
The effect of photolatent catalysts on the exchange kinetics of dual-wavelength 3D printable and photopatternable thiol-click vitrimers†
Elisabeth
Rossegger
,
Usman
Shaukat
,
Khadijeh
Moazzen
 ,
Mathias
Fleisch
,
Michael
Berer
and
Sandra
Schlögl
*
,
Mathias
Fleisch
,
Michael
Berer
and
Sandra
Schlögl
*
Polymer Competence Center Leoben GmbH, Roseggerstrasse 12, A-8700 Leoben, Austria. E-mail: sandra.schloegl@pccl.at
First published on 2nd May 2023
Abstract
The use of photolatent transesterification catalysts provides a unique way to locally control dynamic bond exchange reactions in vitrimers. In the non-illuminated state, the polymer network behaves like a permanent crosslinked duromer. Upon light exposure, the catalysing species is formed on demand facilitating topology arrangements above the network's topological freezing temperature (Tv). For photopolymer networks, photoacid generators provide distinctive advantages as latent catalysts. They are well soluble, comprise a high temperature stability and release strong Brønsted acids upon light exposure, which are able to efficiently catalyse thermo-activated transesterifications. In addition, they are typically transparent in the visible light region, which enables the radical induced curing of photopolymers by light irradiation at 405 nm, without premature release of Brønsted acids. Herein, a library of photoacid generators is comprehensively studied for the design of photocurable vitrimers applicable for dual-wavelength 3D printing and photopatterning. Stress relaxation measurements revealed that the bond exchange rate is affected by the thermal stability of the released Brønsted acid, the size of the counter-anion and the quantum yield of the cleavage reaction. As the photolatent catalysts neither affect pot life nor cure kinetics of the photocurable resins, it is possible to fabricate 3D printed objects via vat photopolymerization. By carrying out the printing at two different wavelengths (405 and 365 nm), the catalyst is locally activated during the layer-by-layer build-up of the 3D structures. In the UV exposed areas, the dynamic network is able to undergo triple shape memory by using the glass transition temperature (Tg) and Tv, whilst in the non-exposed parts it performs like a simple Tg-based shape memory polymer. This enables a controlled locking of defined areas during the thermal programming step and offers exciting ways towards the additive manufacturing of functional devices. In addition, the local activation of the catalyzing species was exploited to inscribe positive-tone microstructures in the photopolymer networks by photolithography. In the exposed areas, the network was selectively depolymerized during the development step in organic solvents at room temperature. Without any opimization, structures with a feature size of around 500 μm could be obtained.
Introduction
Facing a rapidly growing automation and digitalization of traditional manufacturing processes, modern industry is changing significantly. Personalization of products at reasonably low costs has become an important aspect of this industrial change.1 To date, the most efficient method for the fabrication of individual products is additive manufacturing, also known as 3D printing. This method is highly versatile and can produce complex geometries with high intricacy.2 Additive manufacturing yields parts with high performance and is cost-effective.3 Polymers offer numerous advantages as they are conveniently tailorable in terms of mechanical, thermal, electrical, and other functional properties.4 Moreover, polymers can be designed to undergo stimuli-induced changes in shape, property, or functionality over time. These stimuli include temperature, light, humidity, pH value, solvents, electric or magnetic fields, and enable a controlled conversion of the 3D printed part into another predictable structure.5In particular, stereolithography based techniques such as digital light processing (DLP) 3D printing have gained increased attention, as they provide a decent building speed, high resolution and good surface quality of the printed parts. They rely on the local solidification of a liquid resin by photopolymerization, which provides distinctive advantages compared to thermal polymerization routes. The polymer is formed rapidly under mild conditions, the reaction is typically free of solvents and highly volatile organic compounds, which makes it an environmentally friendly processing method. In addition, the mechanical properties can be easily adjusted by the choice of monomers and crosslinkers.
Various strategies are reported in literature, to print stimuli-responsive structures by DLP 3D printing.6,7 By adding multi-walled carbon nanotubes to acrylate monomers, Chiappone et al. successfully fabricated electrically conductive devices, whose shape memory properties were activated by Joule's heating.8 In a recent work, they in situ formed electrically conductive fillers within DLP 3D printed structures during a UV induced post-processing step.9
Following a multi-material approach, Hawker and co-workers printed multi-material soft active devices by Solution Mask Liquid Lithography (SMaLL).10 SMaLL exploits photochromic dyes with reversible absorption characteristics that allow a local activation or deactivation of photoreactions at different wavelengths. By combining a radical curing of acrylate monomers (at 530 nm) with additional activation of the cationic ring opening of epoxy monomers (at 470 nm), they fabricated soft joints and mechanically reinforced “brick-and-mortar” structures with high resolution.
Boydston et al. constructed a DLP printer that operated with two different light engines, which they used to sequentially cure an epoxy-acrylate resin.11 By visible light irradiation, they radically crosslinked the acrylate resin. Exposure with UV light induced the curing of the acrylate as well as the epoxy monomers yielding an interpenetrating network with higher crosslink density. Soft active devices were fabricated, whose activation relied on the differences in network structure and related swelling properties. Recently, we optimized the composition of the hybrid epoxy-acrylate system and introduced a dual-wavelength 3D printer containing light emitting diodes with higher light intensity.12 This allowed a significant decrease in the layer cure time (from 60–120 s to 1–5 s) and thus, a distinctive increase in the build time of multi-material structures was accomplished.
Along with post-processing and the use of orthogonal photoreactions, which are activated at different wavelengths, other concepts towards DLP 3D printing of stimuli-responsive structures with multi-material properties rely on an automatic exchange of vats and a local variation of the crosslink density by grey scale imaging.6,13
Another way for designing stimuli-responsive materials is the preparation of a polymer network that undergoes bond exchange reactions to adapt itself to desired conditions. The dynamic nature of these reactions renders the networks self-healable, weldable and malleable, whilst introducing shape memory properties and enhanced toughness.14 These so-called covalent adaptable networks (CANs) fall into a category between thermosets and thermoplastics. In other words, while having permanent bonds in the form of covalent crosslinks, they undergo stimuli-triggered bond exchange reactions. As a result, the viscoelastic properties of CANs highly depend on the kinetics of these dynamic exchange reactions, which follow either an associative or a dissociative pathway.15 In 2011, Leibler et al. reported on insoluble, yet reprocessable, dynamic networks that could rearrange their topology via thermo-activated associative bond exchange reactions.16 They were termed vitrimers as their viscosity change over temperature followed an Arrhenius-like trend comparable to vitreous silica.17 Besides the glass transition temperature (Tg), vitrimers are characterized by another transition, which is called the topology freezing transition temperature (Tv). Above Tv, the bond exchange reactions become considerably fast and result in a macroscopic flow of the polymeric network.18 In vitrimers, the rate of exchange reactions and the Tv are influenced by a number of factors, including the nature and content of catalyst, availability of functional groups, or the structure and mobility of the network.19 To date, one of the most widely utilized exchange reaction for vitrimers is the thermo-activated transesterification, which requires appropriate catalysts such as Brønsted acids, organometallic complexes, or organic bases to proceed at high rates and at moderate temperature.20 Advancing from traditional processing techniques such as moulding or extrusion, recent research is geared towards the introduction of dynamic hydroxyl ester bonds in 3D printable materials.
However, as a typical thermo-activated dynamic exchange reaction, the catalysed transesterification suffers from a lack of spatial control. Thus, various strategies have been pursued to locally control exchange reactions in thermo-activated vitrimers. On the one hand, nano-sized fillers with photothermal properties are exploited, which efficiently convert light to heat upon irradiation with a NIR laser.21 On the other hand, photolatent transesterification catalysts are applied, which can be selectively switched on by light exposure.22 In previous work, we used the concept of photolatent bases and acids to locally activate the catalysing species in dynamic thiol-acrylate and thiol-epoxy networks simply by light exposure.22–24 In particular, photoacid generators showed superior performance in photopolymer networks as they are well soluble in the resin formulations, benefit from a temperature stability up to 180 °C, do not affect cure kinetics or shelf life and release strong Brønsted acids upon light exposure. We developed a visible light curable thiol-acrylate resin using phenylbis(2,4,6-trimethylbenzoyl) phosphine oxide (BAPO) as radical photoinitiator, which had an absorption in the visible light region. We added a triarylsulfonium hexafluorophosphate as photolatent acid, which was activated in the UV-A spectral region but was transparent at longer wavelengths (>380 nm). The sequence dependent orthogonality of the curing and the release of the catalyst allowed the printing of 3D objects upon visible light exposure (405 nm) without premature release of Brønsted acids. By using a dual-wavelength DLP printer, which contained two light sources (405 and 365 nm), we were able to additionally expose selected layers at 365 nm to activate the catalyst during the printing. Consequently, soft active 3D structures were obtained, which underwent a controlled shape change after a programming step above the network's Tv.24
Advancing from these proof-of-concept studies, the current work addresses structure–property relationships of photolatent catalysts in vitrimeric photopolymers to fabricate soft active devices with fast relaxation kinetics and locally controllable shape memory properties. A particular focus is placed on the structure and absorption characteristics of the photoacid generator, as it is well known that the acidic strength of the released acid depends on the nucleophilicity of the counter-anion, whilst the cation governs the photochemistry.25 The most efficient photolatent catalyst, which additionally shows an onset of absorption at around 365 nm is then used in printing experiments with a dual wavelength DLP 3D printer operating at 405 and 365 nm. During printing, the catalysing species is activated in a spatially-controlled way and soft active devices are fabricated, whose shape memory properties are conveniently varied along the three axes.
In addition, we performed a photopatterning experiment, to demonstrate the versatility and resolution of the developed system. Structures with feature of around 500 μm could be obtained by local activation of the transesterification catalyst and thus, selectively changing the solubility of the network in methanol.
Results and discussion
Introducing a photolatent transesterification catalyst in photocurable CANs opens the way towards a local control of bond exchange reactions in thermo-activated vitrimers. At the same time, the photo-reactive resin formulations can be locally solidified by light exposure and thus, conveniently processed by vat based 3D printing processes.26 For this, the photolatent catalyst has to fulfil several requirements including a high solubility in the resin formulation, a high temperature stability (well above the Tv of the vitrimers), optical transparency in the visible light region (to avoid premature activation during the visible light induced radical curing) and the release of catalysing species, which are able to promote exchange reactions. Our previous work revealed that onium salts meet those requirements and enable a selective formation of strong Brønsted acids, which efficiently catalyse transesterifications in thiol-acrylate vitrimers.24 In particular, triarylsulfonium salts are promising candidates as they benefit from a low reduction potential and do not oxidize photolytically formed radicals.27 However, compared to conventional acid-catalysed transesterifications, whose kinetics are accelerated with rising pKa values of the acid, the use of photolatent acids is far more complex as the formation of the catalysing species is also affected by the photochemistry (e.g. absorption characteristics, quantum yields) of the compound.28Herein, we studied the performance of selected triphenylsulfonium salts with varying chemical structures to get a better understanding of the catalytic activity of photoacid generators in thiol-click vitrimers. Upon direct UV exposure, the heterolytic bond rupture of the sulphur-carbon bond in triphenylsulfonium salts preferably yields reactive carbocations (C+).29 Subsequent reaction of the cations with solvent or monomers leads to the formation of strong Brønsted acids, whose acidity is influenced by the chemical nature of the counter anion. Whilst the size and nucleophilicity of the anion determines the acidity of the generated acid, the structure of the cation controls the photochemistry of the triphenylsulfonium salt, including absorption characteristics, molar extinction coefficients and quantum yields.
Influence of the photolatent catalyst's counter anion on stress relaxation kinetics
In a first step, we evaluated the influence of the counter anion on the stress relaxation kinetics of a thiol-acrylate vitrimer, which consisted of 50 mol% HPPA, 25 mol% GDGDA, 25 mol% TMPMP and 2 wt% BAPO as long wavelength absorbing photoinitiator (Fig. 1). Upon light exposure at 405 nm, a flexible photopolymer network (Tg = 3 °C) was accomplished, which provides ample –OH and ester groups for subsequent thermo-activated transesterification. | ||
| Fig. 1 Chemical structures of acrylate monomers, thiol crosslinker and photoinitiator, employed for the preparation of thiol-acrylate vitrimers. | ||
For a direct comparison of the photolatent catalytic activity, the photoacid catalysts were dissolved in propylene carbonate and their content was kept constant at 1.5 mol% (related to the free –OH groups in the network).
In particular, the catalytic activity of Speedcure 992, a mixture of triphenylsulphonium hexafluorophosphate and its bis-functional derivative, was compared to Speedcure 976s as hexafluoroantimonate counterpart (Fig. 2a). Since the absorption characteristics of onium salts are mainly governed by the organic cation, both derivatives exhibit a comparable UV-Vis absorption spectrum (Fig. 2b). They are transparent in the visible light region and show absorption maxima at 231, 285 and 325 nm. In addition, (4-phenylthiophenyl)-diphenylsulfonium trifluoromethanesulfonate (PDS-triflate) was employed as triflate counterpart. Although it does not contain any bis-functional species and thus, only shows two absorption maxima at 234 and 300 nm, we assume a photochemistry somewhat comparable to the other two salts. The anions of the chosen onium salts are poorly nucleophilic and non-coordinating, which is crucial to prevent termination reactions by anion–cation combination in cationic polymerization or cationic curing of epoxides. Previous work showed that the cure rate and molecular weight of related polymer systems decrease with increasing nucleophilicity of the anions in the order SbF6− > PF6− > CF3SO3−.30 At the same time, the acidity of the released Brønsted acids increases in the same order, which should directly affect the transesterification rate.
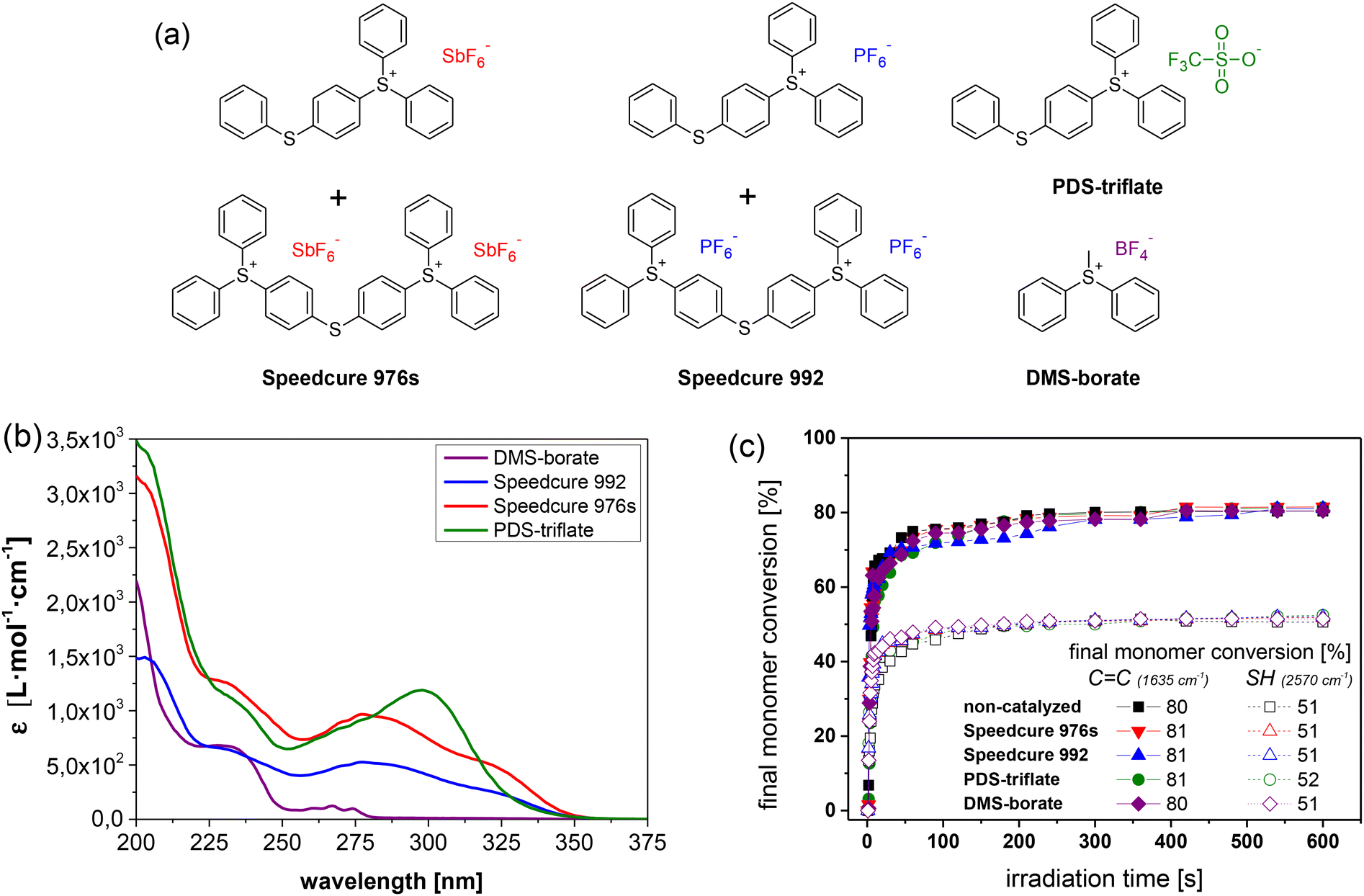 | ||
| Fig. 2 (a) Chemical structures and (b) UV-Vis absorption spectra of triphenylsulfonium salts (1 mol% in acetonitrile) with varying counter anions used in this study. (c) Monitoring the cure kinetics in catalysed and non-catalysed thiol-acrylate formulations as a function of the applied photolatent catalyst by FTIR spectroscopy. The normalized peak area of the IR absorption bands of the acrylate groups (1635 cm−1) and thiol groups (2570 cm−1) is plotted against exposure time. Irradiation (λ = 420–450 nm, 3.3 mW cm−2) was performed under air (the zoom of the data at an irradiation time range between 0 and 50 s is provided in Fig. S3 in ESI†). The lines are a guide for the eye. | ||
As the spectral sensitivity of the photoacid generators under investigation was limited to the short UV wavelength region, they did not compromise the visible light-induced curing reaction of the thiol-acrylate system. The time-dependent depletion of the characteristic FTIR absorption bands of acrylate (1635 cm−1) and thiol groups (2570 cm−1) of the resin formulation with PDS-triflate as photolatent catalyst are provided in ESI (Fig. S1†). Catalysed as well as non-catalysed thiol-acrylate formulations were characterized by a comparable cure rate and final conversion of the monomers (Fig. 2c). However, the final acrylate conversion was higher (80–81%), compared to the thiol one (51–52%), as acrylates are also prone to homopolymerization via a chain growth mechanism under the applied conditions.31 Along with cure kinetics, thermogravimetric analysis further revealed that the photolatent catalysts were not affecting the thermal stability of the photopolymer networks, which were stable well above 180 °C (as an example the TGA data of the thiol-acrylate network containing PDS-triflate as photolatent catalyst is provided in Fig. S5 in ESI† and the TGA data of photolatent catalysts under investigation in Fig. S6 in ESI†).
Once cured, the stress relaxation kinetics of the photopolymers was determined by rheometer measurements using DLP 3D printed discs with a diameter of 10 mm. The stress relaxation of catalysed networks was determined at 180 °C, prior to and after UV exposure with a conventional mercury UV lamp, and compared to a non-catalysed network. Fig. S7 in ESI† shows an example of the thiol-acrylate network with PDS-triflate as photolatent catalyst. In the absence of the photolatent catalyst, a slight stress relaxation is observed, which is related to a thermal release of volumetric shrinkage stresses arisen during network evolution.32 Prior to UV exposure, the catalysed networks exhibit a similar behavior, which indicates that the catalysing species is not prematurely released during the photo-curing process or the high temperature applied in the stress relaxation experiment. This behavior is also observed for the photopolymer networks containing Speedcure 976s and Speedcure 992 as latent catalysts (see Fig. S9 in ESI†).
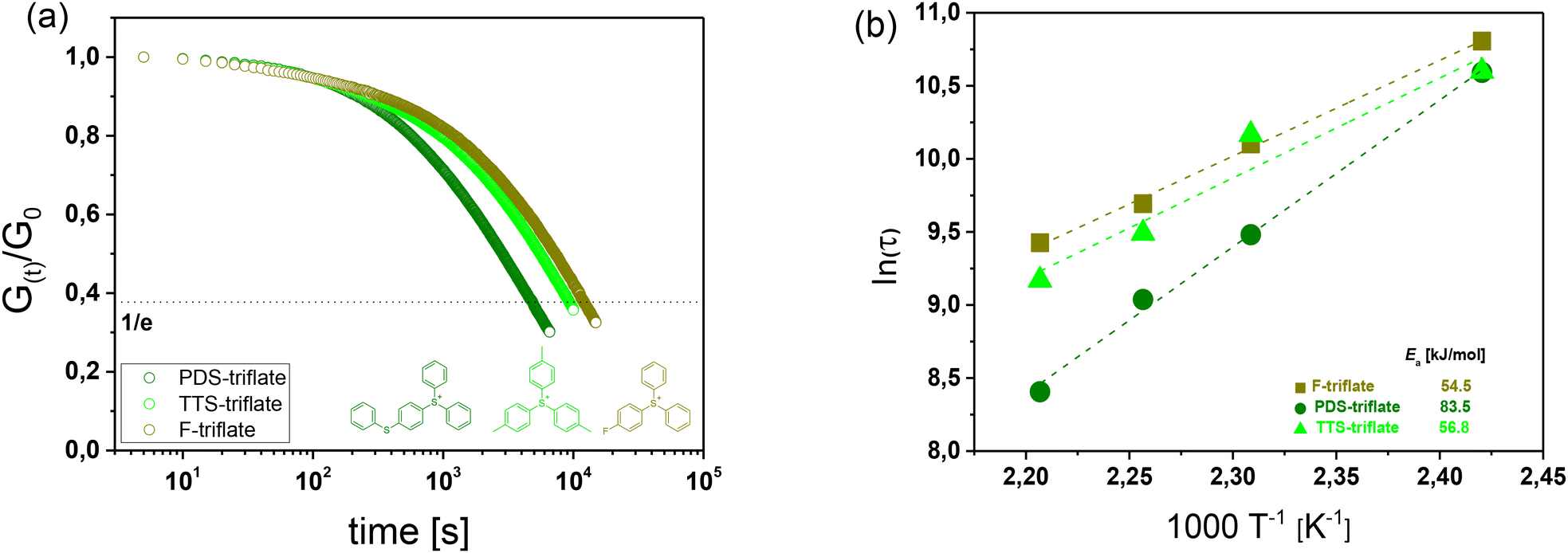
The stress relaxation studies clearly show that all three photolatent catalysts are able to catalyse bond exchanges upon subsequent UV exposure, albeit at different rates (Fig. 3a). Interestingly, the stress relaxation rate corresponding to the rate of bond exchange reactions seems to increase with decreasing acidity of the released “super acid”: HSbF6 < HPF6 < HCF3SO3. However, it should be noted that for HCF3SO3, no decomposition is observed near room temperature, whilst both HSbF6 and HPF6 suffer from a low thermal stability and start to degrade at 25 and 40 °C, respectively.33 The thermolysis yields HF, whose pKa value (3.18) is significantly lower than the pKa value of HCF3SO3 (−14).34 We assume that at the temperatures (140–180 °C) applied in the stress relaxation experiments, the majority of the formed “super acids” have been converted to the lower acidic HF, which explains the superior catalysing performance of PDS-triflate.
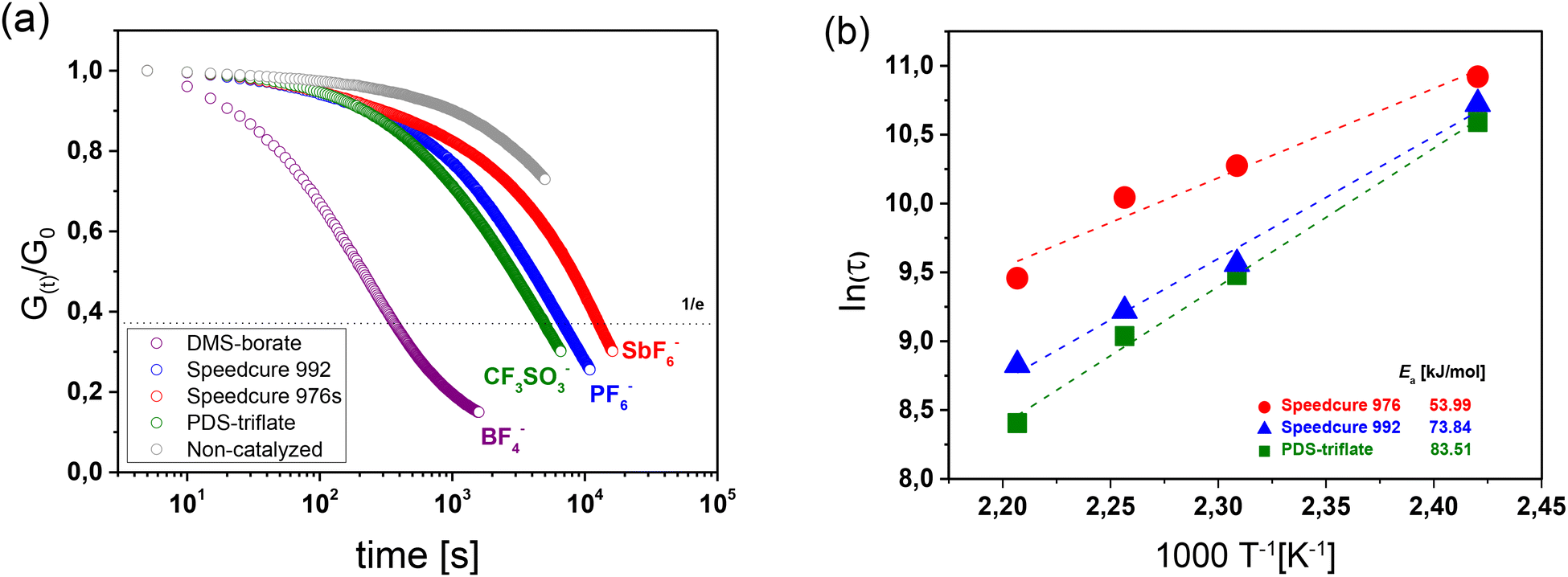 | ||
| Fig. 3 (a) Normalized stress relaxation curves (obtained at 180 °C) of catalysed and non-catalysed thiol-acrylate networks as a function of triphenylsulfonium salts with varying counter anions. Prior to the measurements the films were UV irradiated with a mercury emitter for 10 s (λ = 200–550 nm, 20 mW cm−2). (b) Arrhenius plots derived from the measured relaxation times of catalysed thiol-acrylate networks. | ||
In contrast, the higher reactivity of HPF6 compared to HSbF6 might be attributed to the size of the counter anion. Along with the acidity, the size of the counter anion also influences the migration rate of the released acid. For the design of positive tone photoresists, it was reported that the resolution of the micropatterns increased with rising size of the counter anion of the applied photoacid generator, which was associated with the lower migration rate of the released acid.35
Due to the poor spectral sensitivity, we expect that Brønsted acids were mainly formed at the surface of the printed disc, which then migrated inside the bulk leading to a more homogenous distribution of the photochemically formed catalyst. Thus, the faster migration of HPF6 compared to the bulkier HSbF6 might also explain the more efficient stress relaxation of the sample containing the hexafluorophosphate salt as photolatent acid.
To confirm the crucial role of the size of the counter anion and its migration properties on the stress relaxation kinetics, we repeated the experiments with diphenyl(methyl)sulfonium tetrafluoroborate (DMS-borate). Compared to the other three photolatent acids under investigation, it contained the smallest and least nucleophilic counter anion (BF4−).
Thus, the released acid (HBF4) is highly mobile but suffers from a lower acidity. However, in contrast to HPF6 and HSbF6, it benefits from a significantly higher thermal stability (130 °C).36 It should be noted that the photochemistry of DMS-borate is not directly comparable to the other three investigated onium salts since it differs in the chemical structure of the cation. In particular, with an even more limited UV absorption in the lower wavelength region (onset of absorption occurs at 280 nm), we expect a lower quantum yield under the applied irradiation conditions.
Despite the unfavorable absorption characteristics and lower acidity of the released Brønsted acid, DMS-borate is by far the most efficient catalyst in the current study (Fig. 3a). However, by comparing the relaxation curves of the system prior to and after UV exposure it is obvious that the catalyst is already thermally released (Fig. S8 in ESI†) as the exchange kinetics are similar. This thermal activation together with the higher mobility of the acid catalyst might explain the fast relaxation rate.
In contrast to conventional acid catalysts applied in vitrimers, the results clearly show that the efficiency of photolatent systems for catalysing transesterifications is strongly affected by the thermal stability and migration kinetics of the catalysing species, whilst the acidity of the released Brønsted acid plays a minor role. Thus, by simply varying the counter anion of the photoacid generator, we were able to adjust the relaxation time over two orders of magnitude.
As the stress relaxation is directly related to the bond exchange rate in vitrimers, we further characterized the temperature-dependent relaxation kinetics to determine the activation energy (Ea) of the catalysed networks.37 The stress relaxation curves obtained at different temperatures for the investigated photolatent catalysts are provided in Fig. S10–S14 in ESI.†
In dynamic networks undergoing an associative bond exchange mechanism, the relaxation time (τ*) follows an Arrhenius-type temperature dependency τ* = τ0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp (Ea/RT), in which Ea is the activation energy of the exchange reaction and R is the universal gas constant.16,38 By following the Maxwell Model, τ* is obtained from the measured relaxation data by taking the normalized relaxation modulus (G(t)/G0) required to relax e−1 (37%) of the initial stress.39 In Fig. 3b, τ* is plotted against (1/T) in a semilogarithmic scale and Ea of the catalysed bond exchange reactions was determined, by taking the slope (m = Ea/R) of the straight line fitted to the data. Independent of the applied photoacid generator, the irradiated thiol-acrylate networks' τ* follows a linear trend, which confirms the Arrhenius-type temperature dependence and the vitrimeric nature of the networks.40 Whilst the networks exhibited comparable final monomer conversions (Fig. 2b) and network properties, they significantly differed in their Ea values.
exp (Ea/RT), in which Ea is the activation energy of the exchange reaction and R is the universal gas constant.16,38 By following the Maxwell Model, τ* is obtained from the measured relaxation data by taking the normalized relaxation modulus (G(t)/G0) required to relax e−1 (37%) of the initial stress.39 In Fig. 3b, τ* is plotted against (1/T) in a semilogarithmic scale and Ea of the catalysed bond exchange reactions was determined, by taking the slope (m = Ea/R) of the straight line fitted to the data. Independent of the applied photoacid generator, the irradiated thiol-acrylate networks' τ* follows a linear trend, which confirms the Arrhenius-type temperature dependence and the vitrimeric nature of the networks.40 Whilst the networks exhibited comparable final monomer conversions (Fig. 2b) and network properties, they significantly differed in their Ea values.
In the presence of PDS-triflate, Speedcure 992 and Speedcure 976s, the networks exhibited Ea values between 73.84 and 53.99 kJ mol−1. In the same network, we obtained similar Ea values (42.4–64.5 kJ mol−1) with protic organophosphate catalysts comprising pKa values in the range of 1.29 to 2.11.32,39 The results demonstrate that photolatent catalysts are highly versatile in accelerating dynamic exchange reactions in vitrimeric photopolymers and based on their thermal stability, migration properties and acidity enable a convenient tuning of exchange kinetics.
Influence of the photolatent catalyst's cation on stress relaxation kinetics
By keeping the composition of the thiol-acrylate network constant, we studied the role of the photolatent catalyst's cation in more detail by applying selected triphenylsulfonium triflates with various substituents on the phenyl rings (Fig. 4a). Fig. 4b displays the UV-Vis spectra of the photolatent catalysts under investigation, showing the influence of the cation structure on its absorption characteristics. In contrast to the unsubstituted triphenylsulfonium cation (F-triflate), which exhibits an absorption maximum at 235 nm, the absorption window of the substituted derivatives is extended to longer wavelengths.41 This is in good agreement with the work of Dektar and Hacker, who demonstrated that the absorption coefficients of triphenylsulfonium salts increase with increasing substitution whilst the absorption maxima are shifted to longer wavelengths.27 In particular, PDS-triflate is characterized by the most red-shifted maximum (300 nm), which Dektar and Hacker explained by the resonance stabilization of the electron-donating 4-phenylthio group due to presence of electron-withdrawing sulfonium sulfur moiety.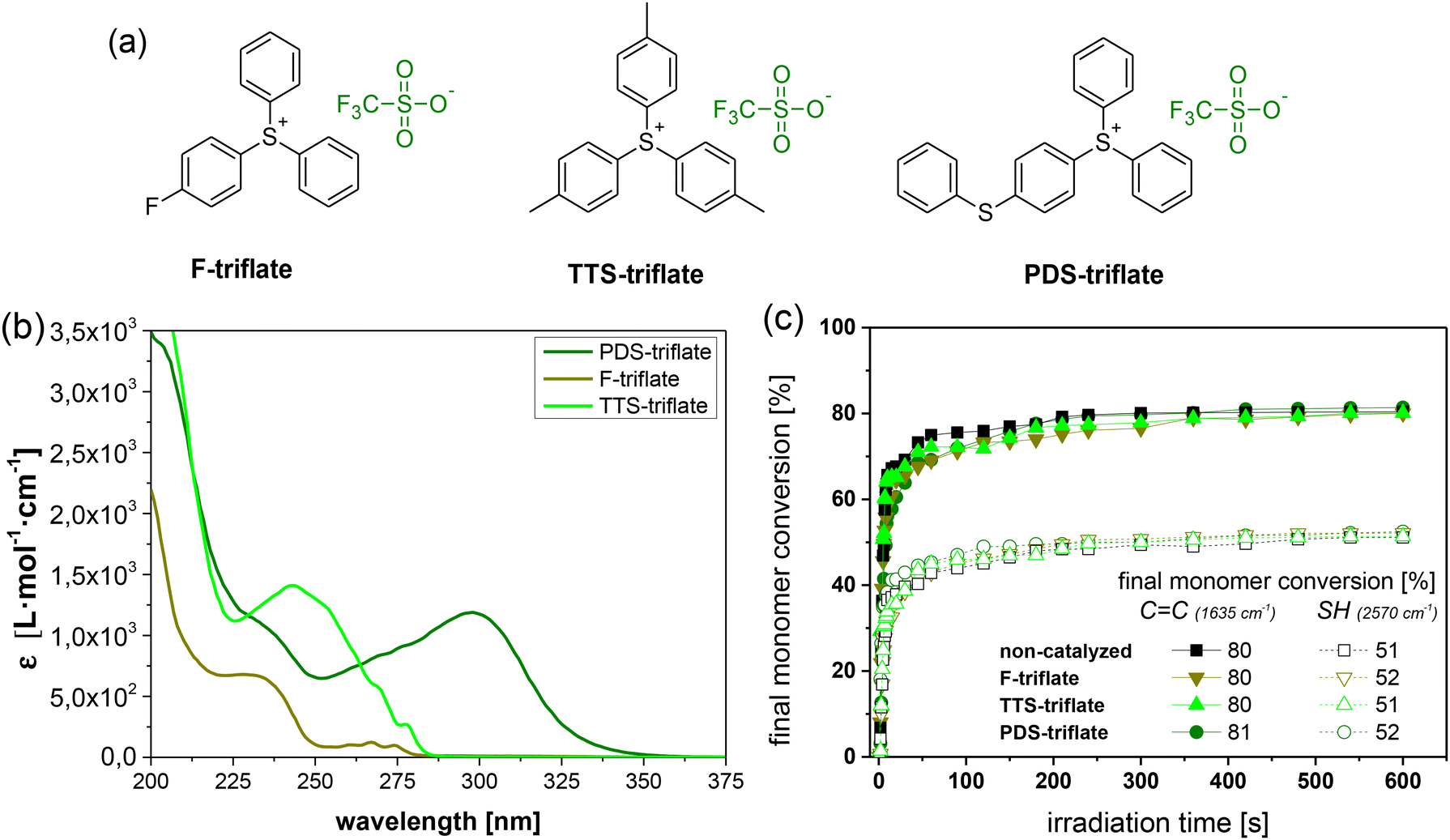 | ||
| Fig. 4 (a) Chemical structures and (b) UV-Vis absorption spectra of the triphenylsulfonium salts (1 mol% in acetonitrile) with varying cations under investigation. (c) Monitoring the cure kinetics in catalysed and non-catalysed thiol-acrylate formulations as a function of the applied photolatent catalyst by FTIR spectroscopy. The normalized peak area of the IR absorption bands of the acrylate groups (1635 cm−1) and thiol groups (2570 cm−1) is plotted against exposure time (the zoom of the data at an irradiation time range between 0 and 50 s is provided in Fig. S4 in ESI†). Irradiation (λ = 420–450 nm, 3.3 mW cm−2) was performed under air. The lines are a guide for the eye. | ||
Along with the absorption characteristics, the substituents also govern the rate of acid formation. Whilst mono-fluoro (F-triflate) substituted and tris-tolyl (TTS-triflate) substituted salts are reported to exhibit about the same rate as the unsubstituted triphenylsulfonium salt, 4-phenylthio substituted derivatives such as PDS-triflate benefit from a significantly higher rate. This can be explained by differences in the direct photolysis of the triphenylsulfonium salts. Whilst photolysis of 4-phenylthio substituted salts proceeds through the triplet state, mono-substituted and the parent triphenylsulfonium salt decompose via the singlet state.27
Introduced as photolatent catalyst in the thiol-acrylate resin formulation, the triphenylsulfonium triflates under investigation do not affect the cure kinetics of the visible light-induced radical curing as evidenced by FTIR experiments (Fig. 4c). Both cure rate and final monomer conversion are comparable to the thiol-acrylate resin containing no catalyst.
Upon subsequent UV exposure, all three triphenylsulfonium salts yield the thermally stable acid HCF3SO3, albeit at different rates.
This is directly affecting the stress relaxation kinetics of the UV irradiated thiol-acrylate networks (Fig. 5a). Whilst the stress relaxation rates of networks with F-triflate and TTS-triflate are slower, with 63% of the initial stress being relaxed within 153 and 196 min (at 180 °C), respectively, the stress relaxation rate is significantly faster in the presence of PDS-triflate (79 min). The results correlate well with the rate of acid formation of the related photolatent catalysts giving rise to faster exchange reactions at higher acid content. This is also in good agreement with previous work on acid catalysed vitrimers relying on transesterification, whose exchange rate increased with a larger amount of catalyst.32,42
 | ||
| Fig. 5 (a) Normalized stress relaxation curves (obtained at 180 °C) of thiol-acrylate networks as a function of triphenylsulfonium salts with varying cations. Prior to the measurements the films were UV irradiated with a mercury emitter for 10 s (λ = 200–550 nm, 20 mW cm−2). (b) Arrhenius plots derived from the measured relaxation times of catalysed thiol-acrylate networks. | ||
Dual-wavelength DLP 3D printing of soft active devices
For the printing experiments, PDS-triflate was applied as catalyst as it was the most efficient derivative for catalysing thermo-activated transesterification in the thiol-acrylate system under investigation. Whilst DMS-borate was even more reactive, it's low thermal stability (thermal release of the catalyst) makes it unsuitable for locally controlling exchange reactions in 3D printed structures.In addition, 0.05 wt% of a light absorbing dye (Sudan II) was added to the formulation to improve the resolution of the printed objects.43 Since viscosity and stability are crucial parameters for 3D printing, the viscosity of a freshly prepared resin and after 6 hours was determined with a shear rate ranging from 1 to 300 s−1. The viscosity increased slightly from 740 mPa s to 1078 mPa s, which is still in a reasonable range for DLP 3D-printing (see Fig. S10 in ESI†).
For the manufacture of soft active devices with locally-controlled shape memory properties, 3D objects were fabricated with a dual-wavelength DLP 3D printer operating at 405 and 365 nm. The schematic set-up of the printer is provided in Fig. 6a. During the printing process, the thiol-acrylate formulation is selectively cured layer-by-layer upon visible light irradiation (405 nm) without premature activation of PDS-triflate. In contrast, if the printing is carried out at 365 nm, radical curing of the photopolymer and the activation of the catalyst occur simultaneously. Consequently, the formation of the catalyst can be controlled in x- and y-direction and along the z-axis. However, it should be considered that the migration of the photo-released acids, particularly during stress relaxation experiments at elevated temperature, can negatively affect the resolution of the differently irradiated domains.
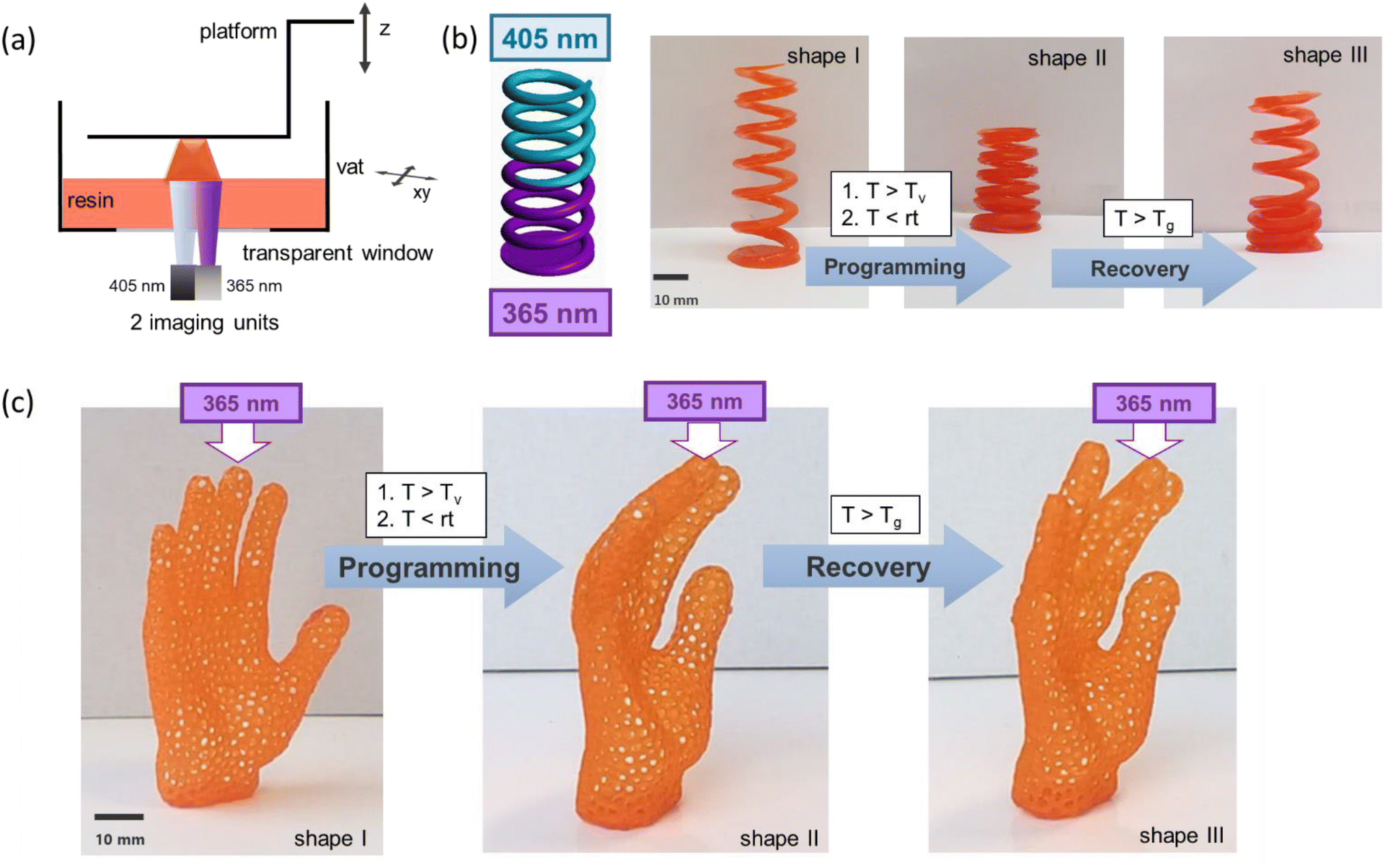 | ||
| Fig. 6 (a) Schematic representation of the dual-wavelength DLP 3D printing of thiol-acrylate vitrimers containing photolatent catalysts. (b) Dual wavelength DLP 3D printed spring (d = 25 mm, h = 60 mm) containing PDS-triflate as photolatent catalyst, which has been activated along the z-axis by printing with 365 nm. Programming and local shape recovery of the printed spring is demonstrated. (c) Hand form obtained by dual wavelength DLP 3D printing with the thiol-acrylate resin containing PDS-triflate as photolatent catalyst. The catalyst has been activated in the area of the middle finger by printing with 365 nm. Programming and selective shape recovery of the fingers illuminated with 405 nm is demonstrated. | ||
ATR FTIR spectra were taken directly after the printing with 405 nm and after a subsequent irradiation with UV light (365 nm). The spectra are provided in Fig. S4 in ESI.† The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H band of the acrylates (1635 cm−1) has already nearly disappeared in the printed test samples, giving rise to a high monomer conversion during the printing process. Additional UV exposure led to a further decrease of the signal, which evidences further crosslinking induced by residual photoinitiator molecules.
C–H band of the acrylates (1635 cm−1) has already nearly disappeared in the printed test samples, giving rise to a high monomer conversion during the printing process. Additional UV exposure led to a further decrease of the signal, which evidences further crosslinking induced by residual photoinitiator molecules.
In first printing experiments, simple test structures involving rectangular shaped samples and open cubes were fabricated, in which selected areas were UV exposed layer-by-layer (Fig. S5 in ESI†). To demonstrate the localized activation of the catalyst and the related topological rearrangements in the 3D structures, shape memory experiments were performed.
For the programming step, the printed test specimens were heated at 160 °C, which was above the network's Tv, and deformed. By applying an external force, the samples were kept in the deformed state for 2 h at 160 °C. During this thermal treatment, the UV exposed areas containing the photo-released catalyst changed from elastic solid to a viscoelastic liquid and thus, could rearrange their network topology. Subsequently, the structures were cooled to room temperature and were able to retain their programmed shape without applying any external force.
The specimens were then heated at 45 °C, which was well above the network's Tg and within 40 min, the domains illuminated at 405 nm were able to move back to their original shape as the elongated polymer chains returned to their entropically more favorable initial state. In contrast, the domains irradiated with 365 nm retained their programmed shape due to the topological rearrangements of the network, catalysed by the photo-released acid.
Advancing to more complex structures, a spring was printed in the next step, in which the catalyst was selectively activated along the z-axis (Fig. 6b). After the programming step, a selective movement of the 405 nm exposed part of the spring was achieved, whilst the 365 nm exposed part remained locked in its programmed position due to the catalysed topological rearrangements.
To demonstrate the potential for the fabrication of elaborate 3D objects with locally controllable shape memory properties, we printed a lattice structure in the form of a hand (Fig. 7c). The middle finger was printed with 365 nm, whilst the other four fingers were printed with 405 nm. During the programming step, the fingers were bent and only the middle finger was able to undergo topological rearrangements at 160 °C. Thus, in the subsequent shape recovery step at 45 °C, the middle finger stayed in its bent shape, whilst the other fingers were moving back to their original position. The results clearly confirm that concept of photolatent catalysts in combination with dual wavelength DLP 3D printing is a promising route towards the personalized fabrication of complex soft active devices.
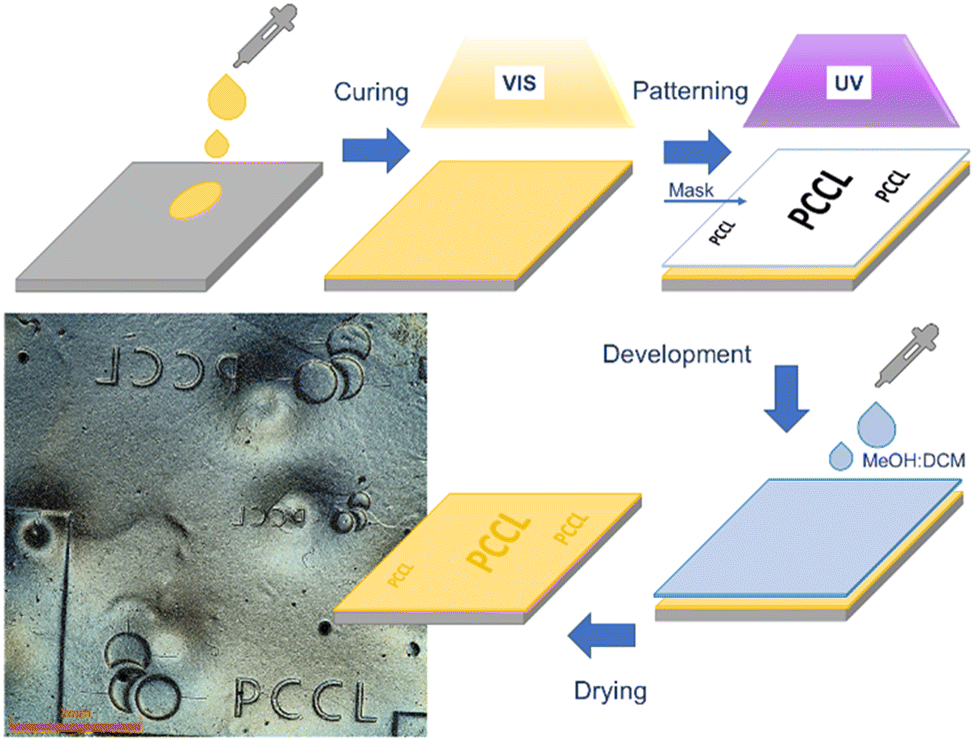 | ||
| Fig. 7 Schematic procedure and positive-tone photopatterns inscribed in a thiol-acrylate photopolymer containing PDS-triflate as latent catalysts. | ||
To show the versatility of the system, we also printed a simple test structure only with 365 nm and performed a shaping-experiment (see Fig. S12 in ESI†).
Inscribing positive-tone micropatterns by photolithography
Along with multi-material 3D printing of soft active devices, we further exploited the local activation of the catalyzing species for inscribing positive-tone micropatterns in thin films. For this, a thin layer of the thiol-acrylate resin containing PDS-triflate as photolatent acid was drop cast on a Si-wafer, which was surface modified with an organosilane to improve the adhesion. The thin film was cured via flood exposure with visible light. Photopatterning was then carried out with UV light (365 nm) using a quartz mask (see Fig. 7) to locally activate the acid in the exposed areas. The patterned film was immersed in a methanol solution containing 2 vol% dichloromethane (DCM), which was added to slightly swell the photopolymer and to facilitate the dissolving process. The exposed areas could be selectively depolymerized by bond exchange reactions of the ester moieties across the –OH groups of the mono-functional alcohol. Due to the high reactivity of the catalyst, depolymerization proceeded already at room temperature and after 2 hours, positive tone patterns could be observed (optical microscopy image is provided in Fig. 7). Without any optimization, micropatterns with a feature size of 500 μm were realized. This significantly enhances the applicability of the developed system in the direction of photoresist technologies. Here, further studies will be dedicated to improve the resolution of the patterns by covalent immobilization of the photolatent catalyst within the photopolymer network.Conclusions
In a comprehensive way, the efficiency of selected photolatent catalysts towards the local activation of topological rearrangements in thermo-activated vitrimers was studied. In particular, triphenylsulfonium salts were applied, which are transparent in the visible light region and release strong Brønsted acids upon UV exposure. However, compared to conventional protic acids applied for catalysing transesterifications in vitrimers, the use of photolatent acids is far more complex, since the formation of the catalysing species is affected by the type of counter anion (acidity of the released acid) as well as the structure of the cation (absorption characteristics and quantum yields of the released acid).Applied in thiol-acrylate vitrimers, it was found that the bond exchange rate did not increase with the size of the counter-anion and thus, the acidity of the released acid. This is explained by the low thermal stability of the generated Brønsted acids (HPF6 and SbF6), which are easily converted to HF at higher temperatures, and by the faster migration of acids featuring a smaller anion. In contrast, stress relaxation experiments revealed that the bond exchange rate correlates well with the quantum yield and rate of acid formation of the corresponding triphenylsulfonium triflates.
Due to the narrow absorption window of the triphenylsulfonium salts under investigation, visible light curing of thiol-acrylate photopolymers was accomplished without premature release of the acid and was furthermore not affected by the presence of the photolatent catalyst. This enabled the spatially controlled activation of the catalyst in 3D objects during dual wavelength 3D printing, which was carried out at two different wavelengths (405 and 365 nm). Owing to the fast stress relaxation and appropriate absorption characteristics, PDS-triflate was used as photolatent catalyst in the printing studies. Local movement of simple and complex 3D structures was achieved after a programming step above the network's Tv, confirming the selective activation of the catalyst during the printing process. Moreover, the local control of the dynamic nature of the photopolymer networks was used to inscribe positive-tone micropatterns in a thin film. Structure sizes of 500 μm could be resolved by immersing the photopatterned (and locally activated) film in a methanol solution containing 2 vol% dichloromethane for 2 hours at room temperature.
Experimental
Materials and chemicals
Trimethylolpropane tri(3-mercaptopropionate) (TMPMP) was kindly provided by Bruno Bock Chemische Fabrik GmbH & Co. KG (Germany). 2-Hydroxy-3-phenoxypropyl acrylate (HPPA), glycerol 1,3-diglycerolate diacrylate (GDGDA), phenylbis(2,4,6-trimethylbenzoyl) phosphine oxide (BAPO), propylene carbonate, (4-phenylthiophenyl)-diphenylsulfonium trifluoro methane sulfonate (PDS-triflate) and (4-fluorphenyl)-diphenylsulfonium trifluormethansulfonate (F-triflate) were purchased from Sigma-Aldrich (USA). Diphenyl(methyl) sulfonium tetrafluoroborate (DMS-borate), tri-p-tolylsulfonium trifluoromethanesulfonate (TTS-triflate) and Sudan II were obtained from Tokyo Chemical Industry Co. (Japan). 4-{[4-(Diphenylsulfanylium)phenyl]sulfanyl}phenyl diphenyl sulfonium bishexafluorophosphate (40 wt%) dissolved in propylene carbonate (Speedcure 992) and (sulfanediyldibenzene-4,1-diyl)bis(diphenylsulfonium) bis(hexa fluoroantimonate) (Speedcure 976s) were supplied by Lambson Co. Chemicals GmbH (UK). All chemicals were used without further purification.Sample preparation
For the preparation of thiol-acrylate formulations, BAPO (2 wt%) was dissolved into a resin containing 50 mol% HPPA, 25 mol% GDGDA and 25 mol% TMPMP. Each photoacid generator was separately dissolved in propylene carbonate (50 wt%) at 50 °C using a magnetic stirrer. 1.5 mol% (related to –OH groups within the formulation) of the photoacid generator in solution was added to the thiol-acrylate resin formulation and dissolved at room temperature by means of a magnetic stirrer.For the printing experiments, 0.05 wt% of Sudan II was added as dye to the formulation to enhance the resolution of the printed objects. The chemical structures of the monomers, crosslinker and photoinitiator are provided in Fig. 1.
Characterization
Cure kinetics was determined with a Vertex 70 FTIR spectrometer (Bruker, United States). Sixteen scans were accumulated in transmission mode between 4000 cm−1 and 700 cm−1 with a resolution of 4 cm−1. The absorption peak areas were calculated utilizing OPUS software. For sample preparation, 1.5 μL of each formulation was drop-casted between two CaF2 discs, and the curing process was initiated by a light-emitting diode lamp (zgood® wireless LED curing lamp) with a power density of 3.3 mW cm−2 (λ = 420–450 nm). The emission spectra of the light sources used in this study are provided in ESI (Fig. S18–S20†). To ensure that the film thickness of each sample was comparable, the intensity of the FTIR bands was evaluated if it reached similar transmission values. The conversion rates were calculated using the normalized intensity of CC (1635 cm−1) and –SH (2570 cm−1) absorption bands with the OPUS software. After the experiments, the CaF2 discs were immersed in acetone for several days. Once the cured resin layer was swollen, the discs were separated from each other, cleaned with acetone and reused for other studies.
Ultraviolet–visible spectrophotometry (UV/Vis) was performed utilizing a UV/VIS Spectrophotometer Cary 50 Conc (Varian Inc.). All photoacid generators were dissolved in acetonitrile (1 mol%), and the absorption characteristics were recorded between 200 and 420 nm.
Thermogravimetric analysis (TGA) was carried out utilizing a Mettler Toledo (United States) TGA thermogravimetric analyser. The measurements were carried out in N2 atmosphere by heating the samples from 30 to 900 °C with a heating rate of 10 °C min−1.
Differential scanning calorimetry (DSC) measurements were conducted on a Mettler-Toledo DSC 821e instrument (United States). The cured networks were heated from −25 to 120 °C with a heating rate of 10 K min−1 in nitrogen atmosphere (nitrogen flow was 20 mL min−1). The glass transition temperature (Tg) was obtained from the second heating run by taking the midpoint in heat flow.
Stress relaxation analysis was performed with a moving die rheometer (Anton Paar, Austria) at 140 °C, 160 °C, 170 °C, and 180 °C. Samples were DLP 3D printed with a diameter of 10 mm, and thickness of 1 mm for such measurements. Before carrying out the measurements, the samples underwent an equilibration step at 20 N at the desired test temperature for 20 min in the rheometer. Subsequently, a 3% step strain was applied, and the decreasing stress was recorded over time.
The viscosity of the formulations was obtained by using a modular compact rheometer MCR 102 from Anton Paar (Austria) with a CP60-0.5/TI cone (49.97 mm diameter and 1.982° opening angle). Each measurement was carried out with 1 mL formulation at room temperature and a shear rate ranging between 0.1 and 300 s−1.
Vat photopolymerization 3D printing
Digital light processing (DLP) 3D printing was carried out on a prototype from way2production (Austria) equipped with two LED light sources (405 and 365 nm). For the preparation of test specimens used for stress relaxation experiments, the bottom layer was exposed at 405 nm (4 mW cm−2) for 8 s and the subsequent layers were illuminated at 405 nm for 6 s. Under these conditions, the catalyst was not activated. After printing, a general post-curing step was performed by using a light emitting diode (zgood® wireless LED curing lamp) with a power density of 3.3 mW cm−2 (λ = 420–450 nm). The samples were then UV irradiated with a mercury emitter (Omnicure S1000, Lumen Dynamics, Canada) to release the transesterification catalyst. The light intensity of UV activation in the sample plane was determined with an integrating radiometer (Powerpuck II, EIT Instrument Markets, United States) and was 20 mW cm−2 (λ = 250–470 nm).For preparing dual-wavelength DLP printed structures, in which the catalyst was selectively activated during the printing, the bottom layer was again exposed at 405 nm (8 mW cm−2) for 8 s whilst the subsequent layers were illuminated at 405 nm for 6 s. The activation of the catalyst was simultaneously carried out by irradiation of desired layers at 365 nm (8 mW cm−2) for 10 s.
FT-IR spectra were taken from printed structures prior to and after activation with UV light (365 nm) using an FT-IR spectrometer (Vertex 70, Bruker, United States) equipped with a reflection diamond attenuated total reflection (ATR) accessory (Platinum ATR). 16 scans were accumulated at 4 cm−1 resolution.
Reshaping experiments
Shape memory experiments were performed using dual-wavelength DLP 3D printed test specimen (rectangular shaped film, open cube, spring with a height of 60 mm and diameter of 25 mm, and a hand built up by a lattice structure). Selected domains of the 3D objects were irradiated by 365 nm light to activate the photocatalyst, whilst the other parts of the printed structures were exposed to 405 nm light. To erase the thermal history, the samples were heated to 160 °C for an hour. To fix the first shape, the sample was heated to 160 °C, deformed by applying an external force, and kept at 160 °C for 2 h. The sample was then cooled down to room temperature and the external force was removed. Selective shape recovery of the areas printed at 405 nm (no photo-induced activation of the transesterification catalyst) was carried out by heating the printed test specimen at 45 °C, which was above the network's glass transition temperature (3 °C).Photopatterning experiment
A thin resin film containing PDMS-triflate as latent catalyst was coated on a modified Si-wafer (silanization was performed according to literature44) by placing 100 μL resin between the wafer and a PTFE foil. By applying some pressure, the resin was spread and a homogenous solid film was obtained after curing with a light-emitting diode lamp (zgood® wireless LED curing lamp) using a power density of 3.3 mW cm−2 (λ = 420–450 nm). Subsequently, a photomask was placed on the film, and UV-light activation of selected parts was carried out with a mercury emitter (Omnicure S1000, Lumen Dynamics, Canada, 20 mW cm−2). To remove the UV exposed and activated areas of the film, the wafer was placed in a methanol solution containing 2 vol% dichloromethane for 2 hours at room temperature. The film was then dried under atmospheric conditions for one hour.Images of the patterned film were taken with an OLYMPUS DSX1000 (Olympus, Japan).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Part of the research work was performed with the “SMART” project. This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement no. 860108. Part of the research work was performed also within the COMET-Module “Chemitecture” (project-no.: 21647048) at the Polymer Competence Center Leoben GmbH (PCCL, Austria) within the framework of the COMET-program of the Federal Ministry for Transport, Innovation and Technology and the Federal Ministry for Digital and Economic Affairs with contributions by the Institute of Chemistry of Polymeric Materials (Montanuniversitaet Leoben, Austria). The PCCL is funded by the Austrian Government and the State Governments of Styria, Upper and Lower Austria.Special thanks go to DI Bettina Schlemmer (Institute for Chemistry and Technology of Materials, Graz University of Technology) for taking the microscope pictures and Daniel Bautista MSc. (Polymer Competence Center Leoben) for carrying out the TGA measurements.
References
- K. Łukasik and T. Stachowiak, Management and Production Engineering Review, 2020 Search PubMed.
- (a) P. K. Rajamani, T. Ageyeva and J. G. Kovács, Polymers, 2021, 13, 309 CrossRef CAS PubMed; (b) Y. Zhang, W. Jarosinski, Y.-G. Jung and J. Zhang, Additive Manufacturing: Materials, Processes, Quantifications and Applications, Elsevier, 2018, pp. 39–51 Search PubMed.
- R. Agrawal, RPJ, 2019, 25, 1045–1060 CrossRef.
- L. J. Tan, W. Zhu and K. Zhou, Adv. Funct. Mater., 2020, 30, 2003062 CrossRef CAS.
- (a) A. Rayate and P. K. Jain, Mater. Today: Proc., 2018, 5, 20474–20484 CAS; (b) M.-Y. Shie, Y.-F. Shen, S. D. Astuti, A. K.-X. Lee, S.-H. Lin, N. L. B. Dwijaksara and Y.-W. Chen, Polymers, 2019, 11, 1864 CrossRef CAS PubMed.
- U. Shaukat, E. Rossegger and S. Schlögl, Polymers, 2022, 14, 2449 CrossRef CAS PubMed.
- R. T. Shafranek, S. C. Millik, P. T. Smith, C.-U. Lee, A. J. Boydston and A. Nelson, Prog. Polym. Sci., 2019, 93, 36–67 CrossRef CAS.
- A. Cortés, A. Cosola, M. Sangermano, M. Campo, S. González Prolongo, C. F. Pirri, A. Jiménez-Suárez and A. Chiappone, Adv. Funct. Mater., 2021, 31, 2106774 CrossRef.
- E. Fantino, A. Chiappone, I. Roppolo, D. Manfredi, R. Bongiovanni, C. F. Pirri and F. Calignano, Adv. Mater., 2016, 28, 3712–3717 CrossRef CAS PubMed.
- (a) N. D. Dolinski, Z. A. Page, E. B. Callaway, F. Eisenreich, R. V. Garcia, R. Chavez, D. P. Bothman, S. Hecht, F. W. Zok and C. J. Hawker, Adv. Mater., 2018, 30, e1800364 CrossRef PubMed; (b) N. D. Dolinski, E. B. Callaway, C. S. Sample, L. F. Gockowski, R. Chavez, Z. A. Page, F. Eisenreich, S. Hecht, M. T. Valentine, F. W. Zok and C. J. Hawker, ACS Appl. Mater. Interfaces, 2021, 13, 22065–22072 CrossRef CAS PubMed.
- J. J. Schwartz and A. J. Boydston, Nat. Commun., 2019, 10, 791 CrossRef CAS PubMed.
- I. Cazin, M. O. Gleirscher, M. Fleisch, M. Berer, M. Sangermano and S. Schlögl, Addit. Manuf., 2022, 57, 102977 CAS.
- (a) J. Odent, S. Vanderstappen, A. Toncheva, E. Pichon, T. J. Wallin, K. Wang, R. F. Shepherd, P. Dubois and J.-M. Raquez, J. Mater. Chem. A, 2019, 7, 15395–15403 RSC; (b) X. Kuang, J. Wu, K. Chen, Z. Zhao, Z. Ding, F. Hu, D. Fang and H. J. Qi, Sci. Adv., 2019, 5, eaav5790 CrossRef CAS PubMed; (c) V. Chan, J. H. Jeong, P. Bajaj, M. Collens, T. Saif, H. Kong and R. Bashir, Lab Chip, 2012, 12, 88–98 RSC; (d) G. I. Peterson, J. J. Schwartz, Di Zhang, B. M. Weiss, M. A. Ganter, D. W. Storti and A. J. Boydston, ACS Appl. Mater. Interfaces, 2016, 8, 29037–29043 CrossRef CAS PubMed.
- (a) N. de Alwis Watuthanthrige, P. Chakma and D. Konkolewicz, Trends Chem., 2021, 3, 231–247 CrossRef CAS; (b) J. Wu, X. Yu, H. Zhang, J. Guo, J. Hu and M.-H. Li, ACS Sustainable Chem. Eng., 2020, 8, 6479–6487 CrossRef CAS.
- (a) M. K. McBride, B. T. Worrell, T. Brown, L. M. Cox, N. Sowan, C. Wang, M. Podgorski, A. M. Martinez and C. N. Bowman, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 175–198 CrossRef CAS PubMed; (b) M. A. Bin Rusayyis and J. M. Torkelson, Polym. Chem., 2021, 12, 2760–2771 RSC.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- A. Perego and F. Khabaz, Macromolecules, 2020, 53, 8406–8416 CrossRef CAS.
- R. Du, Thermal-mechanical behavior of bio-enhanced vitrimers, 2021 Search PubMed.
- M. Guerre, C. Taplan, J. M. Winne and F. E. Du Prez, Chem. Sci., 2020, 11, 4855–4870 RSC.
- W. Alabiso and S. Schlögl, Polymers, 2020, 12, 1660 CrossRef CAS PubMed.
- H. Xie, J. Shao, Y. Ma, J. Wang, H. Huang, N. Yang, H. Wang, C. Ruan, Y. Luo, Q.-Q. Wang, P. K. Chu and X.-F. Yu, Biomaterials, 2018, 164, 11–21 CrossRef CAS PubMed.
- D. Reisinger, S. Kaiser, E. Rossegger, W. Alabiso, B. Rieger and S. Schlögl, Angew. Chem., 2021, 60, 14302–14306 CrossRef CAS PubMed.
- D. Reisinger, K. Dietliker, M. Sangermano and S. Schlögl, Polym. Chem., 2022, 13, 1169–1176 RSC.
- E. Rossegger, K. Moazzen, M. Fleisch and S. Schlögl, Polym. Chem., 2021, 12, 3077–3083 RSC.
- G. Pohlers, J. C. Scaiano, E. Step and R. Sinta, J. Am. Chem. Soc., 1999, 121, 6167–6175 CrossRef CAS.
- S. C. Ligon, R. Liska, J. Stampfl, M. Gurr and R. Mülhaupt, Chem. Rev., 2017, 117, 10212–10290 CrossRef CAS PubMed.
- J. L. Dektar and N. P. Hacker, J. Am. Chem. Soc., 1990, 112, 6004–6015 CrossRef CAS.
- J. L. Self, N. D. Dolinski, M. S. Zayas, J. Read de Alaniz and C. M. Bates, ACS Macro Lett., 2018, 7, 817–821 CrossRef CAS PubMed.
- (a) M. Sangermano, I. Roppolo and A. Chiappone, Polymers, 2018, 10, 1–8 CrossRef PubMed; (b) J. V. Crivello and J. H. W. Lam, J. Polym. Sci., Polym. Chem. Ed., 1979, 17, 977–999 CrossRef CAS.
- (a) S. Shi, C. Croutxé-Barghorn and X. Allonas, Prog. Polym. Sci., 2017, 65, 1–41 CrossRef CAS; (b) N. Zivic, P. K. Kuroishi, F. Dumur, D. Gigmes, A. P. Dove and H. Sardon, Angew. Chem., 2019, 58, 10410–10422 CrossRef CAS PubMed.
- N. B. Cramer, J. P. Scott and C. N. Bowman, Macromolecules, 2002, 35, 5361–5365 CrossRef CAS.
- E. Rossegger, R. Höller, D. Reisinger, J. Strasser, M. Fleisch, T. Griesser and S. Schlögl, Polym. Chem., 2021, 117, 10212 Search PubMed.
- (a) J. A. Rard, D. A. Palmer and J. G. Albright, J. Chem. Eng. Data, 2003, 48, 158–166 CrossRef CAS; (b) L.-P. Ma, S. Dong, M. Chen, W. Ma, D. Sun, Y. Gao, T. Ma, H.-M. Cheng and W. Ren, ACS Appl. Mater. Interfaces, 2018, 10, 40756–40763 CrossRef CAS PubMed; (c) J. P. Sharma, K. Yamada and S. S. Sekhon, Ionics, 2012, 18, 151–158 CrossRef CAS; (d) M. Chidambaram, D. Curulla-Ferre, A. P. Singh and B. G. Anderson, J. Catal., 2003, 220, 442–456 CrossRef CAS.
- (a) H. Fosshaug, P. Askebjer, J. Karlsson, A. Bajramovic, K. Xing, R. Eklund, J. Walford, M. Ekberg, P. Hogfeldt and T. Öström, in Advances in Resist Technology and Processing XXI, ed. J. L. Sturtevant, SPIE, 2004, p. 879 Search PubMed; (b) G. M. Kim, Y. H. Bae and W. H. Jo, Macromol. Biosci., 2005, 5, 1118–1124 CrossRef CAS PubMed.
- J. F. Cameron, S. L. Ablaza, G. Xu and W. Yueh, in Advances in Resist Technology and Processing XVI, ed. W. Conley, SPIE, 1999, p. 785 Search PubMed.
- (a) J.-S. Baek, J.-S. Park, S. S. Sekhon, T.-H. Yang, Y.-G. Shul and J.-H. Choi, Fuel Cells, 2010, 10, 762–769 CrossRef CAS; (b) M. Doyle, S. K. Choi and G. Proulx, J. Electrochem. Soc., 2000, 147, 34 CrossRef CAS.
- X. Yang, L. Guo, X. Xu, S. Shang and H. Liu, Mater. Des., 2020, 186, 108248 CrossRef CAS.
- T. Liu, B. Zhao and J. Zhang, Polymer, 2020, 194, 122392 CrossRef CAS.
- K. Moazzen, E. Rossegger, W. Alabiso, U. Shaukat and S. Schlögl, Macromol. Chem. Phys., 2021, 222, 2100072 CrossRef CAS.
- (a) K. Yu, P. Taynton, W. Zhang, M. L. Dunn and H. J. Qi, RSC Adv., 2014, 4, 48682–48690 RSC; (b) M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS PubMed.
- R. A. Lawson, C.-T. Lee, R. Whetsell, W. Yueh, J. Roberts, L. Tolbert and C. L. Henderson, in Advances in Resist Materials and Processing Technology XXIV, ed. Q. Lin, SPIE, 2007, p. 65191N Search PubMed.
- E. Rossegger, R. Höller, D. Reisinger, M. Fleisch, J. Strasser, V. Wieser, T. Griesser and S. Schlögl, Polymer, 2021, 221, 123631 CrossRef CAS.
- A. Vitale and J. T. Cabral, Materials, 2016, 9, 760 CrossRef PubMed.
- W. Kern, M. Müller, C. Bandl, N. Krempl and M. Kratzer, Polymers, 2022, 14, 4006 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR spectra of curing reaction, TGA and stress relaxation data of thiol-acrylate vitrimers, photographs of 3D printed objects undergoing locally controlled shape change. See DOI: https://doi.org/10.1039/d3py00333g |
| This journal is © The Royal Society of Chemistry 2023 |