
Anion vacancy correlated photocatalytic CO2 to CO conversion over quantum-confined CdS nanorods under visible light†
Qing
Guo
ac,
Shu-Guang
Xia
ac,
Zhi-Kun
Xin
ac,
Yang
Wang
ac,
Fei
Liang
bc,
Xiao-Lei
Nan
ac,
Zhe-Shuai
Lin
 bc,
Xu-Bing
Li
*ac,
Chen-Ho
Tung
ac and
Li-Zhu
Wu
*ac
bc,
Xu-Bing
Li
*ac,
Chen-Ho
Tung
ac and
Li-Zhu
Wu
*ac
aKey Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, People's Republic of China. E-mail: lixubing@mail.ipc.ac.cn; lzwu@mail.ipc.ac.cn
bKey Laboratory of Functional Crystals and Laser Technology, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, Beijing 100190, People's Republic of China
cSchool of Future Technology, University of Chinese Academy of Sciences, Beijing 100049, People's Republic of China
First published on 30th January 2023
Abstract
Surface vacancies in II–VI semiconductors have been confirmed as active sites for photocatalytic CO2 reduction. Here we take quantum-confined CdS nanorod as a model photocatalyst to correlate anion vacancies with photocatalytic CO2 reduction performance. In terms of electronic structure change, CdS nanorods with more surface S vacancies give enhanced CO generation rates, which were confirmed by deliberately introducing S vacancies via a post-treatment.
Production of solar fuels or valuable chemicals from CO2 reduction driven by solar energy remains one of the most promising pathways to relieve the energy crisis and environmental disruption.1–9 Semiconductor nanocrystals containing II–VI elements have emerged as ideal candidates in the field of CO2 photoreduction,10–14 because of their large extinction coefficient, high probability of exciton generation and easy modification of surface properties.15,16 Although most of the reported systems involve external active sites,17–19 such as metal complexes20,21 and surface ligands,22 surface vacancies of II–VI nanocrystals were also found to work as active sites.23–25 For example, zinc vacancies in ZnS QDs were effective binding sites for CO2 photoreduction.26 Anion vacancies on CdSe/CdS QDs could achieve highly efficient and selective CO evolution via photocatalytic CO2 reduction.27 Moreover, S vacancies formed in situ on CdS nanoparticles contributed to electrocatalytic CO2 reduction.28 These results prompted us to understand the inherent correlation of surface anion vacancies and catalytic performance of CO2 photoreduction by semiconductors nanocrystals.
In this contribution, we chose quantum-confined CdS nanorod as a model photocatalyst to explore the anion vacancy–activity relationship toward photocatalytic CO2 reduction. First, a structural model of hexagonal CdS without or with surface S vacancies was built for density functional theory (DFT) calculations,27 see details in the ESI.† Owing to the deficiency of S atoms, the four adjacent Cd atoms deviated from their original positions and demonstrated a small shift toward S vacancies with distinct lattice constriction as compared with pristine CdS (Fig. 1a andb). Electron density calculations (Fig. 1c andd) revealed that the electron redistribution around S vacancies occurred by removing a neutral S atom from the CdS supercell. The two unbound electrons became active and produced a localized electron trapping center because of positively charged S vacancies. As shown in Fig. 1e and f, a linear CO2 molecule bent on a S vacancy site after geometric optimization while remaining in the linear configuration on the perfect CdS(001) facet. The adsorption energy changed from −0.078 eV on the perfect facet to −0.815 eV on the S-vacancy facet. Meanwhile, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond was elongated from 1.17 Å to 1.268 Å and the O–C–O angle reduced from 180° to 122.37°, indicating that the CO2 molecule was highly activated on S vacancies.6,29 In contrast, the geometric structure of the CO2 molecule almost remained unchanged on the perfect CdS surface. The S vacancies changed the local electronic structure and promoted CO2 adsorption and activation.
O bond was elongated from 1.17 Å to 1.268 Å and the O–C–O angle reduced from 180° to 122.37°, indicating that the CO2 molecule was highly activated on S vacancies.6,29 In contrast, the geometric structure of the CO2 molecule almost remained unchanged on the perfect CdS surface. The S vacancies changed the local electronic structure and promoted CO2 adsorption and activation.
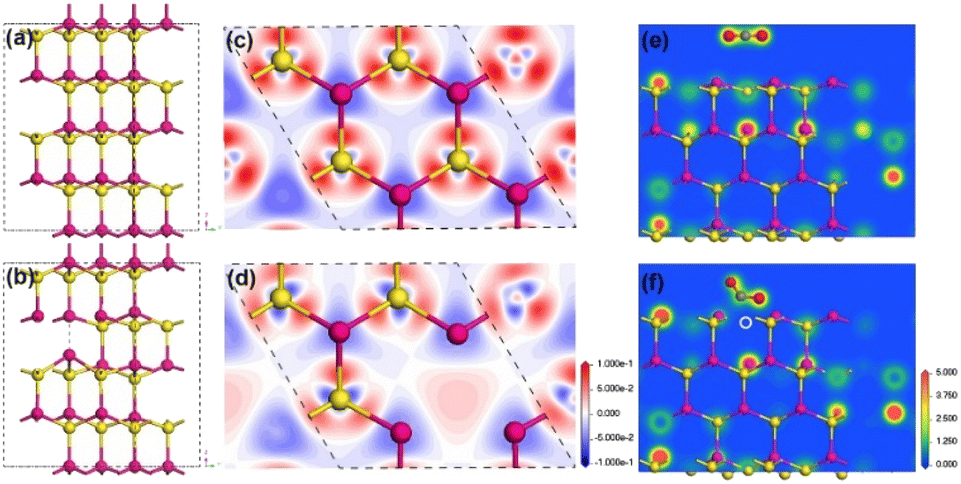 | ||
| Fig. 1 Optimized bulk lattice structure of 2 × 2 × 2 supercell hexagonal CdS without (a) and with S vacancies (b). Charge-density difference maps around the S site at the (001) cross section without (c) and with S vacancies (d). Electron density distribution around the adsorbed CO2 site at the hexagonal CdS(001) facet (e) without and (f) with S vacancies. Cd, S, C and O atoms are represented by pink, yellow, gray and red balls, respectively. The S vacancy is indicated by a white dashed circle. The color level in (c) and (d) goes from a reduction (blue) to a gain in density (red) and the units are e Å−2. | ||
Under the guidance of theoretical studies, we prepared quantum-confined CdS nanorods with different numbers of S-vacancies to work as photocatalysts towards CO2 reduction under visible light. To our delight, with more surface S vacancies, the rate of CO generation was significantly enhanced, indicating the positive correlation between CO evolution and the amount of surface S vacancies. Furthermore, the deliberate introduction of S vacancies on CdS nanorods via a post-treatment procedure of exchanging Cd2+ with Ag+ ions could also improve the rate of CO evolution by about ∼3-fold, further suggesting that the more surface S vacancies on CdS the higher the activity for CO2 photoreduction.
Quantum-confined CdS nanorods with different amounts of S vacancies were synthesized via a modified hot injection method at high temperature,30,31 see synthetic details in the ESI.† The obtained CdS nanorods with varied amounts of S vacancies were denoted as CS-1, CS-2, CS-3 and CS-4, respectively. Transmission electron microscopy (TEM) confirmed that the obtained CdS samples were in the shape of nanorods (Fig. 2a–d), in which the four CdS samples exhibited a similar size distribution with a length of ∼20 nm and a width of ∼4 nm. An enlarged aberration-corrected scanning transmission electron microscopy (STEM) image showed a lattice fringe of 3.29 Å, which was ascribed to the (002) facet of hexagonal CdS (Fig. S1, ESI†). The corresponding elemental mapping of a single CdS nanorod showed the coexistence of Cd and S elements (Fig. 2e), which was further verified by full X-ray photoelectron spectroscopy (XPS) analysis (Fig. S2, ESI†). Moreover, the narrow and sharp peaks of the prepared samples in X-ray diffraction (XRD) patterns perfectly matched the standard pattern of hexagonal CdS (PDF No. 41-1049),31 indicating the successful formation of CdS nanorods with identical shapes and crystalline structures (Fig. 3a).
 | ||
| Fig. 2 TEM images of (a) CS-1, (b) CS-2, (c) CS-3 and (d) CS-4 CdS samples. (e) STEM image of CdS nanorods and the corresponding elemental mapping, respectively. | ||
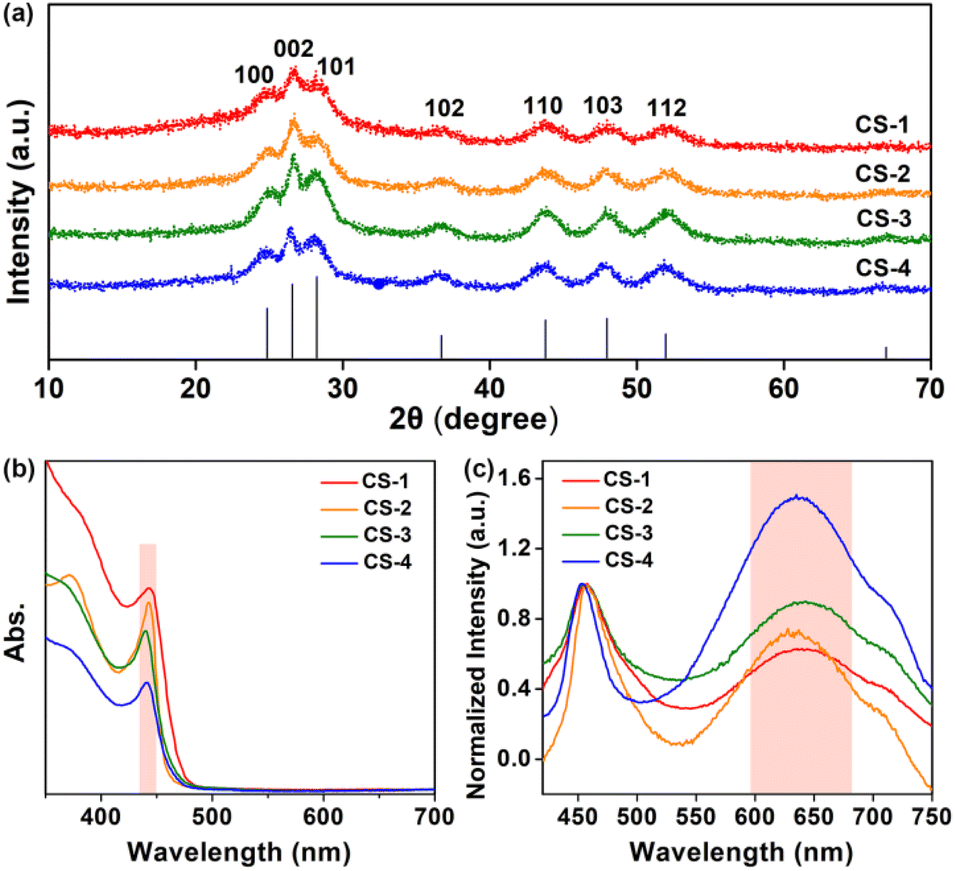 | ||
| Fig. 3 (a) XRD patterns of CS-1, CS-2, CS-3 and CS-4 CdS nanorods. (b) UV-vis absorption spectra and (c) steady-state PL spectra (excitation: 400 nm) of CdS nanorods for CS-1, CS-2, CS-3 and CS-4 in n-hexane at room temperature. | ||
The four CdS samples were then characterized in solution. As shown in Fig. 3b, the first absorption peak was centered at ∼440 nm in ultraviolet-visible (UV-vis) absorption spectra, because of the quantum confinement effects of CdS nanorods in the radial direction. A similar first absorption peak location suggested that these samples of the four CdS nanorods, either in width or length, were well maintained. Subsequently, electron paramagnetic resonance (EPR) was carried out. The observed signal centred at g = 2.0016 (Fig. S3, ESI†) resulted from anion S vacancies, in line with the fact that anion vacancies in II–VI semiconductors gave rise to EPR signals near g = 2.32–35 In addition, the line at g = 2.0016 with slight asymmetry was attributed to S vacancies located at the surface of nanorods because surface defects with an anisotropic Zeeman interaction were actually anisotropic.35 In CdS nanorods, the formation of S vacancies on the surface resulted in excess Cd sites associated with electron traps,36–38 thus leading to the formation of positively charged trap states with paramagnetism.
Given the distinct electron binding energies of inner and surface atoms, XPS analysis, a powerful technique to provide the composition information of nanomaterials, was utilized to reveal the surface stoichiometry of CdS nanorods. Taking the ratio of their respective integrated XPS signals,39,40 we calculated the percentages of surface Cd and S atoms on CdS nanorods, see details in the ESI (Fig. S4 and Table S1, ESI†). The ratios of surface atoms to whole atoms for Cd and S in CdS nanorods were determined from integrated XPS data. As summarized in Table1, the ratios of surface Cd to S atoms for various CdS samples were determined to be 3.24, 3.40, 3.70 and 3.88 from CS-1 to CS-4. Obviously, the surface Cd/S ratios of all samples were greater than 1, indicating surface Cd-rich properties.27 Thus, the existing defects in CdS were electron traps-anion S vacancies,41 and the increased Cd/S ratio from CS-1 to CS-4 suggested the enhanced amounts of S vacancies.
| Samples | Surface Cd (%) | Surface S (%) | Surface Cd/S ratio | VB vs. NHE (V) | CB vs. NHE (V) |
|---|---|---|---|---|---|
| CS-1 | 41.24 | 37.94 | 3.24 | 1.49 | −1.21 |
| CS-2 | 47.59 | 26.84 | 3.40 | 1.53 | −1.21 |
| CS-3 | 47.36 | 39.07 | 3.70 | 1.61 | −1.13 |
| CS-4 | 43.14 | 34.61 | 3.88 | 1.61 | −1.13 |
The steady-state photoluminescence (PL) spectroscopic analysis (Fig. 3c) displayed two emissions including a narrow band at ∼457 nm and a broad band at ∼630 nm, which were radiative recombinations of photogenerated electrons and holes at the band edge and the trap sites, respectively.36,42 To scrutinize the broad emission, the intensity of the narrow bands in the spectra was normalized. The significant enhancement of the trap-state emission at ∼630 nm from CS-1 to CS-4 indicated the increased amounts of surface trap states in these samples.43 Apparently, this trend was well consistent with the surface Cd/S ratio variation from CS-1 to CS-4, implying that the emission peak at ∼630 nm was radiative recombination of electron–hole at S vacancies. In addition, the corresponding PL lifetimes of the four samples decreased from 4.9 ns to 2.6 ns (Fig. S5 and Table S2, ESI†), suggesting the promoted electron–hole recombination at surface S vacancies.
Then, photocatalytic CO2 reduction experiments were carried out under ambient conditions, see details in the ESI.† The initial photocatalytic experiments were performed with triethylamine (TEA) as a sacrificial reagent under visible light (λ = 450 nm). Excitingly, CdS nanorods could produce CO gas as the sole carbon-containing product under visible light as no other reduction products were observed. Moreover, the CO generation rate was gradually improved from ∼295 mmol g−1 to ∼513 mmol g−1 with increased S vacancies from CS-1 to CS-4 under long-time irradiation (Fig. 4a). Meanwhile, negligible change was observed in the selectivity of CO, calculated according to the equation η = [nCO/(nCO + nH2)] × 100%, with variation in S vacancy amounts (Fig. 4b). Obviously, rich surface S vacancies showed vital influence on CO2 reduction, which coincided with our DFT prediction. In order to further validate the origin of generated CO, an isotopic experiment using 13CO2 as the reactant was performed, and the obtained gaseous products were analyzed by gas chromatography-mass spectrometry (GC-MS). A signal at m/z = 29 was observed (Fig. S6, ESI†), which could be assigned to the photogenerated 13CO. This result directly confirmed the photoreduction of CO2 to CO.
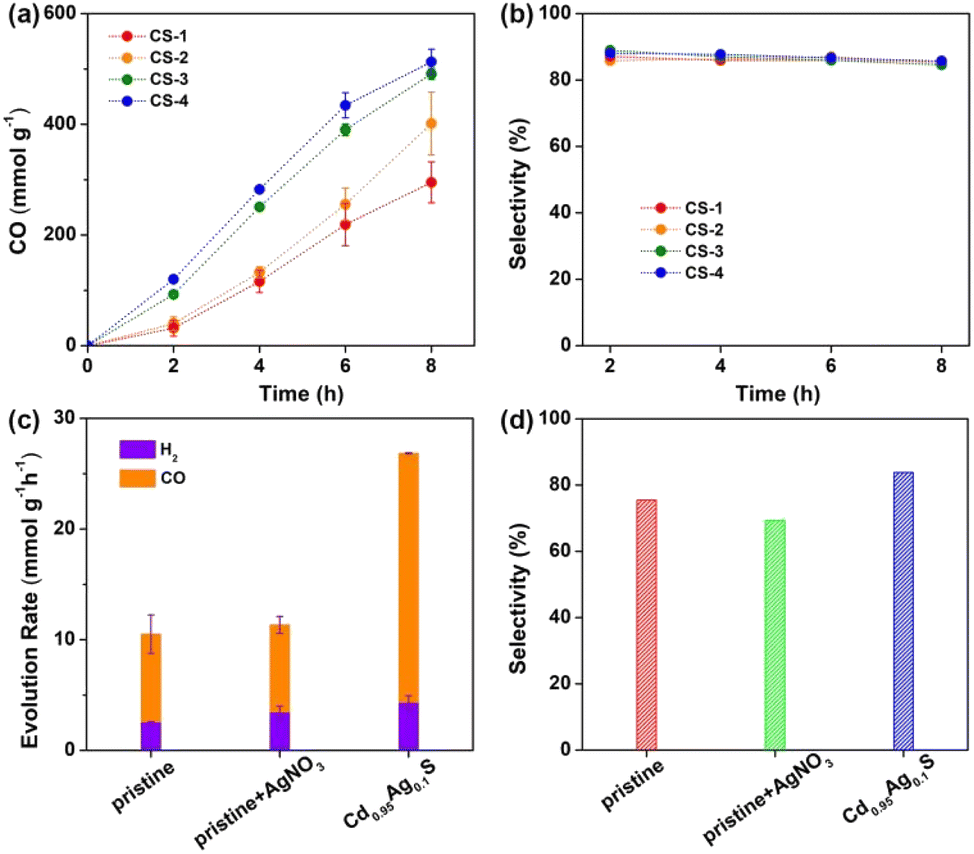 | ||
| Fig. 4 (a) Profile of photocatalytic CO2 reduction activity and (b) the corresponding CO selectivities for CS-1, CS-2, CS-3 and CS-4. (c) The rate of photocatalytic CO2 reduction, and (d) the corresponding selectivity of CO using original CdS (denoted as pristine), a physical mixture of CdS and AgNO3 (denoted as pristine + AgNO3), and CdS after Ag substitution (denoted as Cd0.95Ag0.1S) as the photocatalysts. Error bars represent mean ± s.d. of at least three independent experiments. | ||
The valence-band (VB) XPS analysis (Fig. S7, ESI†) was utilized to determine the VB potentials of CdS, which were +1.49, +1.53, +1.61 and +1.61 V versus the normal hydrogen electrode (NHE) for CS-1, CS-2, CS-3 and CS-4 (Table 1), respectively. The band gaps of the four samples were calculated in the range of ∼2.70 eV to ∼2.74 eV by using the Tauc plot (Fig. S8, ESI†). Thus, the conduction band (CB) positions were calculated to be −1.21 V, −1.21 V, −1.13 V and −1.13 V vs. NHE for CS-1, CS-2, CS-3 and CS-4, respectively. Accordingly, the driving force for CO2 reduction was almost identical with the varied amounts of S vacancies, thus excluding the influence of band-position change on CO2 reduction.
In order to further confirm the above results, anion S vacancies were deliberately introduced on CdS nanorods via a post-treatment method.35,44,45 AgNO3 was here chosen as a Ag+ precursor to realize the cation–exchange reaction under ambient conditions,39 see details in the ESI.† TEM images indicated the unchanged morphology of CdS nanorods after exchanging 0.05% Cd2+ with Ag+ ions, denoted as Cd0.95Ag0.1S (Fig. S9, ESI†). The first absorption peak indicated similar band gaps (Fig. S10, ESI†). However, the emission intensity at ∼630 nm dramatically enhanced (Fig. S11, ESI†), suggesting the increase of trap states in the form of anion S vacancies. These results matched well with those of a previous report,45 in which chemically induced Ag+ ions could achieve electronic tuning of CdS nanorods. Subsequently, photocatalytic CO2 reduction experiments were carried out with pristine CdS and Cd0.95Ag0.1S nanorods under identical conditions. As shown in Fig. 4c, the CO generation rate was improved from ∼7.9 mmol g−1 h−1 to ∼22.5 mmol g−1 h−1 after Ag+ ion exchange, almost a 3-fold enhancement. Meanwhile, the selectivity of CO increased from ∼75% to ∼84% (Fig. 4d), indicating an effective pathway to control CO2 reduction activity and selectivity. DFT simulations also showed that CO2 adsorption occurred at the trap states but not on Ag or Cd sites on the surface, indicating that CO2 activation and reduction still proceeded at the anion vacancies in Cd0.95Ag0.1S nanorods (Table S3 and Fig. S12†). In contrast, a physical mixture of CdS nanorods and AgNO3 (denoted as pristine + AgNO3) exhibited a similar CO generation rate to pristine CdS, further confirming that the improved CO generation was a result of increased anion traps, but not Ag species.
Because of the sluggish thermodynamic and kinetic nature of the linear CO2 molecule, its activation is considered as a key step for further conversion. The anion S vacancies play two crucial roles in this process: one is as active sites for CO2 adsorption and activation (vide ante DFT calculation results). The other is as effective electron trap states, which give rise to energy states energetically localized within the nanorods' energy gap and capture charge carriers from delocalized electronic states. The trap states reduce the mobility and the diffusion length of charge carriers, as illustrated in Fig. 5. In addition, the presence of trap states decreases interfacial charge transfer resistance, which is demonstrated by the smaller arc radius of the electrochemical impedance spectroscopy (EIS) Nyquist plot of vacancy-rich CdS nanorods (Fig. S13, ESI†).38 With visible light irradiation, photogenerated electrons at the CB of CdS are captured by trap states, and reduce adsorbed CO2 molecules at S vacancies on the CdS surface to generate COOH* intermediates through a proton-coupled electron transfer process. Subsequently, the COOH* intermediate is reduced by photoelectrons to generate OH* and CO* intermediates. Finally, OH* combines with a proton to produce H2O and CO* detaches from the surface to evolve the product of CO.25,46
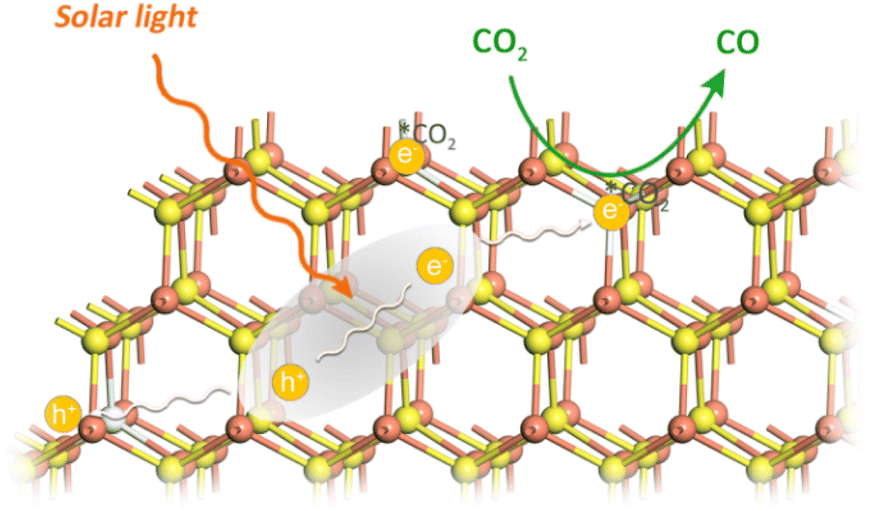 | ||
| Fig. 5 Schematic illustration of CdS nanorods with S vacancies for photocatalytic CO2 to CO conversion. | ||
In summary, we have demonstrated that rich anion S vacancies on quantum-confined CdS nanorods can effectively enhance photocatalytic CO2 reduction activity. CdS nanorods with more S vacancies show enhanced CO2 photoreduction activity. Combined with electronic tuning via the cation–exchange reaction of Ag+ substituting Cd2+ ions, the improved activity is a result of the promoted CO2 adsorption and activation, the facilitated interfacial charge transfer, and reduced mobility and diffusion length of charge carriers in the presence of high density of surface S vacancies. Understanding the influence of surface S vacancies on CdS photocatalytic CO2 reduction activity would provide new insights for tuning CO2 photoreduction performance under mild conditions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Key R&D Program of China (2022YFA1502900, 2022YFA0911900, and 2021YFA1500800), the National Natural Science Foundation of China (22088102 and 21971251), the CAS Project for Young Scientists in Basic Research YSBR-004, the Strategic Priority Research Program of the Chinese Academy of Science (XDB17000000), and the Beijing Natural Science Foundation (2222032).References
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- D. Devasia, A. J. Wilson, J. Heo, V. Mohan and P. K. Jain, Nat. Commun., 2021, 12, 2612 CrossRef CAS PubMed.
- Y. Wang, X. Shang, J. Shen, Z. Zhang, D. Wang, J. Lin, J. C. S. Wu, X. Fu, X. Wang and C. Li, Nat. Commun., 2020, 11, 3043 CrossRef CAS PubMed.
- Y. A. Wu, I. McNulty, C. Liu, K. C. Lau, Q. Liu, A. P. Paulikas, C.-J. Sun, Z. Cai, J. R. Guest, Y. Ren, V. Stamenkovic, L. A. Curtiss, Y. Liu and T. Rajh, Nat. Energy, 2019, 4, 957–968 CrossRef CAS.
- Y.-J. Xu, Angew. Chem., Int. Ed., 2021, 60, 2–25 CrossRef.
- H. Li, J. Zhao, L. Luo, J. Du and J. Zeng, Acc. Chem. Res., 2021, 54, 1454–1464 CrossRef CAS PubMed.
- G. Wang, C. T. He, R. Huang, J. Mao, D. Wang and Y. Li, J. Am. Chem. Soc., 2020, 142, 19339–19345 CrossRef CAS PubMed.
- H. L. Wu, X. B. Li, C. H. Tung and L. Z. Wu, Adv. Mater., 2019, e1900709 CrossRef PubMed.
- J. Albero, Y. Peng and H. García, ACS Catal., 2020, 10, 5734–5749 CrossRef CAS.
- P. Zhang, S. Wang, B. Guan and X. W. D. Lou, Energy Environ. Sci., 2019, 12, 164–168 RSC.
- K. K. Sakimoto, A. B. Wong and P. Yang, Science, 2016, 351, 74–77 CrossRef CAS PubMed.
- X.-B. Li, Z.-K. Xin, S.-G. Xia, X.-Y. Gao, C.-H. Tung and L.-Z. Wu, Chem. Soc. Rev., 2020, 49, 9028–9056 RSC.
- B. Su, L. Huang, Z. Xiong, Y. Yang, Y. Hou, Z. Ding and S. Wang, J. Mater. Chem. A, 2019, 7, 26877–26883 RSC.
- G. Chen, F. Wei, Z. Zhou, B. Su, C. Yang, X. F. Lu, S. Wang and X. Wang, Sustainable Energy Fuels, 2023, 7, 381–388 RSC.
- X.-B. Li, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2019, 58, 10804–10811 CrossRef CAS PubMed.
- J. Wang, T. Xia, L. Wang, X. Zheng, Z. Qi, C. Gao, J. Zhu, Z. Li, H. Xu and Y. Xiong, Angew. Chem., Int. Ed., 2018, 57, 16447–16451 CrossRef CAS PubMed.
- F. Xu, K. Meng, B. Zhu, H. Liu, J. Xu and J. Yu, Adv. Funct. Mater., 2019, 29, 1904256 CrossRef CAS.
- J. Di, C. Chen, C. Zhu, P. Song, M. Duan, J. Xiong, R. Long, M. Xu, L. Kang, S. Guo, S. Chen, H. Chen, Z. Chi, Y.-X. Weng, H. Li, L. Song, M. Wu, Q. Yan, S. Li and Z. Liu, Nano Energy, 2021, 79, 105429 CrossRef CAS.
- Y. L. Men, Y. You, Y. X. Pan, H. Gao, Y. Xia, D. G. Cheng, J. Song, D. X. Cui, N. Wu, Y. Li and S. Xin, J. Am. Chem. Soc., 2018, 140, 13071–13077 CrossRef CAS PubMed.
- M. F. Kuehnel, C. D. Sahm, G. Neri, J. R. Lee, K. L. Orchard, A. J. Cowan and E. Reisner, Chem. Sci., 2018, 9, 2501–2509 RSC.
- M. F. Kuehnel, K. L. Orchard, K. E. Dalle and E. Reisner, J. Am. Chem. Soc., 2017, 139, 7217–7223 CrossRef CAS PubMed.
- Y. -X. Feng, H.-J. Wang, J.-W. Wang, W. Zhang, M. Zhang and T.-B. Lu, ACS Appl. Mater. Interfaces, 2021, 13, 26573–26580 CrossRef CAS PubMed.
- H. Cao, J. Xue, Z. Wang, J. Dong, W. Li, R. Wang, S. Sun, C. Gao, Y. Tan, X. Zhu and J. Bao, J. Mater. Chem. A, 2021, 9, 16339–16344 RSC.
- Z.-W. Wei, H.-J. Wang, C. Zhang, K. Xu, X.-L. Lu and T.-B. Lu, Angew. Chem., Int. Ed., 2021, 60, 16622–16627 CrossRef CAS PubMed.
- P.-P. Luo, X.-K. Zhou, Y. Li and T.-B. Lu, ACS Appl. Mater. Interfaces, 2022, 14, 21069–21078 CrossRef CAS PubMed.
- H. Pang, X. Meng, P. Li, K. Chang, W. Zhou, X. Wang, X. Zhang, W. Jevasuwan, N. Fukata, D. Wang and J. Ye, ACS Energy Lett., 2019, 4, 1387–1393 CrossRef CAS.
- Q. Guo, F. Liang, X.-B. Li, Y.-J. Gao, M.-Y. Huang, Y. Wang, S.-G. Xia, X.-Y. Gao, Q.-C. Gan, Z.-S. Lin, C.-H. Tung and L.-Z. Wu, Chem, 2019, 5, 2605–2616 CAS.
- B. Qin, Y. Li, H. Wang, G. Yang, Y. Cao, H. Yu, Q. Zhang, H. Liang and F. Peng, Nano Energy, 2019, 60, 43–51 CrossRef CAS.
- A. Wagner, C. D. Sahm and E. Reisner, Nat. Catal., 2020, 3, 775–786 CrossRef CAS.
- L. Carbone, C. Nobile, D. M. Giorgi, D. F. Sala, G. Morello, P. Pompa, M. Hytch, E. Snoeck, A. Fiore, R. I. Franchini, M. Nadasan, F. A. Silvestre, L. Chiodo, S. Kudera, R. Cingolani, R. Krahne and L. Manna, Nano Lett., 2007, 7, 2942–2950 CrossRef CAS PubMed.
- J. Yang, J.-H. Zeng, S.-H. Yu, L. Yang, G.-e. Zhou and Y.-t. Qian, Chem. Mater., 2000, 12, 3259–3263 CrossRef CAS.
- J. Schneider and A. Räuber, Solid State Commun., 1967, 5, 779–781 CrossRef CAS.
- B. K. Meyer, P. Omling, E. Weigel and G. Müller-Vogt, Phys. Rev. B, 1992, 46, 15135–15138 CrossRef CAS PubMed.
- S. D. Setzler, M. Moldovan, Z. Yu, T. H. Myers, N. C. Giles and L. E. Halliburton, Appl. Phys. Lett., 1997, 70, 2274–2276 CrossRef CAS.
- A. J. Almeida, A. Sahu, A. Riedinger, D. J. Norris, M. S. Brandt, M. Stutzmann and R. N. Pereira, J. Phys. Chem. C, 2016, 120, 13763–13770 CrossRef CAS.
- H. Fujiwara, H. Hosokawa, K. Murakoshi, Y. Wada and S. Yanagida, J. Phys. Chem. B, 1997, 101, 8270–8278 CrossRef CAS.
- W. N. Nan, Y. A. Niu, H. Y. Qin, F. Cui, Y. Yang, R. C. Lai, W. Z. Lin and X. G. Peng, J. Am. Chem. Soc., 2012, 134, 19685–19693 CrossRef CAS PubMed.
- C. Pu and X. Peng, J. Am. Chem. Soc., 2016, 138, 8134–8142 CrossRef CAS PubMed.
- Z.-J. Li, J.-J. Wang, X.-B. Li, X.-B. Fan, Q.-Y. Meng, K. Feng, B. Chen, C.-H. Tung and L.-Z. Wu, Adv. Mater., 2013, 25, 6613–6618 CrossRef CAS PubMed.
- H. H. Wei, C. M. Evans, B. D. Swartz, A. J. Neukirch, J. Young, O. V. Prezhdo and T. D. Krauss, Nano Lett., 2012, 12, 4465–4471 CrossRef CAS PubMed.
- Y. Gao and X. G. Peng, J. Am. Chem. Soc., 2015, 137, 4230–4235 CrossRef CAS PubMed.
- X. Zhang, Z. Zhao, W. Zhang, G. Zhang, D. Qu, X. Miao, S. Sun and Z. Sun, Small, 2016, 12, 793–801 CrossRef CAS PubMed.
- C. Pu and X. Peng, J. Am. Chem. Soc., 2016, 138, 8134–8142 CrossRef CAS PubMed.
- D. H. Son, S. M. Hughes, Y. Yin and A. Paul Alivisatos, Science, 2004, 306, 1009–1012 CrossRef CAS PubMed.
- P. K. Jain, B. J. Beberwyck, L.-K. Fong, M. J. Polking and A. P. Alivisatos, Angew. Chem., Int. Ed., 2012, 51, 2387–2390 CrossRef CAS PubMed.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta09451g |
| This journal is © The Royal Society of Chemistry 2023 |