
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAncient and modern mechanisms compete in progesterone receptor activation†
Sabab Hasan
Khan
 a,
Namita
Dube
a,
Nishanti
Sudhakar
a,
Olivia
Fraser
a,
Priscilla
Villalona
a,
Sean M.
Braet
b,
Stephanie
Leedom
a,
Erin R.
Reilly
a,
Jacob
Sivak
b,
Kenidee
Crittenden
b and
C. Denise
Okafor
*ab
a,
Namita
Dube
a,
Nishanti
Sudhakar
a,
Olivia
Fraser
a,
Priscilla
Villalona
a,
Sean M.
Braet
b,
Stephanie
Leedom
a,
Erin R.
Reilly
a,
Jacob
Sivak
b,
Kenidee
Crittenden
b and
C. Denise
Okafor
*ab
aDepartment of Biochemistry and Molecular Biology, Pennsylvania State University, University Park, PA 16802, USA. E-mail: cdo5093@psu.edu
bDepartment of Chemistry, Pennsylvania State University, University Park, PA 16802, USA
First published on 15th April 2024
Abstract
The progesterone receptor (PR) belongs to the steroid receptor family of ligand-regulated transcription factors, controlling genes important for development, metabolism, and reproduction. Understanding how diverse ligands bind and modulate PR activity will illuminate the design of ligands that control PR-driven signaling pathways. Here, we use molecular dynamics simulations to investigate how PR dynamics are altered by functionally diverse ligands. Using a library of 33 steroidal ligands that range from inactive to EC50 < 0.1 nM, we reveal an unexpected evolutionary basis for the wide gamut of activation. While other oxosteroid receptors employ an evolutionarily conserved mechanism dependent on a hydrogen bond between the receptor and ligand, extant PR has evolved a preference for activation that is not reliant on this polar interaction. We demonstrate that potent ligands utilize the modern PR mechanism while weaker ligands coopt the defunct ancestral mechanism by forming hydrogen bonds with Asn719. Based on their structures and dynamic signatures, ligands partition into four classes (inactive, weak, moderate and high potency) that interact distinctly with the PR binding pocket. Further, we use luciferase reporter assays and PR mutants to probe the roles of pocket residues in mediating distinct PR mechanisms. This combination of MD simulations and in vitro studies provide insight into how the evolutionary history of PR shapes its response to diverse ligands.
Introduction
The progesterone receptor (PR) belongs to the nuclear receptor superfamily of ligand-regulated transcription factors which regulates a plethora of biological processes such as development, metabolism, reproduction, and inflammation upon binding of cognate ligands.1–5 In association with estrogen receptor (ER), PR functions as a primary regulator of female development and reproduction.6,7 Target genes of PR are important in normal breast development,8,9 embryo attachment, and uterine decidualization in pregnancy10,11 as well as other functions related to menstruation and the endometrium.12,13 In addition to direct roles in breast cancer,8,14 PR signaling is implicated in uterine diseases such as endometrial cancer, endometriosis, and leiomyoma.15–17 Therefore, targeting PR transcriptional activity and signaling is a widely accepted and promising therapeutic approach for many diseases.The transcriptional function of PR is initiated by progesterone binding in the hydrophobic binding pocket of the moderately conserved ligand binding domain (LBD). The binding of progesterone and other steroidal ligands is mediated by substituents on the four-ring steroid scaffold. These pharmacophores lead to distinct interactions with surrounding amino acid residues, allowing for diverse and ligand-specific functional modulation of PR. For example, a small modification to steroid hormones, such as C17-α acetylation or 19 nor-substitution, increases both ligand binding affinity and transcriptional activity in PR.12,18,19 C11-β substituents permit selective PR modulation by differential stabilization of helix 12 (H12);20 this mechanism is observed in ulipristal acetate, a selective PR modulator with a bulky dimethyl-amino phenyl group at C11.19
Previous PR studies identify several LBD residues that are crucial for ligand recognition, including Arg766 on H5 which is conserved across steroid receptors, as well as Gln725 on H3, Met908 and Met909 on H12, and Cys891 and Thr894 on H10 (ref. 20–25). A unique feature of PR is the behavior of Asn719, a conserved helix 3 (H3) residue. In all other oxosteroid receptors (i.e. androgen, glucocorticoid and mineralocorticoid receptors), Asn719 forms hydrogen bonds with the cognate ligands which have been shown by mutagenesis to be crucial for activation (ref. 26 and 27). This interaction is missing in the PR-progesterone complex, and mutation of Asn719 only has a minor effect on progesterone activity, indicating its lesser role in PR.26 Conversely, introducing N705A into AR causes the receptor to behave like PR, increasing its preference for progestins.28 The interaction between ligand and Asn719 also exists in the oxosteroid ancestor AncSR2,29 which suggests that PR diverged from its ancestor, evolving a mechanism to exclude Asn719 in steroid hormone activation. In this work, we aim to gain a mechanistic understanding of how diverse steroidal ligands activate PR by investigating how they impact PR dynamics.
While the plasticity of the PR binding pocket and its ability to accommodate ligands of various sizes is documented,18 there is poor understanding of how ligands modulate PR dynamics to achieve distinct transcriptional profiles. Molecular dynamics (MD) simulations are a valuable tool for studying ligand effects on the conformational dynamics of proteins. Here, we use MD simulations to investigate the dynamic behavior of ligand-bound PR complexes. We have assembled a library of 33 steroidal ligands (Fig. S1 and Table S1, ESI†) with reported EC50 values ranging from 0.1 nM–1000 μM.30–32 Ligands were classified by potency as inactive (EC50 > 10 μM), weak (10 μM to 1 nM) or high (<1 nM). All reported EC50 values are obtained from cellular transcriptional assays in dose–response format. Our structure–activity relationship (SAR) analysis revealed that C11-hydroxylation of high potency agonists weakens EC50, defining a 4th class of ‘moderate’ potency ligands (i.e. 1 nM–10 nM vs. <1 nM). We analyzed dynamics, protein–ligand interactions, binding free energies and solvent accessibility within the binding pocket. Our studies reveal that while high potency PR ligands activate independently of Asn719, this residue drives the activity of both weak and moderate potency ligands via two distinct mechanisms. By forming a hydrogen bond with C17–OH substituents, weak ligands coopt the ancestral steroid receptor mechanism to achieve weak activity in PR. Conversely, hydrogen-bonding between C11-hydroxyl and Asn719 repositions ligands in the pocket, reducing the activity of moderate-potency ligands relative to high potency steroids. Our research demonstrates multiple activation mechanisms in PR, including a residual, diminished mode of activation triggered by C17-hydroxylated steroids. We also reveal the dual role of Asn719 in facilitating and impeding activation, underscoring its pivotal significance in the design of PR ligands.
Results and discussion
Residue communication within the binding pocket groups steroidal PR ligands into four classes
Triplicate MD trajectories were obtained for each complex. Representative structures for simulations reveal that all ligands retain the same relative orientation and position within the PR binding pocket (Fig. S2, ESI†). To reveal a relationship between dynamic traits observed in MD simulations and structural features of ligands, we ordered the ligand library by EC50 and identified the most defining characteristics of each, focusing on the C3, C11, and C17 substituents on each steroid (Fig. 1A and B). Where applicable, C20 and C21 attached to the C17-β substituent were also considered. C17 substitutions are designated as ‘bulky’ if they have a minimum of two carbons, the only exception being promegestone (P0M) with a C17-α methyl, labeled ‘m’. Further, bulky substituents are labeled ‘c’ if they contain the C20-ketone and C21-hydroxyl group that is characteristic of corticosteroids. Also labeled are ligands containing an esterified substitution at C17-α (‘e’). This SAR quickly grouped our library into 3 classes designated ‘inactive’, ‘weak’ and ‘potent’ ligands, with distinguishing features consistent with previous reports.33,34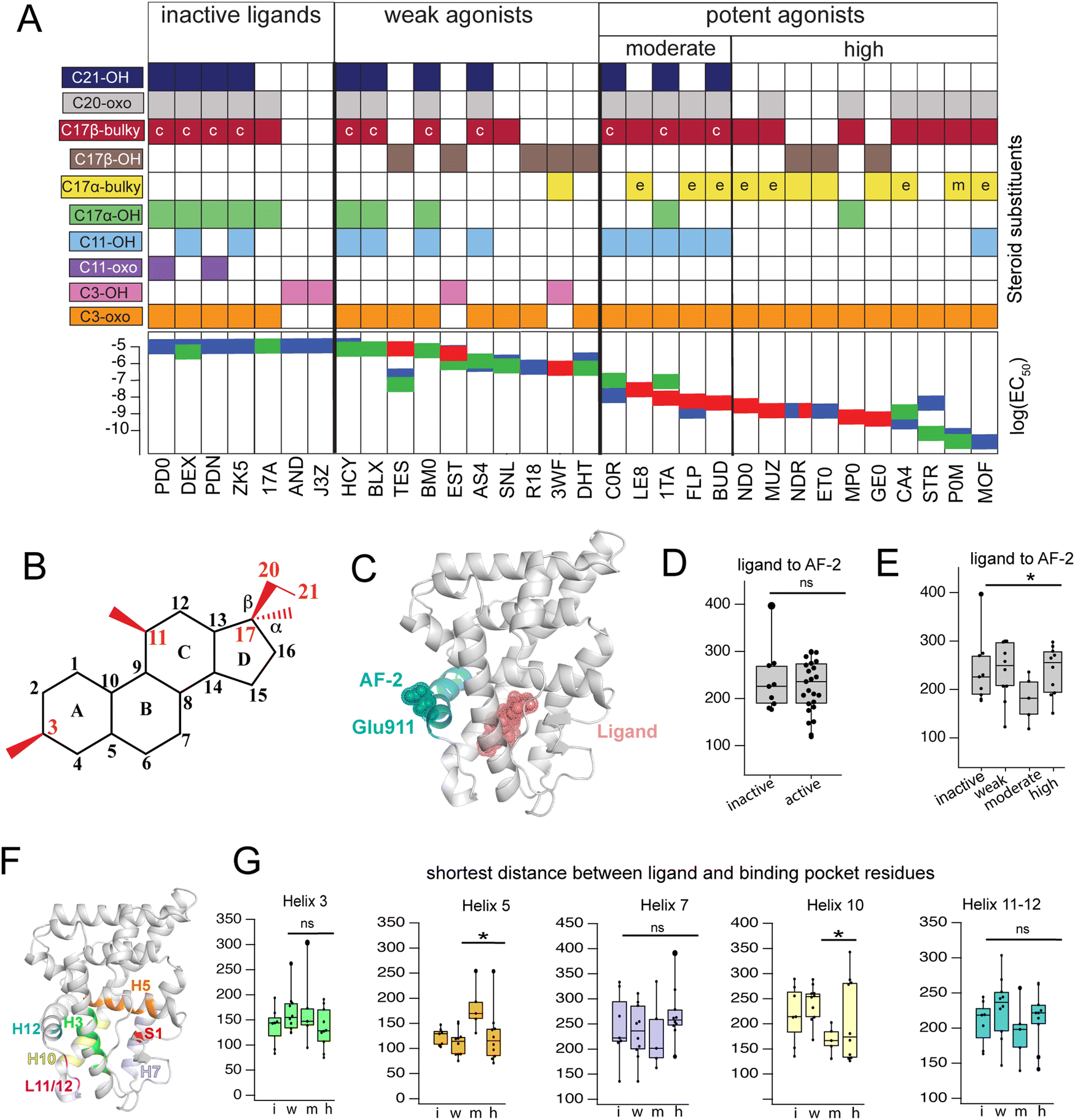 | ||
| Fig. 1 Classification and coupling in PR complexes. (A) Steroidal ligands in this study group are divided into 4 classes based on the potency of PR activation: inactive, weak, moderate, and high. Substituents on C3, C11, C17-α, C17-β, C20 and C21 are defined for the 33 ligands. Bulky substituents at C17-α or β are defined as groups with 2 or more C atoms, excluding promegestone (P0M) which has a methyl C17-α substituent, labeled ‘m’. Ligands with bulky C17-α esterified substituents are labeled ‘e’. Ligands with a C20 ketone and C21 alcohol are labeled ‘c’ for corticosteroids. EC50 values for all ligands are indicated, colored by the reference from which they were obtained: blue,32 green30 and red.31 (B) Atom numbering of the steroidal scaffold. Positions of interest for this study are colored red. (C) Ligand binding pocket and Glu911 at AF-2 are highlighted on PR structure. (D) Comparison of shortest distances between ligand and AF-2 for active and inactive ligands do not reveal significant differences. (E) Comparison of shortest distances between ligand and AF-2 for inactive, weak, moderate, and high potency ligands reveal that moderate potency agonists have significantly lower shortest distances compared to other ligands. (F) Binding pocket residues are highlighted on PR structure. (G) Shortest distances between ligand and binding pocket residues, separated by helix. Shortest distances are averaged for residues in the helix and compared between ligand classes. Only H5 and H10 show significant differences, both observed in moderate potency agonists (*, p < 0.05 by two-tailed, unpaired t-test). | ||
Four of seven inactive ligands are C21 steroids, possessing the C17-β substituent with C20-ketone and C21-hydroxyl groups that is typical of corticosteroids. Glucocorticoids are known to not activate PR.33 C17-α hydroxy substituents are also a trait of inactive ligands, found in five of seven (Fig. 1A). The last two inactive ligands are the only ones in our library with a C11-oxo substitution which may explain their inactivity. Weak ligands exhibit a less distinctive SAR. The weak class has the highest number of ligands with a C17-β hydroxyl (5/10). All ligands without a C17-β hydroxyl contain a bulky C17-β substituent and a C11–OH, with one exception: spironolactone (SNL). The defining feature of potent agonists is a bulky substituent at C17-α and/or C17-β positions with 15/15 having at least one and 8/15 possessing both. This SAR is consistent with reports that PR affinity and activity is optimized in ligands with bulky C17 substituents.18,25
To compare how different ligands affect communication and coupling in PR complexes, we measured the ‘shortest distance’ between: (i) ligand and the activation function surface/AF-2, (ii) ligand and binding pocket residues. This metric uses covariances calculated between proximal amino acids to approximate the strength of communication (see Methods). Calculated from MD trajectories, shortest distance is inversely proportional to strength of communication between two residues.
First, we quantified communication between ligands and AF-2. The activation surface, i.e., the coregulator binding site on the LBD, contains helix 12 (Fig. 1C). Helix 12 (H12) contains Glu911, a conserved residue that serves as a ‘charge clamp’, providing electrostatic stabilization for the LXXLL peptide fragment of coregulators bound at AF-2 (Fig. 1C). To quantify communication between ligands and AF-2, we measured the ‘shortest distance’ between the ligand and Glu911. In comparing shortest distances between active and inactive ligand classes, we did not observe significant differences (Fig. 1D). We further split active ligands into ‘weak’ and ‘high’ potency agonists, which also did not yield statistically significant differences. However, a close analysis revealed that the high potency class of agonists splits into two subgroups, subsequently designated as moderate and high potency ligands. Moderate potency agonists display significantly stronger coupling (i.e., lower shortest distance) with AF-2 (Fig. 1E). The five ligands in the moderate potency class all possess a hydroxyl group at C11 which is not observed in the majority of high potency ligands.
Next, we calculated shortest distances between ligands and the surrounding amino acids that comprise the binding pocket (subsequently referred to as binding pocket residues) (Fig. 1F), including residues on helices 3, 5, 7, 10, 12 as well as the loop between H11 and H12 (L11-12) and S1 β-strand. We observed that moderate potency ligands show significantly weaker communication (higher shortest distance) with H5 residues compared to high potency agonists. These moderate potency agonists also show stronger communication with H10 compared to other ligands. No significant differences were seen in shortest distances between ligands and other helices (Fig. 1G). This analysis suggests that moderate and high potency agonists induce different patterns of coupling within PR, possibly equating to distinct activation mechanisms between the two classes. Furthermore, the findings indicate that the distinction between the two classes involves varying modulation of H5 and/or H10.
PR agonists induce distinct environments in the binding pocket
To quantify the interaction between ligands and PR, we used MM-PBSA and MM-GBSA methods to calculate the binding energy of ligand binding from our trajectories. Because only interaction energies are calculated and entropy is not included, these values are not true free energies. Grouped by class, energies reveal a trend with potency, as inactive ligands have the highest binding energy while high potency agonists have the lowest energy (Fig. 2A and Fig. S3, ESI†). This result suggests that the potency of PR ligands is correlated with the strength of their interaction with PR. To further investigate this pattern, we performed solvent-accessible surface area (SASA) calculations to determine how ligands alter the local environment within the PR binding pocket. Simultaneously, we used energy decomposition to calculate the energetic contributions of the binding pocket residues to the energy of ligand binding. Binding energies are calculated using the MM-PBSA and MM-GBSA methods, followed by per-residue energy decomposition. We examined residues near the D-ring and A-ring ends of the steroids to determine how structural features of the four ligand classes influence SASA and binding energy contributions. All SASA values and binding energies are presented in table (Tables S2 and S3, ESI†) and graph form (Fig. S4 and S5, ESI†).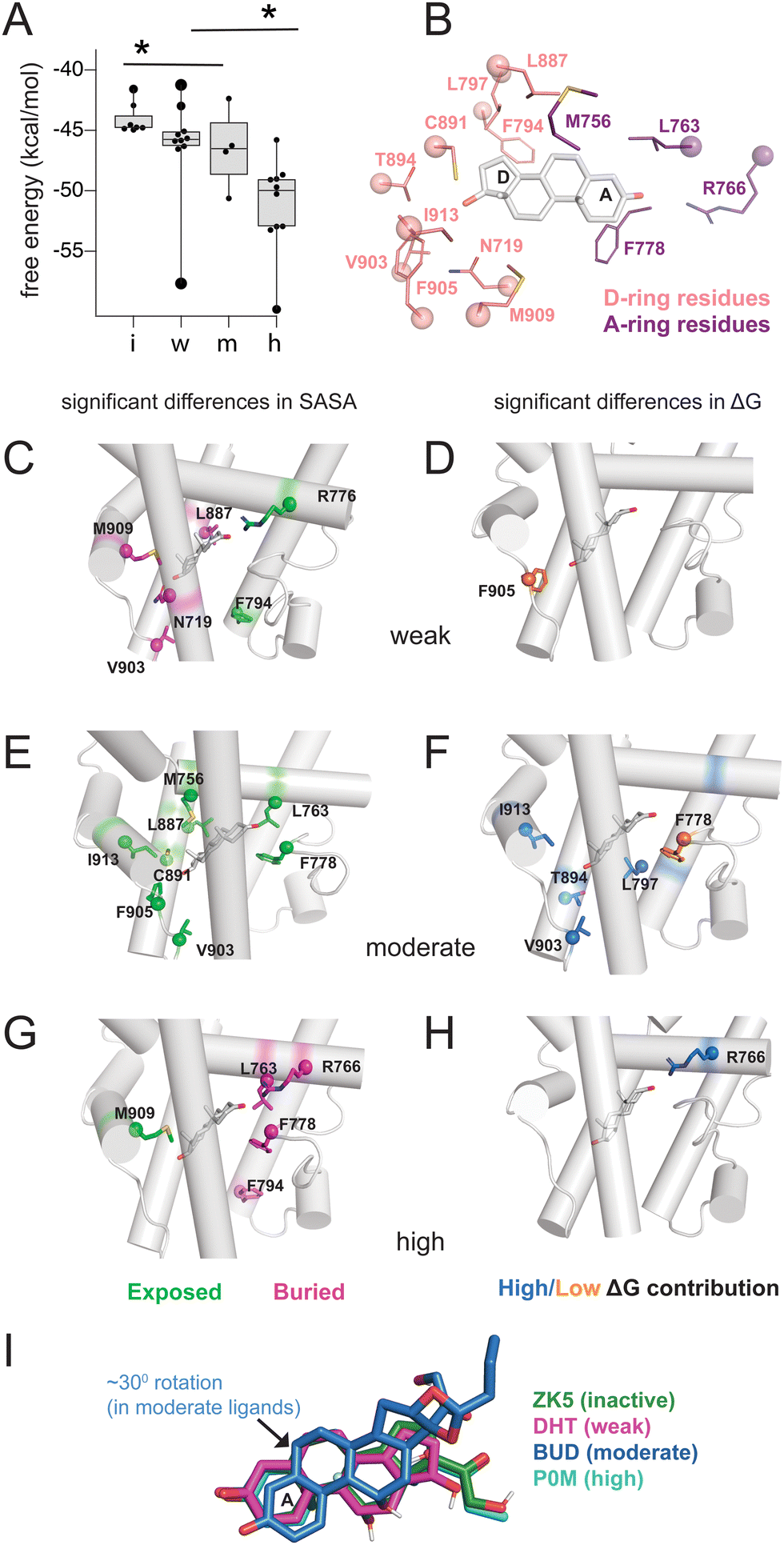 | ||
| Fig. 2 Binding pocket environment of PR ligands. (A) Binding energy calculations using MM-GBSA reveal a relationship between potency and binding energy. Compared to all other ligands, inactive ligands and high potency ligands displayed significantly higher and lower free energies respectively (*, p < 0.05 by two-tailed, unpaired t-test). (B) Binding pocket residues flanking steroidal A-ring and D-rings were analyzed for solvent accessibility and contributions to binding energy. (C, E and G) Ligands are grouped by class and residues identified for which calculated SASA were significantly higher (i.e., more exposed, shown in green) or lower (i.e., more buried, shown in pink) compared to other ligand classes. (D, F and H) Ligands are grouped by class and residues identified whose contribution to binding energy are significantly higher (shown in blue) or lower (shown in orange) compared to other ligand classes. For C–H, significance is defined as p < 0.05 by a two-tailed, unpaired t-test. (I) Ligand orientation is shown for a representative structure from each class. The moderate potency ligand (BUD) reveals a 30° rotation compared to other ligand classes. | ||
On the D-ring end, we examine Phe794, Leu797, Leu887, Cys891, Thr894, Val903, Phe905, Met909, Ile913 and Asn719 (Fig. 2B). Phe794 is the most exposed in weak agonists, with significantly higher SASA values (*, p < 0.05 by two-tailed, unpaired t-test) compared to moderate/high potency agonists (Fig. 2C). This results from the lower tendency of weak agonists to have substituents in the C17-α position (Fig. 2A). Conversely Phe794 is most buried in high potency agonists (Fig. 2G), resulting from the presence of bulky C17-α substituents. We then analyzed exposure of Asn719 on H3 which is physically close to L11–12 residues Val903 and Phe905. Asn719 is conserved in all oxosteroid receptors and forms hydrogen bonds with C21 and C17 and C11 hydroxyl groups. We observe lower SASA (i.e. more buried) with weak agonists (Fig. 3B), suggesting that weak ligands induce a buried conformation around this H3 region and are most likely to interact with Asn719 via hydrogen bonding with C17 groups. We note that 8 of 10 weak ligands have a C17–OH (5 β, 3 α).
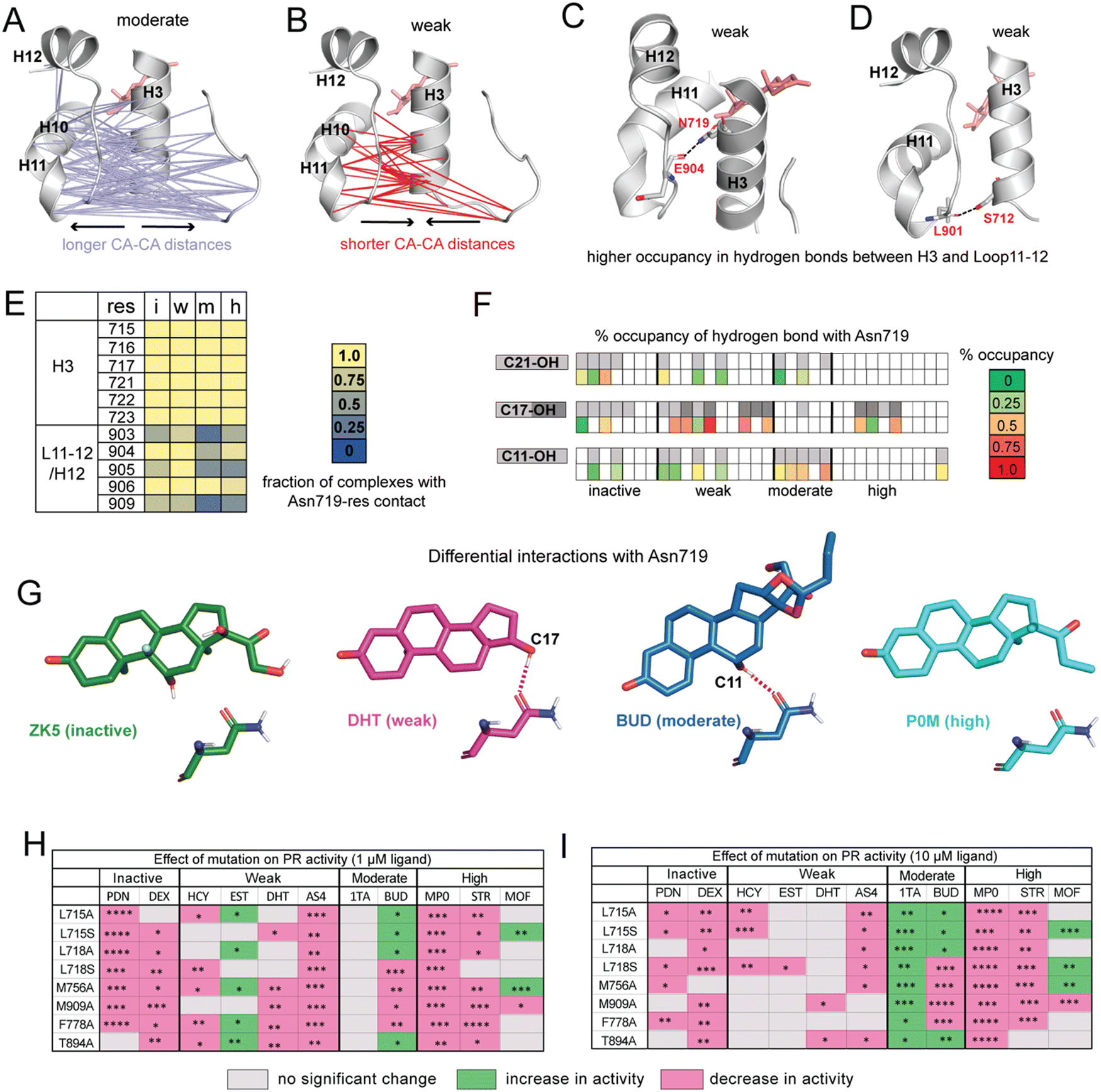 | ||
| Fig. 3 Ligands display distinct hydrogen bonding patterns and response to mutagenesis. (A) Cα distances between H10/H11/L11–12 and H3/L1–2 are significantly longer in moderate potency agonists compared to other ligands. (B) Cα are significantly shorter in weak agonists. (C) Hydrogen bond between Asn719 (H3) and Glu904 (L11–12) has significantly higher occupancy in weak ligand complexes. (D) Hydrogen bond between Ser712 (H3) and Leu901 (L11–12) also has significantly higher occupancy in weak ligand complexes. (E) Interactions between Asn719 (H3) and nearby residues are modulated by ligand identity. Interactions with H3 are maintained across all ligand classes. However, interactions between Asn719 and L11–12/H12 are noticeably reduced in moderate agonist complexes. (F) Hydrogen bonding with Asn719 is dependent on ligand substitutions at C11, C17, and C21. Ligands with C11/C17/C21 substitutions are shown in gray on the top row while bottom row shows occupancy of hydrogen bond between these OH groups and Asn719. For C17–OH substitutions, light gray indicates C17-α substituent while dark gray indicates a C17-β substituent (see Fig. 2A). Moderate agonists are likely to utilize C11–OH while weak agonists are more likely to use C17–OH for hydrogen bonding. (G) Characteristic Asn719 hydrogen bonding patterns for each ligand class. Weak agonists favor Asn719 hydrogen bonding with C17–OH while moderate potency agonists tend to bond with C11–OH. Inactive and highly potent ligands display weak/inconsistent hydrogen bonding patterns which are not represented. (H and I) Transactivation assays reveal the effects of PR mutations on activity for representative ligands from the four ligand classes. Activity is observed to increase (green), decrease (pink), or not change (grey) in mutant PRs. The fold activation with ligand concentrations at 1 μM and 10 μM of wild-type PR-LBD is set as the maximum fold activation response. The ligand activity difference in mutants against wild-type represented as statistically significant values and calculated by two-tailed unpaired t-test in Graph pad Prism 10.1. The statistical significance was estimated from either one or two sets of experiments where each experiment consists of two or three individual replicates. Statistical significance is represented as ****p < 0.0001; ***p < 0.0001; **p < 0.01; *p < 0.05. While observed effects do not appear to differ by mutation (i.e., mutations all tend to have a similar effect on activity for any given ligand), trends are observed by ligand class at both low and high ligand concentrations. For A–D, significance is defined as p < 0.05 by a two-tailed, unpaired t-test. | ||
Non-polar L11–12 residues Val903 and Phe905 are positioned close to the C17-α and/or C17-β substituent, and reveal significantly higher SASA, more exposed, in moderate potency agonists (Fig. 2D). Interestingly, Ile913 located on H12, more distant from the D-ring, shows similar SASA trends to Val903/Phe905, suggesting that these residues form fewer contacts with surrounding residues in moderate potency ligand complexes. This exposure of Val903, Phe905, and Ile913 stabilizes ligand binding, as two of these residues (Val903, Ile913) make higher binding energy contributions in moderate potency agonist complexes than other ligands (Fig. 2E and F). We observed that Thr894 (H10) and Leu797 (H7) also contribute significantly to stabilizing moderate agonist complexes (Fig. 2E). These combined results suggest that moderate potency agonists are uniquely stabilized by the AF-2 and pre-AF-2 regions of PR.
On the A-ring end, we examine Met756, Leu763, Arg766 (all on H5), and Phe778 (S1/S2), with sidechains close to the A/B steroidal rings. Met756 has the highest SASA in moderate potency agonist complexes (Fig. 2D). It is difficult to identify a structural rationale for this observation as Met756 is close to the C19 methyl group which is present in 24 of 33 ligands. However, this observation suggests that separate events may cause a repositioning of moderate agonists in the binding pocket, contributing to a less hydrophobic environment on the β face. Forming a cavity around the A-ring, Leu763 and Phe778 have significantly higher SASA in moderate agonists but are buried in high potency agonists.
Arg766 (H5) is conserved in steroid receptors and positioned to interact with the C3 atom of the A-ring. We observe lower SASA for Arg766 in high potency ligand complexes (Fig. 2G) and higher SASA in weak agonist complexes (Fig. 2C). In addition, Arg766 contributes favorably to the binding energy of high potency ligands (Fig. 2H), suggesting a direct interaction that confers stabilization to the potent agonists. In summary, the SASA and energy decomposition produces some expected findings, such as a clear distinction around the D-ring between high potency and weak agonists based on C17-α substitution. Moreover, four unexpected predictions emerge from this analysis: (i) moderate agonists are uniquely stabilized by the AF-2 and L11–12 region, (ii) weak agonists have higher burial of Asn719 (H3), possibly equating to stronger interaction with this conserved residue, and (iii) moderate agonists appear to be repositioned in the pocket, evidenced by distinct H5 interactions, and (iv) high potency ligands are uniquely stabilized around the A-ring. A comparison of representative structures from each ligand class reveals a reorientation in the moderate potency ligand (BUD) that is absent with other classes (Fig. 2I), providing support for the observations made from binding pocket environment analysis.
Differential interactions with Asn719 drive agonist potencies
To determine whether functional profiles of ligands influence specific inter-residue interactions across the receptor, we calculated the average distance between every pair of Cα atoms. Distances between residues 705–716 (Loop1–3 and H3) and 890–904 (H10, H11, Loop11–12) are significantly longer in moderate agonists and shorter in weak complexes (Fig. 3A and B). This result is particularly striking because the distance between H11 and H3 was previously identified in GR35 and ERα36 as a predictor of functional effects of ligands.To understand the origin of the trends observed in H11–H3 distances, we identified all hydrogen bonds and compared their occupancies between PR complexes. We identified 8 hydrogen bonds that show significant differences in occupancy between ligand classes (Table S4, ESI†). Of these, the highest occupancy is between Glu904–Asn719, present in weak agonist complexes (Fig. 3C). We also observe a hydrogen bond between Leu901–Ser712 in weak agonist complexes (Fig. 3D). Both hydrogen bonds link H3 to the H11/Loop11–12 region of PR, consistent with the shorter Cα–Cα distances observed between H3 and H11 residues in weak complexes (Fig. 3C). Both display significantly reduced occupancy in moderate potency complexes.
This Asn719–Glu904 hydrogen bond is observed in the PR crystal structure, hypothesized to be important for correctly positioning of H12.26 A similar role is proposed for the equivalent Asn residues in AR, MR and GR.27,28,37 This stabilization is believed to play a role in activation of steroid receptors but may be less relevant in PR due to the reduced importance of Asn719. Thus, our simulations suggest that weak ligands (with C17-α OH group) are most likely to coopt this vestigealized mechanism to mediate the stabilizing interaction between Asn719 on H3 and L11–12. This conclusion is consistent with the low SASA observed in Asn719 in weak agonists (Fig. 2C), suggesting higher levels of Asn719 hydrogen bonding. To further investigate the behavior of Asn719 in PR complexes, we examined the interactions between Asn719 and residues within van der Waals contact. We determined that interactions between Asn719 and H11/H12 tend to be highest in weak potency ligands, supporting the notion that weak ligands pull H3 and H11/H12 closer (Fig. 3E).
Conversely, the opposite effect is observed in moderate potency ligands. These ligands reduce interactions between Asn719 and H11/H12, measured by distances (Fig. 3A), hydrogen bond occupancy (Table S4, ESI†) and van der Waals interactions (Fig. 3E). We hypothesize that this unexpected behavior may result from the C11–OH group and which is a characteristic of this ligand class. To test this hypothesis that the C11 hydroxyl group forms unique interactions, we characterized hydrogen bonding between all ligands and Asn719, specifically C11, C17 (α and β) or C21 hydroxyl groups (Fig. 3F). Of all ligands, moderate potency ligands have the highest hydrogen bonding levels (50% and higher) between C11 and Asn719. While some inactive and weak ligands also use the C11–OH for Asn719 hydrogen bonding, these interactions are less frequent (25% and lower). Instead, weak agonists have a higher propensity to interact with Asn719 via C17–OH (50% and higher), while inactive and high potency ligands form C17–OH hydrogen bonds to a lesser extent. These patterns indicate that Asn719 will preferentially interact with C17–OH of PR ligands and in their absence, with the C11–OH. The hydrogen bond with C11–OH is suboptimal and is unable to stabilize the interaction between H3 and H11/H12. Thus, our findings suggest that the unique behavior of moderately potent agonists with Asn719 can be attributed to both the presence of the C11–OH and absence of a C17–OH. We summarize the hydrogen bonding behavior of each class using representative ligands ZK5, DHT, BUD and P0M respectively (Fig. 3G).
Finally, our studies draw a strong distinction between high potency PR agonists and the moderate/weak agonists. High potency agonists function independently of Asn719, as the receptor has evolved a new optimal method for ligand recognition. Conversely, moderate and weak ligand classes both function in relation to Asn719. Moderate ligands have bulky D-ring substitutions that allow for potent PR activation, but display weaker activity than high potency ligands because of the C11–OH, which introduces evolutionary pressure on ligands to revert to the ancient Asn719-dependent activation. Weak ligands generally lack bulky C17-α substituents which makes them incapable of potently activating PR. Their weak activity results from the presence of a C17-β OH group which recognizes Asn719 and stimulates the ancestral steroid receptor activation mechanism. For the handful of weak ligands without a C17-β OH (e.g. HCY, BLX, BM0, Fig. 2A), we note that these tend to also have a C11–OH and/or C17α-OH without a bulky C17-α substituent, suggesting that their weak activity also can be directly linked to activation via Asn719.
Transactivation assays of PR mutants reveal trends within PR ligand classes
Analyses of our MD simulations suggest that mechanisms of PR modulation differ among the ligand classes proposed in this study. To clarify the role of specific amino acids in mediating ligand activity, we combined mutagenesis studies with luciferase reporter-based transactivation assays (Fig. 3H and I, full data is shown in Fig. S6 and S7, ESI†). We tested the effect of reducing the hydrophobicity around C17 by individually mutating H3 residues Leu715 and Leu718 to both alanine and serine in vitro. We also tested the effect of mutating Met756 (H5), Phe778 (S1/S2), Thr894 (H10) and Met909 (H12) to alanine. All residues are in the binding pocket and from our MD analyses determined to be important in mediating activity profiles of the various ligand classes. Thus, we hypothesized that responses to PR mutations would differ by ligand class. While we observe some patterns within classes, an unexpected result was that ligands tended to display the same effects across all mutations. This was observed at both low and high ligand concentrations.In inactive ligands prednisone (PDN) and dexamethasone (DEX), the response was lowered below DMSO control by most mutations as compared to wild-type PR. At higher concentrations, prednisone displayed additional reduction. Weak ligands showed a more variable response. At 1 μM ligand, three out of four ligands showed no significant change in activity for most mutants. At 10 μM, cortisol (HCY), dihydrotestosterone (DHT), and aldosterone (AS4) tended to show reduced activity while estradiol (EST) shows an increase for 5 out of 8 mutations studied. Moderate potency agonist triamcinolone acetonide (1TA) showed increased activity for all mutations while budesonide (BUD) showed either increased or diminished responses. Activity of high potency agonists progesterone (STR) and medroxyprogesterone (MP0) were lowered in all mutants while mometasone furoate (MOF) had more variable responses. Overall, ligand classes reveal similarities, particularly at the low dose, that support our general hypothesis that structural features common to a ligand class will drive similar functional responses to mutations. However, the mutagenesis studies do not provide strong support for the notion of specific amino acids differentially mediating activity of these ligand classes. Instead, by reducing hydrophobicity in the pocket, all mutations regardless of identity tend to confer a similar, ligand-dependent effect resulting in increase, decrease or no change in response.
Conclusion
Ligands targeting PR have therapeutic potential for several women's health ailments. By understanding the molecular features that drive or inhibit potency in PR ligands, we may gain insight that enables the design of new ligands. In this work, we have focused on using MD simulations to learn how steroidal ligands of varying activity levels modulate PR dynamics. We assembled a group of inactive, weak, and potent PR agonists and examined dynamics, intra-receptor communication, binding pocket environment and energetics in PR complexes. Using correlations calculated from MD trajectories, we predicted the strength of communication between ligands, surrounding residues, and AF-2. Patterns observed in our analysis split the group of potent agonists into moderate and high potency ligands, generating four ligand classes.Steroidal ligands were modeled into the agonist-bound PR conformation where H12 is docked against H3 and H4, allowing the formation of a coactivator binding site. Conventional models of nuclear receptor activation suggest that this conformation is not supported in an unliganded or antagonist-bound state.38 As such, the inactive ligand complexes in this study may not represent conformational states that are physiologically plausible. This observation may explain why our MD simulations reveal very few dynamic features that accompany and/or explain the phenotype of the inactive ligand class. Two observations of note were made. First, calculated binding free energies were significantly higher in inactive ligands than other agonists, consistent with published findings that bulky C17-α substituents increase both binding affinity and activity over the C17-α OH substitution that characterizes 5 out of 7 inactive ligands in this study. Second, we do not observe any conformational effects (e.g., unfolding, or increased fluctuations) at AF-2, even in inactive ligand complexes on the timescales of these simulations. This is consistent with prior simulation studies which observed large AF-2 fluctuations only when the antagonistic X-ray structure was modeled.24
Our simulations reveal that weak ligands activate PR by coopting a defunct steroid receptor mechanism involving conserved H3 residue, Asn719 (Fig. 4). Along with Val903 and Met909 on L11–12 and H12 respectively, Asn719 was most buried in weak complexes, suggesting that these ligands push H3 and L11–12 close together. This claim is supported by both interhelical distances and hydrogen bond analyses. Weak agonists also tend to contain a hydroxyl at C17 which is ideally positioned to engage the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the Asn719 sidechain in a hydrogen bond (Fig. 3F). Concurrently, the amide N interacts with and stabilizes the pre-H12 loop and the AF-2 by extension. This mechanism is observed in other oxosteroid receptors and likely existed in their ancestor, but was lost during PR evolution (summarized in Fig. 4).
O of the Asn719 sidechain in a hydrogen bond (Fig. 3F). Concurrently, the amide N interacts with and stabilizes the pre-H12 loop and the AF-2 by extension. This mechanism is observed in other oxosteroid receptors and likely existed in their ancestor, but was lost during PR evolution (summarized in Fig. 4).
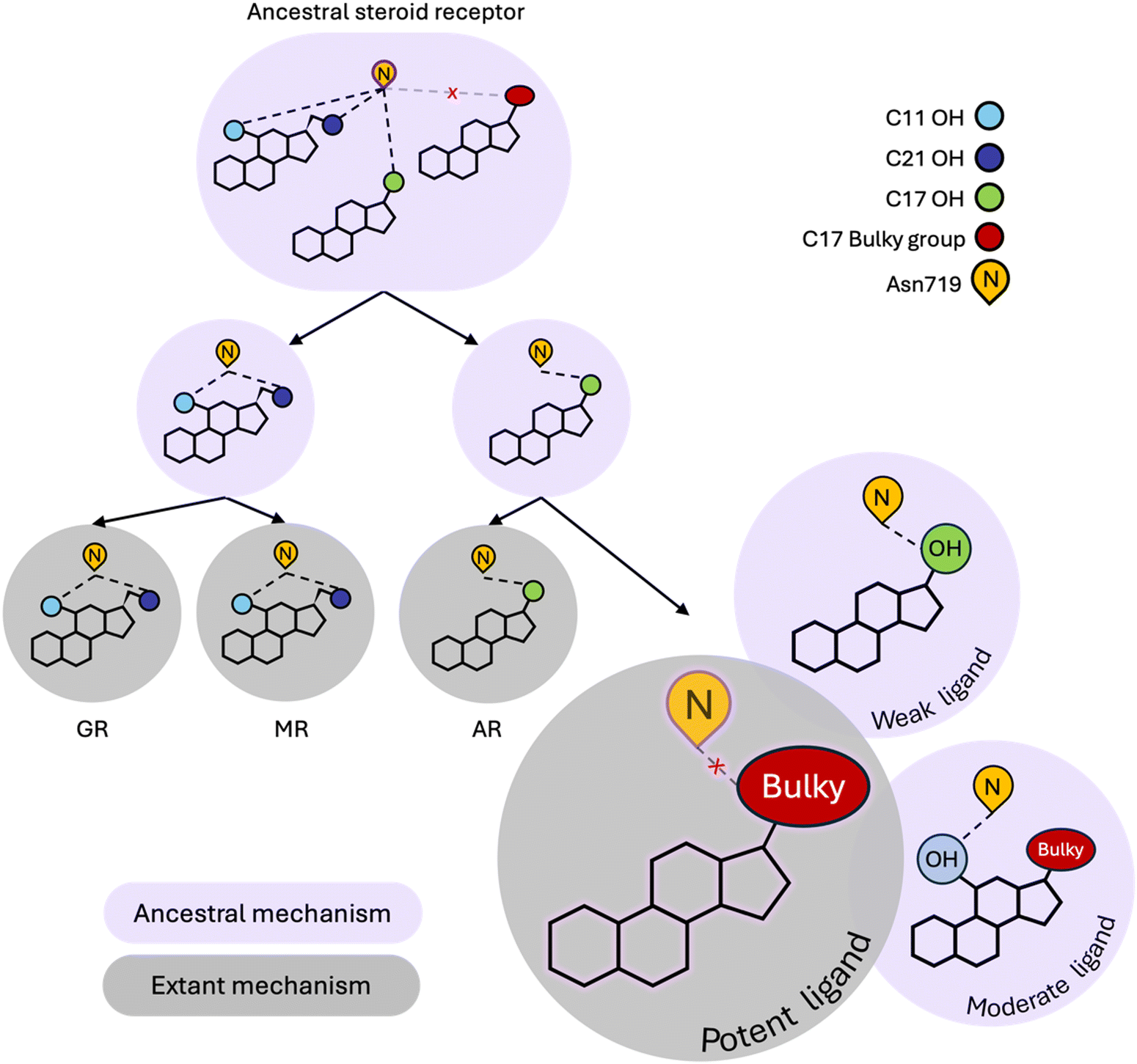 | ||
| Fig. 4 Modern PR utilizes both ancestral and extant steroid receptor (SR) mechanisms. Ancestral SR mechanisms rely on hydrogen bonding with N719, mediated by C17–OH, C11–OH and/or C21–OH in steroids. This mechanism is present in the ancestral oxosteroid receptor (ancestral SR) and conserved in GR, MR and AR. While PR has evolved a unique extant mechanism that is preferentially activated by bulky C17 substituents, it can also undergo N719-dependent activation, representative of a defunct ancestral mechanism. Ligands with C17–OH trigger weak activation of PR via hydrogen bonding with N719. The combination of C11–OH and a bulky C17 substituent triggers the ancestral N719-dependent mechanism which attenuates potency, generating the moderate potency class of ligands. | ||
Instead, the hydrophobic nature of the PR pocket favors ligands with bulky substituents at C17, which characterizes the majority of moderate and high potency ligands in this study. However these two classes reveal dynamic differences which appear to be meditated by the presence of a C11 hydroxyl group that attenuates activity of moderate ligands. Specifically, because they tend to lack an OH group at C17, moderate potency ligands engage in hydrogen bonding with Asn719 via the C11–OH, causing a reorientation of the steroids in the ligand binding pocket, and several effects observed in simulations, including destabilizing the interaction between H3 and H11/H12. High potency ligands are also less likely to form hydrogen bonds with Asn719, confirming that evolution has shaped PR to prefer nonpolar interactions over polar contacts with Asn719. Interestingly, while the Asn719 hydrogen bond with C11–OH is deleterious for PR activity, this interaction improves activity in MR ligands.39 Additionally, GR and MR ligands (i.e. corticosteroids) which are all C11-hydroxylated strongly activate the oxosteroid ancestor,29 suggesting that C11–OH was historically beneficial for steroid receptor activation (Fig. 4). Evolution altered this relationship in the PR/AR lineage, making the C11–OH in steroids detrimental for activity.
In summary, our simulation study reveals that steroidal ligands bound to PR trigger different activation mechanisms, which we unravel by monitoring dynamic signatures. We show that while PR has evolved new preferences for responding to ligand signals, it retains vestiges of a defunct activation mechanism that can be turned on by ligands with the appropriate molecular structure, i.e. a C17 (preferentially β) hydroxyl group that can hydrogen bond with Asn719. Because this mechanism is evolutionarily obsolete, this activity is weak at best. Additionally, when ligand structures enable them to incorporate both the ancestral and extant PR mechanisms, frustration arises leading to attenuated activation, as in the case of moderate potency ligands compared to high potency ligands. We note that the conclusions achieved here would not be possibly by crystallographic studies alone. While they might show the existence of a hydrogen bond in a structure, X-ray structures are limited for quantifying the occupancy of these interactions, which is one of the key results for distinguishing the between PR dynamics of weak and moderate agonists. These results may inform rational drug design strategies, as PR agonism can be modulated by designing ligands to target specific activation mechanisms. Because several aspects of nuclear receptor activation are conserved, it is possible that similar mechanistic features observed in PR might apply to other receptors.
Finally, we note that evolution does not explain all PR activation differences observed in these ligand classes, such as the effect of the A-ring. We observe that our ligand classes exhibit differences around the A-ring which clearly influence activity. Indeed, PR activation studies show that a double bond added into the A-ring of 11β-hydroxydihydroprogesterone (11βOHDHP4) significantly increases activity.40 Additionally, we have not accounted for full versus partial agonistic activity of the ligands here, as we have focused solely on EC50 values. Further work will be required to determine whether there is an evolutionary basis for partial agonistic activity reported for steroidal PR ligands.
Methods
Molecular dynamics simulations
Trajectory processing and analysis
Root mean square deviation (RMSD) values were calculated over all combined trajectories to assess stability and equilibration (Fig. S8, ESI†). The ‘strip’ and ‘trajout’ commands of the CPPTRAJ module50 were used to remove solvent atoms and extract 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 evenly spaced frames from each 500-ns simulation for analysis. For each complex, the three simulations were combined to generate 75000 frames for analysis. Root mean square fluctuations (RMSF) analysis was performed using Cα atoms of protein residues, computed for each frame in the trajectory relative to the initial structure. The ‘hbond’ command in CPPTRAJ was used to identify and quantify all hydrogen bonds in trajectories. The ‘surf’ command was used to calculate the solvent-accessible surface area (SASA) of amino acids in the binding pocket. The ‘dist’ command was used to calculate the distances between all pairs of Cα atoms. To calculate binding free energies of ligands to PR, the MM-PBSA and MM-GBSA methods were utilized.51 For SASA, distances and free energy analyses, we partitioned data by ligand class (i.e. inactive, weak, moderate and high potency ligands) and used t-tests to determine whether the values for any given ligand class were significantly different (in both directions) from those of the other ligands.
000 evenly spaced frames from each 500-ns simulation for analysis. For each complex, the three simulations were combined to generate 75000 frames for analysis. Root mean square fluctuations (RMSF) analysis was performed using Cα atoms of protein residues, computed for each frame in the trajectory relative to the initial structure. The ‘hbond’ command in CPPTRAJ was used to identify and quantify all hydrogen bonds in trajectories. The ‘surf’ command was used to calculate the solvent-accessible surface area (SASA) of amino acids in the binding pocket. The ‘dist’ command was used to calculate the distances between all pairs of Cα atoms. To calculate binding free energies of ligands to PR, the MM-PBSA and MM-GBSA methods were utilized.51 For SASA, distances and free energy analyses, we partitioned data by ligand class (i.e. inactive, weak, moderate and high potency ligands) and used t-tests to determine whether the values for any given ligand class were significantly different (in both directions) from those of the other ligands.
Contact maps and network analysis
The Network View plugin52 in VMD53 and the Carma program54 were used to analyze contacts and produce dynamic networks for each system.55 Residue contact maps were used to determine how dynamic contacts are altered across various complexes. To generate contact maps, protein residues are defined as nodes and edges (or contacts) are created between two non-neighboring nodes if the heavy atoms of the two residues are within 4.5 Å of each other for 75% of the trajectory. To produce dynamic networks, edges in residue contact maps were weighted by covariances calculated from MD simulations (following the protocol described in ref. 55. Edge weights inversely proportional to the calculated pairwise correlation between the nodes. To identify groups of nodes with correlated motions, communities were generated from dynamic networks using the Girvan–Newman algorithm.56 The ligand community was defined as the community containing the ligand.Shortest distance
Communication between the pairs of residues (e.g. ligand and helix 12, ligand and binding pocket residues) was described by quantifying the shortest distance between these sites using the Floyd–Warshall algorithm.57 For distant residues, a communication path is drawn as a chain comprised of nodes and edges that connect the two residues. Due to the inverse correlation between correlation and edge weights, the sum of edges along the path between two distant nodes becomes lower as the strength of communication (i.e., correlation) increases. Among all possible paths between the two residues, the shortest distance refers to the path for which the sum of edge weights is the lowest. Shortest distance is a unitless parameter that quantifies the sum of the edge weights. For closely located residues within contact of one another, the shortest distance is simply calculated as the length of the edge connecting the two nodes.Experimental methods
The plasmid containing full-length PR-B receptor (pcDNA3.0-PRB) was purchased from Addgene, USA. The DNA encoding PR hinge and LBD region (632–933) (UniProt ID: P06401) was PCR amplified using a combination of primers: 5′TATAGAATTCATGGTCCTTGGAGGTCGA-3′ and 5′-TATAAGATCTTCACTTTTTATGAAAGAGAAGG-3′ with 5′-EcoRI and 3′-BglII restriction enzyme sites, respectively. The enzyme-digested PCR insert was cloned in-frame with DBD of pSG5-Gal4-DBD vector with the same restriction sites. This vector (pSG5-Gal4-DBD-PR) was used later as a template to generate different LBD mutants by PCR-based site-directed mutagenesis.The following primers used for the mutagenesis reactions:
| Phe778Ala_FP | 5′-ACATGTCAGTGGGCAGATGCTGTATGCTGCACCTGATCTA-3′ |
| Phe778Ala_RP | 5′-TAGATCAGGTGCAGCATACAGCATCTGCCCACTGACATGT-3′ |
| Thr894Ala_FP | 5′-CATCTGTACTGCTTGAATGCATTTATCCAGTCCCGGG-3 |
| Thr894Ala_RP | 5′-CCCGGGACTGGATAAATGCATTCAAGCAGTACAGATG-3′ |
| Met756Ala_FP | 5′-TAACTCTCATTCAGTATTCTTGGGCGAGCTTAATGGTGTTTGGTCTAG-3′ |
| Met756Ala_RP | 5′-CTAGACCAAACACCATTAAGCTCGCCCAAGAATACTGAATGAGAGTTA-3′ |
| Met909Ala_FP | 5′-CTGAGTGTTGAATTTCCAGAAATGGCGTCTGAAGTTATTGCTGCACAATT-3′ |
| Met909Ala_RP | 5′-AATTGTGCAGCAATAACTTCAGACGCCATTTCTGGAAATTCAACACTCAG-3′ |
| Leu715Ala_FP | 5′-CTGACACCTCCAGTTCTTTGGCGACAAGTCTTAATCAACTAG-3′ |
| Leu715Ala_RP | 5′-CTAGTTGATTAAGACTTGTCGCCAAAGAACTGGAGGTGTCAG-3′ |
| Leu715Ser_FP | 5′-CCTGACACCTCCAGTTCTTTGTCGACAAGTCTTAATCAACTAGG-3′ |
| Leu715Ser_RP | 5′-CCTAGTTGATTAAGACTTGTCGACAAAGAACTGGAGGTGTCAGG-3′ |
| Leu718Ala_FP | 5′-CAGTTCTTTGCTGACAAGTGCTAATCAACTAGGCGAGAGG-3′ |
| Leu718Ala_RP | 5′-CCTCTCGCCTAGTTGATTAGCACTTGTCAGCAAAGAACTG-3′ |
| Leu718Ser_FP | 5′-CCAGTTCTTTGCTGACAAGTAGTAATCAACTAGGCGAGAGGC-3′ |
| Leu718Ser_RP | 5′-GCCTCTCGCCTAGTTGATTACTACTTGTCAGCAAAGAACTGG-3′ |
All mutants were confirmed by DNA sequencing. The hormone-dependent transcription activity of wild-type progesterone receptor and its mutants was assayed in the luciferase reporter system. HeLa cells were grown and maintained in phenol red-free medium MEM-α supplemented with 10% charcoal-dextran stripped FBS. The cells were seeded in 96-well plates at 90% confluency (8000–10000 cells per well) and co-transfected with 50 ng 9x-UAS firefly luciferase reporter, 1 ng Renilla (pRL-SV40), and 5 ng wild-type progesterone receptor (pSG5-Gal4-DBD-PR) or mutant receptor plasmids using FuGene HD (Promega). Twenty-four hours after transfection, cells were treated with DMSO or selected ligands at two different concentrations (1 μM and 10 μM). Firefly and Renilla luciferase activities were measured 24 hours after ligand treatment using the Dual-Glo kit (Promega) using Spectramax iD5 plate reader. Firefly readings normalized over renilla for transfection efficiency check. The graphs were plotted as mutant versus wild type which were already normalized over DMSO.
Conflicts of interest
There are no conflicts to declare.References
- K. W. Reding, et al., Genetic variation in the progesterone receptor and metabolism pathways and hormone therapy in relation to breast cancer risk, Am. J. Epidemiol., 2009, 170(10), 1241–1249 CrossRef PubMed.
- R. J. Collier, et al., A review of endocrine regulation of metabolism during lactation, J. Anim. Sci., 1984, 59(2), 498–510 CrossRef CAS PubMed.
- T. A. Tibbetts, O. M. Conneely and B. W. O'Malley, Progesterone via Its Receptor Antagonizes the Pro-Inflammatory Activity of Estrogen in the Mouse Uterus1, Biol. Reprod., 1999, 60(5), 1158–1165 CrossRef CAS PubMed.
- J. M. Azeez, et al., New insights into the functions of progesterone receptor (PR) isoforms and progesterone signaling, Am. J. Cancer Res., 2021, 11(11), 5214–5232 CAS.
- R. D. Brinton, et al., Progesterone receptors: form and function in brain, Front. Neuroendocrinol., 2008, 29(2), 313–339 CrossRef CAS PubMed.
- A. Christensen, et al., Hormonal regulation of female reproduction, Horm. Metab. Res., 2012, 44(8), 587–591 CrossRef CAS PubMed.
- E. Seli, et al., Minireview: Metabolism of female reproduction: regulatory mechanisms and clinical implications, Mol. Endocrinol., 2014, 28(6), 790–804 CrossRef PubMed.
- A. R. Daniel, C. R. Hagan and C. A. Lange, Progesterone receptor action: defining a role in breast cancer, Expert Rev. Endocrinol. Metab., 2011, 6(3), 359–369 CrossRef CAS PubMed.
- A. Kariagina, M. D. Aupperlee and S. Z. Haslam, Progesterone receptor isoform functions in normal breast development and breast cancer, Crit. Rev. Eukaryot. Gene Exp., 2008, 18(1), 11–33 CrossRef CAS PubMed.
- M. A. Ochoa-Bernal and A. T. Fazleabas, Physiologic Events of Embryo Implantation and Decidualization in Human and Non-Human Primates, Int. J. Mol. Sci., 2020, 21(6), 1973 CrossRef CAS PubMed.
- C. Y. Ramathal, et al., Endometrial decidualization: of mice and men, Semin. Reprod. Med., 2010, 28(1), 17–26 CrossRef CAS PubMed.
- S. J. Lusher, et al., X-ray structures of progesterone receptor ligand binding domain in its agonist state reveal differing mechanisms for mixed profiles of 11beta-substituted steroids, J. Biol. Chem., 2012, 287(24), 20333–20343 CrossRef CAS PubMed.
- F. J. DeMayo and J. Lydon, 90 YEARS OF PROGESTERONE: New insights into progesterone receptor signaling in the endometrium required for embryo implantation, J. Mol. Endocrinol., 2020, 65(1), T1–T14 CAS.
- E. Lim, C. Palmieri and W. D. Tilley, Renewed interest in the progesterone receptor in breast cancer, Br. J. Cancer, 2016, 115(8), 909–911 CrossRef CAS PubMed.
- J. J. Kim, T. Kurita and S. E. Bulun, Progesterone action in endometrial cancer, endometriosis, uterine fibroids, and breast cancer, Endocr. Rev., 2013, 34(1), 130–162 CrossRef CAS PubMed.
- B. Patel, et al., Role of nuclear progesterone receptor isoforms in uterine pathophysiology, Hum. Reprod. Update, 2015, 21(2), 155–173 CrossRef CAS PubMed.
- R. M. Marquardt, et al., Progesterone and Estrogen Signaling in the Endometrium: What Goes Wrong in Endometriosis?, Int. J. Mol. Sci., 2019, 20(15), 3822 CrossRef CAS PubMed.
- K. P. Madauss, et al., Progesterone receptor ligand binding pocket flexibility: crystal structures of the norethindrone and mometasone furoate complexes, J. Med. Chem., 2004, 47(13), 3381–3387 CrossRef CAS PubMed.
- I. Petit-Topin, et al., Molecular determinants of the recognition of ulipristal acetate by oxo-steroid receptors, J. Steroid Biochem. Mol. Biol., 2014, 144 Pt B, 427–435 CrossRef CAS PubMed.
- L. Zheng, K. Xia and Y. Mu, Ligand Binding Induces Agonistic-Like Conformational Adaptations in Helix 12 of Progesterone Receptor Ligand Binding Domain, Front. Chem., 2019, 7, 315 CrossRef CAS PubMed.
- S. P. Williams and P. B. Sigler, Atomic structure of progesterone complexed with its receptor, Nature, 1998, 393(6683), 392–396 CrossRef CAS PubMed.
- J. Zhang, et al., A critical role of helix 3-helix 5 interaction in steroid hormone receptor function, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(8), 2707–2712 CrossRef CAS PubMed.
- I. Petit-Topin, et al., Met909 plays a key role in the activation of the progesterone receptor and also in the high potency of 13-ethyl progestins, Mol. Pharmacol., 2009, 75(6), 1317–1324 CrossRef CAS PubMed.
- L. Zheng, V. C. Lin and Y. Mu, Exploring Flexibility of Progesterone Receptor Ligand Binding Domain Using Molecular Dynamics, PLoS One, 2016, 11(11), e0165824 CrossRef PubMed.
- A. Hillisch, et al., The significance of the 20-carbonyl group of progesterone in steroid receptor binding: a molecular dynamics and structure-based ligand design study, Steroids, 2003, 68(10–13), 869–878 CrossRef CAS PubMed.
- M. Letz, et al., Investigation of the binding interactions of progesterone using muteins of the human progesterone receptor ligand binding domain designed on the basis of a three-dimensional protein model, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1999, 1429(2), 391–400 CrossRef CAS PubMed.
- J. Fagart, et al., Antagonism in the human mineralocorticoid receptor, EMBO J., 1998, 17(12), 3317–3325 CrossRef CAS PubMed.
- N. Poujol, et al., Specific Recognition of Androgens by Their Nuclear Receptor: A STRUCTURE-FUNCTION STUDY, J. Biol. Chem., 2000, 275(31), 24022–24031 CrossRef CAS PubMed.
- G. N. Eick, et al., Evolution of minimal specificity and promiscuity in steroid hormone receptors, PLoS Genet., 2012, 8(11), e1003072 CrossRef CAS PubMed.
- D. Sedlák, A. Paguio and P. Bartůněk, Two panels of steroid receptor luciferase reporter cell lines for compound profiling, Comb. Chem. High Throughput Screening, 2011, 14(4), 248–266 CrossRef PubMed.
- F. Fan, et al., Utilization of human nuclear receptors as an early counter screen for off-target activity: a case study with a compendium of 615 known drugs, Toxicol. Sci., 2015, 145(2), 283–295 CrossRef CAS PubMed.
- J. M. Wilkinson, et al., Compound profiling using a panel of steroid hormone receptor cell-based assays, SLAS Discovery, 2008, 13(8), 755–765 CrossRef CAS PubMed.
- R. M. Hoyte, et al., Synthesis and evaluation of potential radioligands for the progesterone receptor, J. Med. Chem., 1985, 28(11), 1695–1699 CrossRef CAS PubMed.
- H. E. Smith, et al., Binding of steroids to progesterone receptor proteins in chick oviduct and human uterus, J. Biol. Chem., 1974, 249(18), 5924–5932 CrossRef CAS PubMed.
- N. E. Bruno, et al., Chemical systems biology reveals mechanisms of glucocorticoid receptor signaling, Nat. Chem. Biol., 2021, 17(3), 307–316 CrossRef CAS PubMed.
- J. C. Nwachukwu, et al., Predictive features of ligand-specific signaling through the estrogen receptor, Mol. Syst. Biol., 2016, 12(4), 864 CrossRef PubMed.
- X. Liu, et al., Disruption of a key ligand-H-bond network drives dissociative properties in vamorolone for Duchenne muscular dystrophy treatment, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(39), 24285–24293 CrossRef CAS PubMed.
- Y. Li, M. H. Lambert and H. E. Xu, Activation of Nuclear Receptors: A Perspective from Structural Genomics, Structure, 2003, 11(7), 741–746 CrossRef CAS PubMed.
- R. K. Bledsoe, et al., A Ligand-mediated Hydrogen Bond Network Required for the Activation of the Mineralocorticoid Receptor*[boxs], J. Biol. Chem., 2005, 280(35), 31283–31293 CrossRef CAS PubMed.
- R. Gent, et al., C11-hydroxy and C11-oxo C19 and C21 Steroids: Pre-Receptor Regulation and Interaction with Androgen and Progesterone Steroid Receptors, Int. J. Mol. Sci., 2024, 25(1), 101 CrossRef CAS PubMed.
- D. Case, et al., AMBER 2020, University of California, San Francisco, 2020 Search PubMed.
- J. A. Maier, et al., ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB, J. Chem. Theory Comput., 2015, 11(8), 3696–3713 CrossRef CAS PubMed.
- C. Tian, et al., ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution, J. Chem. Theory Comput., 2020, 16(1), 528–552 CrossRef PubMed.
- J. Wang, et al., Antechamber: an accessory software package for molecular mechanical calculations, J. Am. Chem. Soc., 2001, 222, U403 Search PubMed.
- J. Wang, et al., Automatic atom type and bond type perception in molecular mechanical calculations, J. Mol. Graph. Model., 2006, 25(2), 247–260 CrossRef CAS PubMed.
- J. Wang, et al., Development and testing of a general amber force field, J. Comput. Chem., 2004, 25(9), 1157–1174 CrossRef CAS PubMed.
- A. W. Götz, et al., Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born, J. Chem. Theory Comput., 2012, 8(5), 1542–1555 CrossRef PubMed.
- R. Salomon-Ferrer, et al., Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald, J. Chem. Theory Comput., 2013, 9(9), 3878–3888 CrossRef CAS PubMed.
- J.-P. Ryckaert, G. Ciccotti and H. J. Berendsen, Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes, J. Comput. Phys., 1977, 23(3), 327–341 CrossRef CAS.
- D. R. Roe and T. E. Cheatham III, PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data, J. Chem. Theory Comput., 2013, 9(7), 3084–3095 CrossRef CAS PubMed.
- B. R. Miller, et al., MMPBSA.py: An Efficient Program for End-State Free Energy Calculations, J. Chem. Theory Comput., 2012, 8(9), 3314–3321 CrossRef CAS PubMed.
- J. Eargle and Z. Luthey-Schulten, NetworkView: 3D display and analysis of protein·RNA interaction networks, Bioinformatics, 2012, 28(22), 3000–3001 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graphics, 1996, 14(1), 33–38 CrossRef CAS PubMed.
- N. M. Glykos, Carma: a molecular dynamics analysis program, J. Comput. Chem., 2006, 27, 1765–1768 CrossRef CAS PubMed.
- A. Sethi, et al., Dynamical networks in tRNA:protein complexes, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(16), 6620–6625 CrossRef CAS PubMed.
- M. E. Newman, Modularity and community structure in networks, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(23), 8577–8582 CrossRef CAS PubMed.
- R. W. Floyd, Algorithm 97: shortest path, Commun. ACM, 1962, 5(6), 345 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00002a |
| This journal is © The Royal Society of Chemistry 2024 |