
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent progress in polymer nanosheets for photocatalysis
Shunhang
Wei†
 ab,
Lei
Wang†
c,
Jianyong
Yue†
d,
Rong
Wu
d,
Zebo
Fang
*a and
Yuxi
Xu
*b
ab,
Lei
Wang†
c,
Jianyong
Yue†
d,
Rong
Wu
d,
Zebo
Fang
*a and
Yuxi
Xu
*b
aZhejiang Engineering Research Center of MEMS, Shaoxing University, Shaoxing 312000, China. E-mail: csfzb@usx.edu.cn
bSchool of Engineering, Westlake University, Hangzhou 310024, China. E-mail: xuyuxi@westlake.edu.cn
cSchool of Environmental Science and Engineering, Guangzhou University, Guangzhou 510006, China
dSchool of Physics Science and Technology, Xinjiang University, Urumqi 830000, China
First published on 16th October 2023
Abstract
Polymers have been considered as promising photocatalysts for energy conversion and environmental remediation, because of their inherent advantages such as adjustable optical and electronic properties as well as abundant active sites. However, it is difficult for polymers without effective design to achieve ideal photocatalytic performance. Compared with bulk polymers, polymer nanosheets obtained by exfoliation have the thickness of single or multiple atomic layers, which possess numerous unique advantages, such as more accessible active sites, shorter carrier transport distances, a prolonged lifetime of photogenerated carriers and the atomic level regulation of the backbone. Therefore extensive efforts have been focused on photocatalysts based on polymer nanosheets, and remarkable progress has been reported. In this review, the principles of photocatalytic water splitting, pollutant degradation and CO2 reduction are briefly introduced. And then recent advances in polymer nanosheets (conjugated microporous polymers, covalent organic frameworks, conjugated triazine frameworks and carbon nitrides) and their heterojunctions in the above-mentioned fields are highlighted. Specifically, the effects of the modification strategy on their light absorption ability, carrier migration, and photocatalytic activity are discussed. Finally, the important challenges and potential opportunities for developing polymer nanosheets and their heterojunctions as high-performance photocatalysts are envisaged. The insights exhibited in this review can provide a useful guidance for the design and development of polymer nanosheet based photocatalysts.
 Shunhang Wei | Shunhang Wei received his PhD degree from Tongji University (2020) under the supervision of Prof. Xiaoxiang Xu. Currently, he works at Shaoxing University, and his research is mainly focused on low-dimensional materials and their applications in energy storage and conversion. |
 Zebo Fang | Zebo Fang received his PhD degree from Lanzhou University (2006). He then worked as a postdoctoral fellow at Fudan University and a visiting scholar at the City University of Hong Kong, respectively. Currently, he works at Shaoxing University as a professor, and his research is mainly focused on semiconductor materials for energy storage and conversion. |
 Yuxi Xu | Yuxi Xu received his B.S. degree from Wuhan University (2007) and PhD degree from Tsinghua University (2011). He then worked as a postdoctoral fellow at the University of California, Los Angeles. He joined the faculty of Fudan University in 2015 and moved to Westlake University in 2019. His research interests include chemically modified graphene, 2D polymers, and self-assembled functional materials. |
1. Introduction
The energy of sunlight reaching the Earth per hour is enough to meet the energy demand of modern society per year,1,2 thereby the efficient use of solar energy is expected to alleviate the energy crisis and environmental pollution.3–6 Due to some inherent properties of sunlight (low energy density, intermittency, diffusion, etc.), converting solar energy into chemical energy is considered as an effective way to improve solar energy utilization efficiency.7–13 Inspired by photosynthesis in nature through which green plants convert carbon dioxide and water into energy-rich organic matter under mild conditions, artificial photosynthesis based on semiconductor photocatalytic technology is proposed and widely concerned. For example, photocatalytic water splitting to produce clean hydrogen fuel14–20 and photocatalytic CO2 reduction to produce high value-added chemicals (methane, methanol, etc.)21–25 have shown great prospects in alleviating the energy crisis. In addition, solar-driven degradation of pollutants (such as volatile organic compounds, antibiotics and organic dyes) is a powerful technology to solve the problem of environmental pollution.26–31Since the successful application of TiO2 in photocatalytic water splitting,32 a large number of inorganic photocatalysts have been explored, such as oxides (NaTaO3, ZnO, and SrTiO3),33–35 sulfides (In2S3 and ZnIn2S4)36–38 and oxynitrides (Ta3N5, LaTiO2N, and BaTaO2N).39–41 Although a series of advances have been achieved in photocatalysis, they usually exhibit low quantum efficiency due to rapid carrier recombination. In addition, some of them have poor visible-light absorption ability, toxicity, photocorrosion and costly transition metals, which severely limit their large-scale applications. More importantly, the optical and electronic properties of inorganic materials can only be adjusted within a relatively limited range due to their inherent properties.42 Therefore, it remains an urgent need to develop inexpensive, stable, harmless and structurally adjustable photocatalysts to achieve high photocatalytic performance.
Polymer photocatalysts, composed of earth-abundant light elements (C, H, N, S, etc.), possess excellent designability of the structure and functionality and can overcome some shortcomings of inorganic photocatalysts. Most polymer photocatalysts have visible-light absorption ability benefited from their delocalized π-system.43 More importantly, their electronic and optical properties can be effectively regulated via controlling the synthesis conditions. For example, Cooper et al. synthesized a series of conjugated microporous polymers (CMPs) with an optical gap ranging from 1.94 eV to 2.95 eV via changing the monomer compositions involved in the synthesis reaction.44 At present, carbon nitrides, covalent organic frameworks (COFs), conjugated triazine frameworks (CTFs), CMPs and linear conjugated polymers are the main polymer photocatalysts. Among them, although linear conjugated polymers are first reported,45 their photocatalytic activity is generally poor due to the limited electron delocalization.46 Therefore, the evolution of polymer photocatalysts mainly focuses on the other four types. To further improve the photocatalytic performance of polymers, morphology design and construction of heterojunctions are widely adopted. Compared with other morphologies, polymer nanosheets display distinctive advantages including a large specific surface area, abundant active sites and a short carrier migration distance. The construction of heterojunctions can improve various properties such as light absorption and carrier migration. Therefore single-component polymer nanosheets and polymer nanosheet-based heterojunctions are important research directions for polymer photocatalysts.
This work summarizes the research progress of COF, CTF, CMP and carbon nitride nanosheets and their heterojunctions in photocatalytic hydrogen production, CO2 reduction and pollutant degradation (Scheme 1). Firstly, the basic rules of the above three types of photocatalytic processes and the advantages of polymer nanosheets in photocatalysis are briefly discussed. Subsequently, the current advances and optimization methods of single-component polymer nanosheets and polymer nanosheet-based heterojunctions are summarized. Finally, some beneficial guiding opinions of polymer nanosheets for photocatalysis are put forward and expected to obtain better photocatalytic activities.
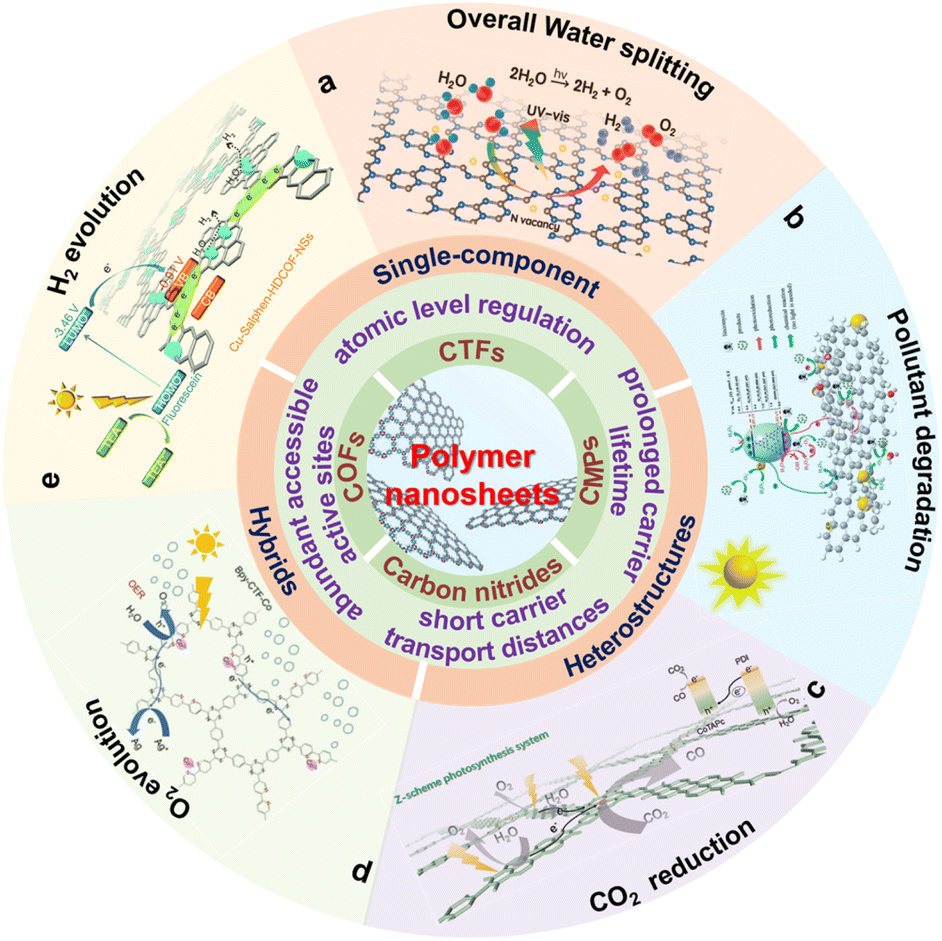 | ||
| Scheme 1 Schematic illustration of the properties and photocatalytic applications for polymer nanosheets. (a) Reproduced with permission from ref. 175, © American Chemical Society 2022. (b) Reproduced with permission from ref. 176, © Wiley-VCH GmbH 2020. (c) Reprinted with permission from ref. 84, © Wiley-VCH GmbH 2022. (d) Reprinted with permission from ref. 177, © Royal Society of Chemistry 2022. (e) Reprinted with permission from ref. 148, © Royal Society of Chemistry 2020. | ||
2. Basic principles for photocatalytic water splitting, pollutant degradation and CO2 reduction
Photocatalytic water splitting and CO2 reduction convert solar energy to chemical energy (such as H2, CH4 and CH3OH), which have attracted enormous attention in alleviating energy shortage and greenhouse effects. In addition, thanks to the green, efficient and secondary pollution free characteristics, photocatalytic degradation shows promising potential in pollutant removal (dye molecules, antibiotics, NOx, etc.). The photocatalytic process needs to meet both dynamic and thermodynamic requirements. Photocatalytic water splitting, degradation and CO2 reduction have the same dynamic process, as shown in Fig. 1a. First, electrons and holes are generated under irradiation. And then, some of the generated carriers will migrate to the surface active sites for a redox reaction. Meanwhile, some carriers will be recombined in the bulk and on the surface of photocatalysts. Inhibiting carrier recombination is an important means to improve photocatalytic activity, and lots of effective methods have been proposed, including doping,47,48 constructing heterojunctions,49–52 and loading cocatalysts.53,54 On the other hand, thermodynamic requirements for the three photocatalytic reactions are different (Fig. 1b). For water splitting, the conduction band minimum (CBM) for H2 production needs to locate at a more negative position than −0.41 V (vs. NHE, pH = 7), and the valence band maximum (VBM) location for O2 production should more positive than 0.82 V (vs. NHE, pH = 7). Generally, the photocatalytic CO2 reduction reaction is more difficult than the photocatalytic water splitting, because the high stability of CO2 molecules requires a significant energy input for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond cleavage (bond enthalpy + 805 kJ mol−1).55,56 The photogenerated electrons and holes reduce CO2 to various products with the corresponding redox potentials (eqn (1)–(5)) and oxidize water to O2, respectively.57–59 For photodegradation, the photogenerated electrons and holes can directly decompose pollutants via redox reactions. Also, they can firstly react with O2 dissolved in water and water molecules to form superoxide radicals (˙O2−, −0.33 V vs. NHE, pH = 7),60 hydroxyl radicals (˙OH, 2.34 V vs. NHE, pH = 7),61etc., and then decompose pollutants.
O bond cleavage (bond enthalpy + 805 kJ mol−1).55,56 The photogenerated electrons and holes reduce CO2 to various products with the corresponding redox potentials (eqn (1)–(5)) and oxidize water to O2, respectively.57–59 For photodegradation, the photogenerated electrons and holes can directly decompose pollutants via redox reactions. Also, they can firstly react with O2 dissolved in water and water molecules to form superoxide radicals (˙O2−, −0.33 V vs. NHE, pH = 7),60 hydroxyl radicals (˙OH, 2.34 V vs. NHE, pH = 7),61etc., and then decompose pollutants.| CO2 + 2H+ + 2e− → HCOOH −0.61 V vs. NHE (pH = 7) | (1) |
| CO2 + 2H+ + 2e− →CO + H2O −0.53 V vs. NHE (pH = 7) | (2) |
| CO2 + 4H+ + 4e− →HCHO + H2O −0.48 V vs. NHE (pH = 7) | (3) |
| CO2 + 6H+ + 6e− →CH3OH + H2O −0.38 V vs. NHE (pH = 7) | (4) |
| CO2 + 8H+ + 8e− →CH4 + 2H2O −0.24 V vs. NHE (pH = 7) | (5) |
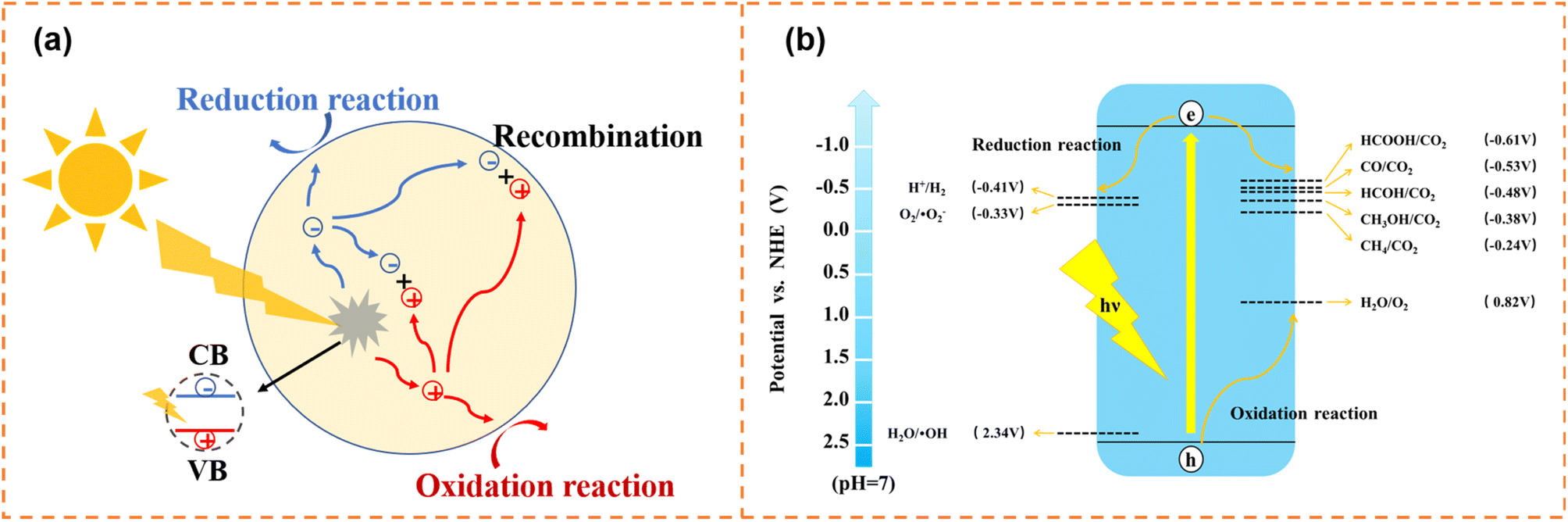 | ||
| Fig. 1 (a) Schematic diagram of the photogenerated carrier transfer process. (b) Schematic illustration of thermodynamic requirements for photocatalysis. | ||
For the three photocatalytic reactions, there are different research focuses in increasing photocatalytic performance. For example, H2 and O2 can combine to form water on the surface of photocatalysts, so the inhibition of the H2–O2 recombination reverse reaction is a research focus for photocatalytic water splitting. Several strategies have been proven to be effective, such as selective cocatalyst deposition14 and introducing amino functional groups on the catalyst surface.62 Some pollutants (antibiotics, etc.) cannot be completely degraded to non-toxic substances, so the design of photocatalysts with high redox ability based on the degradation pathway is a research focus of photocatalytic degradation. The photocatalytic CO2 reduction reaction undergoes various intermediate steps and produces various by-products, resulting in a complex reaction mechanism and low selectivity of products. Therefore achieving efficient CO2 conversion with high selectivity is a research focus of photocatalytic CO2 reduction.
3. Advantages of polymer nanosheets for photocatalysis
For semiconductor photocatalysts, the morphology has an important effect on carrier migration and surface transfer. Therefore photocatalysts with different morphologies are developed to achieve excellent photocatalytic performance. Compared with other morphologies, nanosheets have a cross section that can be as large as micrometers and a thickness as thin as a single atomic layer. The wide cross section is beneficial for receiving solar photons, and the ultra-thin thickness greatly shortens the carrier migration distance. Recently, abundant polymer nanosheets with an adjustable thickness have been prepared by top-down (exfoliation method, etc.) or bottom-up (interface polymerization, solution polymerization, mechanochemistry, etc.) approaches.46,63,64Modification strategies (doping, increasing active sites, etc.) are considered as important routes to enhance the photocatalytic performance of polymer nanosheets. Generally, the modifications in bulk photocatalysts are limited to the surface or the shallow depth below the surface because of the low ratio of surface atoms to entire material. In contrast, the thin polymer nanosheets are mostly composed of surface atoms, which can be modified adequately. More importantly, polymer nanosheets can even realize atomic-level regulation of the backbone and targeted introduction of active sites, resulting in precise and diversified modifications.65 For example, on-surface synthesis (surface as a template to catalyze bond formation) could achieve the atomically precise design of polymer nanosheets.66,67 Song et al. constructed a compact active site layout (boron atoms as the active sites) by adjusting the molecular size of monomers.68 In addition, the band gap and electronic properties of polymers can be effectively regulated by adjusting monomer types and the conjugation degree.69,70 In light of the above-mentioned features, polymer nanosheets show bright application prospects in the field of photocatalysis.
4. Single-component polymer nanosheets for photocatalysis
4.1 CMP photocatalysts
CMP photocatalysts are a class of amorphous polymers with an expanded π-conjugated structure and continuous networks, featuring excellent chemical and thermal stability.71 Because CMPs can be synthesized using thousands of monomers, their band structure and active sites can be flexibly regulated.72 Over the past decade, various CMPs have exhibited outstanding photocatalytic activity, for example, perylene-containing CMPs,73 dibenzothiophene dioxide containing CMPs,74 spirobifluorene-linked CMPs,75 and dibenzothiophene-S,S-dioxide-containing CMPs.76 Typical CMPs usually show a nanoparticle-like morphology because of the highly cross-linked network. In order to enrich the morphologies of CMPs, some effective strategies have been developed. For example, using carbon nanospheres, single-walled carbon nanotubes and reduced graphene oxide as templates can form ball-, stick- and sandwich-like CMPs, respectively.77 Selecting toluene, p-xylene and mesitylene as solvents can obtain CMPs with sphere-, tubular- and plate-like structures, respectively.78 Limited by the synthesis methods of CMP nanosheets, their applications in photocatalysis have been rarely reported so far. Nonetheless, some representative articles are still worth mentioning.4.1.1.1 Photocatalytic H2 production. Jiang et al. used 1,4-benzenediboronic acid bis(pinacol)ester (DBABz), 1,3,6,8-tetrabromopyrene (TBrPy) and 3,7-dibromodibenzothiophene-S,S-dioxide (DBrBTDO) as the π-bridge, donor and acceptor, respectively, to prepare D–π–A CMP photocatalysts (Fig. 2a).79 D–π–A CMP nanosheets with a thickness of around 100 nm (denoted as PyBS-4) were obtained when the ratio of donor to acceptor was 15
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85. The high content of acceptors resulted in high hydrophilicity (Fig. 2b), which was beneficial for the contact between water and the CMP photocatalyst surface. More importantly, thanks to the structural nature of D–π–A polymers, the light-induced electrons could rapidly transfer from the HOMO orbits of the donor to the LUMO orbits of the acceptor along the polymer skeleton, inhibiting photogenerated carrier recombination efficiently (Fig. 2c). Therefore the photocatalytic H2 evolution rate of PyBS-4 reached 0.21 mmol h−1 under visible-light irradiation (λ > 420 nm; its dosage was 25 mg), which was higher than that of D–π–A CMP photocatalysts with other morphologies.80
:85. The high content of acceptors resulted in high hydrophilicity (Fig. 2b), which was beneficial for the contact between water and the CMP photocatalyst surface. More importantly, thanks to the structural nature of D–π–A polymers, the light-induced electrons could rapidly transfer from the HOMO orbits of the donor to the LUMO orbits of the acceptor along the polymer skeleton, inhibiting photogenerated carrier recombination efficiently (Fig. 2c). Therefore the photocatalytic H2 evolution rate of PyBS-4 reached 0.21 mmol h−1 under visible-light irradiation (λ > 420 nm; its dosage was 25 mg), which was higher than that of D–π–A CMP photocatalysts with other morphologies.80
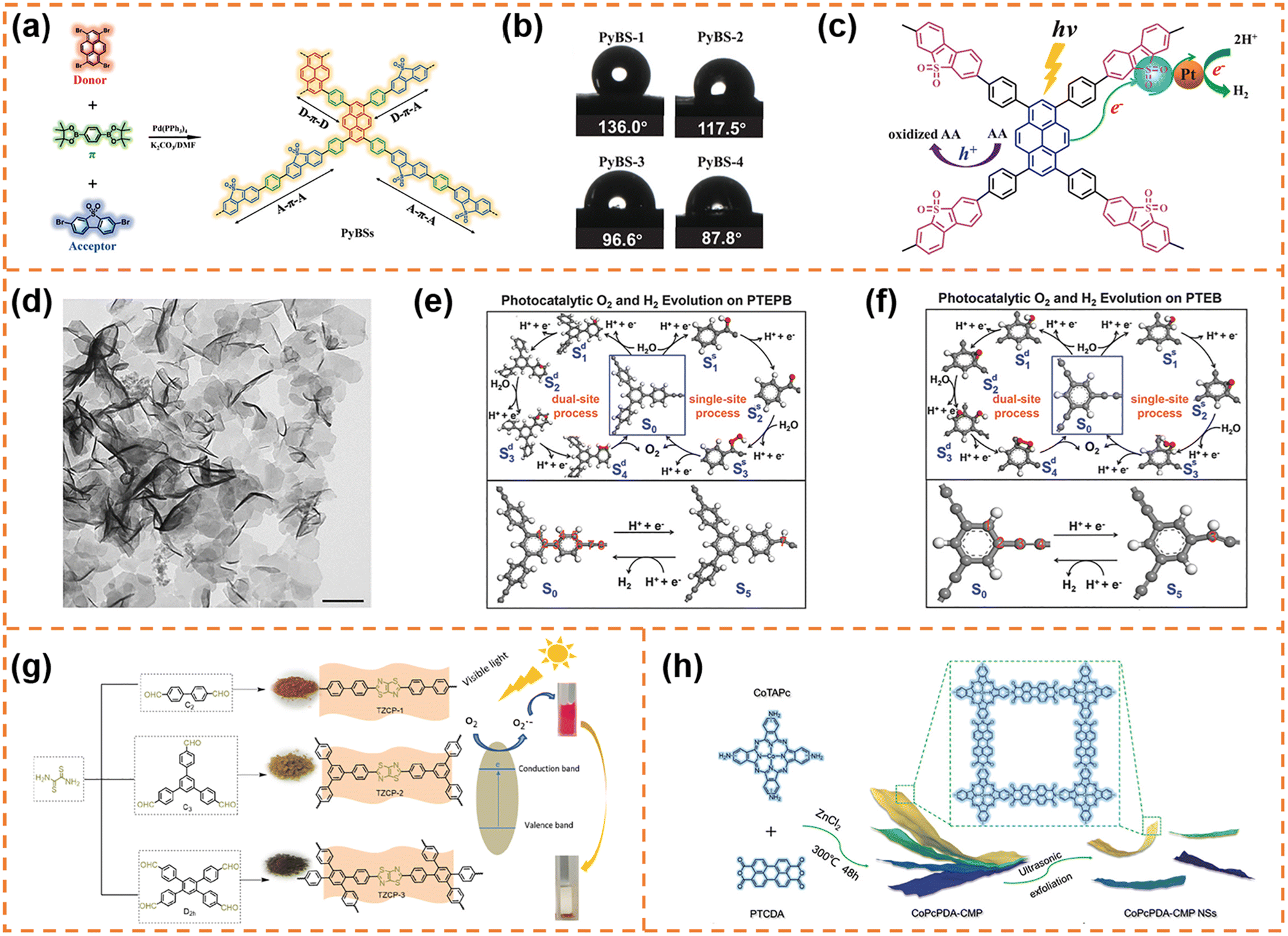 | ||
| Fig. 2 (a) The synthesis of the D–π–A containing copolymer photocatalysts with the notional polymer structure. (b) Water contact angles for the copolymers. (c) The proposed photocatalytic mechanism of the D–π–A copolymer photocatalyst. Reprinted with permission from ref. 79, © Wiley-VCH GmbH 2021. (d) TEM image of PTEPB. Proposed photocatalytic cycles of the oxygen evolution reaction (via single-site and dual-site processes) and hydrogen evolution reaction with the energetically most feasible active sites in both PTEPB (e) and PTEB (f). Reprinted with permission from ref. 82, © Wiley-VCH GmbH 2017. (g) Synthetic routes of the TzTz-based conjugated porous polymers. Reprinted with permission from ref. 83, © American Chemical Society 2019. (h) Schematic illustration of CoPcPDA-CMP NS synthesis. Reprinted with permission from ref. 84, © Wiley-VCH GmbH 2022. | ||
4.1.1.2 Photocatalytic O2 production. Xu et al. successfully synthesized aza-fused CMP (aza-CMP) nanosheets using 1,2,4,5-benzenetetramine tetrahydrochloride and hexaketocyclohexane octahydrate as the raw monomers.81 The band gap of the aza-CMP nanosheets was ∼1.22 eV, and their VBM potential (6.08 eV vs. vacuum level) far exceeded the required potential for water oxidation. Therefore, the aza-CMP nanosheets exhibited photocatalytic O2 production activity under visible-light irradiation (λ > 420 nm; ∼1.0 μmol h−1) and even near infrared light irradiation (λ > 800 nm; ∼0.4 μmol h−1). When the aza-CMP nanosheets were further treated using the liquid exfoliation method, ultrathin aza-CMP nanosheets with a thickness of ∼10 nm were obtained. Benefiting from improved photoresponsivity and more accessible reaction sites, the ultrathin aza-CMP nanosheets exhibited a three-fold enhancement in the photocatalytic O2 production rate under visible-light irradiation.
4.1.1.3 Photocatalytic overall water splitting. Xu et al. employed terminal alkynes (1,3,5-tris-(4-ethynylphenyl)-benzene (TEPB) and 1,3,5-triethynylbenzene (TEB)) as raw materials to fabricate 1,3-diyne-linked CMP nanosheets.82 The obtained CMP nanosheets prepared from TEPB (denoted as PTEPB) exhibited a single-layer or possible double-layer morphology (Fig. 2d). In contrast, the CMP derived from TEB (denoted as PTEB) was prone to form aggregated structures due to its large interlayer interaction energy (0.035 J m−2) and planar molecular structure. Thanks to the role of 1,3-diyne covalent linkage in tuning band structures, both PTEPB and PTEB exhibited narrow band gaps (∼2.85 eV and ∼2.94 eV, respectively). And their VBM and CBM positions satisfied the thermodynamic requirements for water splitting. Therefore they exhibited stable photocatalytic overall water splitting activity under visible-light irradiation. Theoretical calculations further revealed the possible reactive sites for water oxidation (Fig. 2e and f): the O2 evolution for PTEPB could occur spontaneously under visible-light irradiation via a dual-site process, while both single-site and dual-site processes might take place for PTEB. Benefiting from the ultrathin thickness and reactive sites, the average H2 production rate of PTEPB (218 μmol h−1 g−1) was better than that of PTEB (102 μmol h−1 g−1). These findings provided a new insight into designing and synthesizing CMP nanosheets for photocatalytic overall water splitting.
:2), indicating that the unique structure is beneficial for electron–hole separation. When the bulk CoPcPDA-CMPs were exfoliated into nanosheets, the exposed active sites were obviously increased and the electron transfer process was more efficient, so that the CoPcPDA-CMP nanosheets exhibited a faster CO formation rate (14.27 μmol g−1 h−1) and a higher CO selectivity (92%) under visible-light irradiation. This work developed a new high-performance CMP nanosheet photocatalyst for CO2 conversion with H2O as an electron donor, which was competitive to the state-of-the-art visible-light-driven organic photocatalysts for overall CO2 reduction.
4.2 COF photocatalysts
COFs are a class of highly crystalline porous polymers with a high specific surface area, abundant active sites and chemical stability.85 Their light absorption ability can be adjusted by modifying building blocks. In addition, their highly ordered structures and strong interlayer interactions provide unique advantages in separation of photogenerated carriers.86 Therefore, COFs have great application potential in photocatalysis.The preparation strategies of COF nanosheets can be classified into top-down and bottom-up strategies.87 The top-down strategy aims to exfoliate a bulk polymer into single- or multi-layer nanosheets via utilizing external forces to break the weak interlayer interactions. So far, the well-developed exfoliation strategies generally include sonication exfoliation, chemical exfoliation, mechanical exfoliation and self-exfoliation. For example, Dichtel et al. exfoliated a hydrazone-linked COF into bulk quantities of two-dimensional (2D) polymer nanosheets by immersing in several common laboratory solvents (THF, dioxane, MeOH, etc.).88 Tsuru et al. prepared COF-1 nanosheets by ultrasonic exfoliation through dispersing COF-1 powder in dichloromethane.89 Banerjee et al. used a mechanical grinding route to obtain graphene-like layered COF nanosheets.90 The bottom-up methods are usually realized via interfacial synthesis (such as liquid–liquid interface and gas–liquid interface). For example, Zhang et al. prepared an imine-linked covalent organic monolayer at the air–water interface.91 Ma et al. synthesized high-quality COF nanosheets at the interface of two miscible organic solvents.92 It is worth noting that reports of COF nanosheets for photocatalysis are still rare, which may be ascribed to the limited synthesis methods.93–95 Herein, the relevant important developments of COF nanosheets in photocatalysis are discussed.
4.2.1.1 Photocatalytic H2 production. Osakada et al. used a liquid-phase exfoliation method to obtain thin-layered porphyrin-comprised COF nanosheets (denoted as e-CON) in common solvents (dichloromethane, methanol, pyridine, nitrobenzene and H2O).96 Compared with other solvents, the surface energy of H2O (102.8 mJ m−2) was closer to that of bulk porphyrin-comprised COFs (DhaTph) (106.6 mJ m−2), so the porphyrin-comprised COFs could be easily exfoliated in H2O (denoted as e-CON(H2O)). The produced e-CON(H2O) had a thickness of 1–2 nm and a width of 50–60 nm. Using Pt/RGO as the co-photocatalyst and methanol as the sacrificial agent, its photocatalytic H2 yield rate was nearly five times higher than that of unexfoliated DhaTph. Cooper et al. used 3,9-diamino-benzo[1,2-b
:4,5-b′]bis[1]benzothiophene sulfone (FSA) as the monomer to produce FS-COF via a Schiff-base condensation reaction.97 It could be partially exfoliated into thin stacks with thicknesses of 5–25 nm. Using ascorbic acid as the sacrificial agent and Pt as the co-catalyst, its photocatalytic H2 yield rate reached 10.1 mmol g−1 h−1 under visible-light irradiation (λ > 420 nm), which was ∼6.5 times higher that that of TP-COF (synthesised using 4,4′′ diamino-p-terphenyl as the monomer). Besides the longer absorption edge and higher BET surface area, the authors believed that the partial exfoliation of FS-COF also contributed to the improved photocatalytic performance because the nanosheet-like morphology could provide more active sites for hydrogen evolution.98,99
4.2.1.2 Photocatalytic overall water splitting. Lan et al. exfoliated three β-ketoamine COFs (TpBD-COF with biphenyl units, and TpBpy-COF and TpBpy-2-COF with a bipyridine structure) to obtain nanosheets (NSs) with a thickness of 3–4 nm via a “top-down” route, and then in situ incorporated 1.19–1.28 wt% ultra-small Pt nanoparticles (around 1.1 nm) co-catalyst into their open channels (Fig. 3a–c).100 The obtained samples were denoted as Pt@ TpBD-NS, Pt@TpBpy-NS and Pt@TpBpy-2-NS, respectively. Except Pt@TpBD-NS, both Pt@TpBpy-NS and Pt@TpBpy-2-NS exhibited overall water splitting activity. The H2 and O2 evolution amounts in 5 h under visible-light irradiation for Pt@TpBpy-NS were 9.9 and 4.8 μmol, respectively, and those for Pt@TpBpy-2-NS were 3.1 and 1.4 μmol. The different photocatalytic performance indicated that the bipyridine structure and the position of N sites in the heterocyclic ring were important for overall water splitting. Control experiments using bulk COFs without a nanosheet morphology and post-loading Pt on COF NSs confirmed that the nanosheet morphology and ultra-small Pt NPs uniformly distributed in the pores of COFs were key factors for enhancing the water splitting activity. DFT theoretical calculation (Fig. 3d–f) further indicated that the electron transfer from the Bpy to the Tp section was more efficient in TpBpy-NS than that in TpBpy-2-NS, and C2d was the optimal oxygen evolution reaction path.
 | ||
| Fig. 3 (a) β-Ketoamine COFs with the same topology and similar atomic composition. (b) AFM image and (c) TEM image of Pt@TpBpy-NS. (d) The TD-DFT calculated electronic transition of TpBpy-NS. (e) The possible process of the HER on the Tp segment and the OER via a dual-site process on the Bpy segment in TpBpy-NS. (f) The comparison of calculated Gibbs free energy change for C2d paths of the OER for TpBD-NS, TpBpy-2-NS and TpBpy-NS at pH = 7. Reprinted with permission from ref. 100, © The Author(s) 2023. | ||
4.3 CTF photocatalysts
CTFs are composed of covalent and aromatic triazine linkages, possessing high pore density, rich active sites and chemical and thermal stability. Currently, most of the reported CTFs are amorphous or semicrystalline. Although CTFs have an intrinsically 2D layered structure, the π–π interaction between layers generally results in the formation of bulk CTFs. The long charge diffusion distance, abundant defects and boundaries in bulk CTFs seriously restrict photocatalytic performance. In order to synthesize CTF nanosheets, different strategies have been developed, including chemical exfoliation, interfacial polymerization, mechanical exfoliation and monomer-dependent synthesis.101 For example, Fan et al. utilized an acid-base driven intercalation reaction to obtain 1–2 layered CTF nanosheets.102 Xu et al. exfoliated bulk CTFs into micrometer-size few-layer CTF nanosheets with a thickness of 2–3 nm through micromechanical cleavage and liquid sonication.103 Wang et al. used a ball mill strategy to break the interlayer π–π interactions of bulk perfluorinated CTFs and obtain ultrathin nanosheets with a thickness of ∼4.2 nm.1044.3.1.1 Photocatalytic H2 production. Xu et al. prepared layered crystalline CTF powders using terephthalonitrile as the monomer and CF3SO3H as the catalyst.105 The obtained layered crystalline CTFs were treated with sulfuric acid so that they were exfoliated into nanosheets containing amide groups and oxygen-containing groups (o-CTFNSs) through a rapid exothermic effect formed by adding water into the mixture containing sulfuric acid. Finally, these oxygen-containing groups were reduced using hydrazine hydrate to obtain amide-functionalized CTF nanosheets with an ultrathin thickness of ∼1.5 nm (Fig. 4a–c). The amide functional groups served as electron donors, which could adjust the band gap and promote charge separation. The photocatalytic H2 yield rate of the amide-functionalized CTF nanosheets could reach 512.3 μmol h−1 under visible-light irradiation, which was around 17 times higher than that of pristine layered crystalline CTFs (Fig. 4d). More importantly, the amide-functionalized CTF nanosheets have an apparent quantum yield of 11.3% at 420 nm and excellent photostability for photocatalytic H2 evolution. Afterwards, Xu's group focused on scalable production of high-quality CTF nanosheets for photocatalytic H2 evolution.106,107 For example, CF3SO3H catalysts were employed to absorb microwave energy, which could exclusively transfer energy to the nitrile-based monomers (1,3,5-tricyanobenzene, 1,4-dicyanobenzene and 4,4′-dicyano-1,1′-biphenyl) under solvent-free conditions to facilitate a continuous cyclotrimerization reaction (Fig. 4e).106 The microwave-assisted synthetic route can obtain a series of highly crystalline layered CTFs with the hundred-gram level in a short time (within 20 minutes). Abundant crystalline single-layer/few-layer CTF nanosheets with a thickness of 0.6–3 nm could be obtained after simple ball-milling exfoliation of the above bulk layered crystalline CTFs (Fig. 4g and h). The photocatalytic hydrogen evolution rate of the CTF nanosheets reached up to 7971 μmol g−1 h−1, which was nearly five times higher than that of bulk CTFs. Such high photocatalytic performance could be ascribed to the more accessible active sites and higher carrier separation efficiency of exfoliated nanosheets. In the subsequent research, bulk layered crystalline CTFs were further homogeneously mixed with a sodium 1-pyrenebutyrate (PB) aqueous solution.107 The PB molecules could spontaneously adsorb on the surface and interlayer edges of the bulk CTF, and the introduced negative charges enhanced interlayer repulsion of the CTF layers (Fig. 4i). Under mechanical ball-milling treatment, the formed shear force slipped the 2D triazine layers of the bulk CTFs into CTF nanosheets (PB-modified 2D-TP) on a large scale in water (Fig. 4j). The adsorbed PB caused a molecular doping effect on the CTF nanosheets, resulting ineffective carrier separation. The interaction between 2D-TP and PB formed a strong built-in electric field, which also accelerated photogenerated charge separation and transfer. Therefore, the photocatalytic H2 yield rate of the PB-modified 2D-TP nanosheets (50 mg dosage) reached 1249 μmol h−1 under visible-light irradiation, which was 24-fold improvement in comparison with that of the bulk CTFs (Fig. 4k). In addition, they found a new solvent-free salt-catalyzed nitrile cyclotrimerization process, which could result in the formation of atomic-thin CTF nanosheets on a large scale (Fig. 5a).108 Common salts (KCl, etc.) could not only facilitate the formation of a nanosheet morphology but also act as a kind of green solid catalyst to catalyze the cyclotrimerization reaction of carbonitrile monomers. The obtained crystalline CTF nanosheets (CTF NSs) with a thickness of ∼80 nm could be further exfoliated into single-layer crystalline 2D triazine polymers (2D-TPs) with a thickness of ∼0.6 nm via simple liquid ultrasonication. The obtained atomic-thin 2D-TPs with a suitable band structure and rich triazine active groups exhibited an excellent photocatalytic H2 evolution rate (1321 μmol h−1) under visible-light irradiation.
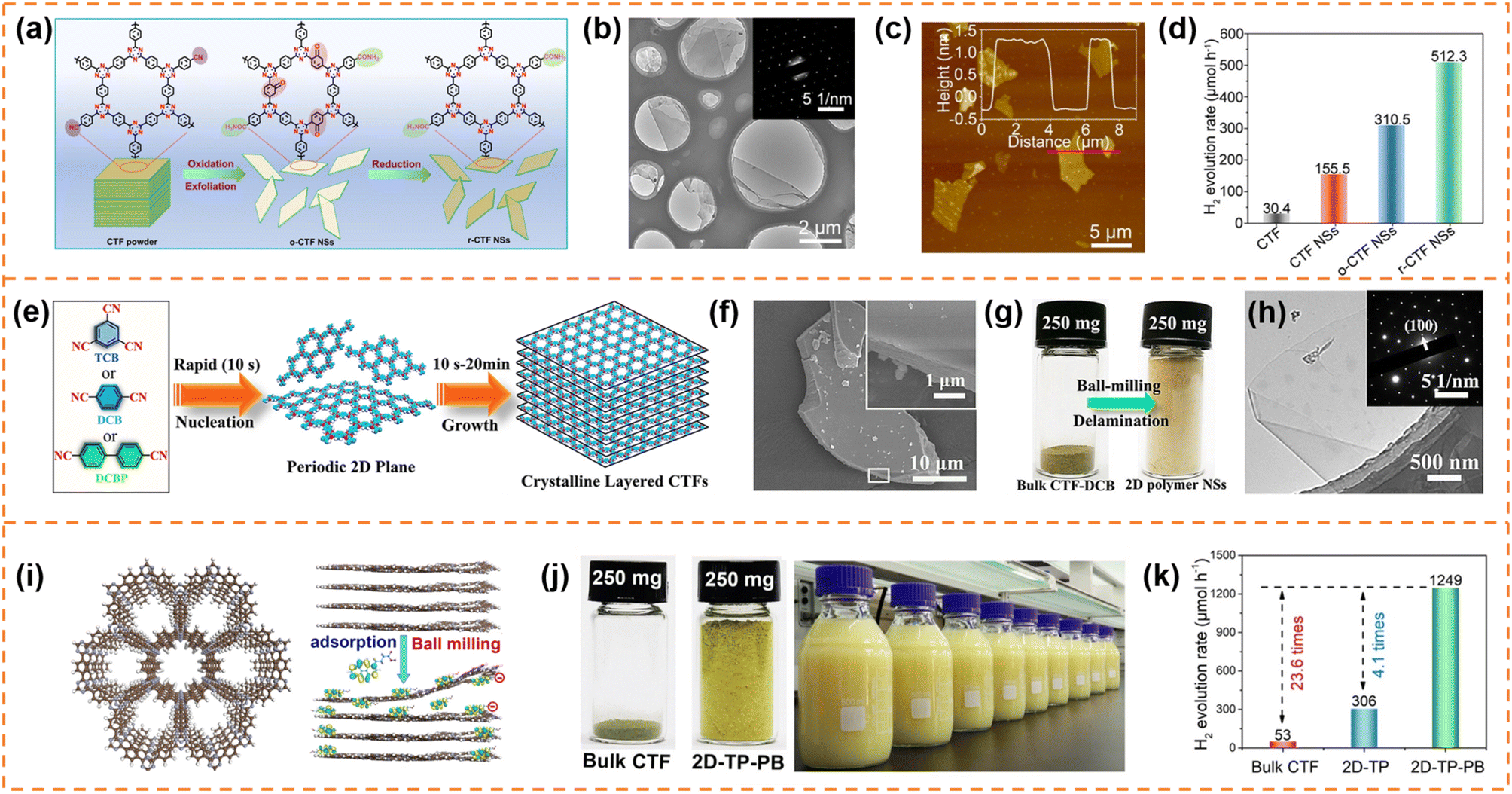 | ||
| Fig. 4 (a) Schematic illustration of the redox exfoliation process. TEM image (b) and AFM image (c) of r-CTF NSs. (d) Photocatalytic H2 evolution rates of the obtained samples. Reprinted with permission from ref. 105, © Wiley-VCH GmbH 2021. (e) The rapid synthetic route and polymerization mechanism. (f) SEM image of layered crystalline CTF-DCB. (g) Photograph of bulk CTF-DCB and the exfoliated 2D triazine polymer NSs. (h) TEM image of 2D triazine polymer NSs. Reprinted with permission from ref. 106, © Wiley-VCH GmbH 2022. (i) Schematic illustration of the adsorption of PB molecules on the surface of the CTF layer and the simultaneous exfoliation process. (j) The photograph of the bulk CTF and the corresponding exfoliated 2D-TP-PB, and scalable aqueous dispersion of 2D-TP-PB nanosheets with a concentration of 5 mg mL−1. (k) Photocatalytic hydrogen evolution rate comparison at a photocatalyst loading of 50 mg. Reprinted with permission from ref. 107, © Wiley-VCH GmbH 2023. | ||
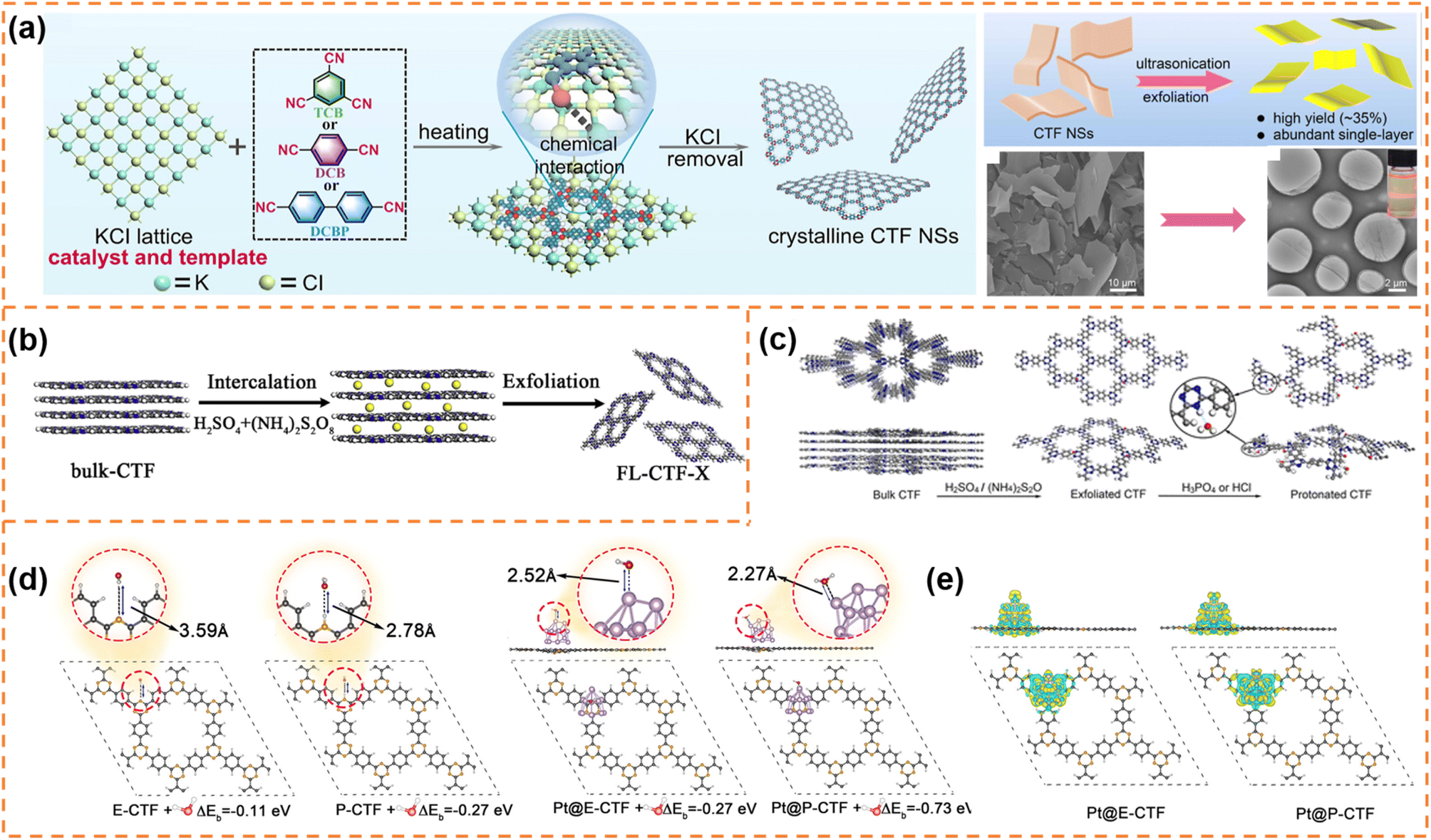 | ||
| Fig. 5 (a) Proposed salt-catalytic method based on the chemical interaction between the salt lattice and carbonitrile monomers during the polymerization process, SEM image of CTF-DCB NSs and TEM image of 2D-TPs with the inset showing the Tyndall effect of 2D-TP dispersion. Reprinted with permission from ref. 108, © American Chemical Society 2023. (b) Illustration of the preparation of exfoliated CTF nanosheets. Reprinted with permission from ref. 109, © Elsevier B.V. 2020. (c) Schematic diagram of protonation modification of CTF-1. (d) Optimized configurations for E-CTF-1, P-CTF, Pt@E-CTF and Pt@P-CTF after binding with a H2O molecule. (e) Differential charge density for Pt@E-CTF and Pt@P-CTF. Reprinted with permission from ref. 110, © American Chemical Society 2022. | ||
Fan et al. synthesized few-layer CTF nanosheets using H2SO4 and (NH4)2S2O8 as an intercalant and mild oxidant, respectively, to intercalate and exfoliate bulk CTFs (Fig. 5b).109 The defect states introduced by exfoliation could improve visible-light absorption and suppress the radiative electron–hole recombination. Besides, the ultrathin structure could shorten the diffusion length of the photoexcited carriers and expose more active sites. Under optimum conditions, the exfoliated CTF nanosheets exhibited a high H2 production rate (1527 μmol h−1 g−1) under visible-light irradiation, which was much better than that of bulk CTFs. Afterwards, Fan et al. firstly obtained thin CTF-1 nanosheets (denoted as E-CTF) using the above exfoliation method, and then obtained protonated CTF-1 nanosheets (P-CTF) through the reaction of E-CTF with H3PO4 (Fig. 5c).110 The protonation could improve the interaction among the P-CTF, H2O molecules and Pt co-catalyst, thus facilitating the transfer of the photogenerated electrons from the Pt co-catalyst to the H2O (Fig. 5d and e). Therefore, in the presence of a Pt (3 wt%) co-catalyst and triethanolamine sacrificial agent, the photocatalytic H2 production rate of P-CTF (6595 μmol g−1 h−1) under AM 1.5 irradiation was higher than that of E-CTF (3972.5 μmol g−1 h−1) and bulk CTF-1 (810 μmol g−1 h−1).
4.3.1.2 Photocatalytic O2 production. The amide-functionalized CTF nanosheets prepared by Xu et al. also showed a better photocatalytic O2 production rate (12.37 μmol h−1) than the corresponding bulk CTFs (0.52 μmol h−1) under visible-light irradiation (Co2+ and AgNO3 served as a co-catalyst and sacrificial agent, respectively).105 Besides the advantages of amide functional groups and morphology, the more positive VBM potential of the amide-functionalized CTF nanosheets resulted in adequate thermodynamic driving force, which was also an important reason for the enhanced photocatalytic water oxidation performance.
4.3.1.3 Photocatalytic overall water splitting. High crystallinity and hydrophilicity are the key factors to obtain photocatalytic overall water splitting activity. Besides the amide-functionalized CTF nanosheets and atomically thin crystalline 2D-TPs prepared by Xu et al.105,108 the crystalline CTF nanosheets with high hydrophilicity (denoted as CTF-HUST-A1) prepared by Jin et al. also exhibited photocatalytic overall water splitting activity.111 The crystalline CTF-HUST-A1 nanosheets were synthesized using p-xylylenediamine as the monomer and a strong base as the catalyst. Among various base reagents (K2CO3, KOH, EtOK and tBuOK), the CTF-HUST-A1 synthesized in tBuOK (denoted as CTF-HUST-A1-tBuOK) showed the highest crystallinity and BET surface area (644 m2 g−1). The CTF-HUST-A1-tBuOK nanosheets had a thickness of around 5 nm. Structures analysis showed that the –C
O groups (formed by the hydrolysis of the –CN– groups or the oxidation of the benzylamine groups under alkaline conditions) and the –C–O–H groups (formed from the –C–NH2 groups or by further hydrolysis of the –CO or –CNH groups under alkaline conditions) were the major reason for the good hydrophilicity. In addition, the high surface area and the layer structure might also partially contribute to the high hydrophilicity. The high crystallinity could inhibit the recombination of photogenerated charge carriers, the thin nanosheets could provide a shortcut for the photo-generated charges to the surface and the high hydrophilicity could promote interactions between the CTF nanosheets and water. Their synergistic roles resulted in a good overall water splitting activity (25.4 mmol g−1 h−1 and 12.9 mmol g−1 h−1) under visible-light irradiation (λ > 420 nm) of CTF-HUST-A1-tBuOK with 4.5 wt% NiPx and 3.0 wt%Pt as dual cocatalysts.
4.4 Carbon nitride photocatalysts
Carbon nitrides are star semiconductors in the field of photocatalysis due to the advantages of easy preparation, low cost, non-toxicity, stable physical and chemical properties and suitable band structure. Carbon nitrides have different allotropes, among which graphitic carbon nitride (g-C3N4) is the most stable.112,113 Recently, g-C3N4 with an amorphous, semi-crystalline or highly crystalline structure has been widely reported. Since Wang's pioneering report showed that g-C3N4 possessed photocatalytic H2 and O2 production activity,114 it has been extensively studied in different photocatalytic applications (CO2 reduction, pollutant degradation, hydrogen peroxide production, etc.).115–118 However, bulk g-C3N4 exhibits low charge carrier separation efficiency and a small surface area, resulting in poor photocatalytic activity.119,120 Therefore g-C3N4 photocatalysts with different morphologies (nanosheets,121,122 hollow spheres,123,124 nanofibers125 and nanotubes126,127) have been developed to enhance photocatalytic performance. Among them, g-C3N4 nanosheets have attracted much attention due to the advantages of a high specific surface area, short diffusion length of photogenerated carriers and abundant accessible active sites.113 g-C3N4 has been extensively reviewed in the field of photocatalysis.53,112,128–135 Here, we provide a brief summary of the photocatalytic properties of exfoliated ultrathin g-C3N4 nanosheets.4.4.1.1 Photocatalytic H2 production. Fang et al. used a liquid-phase ultrasonic exfoliation method to prepare mesoporous few-layer g-C3N4 nanosheets.136 Compared with bulk g-C3N4, the g-C3N4 nanosheets possessed a higher specific surface area, abundant mesopores and a narrower bandgap. Therefore a high hydrogen evolution rate (97.6 μmol h−1 g−1) of the g-C3N4 nanosheets under visible-light irradiation was achieved, which was 3.3 times higher than that of bulk g-C3N4. Kang et al. prepared g-C3N4 nanosheets with a thickness of ∼4 nm by the hydrothermal exfoliation method (NaClO as the exfoliation agent) (Fig. 6a).137 The simultaneous Na+ intercalation and ClO− oxidation resulted in the formation of abundant in-plane holes on g-C3N4 nanosheets, which accelerated the cross-plane diffusion of photogenerated carriers. Thanks to the inherent advantages of exfoliated nanosheets and the holey structure, the hydrogen evolution rate of the g-C3N4 nanosheets reached 0.89 mmol g−1 h−1, which was 9 times higher than that of bulk g-C3N4. The photocatalytic activity can be further enhanced by introducing element doping during the preparation of ultrathin nanosheets. For example, Xu et al. prepared ultrathin g-C3N4 nanosheets co-doped with S, P and O non-metal atoms by a thermal-assisted exfoliation method (Fig. 6b).138 The exfoliation resulted in an increase in the specific surface area (almost 2 times that of bulk g-C3N4) and more exposure of active sites, which was beneficial for photocatalysis. Upon elemental doping, S atoms occupied the interstitial sites, and P and O atoms replaced the C and N atoms, respectively, resulting in the formation of an O–P–C–N2–S–N2′ (or N2′–S–N2–C1–P–O) chain between two adjacent heptazine units (Fig. 6c). The chain as a more favorable charge transfer pathway significantly enhanced photo-induced charge transfer and separation. Therefore, the multi-atom doping and ultrathin nanosheet morphology remarkably enhanced the photocatalytic H2 production rate (2480 μmol g−1 h−1) under visible-light irradiation, which was about 5.3 times that of pure g-C3N4. Zhang et al. firstly prepared ultrathin g-C3N4 nanosheets doped with O atoms (denoted as O–CNS) by the oxygen etching method, and then incorporated P into the basal plane of g-C3N4 (Fig. 6d–g). Finally, oxygen and phosphorus dual-doped ultrathin g-C3N4 nanosheets (denoted as O, P–CNS) with a thickness of about 2.5–3.0 nm were obtained.139 The O, P–CNS with enhanced electronic conductivity and an activated basal plane exhibited a superior H2 generation rate of 8.7 mmol g−1 h−1 under visible-light irradiation.
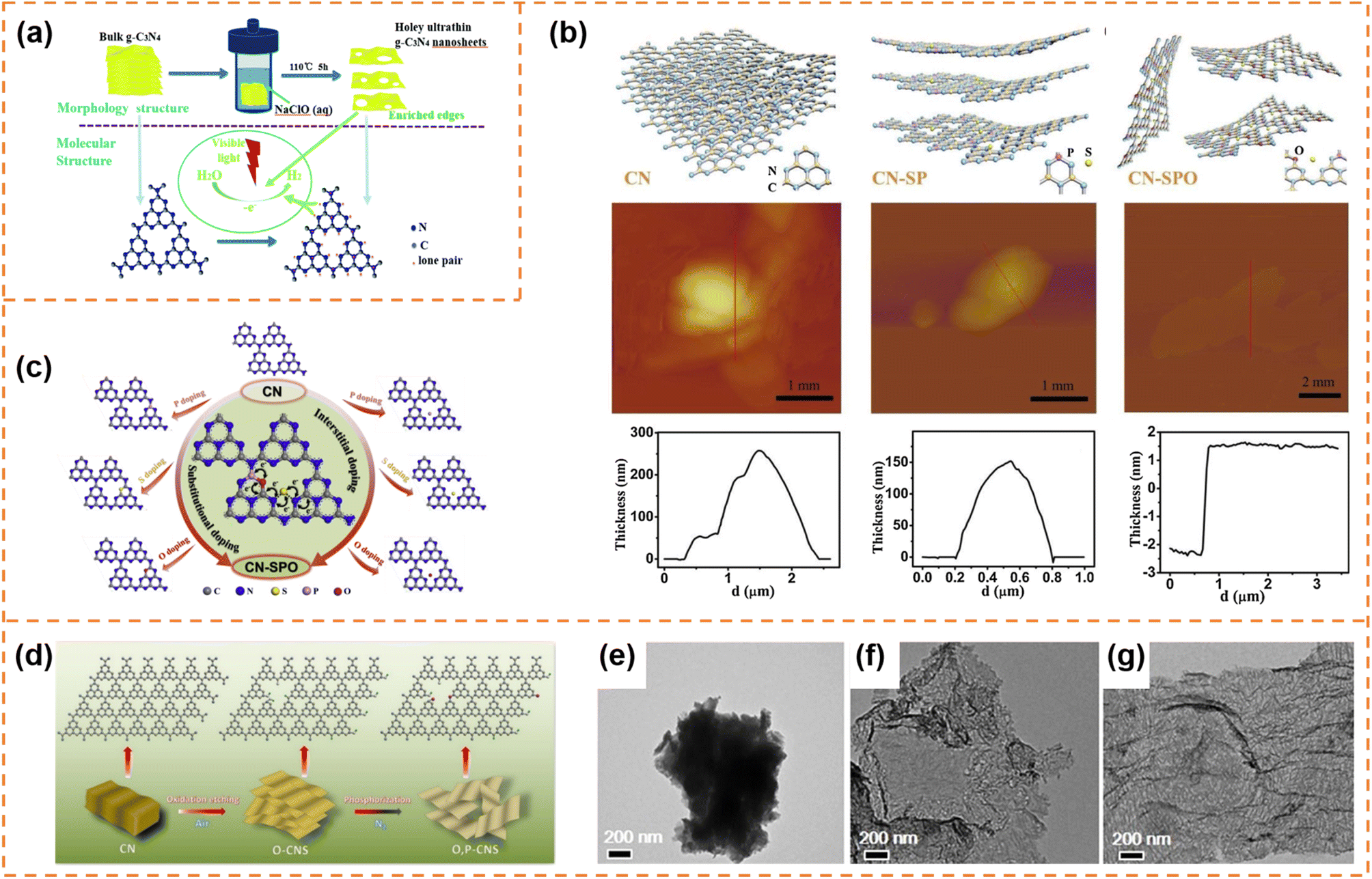 | ||
| Fig. 6 (a) Schematic illustration of the NaClO (aq) hydrothermal exfoliation of bulk g-C3N4 into single-layer nanosheets for the photocatalytic H2 evolution. Reprinted with permission from ref. 137, © Royal Society of Chemistry 2018. (b) Schematic illustration of the exfoliation procedure of g-C3N4 from CN to CN-SP and CN-SPO, and corresponding AFM images and height profile. (c) Schematic illustration of P (or S/O) doped monolayer g-C3N4 atomic structures with substitutional-doped sites (left), interstitial-doped sites (right) and the pathway for electron transport (middle). Reprinted with permission from ref. 138, © Elsevier B.V. 2019. (d) Fabrication process of O, P–CNS. (e–g) TEM images of CN, O–CNS and O, P–CNS. Reprinted with permission from ref. 139, © American Chemical Society 2020. | ||
4.4.1.2 Photocatalytic overall water splitting. Bao et al. used a femtosecond pulsed laser to exfoliate bulk g-C3N4 into ultrathin nanosheets with a thickness of ∼2 nm (Fig. 7a).140 The femtosecond laser pulses broke the strong C–N covalent bonds and cleaved the s-triazine units as well, forming abundant –C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N defects on the ultrathin g-C3N4 nanosheet surface. These cyano (–CN) defects not only favored the anchoring of Pt atoms but also shifted down the band edge positions. Meanwhile, the atomically dispersed Pt provided abundant active sites for the surface reaction and inhibited the back-oxidation of H2. In addition, the more positive valence band edge position enhanced the water oxidation ability of the holes. Therefore the laser-exfoliated g-C3N4 nanosheets with Pt cocatalysts achieved overall water splitting activity (42.6 μmol g−1 h−1 for H2 and 18.7 μmol g−1 h−1 for O2; Fig. 7b).
N defects on the ultrathin g-C3N4 nanosheet surface. These cyano (–CN) defects not only favored the anchoring of Pt atoms but also shifted down the band edge positions. Meanwhile, the atomically dispersed Pt provided abundant active sites for the surface reaction and inhibited the back-oxidation of H2. In addition, the more positive valence band edge position enhanced the water oxidation ability of the holes. Therefore the laser-exfoliated g-C3N4 nanosheets with Pt cocatalysts achieved overall water splitting activity (42.6 μmol g−1 h−1 for H2 and 18.7 μmol g−1 h−1 for O2; Fig. 7b).
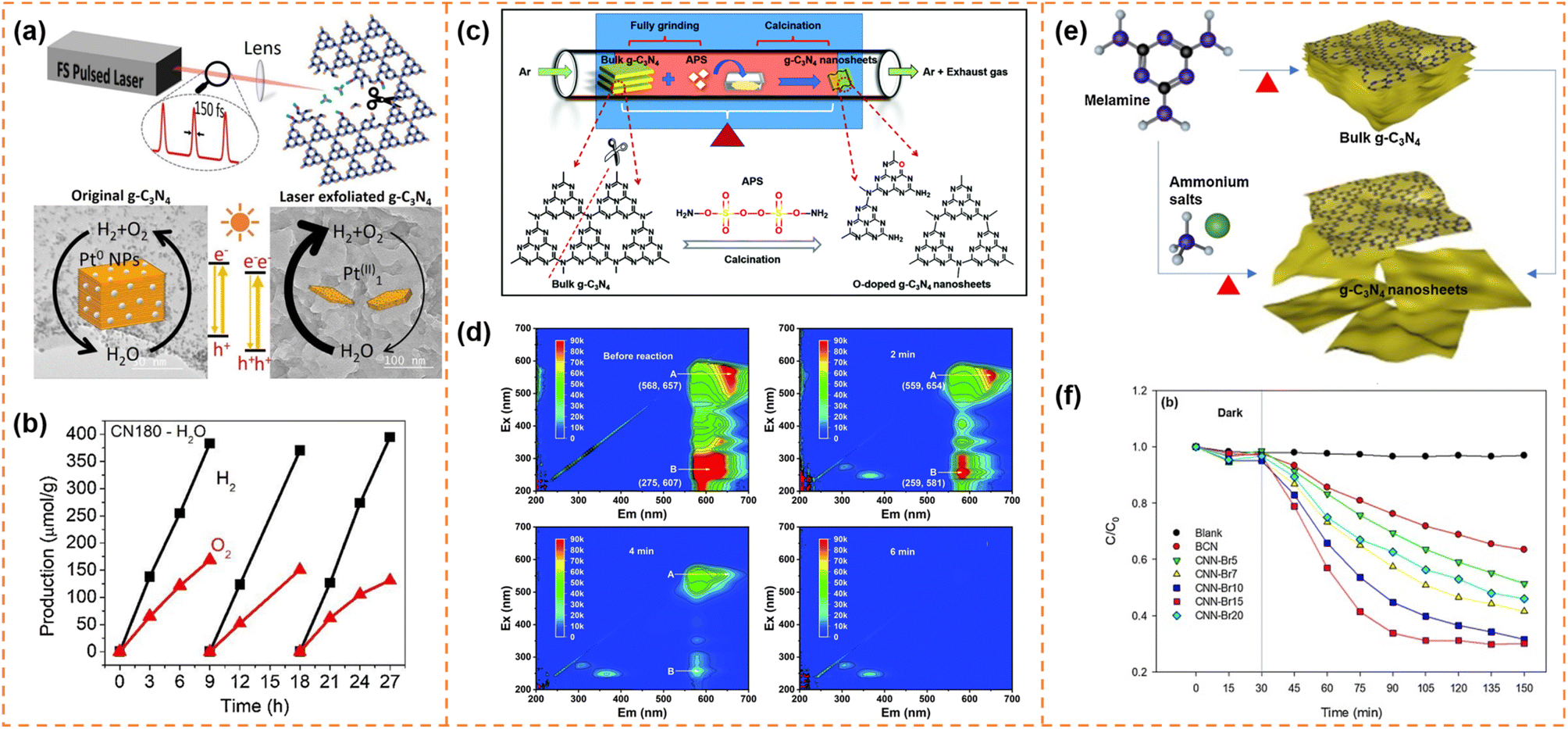 | ||
| Fig. 7 (a) Illustration of the formation process of g-C3N4 nanosheets via the femtosecond laser pulse method and their photocatalytic mechanism. (b) The photocatalytic overall water splitting of CN180 with ∼1.4wt% Pt as the cocatalyst. Reprinted with permission from ref. 140, © Elsevier B.V. 2021. (c) Illustration of the formation process of O-doped g-C3N4 nanosheets (OCNs). (d) EEM fluorescence spectra of the residual RhB solution before reaction and after reaction for 2 min, 4 min and 6 min, respectively. Reprinted with permission from ref. 144, © Royal Society of Chemistry 2022. (e) Synthesis of g-C3N4 nanosheets through post-treatment and one-step exfoliation using ammonium salts. (f) Photocatalytic degradation of OTC with BCN and CNN-Brx samples under visible-light irradiation. Reprinted with permission from ref. 145, © Elsevier B.V. 2019. | ||
Due to the quantum size effect, the bandgap of g-C3N4 nanosheets was generally widened after exfoliation. Exfoliation-induced element doping was an efficient method to enhance visible-light absorption ability and photodegradation activity. For example, Li et al. introduced (NH4)2S2O8 (APS) into the thermal exfoliation approach to prepare O-doped g-C3N4 nanosheets (Fig. 7c).144 The method realized the synchronization control of the electronic structure and morphology. Due to the enlarged specific surface area, more accessible active edges, narrowed bandgap and efficient carrier separation, the O-doped g-C3N4 nanosheets showed 1.6 times higher photocatalytic RhB degradation activity than bulk g-C3N4. More importantly, RhB was completely decomposed into small inorganic salt molecules (Fig. 7d). Kim et al. used ammonium salts (NH4Br) and melamine as raw materials to prepare Br-doped g-C3N4 nanosheets via a one-step exfoliation method (Fig. 7e).145 Br doping increased the visible-light absorbance and inhibited photogenerated carrier recombination, which resulted in an increase in the degradation performance of OTC antibiotics from 30% of bulk g-C3N4 to 76% of Br-doped g-C3N4 nanosheets under visible-light irradiation for 2 h (Fig. 7f).
5. Polymer nanosheet-based heterostructures for photocatalysis
Although single-component polymer nanosheets have displayed conspicuous advantages in the field of photocatalysis, the rapid carrier recombination rate still seriously limits their photocatalytic activity, especially in the photocatalytic overall water splitting and CO2 reduction. Recently, polymer nanosheet-based heterostructures are designed and prepared, which reveal remarkably elevated photocatalytic performance.5.1 CMP nanosheet-based photocatalysts
5.1.1.1 Photocatalytic overall water splitting. Xu et al. synthesized ultrathin aza-CMP and C2N nanosheets by the liquid-phase exfoliation method.147 aza-CMP/C2N van der Waals heterostructures were obtained via annealing the mixture of the two different nanosheets. Their electronic band structures resulted in the formation of Z-scheme photocatalytic systems, in which the aza-CMP and C2N served as O2- and H2-evolving catalysts, respectively (Fig. 8a). The heterostructures facilitated photogenerated charge separation and transfer. When the mass ratio of aza-CMP and C2N was 1
:1, a high STH conversion efficiency (0.23%) was obtained. In contrast, the heterostructures formed from bulk aza-CMP and bulk C2N did not exhibit overall water splitting activity, indicating that the ultrathin sheet-like structure of heterostructures were beneficial for charge utilization. In order to further enhance photocatalytic activity, reduced graphene oxide (RGO) sheets were used as good solid electron mediators to shuffle electrons from aza-CMP nanosheets to C2N nanosheets (Fig. 8b). The photocatalytic overall water splitting rate under visible light irradiation (Fig. 8c) and STH conversion efficiency (0.40%) of the aza-CMP/RGO/C2N heterojunction were nearly twice as much as those of aza-CMP/C2N. When Pt and Co(OH)2 cocatalysts were loaded onto C2N and aza-CMP nanosheets, respectively, the STH conversion efficiency of the aza-CMP/RGO/C2N heterojunction reached up to 0.73%, highlighting the potential of the ultrathin polymer nanosheets for achieving high overall water splitting activity.
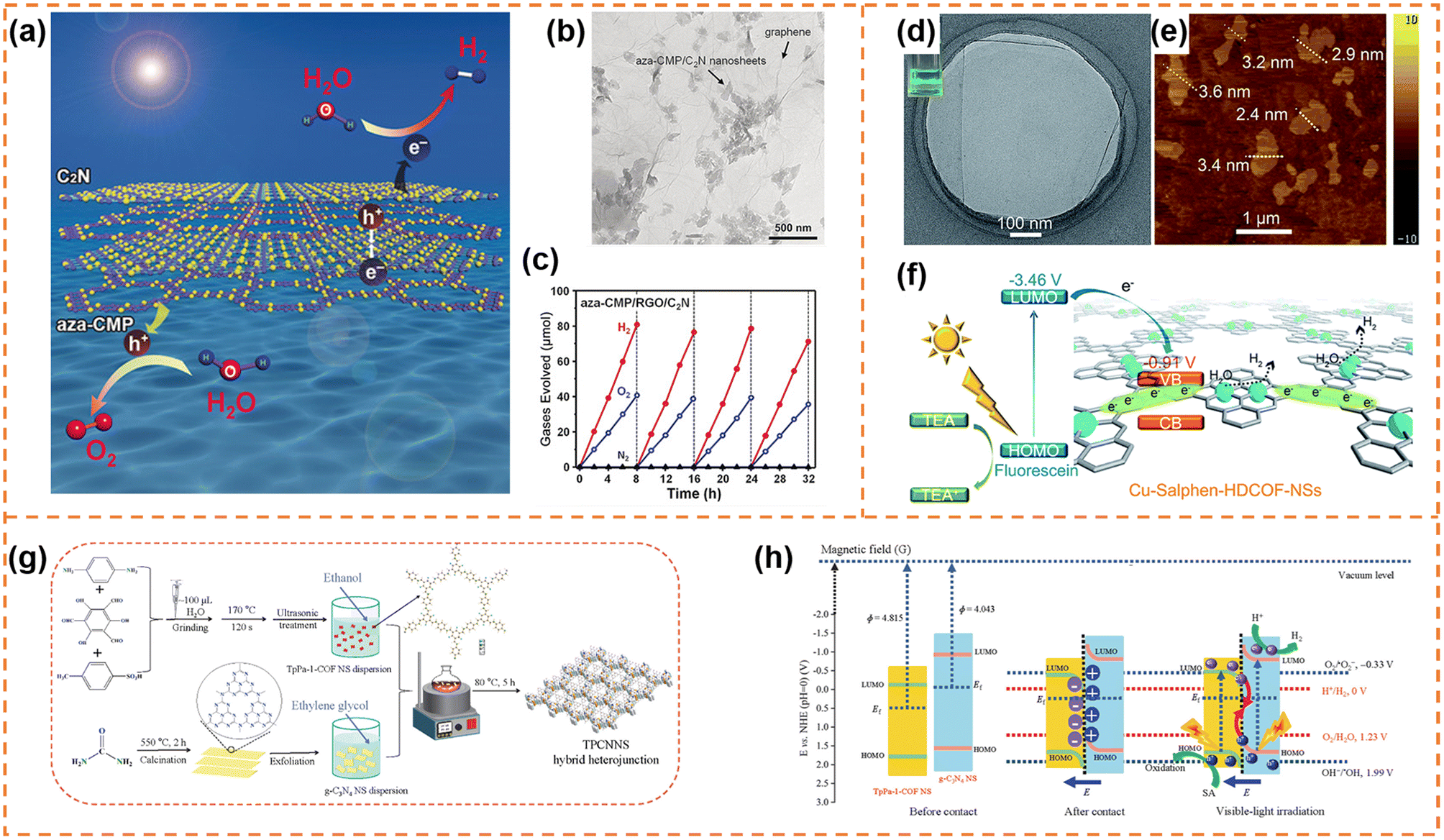 | ||
| Fig. 8 (a) Illustration of the Z-scheme structure of aza-CMP and C2N nanosheets. (b) TEM image of the aza-CMP/RGO/C2N photocatalyst. (c) Typical time course of H2 and O2 production using the aza-CMP/RGO/C2N photocatalyst under visible-light irradiation. Reprinted with permission from ref. 147, © Wiley-VCH GmbH 2018. (d) HR-TEM and (e) AFM images of Cu-salphen-HDCOF-NSs. (f) Schematic of the charge dynamics and hydrogen production mechanism of Cu-salphen-HDCOF-NSs. Reprinted with permission from ref. 148, © Royal Society of Chemistry 2020. (g) Preparation of the TpPa-1-COF/g-C3N4 NS (TPCNNS) hybrid heterojunction. (h) Schematic explanation of the charge migration mechanism in the S-scheme TPCNNS heterostructure before contact, after contact in darkness, and irradiated with visible light. Reprinted with permission from ref. 149, © Elsevier B.V. 2022. | ||
5.2 COF nanosheet-based photocatalysts
5.2.1.1 Photocatalytic H2 production. Zang et al. firstly used 2,3,6,7,10,11-hexaiminotriphenylene (HATP·6HCl), 2,6-diformylphenol (DFP) and Cu(OAc)2 as raw materials to synthesize bulk binuclear Cu-salphen-HDCOF. And then, facile solvent-assisted liquid sonication was adopted to exfoliate it into Cu-salphen-HDCOF nanosheets with a thickness of around 2.4–3.6 nm (Fig. 8d and e),148 in which the isolated Cu served as the active site. The synergistic effect of the π-conjugated framework of the COF and sufficient Cu active sites not only efficiently increased the access of photogenerated charges to the redox reaction centers on the surface of the photocatalyst, but also inhibited photocarrier recombination (Fig. 8f). In addition, the nanosheet morphology was beneficial for enhancing the number of exposed Cu active sites. Therefore, in the presence of the TEA sacrificial reagent and fluorescein photosensitizer, the Cu-salphen-HDCOF nanosheets exhibited an excellent H2 evolution rate (36.99 mmol g−1 h−1) under visible-light irradiation, which was much higher than that of bulk Cu-salphen-HDCOF and salphen-HDCOF nanosheets. Xi et al. employed β-ketoenamine-linked TpPa-1-COF nanosheets and g-C3N4 nanosheets to construct a π–π conjugated S-scheme heterojunction (Fig. 8g and h).149 The formed built-in electric field enhanced charge separation and utilization. When the mass ratio of TpPa-1-COF nanosheets to g-C3N4 nanosheets was 2
:1, the highest photocatalytic hydrogen evolution rate (1153 μmol g−1 h−1) was obtained under visible-light irradiation, which was approximately 2.8 and 4.2 times as high as that for the TpPa-1-COF nanosheets and the g-C3N4 nanosheets, respectively. The TpPa-1-COF nanosheets were also employed by Shi et al. to construct a heterojunction with TiO2 nanosheets via a solvothermal strategy.150 Different from the above S-scheme heterojunction, the TpPa-1-COF nanosheets were used as the photosensitizer in the 2D/2D COF-TpPa-1/TiO2 heterojunction. The photoinduced electrons were transferred from COF-TpPa-1 to TiO2 under visible-light irradiation due to their different band edges. The intimate hetero-interface contact between them enhanced charge migration. Therefore, the COF-TpPa-1/TiO2 heterojunction with a mass ratio of 14/6 achieved the highest hydrogen evolution rate (1.37 mmol g−1 h−1) under visible-light irradiation, which was ∼29 times as high as that of COF-TpPa-1.
In addition, COF nanosheets also can serve as a special modifier to improve the photocatalytic H2 yield rate of other semiconductors. For example, Yan et al. prepared a CNS–COF nanosheet heterojunction via the reaction of graphitic carbon nitride nanosheets (CNS) with 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl) trianiline (TTA) and 1,3,5-tri-formylphloroglucinol (TP). In the heterojunction, the percentage of COF nanosheets was below 2%.151 The small amount of COF nanosheets integrated with the CNS through chemical imine bonding, which not only significantly passivated the surface termination of CNS, but also enhanced charge separation due to the strong electronic coupling between them. Under optimum conditions, the CNS–COF nanosheet heterojunction could achieve a H2 yield rate of 46.4 mmol g−1 h−1 under visible-light irradiation, which was much better than that of the CNS itself.
162 μmol h−1 g−1) and CO selectivity (ca. 78%) under visible-light irradiation (λ ≥ 420 nm). Its CO2-to-CO conversion rate was ∼82 times that of bulk COF-367-Co, highlighting the significant advantage of COF nanosheets in photocatalytic CO2 reduction. More importantly, the CO evolution rate and selectivity for COF-367-Co nanosheets showed almost no decline after six successive cycles (Fig. 9b). Different from one step synthesis of a Co-containing COF by Jiang et al., Copper et al. firstly synthesized FPy-COF via a Schiff base condensation reaction, and then ultrasonicated it in acetonitrile containing CoCl2 to obtain Co-loaded COF nanosheets (denoted as Co-FPy-CON).153 Co-FPy-CON had a thickness of 1.9–4.4 nm and a BET surface area of 238 m2 g−1. Its fluorinated moieties could polarize CO2, resulting in high CO2 adsorption ability. However, it exhibited very low CO2 reduction activity in the presence of a hole scavenger. When the (Ir[dF(CF3)-ppy]2(dtbpy))PF6 dye was used as the photosensitizer (Fig. 9c), the new system worked efficiently, which could produce 10.1 μmol CO over 6 h with a selectivity of 76% under visible-light irradiation (Fig. 9d). The reason was that Co-FPy-CON served as a semiconductor support (transferring charge carriers generated in the dye to the Co centers) rather than as a photocatalyst itself. Its excellent selectivity, durability and cyclability were comparable to those of the most advanced visible-light driven polymers.
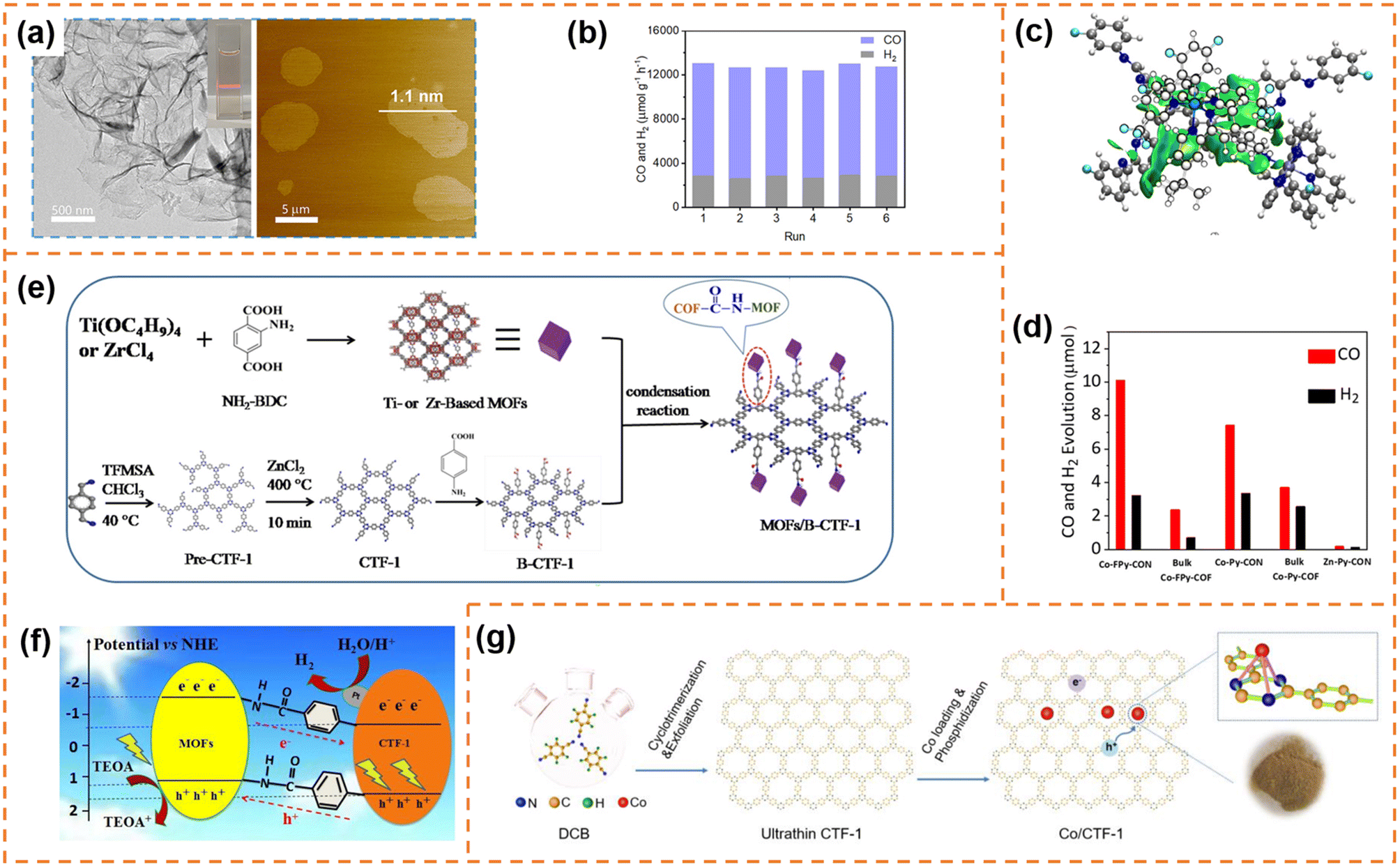 | ||
| Fig. 9 (a)TEM and AFM images of the as-prepared COF-367-Co NSs. (b) Stability tests of COF-367-Co NSs for the photocatalytic reduction of CO2. Reprinted with permission from ref. 152, © American Chemical Society 2019. (c) DFT-optimized adsorption configuration of the dye on the pyrene moiety of Co-FPy-COF. (d) CO and H2 production by the nanosheet and bulk Co-FPy-COF, Co-Py-COF, and Co-Bp-COF, over 6 h under visible light irradiation. Reprinted with permission from ref. 153, © American Chemical Society 2020. (e) Schematic illustration of the formation of the TBC hybrid material. (f) Photocatalytic mechanism of the charge transfer for hydrogen evolution over 15TBC under visible light irradiation. Reprinted with permission from ref. 154, © Elsevier B.V. 2019. (g) Schematic illustration of the preparation process of Co/CTF-1. Reprinted with permission from ref. 155, © Elsevier B.V. 2022. | ||
5.3 CTF nanosheet-based photocatalysts
5.3.1.1 Photocatalytic H2 production. Zou et al. firstly prepared benzoic acid-functionalized CTF-1 nanosheets (denoted as B-CTF-1) by the chemical grafting modification method, and then constructed a type II heterojunction with amino-functionalized Ti-based NH2-MIL-125(Ti) using a step-by-step post-synthetic covalent modification method (Fig. 9e).154 The two components were tightly bound through amide bonds. More importantly, the photogenerated electrons were transferred from the CB of NH2-MIL-125(Ti) to the CB of B-CTF-1 through the amide bridge bonds. Therefore, the amide bonds as a charge transfer mediator promoted photogenerated carrier separation and improved the stability of the photocatalyst (Fig. 9f). Under optimum conditions, the formed heterojunction exhibited a H2 production rate of 360 μmol h−1 g−1 under visible-light irradiation, which was more than twice as high as that of B-CTF-1.
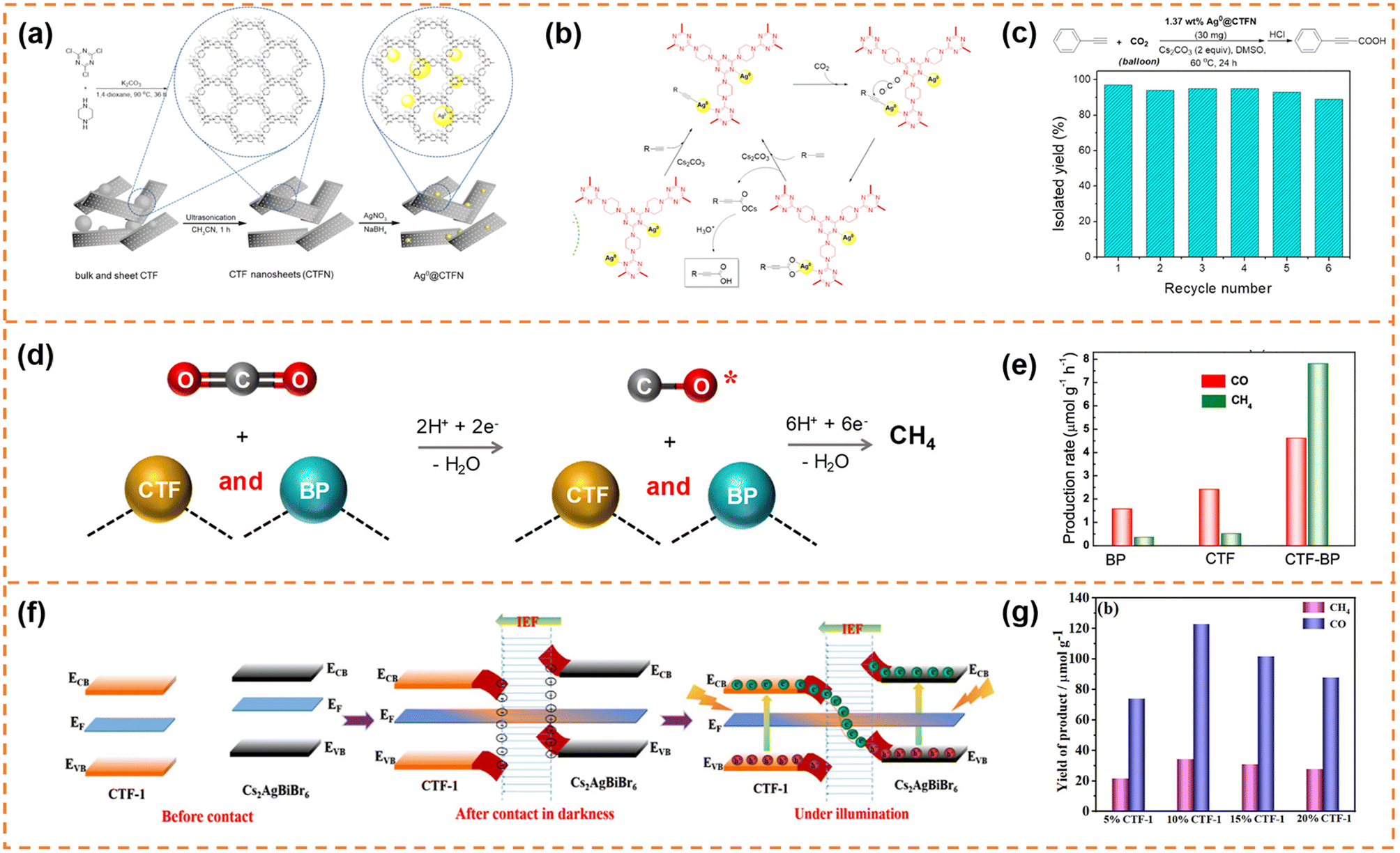 | ||
| Fig. 10 (a) Illustration of the design and preparation procedure of Ag0@CTFN nanocomposites. (b) A proposed mechanism for the carboxylation of terminal alkynes with CO2. (c) Reuse of the Ag0@CTFN catalyst. Reprinted with permission from ref. 156, © American Chemical Society 2018. (d) Photocatalytic reaction mechanism over the CTF-BP heterostructured catalyst. (e) Averaged production rates of CO and CH4. Reprinted with permission from ref. 157, © American Chemical Society 2020. (f) Schematic illustration of the S-scheme charge transfer mechanism between CTF-1 NSs and Cs2AgBiBr6 NSs. (g) CO2 photoreduction performance of the CABB/CTF-1 hybrids with different contents of CTF-1 after 4 h of visible illumination. Reprinted with permission from ref. 158, © American Chemical Society 2022. | ||
The self-assembly method was also a commonly used method to prepare CTF nanosheet-based heterojunctions for CO2 photoreduction.157,158 For example, Zhong et al. synthesized black phosphorus (BP)/CTF nanosheets (denoted as CTF-BP) with a thickness of ∼1.2 nm.157 The strong interaction between CTF and BP resulted in fast charge transfer, and the CO production rate of CTF-BP under visible-light irradiation was 3 and 2 times higher than that of BP and CTF, respectively. More importantly, the strong interaction between CO and CTF-BP could prevent CO desorption from the photocatalyst surface, which was beneficial for photoreducing CO2 to CH4via an 8-electron process (Fig. 10d). Therefore, the CTF-BP catalyst exhibited a high CH4 production rate of 7.81 μmol g−1 h−1 under visible-light irradiation, which was 23 and 16 times higher than that of BP and CTF, respectively (Fig. 10e). Xu et al. prepared Cs2AgBiBr6 nanosheets and CTF-1 nanosheets.158 The ionization of surface capping ligands resulted in Cs2AgBiBr6 with positive charges, and the lone pair electrons of the nitrogen atoms in the triazine units resulted in CTF-1 with negative charges, so the Cs2AgBiBr6/CTF-1 (denoted as CABB/CTF-1) nanosheet heterojunction with a large surface-to-surface contact could be formed through electrostatic self-assembly. The two components formed an S-scheme heterojunction, resulting in efficient electron transfer and high redox capacity (Fig. 10f). The CABB/CTF-1 heterojunction with 10% CTF-1 (Fig. 10g) exhibited the highest photocatalytic CO2 reduction activity under visible-light irradiation (∼30.7 μmol g−1 h−1 for CO and 8.6 μmol g−1 h−1 for CH4), which was much better than that of the Cs2AgBiBr6 nanosheets and CTF-1 nanosheets.
5.4 Carbon nitride nanosheet-based photocatalysts
Carbon nitride nanosheet-based heterojunctions have been widely reported. In this section, heterojunctions formed from exfoliated carbon nitride nanosheets with other zero-dimensional (0D)/one-dimensional (1D)/2D/three-dimensional (3D) photocatalysts are briefly displayed and their photocatalytic performances are discussed.5.4.1.1 Photocatalytic H2 production. Li et al. inserted 0D N-doped carbon dots (NCDs) onto g-C3N4 nanosheets with a thickness of 1 nm via a two-step thermal exfoliation method.159 The introduction of NCDs provided catalytic active sites and shortened the distance of charge transfer to the g-C3N4 surface. Compared with bulk g-C3N4/NCDs, g-C3N4 nanosheet/NCDs possessed a larger number of exposed NCDs, and thus exhibited a better photocatalytic H2 production rate. Under optimal conditions, its H2 production rate reached 3319.3 μmol g−1 h−1, which was about 13 fold that of bulk g-C3N4 and 2.7 fold that of bulk g-C3N4/NCDs. Jia et al. prepared an isotype heterojunction of 1D/2D g-C3N4 through thermal polymerization (melamine grafted with ethylenediamine was used as the precursor) and subsequent HCl-assisted hydrothermal exfoliation (Fig. 11a).160 In the preparation process, the grafted ethylenediamine limited the directional growth of g-C3N4 into nanowires and introduced carbon doping, and the exfoliation resulted in ultrathin g-C3N4 nanosheets. The carbon doping increased visible-light absorption ability. And the ultra-thin nanosheets provided more photogenerated charges to participate in the transfer among isotype heterojunctions. Moreover, the formed built-in electric field between the two components facilitated rapid migration of photogenerated charges at the interface (Fig. 11b). Therefore the photocatalytic H2 evolution activity of the isotype heterojunction under visible-light irradiation was 8.2 times that of pristine g-C3N4. Yu et al. fabricated ultrathin WO3 and g-C3N4 nanosheets with a thickness of 2.5–3.5 nm by electrostatic-assisted ultrasonication of bulk WO3 and thermal-etching exfoliation of bulk g-C3N4, respectively (Fig. 11c).161 The ultrathin WO3/g-C3N4 nanosheets formed an S-scheme heterojunction through electrostatic self-assembly (Fig. 11d). The ultrathin layered heterojunction structure was beneficial for the transfer of photogenerated electrons and holes at the heterogeneous interface to the surface of photocatalysts, and the S-scheme heterojunction inhibited the recombination of useful electrons and holes. Therefore the H2-production activity of the WO3/g-C3N4 nanosheets under xenon lamp irradiation was better than that of pure g-C3N4 and WO3 nanosheets. Parida et al. deposited MgIn2S4 microflowers onto the surface of exfoliated B-doped g-C3N4 nanosheets (e-BCN) to form a p–n heterojunction.162 e-BCN provided abundant active sites to the MgIn2S4 microflowers for water molecule adsorption, and the built-in electric field inhibited carrier recombination. Under the optimal conditions, the heterojunction exhibited an excellent photocatalytic H2 generation rate with an apparent conversion efficiency of 5.27%.
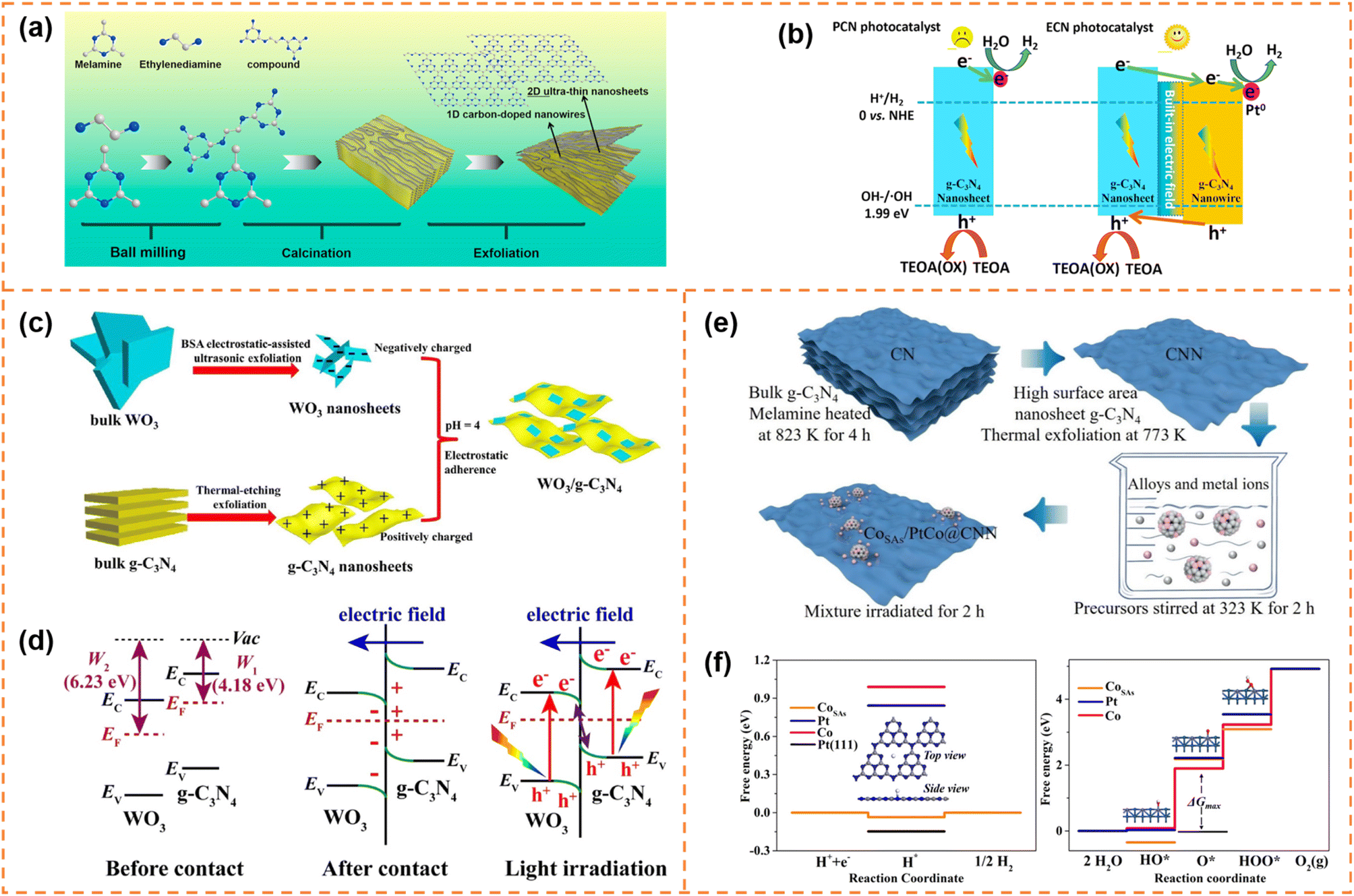 | ||
| Fig. 11 (a) Schematic representation of the preparation process of U-ECN. (b) The migration direction of photogenerated electrons and holes and the possible mechanism of PCN and ECN for photocatalytic H2 evolution. Reprinted with permission from ref. 160, © Elsevier B.V. 2021. (c) The formation schematic diagram of 2D/2D WO3/g-C3N4 heterojunctions by coulomb electrostatic interaction. (d) The S-scheme formation mechanism between WO3 and g-C3N4. Reprinted with permission from ref. 161, © Elsevier B.V. 2019. (e) Schematic illustration of the synthetic strategy of producing the CoSAs/PtCo@CNN sample. (f) Free energy diagram for the HER and the OER under standard conditions for the CoSAs/PtCo@CNN catalyst. Reprinted with permission from ref. 163, © American Chemical Society 2022. | ||
5.4.1.2 Photocatalytic overall water splitting. Yao et al. anchored 0D single-atom Co and PtCo alloy nanoparticles on exfoliated g-C3N4 nanosheets to achieve overall water splitting (Fig. 11e).163 The single-atom Co and PtCo alloys acted as highly active sites for the hydrogen evolution reaction and the oxygen evolution reaction, respectively (Fig. 11f). In addition, the authors suggested that there might be a spillover phenomenon of protons or hydroxyl groups between the single-atom Co centers and PtCo alloy nanoparticles. Ajayan et al. used small amounts of α-Fe2O3 nanosheets to promote exfoliation of g-C3N4.164 Meanwhile, the α-Fe2O3 nanosheets and g-C3N4 nanosheets were tightly bound and formed an all-solid-state Z-scheme heterojunction, which served as an oxidation counterpart and reduction counterpart, respectively. Thanks to the Z-scheme structure and tight interface, electron–hole recombination in both materials was effectively inhibited, which resulted in good overall water splitting activity under visible-light irradiation.
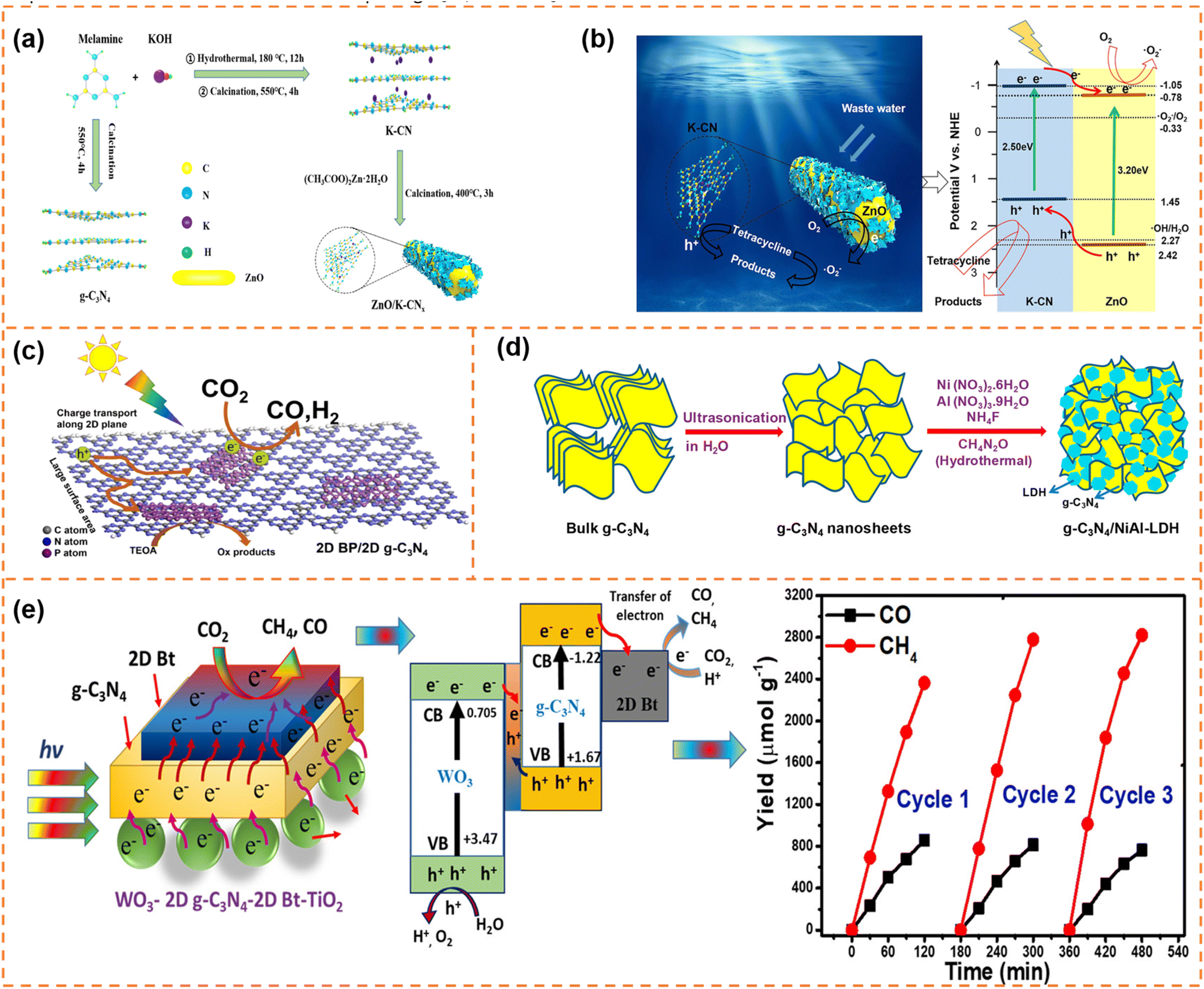 | ||
| Fig. 12 (a) Schematic diagram of the synthesis and assembly route of photocatalyst materials. (b) Schematic diagram of the charge separation and photocatalytic mechanism of type-II ZnO/K-CN0.5 heterojunctions under simulated sunlight. Reprinted with permission from ref. 166, © American Chemical Society 2020. (c) Illustration of the photocatalytic mechanism of the 2D BP/2D g-C3N4 system under visible-light irradiation. Reprinted with permission from ref. 169, © Elsevier B.V. 2020. (d) Schematic illustration of the synthesis process of g-C3N4/NiAl-LDH hybrid heterojunctions. Reprinted with permission from ref. 170, © American Chemical Society 2018. (e) Proposed mechanism and stability analysis of photocatalytic CO2 reduction with H2O over the Bt-mediated WO3/g-C3N4 composite under visible-light irradiation. Reprinted with permission from ref. 172, © American Chemical Society 2020. | ||
6. Conclusions and prospects
We summarized the recent advances in polymer (CMPs, COFs, CTFs and carbon nitrides) nanosheets for photocatalysis, including water splitting, pollutant degradation and CO2 reduction. Besides the conventional advantages of low cost, good visible-light absorption ability and chemical stability, as well as tunable electronic and optical properties, polymer nanosheets have some special virtues, such as a short carrier migration distance, atomic-level regulation of the backbone and targeted introduction of active sites. Therefore polymer nanosheets generally exhibit better photocatalytic activity than bulk polymers and most traditional inorganic semiconductors. In addition, the abundant surface active groups and conjugated porous architecture of polymer nanosheets provide the potential for tightly bonding with organic or inorganic materials to form heterojunctions.Although significant progress has been achieved in these photocatalytic applications, polymer nanosheets and their heterojunctions still face some obvious challenges. (1) The preparation methods of polymer nanosheets have rarely been reported, especially for single-layer/few-layer nanosheets. Meanwhile, the large-scale preparation of polymer nanosheets is also highly challenging. (2) The structural properties of polymer nanosheets like thickness, size and surface active groups have great effects on photocatalytic performance, yet there are few in-depth reports. (3) The effective guidance for obtaining high-performance polymer nanosheets is still lacking. Effective theoretical prediction of structural properties (such as surface active sites and backbone) is encouraged, which can avoid many useless experimental studies. For example, Cooper et al. integrated robotic experimentation with high-throughput computation and discovered new polymers with high H2 production activity.173 (4) Polymer nanosheet-based heterojunctions for photocatalytic application are still rarely reported, especially those formed by in situ growth. Compared with the common self-assembly method, the bonds formed by in situ growth are more stable, which benefit charge transfer and photostability. (5) Due to the poor water oxidation ability caused by slow surface dynamics,174 it is still difficult for polymer nanosheets to achieve overall water splitting. Many inorganic semiconductors with good visible-light absorption ability possess excellent water oxidation ability, so it is very valuable to construct S-scheme or Z-scheme heterojunctions between polymer nanosheets and these inorganic semiconductors. (6) The photocatalytic CO2 reduction activity of polymer nanosheets and their heterojunctions is still low, especially for reducing CO2 to high-value chemicals (CH4etc.). In addition, the reductive half reaction of the CO2 transformation process has been densely reported, while the oxidation half reaction (H2O oxidation, etc.) is rare. (7) The photodegradation of polymer nanosheets and their heterojunctions mainly focuses on dye degradation. It can be extended to more types of pollutants (such as antibiotics and volatile organic compounds) to further improve their practicability.
Despite these challenges, the inherent advantages of polymer nanosheets and their heterojunctions bring about infinite opportunities to achieve high photocatalytic activity. We expect that this review can provide a helpful reference for the rapid development of polymer nanosheets for photocatalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22022510, 51872186, and 22066024).References
- A. J. Carrillo, J. González-Aguilar, M. Romero and J. M. Coronado, Chem. Rev., 2019, 119, 4777–4816 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- Y. Du, Y. Wang, J. Wu, Q. Chen, C. Deng, R. Ji, L. Sun, L. Tan, X. Chen and Y. Xie, InfoMat, 2023, e12431 CrossRef CAS.
- Y. Lin, H. Kuang, S. Zhang, X. Zhang, G. Zhai, X. Lin, D. Xu, J. Jia, X. Li and J. Chen, CCS Chem., 2022, 4, 3482–3490 CrossRef CAS.
- H. Li, C. Chen, H. Hu, Y. Li, Z. Shen, F. Li, Y. Liu, R. Liu, J. Chen and C. Dong, InfoMat, 2022, 4, e12322 CrossRef CAS.
- Y. Jiang, Y. Fan, S. Li and Z. Tang, CCS Chem., 2023, 5, 30–54 CrossRef CAS.
- C. H. Dai and B. Liu, Energy Environ. Sci., 2020, 13, 24–52 RSC.
- Z. Gao, Y. Jian, S. Yang, Q. Xie, C. J. Ross Mcfadzean, B. Wei, J. Tang, J. Yuan, C. Pan and G. Yu, Angew. Chem., Int. Ed., 2023, 62, e202304173 CrossRef CAS PubMed.
- Q. Zuo, R. Cui, L. Wang, Y. Wang, C. Yu, L. Wu, Y. Mai and Y. Zhou, Sci. China Chem., 2023, 66, 570–577 CrossRef CAS.
- Q. Zuo, K. Feng, J. Zhong, Y. Mai and Y. Zhou, CCS Chem., 2021, 3, 1963–1971 CrossRef CAS.
- J. Liu, Y. Guo, Z. Hu, H. Zhao, Z. Yu, L. Chen, Y. Li, G. V. Tendeloo and B. Su, CCS Chem., 2022, 5, 372–384 CrossRef.
- Z. Meng, Z. Qiu, Y. Shi, S. Wang, G. Zhang, Y. Pi and H. Pang, eScience, 2023, 100092 CrossRef.
- A. Li, P. Zhang, E. Kan and J. Gong, eScience, 2023, 100157 CrossRef.
- S. Wei, S. Chang, J. Qian and X. Xu, Small, 2021, 17, 2100084 CrossRef CAS PubMed.
- Y. Bai, K. Hippalgaonkar and R. S. Sprick, J. Mater. Chem. A, 2021, 9, 16222–16232 RSC.
- X. Li, J. G. Yu, J. X. Low, Y. P. Fang, J. Xiao and X. B. Chen, J. Mater. Chem., 2015, 3, 2485–2534 RSC.
- J. Wei, D. Luo, M. Shi, Q. Yuan, M. Wang, Y. Huang and Y. Ni, Inorg. Chem., 2023, 62, 10973–10983 CrossRef CAS PubMed.
- Q. Zuo, T. Liu, C. Chen, Y. Ji, X. Gong, Y. Mai and Y. Zhou, Angew. Chem., Int. Ed., 2019, 58, 10198–10203 CrossRef CAS PubMed.
- G. Zhang, X. Li, M. Wang, X. Li, Y. Wang, S. Huang, D. Chen, N. Li, Q. Xu, H. Li and J. Lu, Nano Res., 2023, 16, 6134–6141 CrossRef CAS.
- D. Zhao, Y. Wang, C. Dong, F. Meng, Y. Huang, Q. Zhang, L. Gu, L. Liu and S. Shen, Nano-Micro Lett., 2022, 14, 223 CrossRef CAS PubMed.
- K. M. Kamal, R. Narayan, N. Chandran, S. Popovic, M. A. Nazrulla, J. Kovac, N. Vrtovec, M. Bele, N. Hodnik, M. M. Krzmanc and B. Likozar, Appl. Catal., B, 2022, 307, 121181 CrossRef CAS.
- F. L. Wang, R. Q. Fang, X. Zhao, X. P. Kong, T. T. Hou, K. Shen and Y. W. Li, ACS Nano, 2022, 16, 4517–4527 CrossRef CAS PubMed.
- K. Li, B. S. Peng and T. Y. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- W. Pan, Z. Wei, Y. Su, Y. Lian, Z. Zheng, H. Yuan, Y. Qin, X. Xie, Q. Bai, Z. Jiao, W. Hua, J. Chen, W. Yang, Z. Deng and Y. Peng, Nano Res., 2023 DOI:10.1007/s12274-023-6083-2.
- C. Ban, Y. Duan, Y. Wang, J. Ma, K. Wang, J. Meng, X. Liu, C. Wang, X. Han, G. Cao, L. Gan and X. Zhou, Nano-Micro Lett., 2022, 14, 74 CrossRef CAS PubMed.
- A. Ahmad, M. Ali, A. G. Al-Sehemi, A. A. Al-Ghamdi, J.-W. Park, H. Algarni and H. Anwer, Chem. Eng. J., 2023, 452, 139436 CrossRef CAS.
- S. Wei, S. Fan, M. Zhang, J. Ren, B. Jia, Y. Wang, R. Wu, Z. Fang and Q. Liang, Mater. Today Sustainability, 2023, 21, 100311 CrossRef.
- S. Wei, Y. Chen, P. Wu, X. Liu, J. Ren, B. Yao, H. Xu, W. Dou, Y. Wang and R. Wu, Appl. Catal., A, 2022, 640, 118675 CrossRef CAS.
- F. He, X. Liu, X. Zhao, J. Zhang, P. Dong, Y. Zhang, C. Zhao, H. Sun, X. Duan and S. Wang, J. Mater. Chem. A, 2022, 10, 20559–20570 RSC.
- J. Luo, Z. Dai, M. Feng, M. Gu and Y. Xie, Nano Res., 2023, 16, 371–376 CrossRef CAS.
- X. Sun, L. Li, S. Jin, W. Shao, H. Wang, X. Zhang and Y. Xie, eScience, 2023, 3, 100095 CrossRef.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- J. Shi, G. Liu, N. Wang and C. Li, J. Mater. Chem., 2012, 22, 18808–18813 RSC.
- W. Yu, J. Zhang and T. Peng, Appl. Catal., B, 2016, 181, 220–227 CrossRef CAS.
- H. Tan, Z. Zhao, W.-b. Zhu, E. N. Coker, B. Li, M. Zheng, W. Yu, H. Fan and Z. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 19184–19190 CrossRef CAS PubMed.
- H. Xu, Y. Wang, X. Dong, N. Zheng, H. Ma and X. Zhang, Appl. Catal., B, 2019, 257, 117932 CrossRef CAS.
- H. Jing, H. Li, J. Yue, S. Fan, B. Yao, S. Liu, Z. Fang, R. Wu, L. Yang and S. Wei, Dalton Trans., 2023, 52, 2924–2927 RSC.
- H. Jing, J. Ren, J. Yue, S. Liu, Q. Liang, R. Wu, Y. Wang, Z. Fang, H. Li and S. Wei, Catal. Sci. Technol., 2023, 13, 226–232 RSC.
- G. Lin, C. Zhang and X. Xu, J. Mater. Sci. Technol., 2023, 154, 241–250 CrossRef.
- F. Zhang, A. Yamakata, K. Maeda, Y. Moriya, T. Takata, J. Kubota, K. Teshima, S. Oishi and K. Domen, J. Am. Chem. Soc., 2012, 134, 8348–8351 CrossRef CAS PubMed.
- S. Wei, S. Chang, F. Yang, Z. Fu, G. Liu and X. Xu, Chem. Commun., 2021, 57, 4412–4415 RSC.
- L. Wang, Y. Zhang, L. Chen, H. Xu and Y. Xiong, Adv. Mater., 2018, 30, 1801955 CrossRef PubMed.
- G. Zhang, Z. A. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712–15727 CrossRef PubMed.
- R. S. Sprick, J.-X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef CAS PubMed.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, J. Chem. Soc., Chem. Commun., 1985, 474–475 RSC.
- Y. Jing, X. Zhu, S. Maier and T. Heine, Trends Chem., 2022, 4, 792–806 CrossRef CAS.
- J. M. Valero, S. Obregón and G. Colon, ACS Catal., 2014, 4, 3320–3329 CrossRef CAS.
- P. Chen, P. Xing, Z. Chen, H. Lin and Y. He, Int. J. Hydrogen Energy, 2018, 43, 19984–19989 CrossRef CAS.
- Y. Bao, S. Song, G. Yao and S. Jiang, Sol. RRL, 2021, 5, 2100118 CrossRef CAS.
- M. Dai, Z. He, P. Zhang, X. Li and S. Wang, J. Mater. Sci. Technol., 2022, 122, 231–242 CrossRef CAS.
- M. Gao, F. Tian, X. Zhang, Z. Chen, W. Yang and Y. Yu, Nano-Micro Lett., 2023, 15, 129 CrossRef CAS PubMed.
- S. F. Ng, J. J. Foo and W. J. Ong, InfoMat, 2022, 4, e12279 CrossRef CAS.
- Q. Zhu, Z. Xu, B. Qiu, M. Xing and J. Zhang, Small, 2021, 17, 2101070 CrossRef CAS PubMed.
- B. He, C. Bie, X. Fei, B. Cheng, J. Yu, W. Ho, A. A. Al-Ghamdi and S. Wageh, Appl. Catal., B, 2021, 288, 119994 CrossRef CAS.
- H. Abdullah, M. M. R. Khan, H. R. Ong and Z. Yaakob, J. CO2 Util., 2017, 22, 15–32 CrossRef CAS.
- D. Li, M. Kassymova, X. Cai, S. Zang and H. Jiang, Coordin. Chem. Rev., 2020, 412, 213262 CrossRef CAS.
- D. P. Tran, M. Pham, X. Bui, Y. Wang and S. You, Sol. Energy, 2022, 240, 443–466 CrossRef CAS.
- V. Nguyen, B. Nguyen, Z. Jin, M. Shokouhimehr, H. W. Jang, C. Hu, P. Singh, P. Raizada, W. Peng and S. S. Lam, Chem. Eng. J., 2020, 402, 126184 CrossRef CAS.
- J. Ran, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- P. M. Wood, Biochem. J., 1988, 253, 287 CrossRef CAS PubMed.
- X. Du, T. Zhao, Z. Xiu, Z. Xing, Z. Li, K. Pan, S. Yang and W. Zhou, Appl. Mater. Today, 2020, 20, 100719 CrossRef.
- X. Zhang, L. Jian, L. Wang, R. Liu, Y. Zhao, Y. Li and C. Wang, Appl. Surf. Sci., 2022, 571, 151304 CrossRef CAS.
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452–2456 CrossRef CAS PubMed.
- F. Wang, Z. Zhang, I. Shakir, C. Yu and Y. Xu, Adv. Sci., 2022, 9, 2103814 CrossRef CAS PubMed.
- T. Banerjee, F. Podjaski, J. Kröger, B. P. Biswal and B. V. Lotsch, Nat. Rev. Mater., 2021, 6, 168–190 CrossRef CAS.
- P. A. Held, H. Fuchs and A. Studer, Chem.–Eur. J., 2017, 23, 5874–5892 CrossRef CAS PubMed.
- S. Clair and D. G. de Oteyza, Chem. Rev., 2019, 119, 4717–4776 CrossRef CAS PubMed.
- L. Hou, G. Tang, H. Huang, S. Yin, B. Long, A. Ali, G. Deng and T. Song, J. Mater. Chem., 2022, 10, 5287–5294 RSC.
- M. Mansha, T. Ahmad, N. Ullah, S. Akram Khan, M. Ashraf, S. Ali, B. Tan and I. Khan, Chem. Rec., 2022, 22, e202100336 CrossRef CAS PubMed.
- Y. Z. Cheng, X. Ding and B. H. Han, ChemPhotoChem, 2021, 5, 406–417 CrossRef CAS.
- A. Hayat, M. Sohail, A. El Jery, K. M. Al-Zaydi, S. Raza, H. Ali, Y. Al-Hadeethi, T. A. Taha, I. U. Din, M. A. Khan, M. A. Amin, E. Ghasali, Y. Orooji, Z. Ajmal and M. Z. Ansari, Mater. Today, 2023, 64, 180–208 CrossRef CAS.
- S. Luo, Z. Zeng, G. Zeng, Z. Liu, R. Xiao, P. Xu, H. Wang, D. Huang, Y. Liu and B. Shao, J. Mater. Chem., 2020, 8, 6434–6470 RSC.
- Y. Xu, N. Mao, S. Feng, C. Zhang, F. Wang, Y. Chen, J. Zeng and J. X. Jiang, Macromol. Chem. Phys., 2017, 218, 1700049 CrossRef.
- Z. Wang, X. Yang, T. Yang, Y. Zhao, F. Wang, Y. Chen, J. H. Zeng, C. Yan, F. Huang and J. Jiang, ACS Catal., 2018, 8, 8590–8596 CrossRef CAS.
- R. S. Sprick, B. Bonillo, M. Sachs, R. Clowes, J. R. Durrant, D. J. Adams and A. I. Cooper, Chem. Commun., 2016, 52, 10008–10011 RSC.
- Y. Zhao, W. Ma, Y. Xu, C. Zhang, Q. Wang, T. Yang, X. Gao, F. Wang, C. Yan and J. Jiang, Macromolecules, 2018, 51, 9502–9508 CrossRef CAS.
- X. Zhuang, D. Gehrig, N. Forler, H. Liang, M. Wagner, M. R. Hansen, F. Laquai, F. Zhang and X. Feng, Adv. Mater., 2015, 27, 3789–3796 CrossRef CAS PubMed.
- D. Tan, W. Fan, W. Xiong, H. Sun, Y. Cheng, X. Liu, C. Meng, A. Li and W. Q. Deng, Macromol. Chem. Phys., 2012, 213, 1435–1440 CrossRef CAS.
- C. Shu, C. Han, X. Yang, C. Zhang, Y. Chen, S. Ren, F. Wang, F. Huang and J. X. Jiang, Adv. Mater., 2021, 33, 2008498 CrossRef CAS PubMed.
- S. Wei, G. Zhang and X. Xu, Appl. Catal., B, 2018, 237, 373–381 CrossRef CAS.
- L. Wang, Y. Wan, Y. Ding, Y. Niu, Y. Xiong, X. Wu and H. Xu, Nanoscale, 2017, 9, 4090–4096 RSC.
- L. Wang, Y. Y. Wan, Y. J. Ding, S. K. Wu, Y. Zhang, X. L. Zhang, G. Q. Zhang, Y. J. Xiong, X. J. Wu, J. L. Yang and H. X. Xu, Adv. Mater., 2017, 29, 1702428 CrossRef PubMed.
- Q. Huang, L. Guo, N. Wang, X. Zhu, S. Jin and B. Tan, ACS Appl. Mater. Interfaces, 2019, 11, 15861–15868 CrossRef CAS PubMed.
- Q. Zhi, J. Zhou, W. Liu, L. Gong, W. Liu, H. Liu, K. Wang and J. Jiang, Small, 2022, 18, 2201314 CrossRef CAS PubMed.
- S. Wei, J. Wang, Y. Li, Z. Fang, L. Wang and Y. Xu, Nano Res., 2023, 16, 6753–6770 CrossRef CAS.
- Y. Li, X. Song, G. Zhang, L. Wang, Y. Liu, W. Chen and L. Chen, ChemSusChem, 2022, 15, e202200901 CrossRef CAS PubMed.
- L. Chen, M. Huang, B. Chen, C. Gong, N. Li, H. Cheng, Y. Chen, Y. Peng and G. Xu, Chin. Chem. Lett., 2022, 33, 2867–2882 CrossRef CAS.
- D. N. Bunck and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 14952–14955 CrossRef CAS PubMed.
- G. Li, K. Zhang and T. Tsuru, ACS Appl. Mater. Interfaces, 2017, 9, 8433–8436 CrossRef CAS PubMed.
- S. Chandra, S. Kandambeth, B. P. Biswal, B. Lukose, S. M. Kunjir, M. Chaudhary, R. Babarao, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 17853–17861 CrossRef CAS PubMed.
- W. Dai, F. Shao, J. Szczerbiński, R. McCaffrey, R. Zenobi, Y. Jin, A. D. Schlüter and W. Zhang, Angew. Chem., Int. Ed., 2016, 128, 221–225 CrossRef.
- Y. Li, M. Zhang, X. Guo, R. Wen, X. Li, X. Li, S. Li and L. Ma, Nanoscale Horiz., 2018, 3, 205–212 RSC.
- E. Jin, Z. Lan, Q. Jiang, K. Geng, G. Li, X. Wang and D. Jiang, Chem, 2019, 5, 1632–1647 CAS.
- Z. Zhou, Y. Xiao, J. Tian, N. Nan, R. Song and J. Li, J. Mater. Chem. A, 2023, 11, 3245–3261 RSC.
- S. Liu, M. Wang, Y. He, Q. Cheng, T. Qian and C. Yan, Coordin. Chem. Rev., 2023, 475, 214882 CrossRef CAS.
- X. Li, T. Goto, K. Nomura, M. Zhu, T. Sekino and Y. Osakada, Appl. Surf. Sci., 2020, 513, 145720 CrossRef CAS.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg and R. S. Sprick, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- K. L. Corp and C. W. Schlenker, J. Am. Chem. Soc., 2017, 139, 7904–7912 CrossRef CAS PubMed.
- K. Schwinghammer, M. B. Mesch, V. Duppel, C. Ziegler, J. R. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2014, 136, 1730–1733 CrossRef CAS PubMed.
- Y. Yang, X. Chu, H.-Y. Zhang, R. Zhang, Y.-H. Liu, F.-M. Zhang, M. Lu, Z.-D. Yang and Y.-Q. Lan, Nat. Commun., 2023, 14, 593 CrossRef CAS PubMed.
- T. Sun, C. Wang and Y. Xu, Chem. Res. Chin. Univ., 2020, 36, 640–647 CrossRef CAS.
- Y. Zhu, X. Chen, Y. Cao, W. Peng, Y. Li, G. Zhang, F. Zhang and X. Fan, Chem. Commun., 2019, 55, 1434–1437 RSC.
- J. Liu, P. Lyu, Y. Zhang, P. Nachtigall and Y. Xu, Adv. Mater., 2018, 30, 1705401 CrossRef PubMed.
- H. Zhang, W. Sun, X. Chen and Y. Wang, ACS Nano, 2019, 13, 14252–14261 CrossRef CAS PubMed.
- C. Wang, H. Zhang, W. Luo, T. Sun and Y. Xu, Angew. Chem., Int. Ed., 2021, 60, 25381–25390 CrossRef CAS PubMed.
- T. Sun, Y. Liang and Y. Xu, Angew. Chem., Int. Ed., 2022, 61, e202113926 CrossRef CAS PubMed.
- T. Sun, S. Li, L. Zhang and Y. Xu, Angew. Chem., Int. Ed., 2023, 62, e202301865 CrossRef CAS PubMed.
- C. Wang, P. Lyu, Z. Chen and Y. Xu, J. Am. Chem. Soc., 2023, 145(23), 12745–12754 CrossRef CAS PubMed.
- L. Li, Y. Zhu, N. Gong, W. Zhang, W. Peng, Y. Li, F. Zhang and X. Fan, Int. J. Hydrogen Energy, 2020, 45, 2689–2698 CrossRef CAS.
- H. Ye, N. Gong, Y. Cao, X. Fan, X. Song, H. Li, C. Wang, Y. Mei and Y. Zhu, Chem. Mater., 2022, 34, 1481–1490 CrossRef CAS.
- S. Zhang, G. Cheng, L. Guo, N. Wang, B. Tan and S. Jin, Angew. Chem., Int. Ed., 2020, 59, 6007–6014 CrossRef CAS PubMed.
- S. Ye, R. Wang, M. Wu and Y. Yuan, Appl. Surf. Sci., 2015, 358, 15–27 CrossRef CAS.
- M. Aggarwal, S. Basu, N. P. Shetti, M. N. Nadagouda, E. E. Kwon, Y.-K. Park and T. M. Aminabhavi, Chem. Eng. J., 2021, 425, 131402 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- M. Reli, P. Huo, M. Sihor, N. Ambrozova, I. Troppová, L. Matejova, J. Lang, L. Svoboda, P. Kustrowski and M. Ritz, J. Phys. Chem. A, 2016, 120, 8564–8573 CrossRef CAS PubMed.
- K. Qi, J. Jing, G. Dong, P. Li and Y. Huang, Environ. Res., 2022, 212, 113405 CrossRef CAS PubMed.
- Y. Shiraishi, S. Kanazawa, Y. Sugano, D. Tsukamoto, H. Sakamoto, S. Ichikawa and T. Hirai, ACS Catal., 2014, 4, 774–780 CrossRef CAS.
- X. Zhang, J. Yan, F. Zheng, J. Zhao and L. Y. S. Lee, Appl. Catal., B, 2021, 286, 119879 CrossRef CAS.
- L. Wang, Y. Hong, E. Liu, X. Duan, X. Lin and J. Shi, Carbon, 2020, 163, 234–243 CrossRef CAS.
- Q. Xu, B. Zhu, B. Cheng, J. Yu, M. Zhou and W. Ho, Appl. Catal., B, 2019, 255, 117770 CrossRef CAS.
- J. Jia, W. Sun, Q. Zhang, X. Zhang, X. Hu, E. Liu and J. Fan, Appl. Catal., B, 2020, 261, 118249 CrossRef CAS.
- W. Xing, W. Tu, Z. Han, Y. Hu, Q. Meng and G. Chen, ACS Energy Lett., 2018, 3, 514–519 CrossRef CAS.
- Z. Huang, Y. Zhang, H. Dai, Y. Wang, C. Qin, W. Chen, Y. Zhou and S. Yuan, J. Catal., 2019, 378, 331–340 CrossRef CAS.
- X. Zhu, J. Luo, Z. Dong, Z. Li, Y. Wu, Z. Cheng, X. Cao, Y. Wang, Y. Liu and Z. Zhang, Sep. Purif. Technol., 2023, 124235 CrossRef CAS.
- M. Tahir, C. Cao, N. Mahmood, F. K. Butt, A. Mahmood, F. Idrees, S. Hussain, M. Tanveer, Z. Ali and I. Aslam, ACS Appl. Mater. Interfaces, 2014, 6, 1258–1265 CrossRef CAS PubMed.
- Z. Jiang, X. Zhang, H. S. Chen, X. Hu and P. Yang, ChemCatChem, 2019, 11, 4558–4567 CrossRef CAS.
- Z. Mo, X. Zhu, Z. Jiang, Y. Song, D. Liu, H. Li, X. Yang, Y. She, Y. Lei and S. Yuan, Appl. Catal., B, 2019, 256, 117854 CrossRef CAS.
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef.
- J. Wen, J. Xie, X. Chen and X. Li, Appl. Surf. Sci., 2017, 391, 72–123 CrossRef CAS.
- F. He, Z. Wang, Y. Li, S. Peng and B. Liu, Appl. Catal., B, 2020, 269, 118828 CrossRef CAS.
- Y. Li, X. Li, H. Zhang, J. Fan and Q. Xiang, J. Mater. Sci. Technol., 2020, 56, 69–88 CrossRef CAS.
- Y. Xing, X. Wang, S. Hao, X. Zhang, X. Wang, W. Ma, G. Zhao and X. Xu, Chin. Chem. Lett., 2021, 32, 13–20 CrossRef CAS.
- S. Patnaik, D. P. Sahoo and K. Parida, Carbon, 2021, 172, 682–711 CrossRef CAS.
- R. Guo, J. Wang, Z. Bi, X. Chen, X. Hu and W. Pan, Chemosphere, 2022, 295, 133834 CrossRef CAS PubMed.
- A. Hayat, M. Sohail, J. Ali Shah Syed, A. G. Al-Sehemi, M. H. Mohammed, A. A. Al-Ghamdi, T. Taha, H. Salem AlSalem, A. M. Alenad and M. A. Amin, Chem. Rec., 2022, 22, e202100310 CrossRef CAS PubMed.
- Y. Liu, S. Shen, Z. Li, D. Ma, G. Xu and B. Fang, Mater. Charact., 2021, 174, 111031 CrossRef CAS.
- L. Cui, Y. Liu, X. Fang, C. Yin, S. Li, D. Sun and S. Kang, Green Chem., 2018, 20, 1354–1361 RSC.
- Q. Liu, J. Shen, X. Yu, X. Yang, W. Liu, J. Yang, H. Tang, H. Xu, H. Li and Y. Li, Appl. Catal., B, 2019, 248, 84–94 CrossRef CAS.
- Q. Zhu, B. Qiu, M. Du, J. Ji, M. Nasir, M. Xing and J. Zhang, ACS Sustainable Chem. Eng., 2020, 8, 7497–7502 CrossRef CAS.
- C. Wu, S. Xue, Z. Qin, M. Nazari, G. Yang, S. Yue, T. Tong, H. Ghasemi, F. C. R. Hernandez and S. Xue, Appl. Catal., B, 2021, 282, 119557 CrossRef CAS.
- M. Zhang, Y. Yang, X. An, J. Zhao, Y. Bao and L. Hou, J. Hazard. Mater., 2022, 424, 127424 CrossRef CAS PubMed.
- Z. Ma, P. Zhou, L. Zhang, Y. Zhong, X. Sui, B. Wang, Y. Ma, X. Feng, H. Xu and Z. Mao, Chem. Phys. Lett., 2021, 766, 138335 CrossRef CAS.
- J. Zhou, J. Xue, Q. Pan, X. Yang, Q. Shen, T. Ma, X. Liu and H. Jia, J. Photochem. Photobiol. A, 2019, 372, 147–155 CrossRef CAS.
- J. Yan, J. Liu, Y. Sun, D. Ding, C. Wang, L. Sun and X. Li, Inorg. Chem. Front., 2022, 9, 1423–1433 RSC.
- J. Hong, D. K. Hwang, R. Selvaraj and Y. Kim, J. Ind. Eng. Chem., 2019, 79, 473–481 CrossRef CAS.
- Y. Zhong, Z. Li, X. Zhao, T. Fang, H. Huang, Q. Qian, X. Chang, P. Wang, S. Yan, Z. Yu and Z. Zou, Adv. Funct. Mater., 2016, 26, 7156–7163 CrossRef CAS.
- L. Wang, X. Zheng, L. Chen, Y. Xiong and H. Xu, Angew. Chem., Int. Ed., 2018, 57, 3454–3458 CrossRef CAS PubMed.
- Y. Zang, R. Wang, P.-P. Shao, X. Feng, S. Wang, S.-Q. Zang and T. C. Mak, J. Mater. Chem. A, 2020, 8, 25094–25100 RSC.
- P. Dong, A. Zhang, T. Cheng, J. Pan, J. Song, L. Zhang, R. Guan, X. Xi and J. Zhang, Chin. J. Catal., 2022, 43, 2592–2605 CrossRef CAS.
- H. Shen, D. Shang, L. Li, D. Li and W. Shi, Appl. Surf. Sci., 2022, 578, 152024 CrossRef CAS.
- M. Luo, Q. Yang, W. Yang, J. Wang, F. He, K. Liu, H. Cao and H. Yan, Small, 2020, 16, 2001100 CrossRef CAS PubMed.
- W. Liu, X. Li, C. Wang, H. Pan, W. Liu, K. Wang, Q. Zeng, R. Wang and J. Jiang, J. Am. Chem. Soc., 2019, 141, 17431–17440 CrossRef CAS PubMed.
- X. Wang, Z. Fu, L. Zheng, C. Zhao, X. Wang, S. Y. Chong, F. McBride, R. Raval, M. Bilton and L. Liu, Chem. Mater., 2020, 32, 9107–9114 CrossRef CAS.
- F. Li, D. Wang, Q. Xing, G. Zhou, S. Liu, Y. Li, L. Zheng, P. Ye and J. Zou, Appl. Catal., B, 2019, 243, 621–628 CrossRef CAS.
- J. Chen, G. Li, N. Lu, H. Lin, S. Zhou and F. Liu, Mater. Today Chem., 2022, 24, 100832 CrossRef CAS.
- X. Lan, C. Du, L. Cao, T. She, Y. Li and G. Bai, ACS Appl. Mater. Interfaces, 2018, 10, 38953–38962 CrossRef CAS PubMed.
- J. Li, P. Liu, H. Huang, Y. Li, Y. Tang, D. Mei and C. Zhong, ACS Sustainable Chem. Eng., 2020, 8, 5175–5183 CrossRef CAS.
- Z. Zhang, Y. Jiang, Z. Dong, Y. Chu and J. Xu, Inorg. Chem., 2022, 61, 16028–16037 CrossRef CAS PubMed.
- Y. Jiao, Y. Li, J. Wang, Z. He and Z. Li, Mol. Catal., 2020, 497, 111223 CrossRef CAS.
- B. Yang, Z. Wang, J. Zhao, X. Sun, R. Wang, G. Liao and X. Jia, Int. J. Hydrogen Energy, 2021, 46, 25436–25447 CrossRef CAS.
- J. Fu, Q. Xu, J. Low, C. Jiang and J. Yu, Appl. Catal., B, 2019, 243, 556–565 CrossRef CAS.
- L. Acharya, G. Swain, B. P. Mishra, R. Acharya and K. Parida, ACS Appl. Energy Mater., 2022, 5, 2838–2852 CrossRef CAS.
- B. Xu, X. Fu, X. You, E. Zhao, F. Li, Z. Chen, Y. Li, X. Wang and Y. Yao, ACS Catal., 2022, 12, 6958–6967 CrossRef CAS.
- X. She, J. Wu, H. Xu, J. Zhong, Y. Wang, Y. Song, K. Nie, Y. Liu, Y. Yang and M. T. F. Rodrigues, Adv. Energy Mater., 2017, 7, 1700025 CrossRef.
- C. Sun, J. Yang, Y. Zhu, M. Xu, Y. Cui, L. Liu, W. Ren, H. Zhao and B. Liang, J. Alloy. Compd., 2021, 871, 159561 CrossRef CAS.
- C. Jin, W. Li, Y. Chen, R. Li, J. Huo, Q. He and Y. Wang, Ind. Eng. Chem. Res., 2020, 59, 2860–2873 CrossRef CAS.
- A. Kundu, S. Sharma and S. Basu, J. Phys. Chem. Solids, 2021, 154, 110064 CrossRef CAS.
- M. Wang, P. Guo, Y. Zhang, T. Liu, S. Li, Y. Xie, Y. Wang and T. Zhu, Appl. Surf. Sci., 2018, 453, 11–22 CrossRef CAS.
- G. Zhou, J. Yang, X. Zhu, Q. Li, Q. Yu, W. El-alami, C. Wang, Y. She, J. Qian and H. Xu, J. Energy Chem., 2020, 49, 89–95 CrossRef.
- S. Tonda, S. Kumar, M. Bhardwaj, P. Yadav and S. Ogale, ACS Appl. Mater. Interfaces, 2018, 10, 2667–2678 CrossRef CAS PubMed.
- M. Sayed, B. Zhu, P. Kuang, X. Liu, B. Cheng, A. A. A. Ghamdi, S. Wageh, L. Zhang and J. Yu, Adv. Sustainable Syst., 2022, 6, 2100264 CrossRef CAS.
- B. Tahir, M. Tahir and M. G. Mohd Nawawi, Energy Fuels, 2020, 34, 14400–14418 CrossRef CAS.
- Y. Bai, L. Wilbraham, B. J. Slater, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, J. Am. Chem. Soc., 2019, 141, 9063–9071 CrossRef CAS PubMed.
- C. Ye, J. Li, Z. Li, X. Li, X. Fan, L. Zhang, B. Chen, C. Tung and L. Wu, ACS Catal., 2015, 5, 6973–6979 CrossRef CAS.
- Q. Zhang, X. Chen, Z. Yang, T. Yu, L. Liu and J. Ye, ACS Appl. Mater. Interface, 2022, 14, 3970–3979 CrossRef CAS PubMed.
- Y. Deng, J. Liu, Y. Huang, M. Ma, K. Liu, X. Dou, Z. Wang, S. Qu and Z. Wang, Adv. Funct. Mater., 2020, 30, 2002353 CrossRef CAS.
- H. Chen, A. M. Gardner, G. Lin, W. Zhao, M. Bahri, N. D. Browning, R. S. Sprick, X. Li, X. Xu and A. I. Cooper, Catal. Sci. Technol., 2022, 12, 5442–5452 RSC.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |