
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Activating a high-spin iron(II) complex to thermal spin-crossover with an inert non-isomorphous molecular dopant†
Malcolm A.
Halcrow
 *a,
Hari Babu
Vasili
b,
Christopher M.
Pask
a,
Alexander N.
Kulak
a and
Oscar
Cespedes
b
*a,
Hari Babu
Vasili
b,
Christopher M.
Pask
a,
Alexander N.
Kulak
a and
Oscar
Cespedes
b
aSchool of Chemistry, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK. E-mail: m.a.halcrow@leeds.ac.uk
bSchool of Physics and Astronomy, University of Leeds, W. H. Bragg Building, Leeds, LS2 9JT, UK
First published on 26th March 2024
Abstract
[Fe(bpp)2][ClO4]2 (bpp = 2,6-bis{pyrazol-1-yl}pyridine; monoclinic, C2/c) is high-spin between 5–300 K, and crystallises with a highly distorted molecular geometry that lies along the octahedral–trigonal prismatic distortion pathway. In contrast, [Ni(bpp)2][ClO4]2 (monoclinic, P21) adopts a more regular, near-octahedral coordination geometry. Gas phase DFT minimisations (ω-B97X-D/6-311G**) of [M(bpp)2]2+ complexes show the energy penalty associated with that coordination geometry distortion runs as M2+ = Fe2+ (HS) ≈ Mn2+ (HS) < Zn2+ ≈ Co2+ (HS) ≲ Cu2+ ≪ Ni2+ ≪ Ru2+ (LS; HS = high-spin, LS = low-spin). Slowly crystallised solid solutions [FexNi1−x(bpp)2][ClO4]2 with x = 0.53 (1a) and 0.74 (2a) adopt the P21 lattice, while x = 0.87 (3a) and 0.94 (4a) are mixed-phase materials with the high-spin C2/c phase as the major component. These materials exhibit thermal spin-transitions at T½ = 250 ± 1 K which occurs gradually in 1a, and abruptly and with narrow thermal hysteresis in 2a–4a. The transition proceeds to 100% completeness in 1a and 2a; that is, the 26% Ni doping in 2a is enough to convert high-spin [Fe(bpp)2][ClO4]2 into a cooperative, fully SCO-active material. These results were confirmed crystallographically for 1a and 2a, which revealed similarities and differences between these materials and the previously published [FexNi1−x(bpp)2][BF4]2 series. Rapidly precipitated powders with the same compositions (1b–4b) mostly resemble 1a–4a, except that 2b is a mixed-phase material; 2b–4b also contain a fraction of amorphous solid in addition to the two crystal phases. The largest iron fraction that can be accommodated by the P21 phase in this system is 0.7 ± 0.1.
Introduction
Metal–organic spin-crossover (SCO) compounds1–3 continue to be widely investigated as switching components in functional materials,4–7 and for a variety of prototype nanoscale and macroscopic device applications.8–12 Such compounds also have a more fundamental interest, as mechanistic probes of phase transitions and other transformations in the crystalline phase.13,14 Iron(II) complexes of 2,6-di(pyrazol-1-yl)pyridine derivatives are some the most studied materials for SCO research.15–17 Their popularity reflects the synthetic versatility of the bpp ligand family, which allows substituents to be appended to any position of the ligand framework. Appropriately positioned substituents can exert a steric or electronic influence on the metal spin state, giving some predictable control over its SCO temperature.18,19 Alternatively, [Fe(bpp)2]2+ switching centres bearing functional or tether group substituents have yielded a variety of multifunctional molecules, materials and nanostructures.20–25However, a disadvantage of [Fe(bpp)2]2+ derivatives for SCO applications is that the high-spin complexes are prone to an angular Jahn–Teller distortion in the solid state.26 This is an evolution of their coordination geometry along the Oh–D3h coordinate, constrained by the geometry of the tridentate bpp ligand.16 The distortion is energetically facile, and a range of distorted geometries are computationally accessible to [Fe(bpp)2]2+ derivatives around room temperature.26–28 Hence, the observation of distorted vs. undistorted geometries in a particular crystalline compound is dictated by packing considerations.29 Materials containing distorted molecules are often inactive towards SCO, since conversion of distorted high-spin to undistorted low-spin molecular structures is kinetically inhibited by the solid lattice.27
Salts of the prototype complex [Fe(bpp)2]2+ are a good example of this. The high-spin form of [Fe(bpp)2][BF4]2 has a regular coordination geometry, that deviates only slightly from idealised D2d symmetry (Fig. 1).26 That allows it to undergo an abrupt thermal spin-transition just below room temperature, making it a useful testbed for new materials applications30–32 and computational method development.33,34 In contrast the ClO4−, PF6−, SbF6−, CF3SO3− and I−/I3− salts of that complex all adopt highly distorted coordination geometries, and remain high-spin on cooling (Fig. 1).26,28,35–37
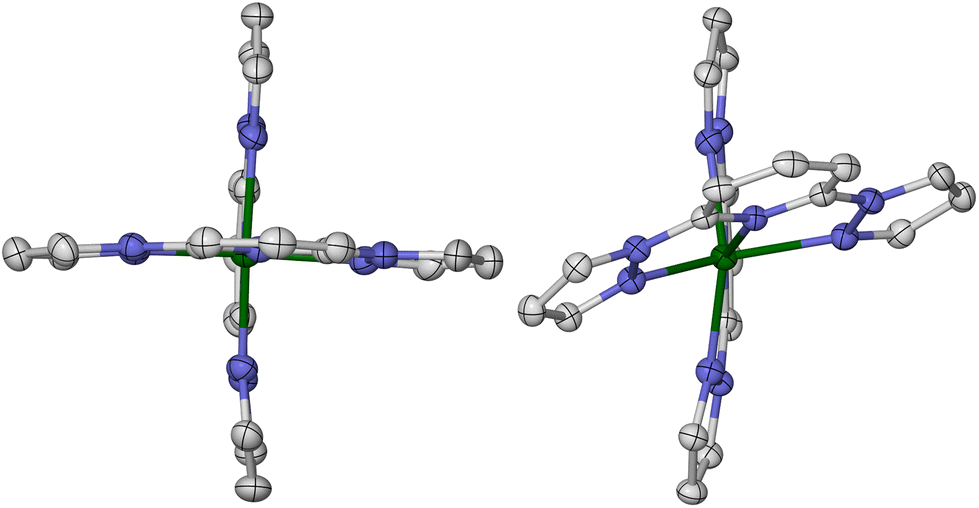 | ||
| Fig. 1 Crystallographic high-spin molecular geometries in SCO-active [Fe(bpp)2][BF4]2 (P21 phase, left),26 and [Fe(bpp)2][ClO4]2 (C2/c phase, right) which does not exhibit SCO.35 H atoms are omitted for clarity. Colour code: C, white; N, blue; Fe, green. | ||
Solid solutions of SCO materials with isomorphous, inert dopants are an established tool for elucidating the lattice energetics of SCO processes.38–55 We, and others, have also investigated doping as a way to introduce new functionality into SCO materials.42,55–60 Two examples are particularly relevant to this work, where doping a complex into a host lattice changes its spin state properties. First, low-spin [Fe(terpy)2][ClO4]2 (terpy = 2,2′:6′,2′′-terpyridine) is activated towards light-induced spin state trapping38 at low temperatures when doped into isomorphous [Mn(terpy)2][ClO4]2.56 Second, two families of solid solutions of different, non-isomorphous SCO complexes exhibit simultaneous, allosteric switching of both their component molecules.57–59 These examples highlight that SCO properties can be manipulated, by changes to the localised chemical pressure exerted on switchable molecules diluted within a host material.
It is known in other contexts, that crystals can be induced to adopt polymorphic forms by the inclusion of impurities or dopants.61 We reasoned that co-crystallising [Fe(bpp)2][ClO4]2 with another [M(bpp)2][ClO4]2 compound might force the iron complex to crystallise in an SCO-active form, if the dopant M2+ is a less plastic metal ion which is more resistant to the angular distortion. That could be a useful route to new SCO materials, if the dopant concentration needed to induce SCO is sufficiently small to retain their cooperative switching properties.38,62 This is a proof-of-principle study of this concept.
Results and discussion
Selection of a suitable dopant molecule
To identify a suitable dopant species, a gas phase DFT study was undertaken at the ω-B97X-D/6-311G** level (we used the same protocol in a recent survey of the structural preferences of the angular distortion).28 Different [M(bpp)2]2+ complexes were minimised at stages along the angular distortion, by fixing the trans-N{pyridyl}-M-N{pyridyl} angle (ϕ) to values between 165 ≤ ϕ ≤ 155°;26,63 the lower limit is the experimental value of ϕ in [Fe(bpp)2][ClO4]2.35 The metal ions listed below were examined because salts of those [M(bpp)2]2+ complexes are known to be isomorphous with one of the iron complex phases shown in Fig. 1.42,47,54,57,64–67The energy penalty associated with the distortion, ΔE(dist), for the different complexes runs as follows (Fig. 2 and ESI;† HS = high-spin):
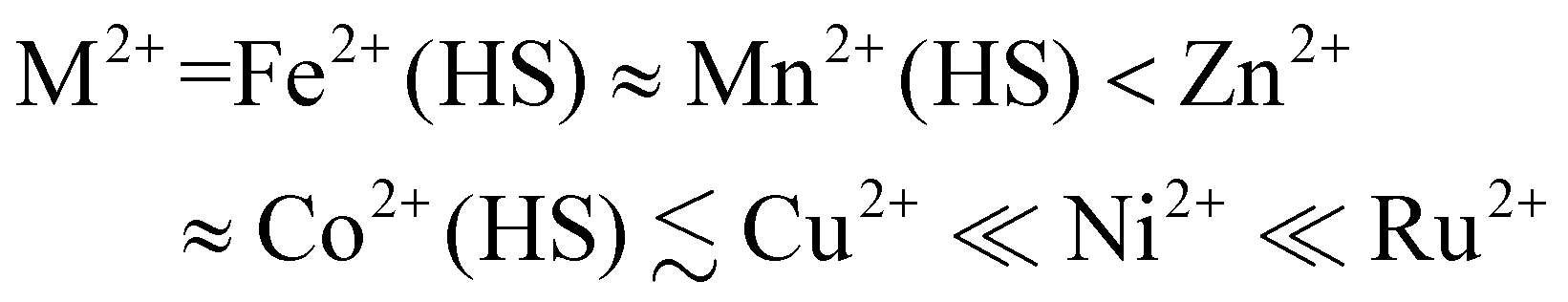
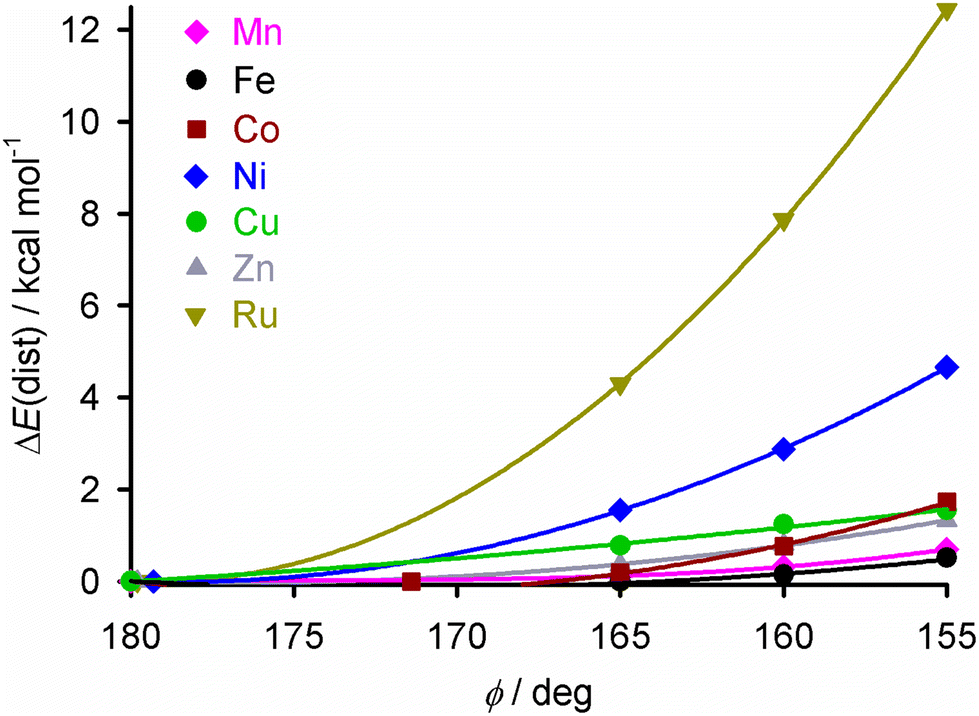 | ||
| Fig. 2 The energy penalty associated with the angular distortion [ΔE(dist)] for different [M(bpp)2]2+ complexes. ΔE(dist) is the energy of each minimisation relative to the corresponding molecule with an undistorted structure; see the ESI† for more details. The data for the iron complex are taken from ref. 28. | ||
The ΔE(dist) values for [Fe(bpp)2]2+ by this protocol are <1 kcal mol−1,28 which is consistent with previous studies of this type using different functionals.26,27,68–70
[Ni(bpp)2]2+ was chosen as the dopant for this study, for three reasons. It shows a higher ΔE(dist) distortion energy than the other potential dopants, except the ruthenium complex. It is more synthetically accessible than its ruthenium analogue. Lastly, doping iron(II) SCO materials with nickel(II) has little effect on their transition temperature (T½), which simplifies the interpretation of these results.40,42–44,48,52,55,71
Crystalline [Ni(bpp)2][ClO4]2 (monoclinic, space group P21, Z = 2) is not isomorphous with [Fe(bpp)2][ClO4]2 (monoclinic, C2/c, Z = 4). However it is isomorphous with both [Ni(bpp)2][BF4]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 64 and the SCO-active salt [Fe(bpp)2][BF4]2,26 which adopt the same P21 phase. The cation in [Ni(bpp)2][ClO4]2 exhibits an undistorted six-coordinate geometry, with no unusual features (Fig. S2†).
64 and the SCO-active salt [Fe(bpp)2][BF4]2,26 which adopt the same P21 phase. The cation in [Ni(bpp)2][ClO4]2 exhibits an undistorted six-coordinate geometry, with no unusual features (Fig. S2†).
Composition and SCO properties of the solid solutions
Preformed [Fe(bpp)2][ClO4]2 and [Ni(bpp)2][ClO4]2 were co-crystallised from acetonitrile, using diethyl ether as antisolvent. Four compositions of the [FexNi1−x(bpp)2][ClO4]2 solid solutions were investigated in two solid forms; as polycrystalline materials grown over 1–2 days by a vapour diffusion method (1a–4a; Table 1), and as rapidly precipitated powders (1b–4b). The analytical compositions of the materials were confirmed by elemental microanalysis, EDX measurements and from their magnetic susceptibility data (Table 1). EDX element maps confirm the iron and nickel content is homogeneously distributed through each material (Fig. S1†).| x | y | ω{P21} | z | T ½↓/K | T ½↑/K | χ M T/cm3 mol−1 K | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| T = 300 K | HScalcb | T = 100 K | LScalcb | |||||||
| a y is the completeness of the spin-transition in the magnetic measurements; ω{P21} is the fraction of the sample adopting the P21 phase [eqn (1)]; and z gives the metal composition of the P21 fraction of the sample [eqn (2)]. b Calculated from x, using these values for the constituent compounds: [Ni(bpp)2][BF4]2, 1.2 cm3 mol−1 K; high-spin [Fe(bpp)2][BF4]2, 3.5 cm3 mol−1 K; low-spin [Fe(bpp)2][BF4]2, 0.42 c Calculated from χMT at 230 K to exclude SCO in any amorphous fraction of the sample at lower temperatures. d This is χMT at 230 K, just below the abrupt partial spin-transition, which is given for the mixed phase samples in the Table. 2b–4b show an additional slow monotonic decrease in χMT on further cooling, which implies a fraction of the material is amorphous (Fig. S6†). | ||||||||||
| Polycrystalline | ||||||||||
| 1a | 0.53 | 0.97 | 0.98 | 0.52 | 249 | 249 | 2.35 | 2.42 | 0.62 | 0.56 |
| 2a | 0.74 | 0.98 | 0.98 | 0.74 | 249 | 251 | 2.87 | 2.90 | 0.37 | 0.31 |
| 3a | 0.87 | 0.32c | 0.40c | 0.68c | 250 | 251 | 3.21 | 3.20 | 2.19 [2.24d] | 0.16 |
| 4a | 0.94 | 0.06c | 0.12c | 0.50c | 250 | 251 | 3.38 | 3.36 | 3.13 [3.15d] | 0.07 |
| Powder | ||||||||||
| 1b | 0.52 | 0.97 | 0.98 | 0.51 | 246 | 246 | 2.29 | 2.39 | 0.63 | 0.58 |
| 2b | 0.74 | 0.64c | 0.73c | 0.64c | 248 | 249 | 2.81 | 2.90 | 0.90 [1.25d] | 0.31 |
| 3b | 0.88 | 0.55c | 0.61c | 0.80c | 248 | 250 | 3.16 | 3.22 | 1.33 [1.52d] | 0.14 |
| 4b | 0.93 | 0.07c | 0.14c | 0.48c | 248 | 248 | 3.33 | 3.34 | 2.97 [3.11d] | 0.08 |
The fractional metal compositions (x) deduced in this way were within experimental error of the synthetic stoichiometry used for all the doped materials. Different samples of slowly crystallised 1a–4a, and rapidly precipitated 1b, showed consistent structural and magnetic properties. However, the crystallinity (by powder diffraction) and magnetic behaviour showed some variability between different samples of 2b–4b. Representative examples of each composition of 2b–4b are described below.
X-ray powder diffraction show the polycrystalline materials adopt purely the P21 phase when x = 0.50 (1a, Table 1) and 0.73 (2a); and form a mixture of the P21 and C2/c phases when x = 0.88 (3a) and 0.94 (4a; Fig. 3). That is, 26% nickel doping is sufficient to produce the pure P21 phase under these conditions. The high-spin C2/c phase is the main component of 3a and 4a, but the presence of a P21 fraction is indicated by a low-angle shoulder at 2θ = 11.1°, for example, which becomes weaker as x approaches 1.
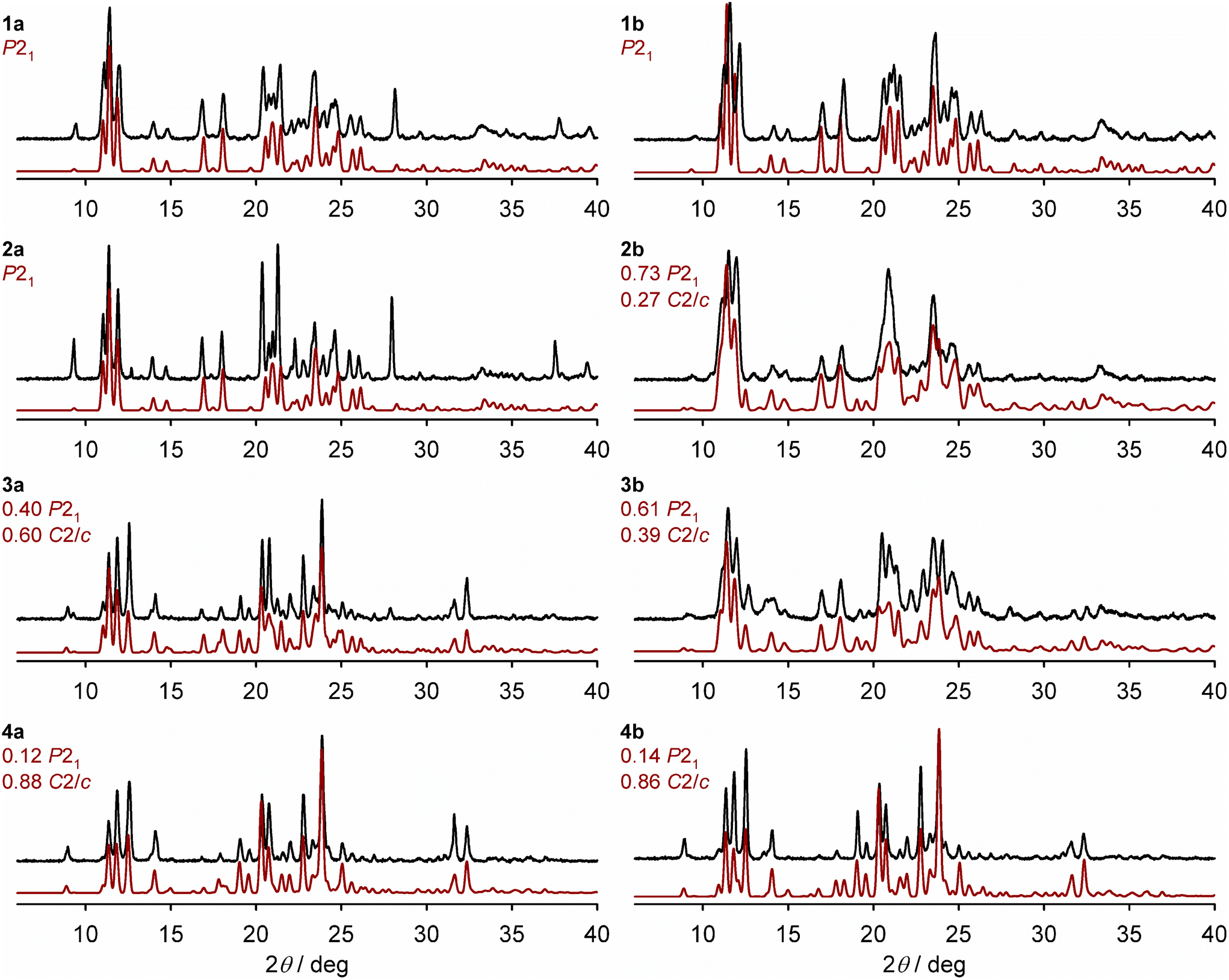 | ||
| Fig. 3 Room temperature X-ray powder patterns for the solid solutions (black), and simulations derived from the single crystal X-ray structures of the precursor materials (red; details in the Experimental section). The composition of phases employed in each simulation is derived from the ω{P21} values in Table 1, although that doesn't account for the presence of amorphous material evident in the magnetic data for 2b–4b (Fig. S6†). Differences in peak intensities between the measured and simulated data arise from preferred orientation effects, which are more pronounced for polycrystalline 1a–4a. | ||
The powder patterns of 1b and 4b are essentially identical to their congeners 1a and 4a (Fig. 3). However, the diffraction peaks for 2b and 3b are broadened, implying those samples have reduced crystallinity. The powder patterns of 2b and 3b can both be simulated as mixed-phase materials, with 3b having a lower fraction of the P21 phase.
All the powder patterns were reproduced by simulations using phase compositions deduced from the magnetic data, as described below (Fig. 3). A previous study of mixed-phase materials comprised of different iron complex salts, proposed that the C2/c phase is the thermodynamic crystallisation product but the P21 phase is the kinetic product which is favoured by rapid precipitation.72 That relationship is less clear in this mixed-metal system, however.
The iron content in all the compounds is high-spin at room temperature, with χMT at 300 K close to the expected values based on their metal composition (Table 1). 1a and 2a exhibit SCO centred at 250 ± 1 K, which is respectively gradual (1a), and abrupt with a small thermal hysteresis (2a; Fig. 4). The completeness of the transition (y, Table 1) is >98% based on their measured iron content. SCO in 3a and 4a resembles 2a but only occurs in ca. 32% (3a) or 6% (4a) of the iron content in the samples, with the rest of their iron remaining high-spin on further cooling.
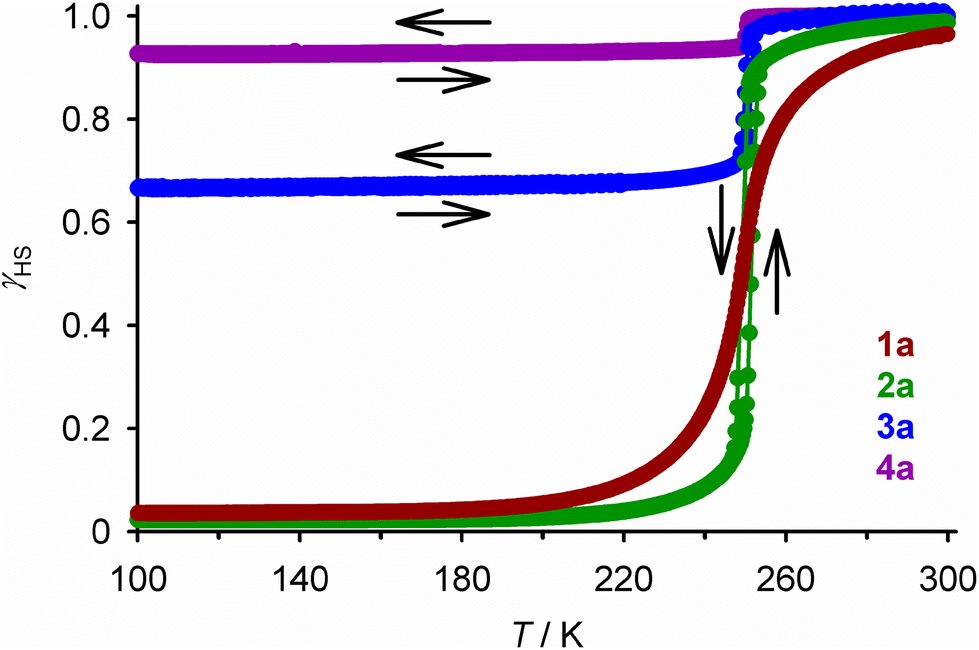 | ||
| Fig. 4 The spin-transitions shown by polycrystalline 1a–4a, from magnetic susceptibility data measured in cooling and warming modes at a scan rate of 2 K min−1. The graph shows the high-spin fraction of the iron content in the sample (γHS) at each temperature, with data for each compound connected by a spline curve for clarity. χMT vs. T plots for all the samples in Table 1 are given in Fig. S6.† | ||
These data are consistent with the phase composition of the samples predicted by powder diffraction. Both 1a and 2a adopt the SCO-active P21 phase, while 3a and 4a are mixed-phase materials. Assuming that the P21 phase contains the nickel (1 − x) plus the SCO-active fraction (y) of the iron content in the sample (x), the fraction of the material adopting the P21 phase (ω{P21}) can be estimated by eqn (1):
| ω{P21} = xy + (1 − x) | (1) |
This predicts P21:C2/c phase ratios of 0.40:0.60 for 3a and 0.12:0.88 for 4a (Table 1); simulated powder patterns based on those compositions are excellent matches for the data (Fig. 3). The metal content of that P21 fraction is then [FezNi1−z] (eqn (2)):
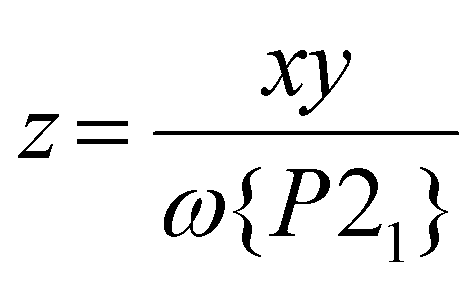 | (2) |
This gives z = 0.68 for 3a and 0.50 for 4a, which are lower than the iron content of the samples as a whole (Table 1). That is, the SCO-active P21 fractions of 3a and 4a are enriched in nickel. In contrast, z for 1a and 2a is equal to their stoichiometric composition x, within experimental error.
The thermal SCO equilibrium in 1b occurs a little more gradually than for 1a, and is centred at slightly lower temperature (Fig. S6†). In other respects, the properties of those two materials are identical within experimental error. However, magnetic data for 2b–4b show more differences from their slowly crystallised counterparts. Each sample undergoes abrupt partial SCO just below 250 K, which is then followed by a very gradual, almost monotonic decrease in χMT on further cooling (Fig. S6†). The latter feature is characteristic of amorphous solids containing SCO centres, where they are less densely packed in heterogeneous chemical environments.38,73,74 The abrupt spin-transitions in 2b and 3b each proceed to ca. 50% completeness based on their iron content, but with 2b exhibiting a more pronounced amorphous tail at lower temperatures. The data imply ω{P21} follows a consistent trend in the rapidly precipitate samples (Table 1):
| 1b (ω{P21} = 0.98) > 2b (0.67) > 3b (0.56) > 4b (0.13) |
The remainder of each of these samples is a mixture of the C2/c phase and a smaller quantity of amorphous material. The SCO-active P21 phase is calculated to be more iron-rich in 3b (z = 0.80) than in 2b (z = 0.64) or 3a (z = 0.68). However the error on that calculation should increase as ω{P21} decreases, and could be as high as ±0.1 for 3a/3b.
The midpoint temperature (T½) for the spin transitions in all these samples is very similar, at 246 ≤ T½ ≤ 251 K (Table 1). That small variation is consistent with other iron(II) compounds containing isomorphous nickel(II) dopants,40,42–44,48,52,55,71 including [FexNi1−x(bpp)2][BF4]2.42 The SCO cooperativity shown by 1a/1b and 3a/3b closely resembles that in [FexNi1−x(bpp)2][BF4]2 materials with comparable compositions. However, the spin transition in 2a is more abrupt than its closest analogue from the BF4− series (Fig. S7†).
The SCO-active perchlorates in this work exhibit T½ 10 ± 1 K below the corresponding BF4− salt materials.42 That should reflect the larger volume of the perchlorate anion,75 which leads to an expansion of the ClO4−-containing P21 lattice. That expansion additionally stabilises the high-spin state of the iron complex, whose molecular volume is 1.4% larger than the low-spin form,76 thus lowering T½. While counter examples exist,77–80 an inverse relationship between T½ and anion size is most commonly found when isomorphous salts of the same complex with weakly interacting anions are compared.77,80–88
Crystallographic characterisation
Single crystals of 1a and 2a (both monoclinic, P21, Z = 2) are isomorphous with [Ni(bpp)2][ClO4]2. Full structure refinements of 1a were achieved in its high- and low-spin states, at 300 and 100 K respectively (Fig. S8†). However crystals of 2a diffracted poorly above ca. 200 K, and did not afford a satisfactory high-spin dataset. That could be a consequence of their containing almost the maximum iron content consistent with the P21 phase. Better data were obtained at lower temperatures, and a precise structure refinement of low-spin 2a was also achieved at 100 K (Fig. S9†). The metal content x of the crystals at 100 K refined as 0.50(2) for 1a and 0.76(3) for 2a.The structure refinements contain a crystallographically ordered [FexNi1−x(bpp)2]2+ molecule, with partial iron and nickel atoms having the same atomic coordinates and displacement parameters. That is the approach taken in other crystallographic studies of doped SCO crystals.55,89–92 Metric parameters from the refinements are consistent with statistical mixtures of the low-spin iron(II) and nickel(II) complexes, in those ratios (Table S6†).54 That is useful confirmation of the crystal metal stoichiometries in the previous paragraph.
Attempts to resolve the fractional [Fe(bpp)2]2+ and [Ni(bpp)2]2+ molecules in 1a and 2a at 100 K required numerous distance and thermal parameter restraints, which made them less useful as probes of molecular structure. Such disorder models were achieved in [FexM1−x(bpp)2][BF4]2 cocrystals using higher resolution datasets collected with synchrotron radiation.54 Synchrotron data were not obtained for 1a and 2a in this work, however.
Variable temperature unit cell measurements demonstrate gradual thermal SCO in the crystal of 1a with T½ = 245 ± 3 K; and, an abrupt spin-transition for the crystal of 2a at T½ = 255 ± 5 K (Fig. 5 and S13, S14†). These behaviours are broadly consistent with the magnetic data for both compounds (Fig. 5 and S6†). The changes to the unit cell dimensions during SCO resemble our previous study of the [FexNi1−x(bpp)2][BF4]2 system (Table S9†).54 The isothermal volume change during high → low-spin SCO at T½ (ΔVSCO) for 1a [−19.0(5) Å3] is identical to that of a [FexNi1−x(bpp)2][BF4]2 crystal with a similar metal content [−18.8(3) Å3],54 within experimental error. However ΔVSCO for 2a [−21.9(6) Å3] is ca. 3 Å3 smaller than expected, based on data from the BF4− salt materials (Fig. S14†).93 Its a, b and c unit cell dimensions all undergo smaller changes during SCO than predicted from its composition (Table S9†). While its low-spin crystal structure is as-expected (Table S6†), a high-spin structure refinement of 2a was not achieved. So, the structural basis for that anomaly is unclear. The small ΔVSCO of 2a has no impact on the abruptness of its spin-transition, however.62
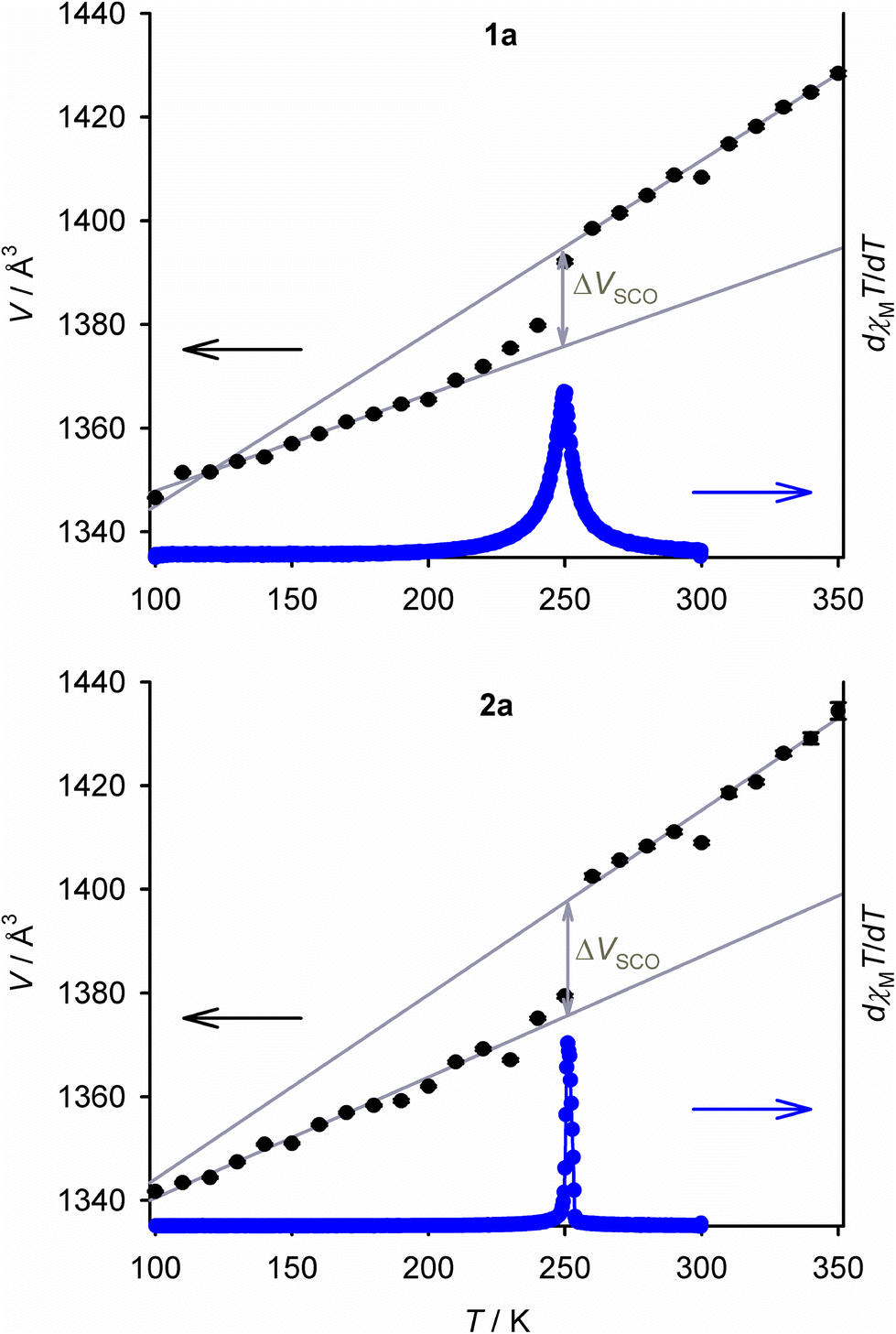 | ||
| Fig. 5 Variable temperature unit cell volumes for 1a (top) and 2a (bottom). The grey lines show the thermal expansion linear regression fits for the volume change at T½ (ΔVSCO). The first derivative of the magnetic susceptibility data for each compound is also plotted to an arbitrary scale, as blue data points connected by a spline curve. | ||
The complex molecules in the P21 phase pack into 2D “terpyridine embrace” layers94 in the (001) plane, through interdigitation of their pyrazolyl arms via face-to-face C–H⋯π and edge-to-face π⋯π contacts (Fig. S10 and S11†).54 Neighbouring cation layers associate more loosely along c, and are separated by the ClO4− ions. [Fe(bpp)2]2+ derivatives with this crystal packing motif often show abrupt thermal spin-transitions with narrow thermal hysteresis;95,96 the magnetic data from 2a and 3a imply the P21 phase of [Fe(bpp)2][ClO4]2 behaves similarly (Fig. 4).
Other salts of [Fe(bpp)2]2+ and [Ni(bpp)2]2+
Solid [Fe(bpp)2][PF6]2 is high-spin, and crystallises in the C2/c crystal phase.26 A crystal structure of [Fe(bpp)2][CF3SO3]2 in the C2/c phase is also available,28 although bulk samples of that salt contain a mixture of phases by powder diffraction (Fig. S16†). Hence, [Ni(bpp)2][PF6]2 and [Ni(bpp)2][CF3SO3]2 were also investigated as potential dopants in this study. However crystals of those compounds were difficult to obtain, and decomposed rapidly outside their mother liquor which prevented their structural characterisation. Powder samples of [Ni(bpp)2][PF6]2 and [Ni(bpp)2][CF3SO3]2 are not isomorphous by X-ray powder diffraction, and neither salt adopts the P21 phase (Fig. S15 and S16†). Since structures of the nickel complex host lattices could not be determined, solid solutions of those iron and nickel complex salts were not pursued further.Conclusions
High-spin [Fe(bpp)2][ClO4]2, which adopts the C2/c crystal phase, can be induced to crystallise in the SCO-active P21 phase by doping with [Ni(bpp)2][ClO4]2. (Poly)crystalline solid solutions [FexNi1−x(bpp)2][ClO4]2 with x = 0.54 (1a) or 0.74 (2a), produced under slow crystallisation conditions, are phase-pure and exhibit gradual (1a) or abrupt (2a) SCO at T½ = 250 ± 1 K. Both transitions proceed to ca. 98% completeness based on their iron content (Fig. 4). Analogous materials with x = 0.87 (3a) and 0.94 (4a) have a mixed phase composition, with the high-spin C2/c phase as the major component. Those samples exhibit partial spin-transitions which resemble 2a, but are incomplete.Rapidly precipitated powders with the same compositions (1b–4b) behave similarly, except that 2b–4b appear to contain an amorphous fraction as well as the two crystalline phases. Hence materials with the best-defined phase compositions and SCO properties are produced by slow crystallisation in this system. On the basis of Table 1, the maximum iron content that can be present in the P21 phase is estimated at z = 0.7 ± 0.1 (eqn (2)). Surplus iron in the more iron-rich samples, which cannot be accommodated within their P21 crystallites, is then present as the C2/c or (for 2b–4b) amorphous material.
The dependence of T½ on the composition of 1a–4a resembles that found in isomorphous [FexNi1−x(bpp)2][BF4]2 (Fig. S14†). The putative P21 phase of [Fe(bpp)2][ClO4]2 itself is predicted to show T½ = 251 ± 1 K, which is 10 K below that of isomorphous BF4− salt.72 That is consistent with the behaviour of most other isomorphous salts of SCO compounds containing those anions.77,80–88 Interestingly, the isothermal unit cell volume change during SCO (ΔVSCO) for 1a and 2a does not mirror the trend shown by the [FexNi1−x(bpp)2][BF4]2 series, with ΔVSCO for 2a being smaller than expected.93 The poor X-ray diffraction exhibited by crystals of 2a at room temperature also contrasts with 1a, and other [M(bpp)2]2+ complex salts exhibiting the P21 phase.26,54,64–66,72 These anomalies may reflect the metal stoichiometry of 2a (x = 0.74), which is around the maximum iron content that the P21 phase can accommodate; that merits further investigation.
In conclusion, 26% nickel(II) doping is sufficient to convert high-spin [Fe(bpp)2][ClO4]2 into a single phase (poly)crystalline material showing a complete, cooperative thermal spin-transition. These are rare examples of SCO solid solutions formed from non-isomorphous constituents.42,57–59 This is also the first demonstration of a new method for producing SCO compounds from nominally inert precursors, which could further expand the range of available SCO materials.
Experimental
2,6-Bis(pyrazol-1-yl)pyridine (bpp),97 [Fe(bpp)2][ClO4]2,35 [Fe(bpp)2][PF6]226 and [Fe(bpp)2][CF3SO3]228 were prepared by the published procedures.
CAUTION We experienced no problems using the perchlorate salts in this study, but metal–organic perchlorates are potentially explosive and should be handled with care in small quantities.
Synthesis of [Ni(bpp)2][ClO4]2
A mixture of Ni[ClO4]2·6H2O (0.22 g, 0.59 mmol) and bpp (0.25 g, 1.2 mmol) in acetonitrile (25 cm3) was stirred at room temperature until all the solid had dissolved. The blue solution was concentrated to ca. 10 cm3, then filtered. Slow diffusion of diethyl ether vapour into the solution yielded large blue crystals of the product complex. Yield 0.31g, 77%. Found C, 38.9; H, 2.53; N, 20.3%. Calcd for C22H18Cl2N10NiO8 C, 38.9; H, 2.67; N, 20.6%.Synthesis of [Ni(bpp)2][PF6]2
A mixture of NiCl2·6H2O (0.14 g, 0.59 mmol), bpp (0.25 g, 1.2 mmol) and AgPF6 (0.30 g, 1.2 mmol) in acetonitrile (25 cm3) was stirred at room temperature for 1 h. The resultant mixture was allowed to settle, then filtered to remove the AgCl precipitate and concentrated to ca. 10 cm3. Addition of excess diethyl ether to the filtrate yielded a blue powder, which was collected and dried in vacuo. Yield 0.33 g, 72%. Found C, 34.1; H, 1.91; N, 18.3%. Calcd for C22H18F12N10NiP2 C, 34.3; H, 2.35; N, 18.2%.Synthesis of [Ni(bpp)2][CF3SO3]2
Method as for [Ni(bpp)2][ClO4]2, using Ni[CF3SO3]2 (0.21 g, 0.59 mmol). The product was a blue powder. Yield 0.31 g, 68%. Found C, 36.9; H, 2.00; N, 18.0%. Calcd for C24H18F6N10NiO6S2 C, 37.0; H, 2.33; N, 18.0%.While [Ni(bpp)2][PF6]2 and [Ni(bpp)2][CF3SO3]2 are tractable as rapidly precipitated powders, attempted crystallisations of both salts gave similar results. Slow diffusion of diethyl ether vapour into solutions of the compounds in common organic solvents sometimes gave crystals with an ingrown plate morphology, if the dehydrating agent triethyl orthoformate was added to the solution. These crystals decomposed rapidly when removed from the original mother liquor, however. Other crystallisations under the same conditions instead afforded blue oils, while recrystallisations using di-iso-propylether or methyl(tert-butyl)ether as the antisolvent also led to oils. Hence, no crystal structure determination was achieved for these salts.
Synthesis of [FexNi1−x(bpp)2][ClO4]2 (1–4)
Preformed [Fe(bpp)2][ClO4]2 and [Ni(bpp)2][ClO4]2 were mixed in different mole ratios, to a combined mass of 0.25 g. The combined solids were stirred in acetonitrile (25 cm3), until all the solid had dissolved. The solutions were concentrated to ca. 10 cm3, then filtered. Slow diffusion of diethyl ether vapour into the filtered solutions over a period of 48 h afforded the polycrystalline materials 1a–4a. Alternatively the powder samples 1b–4b were produced by rapid addition of excess diethyl ether to these same filtered solutions, yielding a fine precipitate which was stored at 255 K for 18 h, then collected and dried in vacuo. Crystallised yields were in the range 80–85%.The materials with high iron content are yellow at room temperature, which progresses towards a green colour as the nickel concentration increases. The compositions and analytical data for the solid solutions are given in Table S1.†
Single crystal X-ray structures
Single crystals of the precursor complexes were obtained by slow diffusion of diethyl ether vapour into solutions of the compounds in nitromethane. Crystals of 1a and 2a were obtained similarly, from acetonitrile solution. Diffraction data were measured with an Agilent Supernova or a Nonius Kappa-CCD diffractometer, which were fitted with an Oxford Cryostream low-temperature device. Monochromated Cu-Kα (λ = 1.5418 Å) or Mo-Kα (λ = 0.7107 Å) radiation was used for different measurements.Experimental details of the structure determinations are given in Table S2.† All the structures were solved by direct methods (SHELXTL98), and developed by full least-squares refinement on F2 (SHELXL-201899). Crystallographic figures and powder pattern simulations were prepared using XSEED,100 and other publication materials were prepared using Olex2.101
CCDC 2332656–2332661† contain the supplementary crystallographic data for this paper.
Other measurements
Elemental microanalyses were performed by the microanalytical service at the London Metropolitan University School of Human Sciences. SEM images and EDX measurements were obtained using an FEI Nova NanoSEM 450 environmental microscope, operating at 3 kV. Magnetic susceptibility measurements were performed on a Quantum Design MPMS-3 VSM magnetometer, with an applied field of 5000 G and a scan rate of 2 K min−1. A diamagnetic correction for the sample was estimated from Pascal's constants;102 a previously measured diamagnetic correction for the sample holder was also applied to the data.X-ray powder diffraction patterns were measured at room temperature using a Bruker D2 phaser diffractometer. Simulations of the solid solution powder patterns in Fig. 3 were based on the structure refinement of [Fe(bpp)2][ClO4]2 at 300 K in this work (C2/c phase); and, a model derived from the published structure of [Fe(bpp)2][BF4]2 at 290 K,26 with the BF4− ions replaced by ClO4− (P21 phase). Some minor differences between measured and simulated 2θ values in Fig. 3 reflect small discrepancies between the unit cell dimensions in that model and the hypothetical P21 phase of [Fe(bpp)2][ClO4]2.
DFT minimizations of [M(bpp)2]2+ were performed using Spartan'20103 with the ω-B97X-D functional and 6-311G** basis set.104 Molecules with S = 0 (M2+ = Zn2+ and Ru2+) were treated as spin-restricted, while molecules with S ≥ ½ (M2+ = Mn2+, Co2+, Ni2+ and Cu2+) were treated as spin-unrestricted. The calculations were performed in the gas phase because solvent gradients for transition ions are not implemented in Spartan'20. Initial models were constructed de novo with ϕ restrained to the appropriate value, then subjected to a preliminary molecular mechanics minimization before the full DFT geometry minimization calculation. The computational results for [Fe(bpp)2]2+ are reproduced from ref. 28.
Data availability
Data supporting this study are available in the ESI,† or at https://doi.org/10.5518/1493.Author contributions
M. A. H. conceived the study, prepared the materials, did the DFT calculations and wrote the publication.H. B. V. performed the magnetic measurements using SQUID magnetometer time provided by O. C. C. M. P. collected and processed the single crystal diffraction data, and A. N. K. did the SEM and EDX analyses. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the University of Leeds. We acknowledge C. A. Kilner (University of Leeds) for collecting the room temperature crystallographic dataset of [Fe(bpp)2][PF6]2 described in the ESI.†References
- Spin Crossover in Transition Metal Compounds I–III: Topics in Current Chemistry, ed. P. Gütlich and H. A. Goodwin, Springer-Verlag, Berlin, 2004, vol. 233–235 Search PubMed.
- Spin-crossover materials – properties and applications, ed. M. A. Halcrow, John Wiley & Sons, Chichester, UK, 2013, p. 568 Search PubMed.
- J. Zarembowitch, F. Varret, A. Hauser, J. A. Real and K. Boukheddaden, C. R. Chim., 2018, 21, 1056–1059 CrossRef CAS.
- W. Huang, X. Ma, O. Sato and D. Wu, Chem. Soc. Rev., 2021, 50, 6832–6870 RSC.
- M. K. Javed, A. Sulaiman, M. Yamashita and Z.-Y. Li, Coord. Chem. Rev., 2022, 467, 214625 CrossRef CAS.
- Y. Sekine, R. Akiyoshi and S. Hayami, Coord. Chem. Rev., 2022, 469, 214663 CrossRef CAS.
- E. Resines-Urien, E. Fernandez-Bartolome, A. Martinez-Martinez, A. Gamonal, L. Piñeiro-López and J. S. Costa, Chem. Soc. Rev., 2023, 52, 705–727 RSC.
- M. D. Manrique-Juárez, S. Rat, L. Salmon, G. Molnár, C. M. Quintero, L. Nicu, H. J. Shepherd and A. Bousseksou, Coord. Chem. Rev., 2016, 308, 395–408 CrossRef.
- S. Rat, M. Piedrahita-Bello, L. Salmon, G. Molnár, P. Demont and A. Bousseksou, Adv. Mater., 2018, 30, 1703862 CrossRef PubMed.
- E. Coronado, Nat. Rev. Mater., 2020, 5, 87–104 CrossRef.
- K. S. Kumar and M. Ruben, Angew. Chem., Int. Ed., 2021, 60, 7502–7521 CrossRef CAS PubMed.
- R. Torres-Cavanillas, M. Gavara-Edo and E. Coronado, Adv. Mater., 2024, 36, 2307718 CrossRef CAS PubMed.
- M. Chergui and E. Collet, Chem. Rev., 2017, 117, 11025–11065 CrossRef CAS PubMed.
- S. Pillet, J. Appl. Phys., 2021, 129, 181101 CrossRef CAS.
- M. A. Halcrow, Coord. Chem. Rev., 2009, 253, 2493–2514 CrossRef CAS.
- L. J. Kershaw Cook, R. Mohammed, G. Sherborne, T. D. Roberts, S. Alvarez and M. A. Halcrow, Coord. Chem. Rev., 2015, 289–290, 2–12 CrossRef CAS.
- M. Attwood and S. S. Turner, Coord. Chem. Rev., 2017, 353, 247–277 CrossRef CAS.
- L. J. Kershaw Cook, R. Kulmaczewski, R. Mohammed, S. Dudley, S. A. Barrett, M. A. Little, R. J. Deeth and M. A. Halcrow, Angew. Chem., Int. Ed., 2016, 55, 4327–4331 CrossRef CAS PubMed.
- M. A. Halcrow, I. Capel Berdiell, C. M. Pask and R. Kulmaczewski, Inorg. Chem., 2019, 58, 9811–9821 CrossRef CAS PubMed.
- B. Schäfer, T. Bauer, I. Faus, J. A. Wolny, F. Dahms, O. Fuhr, S. Lebedkin, H.-C. Wille, K. Schlage, K. Chevalier, F. Rupp, R. Diller, V. Schünemann, M. M. Kappes and M. Ruben, Dalton Trans., 2017, 46, 2289–2302 RSC.
- E. Burzurí, A. García-Fuente, V. García-Suárez, K. Senthil Kumar, M. Ruben, J. Ferrer and H. S. J. van der Zant, Nanoscale, 2018, 10, 7905–7911 RSC.
- F. Moreau, J. Marrot, F. Banse, C. Serre and A. Tissot, J. Mater. Chem. C, 2020, 8, 16826–16833 RSC.
- M. Palacios-Corella, J. Ramos-Soriano, M. Souto, D. Ananias, J. Calbo, E. Ortí, B. M. Illescas, M. Clemente-León, N. Martíın and E. Coronado, Chem. Sci., 2021, 12, 757–766 RSC.
- S.-Q. Su, S.-Q. Wu, Y.-B. Huang, W.-H. Xu, K.-G. Gao, A. Okazawa, H. Okajima, A. Sakamoto, S. Kanegawa and O. Sato, Angew. Chem., Int. Ed., 2022, 61, e202208771 CrossRef CAS PubMed.
- M. Palacios-Corella, V. García-López, J. C. Waerenborgh, B. J. C. Vieira, G. M. Espallargas, M. Clemente-León and E. Coronado, Chem. Sci., 2023, 14, 3048–3055 RSC.
- J. M. Holland, J. A. McAllister, C. A. Kilner, M. Thornton-Pett, A. J. Bridgeman and M. A. Halcrow, J. Chem. Soc., Dalton Trans., 2002, 548–554 RSC.
- S. Vela, J. J. Novoa and J. Ribas-Arino, Phys. Chem. Chem. Phys., 2014, 16, 27012–27024 RSC.
- I. Capel Berdiell, E. Michaels, O. Q. Munro and M. A. Halcrow, Inorg. Chem., 2024, 63, 2732–2744 CrossRef CAS PubMed.
- There are examples of [Fe(bpp)2]2+ derivatives that crystallise in distorted and undistorted polymorphs (ref. 105), or where distorted high-spin and undistorted SCO-active molecules co-crystallise in the same lattice (ref. 36, 106 and 107).
- M. Matsuda, H. Isozaki and H. Tajima, Chem. Lett., 2008, 37, 374–375 CrossRef CAS.
- S. P. Vallone, A. N. Tantillo, A. M. dos Santos, J. Molaison, R. Kulmaczewski, A. Chapoy, P. Ahmadi, M. A. Halcrow and K. G. Sandeman, Adv. Mater., 2019, 31, 1807334 CrossRef PubMed.
- M. Owczarek, M. Lee, S. Liu, E. R. Blake, C. S. Taylor, G. A. Newman, J. C. Eckert, J. H. Leal, T. A. Semelsberger, H.-P. Cheng, W. Nie and V. S. Zapf, Angew. Chem., Int. Ed., 2022, 61, e202214335 CrossRef CAS PubMed.
- N. Suaud, M.-L. Bonnet, C. Boilleau, P. Labèguerie and N. Guihéry, J. Am. Chem. Soc., 2009, 131, 715–722 CrossRef CAS PubMed.
- S. Vela, M. Fumanal and C. Sousa, J. Mater. Chem. C, 2023, 11, 235–243 RSC.
- J. Elhaïk, D. J. Evans, C. A. Kilner and M. A. Halcrow, Dalton Trans., 2005, 1693–1700 RSC.
- J. Elhaïk, C. A. Kilner and M. A. Halcrow, Dalton Trans., 2006, 823–830 RSC.
- Salts of [Fe(bpp)2]2+ with larger metallate or radical anions have also been reported, which show different spin state behaviours (ref. 36 and 108–110).
- P. Gütlich, A. Hauser and H. Spiering, Angew. Chem., Int. Ed. Engl., 1994, 33, 2024–2054 CrossRef.
- A. Hauser, J. Jeftić, H. Romstedt, R. Hinek and H. Spiering, Coord. Chem. Rev., 1999, 190–192, 471–491 CrossRef CAS.
- T. Tayagaki, A. Galet, G. Molnár, M. C. Muñoz, A. Zwick, K. Tanaka, J. A. Real and A. Bousseksou, J. Phys. Chem. B, 2005, 109, 14859–14867 CrossRef CAS PubMed.
- C. Baldé, C. Desplanches, A. Wattiaux, P. Guionneau, P. Gütlich and J.-F. Létard, Dalton Trans., 2008, 2702–2707 RSC.
- C. A. Tovee, C. A. Kilner, J. A. Thomas and M. A. Halcrow, CrystEngComm, 2009, 11, 2069–2077 RSC.
- Z. Yu, T. Kuroda-Sowa, H. Kume, T. Okubo, M. Maekawa and M. Munakata, Bull. Chem. Soc. Jpn., 2009, 82, 333–337 CrossRef CAS.
- A. Rotaru, M. M. Dîrtu, C. Enachescu, R. Tanasa, J. Linares, A. Stancu and Y. Garcia, Polyhedron, 2009, 28, 2531–2536 CrossRef CAS.
- N. Paradis, G. Chastanet, F. Varret and J.-F. Létard, Eur. J. Inorg. Chem., 2013, 968–974 CrossRef CAS.
- N. Paradis, G. Chastanet, T. Palamarciuc, P. Rosa, F. Varret, K. Boukheddaden and J.-F. Létard, J. Phys. Chem. C, 2015, 119, 20039–20050 CrossRef CAS.
- M. A. Halcrow and G. Chastanet, Polyhedron, 2017, 136, 5–12 CrossRef CAS.
- B. R. Mullaney, L. Goux-Capes, D. J. Price, G. Chastanet, J.-F. Létard and C. J. Kepert, Nat. Commun., 2017, 8, 1053 CrossRef PubMed.
- M. S. Sylla, C. Baldé, N. Daro, C. Desplanches, M. Marchivie and G. Chastanet, Eur. J. Inorg. Chem., 2018, 297–304 CrossRef CAS.
- R. Diego, O. Roubeau and G. Aromí, Chem. Sq., 2021, 5, 1 Search PubMed.
- P. Chakraborty, M. Sy, H. Fourati, T. Delgado, M. Dutta, C. Das, C. Besnard, A. Hauser, C. Enachescu and K. Boukheddaden, Phys. Chem. Chem. Phys., 2022, 24, 982–994 RSC.
- C. Das, S. Dey, A. Adak, C. Enachescu and P. Chakraborty, Cryst. Growth Des., 2023, 23, 3496–3508 CrossRef CAS.
- M. Dutta, S. Bisht, P. Ghosh, A. I. Chilug, D. Mann, C. Enachescu, M. Shatruk and P. Chakraborty, Inorg. Chem., 2023, 62, 15050–15062 CrossRef CAS PubMed.
- P. Ghosh, C. M. Pask, H. B. Vasili, N. Yoshinari, T. Konno, O. Cespedes, C. Enachescu, P. Chakraborty and M. A. Halcrow, J. Mater. Chem. C, 2023, 11, 12570–12582 RSC.
- X. Li, D. Zhang, Y. Qian, W. Liu, C. Mathonière, R. Clérac and X. Bao, J. Am. Chem. Soc., 2023, 145, 9564–9570 CrossRef CAS PubMed.
- F. Renz, H. Oshio, V. Ksenofontov, M. Waldeck, H. Spiering and P. Gütlich, Angew. Chem., Int. Ed., 2000, 39, 3699–3700 CrossRef CAS PubMed.
- M. A. Halcrow, Chem. Commun., 2010, 46, 4761–4763 RSC.
- S. V. Tumanov, S. L. Veber, S. Greatorex, M. A. Halcrow and M. V. Fedin, Inorg. Chem., 2018, 57, 8709–8713 CrossRef CAS PubMed.
- C. Bartual-Murgui, C. Pérez-Padilla, S. J. Teat, O. Roubeau and G. Aromí, Inorg. Chem., 2020, 59, 12132–12142 CrossRef CAS PubMed.
- Y.-Y. Wu, Z.-Y. Li, S. Peng, Z.-Y. Zhang, H.-M. Cheng, H. Su, W.-Q. Hou, F.-L. Yang, S.-Q. Wu, O. Sato, J.-W. Dai, W. Li and X.-H. Bu, J. Am. Chem. Soc., 2024, 146, 8206–8215 CrossRef CAS PubMed.
- W. Kras, A. Carletta, R. Montis, R. A. Sullivan and A. J. Cruz-Cabeza, Commun. Chem., 2021, 4, 38 CrossRef CAS PubMed.
- A.-I. Popa, L. Stoleriu and C. Enachescu, J. Appl. Phys., 2021, 129, 131101 CrossRef CAS.
- While undistorted complexes show ϕ = 180°, geometries with 180 ≤ ϕ ≤ 143° have been observed experimentally in high-spin [Fe(bpp)2]2+ derivatives (ref. 16 and 28).
- J. M. Holland, C. A. Kilner, M. Thornton-Pett and M. A. Halcrow, Polyhedron, 2001, 20, 2829–2840 CrossRef CAS.
- M. A. Leech, N. K. Solanki, M. A. Halcrow, J. A. K. Howard and S. Dahaoui, Chem. Commun., 1999, 2245–2246 RSC.
- N. K. Solanki, M. A. Leech, E. J. L. McInnes, F. E. Mabbs, J. A. K. Howard, C. A. Kilner, J. M. Rawson and M. A. Halcrow, J. Chem. Soc., Dalton Trans., 2002, 1295–1301 RSC.
- K. N. Lazarou, I. Stamatopoulos, V. Psycharis, C. Duboc, C. P. Raptopoulou and Y. Sanakis, Polyhedron, 2018, 155, 291–301 CrossRef CAS.
- I. Capel Berdiell, R. Kulmaczewski and M. A. Halcrow, Inorg. Chem., 2017, 56, 8817–8828 CrossRef CAS PubMed.
- J. Nance, D. N. Bowman, S. Mukherjee, C. T. Kelley and E. Jakubikova, Inorg. Chem., 2015, 54, 11259–11268 CrossRef CAS PubMed.
- H.-Y. Kwon, D. C. Ashley and E. Jakubikova, Dalton Trans., 2021, 50, 14566–14575 RSC.
- J.-P. Martin, J. Zarembowitch, A. Dworkin, J. G. Haasnoot and E. Codjovi, Inorg. Chem., 1994, 33, 2617–2623 CrossRef CAS.
- C. Carbonera, C. A. Kilner, J.-F. Létard and M. A. Halcrow, Dalton Trans., 2007, 1284–1292 RSC . This is a study of mixed-anion salts [Fe(bpp)2][BF4]x[ClO4]2−x, (0 < x < 2) which also adopt the P21 phase, the C2/c phase or a mixture of both depending on their composition and the crystallisation conditions.
- S. Bonnet, G. Molnár, J. S. Costa, M. A. Siegler, A. L. Spek, A. Bousseksou, W.-T. Fu, P. Gamez and J. Reedijk, Chem. Mater., 2009, 21, 1123–1136 CrossRef CAS.
- P. Chakraborty, M.-L. Boillot, A. Tissot and A. Hauser, Angew. Chem., Int. Ed., 2013, 52, 7139–7142 CrossRef CAS PubMed.
- D. M. P. Mingos and A. L. Rohl, J. Chem. Soc., Dalton Trans., 1991, 3419–3425 RSC.
- The volume of the complex molecule in [Fe(bpp)2][BF4]2 (ref. 26), which exhibits the P21 lattice, is 349.9(5) Å3 in the high-spin state at 290 K; and 343.3(5) Å3 in the low-spin state at 240 K. These volumes were calculated with Olex2 (ref. 97).
- M. A. Halcrow, Chem. Soc. Rev., 2011, 40, 4119–4142 RSC.
- S. Heider, H. Petzold, G. Chastanet, S. Schlamp, T. Rüffer, B. Weber and J.-F. Létard, Dalton Trans., 2013, 42, 8575–8584 RSC.
- V. García-López, M. Palacios-Corella, A. Abhervé, I. Pellicer-Carreño, C. Desplanches, M. Clemente-León and E. Coronado, Dalton Trans., 2018, 47, 16958–16968 RSC.
- O. Roubeau, Chem. – Eur. J., 2012, 18, 15230–15244 CrossRef CAS PubMed.
- M. Tafili-Kryeziu, M. Weil, T. Muranaka, A. Bousseksou, M. Hasegawa, A. Jun and W. Linert, Dalton Trans., 2013, 42, 15796–15804 RSC.
- R. Mohammed, G. Chastanet, F. Tuna, T. L. Malkin, S. A. Barrett, C. A. Kilner, J.-F. Létard and M. A. Halcrow, Eur. J. Inorg. Chem., 2013, 819–831 CrossRef CAS.
- H. Phan, S. M. Benjamin, E. Steven, J. S. Brooks and M. Shatruk, Angew. Chem., Int. Ed., 2015, 54, 823–827 CrossRef CAS PubMed.
- Y.-Y. Zhu, H.-Q. Li, Z.-Y. Ding, X.-J. Lü, L. Zhao, Y.-S. Meng, T. Liu and S. Gao, Inorg. Chem. Front., 2016, 3, 1624–1636 RSC.
- R. Kulmaczewski, E. Trzop, L. J. Kershaw Cook, E. Collet, G. Chastanet and M. A. Halcrow, Chem. Commun., 2017, 53, 13268–13271 RSC.
- R. Díaz-Torres, W. Phonsri, K. S. Murray, L. Liu, M. Ahmed, S. M. Neville, P. Harding and D. J. Harding, Inorg. Chem., 2020, 59, 13784–13791 CrossRef PubMed.
- I. Capel Berdiell, R. Kulmaczewski, N. Shahid, O. Cespedes and M. A. Halcrow, Chem. Commun., 2021, 57, 6566–6569 RSC.
- N. Suryadevara, A. Mizuno, L. Spieker, S. Salamon, S. Sleziona, A. Maas, E. Pollmann, B. Heinrich, M. Schleberger, H. Wende, S. K. Kuppusamy and M. Ruben, Chem. – Eur. J., 2022, 28, e202103853 CrossRef CAS PubMed.
- J. Kusz, H. Spiering and P. Gütlich, J. Appl. Crystallogr., 2004, 37, 589–595 CrossRef CAS.
- C. Baldé, C. Desplanches, F. Le Gac, P. Guionneau and J.-F. Létard, Dalton Trans., 2014, 43, 7820–7829 RSC.
- H. Wang, C. Baldé, A. Grosjean, C. Desplanches, P. Guionneau and G. Chastanet, Dalton Trans., 2018, 47, 14741–14750 RSC.
- R. Diego, O. Roubeau and G. Aromí, Chem. Sq., 2021, 5, 1 Search PubMed.
- ΔVSCO in SCO materials containing isomorphous dopants has been proposed to show a linear dependence with the dopant concentration (ref. 89). Since [Fe(bpp)2][BF4]2 exhibits ΔVSCO = −30.4(3) Å3 (ref. 54), by extrapolation from 1a ΔVSCO for 2a would be expected to be ca., −25 Å3 (Fig. S14†).
- I. Dance and M. Scudder, CrystEngComm, 2009, 11, 2233–2247 RSC.
- R. Pritchard, C. A. Kilner and M. A. Halcrow, Chem. Commun., 2007, 577–579 RSC.
- E. Michaels, C. M. Pask, I. Capel Berdiell, H. B. Vasili, M. J. Howard, O. Cespedes and M. A. Halcrow, Cryst. Growth Des., 2022, 22, 6809–6817 CrossRef CAS.
- D. L. Jameson and K. A. Goldsby, J. Org. Chem., 1990, 55, 4992–4994 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Barbour, J. Appl. Crystallogr., 2020, 53, 1141–1146 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. J. O'Connor, Prog. Inorg. Chem., 1982, 29, 203–283 CrossRef.
- Spartan'20, Wavefunction Inc., Irvine, CA, 2020 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- M. Haryono, F. W. Heinemann, K. Petukhov, K. Gieb, P. Müller and A. Grohmann, Eur. J. Inorg. Chem., 2009, 2136–2143 CrossRef CAS.
- R. Kulmaczewski, F. Bamiduro, O. Cespedes and M. A. Halcrow, Chem. – Eur. J., 2021, 27, 2082–2092 CrossRef CAS PubMed.
- K. H. Sugiyarto, D. Onggo, H. Akutsu, V. R. Reddy, H. Sutrisno, Y. Nakazawa and A. Bhattacharjee, CrystEngComm, 2021, 23, 2854–2861 RSC.
- M. Nihei, H. Tahira, N. Takahashi, Y. Otake, Y. Yamamura, K. Saito and H. Oshio, J. Am. Chem. Soc., 2010, 132, 3553–3560 CrossRef CAS PubMed.
- Ö. Üngor, E. S. Choi and M. Shatruk, Eur. J. Inorg. Chem., 2021, 4812–4820 CrossRef.
- J. Li, X.-P. Sun, S. Bi, M. Xu, S. Jia, Z. Tang, P. Ma, J. Wang, J. Tao and J. Niu, Inorg. Chem., 2022, 61, 17932–17936 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical data for the solid solutions; SEM images and EDX element maps; crystallographic data and refinement procedures; crystallographic figures and tables; additional X-ray powder diffraction and magnetic susceptibility data; and details of the minimized structures from the DFT calculations. CCDC 2332656–2332661. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00443d |
| This journal is © The Royal Society of Chemistry 2024 |