
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition metal oxide clusters: advanced electrocatalysts for a sustainable energy future
Sanwal
Piracha†
ab,
Yifei
Zhang†
ab,
Ali
Raza
 c and
Gao
Li
*ab
c and
Gao
Li
*ab
aInstitute of Catalysis for Energy and Environment, College of Chemistry and Chemical Engineering, Shenyang Normal University, Shenyang 110034, Liaoning, China
bState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: gaoli@dicp.ac.cn
cDepartment of Physics “Ettore Pancini”, University of Naples Federico II, Piazzale Tecchio, 80, 80125 Naples, Italy
First published on 8th August 2024
Abstract
The comprehensive utilization of sustainable green energy is essential to face the global energy and environmental crisis. The oxygen reduction reaction (ORR), oxygen evolution reaction (OER), and electrocatalytic urea synthesis (EUS) are the pivotal electrocatalytic processes, necessitating the development of low-cost electrocatalysts with high efficiency. Small-sized transition metal oxide (TMO) clusters have attracted a lot of attention because of their exceptional qualities, such as exhibiting a dense array of low-coordinated metal active sites (e.g. abundant metal cation defects and oxygen vacancy), amorphous structures with high surface energy, high atom utilization efficiency, and cost-effectiveness. Furthermore, the synergistic actions between metal clusters and TM–Nx single atom active sites remarkably boost up the electrocatalytic performances, corroborated by density functional theory (DFT). More efforts in this comprehensive feature article are expected to achieve insights into the fundamental understanding of electrocatalytic reaction mechanisms in our lab and serve as a guide for creating cutting-edge electrocatalysts of transition metal oxide clusters.
Introduction
Due to expanding global energy use and growing environmental concerns, there is a growing demand for clean, sustainable energy technologies including fuel cells, solar cells, and metal–air batteries.1–4 The efficiency of catalysts is crucial for the application of these technologies, which rely on catalytic processes, including the oxygen reduction reaction (ORR), the oxygen evolution reaction (OER), and electrocatalytic urea synthesis (EUS) from nitrate ions and CO2.5–7 The high price, lack of stability, and restricted availability of traditional noble metal catalysts like Ir, Pd, and Pt prevent their broad use as energy solutions.5–7 To overcome these obstacles and make renewable energy systems more practical, lots of efforts have gone into creating metal-free or non-Pt catalysts that are just as efficient for the ORR, OER, EUS, and other processes.8–10 Multifunctional electrocatalysts as substitutions, which have recently been developed, may provide several methods simultaneously, opening up new possibilities for integrated energy conversion systems.11–13 There is an alternative for improving electrochemical activity with less expense and resource consumption thanks to new catalysts, such as ultrasmall-sized particles (also named clusters, size of 2–5 nm). Incorporating sophisticated catalysts into comprehensive electrochemical systems may greatly improve energy conversion's lifespan and efficiency, propelling us toward a greener, more sustainable energy future.5,14–17Transition metal oxide (TMO) clusters (particle size of 3–6 nm) have a dense array of low-coordinated metal active sites (e.g. abundant metal cation defects and oxygen vacancies), amorphous structures with high surface energy due to the distorted TM–O bonds, high atom utilization efficiency, cost-effectiveness, a few to name.18–21 These unique geometric characteristics are conducive to electrochemical activity by providing metal–metal contacts and reaction intermediate adsorption sites.10,22,23 TMO clusters may break scaling relations by lowering reaction barriers, regulating the rate-determining step (RDS) in electrocatalysis and improving catalytic performances (including activity and selectivity) in the ORR, OER, EUS, etc. Their ability to create catalysts with co-adsorbates, promoters, ligands, and new alloy structures makes them ideal options for electrocatalysis and energy conversion technology.24–26 Because of the expensive nature and limited geological resources of precious group metal (PGM) electrocatalysts such as RuO2 and IrO2, there has been growing interest in PGM-free catalysts.27–29 Therefore, catalysts based on transition metals that are common on Earth (such as Ni, Co, Fe, Mn, etc.), particularly transition metal hydroxides (TMHs), have attracted interest due to their electrocatalytic activity, affordability, and strong durability as effective and economical alternatives. In recent decades, several30–32 TM sulfides,33–35 nitrides,36 phosphides,30,32 oxides,37,38 and hydroxides39,40 have been explored as promising alternative OER electrocatalysts. And earth-abundant TMHs have attracted attention as promising OER electrocatalysts due to their activity, low cost, excellent stability, and environmentally friendly nature.41 Fe-based NNMEs have exceptional ORR catalytic activity. These comprise a high concentration of iron-based species such as oxides (FexOy), nitrides (FeNx), carbides (FeCx), and sulfides (FeSx).42–46 Species with high electronegativity can attach stable FeNC electrocatalyst active sites. This might potentially modify the adsorption and desorption of O intermediates on the core Fe atoms, affecting the activities of the oxygen reduction reaction (ORR).47–49
To advance the research in TM composites, our group recently explored novel approaches to increase catalytic efficiency in various applications, such as the creation of intrinsically stable MnCoOx solid solutions for long-term water oxidation, the impact of multiple active sites on promotion in Fe-based ORR electrocatalysts, the cooperative role of cobalt clusters–CoNx composites in enhancing electrochemical-oxygen-reduction, and the synergistic effect of Fe clusters and FeNx for electrocatalytic urea synthesis (EUS) from nitrate and CO2, Scheme 1. Furthermore, the enhanced electrocatalytic properties of ultrafine metallic hydroxide nanoparticles and FeNi hydroxide nanoclusters modified by ionic liquids for oxygen evolution processes are emphasized. Additionally, the use of atomically precise metal nanoclusters for active-site engineering in heterogeneous catalysis is investigated, offering insights into customizing catalytic processes for increased efficiency. What's more, the mechanism of all these electrocatalysis is well revealed by experiments combined with theoretical calculations. These results highlight the significance of materials design, synthesis techniques, and active-site modification in developing electrocatalysts for sustainable energy systems. This joint effort shows encouraging directions for creating robust and effective electrocatalytic systems necessary to meet urgent global energy issues. Lastly, we provide our thoughts about upcoming initiatives to create efficient TM electrocatalysts.
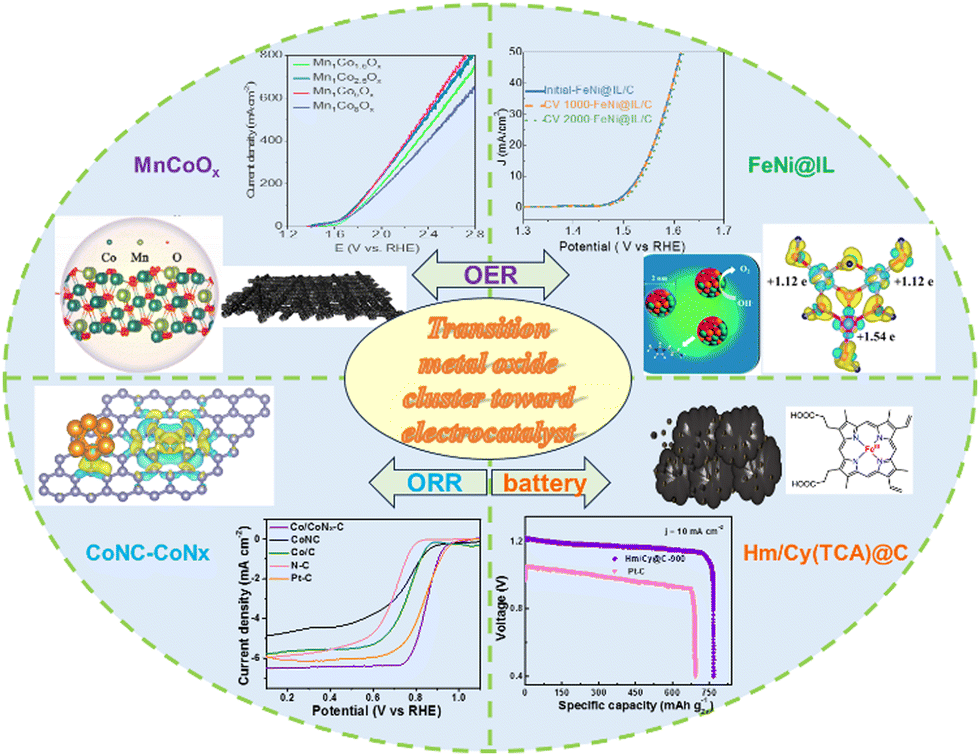 | ||
| Scheme 1 The outline of the articles on metal oxide clusters, including acidic/alkaline OER and alkaline ORR and zinc–air batteries. | ||
Synthetic methods
Pechini method
The Pechini process is a sol–gel route invented by Maggio Pechini in which, suitable metal oxides or salts are chelated with hydroxycarboxylic acid (e.g. citric acid) in aqueous solutions and then reacted with glycol to form large, cross-linked polymeric networks at 150–250 °C, which are eventually charred and achieves small-sized metal oxide solid solutions at temperatures of 300–350 °C.50,51 For example, using the potassium permanganate redox technique, the author researched printing CoMnOx compounds on carbon paper (CP). Mn1Co5Ox produced in research differs significantly from doped compounds. Potassium permanganate oxidizes low-valent cobalt to high-valent cobalt, during which, a uniform solid solution of Mn1Co5Ox is formed, Mn-rich doped Co3O4. The ability of the solid solution's crystal structure to maintain the solvent's crystal structure, resulting in the same material, structure, and characteristics, is fascinating51. This single-phase solid solution has excellent OER catalytic activity. The Mn1Co5Ox catalyst performed well in acid electrolytes, achieving overpotentials of 275 mV@10 mA cm−2 and 569 mV@100 mA cm−2. Additionally, MnCoOx showed exceptional stability during the catalysis for 300 hours at 100 mA cm−2, indicating its potential for electrocatalytic applications. As indicated in Fig. 1a, MnCoOx show strong OER catalytic efficiency in an acidic medium. In addition, it highlights this material's unique design and excellent OER performance.52 DFT results manifested that the Mn dopant can significantly elevate the d-band of Co3O4 (−2.05 eV) to near Fermi level (−1.78 eV) and sharply reduce the overpotential of the OER from 2.79 eV (Co3O4(311)) to 0.82 eV (MnCoOx(311)). Thus, the Mn dopant and Ov can stabilize OER intermediates and then reduce the energy barrier of the OER and improve catalytic activity.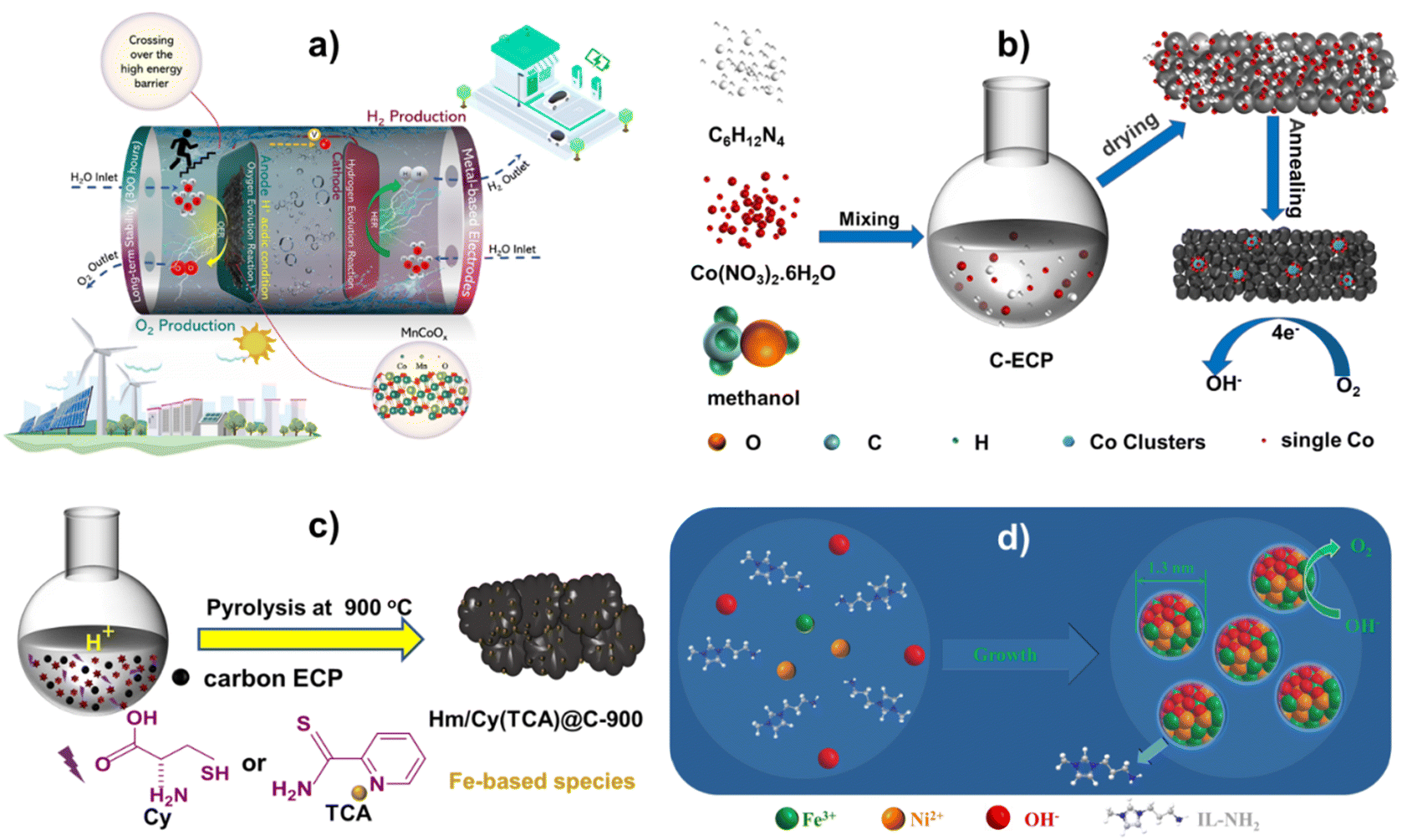 | ||
| Fig. 1 (a) Visual representation of the OER process in acidic environments, elucidating the critical points of this investigation. Reproduced with permission from ref. 52, copyright 2023 Royal Society of Chemistry. (b) The synthesis of Co/CoNx–C. Reproduced with permission from ref. 53, copyright 2022 Royal Society of Chemistry. (c) An illustrated method for producing electrocatalysts using Hm/Cy@C-900 and Hm/TCA@C-900. Redrawn from ref. 54, copyright 2023 Royal Society of Chemistry. (d) Producing amorphous FeNi hydroxide clusters capped with IL. Redrawn from ref. 55, copyright 2020 Royal Society of Chemistry. | ||
Furthermore, Zhang et al.53 developed a straightforward process for mass-producing cobalt-based electrocatalysts by carbonizing at 500 °C as shown in (Fig. 1b). STEM and X-ray absorption showed that pyrolytic treatment merged metallic Co clusters with CoNx sites. In oxygen electroreduction operations, the Co/CoNx–C composite performed better than the commercial Pt/C catalyst, with an onset potential of 0.926 V vs. RHE and a half-wave potential E1/2 of 0.853 V. The enhanced ORR performance of the Co/CoNx–C electrocatalyst is supported using first-principles calculations because of the special integration and ideal synergistic effects of electro-catalytically active CoNx and Co cluster species.55–57 Theoretical results show that the cooperative Co clusters and CoNx have higher electron density compared to bare Co/C and CoNx–C near the Fermi level, thus, Co/CoNx–C exhibits better electrical conductivity. And the RDS over Co/CoNx–C and CoNx–C occurs at the first step of O2 → OOH*, and that for Co/C is the last step of OH* → H2O. Hence, the superior ORR activity of Co/CoNx–C catalysts is attributed to the unique integration and synergistic effect of metallic Co clusters and CoNx species.
Single-step pyrolysis approach
The electrocatalysts were made using a straightforward one-pot method. The N and S precursors, Cy or TCA, were dissolved in an HNO3 solution, and hemin was dissolved in a KOH solution. After that, the alkaline solution and the acidic solution were mixed for thirty minutes using ultrasonic shock. There was additional carbon ECP. An overnight drying process at 80 °C was used to eliminate all of the water following a three-hour reaction in a rotary evaporator. Pyrolysis was used to create electrocatalysts at various temperatures in an argon environment. This was accomplished by using an identical synthesis procedure to Hm/Cy@C but replacing Cy with TCA.54 Notably, producing over 1 g of Fe-based electrocatalysts on a large scale is straightforward (Fig. 1c). M@IL-modified amorphous metallic hydroxide nanoparticles are produced in a single process (Fig. 1d). A IL-NH2 methanol solution was mixed with an aqueous solution of a metal-containing precursor, such as Fe(NO3)3, Co(NO3)2, or Ni(NO3)2, or their combination. Then, while vigorously shaking the mixture of methanol and water, a NaOH aqueous solution was added. Vulcan XC-72R carbon powder was then added to the M@IL colloidal solution.55Mechanistic insights into electrocatalytic activity during the OER/ORR and fuel cell and battery applications
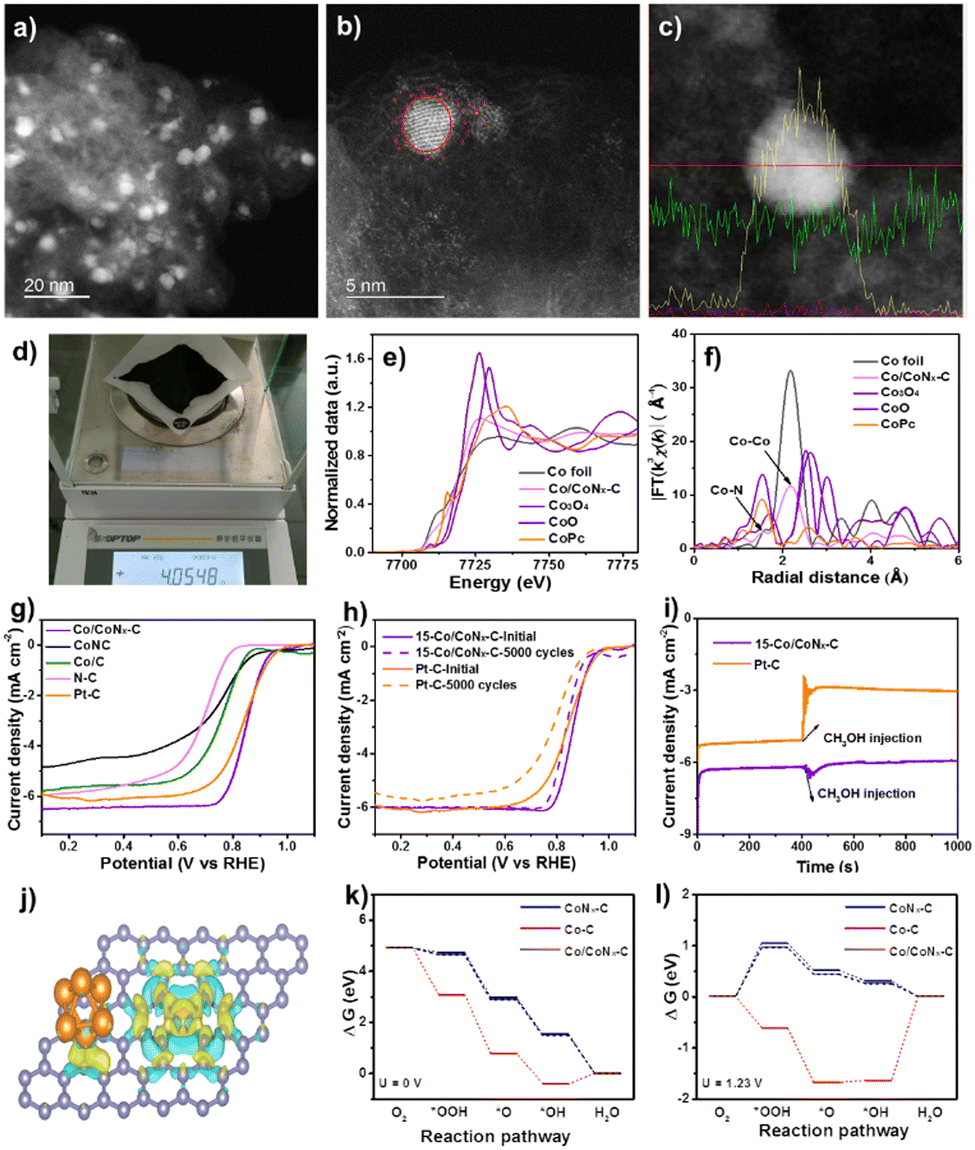 | ||
| Fig. 2 (a)–(c) S/TEM profiles with line scanning of Co/CoNx–C. (d) Quality in one-pot synthesis. (e) FT-EXAFS and (f) normalized K-edge XANES spectra. (g) LSV curves. (h) LSV curves before and after 5000 cycles. (i) I–t responses of Pt/C and Co/CoNx–C after 400s of methanol addition. (j) DFT structure. (k) and (l) Diagram of free energy for the ORR. Reproduced from ref. 53, copyright 2023 Royal Society of Chemistry. | ||
The Co/CoNx–C ORR's catalytically active sites were discovered and identified by analyzing further cobalt-based electrocatalysts of Co/C and CoNC under identical ORR circumstances (Fig. 2g). In terms of E1/2 comparison, ORR activity was as follows: Co/CoNx–C > Co/C > CoNC > N–C. Co species increase ORR activity; the N–C deficient Co species exhibited the lowest ORR activity.58 CoNC had a low restricted current density, suggesting that carbon support improves electrical conductivity. In an O2-saturated 0.1 M KOH solution, Co/CoNx–C was compared to commercial Pt/C for durability. Electrocatalyst LSV curves (Fig. 2h) were analyzed after 5000 cycles at 100 mV s−1. E1/2 losses were ∼18 mV for Co/CoNx–C and 47 mV for Pt/C, indicating the catalyst's stability and robustness. A possible explanation for the slight reduction in catalytic activity might be the partial formation of Co clusters (5.0 ± 1.5 nm) in the spent Co/CoNx–C catalyst. The I–t chronoamperometry in Fig. 2i shows that Co/CoNx–C's current density is relatively steady following methanol injection in an O2-saturated 0.1 M KOH solution, demonstrating a higher resistance to methanol toxicity. The methanol oxidation process (MOR) over the Pt/C catalyst causes the current density to fluctuate following methanol input. CoNC had a low restricted current density, suggesting that carbon support improves electrical conductivity. In the control studies, the Co/CoNx–C ORR activity reduced considerably following acid leaching, which preferentially eliminated metallic Co clusters from the electrocatalyst. Because of the optimal Co/CoNx–C composition of 15 wt%, which promotes excellent dispersion of Co species and the cooperative effects of CoNx sites and Co clusters, the ORR performance is improved.59–62
Integration of Co clusters with CoNx–C in Co/CoNx–C greatly improves stability. The charge density difference indicates several electron clouds surrounding CoNx species and Co clusters (Fig. 2j), indicating substantial electron exchange interaction. According to the Bader charge calculations, Co clusters and CoNx species in Co/CoNx–C transferred 0.35 and 0.96 electrons, respectively; these values are higher than 0.32 and 0.87 electrons transferred in Co/C and CoNx–C. The electrocatalytic activity was increased by the interaction and impact of symbiotic Co clusters and CoNx species in Co/CoNx–C. The rate determining steps for Co/CoNx–C and CoNx–C occur in the first phase (O2 → OOH*), with energy barriers of 0.18 eV and 0.26 eV (U = 0 V), according to the ORR process depicted in Fig. 2k and l. Endothermic water generation results from RDS over Co/C in the last step (OH* → H2O). Three Co sites were assessed for the CoN4 and Co cluster sites because of the Co/CoNx–C's strong ORR performance.63
NNME nanomaterials are potential ORR electrocatalysts.55,64–68 However, the costly and complicated synthetic process will impede NNME expansion. Hemin (Hm) develops ORR-efficient carbon-supported FeNx electrocatalysts.69–71 Iron nitride and sulfide in Hm/Cy@C-900 are confirmed by the overlapping signals of N, S, and Fe species. The different capacities of the N and S-containing functional groups in the Cy and TCA ligands to coordinate could be the cause of the different Fe contents in Hm/Cy@C-900 and Hm/TCA@C-900. The –COOH, –SH, and –NH2 in N, S-containing ligands bind Fe atoms better than the ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S and pyridinic N groups. Next, XRD was used to analyze the electrocatalyst compositions. Carbon ECP exhibited two wide diffraction peaks at ∼20–30° and 40–45°, corresponding to graphitic carbon (002) and (101).72 In contrast, Hm/Cy@C-900 contains Fe3C, FeSx, and metallic Fe. Hm/TCA@C-900 has a novel Fe3O4 XRD pattern, Fig. 3a, unlike Hm/Cy@C-900. Thus, Hm/TCA@C-900 should include Fe3C, FeSx, Fe3O4, and α-Fe nanoparticles. Unlike other iron salts, its metal macrocycle dissolvability permits iron-based species to self-assemble (Fig. 3b). Solubility-induced scattering of Fe nanoparticles with carbon as active sites was done by Shen et al.73
S and pyridinic N groups. Next, XRD was used to analyze the electrocatalyst compositions. Carbon ECP exhibited two wide diffraction peaks at ∼20–30° and 40–45°, corresponding to graphitic carbon (002) and (101).72 In contrast, Hm/Cy@C-900 contains Fe3C, FeSx, and metallic Fe. Hm/TCA@C-900 has a novel Fe3O4 XRD pattern, Fig. 3a, unlike Hm/Cy@C-900. Thus, Hm/TCA@C-900 should include Fe3C, FeSx, Fe3O4, and α-Fe nanoparticles. Unlike other iron salts, its metal macrocycle dissolvability permits iron-based species to self-assemble (Fig. 3b). Solubility-induced scattering of Fe nanoparticles with carbon as active sites was done by Shen et al.73
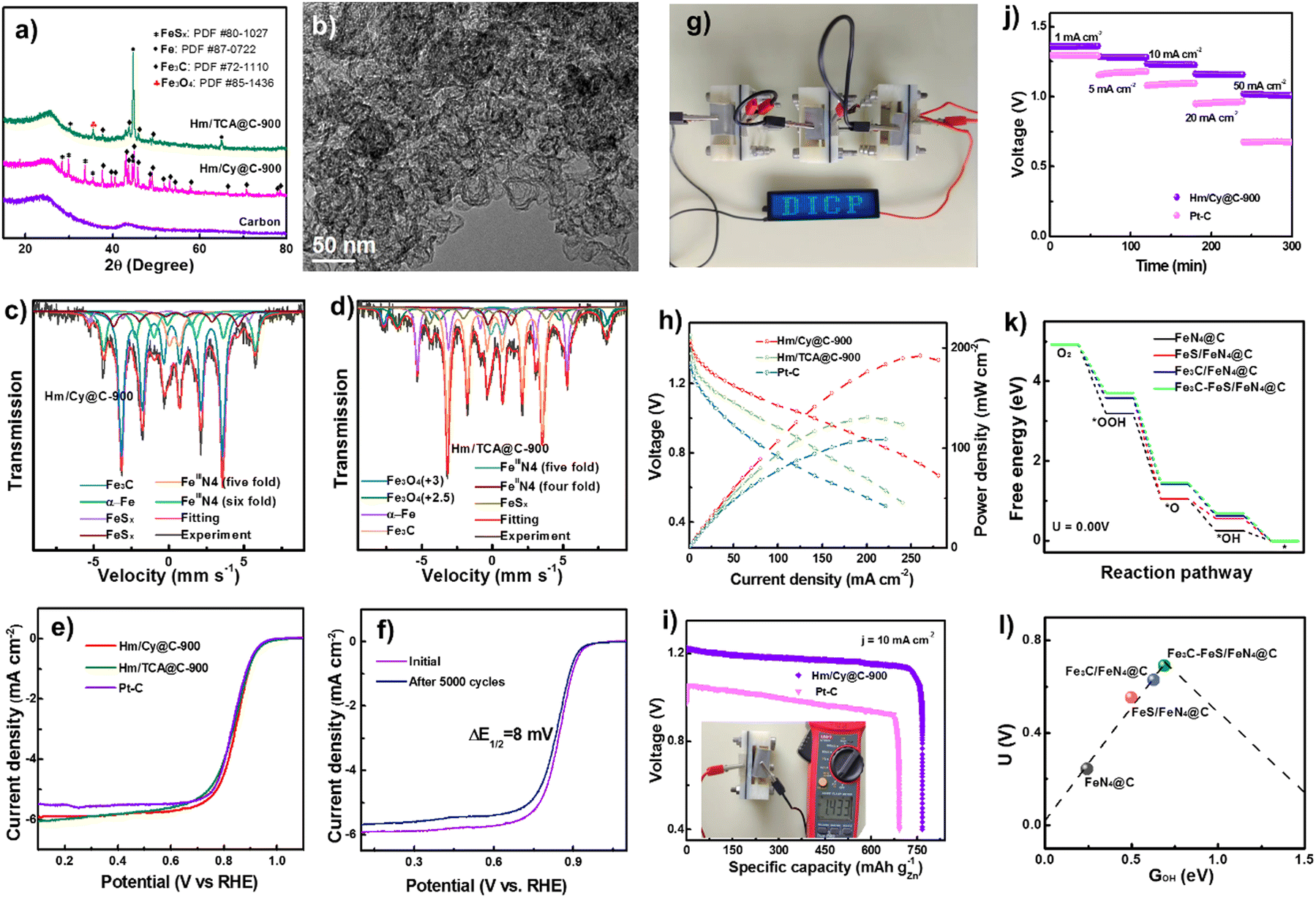 | ||
| Fig. 3 (a) XRD patterns. (b) High resolution TEM of Hm/Cy@C-900. Mössbauer spectra of (d) Hm/Cy@C-900 and (d) Hm/TCA@C-900. (e) LSV curves. (f) LSV curves of Hm/Cy@C-900 before and after 5000 cycles. (g) Image of ZAB turning on an LED. (h) ZAB polarization and power density. (i) Especific Hm/Cy@C-900 and Pt–C electrocatalyst discharging capabilities. (j) Battery discharge curves at various current densities. (k) U 0.00 V's ORR reaction pathway. (l) Free energy volcano diagram with ΔGOH. Reproduced from ref. 54, copyright 2023 Royal Society of Chemistry. | ||
Similarly, Hm/Cy@C-900 and Hm/TCA@C-900 composites have 0.39–0.40 at% S. Hm/Cy@C-900 (39.4%) has more C–S–C species than Hm/TCA@C-900 (32.6%), making it more conducive to carbon-S defects. C–S, C–N, and CN imply that the N and S heteroatoms have been successfully co-doped into the carbon matrixes alongside Fig. 3c's element mapping. The Fe 2p XPS spectrum has low intensity, making it difficult to identify the iron species in the Hm/TCA@C-900 electrocatalyst. So, we used 57Fe Mössbauer spectra to study the iron components in these Fe-based electrocatalysts. Fig. 3f shows the ORR electrocatalyst performance of Hm/Cy@C-T and Hm/TCA@C-900 electrocatalysts with 20 wt% Pt–C. Fig. 3g also shows the Hm/Cy@C-900 electrocatalyst's accelerated durability test. After 5000 cycles, the Hm/Cy@C-900 electrocatalyst showed just an 8 mV change in E1/2, suggesting it may be durable enough for practical applications. A blue LED screen with DICP writing is illuminated by ZABs that have been built from Hm/Cy@C-900 and coupled in series, as shown in Fig. 3h. In comparison to Hm/TCA@C-900 (131 mW cm−2@201.2 mA cm−2) and 20 weight percent Pt–C (109 mW cm−2@221.3 mA cm−2), Fig. 3i demonstrates that Hm/Cy@C-900 had a higher power density of 192 mW cm−2 at 260.7 mA cm−2. These parameters affect the power production and gas diffusion for incorporated electrocatalysts (10 mg cm−2) in Zn–air batteries, especially at elevated current densities. With an open-circuit voltage of 1.433 V (Fig. 3j, inset) and a specific capacity of 766.0 mA h g−1@10 mA cm−2, the Hm/Cy@C-900 electrocatalysts were much more powerful than the Pt–C-assembled battery (691.5 mA h g−1@10 mA cm−2). In contrast to the ZAB built using Pt–C, the Hm/Cy@C-900 battery displays a consistent discharge voltage at 50 mA cm−2 (Fig. 3k). Overall, the Hm/Cy@C-900 electrocatalyst performed best in ZAB studies. Computational simulations show that the reduction step of O* to OH* is the RDS for FeS/FeN4@C in the ORR, but it has a very strong O* chemisorption, leading to a great activation barrier. And the last step of OH* to OH− is the RDS for FeN4@C, Fe3C/FeN4@C, and Fe3C–FeS/FeN4@C, which have a relatively weak O* chemisorption strength and facilitate the ORR process. Furthermore, the offset of d-band center of Fe3C–FeS/FeN4@C was much larger than those of FeN4@C, Fe3C/FeN4@C and FeS/FeN4@C systems, indicating that either FeS or Fe3C clusters synergize with single sites of FeN4 to improve the ORR activity.
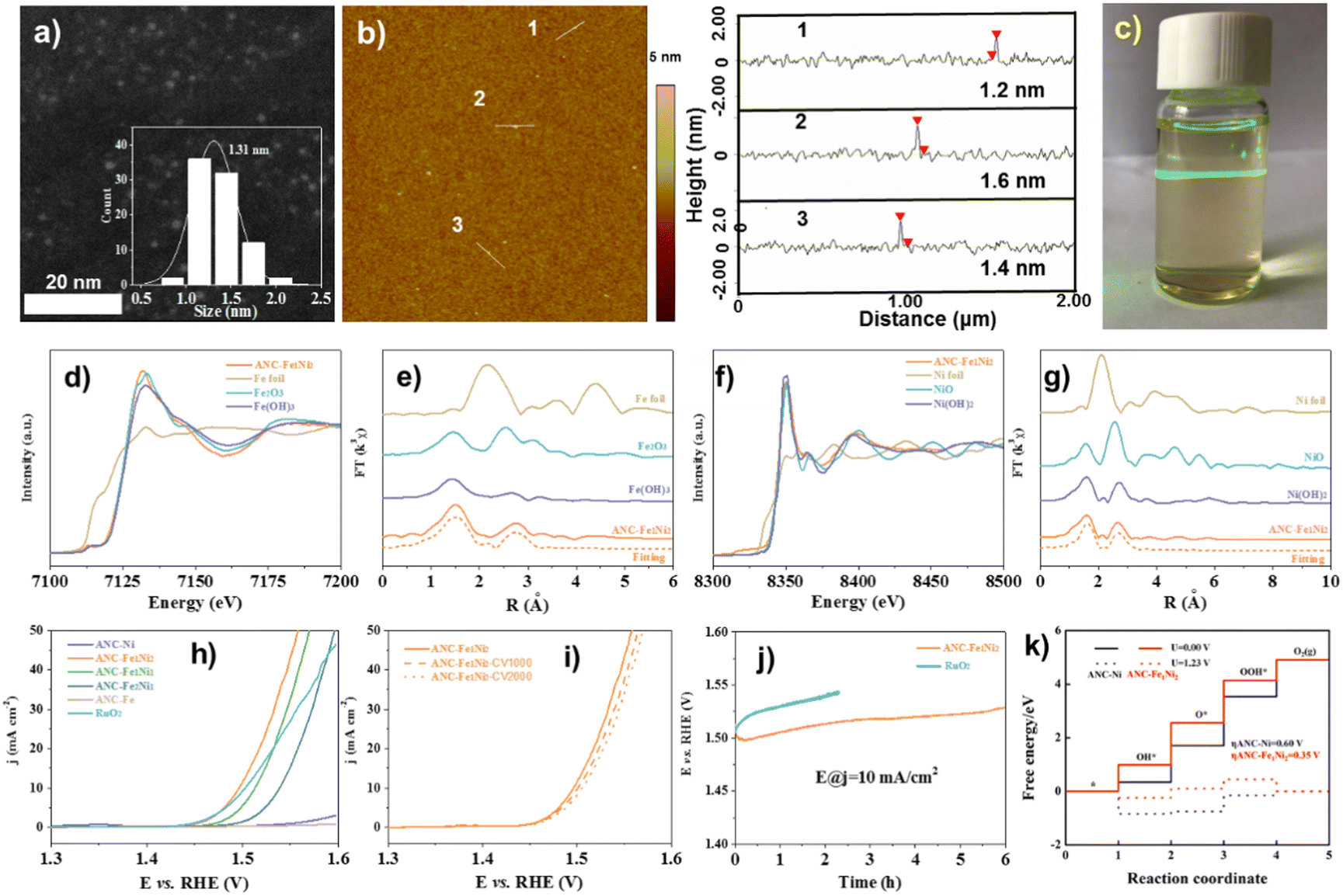 | ||
| Fig. 4 (a) HAADF-STEM, (b) AFM image with height profiles, and (c) Tyndall effect of the ANC-Fe1Ni2 catalyst. (d) Fe K-edge XANES, (e) Fourier-transformed Fe K-edge, (f) Ni K-edge XANES, and (g) Fourier-transformed Ni K-edge of ANC-Fe1Ni2. (h) Tafel plots. (i) LSV curves (before and after 1000/2000 CV cycles) of ANC-Fe1Ni2. (j) Durability tests. (k) Calculated OER free energy diagram. Reproduced from ref. 81, copyright 2022 from Royal Society of Chemistry Royal. | ||
MnCoOx with a cubic structure was determined by XRD (Fig. 5a).82,83 XRD patterns of MnCoOx samples showed no reflections for MnOx species because of the manganese ions' scattering inside the cobaltous matrix, even at high MnOx concentrations (∼40 mol%). The XRD pattern of MnCoOx catalysts with different doping levels showed a small shift towards lower angles around 36.8° for (311) as Mn dopants increased, suggesting that Mn cations were well doped into the Co3O4 lattice to produce solid solutions. We also examined Mn1Co5Ox annealed at various temperatures using XRD. At 200 °C, Co3O4 developed its crystalline phase. High-temperature calcination enhanced the crystallinity. Despite annealing at 500 °C, Mn1Co5Ox composites showed no Mn oxide peaks, suggesting thermal stability. TEM was used to study MnCoOx solid solutions for elemental composition and morphology. TEM scans showed most oxide particles were 6–8 nm and expose Co3O4(220) and (311) planes (Fig. 5b).84–86 Interestingly, the MnOx species has no lattice fringes, suggesting that Mn was doped in Co3O4.
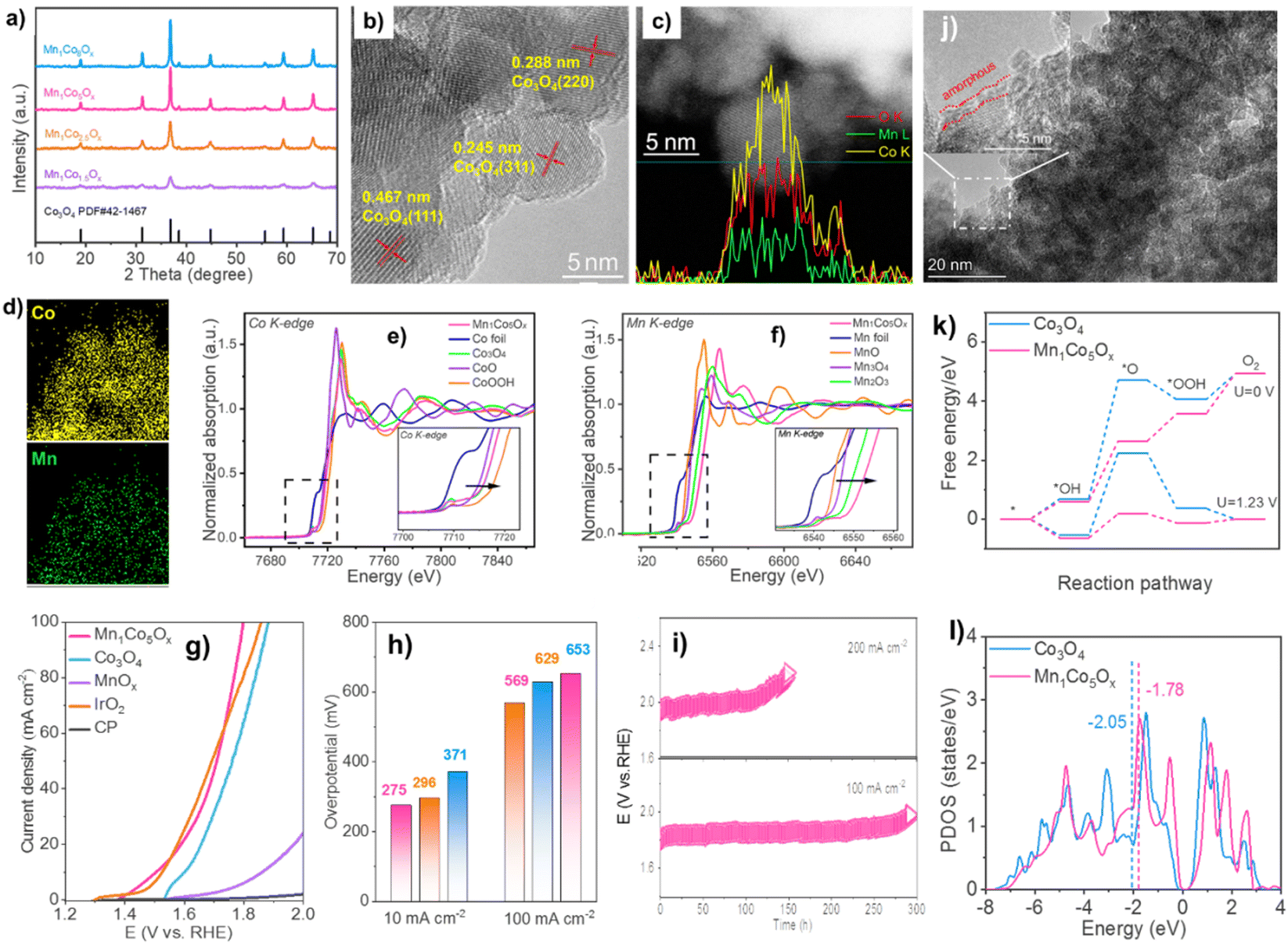 | ||
| Fig. 5 (a) XRD patterns. The linear scanning profiles of catalyst's cross-section (c). (b) and (d) HAADF-STEM picture with elemental mapping. XANES spectra of (e) Co and (f) Mn K-edge. (g) OER polarization curves and (h) overpotentials. (i) Chromatopotentiometric measurements. (j) STEM picture of used Mn1Co5Ox. (k) OER routes' reaction-free energies at 0 and 1.23 volts. (l) Co activity sites' d-band center. Reproduced from ref. 52 copyright 2023 from Royal Society of Chemistry. | ||
The linear-scanning profiles in Fig. 5c showed that Mn atoms were scattered inside Co3O4's lattice. The line sweep region has uniform lattice fringes and a distribution of components that shows that Co and Mn coexist with Co being more abundant. The elemental mapping (Fig. 5d) supports the homogeneity of Co and Mn species in MnCoOx solid solutions. XANES spectra at Co and Mn K-edge were used to study Mn1Co5Ox's electrical structure and local coordination.68 Mn1Co5Ox has a peak intensity equal to Co3O4 but distinct from Co-foil (Fig. 5e). Also, Mn1Co5Ox's pre-line matches Co3O4. This indicates that Mn1Co5Ox's primary Co species valence states are Co2+ and Co3+. In contrast, the XANES spectra at Mn K-edge showed that Mn1Co5Ox has a substantially greater peak intensity of the white line than Mn3O4 and Mn2O3. Mn1Co5Ox's pre-line moved toward higher binding energies than Mn2O3, suggesting that its average Mn species valence should be greater than +3. It also shows potassium permanganate oxidation-related electron shortage around the Mn site. The Mn1Co5Ox catalyst's cooperation context was investigated using an R-space extended X-ray absorption fine structure (EXAFS) fitting analysis at the Co and Mn K-edges, as depicted in Fig. 5g and h. The Co–O bond distance in Mn1Co5Ox increased from 1.81 Å to 1.83 Å against the control group of CoOOH. In contrast, Mn1Co5Ox reduced the Co–O bond distance from 1.91 Å to 1.83 Å compared to Co3O481 Mn1Co5Ox reduced the Mn–O bonding distance from 1.80 Å to 1.67 Å compared to Mn2O3, likely owing to Mn's valence state being greater than +3. The electron density of Mn1Co5Ox's Co–O bonds was comparable to that of Co3O4, but somewhat greater than that of CoOOH and shorter. Compared to Mn2O3, Mn1Co5Ox has a lower electron density in its Mn–O bonds. This difference in electron density facilitated the creation of a solid-solution structure with a roughly uniform M–O bond length and expedited the flow of electrons between the Mn and Co atoms, hence enhancing OER catalytic activity.
The Mn1Co5Ox composite outperformed Co3O4, MnOx, CP support, and IrO2 in terms of catalytic efficiency, as indicated by the OER polarization curves (Fig. 5i). This suggests that the OER took place on the surface. Mn1Co5Ox specifically showed overpotentials of 275 and 569 mV at current densities of 10 and 100 mA cm−2. (Fig. 5j). Mn1Co5Ox's increased catalytic OER performance is due to Mn–Co collaboration. The Mn1Co5Ox catalyst had a ∼15-fold increase in lifetime compared to Co3O4 at 100 mA cm−2 (Fig. 5k), suggesting a 20.9% Mn dopant. The CP support is located at 2θ = 26.5°. The Mn1Co5Ox catalyst's TEM image exhibited an amorphous layer of ∼1 nm thickness (Fig. 5l), confirming Co(OH)x production. XPS was also used to analyze the makeup of the pristine surface and its level of oxidation and used MnCoOx catalysts. Conversely, the intermediate O* has the maximum activation energy, which is thermodynamically unfavorable. After Mn doping,71 the OER overpotential drops from 2.79 eV (Co3O4(311)) to 0.82 eV (MnCoOx (311)). Fig. 5n shows that Mn may raise Co3O4's d-band center position from −2.05 eV to near the Fermi level (−1.78 eV). Interestingly, Ov equilibrates the active site's d-band center (Ed = −1.88 eV). Moderate intermediate species adsorption may reduce the OER reaction energy barrier. Thus, Mn and Ov stabilize reaction intermediates, lowering the OER reaction energy barrier and improving catalytic activity72 Results of theoretical simulations match experimental phenomena.
We created the hybrid MnRuOx catalyst using RuCl3 and KMnO4 as metal precursors and controlled crystallization at different annealing temperatures using a simple Pechini approach (Fig. 6a). Operando X-ray diffraction (XRD) investigation tracked the catalyst's crystal phase shift in response to temperature to determine how temperature affects crystallization, Fig. 6b.87 XRD spectra of MnRuOx calcined at 300–350 °C show an amorphous phase without visible precursor peaks. MnRuOx-300 exhibits no distinguishing peaks due to its homogeneous crystal and amorphous dispersion. XRD patterns showed that the MnRuOx hybrid material crystallizes virtually entirely at temperatures over 400 °C, containing Ru oxides. The MnRuOx particle is ∼4–5 nm in size (Fig. 6c) and exposes RuO2(110) and (101) planes.88 An amorphous phase filled the interstitial spaces between the microcrystalline phases, according to the STEM image. The 3D atomic overlap Gaussian fitting findings from the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) pictures showed diverse atomic configurations in different locations.89 Region 1 had irregular atoms, indicating an amorphous structure (Fig. 6d and e). Region 2 had consistent atom arrangements, suggesting microcrystalline structures (Fig. 6f and g). These findings support the MnRuOx-300 catalyst heterostructures. Thus, the MnRuOx catalyst has both microcrystalline and amorphous structures, with RuO2 crystals in the microcrystalline part. Linear scanning analysis of individual particles revealed a homogenous distribution of Mn and Ru elements, generating a doped phase in the limited size range (Fig. 6h).68 Local nanoscale elemental line scan investigation shows homogeneous Mn and Ru distribution in solid solution. A large-scale elemental mapping study verifies Mn and Ru's uniform distribution in the catalyst sample.55
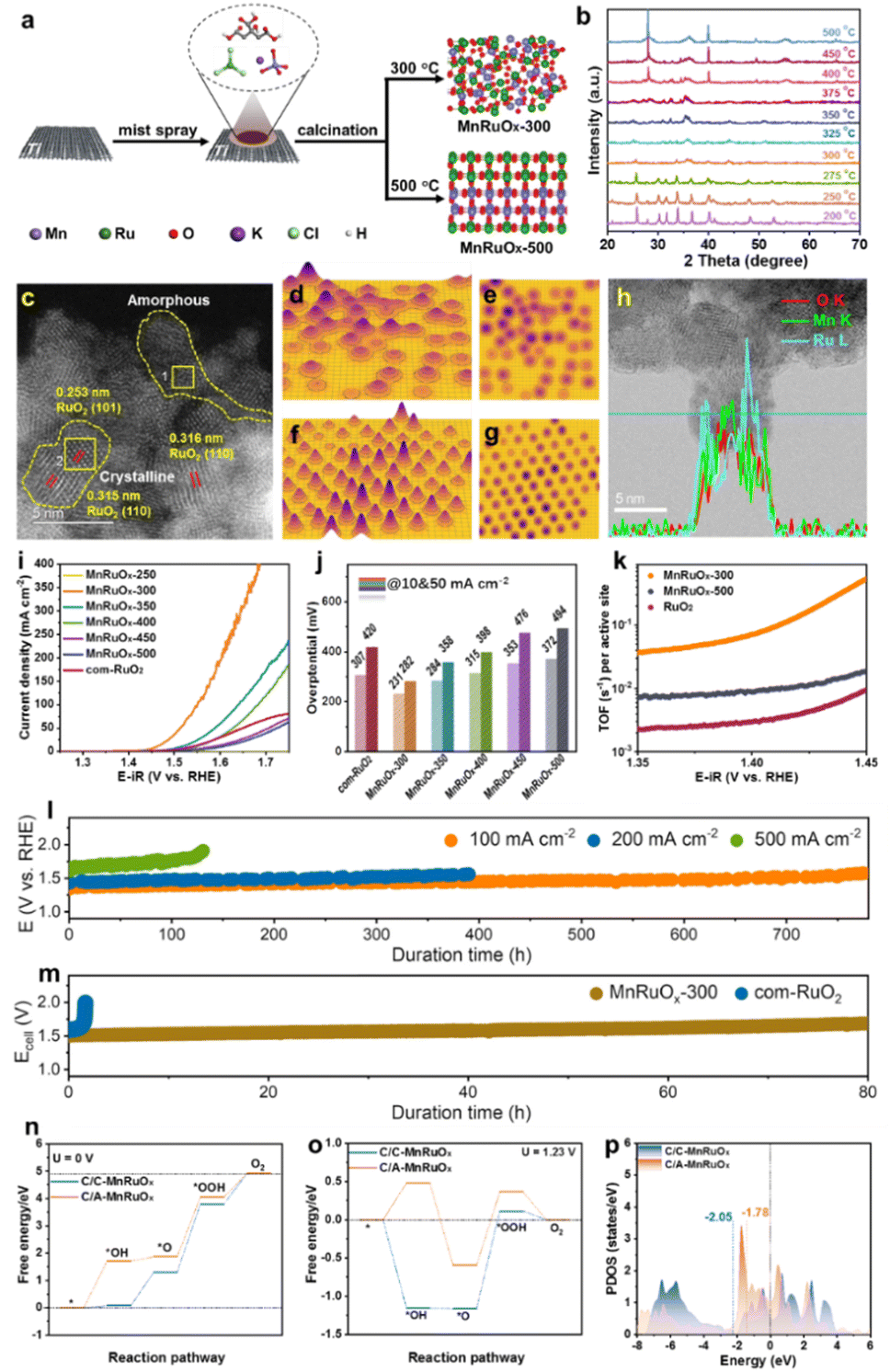 | ||
| Fig. 6 (a) Schematic preparation of the MnRuOx catalyst. (b) Operando XRD pattern of the MnRuOx-150 precursor treated at different temperatures. (c) HAADF-STEM image of MnRuOx-300 showcases the coexistence of microcrystalline and amorphous phases. 3D atomic overlap Gaussian fitting on selected STEM region 1 (d) and (e) and region 2 (f) and (g). (h) HR-TEM and line scan elemental mapping. (i) Polarization curves, (j) Overpotential bar charts, and comparison of turnover frequency (TOF). (l) Durability tests of MnRuOx-300@TFF in an H-type electrolysis cell. (m) Chronopotentiometry testing at 100 mA cm−2 in the PEMWE electrolyser at 80 °C. The energy profile of the OER at (n) U = 0 V and (o) U = 1.23 V. (p) Projected DOS. Reproduced with permission from ref. 90, copyright 2024 from Willey VCH. | ||
MnRuOx samples were tested for linear sweep voltammetry (LSV) under acidic circumstances and compared to com-RuO2 (Fig. 6i). The best OER catalyst was MnRuOx-300. At 10 mA cm−2, RuO2 had an overpotential of 307 mV, consistent with prior results.91,92 However, a bar chart of the samples' overpotentials at various temperatures showed a significant rise with higher annealing temperatures (Fig. 6j). The MnRuOx-300 catalyst, with its amorphous homogenous structure and microcrystalline particles, had the lowest overpotential (231 mV at 10 mA cm−2 and 282 mV at 50 mA cm−2). Additional Tafel slopes were generated to assess catalyst OER kinetics. Comparison of turnover frequencies (TOF) at 1.44 V shows considerable differences between RuO2, MnRuOx-500, and MnRuOx-300 catalysts (Fig. 6k). The TOF of MnRuOx-300 was 0.071 s−1, substantially higher than those of MnRuOx-500 (0.0088 s−1) and RuO2 (0.0030 s−1). The catalyst treated at 300 °C has amorphous and crystalline structures, hence it has better OER activity. Stability experiments were performed on MnRuOx-300@TFF samples in acidic circumstances (pH = 0) at varying current densities using an H-type electrolysis cell. Durability studies of ∼780 hours at 100 mA cm−2, ∼400 h at 200 mA cm−2, ∼130 h at 500 mA cm−2, and ∼30 h at 1.0 A cm−2 (Fig. 6l) indicate good catalyst stability at high current densities. Under 100 mA cm−2 for 80 h, the MnRuOx-300 cell maintained PEMWE performance, but a RuO2 anode only lasts around 1 hour (Fig. 6m). All reaction steps are endothermic at U = 0 V. At the *O formation to the *OOH stage, C/C-MnRuOx and C/A-MnRuOx heterostructures have OER as the RDS. The OER overpotentials of C/A-MnRuOx and C/C-MnRuOx are 1.28 eV and 0.96 eV, respectively, at U = 1.23 V, proving that it decreases it. We estimated the predicted density of states (DOS) for C/C-MnRuOx and C/A-MnRuOx heterostructures (Fig. 6p). The d-band center model describes adsorbate–metal interactions well.93 After amorphization, the DOS at the Fermi level rises, raising the d-band center energy level.
Electrosynthesis of urea with nitrate and carbon dioxide
Simple pyrolysis of FeIII: PVP compounds with carbon ECP at 700 °C in Ar produced the FeNC–Fe1N4/C nanocomposite. Fig. 7a shows that FeNC–Fe1N4 exhibits metallic Fe particles and the carbon support has high defect and disordered carbon matrix population, promoting electroconductibility and activity during electrocatalysis.94 It verified that FeNC–Fe1N4/C contained mostly Fe species and the carbon matrix. STEM using EDX elemental mapping technology also examined the distribution of atomically distributed Fe single atoms and metallic Fe clusters. High-resolution TEM images (Fig. 7b) show metallic Fe(200) facets, and the size of Fe clusters is ∼4–7 nm.95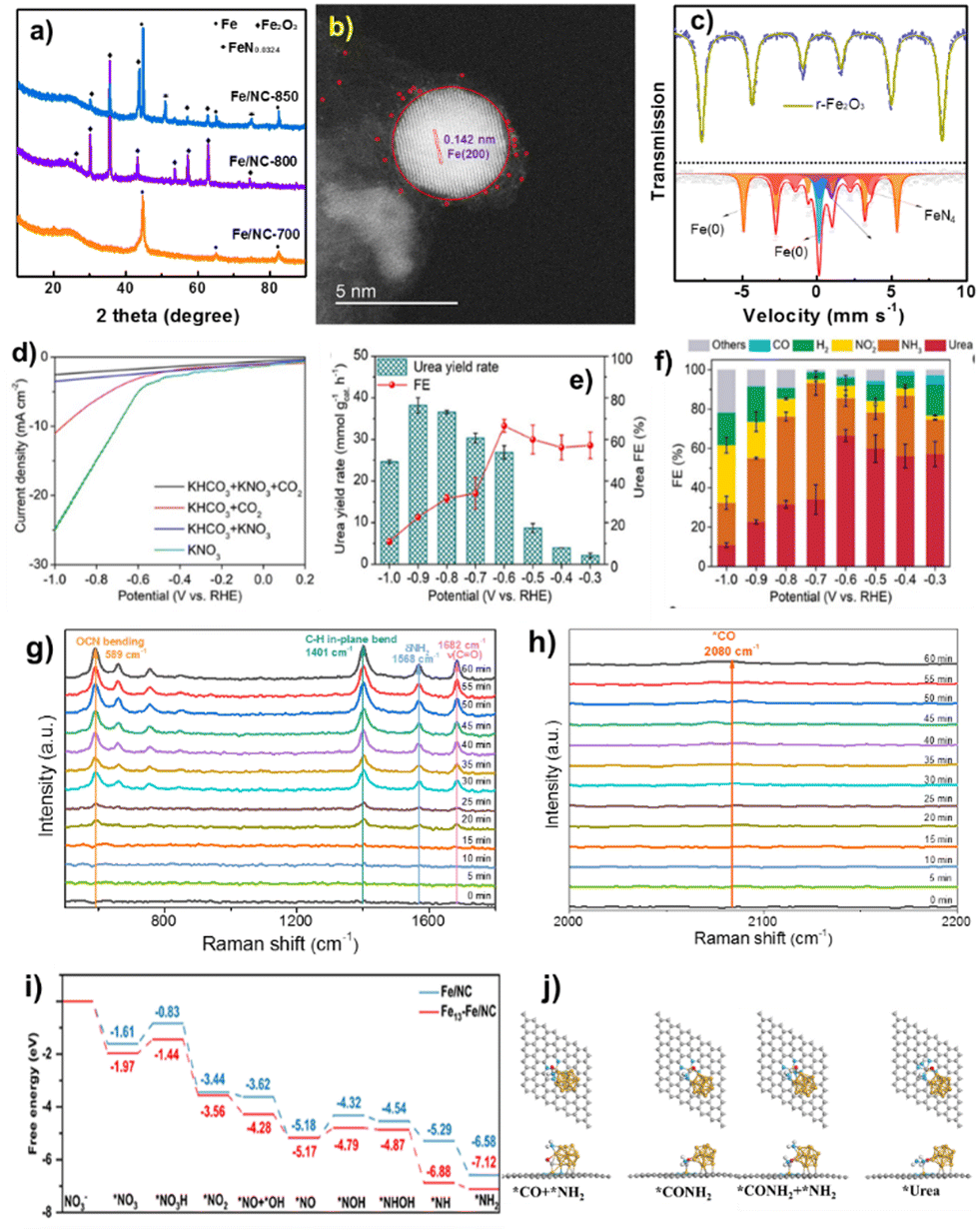 | ||
| Fig. 7 (a) XRD, (b) STEM image, (c) Mössbauer spectra, (d) LSV curves, (e) potential-dependent urea yield rate, and (f) FEs (at −0.6 V) of FeNC–Fe1N4/C. (g) and (h) Time-resolved in situ Raman spectra in urea electrosynthesis at −0.6 V. (i) Free energy diagrams of NO3− reduction to *NH2. (j) Structure diagrams of the C–N coupling process on Fe13–Fe1N4/C. Reproduced from ref. 91, copyright 2024 from Wiley WCH. | ||
A typical three-electrode setup in an H-type cell under continuous CO2 flow with Nafion 117 membranes between the cathode and anode chambers was used to assess urea electrosynthesis performance. Fig. 7d shows FeNC–Fe1N4/C current densities: KNO3 > KHCO3 + CO2 > KHCO3 + KNO3 > KHCO3 + CO2. Research indicates that the co-electrolysis of CO2 and NO3− for urea production has a lower current density than CO2RR and NO3RR. The results show that the CO2RR, NO3RR, and competing HER are effectively regulated, ensuring high urea FE in co-reduction.96Fig. 7e displays FeNC–Fe1N4/C sample urea yield rates: 2.1, 3.9, 8.7, 26.9, 30.3, 36.5, 38.2, and 24.6 mmol gcat.−1 h−1 at −0.3, −0.4, −0.5, −0.6, −0.7, −0.8, −0.9, and −1.0 V, respectively. Urea FEs rise and fall when the applied potential is lowered to −1.0 V. Optimal urea production is 38.2 mmol gcat.−1 h−1 at −0.9 V, and an urea FE of 66.5% at −0.6 V surpasses those of existing electrocatalysts. Fig. 7f shows that urea dominates at 60% between −0.3 and −0.6 V, followed by NH3 and H2 FEs at 20% and 10%, respectively. When the potential is changed downward, FEs of competing NH3, H2, and NO2− rise, corresponding to a rapid increase in current density of about −0.6 V in LSV curves.
To understand the complex C- and N-species in co-electrolysis, it is crucial to identify the reaction route and capture important intermediates in C–N coupling.97Fig. 7g and h show the time-resolved in situ Raman spectra of FeNC–Fe1N4/C obtained at −0.6 V during urea electrosynthesis. Vibration peaks at 589, 1401, 1568, and 1682 cm−1 correspond to OCN bending, adsorbed C–H in-plane bending, δNH2, and CO stretching modes in formamide. However, 1H-NMR spectroscopy shows urea production in electrolyte, not formamide. We infer that *CONH2 is the intermediary in C–N coupling for urea production. As CO is the reduction product in the CO2RR, nucleophilic attack of *CO and *NH2 was used to create C–N coupling. Raman spectra showed the modest signal of *CO at 2080 cm−1, supporting this finding (Fig. 7h). The C–N stretching mode of urea at 1000 cm−1 is not seen in the Raman spectra owing to its feeble vibration signal.98 The free energy diagrams in Fig. 7i depict the chemical pathways of NO3− reduction to *NH2 on Fe13–Fe1N4/C.
The RDS for Fe13–Fe1N4/C and Fe1N4/C is *NO3 to *NO3H and *NO to *NOH, respectively, and the energy barrier of RDS on Fe13–Fe1N4/C is comparatively lower than that on Fe1N4/C, suggesting that nitrate reduction is more favorable on Fe13–Fe1N4/C. Next, there is increased electron transfer between *NO3, *NO3H and *COOH intermediates and Fe13–Fe1N4/C. However, the adsorption of the *COOH intermediate is compromised due to spatial constraints from steric hindrance, as it resides at the interface of the Fe cluster and Fe1N4 site. In all, the Fe cluster and Fe1N4 in Fe13–Fe1N4/C are the primary active sites for nitrate reduction and CO2 reduction and C–N coupling, respectively.
Conclusions and future progress
Research on water oxidation and electrocatalysis shows a variety of ways to increase the catalytic stability and activity. They show the synergistic effect of cobalt clusters–CoNx composites for electrochemical oxygen reduction, investigating the promotion effect of active sites in iron-based oxygen reduction electrocatalysts for Zn–air batteries., and the high activity for sustainable acidic water oxidation through intrinsically robust cubic MnCoOx solid solutions. They also discuss heterogeneous catalysis promoted by ultrafine amorphous metallic hydroxide nanoparticles and oxygen vacancy-rich amorphous FeNi nanoclusters modified by ionic liquids. These findings help develop stable and effective electrocatalysts for energy storage and conversion.Overall, the highlighted study shows how atomically exact metal nanoclusters and metal oxide clusters may revolutionize electrocatalysis. Research on Fe-based oxygen reduction electrocatalysts has demonstrated their potential to greatly enhance the performance of zinc–air batteries. This Li et al. discovery emphasizes the need for several active sites for catalytic effectiveness. Zhang et al. showed that cubic MnCoOx solid solutions are durable and efficient, demonstrating metal oxide clusters' potential for persistent water oxidation and scalable hydrogen production. Zhang et al.'s Investigation of the synergistic effect of cobalt clusters–CoNx composites on electrochemical oxygen reduction highlights their potential for developing customized catalysts. These examples demonstrate the need to properly manage catalyst's shape and composition to increase catalytic activity and lifespan. In addition, catalyst design and production have been successful, but commercializing and scaling up these advanced electrocatalysts has proven difficult. According to Cao et al., optimizing synthesis processes and studying cheap precursor materials would enable large-scale catalytic material fabrication and deployment. The research found that improving integration and electrochemical systems maximize energy conversion efficiency, lifespan, and cost. These studies demonstrate the revolutionary potential of atomically precise metal nanoclusters and metal oxide clusters in electrocatalysis for sustainable energy technologies, which could lead to cleaner, greener energy sources. Future development may need further electrocatalyst design and manufacturing optimization to increase activity, stability, and selectivity for electrochemical reactions such as oxygen reduction, water oxidation, and energy conversion.
Essential areas of attention might be:
1. Nanostructuring and active site engineering: researchers are customizing active sites and improving catalytic activity using nanomaterials and atomically precise metal nanoclusters. Identifying catalytic active sites has fascinated academics for decades and is crucial. Active sites may be single atoms or groups of atoms on metal particles. Future research should focus on developing innovative techniques to accurately observe the intricate steps of catalytic reactions at the catalytic site, achieving spatiotemporal accuracy in milliseconds. This endeavor presents a challenging yet highly promising opportunity. Understanding the electronic structure of TMO oxides via theoretical studies can help explain catalytic processes. In heterogeneous catalysis, learning the existence and structure of active sites is a huge difficulty, but combining experiments and theory should help. High stability of TMO clusters and novel methods for very stable catalysts throughout catalytic processes may aid mechanistic studies.2. Synergism in multifunctional materials: composite materials and multifunctional catalysts are being studied to boost catalytic activity and durability. Examining the oxidation state, atom dispersion, and potential behavior of heteroelements is crucial when examining the catalytic properties of the doped element in an alloy catalyst. This information is hard to get. Thus, future research should combine experimentation and theory and build new tools. The link between multifunctional TMO nanomaterials' geometric and electrical characteristics and catalytic reactivity remains a priority. The fundamental ideas of bimetallic and multimetallic nanocatalysis synergy will influence the composition choices of high-performance nanocatalysts for specific chemical reactions.
3. Understanding reactions using in situ and operando characterization: a deeper understanding of molecular-level reaction mechanisms might influence selectivity and efficiency-enhancing electrocatalyst design.99,100 It is important to have a way to analyze TMO catalysts in real time and real space to obtain a more profound comprehension of the catalytic reaction process, its kinetics, and the occurrence of coking and leaching. Studying the high fluxionality of a few TM atom clusters at elevated temperatures and determining whether the nanocluster structure undergoes reversible or irreversible dynamic changes can be achieved through in situ and operando extended X-ray absorption fine structure (EXAFS) and diffuse reflectance infrared Fourier-transform spectroscopy (DRIFT). Future research should focus on developing advanced in situ and operando technologies, such as high-resolution imaging and spectroscopy, to better understand and address these challenges. An analysis of this nature would be instrumental in exploring the catalytic factors of TM clusters.
4. Studying new high-entropy oxide clusters: investigation of metal–organic frameworks and ionic liquids as electrolytes and electrocatalysts for devices that convert and store energy. When combined with metal–organic frameworks (MOFs) and ionic liquids (ILs), high-entropy oxide (HEO) clusters—which are defined by several metal cations dispersed randomly inside a single-phase lattice—offer promising advances in energy conversion and storage technologies. Because of their large surface area and adjustable porosity, metal oxide frameworks (MOFs) are a great approach to enhance heteroelectrolytes' ion mobility and structural integrity. Additionally, the metal nodes inside MOFs can function as active catalytic sites. In addition to these hybrids, ILs enhance the electrode–electrolyte interactions by offering a favorable ionic environment due to their high ionic conductivity and stability. The cooperation of HEOs, MOFs, and ILs may be used to create novel materials with enhanced catalytic performance and customized characteristics, greatly enhancing the longevity and efficiency of fuel cells, batteries, supercapacitors, and electrolyzers.
5. Single-atom and single-electron tailoring: atomically precise nanochemistry may govern TMO clusters in new ways, opening up fascinating catalytic research options. Quantum-sized clusters' electronic characteristics are subject to atom count. Customized nanocluster catalysts allow atom-by-atom catalytic reactivity tailoring. Accurate nanocatalysts would not be able to produce novel catalytic phenomena like spin effects in chemical processes, but precise regulation of nanocluster attraction at the single-electron level would.
6. Scale-up and commercialization: this involves improving manufacturing methods and adding advanced electrocatalysts to useful products for affordability, scalability, and sustainability.
The area is well-positioned for sustained innovation and interdisciplinary cooperation to tackle global energy concerns and propel the shift towards a more sustainable energy future.
Author contributions
This manuscript was collaboratively written by all authors. The final version of the manuscript has been approved by all authors.Data availability
Data availability is not applicable to this article as no new data were created or analyzed in this study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support by the National Natural Science Foundation of China (22172167).Notes and references
- H. Dong, M. Xue, Y. Xiao and Y. Liu, Sci. Total Environ., 2021, 758, 143688 CrossRef CAS PubMed.
- F. K. Busari, Z. Babar, A. Raza and G. Li, Sustainable Mater. Technol., 2024, 40, e00958 CrossRef CAS.
- H. Bai, D. Chen, Q. Ma, R. Qin, H. Xu, Y. Zhao, J. Chen and S. Mu, Electrochem. Energy Rev., 2022, 5, 24 CrossRef CAS.
- J. Z. Hassan, A. Zaheer, A. Raza and G. Li, Sustainable Mater. Technol., 2023, 36, e00609 CrossRef CAS.
- T. Guo, L. Li and Z. Wang, Adv. Energy Mater., 2022, 12, 2200827 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- Y. Song, C. Zhou and R. Jin, in Crystallization via Nonclassical Pathways Volume 1: Nucleation, Assembly, Observation & Application, ACS Publications, 2020, pp. 47–71 Search PubMed.
- F. Li, G.-F. Han and J.-B. Baek, Acc. Mater. Res., 2021, 2, 147–158 CrossRef CAS.
- J. Gao, H. Tao and B. Liu, Adv. Mater., 2021, 33, 2003786 CrossRef CAS PubMed.
- R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Chem. Rev., 2020, 121, 567–648 CrossRef PubMed.
- Z. Yang, J. Zhang, M. C. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- Y. Zhang, Z. Li, J. Zhang, L. Xu, Z. Han, A. Baiker and G. Li, Nano Res., 2023, 16, 8919 CrossRef CAS.
- S. Lu, J. Liang, H. Long, H. Li, X. Zhou, Z. He, Y. Chen, H. Sun, Z. Fan and H. Zhang, Acc. Chem. Res., 2020, 53, 2106–2118 CrossRef CAS PubMed.
- Y. Guo, S. Mei, K. Yuan, D.-J. Wang, H.-C. Liu, C.-H. Yan and Y.-W. Zhang, ACS Catal., 2018, 8, 6203–6215 CrossRef CAS.
- Z. Li, L. Xu, Z. Babar, A. Raza, Y. Zhang, X. Gu, Y. Miao, Z. Zhao and G. Li, Nano Res., 2024, 17, 4729 CrossRef CAS.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2019, 120, 526–622 CrossRef PubMed.
- Q. Shi, Y. Zhang, Z. Li, Z. Han, L. Xu, A. Baiker and G. Li, Nano Res., 2023, 16, 6951 CrossRef CAS.
- S. Barkaoui, Y. Wang, Y. Zhang, X. Gu, Z. Li, B. Wang, G. Li and Z. Zhao, iScience, 2024, 110255 CrossRef CAS PubMed.
- Z. Li, J. Zhang and G. Li, Chin. J. Struct. Chem., 2024, 43, 100300 CrossRef CAS.
- Z. Qin, Z. Li, S. Sharma, Y. Peng, R. Jin and G. Li, Research, 2022, 0018 CrossRef CAS.
- Y. Sun, Q. Shi, X. Gu, B. Wang, B. Lumbers and G. Li, J. Colloid Interface Sci., 2024, 662, 76 CrossRef CAS PubMed.
- Q. Shi, X. Zhang, Z. Li, A. Raza and G. Li, ACS Appl. Mater. Interfaces, 2023, 15, 30161 CrossRef CAS PubMed.
- K. Kwak and D. Lee, Acc. Chem. Res., 2018, 52, 12–22 CrossRef PubMed.
- P. Sanwal, A. Raza, Y.-X. Miao, B. Lumbers and G. Li, Polyoxometalates, 2024, 3, 9140057 CrossRef.
- C. Yao, N. Guo, S. Xi, C.-Q. Xu, W. Liu, X. Zhao, J. Li, H. Fang, J. Su and Z. Chen, Nat. Commun., 2020, 11, 4389 CrossRef CAS PubMed.
- Y. Chen, X. Gu, S. Guo, J. Zhang, S. Barkaoui, L. Xu and G. Li, ChemSusChem, 2024, 17, e202400309 CrossRef PubMed.
- X. F. Lu, S. L. Zhang, E. Shangguan, P. Zhang, S. Gao and X. W. Lou, Adv. Sci., 2020, 7, 2001178 CrossRef CAS PubMed.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M.-J. Cheng, D. Sokaras, T.-C. Weng and R. Alonso-Mori, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- Y. Yan, T. He, B. Zhao, K. Qi, H. Liu and B. Y. Xia, J. Mater. Chem. A, 2018, 6, 15905–15926 RSC.
- M. Ledendecker, S. Krick Calderón, C. Papp, H. P. Steinrück, M. Antonietti and M. Shalom, Angew. Chem., Int. Ed., 2015, 54, 12361–12365 CrossRef CAS PubMed.
- J. Zhao, X. Li, M. Zhang, Z. Xu, X. Qin, Y. Liu, L. Han and G. Li, Nanoscale, 2023, 15, 4612 RSC.
- J. Wang, J. Chen, P. Wang, J. Hou, C. Wang and Y. Ao, Appl. Catal., B, 2018, 239, 578–585 CrossRef CAS.
- H. Q. Fu, L. Zhang, C. W. Wang, L. R. Zheng, P. F. Liu and H. G. Yang, ACS Energy Lett., 2018, 3, 2021–2029 CrossRef CAS.
- Y. Liu, Q. Li, R. Si, G. D. Li, W. Li, D. P. Liu, D. Wang, L. Sun, Y. Zhang and X. Zou, Adv. Mater., 2017, 29, 1606200 CrossRef PubMed.
- Y. Lin, G. Chen, H. Wan, F. Chen, X. Liu and R. Ma, Small, 2019, 15, 1900348 CrossRef PubMed.
- P. Chen, T. Zhou, L. Xing, K. Xu, Y. Tong, H. Xie, L. Zhang, W. Yan, W. Chu and C. Wu, Angew. Chem., 2017, 129, 625–629 CrossRef.
- R. D. Smith, M. S. Prévot, R. D. Fagan, S. Trudel and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 11580–11586 CrossRef CAS PubMed.
- Y. Huang, R. Yang, G. Anandhababu, J. Xie, J. Lv, X. Zhao, X. Wang, M. Wu, Q. Li and Y. Wang, ACS Energy Lett., 2018, 3, 1854–1860 CrossRef CAS.
- C. Liang, P. Zou, A. Nairan, Y. Zhang, J. Liu, K. Liu, S. Hu, F. Kang, H. J. Fan and C. Yang, Energy Environ. Sci., 2020, 13, 86–95 RSC.
- J. Nai, H. Yin, T. You, L. Zheng, J. Zhang, P. Wang, Z. Jin, Y. Tian, J. Liu and Z. Tang, Adv. Energy Mater., 2015, 5, 1401880 CrossRef.
- A. Dutta, A. K. Samantara, S. K. Dutta, B. K. Jena and N. Pradhan, ACS Energy Lett., 2016, 1, 169–174 CrossRef CAS.
- Y. Xie, H. Li, C. Tang, S. Li, J. Li, Y. Lv, X. Wei and Y. Song, J. Mater. Chem. A, 2014, 2, 1631–1635 RSC.
- W. Wang, Q. Jia, S. Mukerjee and S. Chen, ACS Catal., 2019, 9, 10126–10141 CrossRef CAS.
- C. Xu, C. Guo, J. Liu, B. Hu, J. Dai, M. Wang, R. Jin, Z. Luo, H. Li and C. Chen, Energy Storage Mater., 2022, 51, 149–158 CrossRef.
- H. Xu, D. Wang, P. Yang, A. Liu, R. Li, L. Xiao, J. Zhang, Z. Qu and M. An, Sustainable Energy Fuels, 2021, 5, 2695–2703 RSC.
- Y. Lee, J. H. Ahn, H. Jang, J. Lee, S. Yoon, D.-G. Lee, M. G. Kim, J. H. Lee and H.-K. Song, J. Mater. Chem. A, 2022, 10, 24041–24050 RSC.
- M. Lefèvre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71–74 CrossRef PubMed.
- Y. He, S. Liu, C. Priest, Q. Shi and G. Wu, Chem. Soc. Rev., 2020, 49, 3484–3524 RSC.
- S. N. Zhao, J. K. Li, R. Wang, J. Cai and S. Q. Zang, Adv. Mater., 2022, 34, 2107291 CrossRef CAS PubMed.
- Y. Wang, Y. Zhang, Q. Jiang, S. Guo, A. Baiker and G. Li, ChemCatChem, 2022, 14, e202200203 CrossRef CAS.
- Y. Wang, Q. Jiang, L. Xu, Z.-K. Han, S. Guo, G. Li and A. Baiker, ACS Appl. Mater. Interfaces, 2021, 13, 61078–61087 CrossRef CAS PubMed.
- J. Zhang, A. Raza, Y. Zhao, S. Guo, Z. U. D. Babar, L. Xu, C. Cao and G. Li, J. Mater. Chem. A, 2023, 11, 25345–25355 RSC.
- J. Zhang, Y. Xie, Q. Jiang, S. Guo, J. Huang, L. Xu, Y. Wang and G. Li, J. Mater. Chem. A, 2022, 10, 16920–16927 RSC.
- Z. Li, Y. Xie, J. Gao, X. Zhang, J. Zhang, Y. Liu and G. Li, J. Mater. Chem. A, 2023, 11, 26573–26579 RSC.
- Y. Cao, S. Guo, C. Yu, J. Zhang, X. Pan and G. Li, J. Mater. Chem. A, 2020, 8, 15767–15773 RSC.
- X. Wu, H. Zhang, S. Zuo, J. Dong, Y. Li, J. Zhang and Y. Han, Nano-Micro Lett., 2021, 13, 136 CrossRef CAS PubMed.
- J. Hu, W. Liu, C. Xin, J. Guo, X. Cheng, J. Wei, C. Hao, G. Zhang and Y. Shi, J. Mater. Chem. A, 2021, 9, 24803–24829 RSC.
- C. Dominguez, F. J. Perez-Alonso, M. A. Salam, J. L. Gomez de la Fuente, S. A. Al-Thabaiti, S. N. Basahel, M. A. Pena, J. L. G. Fierro and S. Rojas, Int. J. Hydrogen Energy, 2014, 39, 5309–5318 CrossRef CAS.
- Z. Wang, C. Zhu, H. Tan, J. Liu, L. Xu, Y. Zhang, Y. Liu, X. Zou, Z. Liu and X. Lu, Adv. Funct. Mater., 2021, 31, 2104735 CrossRef CAS.
- L. Zhuang, Z. Jia, Y. Wang, X. Zhang, S. Wang, J. Song, L. Tian and T. Qi, Chem. Eng. J., 2022, 438, 135585 CrossRef CAS.
- W. Zhou, H. Su, Z. Wang, F. Yu, W. Wang, X. Chen and Q. Liu, J. Mater. Chem. A, 2021, 9, 1127–1133 RSC.
- X. Yang, Y. Wang, X. Wang, B. Mei, E. Luo, Y. Li, Q. Meng, Z. Jin, Z. Jiang, C. Liu, J. Ge and W. Xing, Angew. Chem., Int. Ed., 2021, 60, 26177–26183 CrossRef CAS PubMed.
- H. Yao, X. Wang, K. Li, C. Li, C. Zhang, J. Zhou, Z. Cao, H. Wang, M. Gu, M. Huang and H. Jiang, Appl. Catal., B, 2022, 312, 121378 CrossRef CAS.
- X. Jiang, J. Chen, F. Lyu, C. Cheng, Q. Zhong, X. Wang, A. Mahsud, L. Zhang and Q. Zhang, J. Energy Chem., 2021, 59, 482–491 CrossRef CAS.
- Z. Cui and X. Bai, ACS Appl. Mater. Interfaces, 2022, 14, 9024–9035 CrossRef CAS PubMed.
- X. Ao, Y. Ding, G. Nam, L. Soule, P. Jing, B. Zhao, J. Hwang, J. Jang, C. Wang and M. Liu, Small, 2022, 18, 2203326 CrossRef CAS PubMed.
- X. Xu, X. Zhang, Z. Xia, R. Sun, H. Li, J. Wang, S. Yu, S. Wang and G. Sun, J. Energy Chem., 2021, 54, 579–586 CrossRef CAS.
- J. Zhang, Y. Xie, Q. Jiang, S. Guo, J. Huang, L. Xu, Y. Wang and G. Li, J. Mater. Chem. A, 2022, 10, 16920–16927 RSC.
- Z. Liang, H. Song and S. Liao, J. Phys. Chem. C, 2011, 115, 2604–2610 CrossRef CAS.
- L. Li, S. Shen, G. Wei and J. Zhang, Acta Phys.-Chim. Sin., 2021, 37, 1911011 Search PubMed.
- C. Shu, Q. Tan, C. Deng, W. Du, Z. Gan, Y. Liu, C. Fan, H. Jin, W. Tang, X. D. Yang, X. Yang and Y. Wu, Carbon Energy, 2021, 4, 1–11 CrossRef.
- A. Raza, A. A. Rafi, J. Z. Hassan, A. Rafiq and G. Li, Appl. Surf. Sci. Adv., 2023, 15, 100402 CrossRef.
- S. Shen, Z. Zhai, J. Qin, X. Zhang and Y. Song, J. Porphyrins Phthalocyanines, 2019, 23, 1013–1019 CrossRef CAS.
- H. Abroshan, G. Li, J. Lin, H. J. Kim and R. Jin, J. Catal., 2016, 337, 72 CrossRef CAS.
- J. Sun, N. Guo, Z. Shao, K. Huang, Y. Li, F. He and Q. Wang, Adv. Energy Mater., 2018, 8, 1800980 CrossRef.
- M. Antonietti, D. Kuang, B. Smarsly and Y. Zhou, Angew. Chem., Int. Ed., 2004, 43, 4988–4992 CrossRef CAS PubMed.
- H. Park, Y. S. Choi, Y. Kim, W. H. Hong and H. Song, Adv. Funct. Mater., 2007, 17, 2411–2418 CrossRef CAS.
- X. Gu, S. Guo, Y. Zhang, J. Zhang, P. Sanwal, L. Xu, Z. Zhao, R. Jin and G. Li, Nano Res. Energy, 2024, 3, e9120134 Search PubMed.
- D. Yan, Y. Li, J. Huo, R. Chen, L. Dai and S. Wang, Adv. Mater., 2017, 29, 1606459 CrossRef PubMed.
- F. Sun, C. Li, B. Li and Y. Lin, J. Mater. Chem. A, 2017, 5, 23103–23114 RSC.
- Y. Cao, Y. Su, L. Xu, X. Yang, Z. Han, R. Cao and G. Li, J. Energy Chem., 2022, 71, 167–173 CrossRef CAS.
- T. Cai, H. Huang, W. Deng, Q. Dai, W. Liu and X. Wang, Appl. Catal., B, 2015, 166–167, 393–405 CrossRef CAS.
- J. Li, X. Liang, S. Xu and J. Hao, Appl. Catal., B, 2009, 90, 307–312 CrossRef CAS.
- Q. Shi, Z. Li, C. Cao, G. Li and S. Barkaoui, Nanoscale Adv., 2023, 5, 5385 RSC.
- Y. Sun, Q. Wang and Z. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 43508–43516 CrossRef CAS PubMed.
- S. Barkaoui, Z. Li, C. Cao, X. Gu, Q. Zeng, B. Lumbers and G. Li, New J. Chem., 2024, 48, 631 RSC.
- Y. Qin, T. T. Yu, S. H. Deng, X. Y. Zhou, D. M. Lin, Q. Zhang, Z. Y. Jin, D. F. Zhang, Y. B. He, H. J. Qiu, L. H. He, F. Y. Kang, K. K. Li and T. Y. Zhang, Nat. Commun., 2022, 13, 3784 CrossRef CAS PubMed.
- Q. Liang, A. Bieberle-Hütter and G. Brocks, J. Phys. Chem. C, 2022, 126, 1337–1345 CrossRef CAS.
- Y. Hu, G. Luo, L. Wang, X. Liu, Y. Qu, Y. Zhou, F. Zhou, Z. Li, Y. Li, T. Yao, C. Xiong, B. Yang, Z. Yu and Y. Wu, Adv. Energy Mater., 2020, 11, 2002816 CrossRef.
- J. Zhang, L. Xu, X. Yang, S. Guo, Y. Zhang, Y. Zhao, G. Wu and G. Li, Angew. Chem., Int. Ed., 2024, 136, e202405641 CrossRef.
- Y. Yao, S. Hu, W. Chen, Z. Huang, W. Wei, T. Yao, R. Liu, K. Zang, X. Wang, G. Wu, W. Yuan, T. Yuan, B. Zhu, W. Liu, Z. Li, D. He, Z. Xue, Y. Wang, X. Zheng, J. Dong, C. Chang, Y. Chen, X. Hong, J. Luo, S. Wei, W. Li, P. Strasser, Y. Wu and Y. Li, Nat. Catal., 2019, 2, 304–313 CrossRef CAS.
- Z. L. Zhao, Q. Wang, X. Huang, Q. Feng, S. Gu, Z. Zhang, H. Xu, L. Zeng, M. Gu and H. Li, Energy Environ. Sci., 2020, 13, 5143–5151 RSC.
- C. Hitz and A. Lasia, J. Electroanal. Chem., 2001, 500, 213–222 CrossRef CAS.
- G. Panomsuwan, N. Saito and T. Ishizaki, ACS Appl. Mater. Interfaces, 2016, 8, 6962–6971 CrossRef CAS PubMed.
- Z. Li, M. Xu, J. Wang, Y. Zhang, W. Liu, X. Gu, Z. K. Han, W. Ye and G. Li, Small, 2024, 2400036 CrossRef PubMed.
- X. Zhang, X. Zhu, S. Bo, C. Chen, M. Qiu, X. Wei, N. He, C. Xie, W. Chen, J. Zheng, P. Chen, S. P. Jiang, Y. Li, Q. Liu and S. Wang, Nat. Commun., 2022, 13, 5337 CrossRef CAS PubMed.
- P. Hildebrandt, M. Tsuboi and T. G. Spiro, J. Phys. Chem., 1990, 94, 2274–2279 CrossRef CAS.
- Y. Zhao, X. Zhang, N. Bodappa, W. Yang, Q. Liang, P. M. Radjenovica, Y. Wang, Y. Zhang, J. Dong, Z. Tian and J. Li, Energy Environ. Sci., 2022, 15, 3968–3977 RSC.
- Z. Liu, Z. Qin, C. Cui, Z. Luo, B. Yang, Y. Jiang, C. Lai, Z. Wang, X. Wang, X. Fang, G. Li, F. Wang, C. Xiao and X. Yang, Sci. China: Chem., 2022, 65, 1196 CrossRef CAS.
- W. Zhang, Y. Zhou, W. Chen, T. Wang, Z. Qin, G. Li, Z. Ren, X. Yang and C. Zhou, Chin. J. Chem. Phys., 2023, 36, 249 CrossRef CAS.
Footnote |
| † P. S. and Y. Z. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |