DOI:
10.1039/D4DT01112K
(Paper)
Dalton Trans., 2024,
53, 10446-10452
A Bis(silylene)silole – synthesis, properties and reactivity†
Received
15th April 2024
, Accepted 24th May 2024
First published on 29th May 2024
Abstract
A 1,1-bis(silylene)silole has been synthesised by a double salt-metathesis reaction from potassium silacyclopentadienediide, K2[1], and an amidinato-stabilized silylene chloride in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio. The red colour of the title compound is due to the lp(Si)/π*(silole) transition. This band is bathochromically shifted compared to that of other 1,1-bissilylsiloles suggesting enhanced conjugation between the silole π-system and the newly formed Si(II)–Si(IV)–Si(II) group. The bissilylene is easily oxidised by the elemental chalcogens S, Se, and Te and forms a bissilaimide by reaction with an arylazide.
:2 ratio. The red colour of the title compound is due to the lp(Si)/π*(silole) transition. This band is bathochromically shifted compared to that of other 1,1-bissilylsiloles suggesting enhanced conjugation between the silole π-system and the newly formed Si(II)–Si(IV)–Si(II) group. The bissilylene is easily oxidised by the elemental chalcogens S, Se, and Te and forms a bissilaimide by reaction with an arylazide.
Introduction
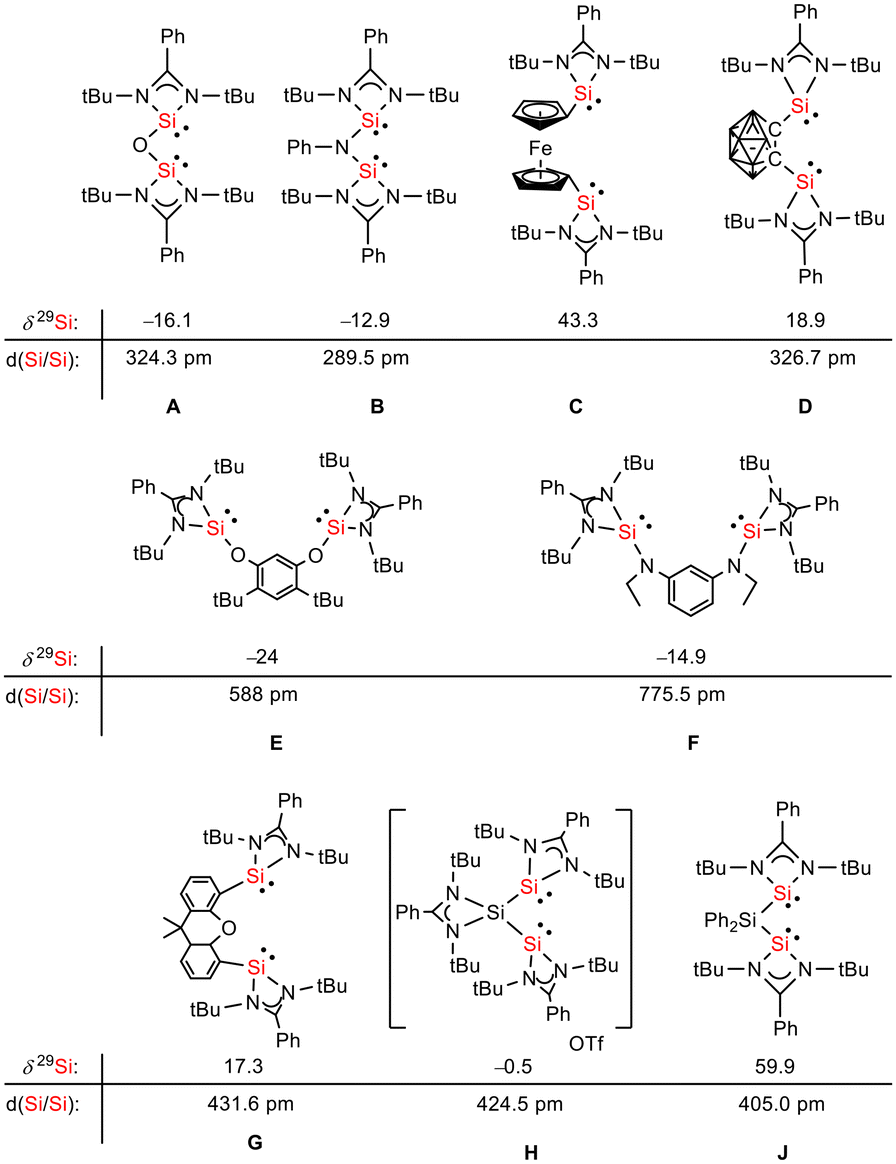
Siloles and amidinato silylenes represent important branches in the growing tree of organosilicon chemistry. The unique photophysical properties of siloles and their derivatives have opened new perspectives for materials science.1 The discovery of the aggregation induced emission (AIE) effect is here only one of the most prominent developments.2 Amidinato silylenes with tricoordinated silicon atoms show unique nucleophilic properties. Since the first report on their synthesis by H. Roesky and coworkers in 2006,3 their use as stabilizing ligands in main group and transition metal chemistry has strongly influenced the area of small molecule and bond activation.4–7 In this respect the work of Driess and coworkers is of particular interest.8–13 This group developed a series of bissilylenes A–G (Fig. 1)11,14–19 that show interesting cooperative reactivity that is useful for the synthesis of unusual main group compounds and makes these compounds valuable ligands in coordination chemistry. The groups of So and Roesky/Stalke added to this series bissilylenes H and J.20,21 In these two examples the silylenes are separated by tetracoordinated silicon atoms and the resulting Si–Si bonds are unusually long (241–245 pm). This suggests relatively small Si–Si bond energies and therefore small separations between σ- and σ*-orbitals with favourable preconditions for σ-conjugation.22 Before the background of these considerations, our motivation to combine the silole group with tricoordinated amidinato silylenes was twofold. We were interested in the influence of the two Si(II) substituents on the electronic situation of the silole ring and we were curious to study whether cooperative reactivity between the two nucleophilic Si(II) centres is possible when held together by the silole group.
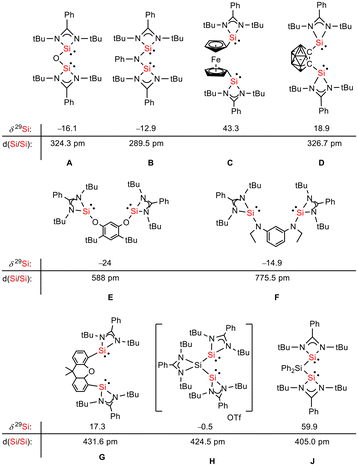 |
| | Fig. 1 Examples of chelating bissilylene ligands A–J, their 29Si NMR chemical shifts, δ29Si, and their Si/Si separation, d(Si/Si). | |
Results and discussion
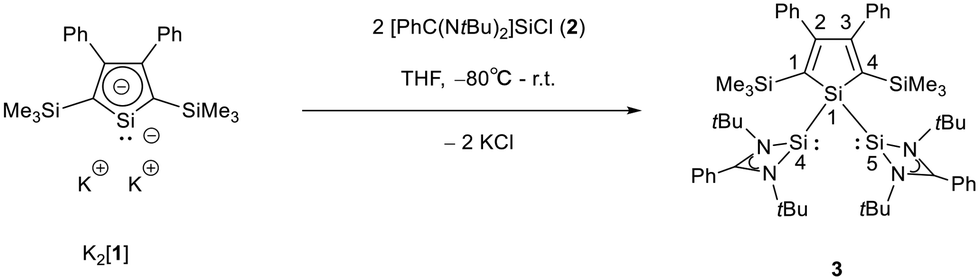
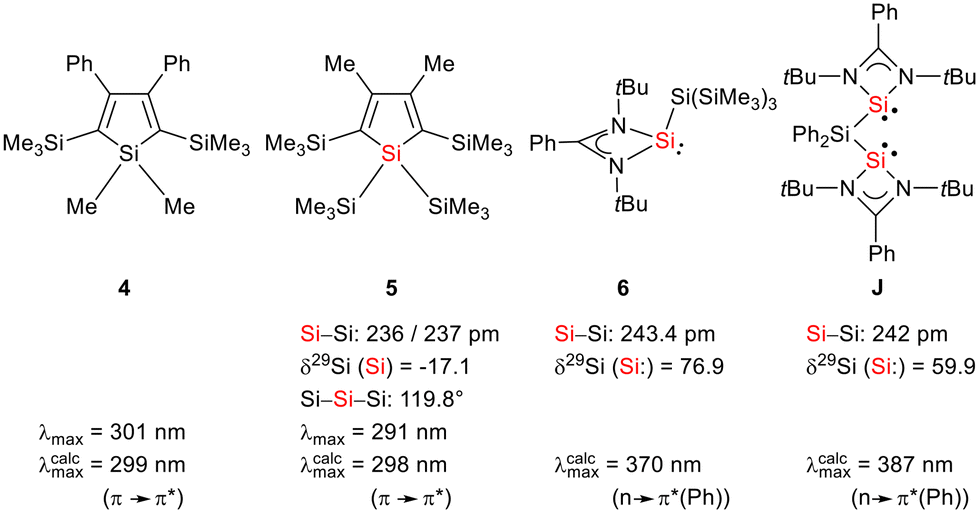
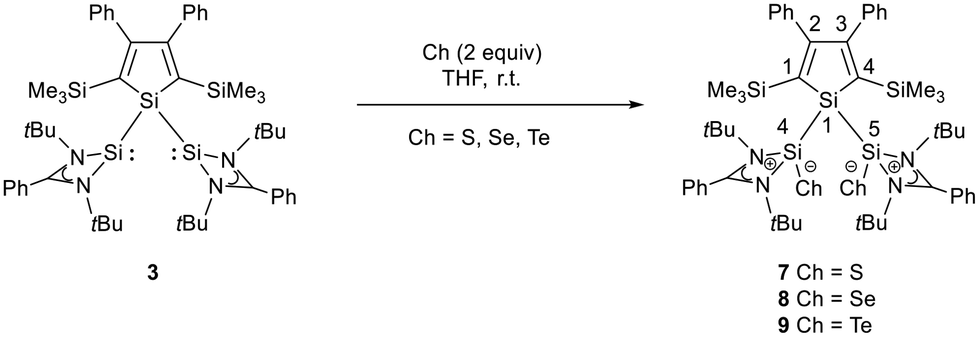
The starting materials of choice were silole dianions [1]2− and amidinate-stabilized silylene chloride RSi(:)Cl (R = PhC(NtBu)2), 2.3,23 Treatment of dipotassium silacyclopentadienediide K2[1] with silylene chloride 2 in a molar ratio of 1:2 in THF gives the expected bis(silylene)silole 3 in 65% yield (Scheme 1). The formation of silole 3 was confirmed by NMR spectroscopy. The 1H NMR spectrum of silole 3 reveals two singlets, one for the tBu groups and one corresponding to the trimethylsilyl groups, as well as one set of resonances for the phenyl groups of the amidinate ligand and of the silole ring (see the ESI†). In the 29Si NMR spectrum, three signals were detected at δ29Si = −10.6, 6.5, and 65.6, which were assigned to the trimethylsilyl group, the silole (Si1) and silylene silicon atoms (Si4, Si5), respectively. The assignment is supported by the detection of a 1J(Si1–Si4/5) coupling constant of 62 Hz for the two resonances at high frequencies. The 29Si NMR resonance of the silylene silicon atoms Si4/5 appears downfield shifted from those of the starting material, amidinato silylene chloride 2 (δ29Si = 14.6),3 but in the same chemical shift region as those of the tris(trimethylsilyl)-substituted benzamidinato silylene 6 and the diphenylsilylene separated bissilylene J (Fig. 1 and 2),24 confirming the deshielding effect of the silicon substituent for this class of silylenes. While the silole silicon atom is strongly shielded by Δδ29Si = −142 compared to that of K2[1] (δ29Si = 148.5),23 its 29Si NMR resonance appears low field shifted compared to disilyl-substituted siloles, such as bissilylsilole 5 (δ29Si = −17.1).25 The origin of the red colour of a solution of silole 3 in n-hexane is a long wave absorption band at λ = 474 nm. In contrast, structurally strongly related siloles such as 1,1-dimethylsilole 4 and 1,1-bis(trimethylsilyl)silole 5 are colourless and show long-wave absorptions in the near UV-region (Fig. 2).26 The colours of crystals of tris(trimethylsilyl)silyl substituted amidinato silylene 6 and of bissilylene J are yellow, which suggests an absorption band around 400 nm.21,24 This is in qualitative agreement with calculated long-wave UV-absorption bands at λmax = 370 nm (6) and at λmax = 387 nm (J) (M06-2X/6-311+G(d,p), Fig. 2).27,28 This comparison indicates that the combination of the silole ring with two amidinato silylene substituents leads to a significant bathochromic shift of the long-wave absorption in bis(silylene)silole 3.
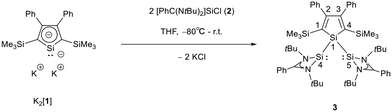 |
| | Scheme 1 Synthesis of silole bridged bissilylene complex 3. | |
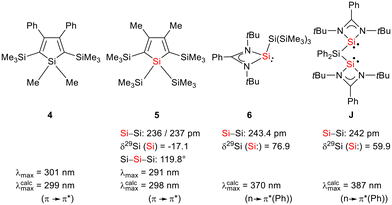 |
| | Fig. 2 Structural and spectroscopic data of selected siloles and silicon(II) amidinates. | |
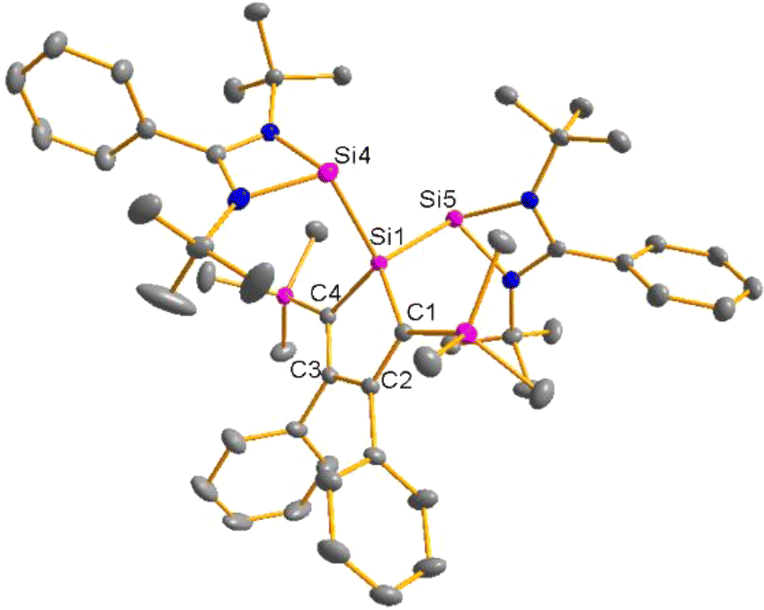
Red crystals of bis(silylene)silole 3 suitable for single crystal X-ray diffraction (sc-XRD) analysis were obtained from a saturated Et2O solution at 5 °C and its molecular structure is pictured in Fig. 3. The central silacyclopentadiene ring shows the expected planar structure (the distance of Si1 from the least square plane of the four carbon atoms is 1.9 pm) with a short/long/short C–C bond length variation of the butadiene part and regular Si1–C1 and Si1–C4 bond lengths (Fig. 2). The silicon atom Si1 is tetracoordinated and the plane spanned by Si1 and the two silylene silicon atoms Si4 and Si5 is almost orthogonally oriented to the silole ring (the dihedral angle between both planes is 80.7°). The Si–Si bonds are long (245.9 and 246.3 pm) compared to exocyclic Si–Si bonds in siloles (e.g. silole 5)25 but are in the expected region for exocyclic Si–Si bonds of amidinate-stabilized silylenes (e.g. silylene 6 and bissilylenes H and J (Si–Si = 240–245 pm)).20,24 The Si4–Si1–Si5 bond angle is 108.9°, close to the ideal tetrahedral angle. The coordination environment of the two tricoordinated silicon atoms Si4 and Si5 is trigonal pyramidal (sum of the bond angles α: ∑α(Si4) = 281.1° and ∑α(Si5) = 284.3°), which is consistent with the presence of a stereoactive lone pair at each silicon centre. The separation between both tricoordinated silicon atoms is 400.5 pm, in the range of related Si/Si distances in bissilylenes A–E and G–J (326–588 pm) (Fig. 1). The amidinate rings are oriented in an anti-configuration with regard to the Si4Si1Si5 plane. Therefore, the lone pairs at the silylene silicon atoms point in opposite directions.
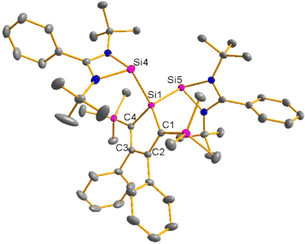 |
| | Fig. 3 Molecular structure of bis(silylene)silole 3 in the crystal (hydrogen atoms are omitted; thermal ellipsoids at 50% probability). Selected atom distances [pm] and angles [°]: Si1–Si4 245.90(6), Si1–Si5 246.35(5), Si1–C1 189.11(17), Si1–C4 189.48(15), C1–C2 137.09(22), C2–C3 147.64(23), C3–C4 137.26(19), Si4–Si1–Si5 108.91(19). | |
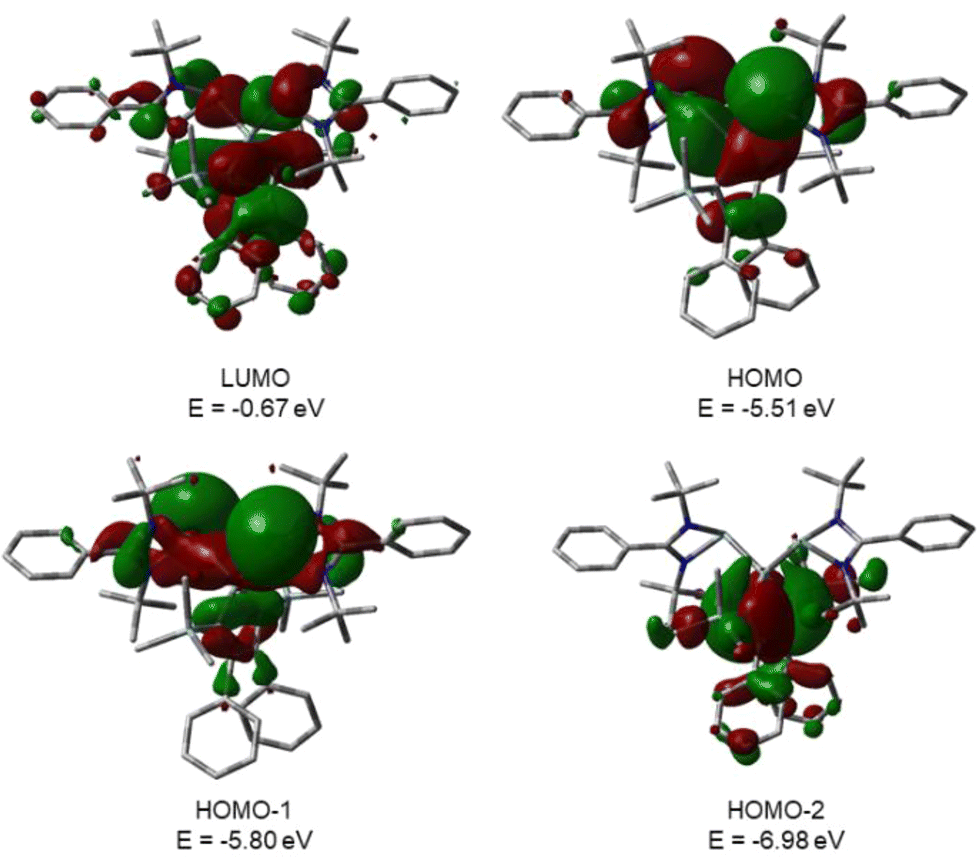
This is supported by the results of quantum mechanical calculations at the M06-2X/6-311+G(d,p) level of theory.27 The molecular structure of silole 3 at this level of theory agrees well with data that was obtained from XRD analysis. The smallest deviation between important structural parameters is less than 2% (see the ESI†). This good agreement allows a closer inspection of the electronic situation in bissilylene silole 3 by this DFT method. The HOMO−2 is dominated by contributions of the highest π-orbital of the butadiene part of the silole ring (Fig. 4). As expected, the HOMO and HOMO−1 are combinations of the lone pairs at the tricoordinated silicon atoms with significant contributions of the σ-Si–Si bonds (see Fig. 4) with the lone pairs pointing in opposite directions. As is typical for siloles, the LUMO is a combination of the π* orbitals of the butadiene part of the silole ring and the σ*-Si–Si bonds (π*/σ* conjugation).29 Different to other siloles (e.g.4 and 5, Fig. 2 and the ESI†) the long wave transition is a lp(Si)/π*(silole) transition. The HOMO/LUMO energy gap is relatively small (2.18 eV) and the lowest absorption band, which is assigned to the HOMO/LUMO transition, is calculated at 425 nm (M06-2X/6-311+G(d,p)), close to the experimental value of 474 nm. Therefore, the substitution of the silole ring with two tricoordinated silylenes leads to a change in the nature of the long wave absorption and to a significant bathochromic shift.
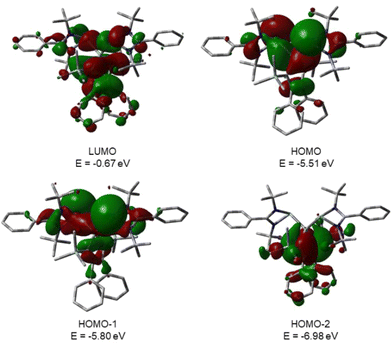 |
| | Fig. 4 Calculated surface diagrams of molecular orbitals of bissilylene silole 3 (M06-2X/6-311+G(d,p), isodensity value 0.02). | |
We investigated the reactivity of several transition metal complexes (such as Fe(0), Ni(0), Ni(II), Pd(0), Pd(II), Au(I), and Zn(II) complexes) applying different conditions and stoichiometries to probe the coordination ability of silole 3. However, in all investigated cases the reaction was not selective, and we were not able to isolate a metal complex of bis(silylene)silole 3. The reason for this failure is not clear but the unfavourable anti-orientation of the two silylene groups in the conformational ground state might contribute. Due to the lack of defined reactivity versus metal complexes, we tested the reactivity of bis(silylene)silole 3versus standard reagents such as elemental chalcogens (Scheme 2) and arylazides (Scheme 3).
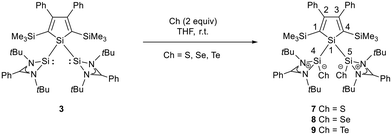 |
| | Scheme 2 Reaction of bis(silylene)silole 3 with chalcogens S, Se, and Te. | |
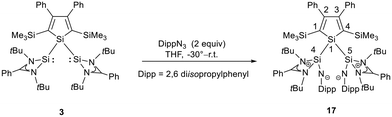 |
| | Scheme 3 Reaction of bis(silylene)silole 3 with 2,6-diisopropylphenylazide. | |
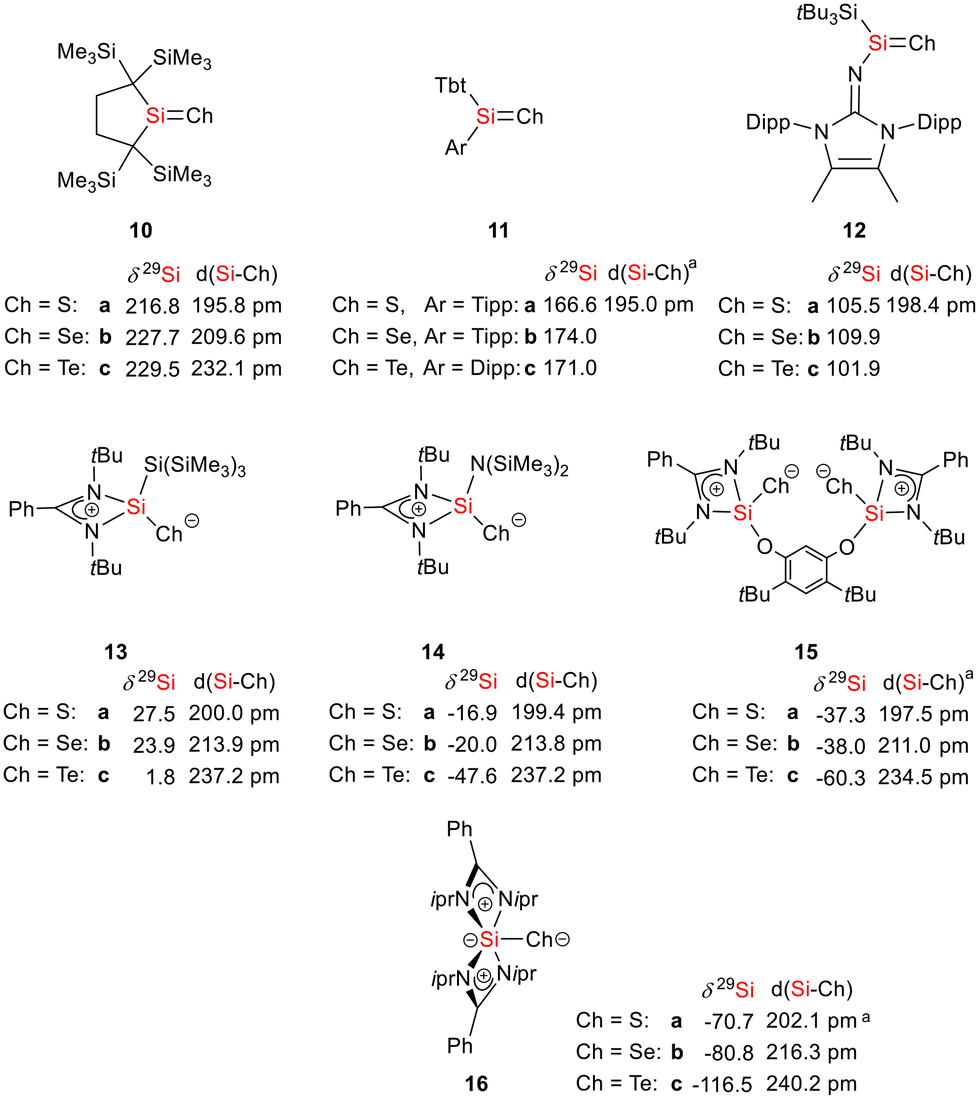
As expected bis(silylene)silole 3 is unstable in air and the controlled reactions with dioxygen (air) or oxygen atom donors (dinitrogen monoxide) are not selective. In contrast, two equivalents of the heavier chalcogens sulphur, selenium and tellurium react smoothly with silole 3 in THF at room temperature to yield the dichalcogenides 7–9 in acceptable to good yields (66–83%, Scheme 2). The identity and structure of these compounds were confirmed by NMR spectroscopy and sc-XRD. The 29Si NMR spectra of chalcogenides 7–9 display for the silylene silicon atom signals at δ29Si = 19.7 (7), 15.0 (8), and −11.7 (9), which show the expected low frequency shift going from sulphide to telluride known for tetra- and pentacoordinated silicon chalcogenides.24,30–32 These 29Si NMR signals are in the typical region of tetracoordinated amidinate stabilized silicon chalcogenides bracketed by the values for the corresponding hypersilyl- and bissilylamino-substituted chalcogenides 13 and 14 (see Fig. 5).24,33 Compared to tricoordinated silicon chalcogenides (heavy silaketones) such as 10–12, the 29Si resonances of 7–9 are significantly shifted to lower frequencies34–39 but they are clearly distinguished from those of the pentacoordinated silicon chalcogenides 16 (Fig. 5).31 Based on this assessment of the 29Si NMR chemical shifts, we place the electronic effects of the silole ring in chalcogenides 7–9 between the electron donating hypersilyl group (in 13) and the electronegative amino substituent (in 14) or the even more electronegative phenoxy substituent in the bis(silicon chalcogenide) 15.24,30,33 The silicon selenide 8 shows Si/Se satellites that are separated by 1J(SiSe) = 292 Hz, in the typical range for tetracoordinated silicon selenides (e.g. 284 Hz (14b); 247 Hz in Ter*(H)(Me4Im)Si(Se), Ter* = 2,6-bis-(2,4,6-triisopropylphenyl)phenyl) and significantly larger than is found for silylselenoethers such as (Me3Si)2Se (1JSiSe = 107 Hz).32,33 In addition, the 29Si NMR data gives no indication of any intramolecular interaction between the two SiCh groups.
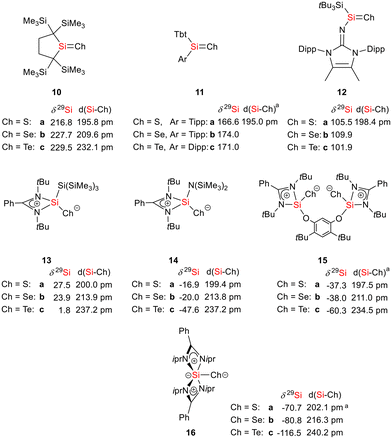 |
| | Fig. 5 Selected structural and NMR parameters of silicon chalcogenides with the silicon atom in the coordination number 3–5 (amean values). | |
The silicon selenide 8 exhibits a resonance in the 77Se{1H} NMR spectrum at δ77Se = −184.3 and the telluride 9 is characterized by one signal at δ125Te = −692.5 in the 125Te{1H} NMR spectrum. This is for both nuclei a significant shift to low frequencies compared to the tricoordinated analogues 11b (δ77Se = 635) and 11c (δ125Te = 731).37 Relative to typical tetracoordinated silicon selenides and tellurides such as 14b,c (δ77Se = −451, δ125Te = −1093)33 and 15b,c (δ77Se = −514, δ125Te = −1242)30 it is however significantly shifted to high frequencies. Although the 77Se and 125Te NMR chemical shift data for silicon chalcogenides in different coordination states are widely spread over large chemical shift ranges (δ77Se = −535 to +635; δ125Te = −1236 to +731), the here reported data are consistent as 77Se and 125Te NMR chemical shifts for this class of compounds are almost linearly correlated including compounds 8 and 9 from the present study (see the ESI† for more detailed data).40
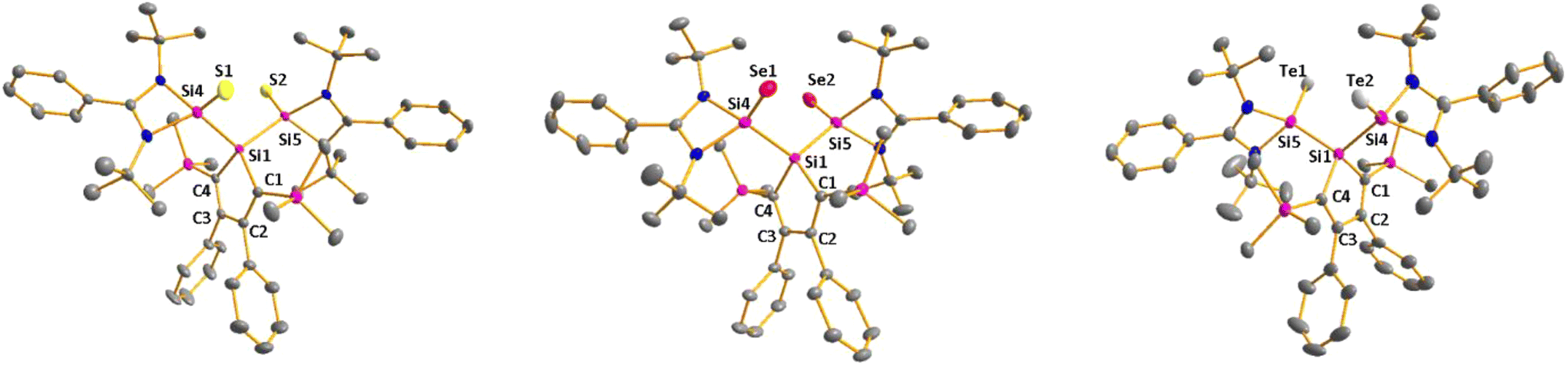
Sc-XRD analysis of suitable crystals of silicon chalcogenides 7, 8 and 9 (Fig. 6) revealed molecular structures that are close to that of the bissilylene silole 3. In all three cases, the central Si1Si4Si5 planes are oriented almost orthogonally to the silole ring. The increase of the coordination number of the silicon atoms Si4 and Si5 by oxidation of the bissilylene silole 3 resulted in slightly shorter Si1–Si4/5 bonds in the chalcogenides 7–9 (Si1–Si4/5 = 240–241 pm) compared to those in the bissilylene 3. These bonds are however still long compared to the regular bonds between tetracoordinated silicon atoms (Si–Simean = 235.8 pm). All three silicon chalcogenides 7–9 show an anti-orientation of the chalcogen substituents relative to the central Si4–Si1–Si5 plane. The lengths of the silicon chalcogen bonds (7: Si–S = 199–200 pm; 8: Si–Se = 213–214 pm; 9: Si–Te = 237 pm) are in the typical range for tetracoordinated silicon chalcogenides (e.g.13–15, Fig. 5),24,30,33 and are placed between those reported for tricoordinated (e.g.10 and 11)34,35 and pentacoordinated silicon chalcogenides (e.g.16).31 As a result of the high polarity of the Si–Ch bonds and significant contributions from negative hyperconjugation, these bonds are all short, close to the theoretically expected value for Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ch double bonds (SiS = 201 pm; SiSe = 214 pm; SiTe = 235 pm).30,32 A short discussion of the bonding of the Si–S linkage in compounds 7 and 10(S) (for comparison) supporting the use of ylidic Lewis representations is provided in the ESI (pp. 30–32†).
Ch double bonds (SiS = 201 pm; SiSe = 214 pm; SiTe = 235 pm).30,32 A short discussion of the bonding of the Si–S linkage in compounds 7 and 10(S) (for comparison) supporting the use of ylidic Lewis representations is provided in the ESI (pp. 30–32†).
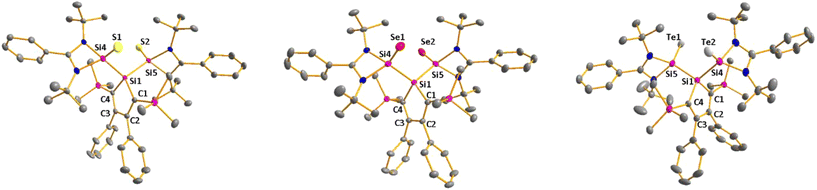 |
| | Fig. 6 Molecular structure of bissilicon sulphide 7 (left), bissilicon selenide 8 (middle) and bissilicon telluride 9 (right) in the crystal (hydrogen atoms are omitted; thermal ellipsoids at 50% probability). Selected atom distances [pm] and angles [°]: 7 Si1–Si4 239.19(7), Si1–Si5 239.76(6), Si4–S1 199.33(7), Si5–S2 199.58(7), Si1–C1 188.85(20), Si1–C4 189.34(15), C1–C2 136.41(27), C2–C3 149.48(24), C3–C4 136.68(24), Si4–Si1–Si5 117.89(2); 8 Si1–Si4 239.59(7), Si1–Si5 239.98(6), Si4–Se1 213.29(7), Si5–Se2 213.61(6), Si1–C1 188.94(13), Si1–C4 189.00(12), C1–C2 136.67(16), C2–C3 149.65(16), C3–C4 136.22(18), Si4–Si1–Si5 119.51(19); 9 Si1–Si4 241.14(12), Si1–Si5 241.62(13), Si4–Te1 237.12(11), Si5–Te2 236.85(11), Si1–C1 189.43(38), Si1–C4 189.38(36), C1–C2 136.29(52), C2–C3 149.86(46), C3–C4 136.08(54), Si4–Si1–Si5 118.98(5). | |
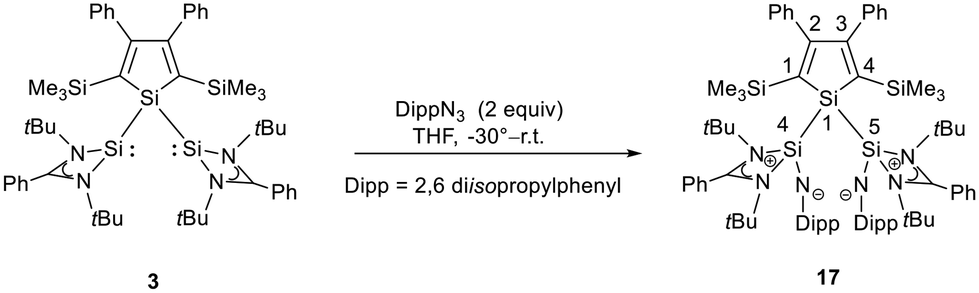
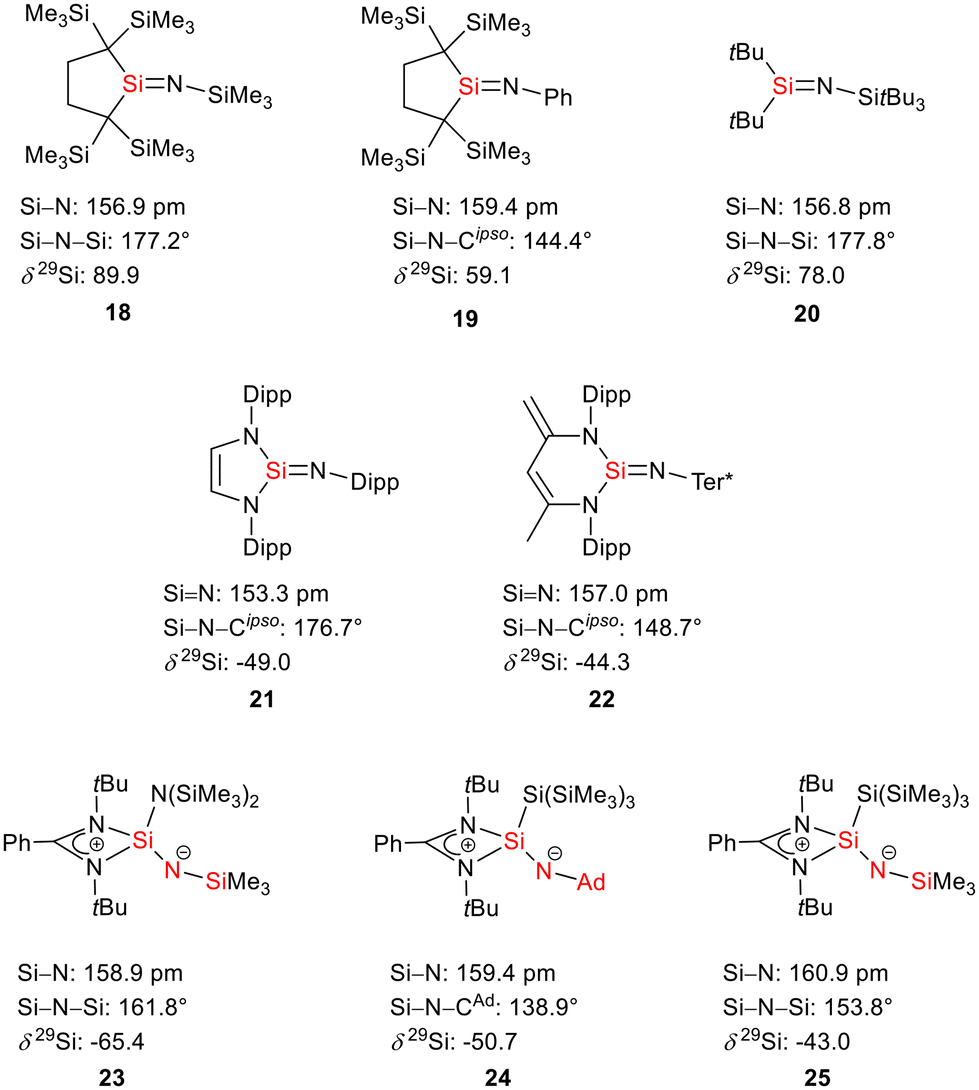
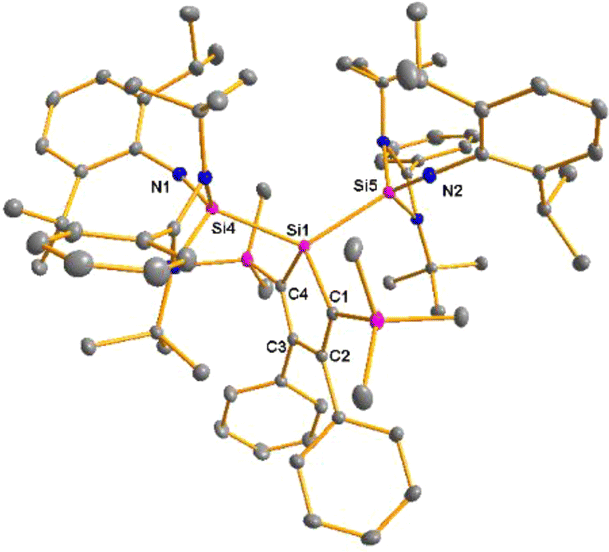
The bissilaimine 17 is formed by the reaction of two equivalents of diisopropylphenylazide, DippN3, with bissilylene 3 in THF in 71% yield (Scheme 3). It is soluble in aromatic hydrocarbons, ethers and chlorinated hydrocarbons. Crystals suitable for sc-XRD analysis were obtained from benzene solution. The 29Si{1H} NMR spectra of bissilaimine 17 display three signals at δ29Si = −9.0 (SiMe3), −20.3 (silole Si), and −73.1 (imide Si). The imide silicon nuclei are significantly shielded compared to those of silaimides with tricoordinated silicon atoms (Fig. 7, cpds 18–20).41,42 Its 29Si NMR signal is even at the low frequency end of the typical region for tricoordinated silaguanidinium derivatives 21 and 2243,44 and of tetracoordinated amidinate stabilized silicon imides (Fig. 7, cpds 23–25).44,45 Interestingly, the trimethylsilyl groups of the silole ring show at room temperature in the 1H NMR spectrum extreme line broadening (1H NMR line width at half height: ω ≈ 300 Hz), indicating the steric overloading of the silaimine 17. Crystals suitable for sc-XRD analysis were obtained from benzene solution. The molecular structure of bissilaimine 17 is of C2-symmetry with the consequence that both silaimine units have the same metrics (Fig. 8). Its main features (localized silole ring, almost orthogonally oriented silole ring and Si4/Si1/Si5 plane) are very similar to the structures of the bischalcogenides 7–9. The central Si1–Si4/Si5 bonds (245.4 pm) are of the same length as in the bissilylene 3. The Si4–N1/Si5–N2 bonds are short (159.8 pm) and compare very well with those of silaimines with tricoordinated and tetracoordinated silicon atoms (Fig. 7). The Si4–N1–Cipso/Si5–N2–Cipso bond angles (166.2°) are wider than are usually found for N-aryl/alkyl substituted silaimines (e.g. cpds 19 and 24) and approach the values found for N-silyl substituted silaimines (Fig. 7).42,45,46
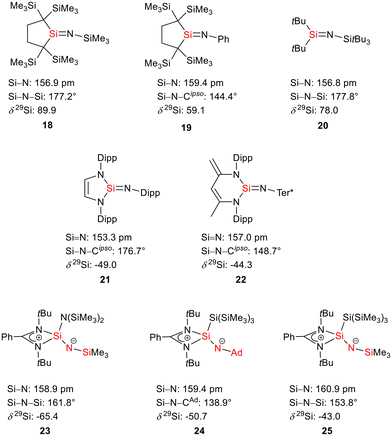 |
| | Fig. 7 Silaimines and silaguanidines with tricoordinated and tetracoordinated silicon atoms and dicoordinated nitrogen atoms (selected atom distances [pm] and angles [°]). | |
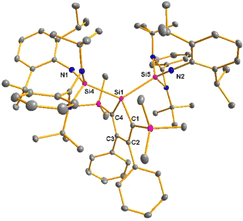 |
| | Fig. 8 Molecular structure of bissilaimine 17 in the crystal (hydrogen atoms are omitted; thermal ellipsoids at 50% probability). Selected atom distances [pm] and angles [°]: Si1–Si4 245.42(4), Si4–N1 159.83(6), Si1–C1 190.08(6), C1–C2 136.97(9), C2–C3 149.96(8), Si4–Si1–Si5 125.76(14), Si1–Si4–N1 156.16(6), Si4–N1–Cipso 166.16(2). | |
Conclusions
A 1,1-bisamidinatosilylene-silole 3 has been synthesized and characterized. In contrast to other 1,1-disubstituted siloles with a similar substitution pattern such as 4 and 5, bissilylene silole 3 is strongly coloured due to a bathochromic shift of the HOMO/LUMO transition. The nature of the long-wave absorption band changes from the regular π(silole)/π*(silole) transition of the butadiene parts of siloles 4 and 526,29 to a lp(Si)/π*(silole) transition in bissilylene silole 3 with a smaller energy separation. The stabilization of the LUMO due to more enhanced σ*/π* conjugation also contributes to the smaller HOMO/LUMO gap in silole 3. Although the distance between the molecular structure of the bissilylenesilole 3 is 400 pm, close to Si/Si distances in related bissilylenes A–E and G–J (Fig. 1), we noticed no cooperative reactivity of the two nucleophilic silicon centres. We attribute this lack of cooperativity in silole 3 to the very unfavourable anti-orientation of the silicon lone pairs in its conformational ground state in combination with the high steric crowding around the central Si4–Si1–Si5 unit which hinders the adoption by bissilylene 3 of a conformation that is more prone to cooperative interactions between the two silicon lone pairs. Bissilylene silole 3 can be easily oxidized by elemental chalcogenides (S, Se, and Te) and by reaction with Dipp-azide. The obtained amidinato silicon chalcogenides 7–9 and the amidinato silaimine 17 adopt structures that are typical for tetracoordinated amidinato silicon derivatives with short silicon chalcogen and silicon nitrogen bonds due to their high polarity and partial double bond character due to hyperconjugation.
Author contributions
Investigation, data curation, formal analysis, validation: C. L.; conceptualization, writing, visualization, funding acquisition: C. L. and T. M.; project administration, supervision, methodology, resources: T. M.; XRD: M. S.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) (MU-1440/13 and INST 184/227-1). Computations were done at the HPC Cluster CARL, University of Oldenburg, funded by the DFG (INST 184/108-1 FUGG) and the Ministry of Science and Culture (MWK) of the Lower Saxony State. C. L. thanks the China Scholarship Council for support CSC no.: 202108330061.
Notes and references
- K. Tamao, M. Uchida, T. Izumizawa, K. Furukawa and S. Yamaguchi, Silole Derivatives as Efficient Electron Transporting Materials, J. Am. Chem. Soc., 1996, 118, 11974–11975 CrossRef CAS.
- Z. Zhao, B. He and B. Z. Tang, Aggregation-induced emission of siloles, Chem. Sci., 2015, 6, 5347–5365 RSC.
- C.-W. So, H. W. Roesky, J. Magull and R. B. Oswald, Synthesis and Characterization of [PhC(NtBu)2]SiCl: A Stable Monomeric Chlorosilylene, Angew. Chem., Int. Ed., 2006, 45, 3948–3950 CrossRef CAS PubMed.
- M. Nazish, C. M. Legendre, R. Herbst-Irmer, S. Muhammed, P. Parameswaran, D. Stalke and H. W. Roesky, Synthesis and Characterization of Substituted Phosphasilenes and its Rare Homologue Stibasilene >Si=Sb–, Chem. – Eur. J., 2023, 29, e202300791 CrossRef CAS.
- M. Nazish, C. M. Legendre, N. Graw, R. Herbst-Irmer, D. Stalke, S. Sankar Dutta, U. Lourderaj and H. W. Roesky, Coordination and Stabilization of a Lithium Ion with a Silylene, Chem. – Eur. J., 2023, 29, e202203528 CrossRef CAS.
- S. S. Sen, H. W. Roesky, D. Stern, J. Henn and D. Stalke, High Yield Access to Silylene RSiCl (R = PhC(NtBu)2) and Its Reactivity toward Alkyne: Synthesis of Stable Disilacyclobutene, J. Am. Chem. Soc., 2010, 132, 1123–1126 CrossRef CAS.
- Z. Hendi, M. K. Pandey, K. Rachuy, M. K. Singh, R. Herbst-Irmer, D. Stalke and H. W. Roesky, Synthesis, Reactivity, and Complexation with Fe(0) of a Tight-bite Bis(N-heterocyclic silylene), Chem. – Eur. J., 2024, e202400389 CrossRef CAS PubMed.
- J. Xu, S. Pan, S. Yao, G. Frenking and M. Driess, The Heaviest Bottleable Metallylone: Synthesis of a Monatomic, Zero-Valent Lead Complex (“Plumbylone”), Angew. Chem., Int. Ed., 2022, 61, e202209442 CrossRef CAS.
- Y. Xiong, S. Dong, S. Yao, J. Zhu and M. Driess, Unexpected White Phosphorus (P4) Activation Modes with Silylene-Substituted o-Carboranes and Access to an Isolable 1,3-Diphospha-2,4-disilabutadiene, Angew. Chem., Int. Ed., 2022, 61, e202205358 CrossRef CAS.
- X. Chen, H. Wang, S. Du, M. Driess and Z. Mo, Deoxygenation of Nitrous Oxide and Nitro Compounds Using Bis(N-Heterocyclic Silylene)Amido Iron Complexes as Catalysts, Angew. Chem., Int. Ed., 2022, 61, e202114598 CrossRef CAS PubMed.
- Y. Xiong, S. Dong, S. Yao, C. Dai, J. Zhu, S. Kemper and M. Driess, An Isolable 2,5-Disila-3,4-Diphosphapyrrole and a Conjugated Si=P–Si=P–Si=N Chain Through Degradation of White Phosphorus with a N,N-Bis(Silylenyl)Aniline, Angew. Chem., Int. Ed., 2022, 61, e202209250 CrossRef CAS PubMed.
- S. Yao, A. Kostenko, Y. Xiong, C. Lorent, A. Ruzicka and M. Driess, Changing the Reactivity of Zero- and Mono-Valent Germanium with a Redox Non-Innocent Bis(silylenyl)carborane Ligand, Angew. Chem., Int. Ed., 2021, 60, 14864–14868 CrossRef CAS PubMed.
- S. Yao, T. Szilvási, Y. Xiong, C. Lorent, A. Ruzicka and M. Driess,
Bis(silylene)-Stabilized Monovalent Nitrogen Complexes, Angew. Chem., Int. Ed., 2020, 59, 22043–22047 CrossRef CAS.
- W. Wang, S. Inoue, S. Yao and M. Driess, An Isolable Bis-Silylene Oxide (“Disilylenoxane”) and Its Metal Coordination, J. Am. Chem. Soc., 2010, 132, 15890–15892 CrossRef CAS PubMed.
- D. Gallego, S. Inoue, B. Blom and M. Driess, Highly Electron-Rich Pincer-Type Iron Complexes Bearing Innocent Bis(metallylene)pyridine Ligands: Syntheses, Structures, and Catalytic Activity, Organometallics, 2014, 33, 6885–6897 CrossRef CAS.
- Y. Wang, A. Kostenko, S. Yao and M. Driess, Divalent Silicon-Assisted Activation of Dihydrogen in a Bis(N-heterocyclic silylene)xanthene Nickel(0) Complex
for Efficient Catalytic Hydrogenation of Olefins, J. Am. Chem. Soc., 2017, 139, 13499–13506 CrossRef CAS.
- Y.-P. Zhou, S. Raoufmoghaddam, T. Szilvási and M. Driess, A Bis(silylene)-Substituted ortho-Carborane as a Superior Ligand in the Nickel-Catalyzed Amination of Arenes, Angew. Chem., Int. Ed., 2016, 55, 12868–12872 CrossRef CAS.
- W. Wang, S. Inoue, S. Enthaler and M. Driess,
Bis(silylenyl)- and Bis(germylenyl)-Substituted Ferrocenes: Synthesis, Structure, and Catalytic Applications of Bidentate Silicon(II)–Cobalt Complexes, Angew. Chem., Int. Ed., 2012, 51, 6167–6171 CrossRef CAS.
- W. Wang, S. Inoue, E. Irran and M. Driess, Synthesis and Unexpected Coordination of a Silicon(II)-Based SiCSi Pincerlike Arene to Palladium, Angew. Chem., Int. Ed., 2012, 51, 3691–3694 CrossRef CAS.
- H.-X. Yeong, H.-W. Xi, Y. Li, K. H. Lim and C.-W. So, A Silyliumylidene Cation Stabilized by an Amidinate Ligand and 4-Dimethylaminopyridine, Chem. – Eur. J., 2013, 19, 11786–11790 CrossRef CAS.
- S. K. Kushvaha, P. Kallenbach, S. M. N. V. T. Gorantla, R. Herbst-Irmer, D. Stalke and H. W. Roesky, Preparation of a Compound with a SiII–SiIV–SiII Bonding Arrangement, Chem. – Eur. J., 2024, 30, e202303113 CrossRef CAS.
- A. Bande and J. Michl, Conformational Dependence of σ-Electron Delocalization in Linear Chains: Permethylated Oligosilanes, Chem. – Eur. J., 2009, 15, 8504–8517 CrossRef CAS PubMed.
- Z. Dong, C. R. W. Reinhold, M. Schmidtmann and T. Müller, Trialkylsilyl-Substituted Silole and Germole Dianions, Organometallics, 2018, 37, 4736–4743 CrossRef CAS.
- M. K. Bisai, V. S. V. S. N. Swamy, T. Das, K. Vanka, R. G. Gonnade and S. S. Sen, Synthesis and Reactivity of a Hypersilylsilylene, Inorg. Chem., 2019, 58, 10536–10542 CrossRef CAS.
- C. R. W. Reinhold, Z. Dong, J. M. Winkler, H. Steinert, M. Schmidtmann and T. Müller, A One-Step Germole to Silole Transformation and a Stable Isomer of a Disilabenzene, Chem. – Eur. J., 2018, 24, 848–854 CrossRef CAS.
- S. Yamaguchi, R.-Z. Jin and K. Tamao, Modification of the electronic structure of silole by the substituents on the ring silicon, J. Organomet. Chem., 1998, 559, 73–80 CrossRef CAS.
- The Gaussian 16 program was used, see the ESI† for details.
- A. Pöcheim, G. A. Özpınar, T. Müller, J. Baumgartner and C. Marschner, The Combination of Cross-Hyperconjugation and σ-Conjugation in 2,5-Oligosilanyl Substituted Siloles, Chem. – Eur. J., 2020, 26, 17252–17260 CrossRef.
- S. Yamaguchi and K. Tamao, Silole-containing σ- and π-conjugated compounds, J. Chem. Soc., Dalton Trans., 1998, 3693–3702, 10.1039/A804491K.
- M. Ghosh, P. Panwaria, S. Tothadi, A. Das and S. Khan,
Bis(silanetellurone) with C–H⋯Te Interaction, Inorg. Chem., 2020, 59, 17811–17821 CrossRef CAS.
- K. Junold, J. A. Baus, C. Burschka, D. Auerhammer and R. Tacke, Stable Five-Coordinate Silicon(IV) Complexes with SiN4X Skeletons (X=S, Se, Te) and SiX Double Bonds, Chem. – Eur. J., 2012, 18, 16288–16291 CrossRef CAS.
- D. Lutters, A. Merk, M. Schmidtmann and T. Müller, The Silicon Version of Phosphine Chalcogenides: Synthesis and Bonding Analysis of Stabilized Heavy Silaaldehydes, Inorg. Chem., 2016, 55, 9026–9032 CrossRef CAS PubMed.
- Y.-C. Chan, Y. Li, R. Ganguly and C.-W. So, Acyclic Amido-Containing Silanechalcogenones, Eur. J. Inorg. Chem., 2015, 3821–3824 CrossRef CAS.
- T. Iwamoto, K. Sato, S. Ishida, C. Kabuto and M. Kira, Synthesis, Properties, and Reactions of a Series of Stable Dialkyl-Substituted Silicon–Chalcogen Doubly Bonded Compounds, J. Am. Chem. Soc., 2006, 128, 16914–16920 CrossRef CAS.
- H. Suzuki, N. Tokitoh, S. Nagase and R. Okazaki, The First Genuine Silicon-Sulfur Double-Bond Compound: Synthesis and Crystal Structure of a Kinetically Stabilized Silanethione, J. Am. Chem. Soc., 1994, 116, 11578–11579 CrossRef CAS.
- H. Suzuki, N. Tokitoh, R. Okazaki, S. Nagase and M. Goto, Synthesis, Structure, and Reactivity of the First Kinetically Stabilized Silanethione, J. Am. Chem. Soc., 1998, 120, 11096–11105 CrossRef CAS.
- N. Tokitoh, T. Sadahiro, K. Hatano, T. Sasaki, N. Takeda and R. Okazaki, Synthesis of Kinetically Stabilized Silaneselone and Silanetellone, Chem. Lett., 2002, 31, 34–35 CrossRef.
- A. V. Protchenko, P. Vasko, D. C. H. Do, J. Hicks, M. Á. Fuentes, C. Jones and S. Aldridge, Reduction of Carbon Oxides by an Acyclic Silylene: Reductive Coupling of CO, Angew. Chem., Int. Ed., 2019, 58, 1808–1812 CrossRef CAS.
- H. Zhu, F. Hanusch and S. Inoue, Facile Bond Activation of Small Molecules by an Acyclic Imino(silyl)silylene, Isr. J. Chem., 2023, 63, e202300012 CrossRef CAS.
- Correlation between 77Se and 125Te compounds of equivalent compounds, see: H. C. E. McFarlane and W. McFarlane, J. Chem. Soc., Dalton Trans., 1973, 2416–2418 RSC.
- N. Wiberg, K. Schurz and G. Fischer, Isolation of the Stable Silaketimine tBu2Si=N-SitBu3, Angew. Chem., Int. Ed. Engl., 1985, 24, 1053–1054 CrossRef.
- T. Iwamoto, N. Ohnishi, Z. Gui, S. Ishida, H. Isobe, S. Maeda, K. Ohno and M. Kira, Synthesis and structure of stable base-free dialkylsilanimines, New J. Chem., 2010, 34, 1637–1645 RSC.
- L. Kong and C. Cui, Synthesis and Reactivity of a Base-Free N-Heterocyclic Silanimine, Organometallics, 2010, 29, 5738–5740 CrossRef CAS.
- P. P. Samuel, R. Azhakar, R. S. Ghadwal, S. S. Sen, H. W. Roesky, M. Granitzka, J. Matussek, R. Herbst-Irmer and D. Stalke, Stable Silaimines with Three- and Four-Coordinate Silicon Atoms, Inorg. Chem., 2012, 51, 11049–11054 CrossRef CAS.
- M. K. Bisai, V. Sharma, R. G. Gonnade and S. S. Sen, Reactivities of Silaimines with Boranes: From Cooperative B–H Bond Activation to Donor Stabilized Silyl Cation, Organometallics, 2021, 40, 2133–2138 CrossRef CAS.
- N. Wiberg and K. Schurz, Zur Kenntnis des stabilen Silanimins tBu2Si=N-SitBu3 und seiner Donoraddukte, Chem. Ber., 1988, 121, 581–589 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Marc
Schmidtmann
and
Thomas
Müller
,
Marc
Schmidtmann
and
Thomas
Müller