
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reaction of size-selected iron-oxide cluster cations with methane: a model study of rapid methane loss in Mars’ atmosphere†
Masashi
Arakawa‡
 *a,
Satoshi
Kono
a,
Yasuhito
Sekine
b and
Akira
Terasaki
*a
*a,
Satoshi
Kono
a,
Yasuhito
Sekine
b and
Akira
Terasaki
*a
aDepartment of Chemistry, Faculty of Science, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: arakawa@chem.kyushu-univ.jp; terasaki@chem.kyushu-univ.jp
bEarth-Life Science Institute (ELSI), Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8550, Japan. E-mail: sekine@elsi.jp
First published on 27th April 2024
Abstract
We report gas-phase reactions of free iron-oxide clusters, FenOm+, and their Ar adducts with methane in the context of chemical processes in Mars’ atmosphere. Methane activation was observed to produce FenOmCH2+/FenOmCD2+ and FenOmC+, where the reactivity exhibited size and composition dependence. For example, the rate coefficients of methane activation for Fe3O+ and Fe4O+ were estimated to be 1 × 10−13 and 3 × 10−13 cm3 s−1, respectively. Based on these reaction rate coefficients, the presence of iron-oxide clusters/particles with a density as low as 107 cm−3 in Mars’ atmosphere would explain the rapid loss of methane observed recently by the Curiosity rover.
1 Introduction
Clusters of metals and metal compounds are a group of substances with a variety of potential applications taking advantage of their size-specific properties. Among the numerous cluster studies, one of the most intriguing topics is cluster chemistry in space. Small mineral particles such as silicate (e.g., pyroxene (Mg, Fe)SiO3 and olivine (Mg, Fe)2SiO4) are among the most abundant materials in space.1,2 It is a prevalent hypothesis that such materials contribute to chemical processes in the planet-forming regions, e.g., formation of organic molecules.3 Size-selected gas-phase clusters provide a good model for this chemistry because it is possible to investigate reactions step by step with precise control over the number of atoms and molecules involved in the reaction,4–6 providing molecular-level insights into metal- and metal compound-mediated catalytic reactions.7 Furthermore, spectroscopic studies in laboratories have been reported for silica and silicate clusters,8–10 which were recently suggested to be highly abundant in the interstellar medium.8,9 Therefore, clusters themselves may play important roles in chemical processes in space. In this context, we have reported reactions of gas-phase free silicate, MglSiOm−, and silica, SinOm−, cluster anions with CO and H2O molecules.11,12 In addition, coadsorption and subsequent reaction of CO and H2 molecules on cobalt cluster cations, Con+, have been examined to discuss the formation of organic molecules on the clusters.13Another chemical process among recent topics in space is rapid methane loss in the atmosphere of Mars.14 Observation by the Curiosity rover found temporary spikes of methane and its rapid loss, but the mechanism of the loss has not been elucidated yet. Although some previous studies suggested a very local, small methane source near the landing site of the Curiosity rover,15,16 an alternative possibility is the effective loss of methane via chemical reaction that is not considered in photochemical models. Mars’ soil is rich in iron oxides, where storms of iron-oxide particles (dust devils) occur very frequently.17 Hypervelocity impacts of small bodies on Mars would have also evaporated or made molten the surface materials.18 These surface processes may have formed very fine iron-bearing condensates on Mars. We thus hypothesize that iron-oxide particles/clusters, which are speculated to be abundant on Mars, are responsible for the rapid loss because the diiron-oxide cluster, Fe2O2, is known as an active center of methane monooxygenase (sMMO),19–23 an enzyme that oxidizes C–H bonds in methane.
The C–H dissociation energy of methane is much larger than those of other alkanes as can be expected from its short bond length and the large HOMO–LUMO gap.24 Numerous theoretical studies on the interaction of methane with model complexes involving a diiron-oxide core have been reported because methane activation is receiving considerable interest in modern chemistry.25–36 These theoretical studies have revealed that the diiron-oxide core in sMMO plays an important role in C–H bond cleavage. Several mechanisms have been proposed for the hydroxylation of CH4,24,34,35 such as a mechanism via a CH3 radical,28,29,31 one not via radicals26,27,33,35 and a non-synchronous concerted one.32
As for experimental studies, gas-phase free FeO+ was reported to mediate the activation of methane.37–41 Hydroxylation of methane by FeO+ was suggested to occur via a hydroxy intermediate HO–Fe+–CH3.37,38 This reaction mechanism has been supported by theoretical studies, where the pathway via the hydroxy intermediate is energetically more favorable than the one via a methoxy intermediate H–Fe+–OCH3.42,43 The methoxy intermediate was investigated experimentally by vibrational spectroscopy.44 The reaction was also reported to be accompanied by spin inversion.45–49 An activation mechanism of methane by neutral FeO and anionic FeO− as well as cationic FeO+ was theoretically reported recently.50
As for multinuclear species, an iron-oxide cluster, Fe2O3,51 as well as iron clusters, Fe2,52 Fe2+53 and Fe4,54 has been reported to show methane activation by theory. Furthermore, a number of theoretical studies present geometric and electronic structures of iron-oxide clusters such as FeO+,55,56 FeO2−,57 FeO2+/0/−,58 FeOn+/−,59 FenOm− (n = 3, 4),60 FenOm− (n = 1, 2, m ≤ 6),61 FenOm (n = 1–5),62 (FeO)n (n = 2–5),63 (FeO)n (n >10),64 (Fe2O3)n (n = 1–6, 10),65–67 (Fe3O4)n (n = 1–5),68 FenOm+ (n = 3–6),69 FenOm (n = 33, 45, 113),70 Fe3O18,71–73 Fe25O30 and Fe33O32.73 However, in contrast to these extensive theoretical studies, experimental reports are limited: geometric-structure studies of (FeO)n+ (n = 2–9) by ion mobility mass spectrometry74 and stability studies by photodissociation,75 collision-induced dissociation76 and mass spectrometry.65,71 In particular, reaction with methane has been examined by experiment only for Fe2O2+.77 Note that, because methane activation has been attracting strong attention, a significant number of methane-reaction experiments have been reported for metal and metal-compound clusters, including Fen+ (n = 2–15),78 Nin+ (n = 2–15),79 Rhn+,80 Pdn+,81–83 Tan+,84 Au2+,81,82,85,86 FeC6−,87 WnCm+ (n =1–5, m ≤ 5),88 WnNm+ (n = 1–6, m ≤ 2),88 Al2O7+,89 Al4O6+ and NiAl3O6+,90 as well as vibrational spectroscopy of methane complexes with Fen+,91–93 Cun+,94 Ta4+,95 Ptn+96 and Au2+.97
On the basis of these backgrounds, the present study reports gas-phase reaction of size-selected iron-oxide cluster cations, FenOm+, with methane, CH4, and deuterated methane, CD4, to verify our hypothesis that iron-oxide particles/clusters are responsible for the rapid loss of methane in Mars’ atmosphere.
2 Methods
Experiments were performed using the setup described in detail previously.98,99 Briefly, FenOm+ (n = 2–4, 0 ≤ m ≤ n) were generated by a magnetron-sputter cluster-ion source,100 with sputtering of an iron plate (Kojundo Chem. Lab. Co., Ltd, 99.99%) in the presence of oxygen (99.99995%). After thermalization by collisions with helium gas at liquid-nitrogen temperature, the generated cluster cations were mass-selected with a quadrupole mass filter (MAX-4000, Extrel CMS, LLC); among the isotopologues for a given composition, the one with the highest abundance was selected. Note that the reactant cluster cations thus mass-selected contained a trace amount of an Ar adduct Fen−1Om+1Ar+ as well, which has the same mass as FenOm+. The cluster cations were introduced into a reaction cell with a linear 40-cm-long rf quadrupole ion guide, where methane (CH4 or deuterated CD4) gas was continuously flowed at 298 K. Ions produced by reaction while passing through the cell were identified by a second quadrupole mass analyzer (MAX-4000, Extrel CMS, LLC) employing a channel electron multiplier (Channeltron 4720, Burle Electro-Optics, Inc.) for an ion detector to measure the yield of each product ion.The partial pressure of CH4 and CD4 in the reaction cell was adjusted to be 0.1 Pa; the pressure was monitored outside the reaction cell by a residual gas analyzer (RGA100, Stanford Research Systems, Inc.) and was converted to the pressure inside the reaction cell in a way reported previously by referring to the reaction cross section of Cr2+ with O2.101,102 The pressure corresponds to the collision rate of 2 × 104 s−1, where the collisional cross section is estimated by the Langevin–Gioumousis–Stevenson model,103
| σ = πe{2α/(4πε0)2E}1/2 | (1) |
3 Results and discussion
3.1 Reaction products
Fig. 1 shows mass spectra of ions upon reaction of Fe3Om+ (m = 0–3) with deuterated methane, CD4; note that Fe2Om+1Ar+, which is coincident in mass with Fe3Om+, coexists in the reactant in our present experimental conditions. For m = 0, 1 and 3, mass spectra of ions upon reaction with CH4 are also shown (panels a′, b′ and d′). The abscissa shows a mass shift, ΔM, from Fe3Om+; the peaks observed at ΔM ≥ 0 are assigned to ions produced from Fe3Om+, whereas those at ΔM < 0 are from Fe2Om+1Ar+ subject to desorption of an Ar atom. Note also that the mass spectra were not recorded in the vicinity of ΔM = 0 because the intensity of the reactant ion was much higher than those of product ions, which caused saturation in the ion detector; the peak at ΔM = −40, which should have been observed in panels a, a′, b and b′, was also excluded from recording by the same reason.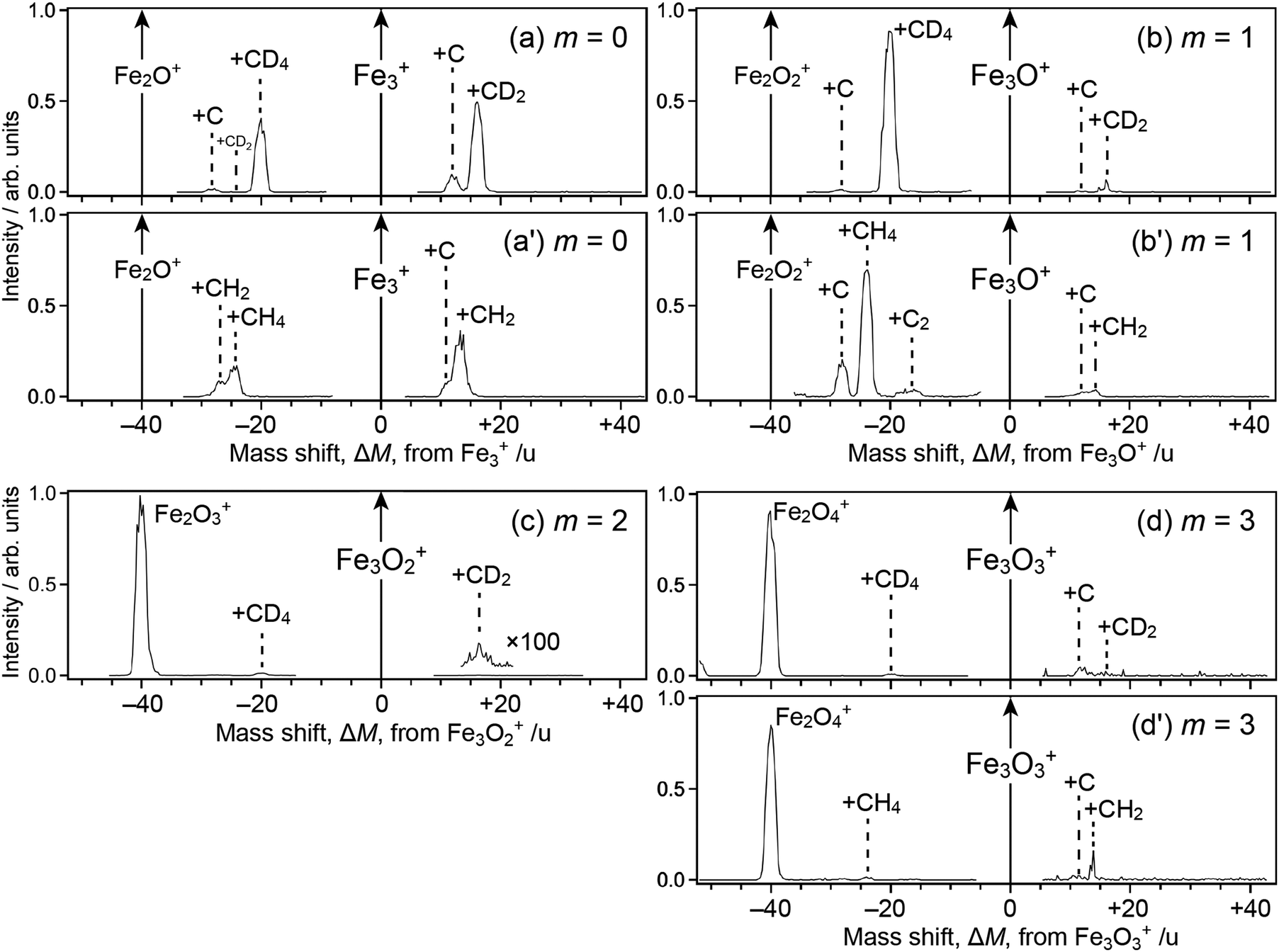 | ||
| Fig. 1 Mass spectra of ions produced from the reaction of Fe3Om+ and Fe2Om+1Ar+ with (a)–(d) CD4 or (a′, b′ and d′) CH4. Reactant ions are m = 0, 1, 2 and 3 for (a) and (a′), (b) and (b′), (c) and (d) and (d′), respectively. The abscissa shows a mass shift, ΔM, from Fe3Om+. Peaks are assigned to adducts to Fe3Om+ in the ΔM ≥ 0 region, while adducts to Fe2Om+1+ after desorption of Ar in the ΔM < 0 region. Note that the following ions are excluded from recording due to saturation: Fe3+ and Fe2O+ in panels (a) and (a′), Fe3O+ and Fe2O2+ in (b) and (b′), Fe3O2+ in (c) and Fe3O3+ in (d) and (d′). | ||
For the ΔM ≥ 0 region manifesting reaction channels from Fe3Om+, peaks at ΔM = 12 and 16 are observed upon reaction with CD4 as shown in Fig. 1a–d (m = 0–3), while ΔM = 12 and 14 are observed for CH4 as shown in Fig. 1a′, b′ and d′ (m = 0, 1 and 3). These results suggest that Fe3OmC+ and Fe3OmCD2+/Fe3OmCH2+ are produced upon reaction, i.e., dehydrogenation from methane proceeds. Mass spectra upon the reaction of Fe2Om+ (m = 0–2) and Fe4Om+ (m = 0–3) with CD4 are shown in the ΔM ≥ 0 region of Fig. 2, where methane activation to produce FenOmC+ and/or FenOmCD2+ is observed as in the case of Fe3Om+. Note that Fe2O+ and Fe2O2+ are exceptions; reaction products are hardly identified. In the reaction of Fe4O2+ and Fe4O3+, partially dehydrogenated products, Fe4OmCD3+, are observed.
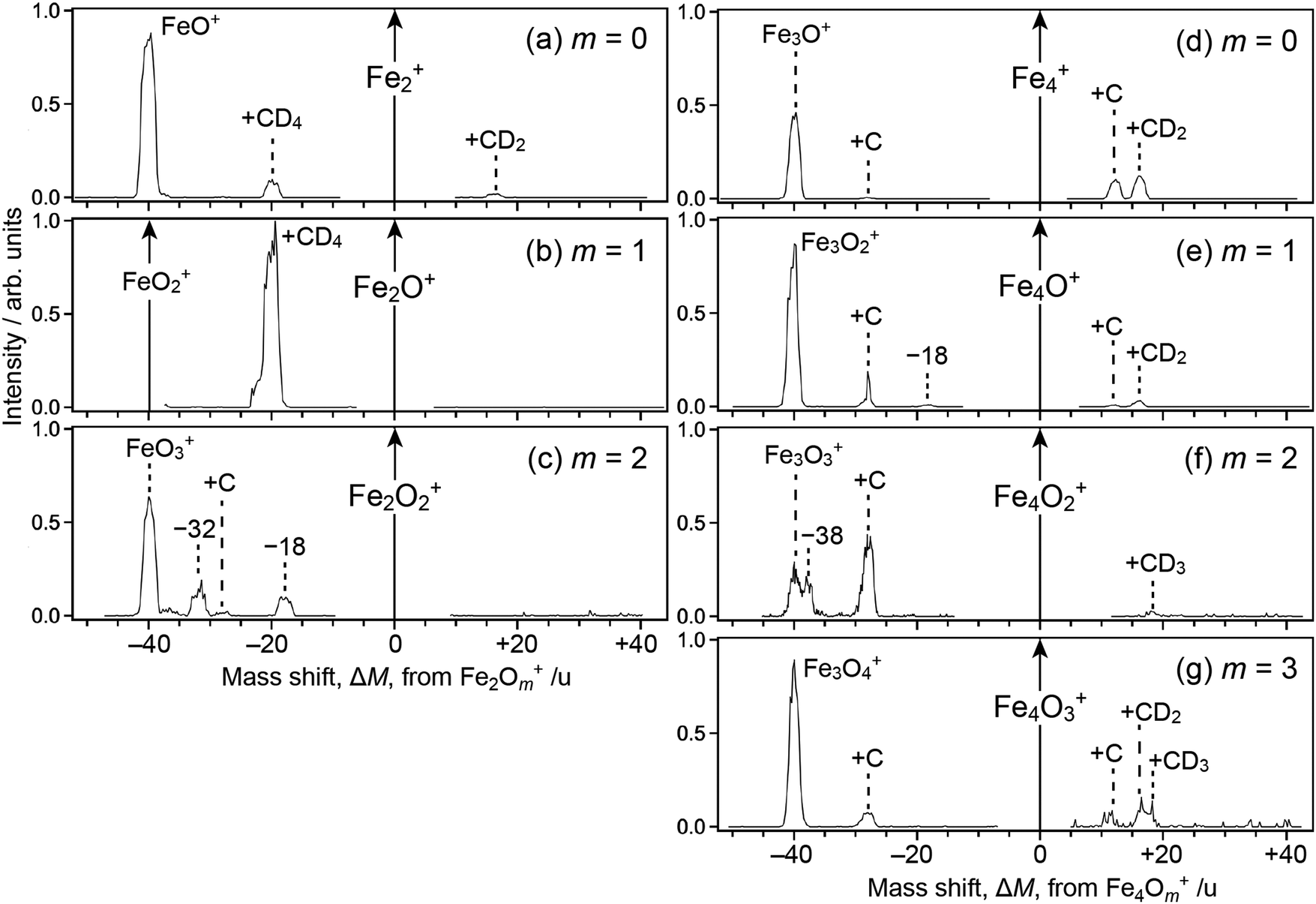 | ||
| Fig. 2 Mass spectra of ions produced from the reaction of FenOm+ and Fen−1Om+1Ar+ (n = 2 and 4) with CD4. Reactant ions are (a)–(c) n = 2, m = 0–2; (d)–(g) n = 4, m = 0–3. The abscissa shows a mass shift, ΔM, from FenOm+. Peaks are assigned to adducts to FenOm+ in the ΔM ≥ 0 region, while adducts to Fen−1Om+1+ after desorption of Ar in the ΔM < 0 region. Note that the following ions are excluded from recording due to saturation: Fe2+ in panel (a), Fe2O+ and FeO2+ in (b), Fe2O2+ in (c), Fe4+ in (d), Fe4O+ in (e), Fe4O2+ in (f), and Fe4O3+ in (g). Several peaks marked by the value of ΔM are not simply assignable, originating probably from reactant ions containing more than two Ar atoms. | ||
As for the ΔM < 0 region in Fig. 1, product ions from Fe2Om+1Ar+ are observed, where the major reaction channel is methane adsorption without dehydrogenation. For example, Fe2Om+1CD4+ along with a small amount of Fe2Om+1C+ is observed in the reaction of Fe2Om+1Ar+ (m = 0, 1), as shown in Fig. 1a and b. Adsorption of CD4 is accompanied by desorption of an Ar atom to form Fe2Om+1CD4+. This is in contrast to the fact that no products are observed in the reaction of Fe2O+ and Fe2O2+ (see Fig. 2b and c). The results suggest that the adsorption energy of CD4 was released by the desorption of Ar, which stabilized the CD4 adducts to survive longer. The peak at ΔM = −20 observed in Fig. 2a and b is also assigned to CD4 adducts accompanied by Ar desorption from FeOAr+ and FeO2Ar+. In particular, the result of FeOAr+ is in contrast to a previous study, which reported that the reaction of FeO+ with CD4 proceeds along two reaction channels: either to FeOD+ + CD3 or to Fe+ + CD3OD.37 This would also be due to stabilization of the CD4 adduct by desorption of Ar as discussed in the previous reaction study of Rhn+Arm with CH4;80 it was reported that the CH4 adducts do not have a sufficient energy for activation of the C–H bond due to energy release by ligand exchange.
3.2 Rate coefficients of methane activation
The reaction-rate coefficients to produce the ions described above were estimated in the same way as reported previously12 by assuming pseudo-first-order reaction kinetics because the density of methane can be treated as constant during the reaction. However, the peak intensity of the reactant is necessary to estimate the rate coefficients, which is missing in the present measurement due to saturation. We therefore performed additional recording of mass spectra with lower sensitivity of the ion detector, where only major peaks, such as those at ΔM = 0 and −40, were observed (see Fig. S1 of the ESI†). The rate coefficients were estimated by combining the relative intensities of the reactant and product ions obtained in this low-sensitivity mode with the intensity ratios of the product ions in Fig. 1 and 2 obtained in a high-sensitivity mode.Fig. 3 shows rate coefficients thus evaluated for methane dehydrogenation of FenOm+ in the size range of n = 2–4, which summed up all the reaction channels to the formation of C, CD2 and CD3 adducts. The result suggests that larger and oxygen-poor clusters tend to exhibit higher reactivity. For example, the rate coefficients of O-free iron clusters, Fe3+ and Fe4+, are estimated to be 3 × 10−12 and 4 × 10−12 cm3 s−1, respectively. These values are consistent with the previous report;78 note that the collision energies in the present experimental conditions for Fe3+ and Fe4+ are 1.0 and 0.8 eV in the center-of-mass frame, respectively. On the other hand, the present results estimate that the rate coefficients of monoxide clusters, Fe3O+ and Fe4O+, are 1 × 10−13 and 3 × 10−13 cm3 s−1, respectively; these values are an order of magnitude smaller than those of O-free iron clusters. The rate coefficients of Fe3O2+, Fe3O2+, Fe4O2+ and Fe4O3+, are even smaller by an order of magnitude than those of monoxide clusters. The extremely low reactivity of Fe2O2+ is consistent with the previous report.77 The rate coefficients obtained for CH4 are shown in Fig. S2 of the ESI,† which are in the same order of magnitude as those for CD4.
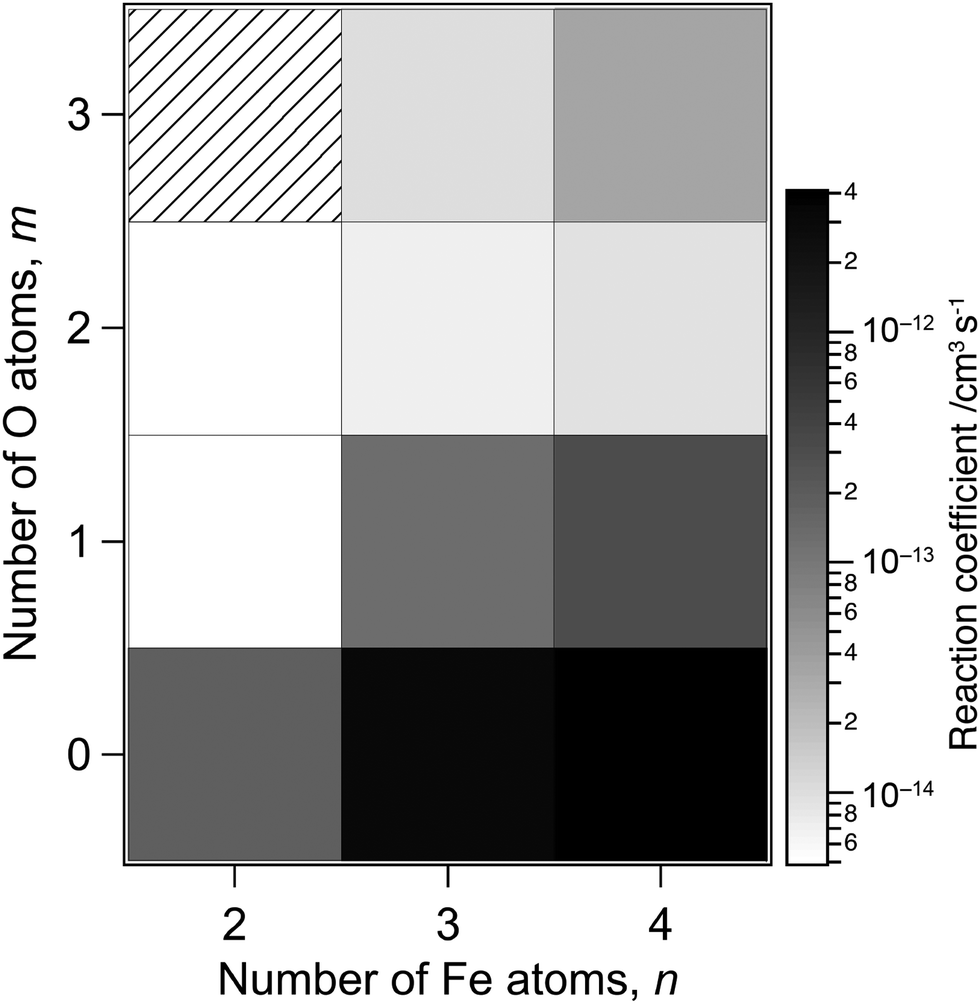 | ||
| Fig. 3 Rate coefficients of activation of CD4 by FenOm+ as a function of the numbers of iron, n, and oxygen atoms, m. White pixels at (n,m) = (2,1) and (2,2) indicate the compositions of the clusters, for which dehydrogenation of methane was not observed. The hatched pixel at (n,m) = (2,3) represents an unexamined composition. | ||
The rate coefficients of FenOmAr+ obtained from the ΔM < 0 region are shown in Fig. S3 of the ESI,† which were examined for CD4; Fig. S3a and b (ESI†) show rate coefficients of dehydrogenation and those of CD4 adsorption, respectively. The rate coefficients of CD4 adsorption are about two orders of magnitude larger than those of the corresponding compositions shown in Fig. 3. This would be due to the effect of ligand exchange as discussed in Section 3.1. The rate coefficients of dehydrogenation are also higher than those of the corresponding compositions. This result is also explainable by the effect of ligand exchange as discussed in the previous reaction study of Rhn+Arm with CH4.80 The presence of Ar encourages CD4 adsorption, causing dehydrogenation to proceed efficiently if the cluster–CD4 complex retains a sufficient energy for methane activation.
3.3 Rapid methane loss in Mars’ atmosphere
As described in Section 3.2, the rate coefficients of dehydrogenation by iron and iron-oxide clusters were obtained from the present reaction experiment. Here we consider the case that methane with an initial concentration of 2 × 109 cm−3 decreases by an order of magnitude in seven days (≈6 × 105 s).14 We also assume pseudo-first-order reaction kinetics by regarding the density of the iron-oxide clusters/particles as constant; we focus here on iron-oxide clusters because Mars’ soil is rich in iron oxides. The pseudo-first-order rate equation, −d[CH4]/dt = kn[CH4], is thus employed to estimate the number density n of the iron-oxide cluster from the rate coefficients k of methane dehydrogenation. Based on the value of k = 1 × 10−13 cm3 s−1 obtained for Fe3O+ in the present study, we estimate that the presence of iron-oxide clusters/particles of 4 × 107 cm−3 (≈10−7 Pa at −50 °C) would explain the mysterious loss of methane in Mars’ atmosphere.4 Conclusions
Gas-phase reaction experiments were performed on size-selected free iron and iron-oxide cluster cations, FenOm+ (n = 2–4, 0 ≤ m ≤ n), with methane. Dehydrogenation of methane was observed; larger and oxygen-poor clusters tend to exhibit higher reactivity. We also revealed that the presence of Ar as a weakly bound ligand promotes the adsorption and dehydrogenation of methane. Based on the rate coefficient of methane dehydrogenation determined in the present study, e.g., 1 × 10−13 cm3 s−1 for Fe3O+, it is speculated that the presence of such iron-oxide clusters/particles in Mars’ atmosphere at a density as low as 4 × 107 cm−3 (≈10−7 Pa at −50 °C) is sufficient to explain the rapid loss of methane observed recently by the Curiosity rover. If ligand exchange with, for example, Ar were plausible, the required cluster/particle density could be even lower by about two orders of magnitude. The chemical process mediated by iron-oxide clusters/particles may play important roles in chemistry not only on Mars but also widely in space because most iron exists as compounds, typically iron oxides.104Author contributions
M. A. contributed to conceptualization, investigation, formal analysis, writing – original draft, funding acquisition and project administration. S. K. contributed to investigation and formal analysis. Y. S. contributed to conceptualization and writing – review & editing. A. T. contributed to writing – review & editing and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present study was supported by Grants-in-Aid for Scientific Research on Innovative Areas (JP17H06456) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), and for Scientific Research (B) (JP22H01288) from the Japan Society for Promotion of Science (JSPS). This work was also supported by the GIMRT Program of the Institute for Materials Research, Tohoku University (Proposal No. 202212-CRKKE-0013 and 202212-SCKXX-0009).References
- T. M. Steel and W. W. Duley, Astrophys. J., 1987, 315, 337–339 CrossRef CAS.
- R. van Boekel, M. Min, Ch Leinert, L. B. F. M. Waters, A. Richichi, O. Chesneau, C. Dominik, W. Jaffe, A. Dutrey, U. Graser, Th Henning, J. de Jong, R. Köhler, A. de Koter, B. Lopez, F. Malbet, S. Morel, F. Paresce, G. Perrin, Th Preibisch, F. Przygodda, M. Schöller and M. Wittkowski, Nature, 2004, 432, 479–482 CrossRef CAS PubMed.
- J. A. Nuth III, N. M. Johnson and S. A. Manning, Astrophys. J., 2008, 673, L225–L228 CrossRef.
- M. Arakawa, K. Kohara and A. Terasaki, J. Phys. Chem. C, 2015, 119, 10981–10986 CrossRef CAS.
- M. Arakawa, K. Ando, S. Fujimoto, S. Mishra, G. Naresh Patwari and A. Terasaki, Phys. Chem. Chem. Phys., 2018, 20, 13974–13982 RSC.
- M. Arakawa, M. Horioka, K. Minamikawa, T. Kawano and A. Terasak, J. Phys. Chem. C, 2020, 124, 26881–26888 CrossRef CAS.
- S. M. Lang and T. M. Bernhardt, Phys. Chem. Chem. Phys., 2012, 14, 9255–9269 RSC.
- A. C. Reber, S. Paranthaman, P. A. Clayborne, S. N. Khanna and A. W. Castleman, Jr., ACS Nano, 2008, 2, 1729–1737 CrossRef CAS PubMed.
- J. M. Guiu, B.-A. Ghejan, T. M. Bernhardt, J. M. Bakker, S. M. Lang and S. T. Bromley, ACS Earth Space Chem., 2022, 6, 2465–2470 CrossRef PubMed.
- T. Studemund, K. Pollow, M. Förstel and O. Dopfer, Phys. Chem. Chem. Phys., 2023, 25, 17609–17618 RSC.
- M. Arakawa, R. Yamane and A. Terasaki, J. Phys. Chem. A, 2016, 120, 139–144 CrossRef CAS PubMed.
- M. Arakawa, T. Omoda and A. Terasaki, J. Phys. Chem. C, 2017, 121, 10790–10795 CrossRef CAS.
- M. Arakawa, D. Okada, S. Kono and A. Terasaki, J. Phys. Chem. A, 2020, 124, 9751–9756 CrossRef CAS PubMed.
- C. R. Webster, P. R. Mahaffy, S. K. Atreya, J. E. Moores, G. J. Flesch, C. Malespin, C. P. McKay, G. Martinez, G. L. Smith and J. Martin-Torres, Science, 2018, 360, 1093–1096 CrossRef CAS PubMed.
- Y. Luo, M. Mischna, J. Lin, B. Fasoli, X. Cai and Y. Yung, Earth Space Sci., 2021, 8(11), e2021EA001915 CrossRef CAS PubMed.
- A. A. Pavlov, J. Johnson, R. Garcia-Sanchez, A. Siguelnitzky, C. Johnson, J. Davis, S. Guzewich and P. Misra, J. Geophys. Res. Planets, 2024, 129, e2023JE007841 CrossRef CAS.
- P. L. Whelley and R. Greeley, J. Geophys. Res., 2008, 213, E07002 Search PubMed.
- C. Ganino, G. Libourel, A. M. Nakamura, S. Jacomet, O. Tottereau and P. Michel, Meteorit. Planet. Sci., 2018, 53(11), 2306–2326 CrossRef CAS.
- A. C. Rosenzweig, P. Nordlund, P. M. Takahara, C. A. Frederick and S. J. Lippard, Nature, 1993, 366, 537–543 CrossRef CAS PubMed.
- A. C. Rosenzweig, P. Nordlund, P. M. Takahara, C. A. Frederick and S. J. Lippard, Chem. Biol., 1995, 2, 409–418 CrossRef CAS PubMed.
- M. Merkx, D. A. Kopp, M. H. Sazinsky, J. L. Blazyk, J. Müller and S. J. Lippard, Angew. Chem. Int. Ed., 2001, 40, 2782–2807 CrossRef CAS.
- L. Shu, J. C. Nesheim, K. Kauffmann, E. Münck, J. D. Lipscomb and L. Que Jr., Science, 1997, 275, 515–518 CrossRef CAS PubMed.
- B. J. Brazeau, R. N. Austin, C. Tarr, J. T. Groves and J. D. Lipscomb, J. Am. Chem. Soc., 2001, 123, 11831–11837 CrossRef CAS PubMed.
- K. Yoshizawa, Acc. Chem. Res., 2006, 39, 375–382 CrossRef CAS PubMed.
- K. Yoshizawa and R. Hoffmann, Inorg. Chem., 1996, 25, 2409–2410 CrossRef PubMed.
- K. Yoshizawa, T. Ohta, T. Yamabe and R. Hoffmann, J. Am. Chem. Soc., 1997, 26, 12311–12321 CrossRef.
- K. Yoshizawa, T. Ohta, Y. Shiota and T. Yamabe, Chem. Lett., 1997, 1213–1214 CrossRef CAS.
- P. E. M. Siegbahn and R. H. Crabtree, J. Am. Chem. Soc., 1997, 119, 3103–3113 CrossRef CAS.
- P. E. M. Siegbahn, J. Biol. Inorg. Chem., 2001, 6, 27–45 CrossRef CAS PubMed.
- K. Yoshizawa, T. Ohta and T. Yamabe, Bull. Chem. Soc. Jpn., 1998, 119, 1899–1909 CrossRef.
- H. Basch, K. Mogi, D. G. Musaev and K. Morokuma, J. Am. Chem. Soc., 1999, 121, 7249–7256 CrossRef CAS.
- B. D. Dunietz, M. D. Beachy, Y. Cao, D. A. Whittington and S. J. Lippard, J. Am. Chem. Soc., 2000, 122, 2828–2839 CrossRef CAS.
- K. Yoshizawa and T. Yumura, Chem. – Eur. J., 2003, 9, 2347–2358 CrossRef CAS PubMed.
- S.-P. Huang, Y. Shiota and K. Yoshizawa, Dalton Trans., 2013, 42, 1011–1023 RSC.
- K. Yoshizawa, Bull. Chem. Soc. Jpn., 2013, 86, 1083–1116 CrossRef CAS.
- M. Barona, C. A. Gaggioli, L. Gagliardi and R. Q. Snurr, J. Phys. Chem. A, 2020, 124, 1580–1592 CrossRef CAS PubMed.
- D. Schröder and H. Schwarz, Angew. Chem., Int. Ed. Engl., 1990, 29, 1433–1434 CrossRef.
- D. Schröder, A. Fiedler, J. Hrušák and H. Schwarz, J. Am. Chem. Soc., 1992, 114, 1215–1222 CrossRef.
- D. Schröder and H. Schwarz, Angew. Chem., Int. Ed. Engl., 1995, 34, 1973–1995 CrossRef.
- D. Schröder, H. Schwarz, D. E. Clemmer, Y. Chen, P. B. Armentrout, V. I. Baranov and D. K. Böhme, Int. J. Mass Spectrom. Ion Processes, 1997, 161, 175–191 CrossRef.
- D. Schröder and H. Schwarz, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18114–18119 CrossRef PubMed.
- K. Yoshizawa, Y. Shiota and T. Yamabe, Chem.–Eur. J., 1997, 3, 1160–1169 CrossRef CAS.
- K. Yoshizawa, Y. Shiota and T. Yamabe, J. Am. Chem. Soc., 1998, 120, 564–572 CrossRef CAS.
- G. Altinay, M. Citir and R. B. Metz, J. Phys. Chem. A, 2010, 114, 5104–5112 CrossRef CAS PubMed.
- K. Yoshizawa, Y. Shiota and T. Yamabe, Organometallics, 1998, 17, 2825–2831 CrossRef CAS.
- K. Yoshizawa, Y. Shiota and T. Yamabe, J. Chem. Phys., 1999, 111, 538–545 CrossRef CAS.
- Y. Shiota and K. Yoshizawa, J. Am. Chem. Soc., 2000, 122, 12317–12326 CrossRef CAS.
- K. Yoshizawa, Y. Shiota, Y. Kagawa and T. Yamabe, J. Phys. Chem. A, 2000, 104, 2552–2561 CrossRef CAS.
- Y. Shiota and K. Yoshizawa, J. Chem. Phys., 2003, 118, 5872–5879 CrossRef CAS.
- Y. Wang, X. Sun, J. Zhang and Jilai Li, J. Phys. Chem. A, 2017, 121, 3501–3514 CrossRef CAS PubMed.
- T. Zubatiuk, G. Hill, D. Leszczynska, M. Fan, A. H. Rony and J. Leszczynski, Chem. Phys. Lett., 2018, 706, 708–714 CrossRef CAS.
- Q. Sun, Z. Li, A. Du, J. Chen, Z. Zhu and S. C. Smith, Fuel, 2012, 96, 291–297 CrossRef CAS.
- S. Chiodo, I. Rivalta, M. del Carmen Michelini, N. Russo, E. Sicilia and J. M. Ugalde, J. Phys. Chem. A, 2006, 110, 12501–12511 CrossRef CAS PubMed.
- Q. Sun, Z. Li, M. Wang, A. Du and S. C. Smith, Chem. Phys. Lett., 2012, 550, 41–46 CrossRef CAS.
- A. Fiedler, J. Hrušák, W. Koc and H. Schwarz, Chem. Phys. Lett., 1993, 211, 242–248 CrossRef CAS.
- A. Fiedler, D. Schröder, S. Shaik and H. Schwarz, J. Am. Chem. Soc., 1994, 116, 10734–10741 CrossRef CAS.
- Z. Cao, M. Duran and M. Solà, Chem. Phys. Lett., 1997, 274, 411–421 CrossRef CAS.
- A. T. García-Sosa and M. Castro, Int. J. Quantum Chem., 2000, 80, 307–319 CrossRef.
- G. L. Gutsev, S. N. Khanna, B. K. Rao and P. Jena, J. Phys. Chem. A, 1999, 103, 5812–5822 CrossRef CAS.
- H. Shiroishi, T. Oda, I. Hamada and N. Fujima, Polyhedron, 2005, 24, 2472–2476 CrossRef CAS.
- N. M. Reilly, J. U. Reveles, G. E. Johnson, S. N. Khanna and A. W. Castleman, Jr., J. Phys. Chem. A, 2007, 111, 4158–4166 CrossRef CAS PubMed.
- H. Shiroishi, T. Oda, I. Hamada and N. Fujima, Eur. Phys. J. D, 2003, 24, 85–88 CrossRef CAS.
- N. O. Jones, B. V. Reddy, F. Rasouli and S. N. Khanna, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 165411 CrossRef.
- G. L. Gutsev, K. G. Belay, L. G. Gutsev and B. R. Ramachandra, Comput. Mater. Sci., 2017, 137, 134–143 CrossRef CAS.
- S. Yin, W. Xue, X.-L. Ding, W.-G. Wang, S.-G. He and M.-F. Ge, Int. J. Mass Spectrom., 2009, 281, 72–78 CrossRef CAS.
- X.-L. Ding, W. Xue, Y.-P. Ma, Z.-C. Wang and S.-G. He, J. Phys. Chem., 2009, 130, 014303 CrossRef PubMed.
- A. Erlebach, C. Hühn, R. Jana and M. Sierka, Phys. Chem. Chem. Phys., 2014, 16, 26421–26426 RSC.
- R. Mejia-Olvera, J. U. Reveles, S. M. Pacheco-Ortín and J. Santoyo-Salazar, Chem. Phys. Lett., 2018, 706, 494–500 CrossRef CAS.
- R. Logemann, G. A. de Wijs, M. I. Katsnelson and A. Kirilyuk, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 144427 CrossRef.
- S. López, A. H. Romero, J. Mejía-López, J. Mazo-Zuluaga and J. Restrepo, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 085107 CrossRef.
- Q. Wang, Q. Sun, M. Sakurai, J. Z. Yu, B. L. Gu, K. Sumiyama and Y. Kawazoe, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 12672–12677 CrossRef CAS.
- Q. Sun, Q. Wang, K. Parlinski, J. Z. Yu, Y. Hashi, X. G. Gong and Y. Kawazoe, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 5781–5785 CrossRef CAS.
- K. Palotás, A. N. Andriotis and A. Lappas, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 075403 CrossRef.
- K. Ohshimo, T. Komukai, R. Moriyama and F. Misaizu, J. Phys. Chem. A, 2014, 118, 3899–3905 CrossRef CAS PubMed.
- K. S. Molek, C. Anfuso-Cleary and M. A. Duncan, J. Phys. Chem. A, 2008, 112, 9238–9247 CrossRef CAS PubMed.
- M. Li, S.-R. Liu and P. B. Armentrout, J. Phys. Chem., 2009, 131, 144310 CrossRef PubMed.
- P. Jackson, J. N. Harvey, D. Schröder and H. Schwarz, Int. J. Mass Spectrom., 2001, 204, 223–245 CrossRef.
- R. Liyanage, X.-G. Zhang and P. B. Armentrout, J. Chem. Phys., 2001, 115, 9747–9763 CrossRef CAS.
- F. Liu, X.-G. Zhang, R. Liyanage and P. B. Armentrout, J. Chem. Phys., 2004, 121, 10976–10990 CrossRef CAS PubMed.
- G. Albert, C. Berg, M. Beyer, U. Achatz, S. Joos, G. Niedner-Schatteburg and V. E. Bondybey, Chem. Phys. Lett., 1997, 268, 235–241 CrossRef CAS.
- S. M. Lang and T. M. Bernhardt, Faraday Disc., 2011, 152, 337–351 RSC.
- S. M. Lang, A. Frank and T. M. Bernhardt, Catal. Sci. Technol., 2013, 3, 2926–2933 RSC.
- S. M. Lang, A. Frank and T. M. Bernhardt, J. Phys. Chem. C, 2013, 117, 9791–9800 CrossRef CAS.
- J. F. Eckhard, T. Masubuchi, M. Tschurl, R. N. Barnett, U. Landman and U. Heiz, J. Phys. Chem. A, 2021, 125, 5289–5302 CrossRef CAS PubMed.
- S. M. Lang, T. M. Bernhardt, R. N. Barnett and U. Landman, Angew. Chem., Int. Ed., 2010, 49, 980–983 CrossRef CAS PubMed.
- S. M. Lang, T. M. Bernhardt, R. N. Barnett and U. Landman, J. Phys. Chem. C, 2011, 115, 6788–6795 CrossRef CAS.
- H.-F. Li, Z.-Y. Li, Q.-Y. Liu, X.-N. Li, Y.-X. Zhao and S.-G. He, J. Phys. Chem. Lett., 2015, 6, 2287–2291 CrossRef CAS PubMed.
- S. Hirabayashi and M. Ichihashi, J. Phys. Chem. A, 2020, 124, 5274–5279 CrossRef CAS PubMed.
- Z.-C. Wang, T. Weiske, R. Kretschmer, M. Schlangen, M. Kaupp and H. Schwarz, J. Am. Chem. Soc., 2011, 133, 16930–16937 CrossRef CAS PubMed.
- C.-M. Sun, G.-P. Wei, Y. Yang and Y.-X. Zhao, J. Phys. Chem. A, 2024, 128, 1218–1225 CrossRef CAS PubMed.
- M. Citir, G. Altinay, G. Austein-Miller and R. B. Metz, J. Chem. Phys. A, 2010, 114, 11322–11329 CrossRef CAS PubMed.
- M. A. Ashraf, Ch. W. Copeland, A. Kocak, A. R. McEnroe and R. B. Metz, Phys. Chem. Chem. Phys., 2015, 17, 25700–25704 RSC.
- C. W. Copeland, M. A. Ashraf, E. M. Boyle and R. B. Metz, J. Phys. Chem. A, 2017, 121, 2132–2137 CrossRef CAS PubMed.
- O. V. Lushchikova, S. Reijmer, P. B. Armentrout and J. M. Bakker, J. Am. Soc. Mass Spectrom., 2022, 33, 1393–1400 CrossRef CAS PubMed.
- J. Lengyel, N. Levin, F. J. Wensink, O. V. Lushchikova, R. N. Barnett, U. Landman, U. Heiz, J. M. Bakker and M. Tschurl, Angew. Chem., Int. Ed., 2020, 59, 23631–23635 CrossRef CAS PubMed.
- D. J. Harding, C. Kerpal, G. Meijer and A. Fielicke, Angew. Chem., Int. Ed., 2012, 51, 817–819 CrossRef CAS PubMed.
- S. M. Lang, T. M. Bernhardt, V. Chernyy, J. M. Bakker, R. N. Barnett and U. Landman, Angew. Chem., Int. Ed., 2017, 56, 13406–13410 CrossRef CAS PubMed.
- A. Terasaki, T. Majima and T. Kondow, J. Chem. Phys., 2007, 127, 231101 CrossRef PubMed.
- M. Arakawa, K. Kohara, T. Ito and A. Terasaki, Eur. Phys. J. D, 2013, 67, 80 CrossRef.
- H. Haberland, M. Karrais, M. Mall and Y. Thurner, J. Vac. Sci. Technol., A, 1992, 10, 3266 CrossRef CAS.
- T. Ito, K. Egashira, K. Tsukiyama and A. Terasaki, Chem. Phys. Lett., 2012, 538, 19–23 CrossRef CAS.
- J. B. Griffin and P. B. Armentrout, J. Chem. Phys., 1998, 108, 8062–8074 CrossRef CAS.
- G. Gioumousis and D. P. Stevenson, J. Chem. Phys., 1958, 29, 294–299 CrossRef CAS.
- Y. Kimura, K. K. Tanaka, T. Nozawa, S. Takeuchi and Y. Inatomi, Sci. Adv., 2017, 3, e1601992 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp01337a |
| ‡ Present address: Department of Earth and Planetary Sciences, Faculty of Science, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: arakawa@geo.kyushu-u.ac.jp |
| This journal is © the Owner Societies 2024 |