. Ehrling, I. Senkovska, S. Kaskel, F. M. Wisser and G. Pintacuda, Angew. Chem., Int. Ed., 2021, 60, 21778–21783 CrossRef CAS PubMed.
- I. G. Shenderovich, J. Phys. Chem. A, 2023, 127, 5547–5555 CrossRef CAS.
- I. G. Shenderovich and G. S. Denisov, Molecules, 2021, 26, 1283 CrossRef CAS.
- M. S. Solum, K. L. Altmann, M. Strohmeier, D. A. Berges, Y. Zhang, J. C. Facelli, R. J. Pugmire and D. M. Grant, J. Am. Chem. Soc., 1997, 119, 9804–9809 CrossRef CAS.
- P. Lorente, I. G. Shenderovich, N. S. Golubev, G. S. Denisov, G. Buntkowsky and H.-H. Limbach, Magn. Reson. Chem., 2001, 39, S18–S29 CrossRef CAS.
- S. Sharif, I. G. Shenderovich, L. González, G. S. Denisov, D. N. Silverman and H.-H. Limbach, J. Phys. Chem. A, 2007, 111, 6084–6093 CrossRef CAS.
- N. Juranić, Coord. Chem. Rev., 1989, 96, 253–290 CrossRef.
- S. C. F. Au-Yeung and D. R. Eaton, J. Magn. Reson., 1983, 52, 366–373 CAS.
- A. Yamasaki, J. Coord. Chem., 1991, 24, 211–260 CrossRef CAS.
- C. Chizallet, H. Petitjean, G. Costentin, H. Lauron-Pernot, J. Maquet, C. Bonhomme and M. Che, J. Catal., 2009, 268, 175–179 CrossRef CAS.
- A. T. F. Batista, D. Wisser, T. Pigeon, D. Gajan, F. Diehl, M. Rivallan, L. Catita, A.-S. Gay, A. Lesage, C. Chizallet and P. Raybaud, J. Catal., 2019, 378, 140–143 CrossRef CAS.
- S. Glante, D. Wisser, M. Hartmann, M. Joos, R. E. Dinnebier and S. Bette, Cryst. Growth Des., 2022, 22, 3795–3807 CrossRef CAS.
- N. S. McCool, D. M. Robinson, J. E. Sheats and G. C. Dismukes, J. Am. Chem. Soc., 2011, 133, 11446–11449 CrossRef CAS PubMed.
- A. Genoni, G. La Ganga, A. Volpe, F. Puntoriero, M. Di Valentin, M. Bonchio, M. Natali and A. Sartorel, Faraday Discuss., 2015, 185, 121–141 RSC.
- P. F. Smith, C. Kaplan, J. E. Sheats, D. M. Robinson, N. S. McCool, N. Mezle and G. C. Dismukes, Inorg. Chem., 2014, 53, 2113–2121 CrossRef CAS.
- A. I. Nguyen, J. Wang, D. S. Levine, M. S. Ziegler and T. D. Tilley, Chem. Sci., 2017, 8, 4274–4284 RSC.
- M. A. Fávaro, J. Yang, D. Ditz, H. Küçükkeçeci, M. H. Alkhurisi, S. Bergwinkl, A. Thomas, E. A. Quadrelli, R. Palkovits, J. Canivet and F. M. Wisser, ChemCatChem, 2023, 15, e202300197 CrossRef.
- Z. Luo, Y. Hou, J. Zhang, S. Wang and X. Wang, Beilstein J. Org. Chem., 2018, 14, 2331–2339 CrossRef CAS PubMed.
- F. M. Wisser, P. Berruyer, L. Cardenas, Y. Mohr, E. A. Quadrelli, A. Lesage, D. Farrusseng and J. Canivet, ACS Catal., 2018, 8, 1653–1661 CrossRef CAS.
- F. M. Wisser, Y. Mohr, E. A. Quadrelli, D. Farrusseng and J. Canivet, ChemCatChem, 2018, 10, 1778–1782 CrossRef CAS.
- F. M. Wisser, Y. Mohr, E. A. Quadrelli and J. Canivet, ChemCatChem, 2020, 12, 1270–1275 CrossRef CAS.
- A. S. Tatton, T. N. Pham, F. G. Vogt, D. Iuga, A. J. Edwards and S. P. Brown, CrystEngComm, 2012, 14, 2654–2659 RSC.
Footnotes |
| † Raw data available on Zenodo: https://doi.org/10.5281/zenodo.10683598. |
| ‡ Electronic supplementary information (ESI) available: Synthesis, additional characterization, catalysis and DFT calculations. CCDC 2239008. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01273a |
| § Present address: Bavarian Polymer Institute and Department of Chemistry, University of Bayreuth, Bayreuth 95447, Germany. |
| ¶ Present address: University of Augsburg, Institute of Physics, Chair of Solid State and Materials Chemistry, Universitätsstraße 1, 86159 Augsburg, Germany. |
| || Crystallographic data for Co4O4(OAc)4Py4 are provided as cif file (CCDC 2239008‡). |
| This journal is © The Royal Society of Chemistry 2024 |