
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-affinity single and double helical pseudofoldaxanes with cationic guests†
Yulong
Zhong‡
 a,
Thomas A.
Sobiech‡
a,
Brice
Kauffmann
b,
Bo
Song
c,
Xiaopeng
Li
d,
Yann
Ferrand
e,
Ivan
Huc
f and
Bing
Gong
*a
a,
Thomas A.
Sobiech‡
a,
Brice
Kauffmann
b,
Bo
Song
c,
Xiaopeng
Li
d,
Yann
Ferrand
e,
Ivan
Huc
f and
Bing
Gong
*a
aDepartment of Chemistry University at Buffalo, The State University of New York Buffalo, New York 14260, USA. E-mail: bgong@buffalo.edu
bIECB, UAR3033 Univ. Bordeaux, CNRS, IN-SERM 2 rue Robert Escarpit, Pessac, 33600, France
cDepartment of Chemistry, Northwestern University Evanston, IL 60208, USA
dCollege of Chemistry and Environmental Engineering, Shenzhen University, Shenzhen, Guangdong 518060, China
eInstitut de Chimie et Biologie des Membranes et des Nano-objets, UMR 5248 CNRS, Universite de Bordeaux, F33600 Pessac, France
fDepartment Pharmazie, Ludwig-Maximilians-Universität München, D-81377 Munich, Germany
First published on 31st March 2023
Abstract
Two aromatic oligoamides, the 8-residue H8 and 16-residue H16, that adopt stable, cavity-containing helical conformations were examined for their complexation of a rodlike dicationic guest, octyl viologen (OV2+) and para-bis(trimethylammonium)benzene (TB2+). Studies based on 1D and 2D 1H NMR, isothermal titration calorimetry (ITC), and X-ray crystallography demonstrated that H8 and H16 wraps around two OV2+ ions as a double helix and a single helix, respectively, resulting in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and 1:2 complexes. Compared to H8, the longer H16 binds the OV2+ ions with much higher binding affinity and with extraordinary negative cooperativity. In contrast to its 1:2 binding with OV2+, the binding of helix H16 with the bulkier guest TB2+ shows a 1:1 ratio. Host H16 also selectively binds OV2+ in the presence of TB2+. This novel host–guest system features pairwise placement of the otherwise strongly repulsive OV2+ ions in the same cavity, strong negative cooperativity, and mutual adaptability of hosts and guests. The resultant complexes are highly stable [2]-, [3]-, and [4]pseudo-foldaxanes with few known precedents.
:2 and 1:2 complexes. Compared to H8, the longer H16 binds the OV2+ ions with much higher binding affinity and with extraordinary negative cooperativity. In contrast to its 1:2 binding with OV2+, the binding of helix H16 with the bulkier guest TB2+ shows a 1:1 ratio. Host H16 also selectively binds OV2+ in the presence of TB2+. This novel host–guest system features pairwise placement of the otherwise strongly repulsive OV2+ ions in the same cavity, strong negative cooperativity, and mutual adaptability of hosts and guests. The resultant complexes are highly stable [2]-, [3]-, and [4]pseudo-foldaxanes with few known precedents.
Introduction
A major aim in molecular recognition is the development of structurally and/or functionally tunable hosts capable of tailoring ionic and molecular guests.1 Since the discovery of crown ethers, a bewildering array of disc-like macrocyclic hosts containing two-dimensional (2D) binding cavities have been created.2 Hosts such as cryptands,3 cavitands,4 and various cages5 containing deepened cavities are also known to have drastically enhanced binding affinity and selectivity for various guest species.Tube-like macrocycles such as cyclodextrins,6 calixarenes,7 cucurbit[n]urils,8 pillar[n]arenes,9 and other systems10 provide a class of hosts offering three-dimensional (3D) cavities with legs or walls defining their inner surfaces.11 Many such hosts, especially those with covalently locked, overall rigid conformations and non-deformable cavities, show spectacular recognition capability. For example, cucurbit[n]urils exhibit remarkably tight binding affinities (up to 1017 M−1 in water) for rigid cationic guests.12
Tubular structures with cylindrical cavities of adjustable lengths (or depths) may serve as hosts with unique binding and transport capability, leading to new understanding of host–guest interactions. For example, hosts with deep cavities may accommodate long, rodlike guests and provide fundamental understanding of the role played by multiple non-covalent forces in host–guest binding. Elongated cavities spanning the lipid bilayers can serve as transmembrane channels that facilitate mass transport with selectivity and large flux,13,14 and allow the identification of guest species without relying on specific guest binding. Several self-assembling organic nanotubes are known.15–21 The majority of such nanotubes, while showing many interesting binding and transport properties, have undefined length, deformable shapes, and low stability that limit their study and applications. Molecular tubes with non-deformable inner cavities reminiscent of those of carbon nanotubes may be constructed by extending the covalent frameworks of rigid tubular macrocycles such as cucurbit[n]urils. Such a possibility, while fascinating, remains to be realized until daunting synthetic challenges are addressed.
A conceptually feasible strategy for constructing molecular tubes is the folding of synthetically accessible oligomers into helices.22 With helical cavities, such foldamers can serve as hosts for recognizing ionic and neutral guests. Examples of foldamer-based hosts for neutral small molecules were reported by Lehn,23 Moore,24 Inouye,25 Li,26 Huc,27 and Jeong;28 those for recognizing cations were described by Lehn,29 Chen,30 Fox,31 Gong,32 and Zeng;33 and hosts for anions were created by Jeong,34 Craig,35 Flood,36 Guichard,37 Berryman,38 and Gong.39 Except for a few systems,26,27,32,33 the majority of foldamer hosts known thus far are those undergoing guest-induced folding driven by binding enthalpy. Association constants (K) between such foldamers and guests including ions and small molecules are typically from 102 to 104 M−1 (up to 107 M−1) in organic solvents such as chloroform and acetonitrile; and from 103 to 104 M−1 (up to 106 M−1) in water-containing solvents with the binding driven by hydrophobic effects. As noncyclic hosts, helical foldamers exhibit unique binding behavior. For example, they can wind around rod-like dumbbell-shaped guests to give host–guest complexes, dubbed by Huc and Ferrand as foldaxanes, that possess properties both similar to and different from traditional rotaxanes.40
While a few short foldamers adopting stably folded conformations are known,26,27,32,33 multi-turn helical foldamers with stable, preorganized cavities capable of binding common organic guests, i.e., those with a size larger than the diameter (∼4 Å) of linear alkyl chains,41 are rare.42,44,45 One class of foldamers with highly stable helical conformations and a sufficiently large cavity are represented by general structure H2n (Fig. 1a, top), which we first proposed and subsequently established.22 With their backbones being constrained by highly favorable three-center hydrogen bonds,43 oligoamides H2n of different lengths, such as the 8-residue H8 and the 16-residue H16 (Fig. 1a), all fold into cavity-containing, “hollow” helices.44 Oligoamides folding into helices of up to 3 turns were synthesized recently, with the crystal structure of H16 revealing a compact helix having ∼6.6 residues per turn and a non-deformable inner pore that is ∼9 Å across.45 Independent of their lengths, helices H2n were found to be stable in solvents of different polarities and at elevated temperatures.44d Thus, by synthetically adjusting the length of oligoamides H2n, stable hollow helices with tunable but defined lengths and inner cavities of a fixed diameter are obtained. With inward-pointing amide oxygens decorating their inner walls, the cylindrical-shaped cavities of helices H2n are highly electronegative and strongly H-bonding, which are distinctly different from the hydrophobic cavities of known high-affinity hosts such as cucurbit[n]urils. As molecular tubes with electronegative cavities having multiple preorganized amide oxygen atoms, our hollow helices are expected to strongly bind cationic guests of suitable sizes.
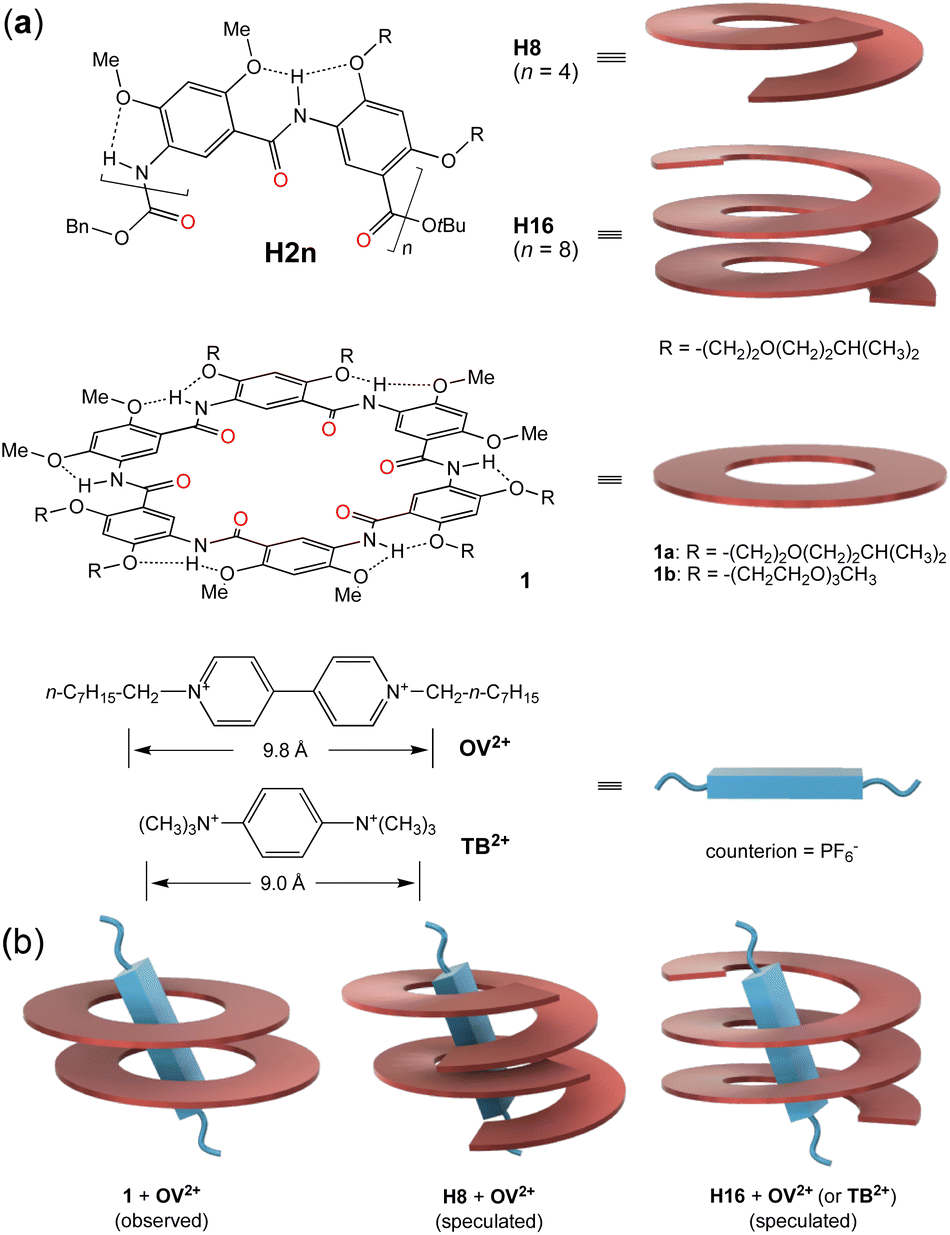 | ||
| Fig. 1 (a) Structures of aromatic oligoamide H2n, macrocycle 1, and guests OV2+ and TB2+. Cartoons on the right illustrate the helical, cyclic, and rodlike shapes of H8, H16, 1, and the two cationic guests. (b) Illustrations of the observed 2:1 complex between 1 and OV2+, the assumed 2:1 complex between H8 and OV2+, and the 1:1 complex between H16 and OV2+ or TB2+. | ||
Sharing the same rigid backbone with oligoamides H2n, six-residue macrocycles 1 (Fig. 1a, middle) were found to strongly bind octyl viologen (OV2+) (Fig. 1a, bottom) in their preorganized electronegative cavity in a highly polar solvent like DMSO or DMF.46 Since the cavity of one molecule of 1 is too “thin” to match the length of the bipyridinium segment of guest OV2+, two molecules of 1 were observed to stack to provide a cavity with a sufficient depth for binding the cationic rod (Fig. 1b, left). The cationic guest threads through the cavities of the two stacked macrocycles, leading to a 2:1 complex 12·OV2+ with overall association constants (Ktotal) of ∼1011 M−2 in DMSO/CHCl3 (1/1, v/v) and ∼108 M−2 in DMF.
The high affinity of macrocycles 1 for guest OV2+ suggests that helical oligoamides H2n, with their electronegative cavities, should also strongly bind this and other rodlike cationic guests. To match the bipyridinium rod of guest OV2+, a short (∼1 turn) helix needs to stack into a self-assembling host consisting of two or more helical molecules. In contrast, a long helix providing a sufficiently deep cavity that matches the cationic segment will serve as a unimolecular host. Here we show that helical oligoamides H2n form extremely stable complexes with OV2+ guests to generate [2]-, [3]-, and [4]-pseudofoldaxanes. Binding affinities are so strong that they lead to the unusual stacking of the dicationic guests in the electronegative interior of the helices.
Results and discussion
Design consideration
To probe the possibility of tailoring the size (length) of guests with helical oligoamides H2n, oligoamides H8 and H16 were studied for their binding with guest OV2+. Based on the 2:1 binding of macrocycles 1 with OV2+, oligoamide H8, as a helix of ∼1.2 turns, was assumed to bind guest OV2+ to give a 2:1 complex (Fig. 1b, middle). The ∼2.5-turn helix H16 provides a cavity with a depth (∼9 Å) that largely matches the length of the bipyridinium segment (∼9.8 Å, between the two N+CH2-carbons), and was conjectured to bind guest OV2+ in a 1:1 ratio (Fig. 1b, right). As a unimolecular host, H16 was expected to bind OV2+ with lowered entropic cost and thus higher binding affinity than that between helix H8 and the same guest. To further demonstrate the role of positive charges and the bulkiness of the guest in host–guest binding, cationic guest 1,4-bis(trimethylammonium)benzene (TB2+) (Fig. 1a, bottom), with a length (∼9.0 Å, between the hydrogens of the two N+(CH3)3 groups) that is the same as the depth of the cavity of helix H16, is designed and examined for its interaction with H16. Consistent with our expectation, our studies demonstrate that helix H8 assembles into dimeric (double helical) host (H8)2 for guests OV2+, while helix H16 serves as a unimolecular host for guests OV2+ and TB2+. Surprisingly, the interiors of double helix (H8)2 and helix H16 are able to accommodate two otherwise highly repulsive OV2+ ions.
Binding process and stoichiometry
The host–guest interactions between oligoamides H8 or H16 and OV2+ were first probed with 1H NMR titration experiments. Since the 1H-NMR signals of H8 or H16 and its complex(es) with OV2+ are broadened at room temperature, this prevents the assignment of the 1H NMR resonances and hampers 2D NMR studies. 1H NMR studies were performed at 45 °C at which the 1H NMR signals turn sharp, allowing all signals to be properly assigned. In DMSO-d6/CDCl3 (3/7, v/v), titrating H8 with 0 to 2 equiv. of OV2+ resulted in a downfield shift of the resonances of aromatic protons b1 (Fig. S1†), b2 through b7 of H8, and protons α and β of OV2+ (Fig. 2a), along with the upfield shift of the signal of proton b8 (Fig. 2a). Among the signals of aromatic protons b2 through b8, those of protons b3, b5, b7, and b8 remained well dispersed with an increasing proportion of OV2+ (Fig. 2a). With more than one equivalent of OV2+, the resonances of aromatic protons b2 through b8 (Fig. 2a) show insignificant shifts, while the signals of protons α and β of OV2+ continue to move upfield, approaching those of a free OV2+ ion with increasing proportions of the guest.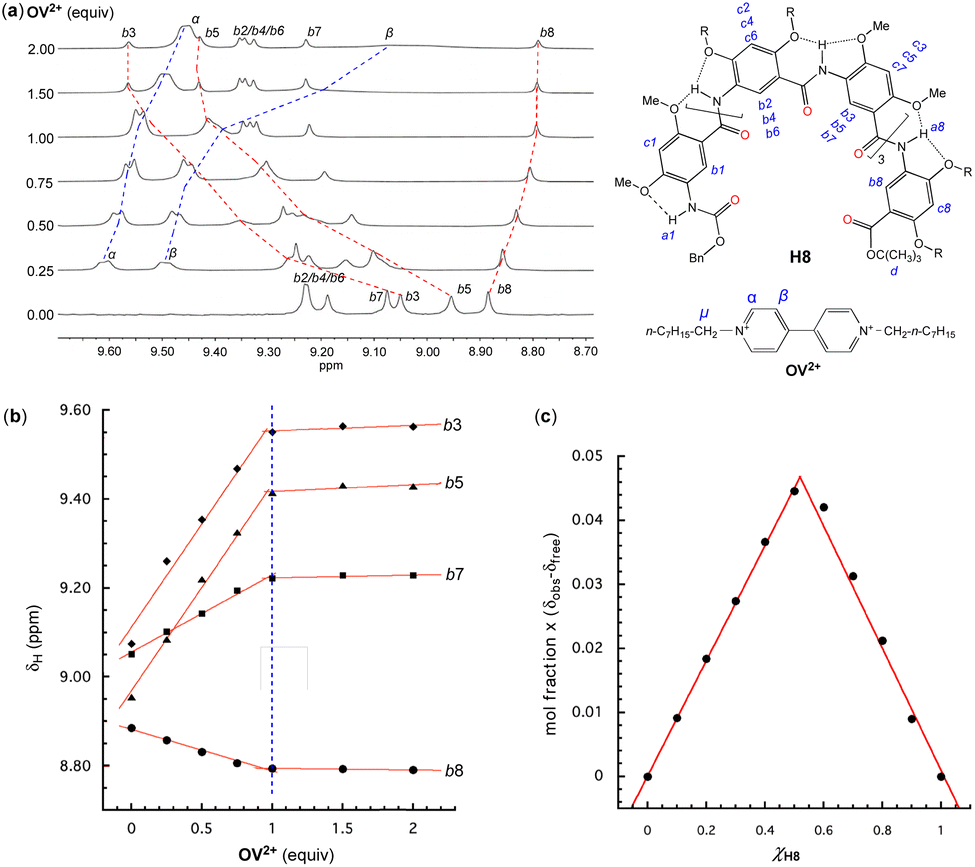 | ||
| Fig. 2 Oligoamide H8 (1 mM) titrated with 0 to 2 equiv. of guest OV2+·(PF6−)2 in DMSO-d6/CDCl3 (3/7, v/v) at 45 °C. (a) 1H NMR spectra (400 MHz), (b) changes in chemical shifts (δH) of aromatic protons b3, b5, b7, and b8 of H8vs. the equiv. of OV2+·(PF6−)2 and, (c) Job's plot based on the chemical shifts of proton b8 of H8 in the presence of different ratios of OV2+·(PF6−)2. The assignment of 1H resonances was assisted with 2D (NOESY) spectra. | ||
Plotting the chemical shifts of protons b3, b5, b7, and b8 of H8 against the ratio of OV2+ reveals a linear dependence that changes abruptly at one equiv. of OV2+ (Fig. 2b), indicating that H8 and OV2+ bind in a 1:1 ratio that is corroborated by a Job plot (Fig. 2c). The observation of only one set of 1H NMR signals with varying proportions of OV2+ suggests that the free and bound host and guest undergo rapid exchange on the 1H NMR time scale. Since the 1H resonances of H8 exhibit an insignificant shift with ≥1 equiv. of OV2+, the equilibrium must have shifted toward the presumable 1:1 complex as the dominant species.
Titrating H16 with OV2+ led to significant changes in the aromatic and amide region from 5.9 to 10.4 ppm (Fig. 3). With <1 equiv. of OV2+, the region containing the resonances of internal aromatic protons b1 through b16 is found to contain many (∼32 aromatic Hs) signals that indicate the presence of free H16 and the 1:1 complex of H16 and OV2+ in slow exchange. With 1 equiv. of OV2+, only the 16 new signals attributed to the 1:1 complex remain. With more than one but less than two equiv. of OV2+, the 16 peaks attributed to the 1:1 complex and another set of ∼16 new peaks corresponding to the 1:2 complex are found in this region, indicating that the 1:1 and 1:2 complexes are in slow exchange; with >2 equiv. of OV2+, the second set of 16 new peaks remain and the peaks of the 1:1 complex completely disappear, suggesting the presence of only the 1:2 complex of H16 and OV2+. In addition, the signals of protons α and β belonging to free OV2+ at 9.4 and 8.7 ppm are observed with >2 equiv. of OV2+. These observations indicate that the binding of H16 with OV2+ happens stepwise. The formation of the 1:1 complex occurs first as up to 1 equiv. of OV2+ is added, followed by the appearance of the 1:2 complex with ≥1 equiv. of OV2+. The presence of ≥2 equiv. of OV2+ completely drives the equilibrium toward the side of the 1:2 complex. In the presence of >2 equiv. of OV2+, the unchanged signals of H16 and the simultaneous presence of both the free and bound OV2+ suggest that H16 strongly binds OV2+, with the free and bound OV2+ undergoing no or slow exchange on the NMR time scale.
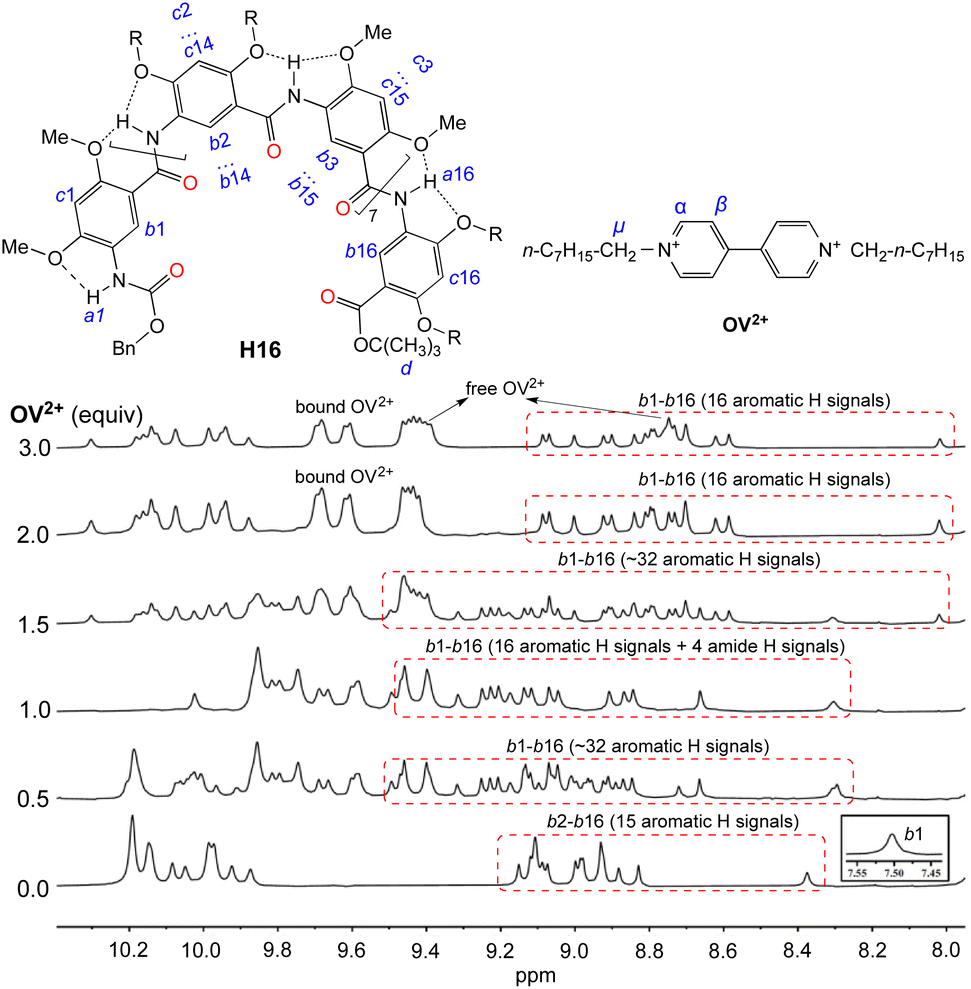 | ||
| Fig. 3 1H NMR spectra of H16 (1 mM) titrated with 0 to 3 equiv. of OV2+·(PF6−)2 in DMSO-d6/CDCl3 (2/3, v/v) at 45 °C. The assignment of 1H resonances was assisted with 2D (NOESY) spectra. | ||
To provide additional evidence for the binding stoichiometry between H8 or H16 with OV2+, mixtures of H8 or H16 and guest OV2+ in different ratios were examined with electrospray-ionization quadrupole time-of-flight mass spectrometry (ESI-QTOF). The mass spectrum of the 2:1 mixture of H8 and OV2+ contains two peaks given by the 2:1 complex (H8)2·OV2+, along with a third peak corresponding to the 1:1 complex H8·OV2+ (Fig. S2a†). The mass spectrum of the 1:1 mixture of H8 and OV2+ (Fig. S2b†) reveals a major peak for H8·OV2+, the 1:1 complex, and another peak of (H8)2·OV2+, the 2:1 complex. Surprisingly, a peak corresponding to (H8)2·(OV2+)2, the 2:2 complex, which cannot be distinguished from the 1:1 complex by NMR, is also observed. In the spectrum of the 1:2 mixture of H8 and OV2+ (Fig. S2c†), the ions of the 1:1 complex H8·OV2+ (dominant), 2:1 complex (H8)2·OV2+ (much weaker), and 2:2 complex (H8)2·(OV2+)2 are detected. In contrast, the 1:2 complex H8·(OV2+)2 could not be clearly detected in the spectra of the mixtures. These observations suggest that H8 can bind with OV2+ in both 1:1 and 2:2 ratios. Complex (H8)2·OV2+, which was observed in the mass spectra of all three mixtures, seems to be the intermediate between the 1:1 and 2:2 complexes.
The ESI-QTOF spectrum of the 1:1 mixture of H16 and OV2+ reveals the presence of only the 1:1 complex H16·OV2+ (Fig. S3a†). With the proportion of OV2+ being doubled, the 1:2 mixture of H16 and OV2+ gives a mass spectrum containing peaks of both the 1:1 complex H16·OV2+ and 1:2 complex H16·(OV2+)2 (Fig. S3b†), which suggests that increasing the ratio of OV2+ drives the complexation of the second guest OV2+ into the cavity of H16.
The results from studies mainly based on mass spectrometry and confirmed by single crystal structures (see Fig. 5 below) indicate that the originally expected complexation stoichiometry and processes of H8 and H16 with OV2+ shown in Fig. 1 need to be revised. As shown in Fig. 4a, the complexation of H8 for OV2+ involves the initial formation of the 1:1 complex, followed by the binding of the second molecule of H8 to give the 2:1 complex which, by binding the second OV2+, yields the 2:2 complex (Fig. 4a). The 1:1, 2:1, and 2:2 complexes of H8 and OV2+ undergo rapid exchange as shown by the observation of only one set of signals throughout the 1H NMR titration (Fig. 2a), making it impossible to distinguish the complexes detected by ESI-TQF in solution. Thus, the major species in solution might be the 1:1 complex, a possibility that is not supported by evidence from mass spectrometry and X-ray structure, which clearly indicate the presence of the 2:2 complex.
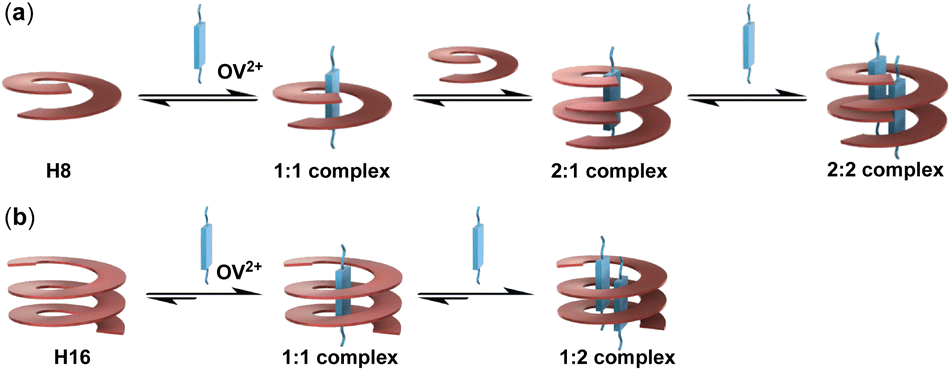 | ||
| Fig. 4 Schematic illustrations of the complexation processes of (a) oligoamide H8 and (b) oligoamide H16 with guest OV2+. | ||
In solution, the formation of the (H8:OV2+) 2:1 complex from the binding of the 1:1 complex with another molecule of H8 is very likely accompanied by positive cooperativity that is promoted by the favorable stacking interactions between the two molecules of H8, along with additional C–H⋯O interactions in the 2:1 complex (H8)2·OV2+. Such favorable (positive) binding cooperativity was directly observed in the binding of macrocycle 1 (Fig. 1), which shares the same aromatic backbone with H8 or H16, with OV2+. However, one OV2+ could only interact with about half of the binding sites (amide carbonyls) in the cavity of H8 or its dimer, which would lead to ineffective binding interaction. Binding the second OV2+ ion to the 2:1 complex (H8)2·OV2+ to give the 2:2 complex should be a negative cooperative process due to the repulsion resulted from stuffing two OV2+ ions into the cavity of the H8 dimer. This unfavorable, negative cooperative process offsets the favorable electrostatic and C–H⋯O interactions the OV2+ ions experienced in the electronegative, strongly H-bonding cavity of the H8 dimer, something that is also exhibited by H16. Based on these considerations, in solution, the 2:2 complex is more likely to be the major species.
The binding of H16 with OV2+ follows a clear stepwise path, with the 1:1 and 1:2 complexes, and the unbound H16 and OV2+ undergoing slow exchange (Fig. 4b). The 1:1 complex exists as the only host–guest species with up to 1 equiv. of OV2+, and the 1:2 complex as the only complex with >2 equiv. of OV2+.
Binding strength and thermodynamic parameters
The binding of OV2+ with H8 and H16 was then probed with isothermal titration calorimetry (ITC) which, in addition to determining the association constants (K), also provides the corresponding thermodynamic parameters including changes of enthalpy (ΔH), entropy (ΔS), and free energy (ΔG) as shown in Table 1. In MeOH/CHCl3 (3/7, v/v) at 35 °C, the ITC thermogram (Fig. S4a(i)†) for the binding of H8 with OV2+ is consistent with a 1:1 binding ratio, with an association constant over 106 M−1 that reflects both the 1:1 and 2:2 binding modes. In the same solvent, the complexation of H16 for OV2+ gives an association constant (K1) over 108 M−1 for the first binding event, followed by that (K2) of the second binding event that is three orders of magnitude smaller than K1 (Fig. S4b†). The high affinity of the first binding event approaches the upper limit of ITC measurements, leading to K1 with a significant error. In the more polar DMSO/CHCl3 (1/1, v/v) at 45 °C, the binding of H8 with OV2+ gives an apparent association constant of ∼105 M−1 (Fig. S4(ii)†), while the affinities of H16 and OV2+ are also reduced (Fig. S5†), with K1 over 107 M−1 and K2 over 104 M−1 that are in the range allowing accurate ITC measurements.
| Host | Guest | Solvent | Temp. (°C) | −ΔH (kcal mol−1) | TΔS (kcal mol−1) | K (or K1, K2) (M−1) | K total (M−2) | α |
|---|---|---|---|---|---|---|---|---|
| a Interaction factor α = 4K2/K1. (α > 1: positive cooperativity; α < 1: negative cooperativity; α = 1 no cooperativity). b K total = K1 × K2. | ||||||||
| H8 | OV2+ | MeOH/CHCl3, (3/7, v/v) | 35 | 8.2 ± 0.1 | 1.3 ± 0.1 | (5.5 ± 1.0) × 106 | — | — |
| H16 | OV2+ | MeOH/CHCl3, (3/7, v/v) | 35 | 2.7 ± 0.1 (ΔH1) | 9.4 ± 0.4 (ΔS1) | (3.6 ± 2.0) × 108 (K1) | (1.7 ± 1.1) × 1014 | 0.005 |
| 6.1 ± 0.1 (ΔH2) | 1.9 ± 0.1 (ΔS2) | (4.6 ± 0.3) × 105 (K2) | ||||||
| H8 | OV2+ | DMSO/CHCl3, (1/1, v/v) | 45 | 1.9 ± 0.1 | 5.3 ± 0.1 | (1.0 ± 0.1) × 105 | — | — |
| H16 | OV2+ | DMSO/CHCl3, (1/1, v/v) | 45 | 9.1 ± 0.1 (ΔH1) | 1.8 ± 0.2 (ΔS1) | (3.2 ± 0.6) × 107 (K1) | (5.2 ± 1.9) × 1011 | 0.002 |
| 4.6 ± 0.1 (ΔH2) | 1.6 ± 0.1 (ΔS1) | (1.6 ± 0.3) × 104 (K2) | ||||||
| H16 | TB2+ | DMSO/CHCl3, (1/1, v/v) | 45 | 13.0 ± 0.1 | 1.9 ± 0.5 | (3.2 ± 0.8) × 107 | — | — |
For the complexation of H16 and OV2+, the much stronger first binding step compared to the second step precluded the determination of the binding parameters with one ITC titration. Instead, in DMSO/CHCl3 (1/1), K1, along with ΔS1 and ΔH1, was obtained by first titrating H16 (50 μM) with OV2+·PF6− (0.5 mM) (Fig. S5a†); the much smaller K2, along with ΔS2 and ΔH2, was obtained by titrating the 1:1 mixture of H16 (3 mM) and OV2+·PF6− (3 mM) with OV2+·PF6− (30 mM) (Fig. S5b†). The binding of H16 for OV2+, with its second binding event being three orders of magnitude weaker than the first one, gives interaction factors α (ref. 47) of 0.005 in MeOH/CHCl3 (3/7) and 0.002 in DMSO/CHCl3 (1/1). Such remarkable negative cooperativity47 reflects the unfavorable stuffing of the second OV2+ guest into the cavity of H16. These observations demonstrate that the inner cavity of H16 offers a highly electronegative environment that not only overcomes the repulsion between the two cationic guests but also provides additional driving force for the formation of the 1:2 complex.
In MeOH/CHCl3 (3/7), the first binding event of H16 and OV2+ is entropically driven, which reflects the desolvation of, i.e., the release of methanol molecules from the cavity of H16 upon binding the first OV2+. The second binding event is enthalpically driven, due to the electrostatic attraction that drives the binding of the second OV2+ to the desolvated cavity. In contrast, in DMSO/CHCl3 (1/1), both first and second binding events of H16 and OV2+ are enthalpically driven. As an aprotic solvent, DMSO is not able to effectively solvate the electronegative cavity of H16. The poorly solvated cavity of H16, with multiple amide carbonyl groups as preorganized binding sites, is amenable to accommodating the cationic guest via attractive electrostatic interaction.
The dominant role played by electrostatic interaction is verified by the binding of rodlike guest TB2+ which, like guest OV2+, is rigid and carries two positive charges. ITC shows that (Fig. S6†) in DMSO/CHCl3 (1/1), the 1:1 binding of H16 and TB2+, with an association constant (K) over 107 M−1 that is the same as that of binding the first OV2+ with H16, is driven predominantly by a favorable (negative) enthalpy change that results from the strong electrostatic interaction between the two positive charges of guest TB2+ and the negative cavity of H16 (Table 1). Unlike the pairwise binding of OV2+, the binding of a second TB2+ ion with H16 was not observed. The bulkiness of the two trimethylammonium groups and the small aromatic surface of TB2+ are the most likely reasons that hinder the cramming of two guests TB2+ inside the cavity of H16. Thus, by performing structural tuning on the guest, the binding stoichiometry involving host H16 can be adjusted and controlled.
Binding selectivity probed with 1H NMR competition experiments
To examine the binding selectivity of H16, the binding of H16 to OV2+ in the presence of TB2+ and vice versa was compared with 1H NMR titration experiments performed in DMSO-d6/CDCl3 (2/3, v/v) at 45 °C. Titrating the 1:2 mixture of H16 and OV2+(PF6−)2 with 0–1.0 equiv. of TB2+(PF6−)2 failed to change the position of the bound OV2+ ions, with the signal of aromatic protons of TB2+ remaining the same as that of the free (unbound) TB2+ ion (Fig. S7a†). In contrast, titrating the 1:1 mixture of H16 and TB2+(PF6−)2 with 0–3.0 equiv. of OV2+(PF6−)2 led to an upfield shift of the aromatic proton signal of TB2+ (Fig. S7b†). With 2 equiv. of OV2+(PF6−)2, the signal of the TB2+ ion is very close to the position of the signal given by the free TB2+ ion. With 3 equiv. of OV2+(PF6−)2, the signal of the TB2+ ion is at the same position of that of the free TB2+ ion. These observations suggest that between OV2+ and TB2+, H16 shows a clear preference for the former despite the higher entropic cost for binding two OV2+ ions than binding one TB2+ ion.
Crystal structures of complexes (H8)2·(OV2+)2 and H16·(OV2+)2
Single crystals of the complexes of OV2+ with H8 and H16 were obtained via liquid–liquid diffusion of methanol into a dichloromethane solution in an NMR tube, which revealed the existence of the 2:2 complex (H8)2·(OV2+)2 and 1:2 complex H16·(OV2+)2 in the solid state, consistent with solution data. The structure of complex (H8)2·(OV2+)2 shows that the two molecules of H8 form a double helix in which the two helical strands pair in an antiparallel orientation, i.e., the N end of one strand aligns with the C end of the other strand, with the C ends of the two H8 molecules being placed in the middle of the double helix. In each helix, the phenyl ring of the terminal benzyl group engages in intramolecular edge-to-face interaction with the oligomide backbone.
The bipyridinium segments of the two OV2+ ions bind to the cavity of the double helix (Fig. 5a, top) and engage in C–H⋯O interactions involving 14 of the 16 aromatic C–H groups of the two OV2+ ions and the amide carbonyls of H8, with an average H⋯O distance of 2.45 Å. N+⋯O distances of 3.02, 3.20, 3.65, and 3.98 Å are found between each of the pyridinium N+ atoms and its nearest amide carbonyl oxygen of H8, indicative of strong charge–dipole interactions. To reduce the repulsion between the two OV2+ ions, the two bipyridinium segments in the cavity of the double helix align in an offset way, with their long axes crossing over one another with an angle of ∼27° and the N+⋯N+ distances between the ends of the two bipyridinium units being 5.52 Å and 5.98 Å (Fig. 5a, bottom). Two of the four pyridinium rings of the OV2+ ions face each other with an average distance of ∼4.3 Å, indicative of very weak, if any, stacking interaction.
 | ||
| Fig. 5 Crystal structures of (a) complex (H8)2·(OV2+)2 (top), along with the two OV2+ ions in the cavity of the double helix of H8 (bottom) and, (b) complex H16·(OV2+)2 (top), along with the two OV2+ ions in the cavity of helix H16 (bottom). The N+⋯N+ distances between the termini of the two OV2+ ions in each complex are highlighted with red dashed lines. For clarity, all side chains are replaced with methyl groups; the PF6− counterions and included solvent molecules are omitted. | ||
Although double helices have been found in a number of other oligoamides,48 the formation of a double helix shown with oligoamide H8 is the first example for this series of oligoamides. The capability of oligoamides H2n to assemble in such a way was unknown, which indicates the possibly similar behavior in the absence or presence of guests.
Similar to that of OV2+ with H8, the binding of OV2+ to the cavity of H16 is driven by C–H⋯O interactions involving 14 of the 16 aromatic CH groups of the OV2+ ions and the amide carbonyls of H16, with an average H⋯O distance of 2.39 Å that is shorter than that (2.45 Å) in complex (H8)2·(OV2+)2 (Fig. 5b, top). N+⋯O distances of 2.97, 3.10, 3.12, and 4.30 Å, which are overall shorter than those in complex (H8)2·(OV2+)2, are found between each of the pyridinium N+ atoms and its nearest amide carbonyl oxygen of H16, indicative of charge–dipole interactions that are stronger than those in complex (H8)2·(OV2+)2. Unlike the offset alignment of the bipyridinium segments of the two OV2+ ions in the cavity of double helix (H8)2, the bipyridinium segments in the cavity of H16 slide much less along their long axes and show a larger extent of overlap. The two aligned cationic rods have their four aromatic rings being partially stacked with an average stacking distance of 4.0 Å, indicating weak but noticeable stacking interaction (Fig. 5b, bottom). To avoid the N+ atoms directly facing one another, the bipyridinium segments cross over one another, with an angle of ∼42° between their long axes. The N+⋯N+ distances of 4.31 and 4.43 Å between the ends of the two bipyridinium units are much shorter than those than in complex (H8)2·(OV2+)2. This arrangement would explain the negative cooperativity between the first and second OV2+ binding events.
The crystal structures of complexes (H8)2·(OV2+)2 and H16·(OV2+)2 reveal that the two guests OV2+ in each complex adjust their alignment to optimize their interaction with each host. Compared to that in complexes (H8)2·(OV2+)2, the cationic segments of OV2+ ions in complex H16·(OV2+)2, being confined in the more compact cavity of H16, have their ends being placed more closely as shown by shorter N+⋯N+ distances, leading to a quadruply charged, shorter dimer that engages in stronger interactions with the host as shown by the shorter C–H⋯O and N+⋯O distances. Additional insights into this unique host–guest system are gained by comparing the crystal structure of H16 alone45 with that in complex H16·(OV2+)2, which indicates that the helical host fine-tunes its folded structure upon binding OV2+. Compared to the ∼9 Å diameter of the cavity of helix H16 alone,45 the cavity diameter of helix H16 in complex H16·(OV2+)2 increases to over 9.5 Å, presumably to better match the size of the bound OV2+ dimer. Such host–guest mutual adaption is made possible by the self-assembling nature of the dimeric guest and the limited conformational flexibility of the host, leading to enhanced host–guest interaction shown by the high binding affinities of H16 for OV2+ and TB2+.
Thus, by overcoming the otherwise strong electrostatic repulsion between guests OV2+, the electronegative cavities of double helix (H8)2 and helix H16 are able to bind a pair of guest OV2+ in high affinity. The pairwise binding of the dicationic guests shown by H8 and H16 is precedented by few known host–guest systems. In fact, few hosts are known to allow two or more viologen-based guests, i.e., four or more positive charges, to be placed in close proximity in the same cavity. Cucurbit[8]uril (CB[8]), although having a cavity large enough to accommodate two molecules or ions with sizes comparable to that of the bipyridinium segment of viologens, was reported to form a 1:1 complex with methyl viologen (MV2+). The complexation of one CB[8] for two MV2+ ions and similar guests has not been realized, presumably because of the strong electrostatic repulsion between the two dicationic guests in the cavity.49 To the best of our knowledge, the only known 1:2 complexes having a dimer of a viologen-based guest residing in the cavity of a host were reported by Chen et al.50 With the binding affinities being presented as “average association constants” (Kav) of ∼103 M−1 in CD3CN/CDCl3 (1/1, v/v), these 1:2 complexes involve a host based on a triptycene-based macrotricycle that provides a partially rigid binding cavity deeper than those of common macrocycles. The guests are viologens carrying β-hydroxyethyl or γ-hydroxypropyl end chains. In this system, the terminal hydroxyl groups of these guests seemed to play a critical role in driving the formation of the 1:2 complex since other guests based on dialkyl viologens could only form 1:1 complexes with this host.
Solution structures of the complexes probed with two-dimensional NMR spectroscopy
As revealed by 1D 1H NMR spectroscopy, ITC measurements, and X-ray crystallography, the binding of guest OV2+ with oligoamides H8 and H16 leads to 2:2 complex(H8)2·(OV2+)2 and 1:2 complex H16·(OV2+)2, respectively. With the assignment of 1D 1H NMR spectra being assisted by COSY and NOESY spectra, the solution structures of these complexes were revealed with two-dimensional (NOESY) 1H NMR experiments.
Fig. 6 shows the partial NOESY spectra of H8 and OV2+ (1:1) recorded in DMSO-d6/CDCl3 (7/3, v/v) at 45 °C. Strong NOEs between aromatic protons b2 through b8 of H8 and aromatic protons α and β (Fig. 6a), and methylene protons μ (Fig. 6b) of OV2+, are revealed. NOEs between protons b1 and α, β, and μ are also detected (Fig. S8†). In contrast, no NOE is observed between protons α, β, or μ of OV2+ and aromatic protons c1–c8, i.e., the “exterior” protons of H8. These observations demonstrate that the OV2+ ions reside in the cavity of the double helix. A NOE between protons d of H8 and proton b2 confirms that H8 remains folded when binding guests OV2+ (Fig. 6c). Another NOE involving protons d and β is also noticed (Fig. 6c), while no NOE can be found between protons d and α, which suggests that the two molecules of H8 have their C-termini placed in the interior and their N-termini located at the two ends of double helix (H8)2. This observation is consistent with what is revealed by the crystal structure of complex (H8)2·(OV2+)2, i.e., in solution, the two molecules of H8 constitute a double helix with an average C2 symmetry in which the C-terminal residues are placed near each other (Fig. 6d).
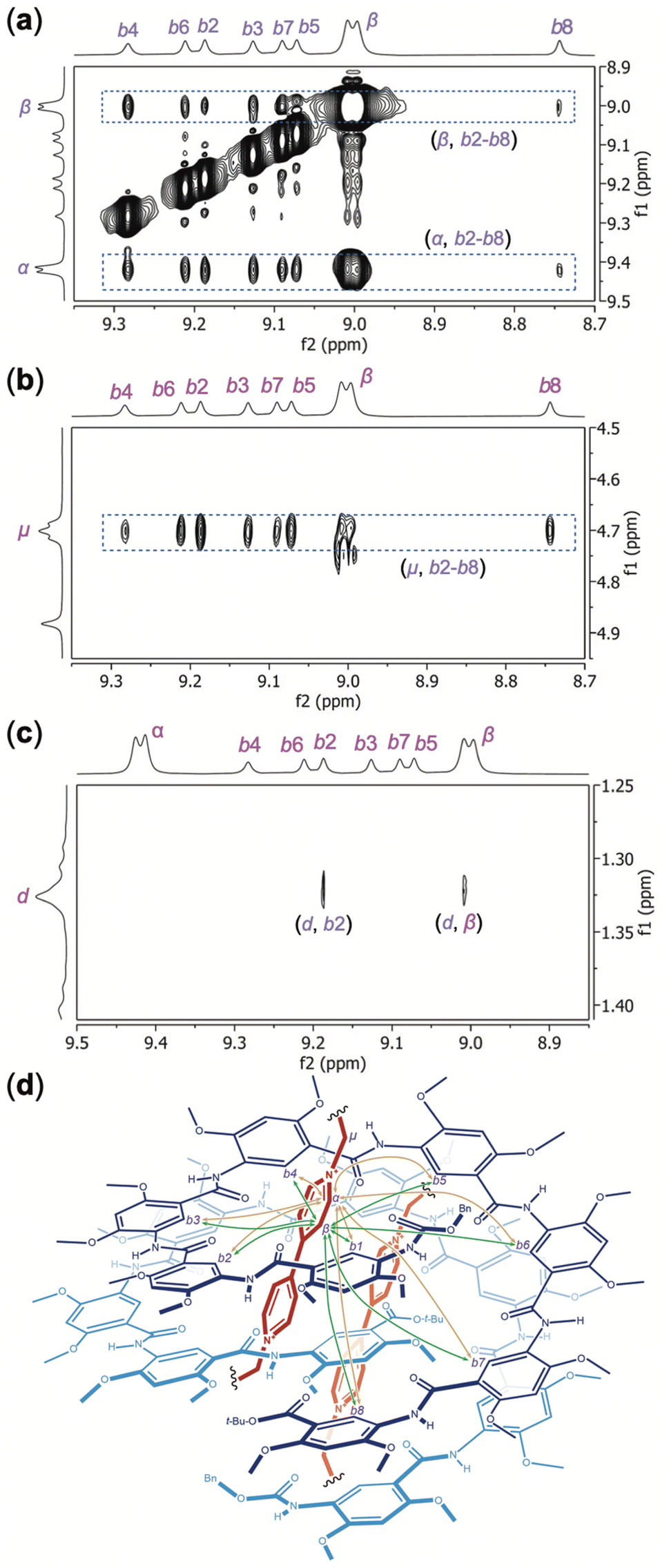 | ||
| Fig. 6 Partial NOESY spectra (500 MHz, mix time: 500 ms) of H8 (5 mM) and OV2+·(PF6−)2 (5 mM) in DMSO-d6/CDCl3 (7/3, v/v) recorded at 45 °C reveal NOEs between (a) protons α and β of OV2+ and protons b2 through b8 of H8, (b) protons μ of OV2+ and protons b2 through b8 of H8, and (c) protons d and b2, and protons d and β. (d) Illustration of the 2:2 complex of H8 and OV2+, with the major NOEs between the internal aromatic protons b1 through b8 of H8 and protons α (shown in yellow) and β (shown in green) of OV2+ being indicated with double-headed arrows. | ||
The backbone of H16 follows an N-to-C direction, leading to an unsymmetrical cavity for helix H16. Upon binding with H16, the two groups of otherwise equivalent aromatic protons of OV2+ give four discrete 1H NMR peaks, suggesting that the two OV2+ ions in the cavity of H16 are desymmetrized. Likewise, protons μ, i.e., those of the methylene groups directly attached to the N+ atoms of OV2+, also give multiple signals. Assisted by COSY and NOESY spectra, the aromatic protons of OV2+ corresponding to the four 1H NMR signals are labeled as α, β, γ, and δ. Four 1H resonances can be attributed to methylene protons μ of OV2+, which is due to the desymmetrization of the bound OV2+ ions and the methylene protons being diastereotopic in a helical cavity. The methylene protons corresponding to these four resonances are labeled as μ1, μ1′, μ2, and μ2′. The two halves of the desymmetrized OV2+ ion are defined by protons α, β, and μ1/μ1′, and protons γ, δ and μ2/μ2′, respectively.
The NOESY spectrum of H16 and OV2+ (1:2) recorded in DMSO-d6/CDCl3 (4/6, v/v) at 45 °C reveals different NOEs between aromatic protons b1 (Fig. S9†), b2 through b16 of H16 and protons α, δ (Fig. 7a), β, γ (Fig. 7b), and μ1/μ1′, μ2/μ2′ (Fig. 7c) of OV2+. Strong NOEs are found between protons α and protons b2, b3, b4, b5, b6, and b7 (Fig. 7a) and b1 (Fig. S9†) that belong to the N-terminal half of H16. The intensities of NOEs involving protons α and protons b8 to b14 follow a descending order, with no NOEs being detected between protons α and protons b15 or b16. Compared to protons α, protons δ exhibit the opposite trend in the strength of their NOEs with aromatic protons b1–b16 of H16 (Fig. 7a). Strong NOEs are found between protons δ and protons b9 through b16 that belong to the C-terminal half of H16, with NOEs involving protons δ decreasing consecutively from protons b8 to b3, and disappearing with protons b2 (Fig. 7a) and b1 (Fig. S9†). The NOEs observed between protons α and δ of OV2+, and protons b1 through b16, along with the absence of NOE between the aromatic protons of OV2+ and protons c1–c16 of H16 suggest that, similar to what is revealed by the crystal structure of complex H16·(OV2+)2, in solution, the two OV2+ ions are aligned along the long axis of the cylindrical cavity of helix H16.
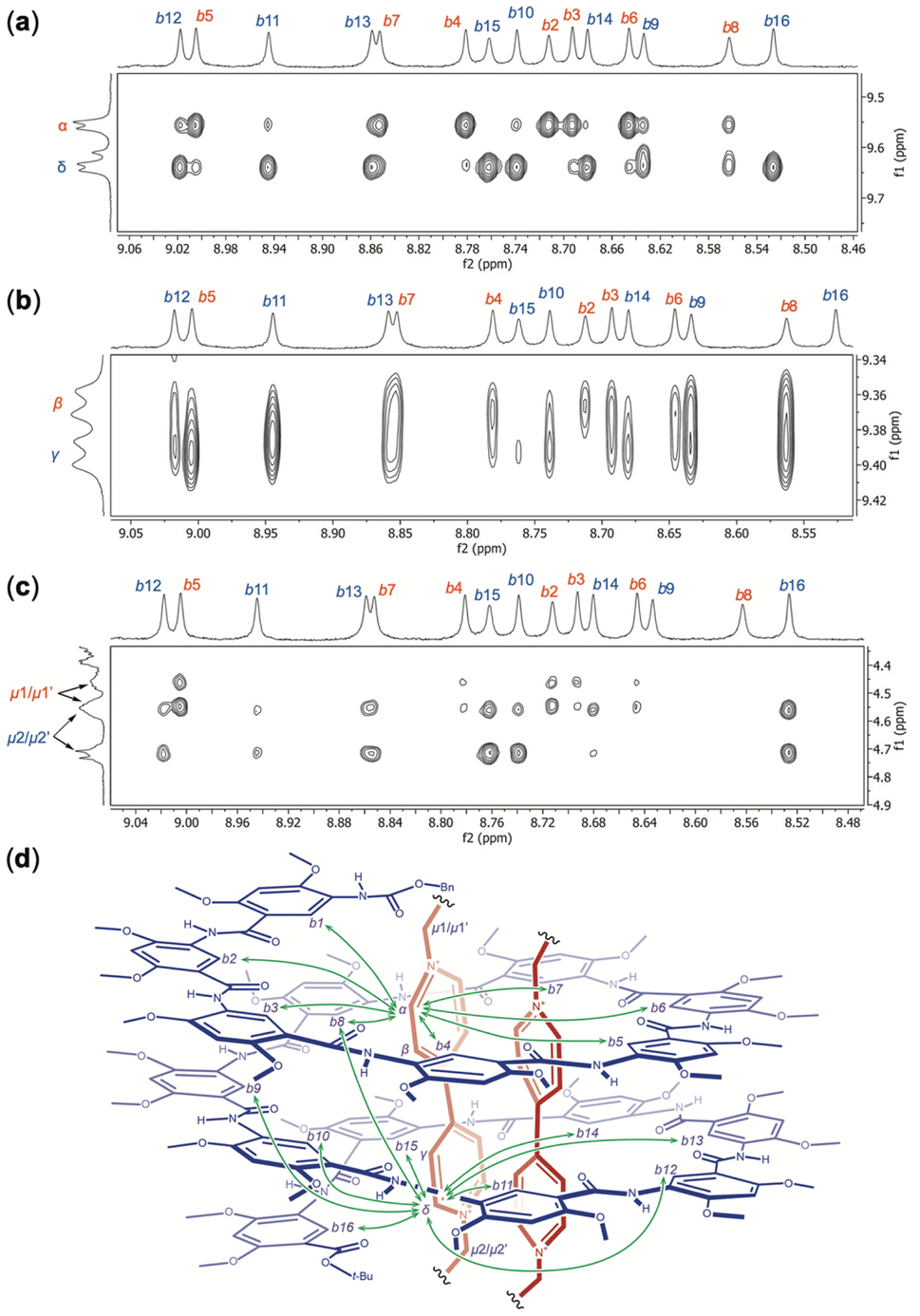 | ||
| Fig. 7 Partial NOESY spectra (500 MHz, mixing time: 500 ms) of H16 (3 mM) and OV2+·(PF6−)2 (6 mM) recorded in DMSO-d6:CDCl3 (4/6, v/v) at 45 °C reveal NOEs between protons b2 through b16 of H16 and protons (a) α and δ, (b) β or γ, and (c) μ1/μ1′ and μ2/μ2′ of OV2+. (d) Illustration of the 1:2 complex of H16 and OV2+, with the major NOEs between the internal aromatic protons b1 through b16 of H16 and protons α and δ of OV2+ being indicated with double-headed arrows. | ||
In addition to NOEs between the aromatic protons of guests OV2+ and those of H16, methylene protons μ also show NOE contacts that corroborate the solution structure of complex H16·(OV2+)2. Protons μ1/μ1′ have obvious NOE contacts with protons b1 (Fig. S9†) and b2–b7 (Fig. 7c), while NOEs between protons μ2/μ2′ and protons b10–b16 are noticeable (Fig. 7c), which indicate that protons μ1/μ1′ and μ2/μ2′ are placed near the N- and C-termini of H16, respectively.
Therefore, the two ends of the OV2+ ion, as represented by protons α and μ1/μ1′, and δ and μ2/μ2′, are placed near the N- and C-termini, respectively, of H16. In such a complex, protons β and γ of OV2+ must be placed near the middle of helix H16. Indeed, protons β and γ have NOE contacts of similar strength with the most internal aromatic protons b of H16 (Fig. 7b). The absence of NOE between protons β and protons b15 and b16, and between protons γ and protons b2 and b16 (Fig. 7b) provides additional evidence supporting the alignment of the OV2+ ions in the cavity of helix H16 in solution (Fig. 7d).
The NOESY spectrum clearly demonstrates the binding of H16 and guest TB2+ (Fig. S10†). Strong NOEs between protons b1 through b16 of H16, and aromatic protons α and methylene protons μ of TB2+ are detected, confirming that the cationic guest resides in the cavity of H16.
Conclusions
Aromatic oligoamides H8 and H16 fold into hollow helices with rigid (yet capable of slight induced-fit adjustments) electronegative cavities that strongly bind rodlike dicationic guests OV2+ and TB2+. The 8-residue H8, a helix of ∼1.2 turns, assembles into double helix (H8)2 with a cavity capable of accommodating two OV2+ ions that align in an offset fashion. The resultant 2:2 complex, (H8)2·(OV2+)2 undergoes rapid exchange with the unbound host and guest. The 16-residue H16, which can be regarded as a covalent dimer of H8, serves as a unimolecular host with a 3D cavity that strongly binds guest OV2+. The high stabilities of the resultant 1:1 complex H16·OV2+ and 1:2 complex H16·(OV2+)2 are reflected by the slow exchange on the 1H NMR time scale between the complexes and the unbound host and guest. The strong binding of H16 for OV2+ is confirmed by ITC. The extraordinary affinity of H16 for the first OV2+ ion and much weaker yet significant binding for the second OV2+ ion point to highly negative cooperativity. In contrast, helix H16 binds only one bulky guest TB2+ with the same affinity as that of H16 with the first OV2+ ion, indicating that binding stoichiometry can be adjusted by tuning the structure (bulkiness) of the guest.
X-ray crystallography provides atomic details for complexes (H8)2·(OV2+)2 and H16·(OV2+)2, which reveals the mutual adaption of the helical hosts and the dimeric guest. Double helix (H8)2 provides a cavity that fully accommodates the two OV2+ ions. The crystal structure of complex H16·(OV2+)2 reveals that the two bound OV2+ ions undergo more compact alignment than those in (H8)2·(OV2+)2, perhaps to better match the cavity of helix H16. In comparison to that of helix H16 alone, the cavity of H16 in complex H16·(OV2+)2 is slightly enlarged to better accommodate the two OV2+ ions.
In solution, the binding of OV2+ ions in the cavity of double helix (H8)2 or helix H16, the head-on alignment of H8 in duplex (H8)2, and the desymmetrization of the otherwise symmetrical guest OV2+ in the cavity of H16 are clearly demonstrated by two-dimensional (NOESY) 1H NMR spectra. The binding of H16 and guest TB2+ in solution is also been confirmed by the NOESY spectrum.
Complexes (H8)2·(OV2+)2, H16·(OV2+)2 and H16·TB2+ are hitherto unknown double and single helical, [2]-, [3]-, and [4]pseudo-foldaxanes40 featuring two and one axle components, respectively, with exceptional stabilities and a high degree of sophistication found with few rotaxanes and pseudo-rotaxanes. The observed tunability in binding stoichiometry is unusual among host–guest complexes. The presence of the discrete complex H16·(OV2+)2 demonstrates that hollow helices, as high-affinity hosts with 3D binding pockets, lead to host–guest complexes with significantly enhanced stability. The selective binding of H16 for the OV2+ ion indicates that this host is capable of achieving optimum interactions despite the entropically unfavorable nature of the complexation. The resultant pseudo-foldaxanes, with foldaxane-like stability, are formed by simple mixing of the molecular components. By being able to tailor the size (length) and/or shape of guests, hollow helices, with their ready synthetic tunability, are new hosts that are uniquely different from hosts based on most macrocycles, resulting in host–guest complexes with adjustable sizes, stability, and binding stoichiometry. A largely unexplored aspect of our hollow helices involves the inherent chirality of these molecules. Resolving the racemic helices into optically pure left- and right-handed helices will also open a wide door allowing the exploration of a variety of chiral molecular recognition and transport processes.
Data availability
All relevant data supporting this article have been included in the main text and the ESI.†Author contributions
Y. L. Z. and T. A. S. performed the major part of the experimental work including NMR data collection and ITC measurements, along with data analysis. Y. L. Z., B. S. and X. P. L. performed mass spectral studies. B. K. and Y. F. performed crystal growth, X-ray data collection, and crystal structure elucidation and refinement. B. G. conceptualised and supervised the study. I. H. contributed to the X-ray study and helped revise the manuscript. B. G. and Y. L. Z. wrote the manuscript. All authors commented on the data and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the US National Science Foundation (CHE-1905094 and 2108538 to B. G.).Notes and references
- (a) J. M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, VCH, Weinheim, 1995 CrossRef; (b) K. N. Houk, A. G. Leach, S. P. Kim and X. Zhang, Angew. Chem., Int. Ed., 2003, 42, 4872–4897 CrossRef CAS PubMed.
- (a) C. J. Pedersen and H. K. Frensdorff, Angew. Chem., Int. Ed. Engl., 1972, 11, 16–25 CrossRef CAS PubMed; (b) G. A. Melson, Coordination Chemistry of Macrocyclic Compounds, Plenum Press, New York, 1979 CrossRef; (c) J. M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, VCH, Weinheim, 1995 CrossRef.
- B. Dietrich, in Comprehensive Supramolecular Chemistry, ed. G. W. Gokel, Elsevier, Oxford, 1999, vol. 1, pp. 153–211 Search PubMed.
- (a) D. J. Cram, Science, 1983, 219, 1177–1183 CrossRef CAS PubMed; (b) A. Wishard and B. C. Gibb, Calixarenes and Beyond, 2016, pp. 195–234 Search PubMed.
- (a) L. R. MacGillivray and J. L. Atwood, Angew. Chem., Int. Ed., 1999, 38, 1018–1033 CrossRef CAS; (b) D. Fujita, K. Suzuki, S. Sato, M. Yagi-Utsumi, Y. Yamaguchi, N. Mizuno, T. Kumasaka, M. Takata, M. Noda, S. Uchiyama, K. Kato and M. Fujita, Nat. Commun., 2012, 3, 1093 CrossRef PubMed.
- R. Breslow and S. D. Dong, Chem. Rev., 1998, 98, 1997–2012 CrossRef CAS PubMed.
- C. D. Gutsche, Calixarenes, Royal Society of Chemistry, Cambridge, 1989 Search PubMed.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS PubMed.
- M. Xue, Y. Yang, X. Chi, Z. Zhang and F. Huang, Acc. Chem. Res., 2012, 45, 1294–1308 CrossRef CAS PubMed.
- (a) K. Yazaki, L. Catti and M. Yoshizawa, Chem. Commun., 2018, 54, 3195–3206 RSC; (b) P. C. Kearney, L. S. Mizoue, R. A. Kumpf, J. E. Forman, A. McCurdy and D. A. Dougherty, J. Am. Chem. Soc., 1993, 115, 9907–9919 CrossRef CAS; (c) Y.-X. Wang, Y.-M. Zhang, Y.-L. Wang and Y. Liu, Chem. Mater., 2015, 27, 2848–2854 CrossRef CAS; (d) Y.-J. Ghang, J. J. Lloyd, M. P. Moehlig, J. K. Arguelles, M. Mettry, X. Zhang, R. R. Julian, Q. Cheng and R. J. Hooley, Langmuir, 2014, 30, 10161–10166 CrossRef CAS PubMed; (e) W. Liu, E. M. Peck, K. D. Hendzel and B. D. Smith, Org. Lett., 2015, 17, 5268–5271 CrossRef CAS PubMed.
- K. Ariga and T. Kunitake, Supramolecular Chemistry—Fundamentals and Applications. Springer, New York, 2006 Search PubMed.
- M. V Rekharsky, T. Mori, C. Yang, Y. H. Ko, N. Selvapalam, H. Kim, D. Sobransingh, A. E. Kaifer, S. Liu, L. Isaacs, W. Chen, S. Moghaddam, M. K. Gilson, K. Kim and Y. Inoue, Proc. Natl. Acad. Sci., 2007, 104, 20737–20742 CrossRef PubMed.
- R. B. Schoch, J. Han and P. Renaud, Rev. Mod. Phys., 2008, 80, 839–883 CrossRef CAS.
- B. Hinds, Curr. Opin. Solid State Mater. Sci., 2012, 16, 1–9 CrossRef CAS.
- (a) D. T. Bong, T. D. Clark, J. R. Granja and M. R. Ghadiri, Angew. Chem., Int. Ed., 2001, 40, 988–1011 CrossRef CAS; (b) W. S. Childers, R. Ni, A. K. Mehta and D. G. Lynn, Curr. Opin. Chem. Biol., 2009, 13, 652–659 CrossRef CAS PubMed; (c) B. Gong and Z. Shao, Acc. Chem. Res., 2013, 46, 2856–2866 CrossRef CAS PubMed; (d) L. S. Shimizu, S. R. Salpage and A. A. Korous, Acc. Chem. Res., 2014, 47, 2116–2127 CrossRef CAS PubMed; (e) A. Ghorai, B. Achari and P. Chattopadhyay, Tetrahedron, 2016, 72, 3379–3387 CrossRef CAS; (f) A. Nitti, A. Pacini and D. Pasini, Nanomaterials, 2017, 7, 167 CrossRef PubMed.
- M. A. B. Block, C. Kaiser, A. Khan and S. Hecht, Top. Curr. Chem., 2005, 245, 89–150 CrossRef CAS.
- J. G. Moralez, J. Raez, T. Yamazaki, R. K. Motkuri, A. Kovalenko and H. Fenniri, J. Am. Chem. Soc., 2005, 127, 8307–8309 CrossRef CAS PubMed.
- P. Jonkheijm, A. Miura, M. Zdanowska, F. J. M. Hoeben, S. De Feyter, A. P. H. J. Schenning, F. C. De Schryver and E. W. Meijer, Angew. Chem., Int. Ed., 2004, 43, 74–78 CrossRef PubMed.
- (a) D. L. Gin, W. Gu, B. A. Pindzola and W.-J. Zhou, Acc. Chem. Res., 2001, 34, 973–980 CrossRef CAS PubMed; (b) M. Zhou, P. R. Nemade, X. Lu, X. Zeng, E. S. Hatakeyama, R. D. Noble and D. L. Gin, J. Am. Chem. Soc., 2007, 129, 9574–9575 CrossRef CAS PubMed.
- V. Percec, A. E. Dulcey, V. S. K. Balagurusamy, Y. Miura, J. Smidrkal, M. Peterca, S. Nummelin, U. Edlund, S. D. Hudson, P. A. Heiney, H. Duan, S. N. Magonov and S. A. Vinogradov, Nature, 2004, 430, 764–768 CrossRef CAS PubMed.
- J. M. Schnur, Science, 1993, 262, 1669–1676 CrossRef CAS PubMed.
- B. Gong, Chem.–Eur. J., 2001, 7, 4336–4342 CrossRef CAS PubMed.
- V. Berl, M. J. Krische, I. Huc, J.-M. Lehn and M. Schmutz, Chem.–Eur. J., 2000, 6, 1938–1946 CrossRef CAS PubMed.
- (a) J. C. Nelson, J. G. Saven, J. S. Moore and P. G. Wolynes, Science, 1997, 277, 1793–1796 CrossRef CAS PubMed; (b) R. B. Prince, S. A. Barnes and J. S. Moore, J. Am. Chem. Soc., 2000, 122, 2758–2762 CrossRef CAS.
- H. Abe, N. Masuda, M. Waki and M. Inouye, J. Am. Chem. Soc., 2005, 127, 16189–16196 CrossRef CAS PubMed.
- J.-L. Hou, X.-B. Shao, G.-J. Chen, Y.-X. Zhou, X.-K. Jiang and Z.-T. Li, J. Am. Chem. Soc., 2004, 126, 12386–12394 CrossRef CAS PubMed.
- (a) J. Garric, J.-M. Léger and I. Huc, Angew. Chem., Int. Ed., 2005, 44, 1954–1958 CrossRef CAS PubMed; (b) C. Bao, B. Kauffmann, Q. Gan, K. Srinivas, H. Jiang and I. Huc, Angew. Chem., Int. Ed., 2008, 47, 4153–4156 CrossRef CAS PubMed; (c) P. Mateus, B. Wicher, Y. Ferrand and I. Huc, Chem. Commun., 2018, 54, 5078–5081 RSC.
- J. Y. Hwang, H.-G. Jeon, Y. R. Choi, J. Kim, P. Kang, S. Lee and K.-S. Jeong, Org. Lett., 2017, 19, 5625–5628 CrossRef CAS PubMed.
- A.-M. Stadler, N. Kyritsakas and J.-M. Lehn, Chem. Commun., 2004, 2024–2025 RSC.
- J.-L. Hou, M.-X. Jia, X.-K. Jiang, Z.-T. Li and G.-J. Chen, J. Org. Chem., 2004, 69, 6228–6237 CrossRef CAS PubMed.
- F. Zhang, S. Bai, G. P. A. Yap, V. Tarwade and J. M. Fox, J. Am. Chem. Soc., 2005, 127, 10590–10599 CrossRef CAS PubMed.
- K. Yamato, L. Yuan, W. Feng, A. J. Helsel, A. R. Sanford, J. Zhu, J. Deng, X. C. Zeng and B. Gong, Org. Biomol. Chem., 2009, 7, 3643–3647 RSC.
- J. Shen, C. Ren and H. Zeng, J. Am. Chem. Soc., 2017, 139, 5387–5396 CrossRef CAS PubMed.
- (a) K.-J. Chang, B.-N. Kang, M.-H. Lee and K.-S. Jeong, J. Am. Chem. Soc., 2005, 127, 12214–12215 CrossRef CAS PubMed; (b) J. Suk and K.-S. Jeong, J. Am. Chem. Soc., 2008, 130, 11868–11869 CrossRef CAS PubMed.
- H. Juwarker, J. M. Lenhardt, D. M. Pham and S. L. Craig, Angew. Chem., Int. Ed., 2008, 47, 3740–3743 CrossRef CAS PubMed.
- Y. Hua, Y. Liu, C.-H. Chen and A. H. Flood, J. Am. Chem. Soc., 2013, 135, 14401–14412 CrossRef CAS PubMed.
- V. Diemer, L. Fischer, B. Kauffmann and G. Guichard, Chem.–Eur. J., 2016, 22, 15684–15692 CrossRef CAS PubMed.
- (a) C. J. Massena, N. B. Wageling, D. A. Decato, E. Martin Rodriguez, A. M. Rose and O. B. Berryman, Angew. Chem., Int. Ed., 2016, 55, 12398–12402 CrossRef CAS PubMed; (b) E. A. John, C. J. Massena and O. B. Berryman, Chem. Rev., 2020, 120, 2759–2782 CrossRef CAS PubMed.
- R. Cao, R. B. Rossdeutcher, B. Gong and X. Wu, Org. Lett., 2020, 22, 7496–7501 CrossRef CAS PubMed.
- V. Koehler, A. Roy, I. Huc and Y. Ferrand, Acc. Chem. Res., 2022, 55, 1074–1085 CrossRef CAS PubMed.
- R. Suresh, N. Venkataraman, S. Vasudevan and K. V Ramanathan, J. Phys. Chem. C, 2007, 111, 495–500 CrossRef CAS.
- N. Chandramouli, Y. Ferrand, G. Lautrette, M. Laguerre, I. Huc, B. Kauffmann, C. D. Mackereth and D. Dubreuil, Nat. Chem., 2015, 7, 334–341 CrossRef CAS PubMed.
- R. D. Parra, H. Zeng, J. Zhu, C. Zheng, X. C. Zeng and B. Gong, Chem.–Eur. J., 2001, 7, 4352–4357 CrossRef CAS PubMed.
- (a) B. Gong, Acc. Chem. Res., 2008, 41, 1376–1386 CrossRef CAS PubMed; (b) J. Zhu, R. D. Parra, H. Zeng, E. Skrzypczak-Jankun, X. C. Zeng and B. Gong, J. Am. Chem. Soc., 2000, 122, 4219–4220 CrossRef CAS; (c) B. Gong, H. Zeng, J. Zhu, L. Yua, Y. Han, S. Cheng, M. Furukawa, R. D. Parra, A. Y. Kovalevsky, J. L. Mills, E. Skrzypczak-Jankun, S. Martinovic, R. D. Smith, C. Zheng, T. Szyperski and X. C. Zeng, Proc. Natl. Acad. Sci., 2002, 99, 11583–11588 CrossRef CAS PubMed; (d) L. Yuan, H. Zeng, K. Yamato, A. R. Sanford, W. Feng, H. S. Atreya, D. K. Sukumaran, T. Szyperski and B. Gong, J. Am. Chem. Soc., 2004, 126, 16528–16537 CrossRef CAS PubMed.
- Y. Zhong, B. Kauffmann, W. Xu, Z.-L. Lu, Y. Ferrand, I. Huc, X. C. Zeng, R. Liu and B. Gong, Org. Lett., 2020, 22, 6938–6942 CrossRef CAS PubMed.
- T. A. Sobiech, Y. L. Zhong, L. S. Sánchez, B. Kauffmann, J. K. McGrath, C. Scalzo, D. P. Miller, I. Huc, E. Zurek, Y. Ferrand and B. Gong, Chem. Commun., 2021, 57, 11645–11648 RSC.
- (a) C. A. Hunter and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 7488–7499 CrossRef CAS PubMed; (b) L. K. S. von Krbek, C. A. Schalley and P. Thordarson, Chem. Soc. Rev., 2017, 46, 2622–2637 RSC.
- (a) V. Berl, I. Huc, R. G. Khoury, M. J. Krische and J.-M. Lehn, Nature, 2000, 407, 720–723 CrossRef CAS PubMed; (b) C. Bao, Q. Gan, B. Kauffmann, H. Jiang and I. Huc, Chem.–Eur. J., 2009, 15, 11530–11536 CrossRef CAS PubMed; (c) Y. Ferrand, Q. Gan, B. Kauffmann, H. Jiang and I. Huc, Angew. Chem., Int. Ed., 2011, 50, 7572–7575 CrossRef CAS PubMed.
- H.-J. Kim, J. Heo, W. S. Jeon, E. Lee, J. Kim, S. Sakamoto, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 1526–1529 CrossRef CAS PubMed.
- J.-M. Zhao, Q.-S. Zong, T. Han, J.-F. Xiang and C.-F. Chen, J. Org. Chem., 2008, 73, 6800–6806 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1D and 2D 1H NMR spectra, ESI-TOF spectra, ITC isotherms and procedures, crystallogenesis and X-ray data. CCDC 2103075 and 2103076. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00524k |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |