
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Introducing lignin as a binder material for the aqueous production of NMC111 cathodes for Li-ion batteries†
Silje Nornes
Bryntesen
 a,
Ignat
Tolstorebrov
a,
Ann Mari
Svensson
b,
Paul
Shearing
ac,
Jacob Joseph
Lamb
a and
Odne Stokke
Burheim
*a
a,
Ignat
Tolstorebrov
a,
Ann Mari
Svensson
b,
Paul
Shearing
ac,
Jacob Joseph
Lamb
a and
Odne Stokke
Burheim
*a
aDepartment of Energy and Process Engineering, Norwegian University of Science and Technology, Kolbjørn Hejes Vei 1B, N-7491 Trondheim, Norway. E-mail: odne.s.burheim@ntnu.no; silje.n.bryntesen@ntnu.no
bDepartment of Materials Science and Engineering, Norwegian University of Science and Technology, Sem Sælands vei 12, N-7491 Trondheim, Norway
cUniversity College London, Gower St, London, England, UK
First published on 4th November 2022
Abstract
By enabling water-based cathode processing, the energy-intensive N-methyl-2-pyrrolidone (NMP) recovery step can be eliminated, reducing the cost and environmental impact of LIBs. Aqueous processing of high capacity Ni-containing LiNixMn1−x−yCoyO2 (NMC) cathodes is problematic due to lithium-ion(Li+) leaching, corrosion of the aluminum (Al) current collector, and the lack of aqueous soluble bio-derived binders. The present study investigates the potential of substituting and fully replacing the commonly used polyvinylidene fluoride (PVDF) and carboxymethyl cellulose (CMC) binders with abundant, bio-derived kraft lignin. This paper gives a holistic overview of the optimal conditions when employing these binders. For the first time, we demonstrate that NMC111 cathodes of comparable specific capacities to NMP/PVDF-based ones over 100 cycles or at high C-rates (5C) can be formulated in water using lignin or CMC/lignin as binder materials. Cyclic voltammetry (CV) revealed that kraft lignin undergoes a redox reaction with the electrolyte between 2.8 and 4.5 V, which diminishes upon subsequent cycles. Differential scanning calorimetry (DSC) revealed that lignin is thermally stable up to 152 °C. Rheology measurements showed that replacing NMP with water allows for a solvent reduction. The cathodes fabricated using an aqueous slurry should be dried at 50 °C, as extensive surface cracks detected using scanning electron microscopy (SEM) diminish. Li+ leaching from NMC111 and NaOH species from kraft lignin caused an increase in pH during aqueous slurry fabrication. A carbon-coated Al foil (C-Al) prevented Al corrosion and increased the lignin cathode's mechanical strength revealing lignin's exceptional binding abilities to carbon. The electrolyte wettability decreased for calendered lignin-containing cathodes with low porosity and a large carbon black/lignin matrix.
1 Introduction and outline
Efforts to de-carbonize the transport sector are driving the shift from fossil-fuelled vehicles to electric mobility. Being the leading battery technology for mobile devices and battery electric vehicles (BEVs), recent advances in lithium-ion battery (LIB) technology have driven a rise in production demand. However, the carbon footprint of LIB production contributes significantly to the greenhouse gas emissions of BEVs.1,2 Therefore, the sustainability and cost of production must be improved without compromising the battery energy density.3LIB graphite anodes are usually fabricated using aqueous slurries, whereas cathodes are mainly produced using the toxic N-methyl-2-pyrrolidone (NMP) solvent. NMP introduces an expensive recovery step and requires high airflow during electrode drying;4 thus, an aqueous cathode processing route will lower the energy consumption and cost of production.5 Additionally, replacing the costly polyvinylidene fluoride (PVDF) cathode binder with water-soluble binders will simplify the recycling process for end-of-life LIBs by mitigating the conversion from solid-coated cathodes to aqueous black mass.6
However, several challenges have been reported when transitioning to the aqueous processing of Ni-containing cathodes. Issues are related to the poor water-compatibility of the active material, high basicity of water leading to corrosion of the aluminum(Al)-foil current collector,7,8 cracking of the cathode coating due to increased capillary pressure during electrode drying,9 and finding a binder material providing sufficient cohesion and adhesion forces between the coated particles and the Al-foil,10 respectively. Therefore, the search for aqueous compatible and electrochemically inactive binders providing sufficient mechanical strength has become an area of significant scientific interest.4,10–14
1.1 Aqueous binder materials
Bresser et al.13 reviewed alternative bio-derived aqueous binders for LIB electrodes. A typical example is a water-soluble and/or dispersible carboxymethyl cellulose/styrene-butadiene rubber (CMC/SBR) binder currently used in the production of the graphite anodes and water-compatible olivine structured LiFePO4 (LFP) cathodes.13 CMC is a linear polymeric derivative of cellulose and has been widely used as a thickener, binder, and stabilizer in various industries.15 Furthermore, CMC has contributed to making the slurry homogeneous as it suppresses the re-agglomeration of the dispersed CB particles.16 Understanding the interaction between the binder and the other electrode components (i.e., the active material and the conductive additive) is vital for the coating's homogeneity, consequently optimizing the electrochemical performance.Lignin is another promising bio-derived binder material that becomes water soluble when the phenolic hydroxy group (Fig. 1) is activated at high pH.17 Being a waste material from the pulp industry, it has low cost, and high abundance.18,19 It has been researched as a precursor in the production of several battery components,13 including the separator, conductive additive, activated carbon for capacitors,20 electrolyte,21 and binder material.3,22–25 The lignin molecule is complex and consists of the monolignol monomer species shown in Fig. 1.19,26 The molecular characteristics depend on the pre-treatment method (kraft, soda, or steam exploded) and whether it is derived from softwood (poplar) or hardwood (pine). The molecular masses and glass transition temperature (Tg) of lignin isolated from different resources have been reported to be in the range of 1000–20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, and 91–174 °C, respectively.19,27 Additionally, it has a higher electrical conductivity than that of the commercial PVDF binder.
000 g mol−1, and 91–174 °C, respectively.19,27 Additionally, it has a higher electrical conductivity than that of the commercial PVDF binder.
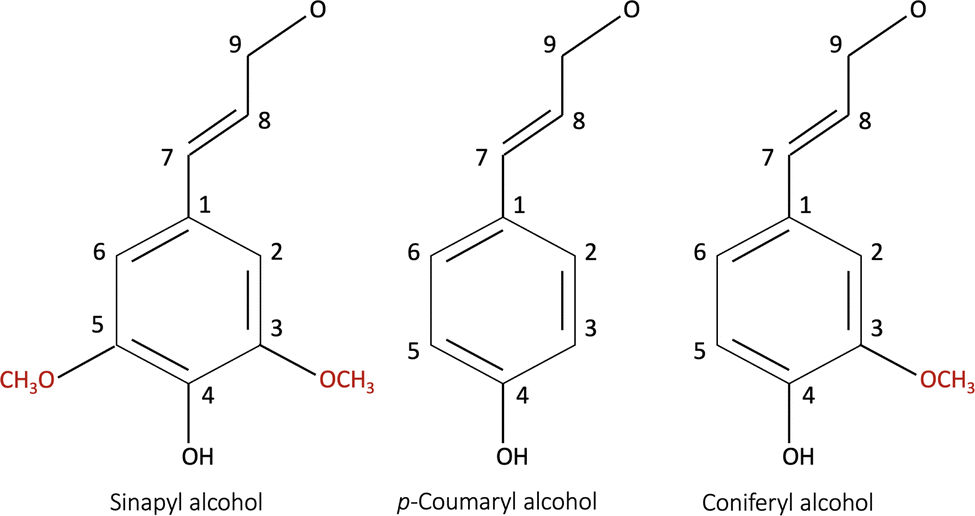 | ||
| Fig. 1 The monomer species building up a lignin molecule: (a) p-coumaryl alcohol (4-hydroxyl phenyl, H), (b) coniferyl alcohol (guaiacyl, G), and (c) sinapyl alcohol (syringyl, S).19,26 | ||
Lu et al.22 studied lignin as a binder material for the aqueous production of LFP cathodes and graphite anodes and reported a high reversible capacity of 148 and 305 mA h g−1, respectively. Pore removal through calendering decreased the cathode's specific capacity due to polarisation.22 They also emphasized the need for pre-treatment of the lignin to remove small molecular structures which supposedly diffused into the electrolyte.
Ponnuchamy et al.23 recently compared the binding ability of lignin, other sustainable cellulose-based binders, and unsustainable binders to graphene sheets. Subsequently, and independently of this work, they reported that lignin offered the highest binding strength amongst all the most common binders. This makes lignin a suitable binder for both the graphite anodes and the cathodes due to the presence of the conductive carbon additive in the latter. Others10 have reported that the CMC binder binds more strongly to the Al current collector than the PVDF binder. Meanwhile, little research is conducted on lignin's binding ability to the active cathode materials or the Al current collector. However, a protective carbon-coated Al (C-Al) current collector may provide strong adhesion between the current collector and lignin-based electrode coating.8,13 Such carbon coating may also prevent poor Al-foil wetting caused by the high surface tension of water28 and protect Al against corrosion8,13 during the casting step of an aqueous slurry.8
1.2 Solvent evaporation and crack propagation
When replacing the NMP with water, the slurry's wettability to the Al-foil during coating decreases, creating an electrode coating with lower adhesion and more extensive cracking. According to Du et al.,28 this is caused by the high surface tension of water (72.80 mN m−1, 20 °C) compared to NMP (40.79 mN m−1, 20 °C). During solvent evaporation, a high capillary pressure build-up leads to crack propagation in the electrode coating.9,28 Others claim that the extensive cracking of the aqueous processed cathode coatings is a result of H2 release during drying, owing to the corrosion of the Al current collector.29 Man and Russel30 found that the critical pressure for crack onset increased with decreasing electrode thickness. Zheng et al.31 reported that mechanical issues increased proportionally to the coating thickness of NMC111 and LFP cathodes.Moreover, Buss et al.32 reported that dissolved or dispersed non-volatile species with a larger density than the volatile solvent can migrate during solvent evaporation and form a “sediment” layer at the electrode surface. Inhomogeneity decreases the adhesion strength of the coating and; consequently, its electrochemical performance. This can be avoided by implementing a two-stage NMP evaporation process33 or lowering the drying temperatures.28 Therefore, electrode drying should be carefully considered to avoid cracking and particle migration during solvent evaporation.
Crack propagation and fractures in the electrode coating result in poor mechanical strength, isolation of active materials, and disruption of the electronic and ionic conductive network. During cycling, the cracks in these mechanically weak coatings will further propagate and create new surfaces. This allows for more electrode–electrolyte side reactions, further growth of the cathode electrolyte interphase (CEI) or solid electrolyte interphase (SEI) surface layers, and surface phase transformations in NMC111 (from layered R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m to disordered spinel Fm and NiO rock salt (Fmm structure), and dissolution of TMs.28,34 This inevitably manifests as impedance growth and capacity deterioration over multiple cycles.34,35 It should be mentioned that a decreasing rate capability with increased mass loading is often ascribed to the Li+-diffusion limitations and not necessarily crack propagation.36,37
m to disordered spinel Fm and NiO rock salt (Fmm structure), and dissolution of TMs.28,34 This inevitably manifests as impedance growth and capacity deterioration over multiple cycles.34,35 It should be mentioned that a decreasing rate capability with increased mass loading is often ascribed to the Li+-diffusion limitations and not necessarily crack propagation.36,37
The term capacity retention varies across scientific papers38,39 depending on their scope of work. This work defines capacity retention as the fraction of capacity relative to the initial discharge capacity, which is useful when defining the cyclability of real devices.40
1.3 Li-ion leaching in Ni-containing cathodes
A suitable cathode chemistry for EVs is nickel(Ni)-containing LiNixMn1−x−yCoyO2 (NMC) cathodes due to their high specific capacity. However, additional challenges have been reported when transitioning to aqueous processing of Ni-containing cathodes such as LiNi0.33Mn0.33Co0.33O2 (NMC111) and LiNi0.8Mn0.1Co0.1O2 (NMC811).41 During moisture exposure, active oxygen (O) species are generated at the particle surface. These combine with water (H2 O) and carbon dioxide (CO2) before they further react with Li+ to form Li hydroxide (LiOH) and eventually Li carbonate (Li2CO3) species at the NMC particle surface.41,42 Upon water exposure during the slurry processing step, Li+ leaches out from the NMC111 surface as displayed in Fig. 2, leaving the outer layers delithiated.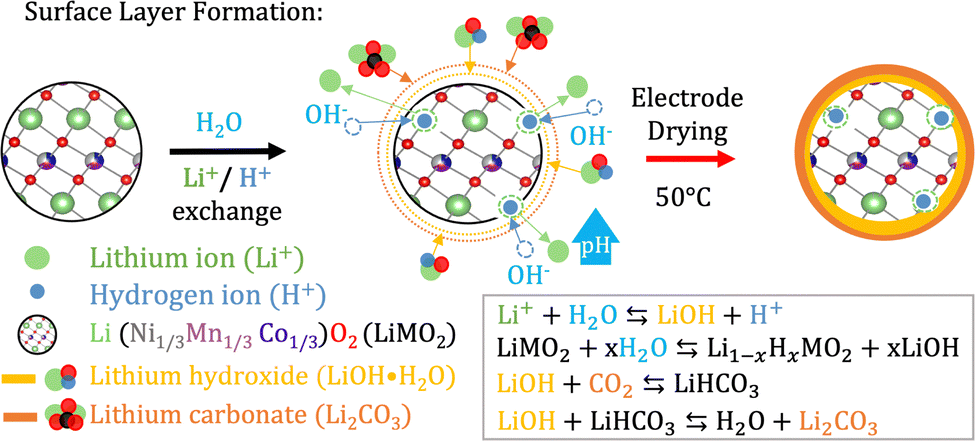 | ||
| Fig. 2 When the NMC material is exposed to water Li+ starts leaching out from its structure while H+ from the water intercalates with the structure.44 OH− in the solution steadily increases the pH and combines with Li+ to form LiOH as the water evaporates during electrode drying. Furthermore, LiOH reacts with CO2 to form LiHCO3 and eventually Li2CO3. | ||
The mechanism related to the charge compensation following the Li+-loss in the near-surface region of the NMC111 is disputed in the literature.29 Two scenarios have been widely discussed; one suggesting the spontaneous reduction of Ni3+ to the more thermodynamically stable Ni2+,43 and the other suggesting a H+/Li+-exchange mechanism illustrated in Fig. 2.29,44 The reduction of Ni-ions is aggravated in more Ni-rich NMC cathodes as the ratio between Ni3+ and Ni2+ is high, and recent articles suggest that this type of charge compensation occurs upon long-term humidity exposure (over several months), whereas the H+/Li+ exchange is dominant during the initial stage of exposure to humidity or water.29
Zhang et al.45 exposed NMC111 particles to water and humidity (ambient atmosphere, 20 °C, 50% relative humidity) and reported that Li+ leaching took place during the first 15 minutes, forming a 10 nm thick surface layer of LiOH and Li2CO3. The humidity-exposed NMC111 was cycled in coin cells (versus a Li-metal anode), and the capacity decreased by 10 mA h g−1 at 1C. The initial discharge capacity and the capacity retention after 30 cycles were 139 mA h g−1 and 95%, respectively. This capacity loss was confirmed by Cetinel et al.16 during aqueous production of NMC111 tested in full cells using graphite as an anode.
On the other hand, Jung et al.43 reported that a 10 nm thick surface layer was found on humidity-stored Ni-rich NMC811; however, no significant changes were detected on the NMC111 surface. Shkrob et al.44 reported that the initial capacity loss from Li+ leaching was regained during the first few cycles. Doberdo et al.8 also detected an initial capacity decrease, but explained that this was likely caused by the corrosion of the Al current collector since the dissolution of LiOH and Li2CO3 raised the slurry pH above the stability window of the Al-foil (pH = 4.5–8.5).
This report investigates lignin as an alternative binder material for the aqueous processing of NMC111 cathodes. First, we gradually replace the standard PVDF/NMP binder/solvent with lignin/NMP, resulting in four cathode coatings with PVDF:lignin binder ratios of 100:0, 75:25, 50:50, and 0:100. Thereafter, the NMP solvent is replaced with water, and CMC/lignin or lignin are tested as binders. The electrochemical and thermal stabilities of lignin are revealed through cyclic voltammetry (CV) and differential scanning calorimetry (DSC), respectively. The slurry rheology for different powder (NMC111:binder:CB) to liquid (NMP or water) ratios (P:L) is measured to reveal the dispersion network and ensure a proper viscosity for the coating. Furthermore, two different drying temperatures (50 °C and 90 °C) and doctor blade coating thicknesses (150 μm and 200 μm), calendering pressures, and carbon-coated Al-foil (C-Al) are tested, aiming to obtain a coating with high mechanical strength. The electrochemical performance of aqueous-produced cathodes using CMC/lignin or pure lignin as binders is compared to PVDF/NMP-based cathodes at high discharge C-rates (5C) and over 100 cycles.
2 Experimental
Table 1 presents an overview of all parameters tested during slurry fabrication and coating. This includes binders, solvents, different NMC111:CB:binder ratios, wet coating thickness, and current collectors. The slurry viscosity, electrochemical and thermal stability, surface cracking, binder migration, mechanical strength, specific capacity, and capacity retention were measured.
:liquid ratio (P:L), plain aluminum (Al) or carbon-coated (C-Al) current collector (CC), and doctor blade coating gap (DBG) for the electrodes tested
| Solvent | Binder(s) [wt%] | NMC111:CB:binder [wt%] |
DT [°C] | P:L [wt%] |
CC | DBG [μm] |
|---|---|---|---|---|---|---|
| NMP | PVDF:lignin |
|||||
| 100:0 |
85:10:5 |
90 | 1:2 |
Al, C-Al | 150, 200 | |
| 75:25 |
85:10:5 |
90 | 1:2 |
Al | 150 | |
| 50:50 |
85:10:5 |
90 | 1:2 |
Al | 150 | |
| 0:100 |
85:10:5 |
90 | 1:2 |
Al | 150 | |
| 0:100 |
85:10:5 |
50 | 1:2 |
Al | 150 | |
| 0:100 |
80:11:9 |
50 | 1:1.7 |
Al, C-Al | 150, 200 | |
| 0:100 |
90:5:5 |
50 | 1:1.3 |
C-Al | 200 | |
| Water | CMC:lignin |
|||||
| 2:7 |
80:11:9 |
50 | 1:2.4 |
Al, C-Al | 150, 200 |
2.1 Cathode perparation
Three pre-mixed binder:liquid solutions of (PVDF:NMP (1:20 wt%), lignin:NMP (1:10 wt%), or lignin:(water + 10 wt% ethanol) (1:10 wt%) were stirred with a magnetic stirrer overnight at 40 °C to dissolve the binder. Details of the chemicals used are presented in Table S1 (ESI†). The dissolved binder was mixed with NMC111 (Targray) and carbon black (CB) (Imerys and Carbon, C45) to a wt% of NMC111:CB:binder and additional solvent to reach a specified powder:liquid wt% (P:L). The slurry chemistry ratio, binder(s) chemistry, electrode drying temperature (°C), current collectors, and doctor blade gap for tape-casting are presented in Table 2.
:CB:binder wt% was 80:11:9, *85:10:5 or **90:5:5. The drying temperature T = 50 °C (water) and 90 °C (NMP) unless specified
| Solvent | Binders [wt%] | DBG/CG [μm] | Thick. b/a cal. [μm] | Por. b/a cal [%] | ML [mg cm−2] | IDC [mA h g−1] | CC | Plot |
|---|---|---|---|---|---|---|---|---|
| NMP 90 °C | PVDF:Lignin |
— | ||||||
| 100:0* |
200/45 | 63 ± 7/33 ± 3 | 53 ± 4/9 ± 1 | 6.6 ± 0.6 | 150 | C-Al | Fig. S9a | |
| 100:0* |
200/45 | 54 ± 4/40 ± 3 | 56 ± 4/30 ± 3 | 6.1 ± 0.8 | 154 | Fig. S9a | ||
| 75:25* |
150/45 | 43 ± 6/28 ± 3 | 53 ± 6/27 ± 4 | 4.4 ± 0.2 | 153 | Fig. 8 | ||
| 50:50* |
150/45 | 50 ± 1/39 ± 1 | 57 ± 1/25 ± 4 | 4.8 ± 0 | 132 | Al | Fig. 8 | |
| Water 50 °C | 0:100* |
150/45 | 42/— | 51/— | 4.4 | 83 | Fig. 8 | |
| 0:100* (50 °C) |
150/45 | 49/— | 57/– | 4.5 | 141 | Fig. 8 | ||
| 0:100 (90 °C) |
150/45 | 74 ± 5/17 | 71 ± 14/- | 4.7 | 128 | Fig. 8 | ||
| 0:100 |
200/45 | 80 ± 5/41 ± 2 | 44 ± 1/0 ± 16 | 9.7 ± 0.2 | 120/125 | Fig. 10c | ||
| — | 200/60 | 77 ± 11/42 ± 2 | 40/16 ± 5 | — | 136 | Fig. 10c | ||
| — | 200/80 | 77 ± 11/64 ± 2 | 38/32 | — | 148 | C-Al | Fig. 10c | |
| 0:100 |
150/45 | 74 ± 9/40 ± 4 | 46 ± 7/15 ± 1 | 7.4 ± 0.3 | 143/127 | Fig. 10b | ||
| — | 150/60 | 65 ± 15/34 ± 5 | 44/22 ± 17 | — | 125 | Fig. 10b | ||
| — | 150/80 | 72 ± 3/54 ± 3 | 50/37 | — | 139 | Fig. 10b | ||
| 0:100** |
200/80–60 | /62 ± 3 | /31 | 7.5 ± 0.2 | 154 | Fig. 12 | ||
| CMC:lignin |
— | |||||||
| 2:7 |
200/40 | 45 ± 3/36 ± 1 | 46 ± 1/32 ± 2 | 5.1 ± 0.4 | 147/151 | Al | Fig. 9 | |
| 2:7 |
200/40 | 46 ± 2/35 ± 1 | 44 ± 2/19 ± 2 | 5.6 ± 0.2 | 135/142 | Fig. 9 | ||
| — | 200/60 | 55 ± 3/47 ± 3 | 51 ± 0/10 ± 0 | — | 141 | — | ||
| — | 200/80 | 55 ± 3/46 ± 3 | 51 ± 1/21 ± 1 | — | 143 | C-Al | — | |
| — | 150/60 | 45/41 ± 3 | 51 ± 0/27 ± 1 | 4.5 ± 0.2 | — | — | ||
| — | 150/80 | 45 ± 1/41 ± 2 | 53 ± 2/30 ± 4 | — | 150/151 | — |
A non-contact planetary mixer was used for slurry mixing (THINKY ARE250CE). The NMP/PVDF-based slurries were mixed at 1500 rpm for 25 minutes (min), followed by 2000 rpm for 5 min. The water-based slurries were pre-mixed with the binder and CB for 10 min at 1000 rpm, 2000 rpm, and 1000 rpm, respectively, before NMC111 was introduced and the mixing procedure was repeated. The rheology and pH of these slurries were measured.
The slurries were coated onto an Al-foil or C-coated Al-foil (C-Al) with a doctor blade gap of 150 or 200 μm using a tape caster (MSK-AFA-HC100, MTI Corp.). The electrodes were dried at 50 or 90 °C overnight in a vacuum, calendered (Electric Hot Press, MSK-HRP-01, MTI corp.) at different gap sizes (40, 45, 60, or 80 μm) four times, and punched to 15 mm diameter discs (MSK-T-12, MTI Corp.). The dried electrodes were weighed, the thickness was measured before and after calendering using a micrometer (VWR ± 0.001 μm), and the resulting porosity was calculated from the material densities using equation 1 in ref. 46. The theoretical material densities used for NMC111 (Targray), CB (Imerys), lignin (Sigma Aldrich), and PVDF (Sigma Aldrich) were 2.3, 1.9, 1.3, and 1.8 (g cm−3) at 25 °C, respectively.
The coated electrodes were stored in a desiccator filled with silica gel and post-dried at 120 °C for a minimum of 4 h to remove moisture before being introduced into an argon-filled glove box (O2 < 0.1 ppm, H2 O < 0.1 ppm) for coin half-cell (CR2032, MTI Corp.) assembly. Pre-cut Li-metal chips (15.6 mm × 0.25 mm diameter × thickness), 99.9%, Xiamen Tmax Battery Equipment) were cleaned to remove surface impurities and used as anode for the coin half-cells. A polypropylene (Celgard 2320) disc (17 mm diameter) was used to separate the coated cathode and the Li-metal anode. A pre-made salt solution of 1 M lithium hexafluorophosphate (LiPF6) dissolved in ethylene carbonate/ethyl-methyl carbonate/diethyl carbonate (EC:DMC:DEC 1:1:1, Sigma Aldrich) was used as the electrolyte. 20 μL of the electrolyte was added to each side of the separator to ensure proper wetting. To achieve good contact and obtain uniform current distribution, a stainless steel spacer and spring were placed on top of the Li-metal anode before crimping the cell (MSK-PN110-S, MTI Corp.) by applying a pressure of 120 psi.
2.2 Characterisation methods
The conventional DSC scanning used for lignin was conducted in one step. The samples were equilibrated at 30.0 °C for 5 min and heated up to 200 °C at a rate of 10.0 °C min−1. The modulated DSC (MDSC) procedure used for the PVDF sample was used to clarify the thermal processes in polymers. The modulation amplitude was ±1.0 °C min−1, and the heating rate was 3.0 °C min−1. The MDSC method helped clarify the melting and re-crystallization processes and glass transition. This method measured the differences in the heat flow between a sample and a reference as a function of time and temperature, but the heating profile differed from that of conventional DSC. The sample temperature continuously increased with time but in a sinusoidal trend. The total heat flow could therefore be split into the heat capacity component (reversing heat flow) and kinetic component (non-reversing heat flow).47 The reversing heat flow denoted melting and glass transition, while the non-reversing heat flow denoted the recrystallization or crystalline structure perfection. The comparison between the reversing and non-reversing heat flow allows the splitting of the exothermic recrystallization peaks when overlapping the endothermic melting peaks or glass transition.
The onset (To) and end (Te) points of melting were defined as the temperatures at which a change in the slope of the heating curve occurs. It should be noted that the glass transition in some substances like cellulose and polymers can be weak. Thus, the inflection point (Ti) was determined as a negative peak of the derived heat flow curve. The melting energy was obtained by integrating the DSC melting peaks (the linear baseline function integration), and the glass transition was determined with TA Universal Analysis 2000 version 4.5A software (TA Instruments, USA) and characterized by the To, Te, and Ti.
Cyclic voltammetry (Biologic BCS-805) was used to investigate the electrochemical stability of the Al-foil, carbon-coated Al (C-Al), carbon black, and carbon black/lignin (50/50 wt%) in coin cells with Li as a counter electrode (two-electrode configuration) and scanned between 2.8 and 4.5 V vs. Li+/Li using a sweep rate of 1 mV s−1. The carbon black and lignin/carbon black were mixed with water in a 1:1.3 powder:liquid ratio using the frame procedure for the slurry mixing and coated onto a C-Al using a 150 μm doctor blade gap. This resulted in a 30 μm thick carbon black coating with a mass loading of 2.9 mg cm−2, and a 20 thick lignin/carbon black coating with a mass loading of 10.6 mg cm−2.
2.3 XRD
The XRD measurements were performed on a Bruker D8 DaVinchi diffractometer using a Cu Kα radiation (λ = 1.54 Å) source, and a fixed slit size of 0.2. The XRD scan was scanned between 2θ values of 10° and 80°. The analysis of the XRD profiles was performed using DIFFRAC.EVA software (Bruker). The reference scans used for matching were found in PDF 2019+ database, and are found in Table S4 (ESI†).Rietveld refinement was used for structural analysis and was performed on DIFFRAC.TOPAS (V5.0, Bruker AXS). They were based on a rhombohedral α-NaFeO2 structural model, with a Rm space group and Li placed in the 3a position, Ni, Mn, and Co in the 3b position, and O in the 6c position. The refinements assumed fully stoichiometric samples and a model in which Li and Ni were free to switch positions (cation mixing) while Mn and Co were confined to the TM layer. The a and c lattice parameters and the Oz coordinate were treated as variables. The line widths were modeled within the fundamental parameters approach assuming microstrain-type broadening.
3 Results and discussion
Since little research has been conducted on cathode fabrication using lignin as a binder, this article aims to give the reader a holistic overview of the optimal fabrication parameters needed to obtain an electrochemical performance comparable to that of commercialized PVDF binders.The four different binder compositions tested (PVDF (85:10:5), PVDF/lignin (85:10:5), and CMC/lignin (80:11:9) and lignin (80:11:9 and 90:5:5) shown in Table 2 will be discussed in the following chapters. Different coating thicknesses, porosities, and mass loadings were tested to find the optimal cathode fabrication route. Only cathodes with comparable mass loading will be compared in the microstructural and electrochemical analyses.
A higher mass loading was obtained for all coatings when increasing the doctor blade gap (DBG) during tape casting from 150 μm to 200 μm. The electrode's mass loading was also dependent on the NMC111:CB:binder composition ratio and binder chemistries used. For example, the mass loading decreased from 9.7 mg cm−2 to 5.5 mg cm−2 when changing the binder from pure lignin to CMC/lignin because of the CMC's thickening effect. The porosity and thickness are, therefore, carefully considered and reported for each cell rather than only the final electrode density (g cm−3). This is especially important when considering the rate capability, as this is kinetically limited by the coating's electronic- and ionic conductivity.36
The calendering process decreased the porosity at the expense of thickness and made the coatings more uniform as indicated by their lower standard deviations. A small calendering gap (40–45 μm) resulted in high pressures (Phigh) which extensively reduced the porosity. A medium calendering pressure (Pmedium, 60–80 μm gap) generally resulted in higher porosity. It should be mentioned that porosity of 0% was defined according to the formula in Equation 1 and the complete removal of pores was unlikely.
Table 2 sums up the initial discharge capacity (IDC) of the cathodes measured through galvanostatic cycling at C/10. The PVDF/NMP-based cathode obtained the specific capacity (154 mA h g−1) expected from the Targray supplier (∼150 mA h g−1). Large variations were detected for lignin-based cathodes depending on the binder (lignin or CMC/lignin), solvent (NMP or water), and current collector (C-Al or Al). The reasons for these discrepancies will be discussed below. An interesting trend is that the lignin/water-based cathodes calendered under Pmedium pressures generally showed a higher initial capacity than those uncalendered or calendered with high pressure, suggesting that Pmedium provided a good combination of electronic conductivity and high porosity. It should also be mentioned that uncalendered lignin/water-based cathodes coated onto pure Al may suffer from larger errors in the measured IDC, since the cutting of these cathodes resulted in more material loss from the edges, Fig. S2b (ESI†).
3.1 Thermal stability of binders
The electrode materials should have high thermal stability to avoid degradation or mass loss during cell fabrication and operation. Cho et al.48 performed DSC analysis on different NMC materials and found that NMC111 is thermally stable below 223 °C. Al-Shroofy et al.46 used thermal gravimetric analysis (TGA) and found that PVDF and CB did not undergo mass loss until reaching temperatures of 400–460 °C and 500–600 °C, respectively. Loeffler et al.49 reported that CMC was not affected by thermal treatments below 200 °C.The DSC analysis revealing the thermal stability of PVDF and lignin is presented in Fig. 3a and b, respectively. The melting peak of lignin was found to be 152 °C, and the glass transition temperature (Tg) expected for the amorphous kraft lignin was 124–174 °C.19,22 According to Fig. 3a, PVDF had a wide melting temperature with a melting peak at 156 °C, and the heat of fusion (ΔHmelting) was 36.6 J g−1. By using ΔHmelting for a 100% crystalline PVDF (104.7 J g−150), the crystallinity of the PVDF was calculated to be 35%. The crystallinity and melting temperature for PVDF usually vary between 35 and 70%50 and 150 and 177 °C, respectively.
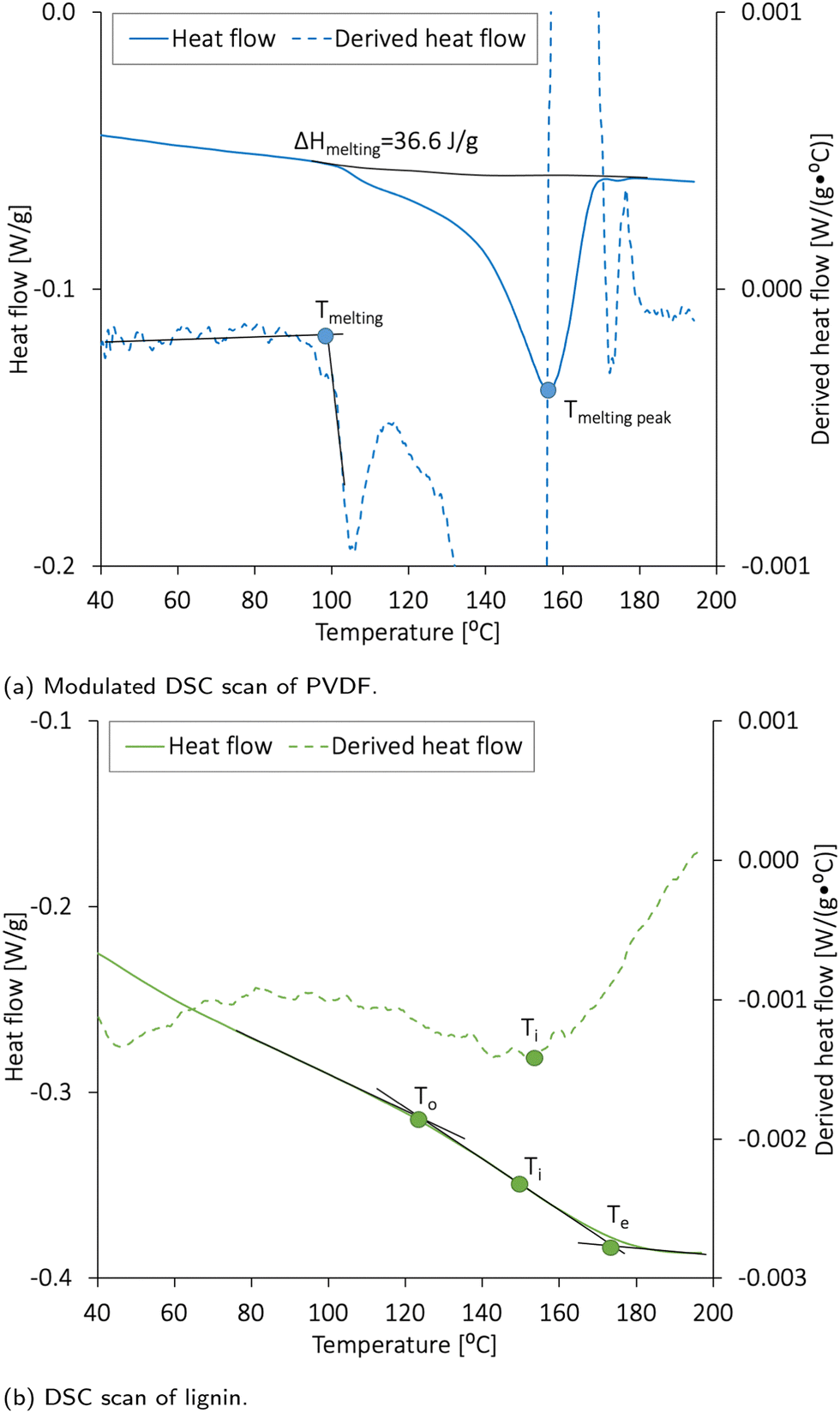 | ||
| Fig. 3 Differential scanning calorimetry (DSC) measurement of (a) PVDF and (b) lignin raw materials between 30 and 300 °C. ΔHmelting = heat of fusion, T = temperature, To = onset, Ti =inflection, and Te = end. | ||
The drying temperature used for cathode vacuum drying at laboratory-scale is generally between 80 and 130 °C.3,51–54 The solid components used in these cathodes are, therefore, expected to be thermally stable within the temperature range used in this report (50–120 °C). The PVDF partly melted during the post-drying step at 120 °C; however, such thermal activation has previously been reported to increase the bonding strength between PVDF, NMC, and CB and maintain the electrode's porosity in dry-coated electrodes.55
3.2 Electrochemical stability of Al, carbon, and lignin
Cyclic voltammetry (CV) is used to investigate the electrochemical stability of the aluminum (Al) current collector, the carbon-coated (1 μm) Al current collector (C-Al), a 2.9 mg carbon black (CB) coating (20 μm thick) on C-Al, and a 10.6 mg CB/lignin (50/50 wt%) coating (30 μm thick) on C-Al. The resulting scans are presented in Fig. 4 and Fig. S14 (ESI†). Lignin was mixed with CB as it has an absolute low conductivity. The current peaks (Ip) increased with the amount of CB and lignin present, indicating that these species underwent oxidation and reduction (redox) reactions within the potential window of 2.8–4.5 V. The Al current collector in Fig. S14a (ESI†) underwent an irreversible oxidation reaction at 4.2 V which contributed to an oxidation current peak (Ipo) of 55 μA at cycle 8, before the oxidation reaction diminished. The same trend was detected for the C-Al-foil in Fig. S14b (ESI†); however, the additional 1 μm carbon coating increased the Ipo by 10 μA. Additionally, the carbon contributed to a reduction reaction at around 4.1 V.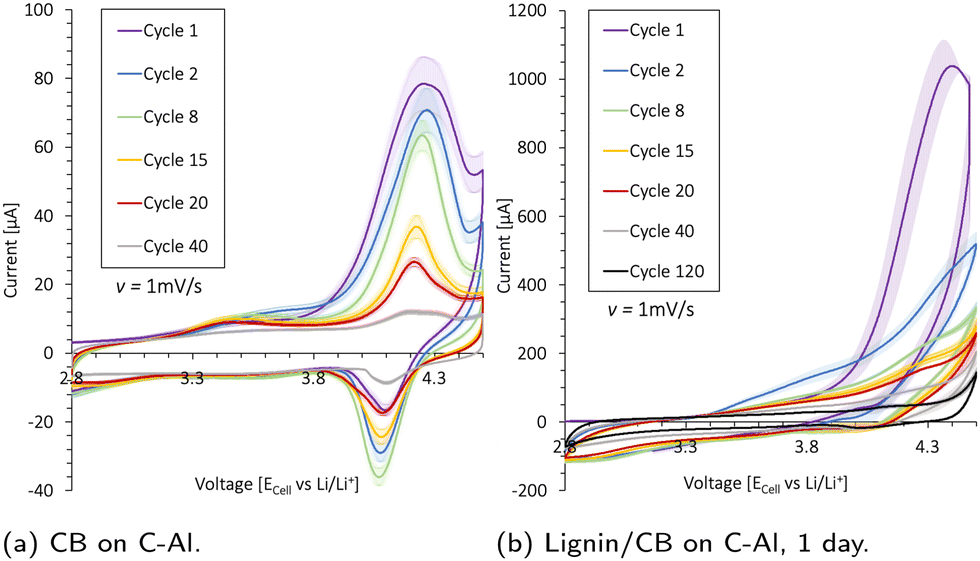 | ||
| Fig. 4 The electrochemical activity of the (a) carbon black (CB) coated on C-Al, and (b) lignin/CB (50/50 wt%) coated on C-Al scanned after 1 day wetting. The coin cells were scanned between 2.8 and 4.5 V (vs. Li/Li+) using a Li-metal anode, LiPF6 in ED/DMC/DEC electrolyte, and a scan rate of ν = 1 mV s−1 at 22 °C. | ||
When increasing the carbon content by coating C-Al with ∼2.9 mg CB as shown in Fig. 4a, there was no peak shift which indicated that the same redox reactions took place, but the Ipo of the first cycle increased with 50 μA. Thus, the CB contributed with ∼17.2 μA mg−1 to the oxidation peak at 4.3 V during the first cycle. The CB underwent its most extensive oxidation reaction during the initial cycle, and the Ipo declined steadily with cycle number.
When introducing lignin into the system by coating C-Al with a 10.6 mg CB/lignin (50/50 wt%) coating, there was a pronounced increase in the overall oxidation current in Fig. 4b and Fig. S14c (ESI†). The Ipo shifted to >4.3 V insinuating that lignin underwent different oxidation reactions than CB. However, during the first cycle, the Ipo for CB and lignin was found at 4.3 V. By deconvoluting the CB current during the first cycle (∼17.2 μA mg−1), lignin contributed to 10 times the amount of current per mass (∼172 μA mg−1). The Ipo was halved for the second cycle, accompanied by a peak shift to 4.5 V, which was above the typical upper cut-off voltage for NMC cathodes used for galvanostatic cycling. During the following cycles, all peak currents slowly diminished and the oxidation reaction stabilized. There was also a reduction reaction taking place with a peak height at 2.8 V, but this current also decreased over subsequent cycles. The results insinuate that the redox side reactions between the lignin and electrolyte were large during the first cycle, but subsided upon cycling.
When incorporating lignin in a cathode and cycling this galvanostatically in a coin cell against the Li metal, the apparent oxidation side reaction in the first cycle may be visible at high voltages during the first charge cycle, while the reduction reaction would appear at low voltages during discharge. In fact, the prominent initial irreversible oxidation reaction of lignin conceivably contributed to the elongated voltage profile >3.8 V and the high initial charge capacity during the first formation cycle of an 80:11:9 lignin/water-based cathode in Fig. 13.
There was no apparent trace of these side reactions after the first charge cycle in the voltage curves. The irreversible side reactions stabilized over subsequent cycles according to the CV, and the discharge curve originated to its expected NMC shape; thus, they did not appear to impede the overall capacity contribution after the first formation cycle. However, coulombic efficiency (CE) can be used to determine the efficiency of Li+-(de)intercalation and is a sign of irreversible side reactions. The average CE over 100 cycles at C/2 was generally lower for all lignin/water-based cathodes (96.6–98.2%) compared with the PVDF/NMP-based cathodes (99.2%) (Fig. S13, ESI†). Thus, the charge capacity was higher than the discharge capacity, which may suggest that additional oxidation reactions took place.
Since the mass loading of lignin for the CV scans was relatively high (∼5.3 mg cm−2) compared to the content in the cathodes (0.4–1.0 mg cm−2), the side reactions associated with lignin were 5–13 times lower than in the CV scan depending on the NMC111:CB:binder ratio and the mass loading of the cathode. Although they may be associated with the sodium (Na) or sulfur (S) in the lignin (EDS and XRD in Table S2 and Fig. S16b, ESI†), investigation on the mechanism behind these redox-reactions is outside the scope of this study.
As there was no apparent change in the reactions after a longer electrolyte exposure time (6 days in Fig. S14c, ESI†), these side reactions were likely not causing the capacity drop seen in the first cycles in Fig. 12. This drop is likely due to the poor wetting of the electrodes as explained in Subsubsection 3.6.2.
3.3 Slurry rheology
Rheology measurements were conducted on the NMP- and water-based slurries to reveal the optimal powder:liquid (P:L) ratio needed to obtain a proper viscosity for the tape casting, Fig. 5.
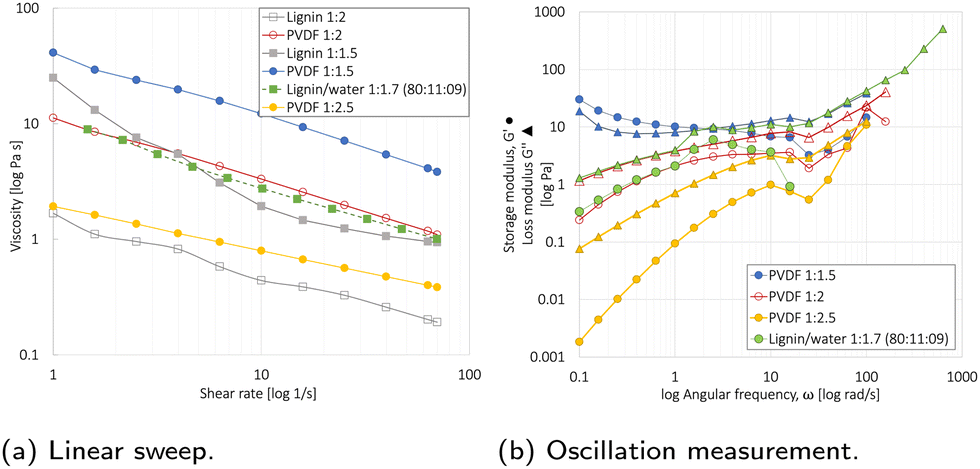 | ||
| Fig. 5 Rheology measurement revealing the viscosity for lignin/water or PVDF/NMP based slurries with different powder (NMC111:CB:binder):liquid (P:L) ratios. Unless specified, the powder ratio is 85:10:5 wt% (relative to wt% NMC111). (a) The linear sweep with increasing share rate from 1–70 1/s. (b) An oscillation measurement angular frequencies 680–1 rad s−1 revealing the storage modulus (G′, •) and loss modulus (G′′, Δ). | ||
The linear sweep measurement in Fig. 5a showed that the viscosity decreased with an increasing solvent and shear rate (1/s). For the 85:10:5 PVDF/NMP-based slurries, a 1:2 P:NMP ratio provided the optimal viscosity for tape casting. For the 80:11:9 lignin/water-based slurries, a 1:1.7 P:water ratio resulted in the same optimal viscosity. This was attributed to the lower viscosity of water compared to NMP,56 and an additional 1 wt% CB in the lignin/water slurry (80:11:9) relative to the PVDF/NMP slurry (85:10:5). Since 50% of the drying time is used to extract the last 10% of the solvent, the solvent reduction will not significantly affect the energy consumption used for evaporation during drying,57 and the elimination of the NMP recovery step will therefore be the main contributor.
The oscillatory mode test shown in Fig. 5b is used to reveal the interaction between PVDF binder, NMC111, and CB through the storage modulus (G′) and loss modulus (G′′).46 The NMP/PVDF-based slurries with powder:liquid ratios of 1:2 and 1:2.5 showed a liquid-like behavior (G′< G′′), whereas the 1:1.5 slurry showed gel-like behavior (G′ > G′′). In agreement with Al-Shroofy et al.,46 these gel-like properties are undesirable for slurry tape casting. This agreed with these results as the 1:2.5 slurry resulted in poor mixing and an uneven slurry coating. The lignin/water 1:1.7 and PVDF/NMP 1:2 slurry behaved almost identically during the oscillatory test, both having favorable liquid-like behavior.
3.4 Micro-structural analysis
The commercial cathode fabricated using PVDF/NMP as binder/solvent was gradually replaced with lignin/NMP, and entirely replaced with lignin/water. The cathodes were dried at 90 °C or 50 °C in vacuum and the resulting binder migration, surface cracking, and particles were analyzed in SEM.The surface of lignin/NMP-based and lignin/water-based cathodes dried at temperatures of 50 °C and 90 °C were analyzed in SEM. The accumulation of lignin binder on the electrode surface was classified according to the percentage of NMC111 particles covering the surface (Fig. S4, ESI†). An extensive surface layer (2% NMC111 surface coverage) was detected for the lignin/NMP-cathodes due to the migration of the undissolved low-density lignin molecules. The lignin binder migration in lignin/NMP cathodes was mitigated when drying at 50 °C, revealing a 12% NMC111 coverage, due to a low NMP evaporation rate.22,58 When replacing the NMP solvent with water, the lignin binder migration further diminished (19% NMC111 surface coverage), which was close to values expected for the standard PVDF/NMP cathodes (22%) with the same NMC:CB:binder ratio. The solubility of lignin molecules was higher in water relative to NMP, and the undissolved particles migrated more extensively during NMP solvent evaporation.59
The number of micro-cracks on the cathode's surface was higher for lignin/water-based cathodes compared with the PVDF/NMP-based cathodes in Fig. 6. Although the cracks diminished on the surface of the lignin/water-cathodes by lowering the drying temperature from 90 °C to 50 °C (Fig. S2a, ESI†), more cracks were still detected when using water as the solvent. Du et al.28 also reported extensive crack formations at drying temperatures >50 °C for aqueous processed electrodes. This was caused by the high surface tension of water relative to that of NMP, which increased the capillary pressure build-up during drying.9,28
 | ||
| Fig. 6 SEM surface crack analysis of an uncalendered PVDF/NMP-based cathode dried at 90 °C (left) and a lignin/water-based (right) cathode dried at 50 °C with a similar mass loading (7.4 mg cm−2 ±0.2). | ||
Scratch tests (Fig. S3, ESI†) and pen tests were used to reveal the cathode sheet's mechanical strength. Generally, the above-mentioned binder migration and cracking decreased the cathode coating's adhesion to the Al current collector and its overall mechanical strength.60
Interestingly, all lignin-based cathode coatings delaminated from the Al current collector in flakes (Fig. S2a, ESI†), which indicated that lignin formed sufficiently strong cohesion forces to NMC111 and CB. This was confirmed when replacing the plain Al current collector with a carbon-coated Al (C-Al) current collector. Fig. S2b (ESI†) shows an image of the cathode surface after cutting out the electrode discs from a PVDF/NMP cathode coated on a pure Al-foil, and a lignin/water-based cathode coated on an Al-foil or a C-Al-foil. The C-Al increased the lignin/water-based cathode's mechanical strength.
Lignin has several functional groups,61 and the classification of the chemical bonds between lignin-CB-NMC111 is out of the scope of this paper and should be further investigated elsewhere.23,62 Additionally, the problems associated with a poor wetting of the Al-foil during the coating-step due to the high surface tension of water,63,64 and the corrosion of the Al-foil,49 was eliminated when using C-Al as current collector.
A SEM analysis of uncalendared and calendered cathode surfaces under different calendering pressures is presented in Fig. 7. These cathodes were fabricated using water as the solvent and lignin as a binder and dried at 50 °C. The uncalendered cathodes show extensive surface cracking and a poor connection between the NMC111 and the CB/binder-matrix. By applying medium (Pmedium = 60–80 μm calendering gap) and high (Phigh = 40–45 μm calendering gap) calendering pressures, the surface cracks were removed and a good NMC111-to-CB/binder matrix connection was provided. However, at high calendering pressures (Phigh), NMC111 particle deformation occurred. Therefore, the medium calendering pressure was preferred to provide a mechanically strong coating with good electronic conduction, while maintaining the NMC111 particle integrity.
 | ||
| Fig. 7 SEM surface analysis of lignin/water cathode coatings with a similar mass loading (7.4 ± 0.3 mg cm−2) which was uncalendered, calendered at medium pressure using a 60 μm calendering gap (Pmedium), or calendered at high pressure using a 45 μm calendering gap (Phigh) (magnitude: 1000×). The NMC111 particles are shown in the top right corner (magnitude: 10000×). The rate capability of these cathodes is shown in Fig. 10b. | ||
To conclude, the reduction of surface cracks through calendering (Fig. 7), drying temperature (from 90 to 50 °C), and the application of a C-Al current collector provided lignin/water-cathodes with high mechanical strength. The calendered lignin/water-cathodes using C-Al, dried at 50 °C, obtained comparable mechanical strength to PVDF/NMP-cathodes using plain Al dried at 90 °C according to pen tests.23
3.5 Electrochemical performance
The following section will present the electrochemical performance of NMC111 cathode coatings fabricated using the following binder/solvent systems: lignin/NMP, PVDF/NMP, (CMC/lignin)/water, and lignin/water. The use of Al-foil and C-Al-foil current collectors is also investigated.For some of the electrodes, different mass loadings and calendering pressures are tested under different discharging rates to understand the effect of electronic and ionic conductivity on the rate performance. Finally, different electrolyte wetting times are presented for the lignin/water-based cathodes with high (90%) and low NMC111 (80%) contents.
:10:5 NMC111:CB:PVDF ratio were fabricated without being exposed to humidity- or water, and used as a baseline. These cathodes showed expected initial capacity (154 mA h g−1), the voltage plateau obtained the expected NMC111 shape, and was stable over 100 cycles as shown in Fig. S9 (ESI†).
A plain Al-foil and a carbon-coated Al-foil (C-Al) current collector were tested on PVDF/NMP-based cathodes (mass loading: 6.1–6.6 mg cm−2). The rate capability for uncalendered and calendered cathodes is presented in Fig. S9a (ESI†).
The capacity of PVDF/NMP-cathodes with plain Al-foil was similar to that of those reported earlier.65 The calendered PVDF/NMP cathode coated onto plain Al with 30% porosity showed higher capacity retention at 5C (85%) compared to the uncalendered cathode with 56% porosity (75%). The low capacity at high C-rates for uncalendered cathodes caused poor electronic connections within the high porosity coating. Earlier reports suggest that the porosity for PVDF/NMP-based cathodes should be between 30 and 40%53 to create a coating with optimal electronic and ionic conductivity.
The application of C-Al did not impact the cell's capacity at low C-rates (<1C); however, at higher rates (1–5C), the capacity retention decreased for the cathodes using C-Al. The uncalendered C-Al cathode obtained 23% of the initial discharge capacity at 5C, compared to 69% for the calendered C-Al cathode.66 The pen tests revealed that the C-Al current collector did not visibly improve the already high mechanical strength of the PVDF/NMP cathodes. According to the supplier of the C-Al current collector (MTI67), the C-Al was resistant to NMP but dissolved in water. This may cause the poor compatibility of C-Al and NMP-based slurries relative to water-based slurries.
:10:5 cathode was gradually replaced with lignin/NMP, resulting in three different (lignin/PVDF)/NMP cathodes with different 25%, 75%, or 100% lignin contents. These cathodes were coated onto a plain Al-foil.
The specific discharge capacity obtained during the third formation cycle is shown in Fig. 8a. The discharge capacity decreased with increasing lignin content, as the expected voltage plateau at 3.8 V seen for the baseline PVDF/NMP cathode gradually diminished due to a high overpotential. According to Fig. 8b, the voltage plateau re-appeared when reducing the drying temperature from 90 °C to 50 °C or by replacing the NMP solvent with water. Thus, the capacity decreased in accord with the accumulated lignin surface layer discussed above (Fig. S4, ESI†).
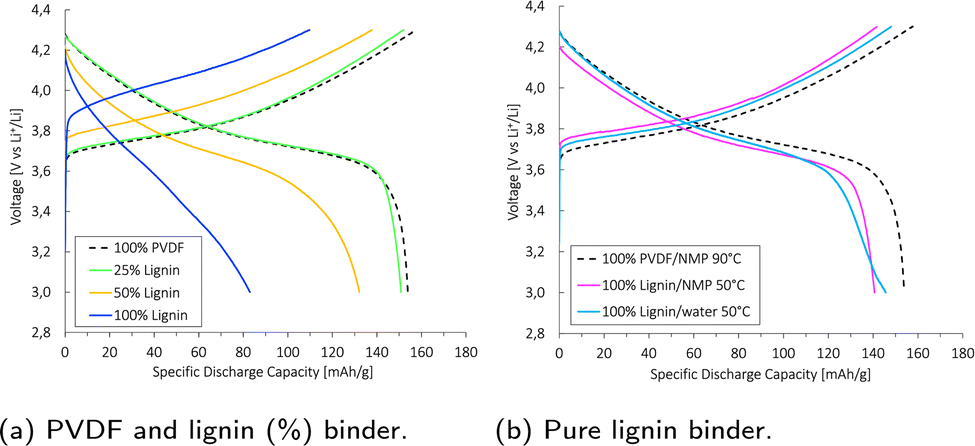 | ||
| Fig. 8 Specific discharge capacity during the third formation cycle at C/10 (1C = 160 mA g−1). The cathodes were coated onto Al-foil with a mass loading of 4.4–4.8 mg cm−2, and calendered at Pmedium. A PVDF/NMP-based cathode dried at 90 °C (black) was used as a baseline. The cathodes were cycled between 3.0 and 4.3 V (vs. Li+/Li) against a Li-metal anode in a coin cell at 22 °C. | ||
A post-mortem SEM analysis of the (lignin/PVDF)/NMP-based cathodes (Fig. S6, ESI†) revealed that the accumulated lignin on the cathode surface reacted with the electrolyte and created a surface layer. This not only resulted in the low capacity during the third formation cycle but also a declining rate capability (Fig. S9b, ESI†). We believe this layer created a barrier for Li+-transport which increased the intercalation resistance for cathodes with increasing lignin content when employing NMP as the solvent.
Qiao et al.68 also detected a surface layer on NMC111 particles in SEM after rate tests and assigned this to an unstable CEI formation associated with the LiPF6 salt. By switching to a moist scavenger sulfonimide salt (Li[N(SO2CF2H)(SO2CF3)], LiDFTFSI), the surface layer was no longer detected. However, the extent of the accumulated layer detected in this report was severe in comparison, suggesting that the accumulated lignin on the surface was the main issue and should be avoided by properly dissolving lignin in another solvent than NMP.
:7 wt%) cathode. Due to the very distinct effect on the viscosity of the paste, the acceptable amount of CMC was limited to 2%.49
The rate capability and coulombic efficiency (%) for (CMC/lignin)/water based cathodes with a mass loading between 5.1 and 5.6 mg cm−2 are presented in Fig. 9. Both a plain Al-foil and a carbon-coated Al-foil (C-Al) were tested as current collectors. The average initial discharge capacity was between 145 and 150 mA h g−1, and the capacity gradually dropped with increasing C-rates. No significant difference was detected for cathodes coated on plain Al and C-Al current collectors. At high C-rates (5C), the capacity retention for both the Al and C-Al was as high as ∼60%. This indicated that the combination of the CMC/lignin binder mix might provide sufficient electronic and ionic pathways and a proper electrical connection to the Al-foil.
 | ||
| Fig. 9 The average rate capability of three cells with aqueous produced NMC111 cathodes using CMC/lignin (green diamond) coated on plain Al-foil (empty) or carbon coated-Al-foil (C-Al) (filled). T = electrode coating thickness, P = porosity. The cathodes were cycled at different [C-rates] at 1C = 160 mA g−1 between 3.0 and 4.3 V (vs. Li+/Li) using a coin cell and Li-metal anode at 22 °C. Calendered at Pmedium. | ||
The long-term cycling, rate capability, and coulombic efficiency (%) for these (CMC/lignin)/water-based cathodes are shown in Fig. S12 (ESI†). The cathode using C-Al as a current collector obtained a higher long-term cyclability compared with the cathode coated onto a plain Al-foil. This can be explained by the larger cracks found for the water-based coatings coated on the Al-foil relative to the C-Al-foil in Fig. S5 (ESI†), and the more extensive corrosion taking place when using plain Al-foil (Fig. S17, ESI†).
Pastor-Fernández et al.69 systematically summed up the aging mechanisms within a cell, and pointed to the binder decomposition, contact loss, and corrosion of the Al-foil as influential factors for impedance growth and capacity loss over multiple cycles.
We conclude that although 2% CMC contributed to improved adhesion to Al for the lignin/water-cathodes, it was not enough to compensate for the poor adhesion forces between lignin and Al when exposed to volume changes over multiple cycles. This, in combination with the corrosion of the Al-foil, contributed to the decrease in the long-term cycling performance of the aqueous-produced lignin/CMC-based cathodes. Thus, a carbon-coated Al-foil was preferred as the current collector and will be used for the lignin/water-based cells presented in the next section.
3.6 Lignin binder/water solvent
Fig. 10 shows the rate capability of lignin/water cathodes with mass loadings of 7.4 ± 0.4 mg cm−2 (Fig. 10a and b) and 9.7 ± 0.2 mg cm−2 (Fig. 10c).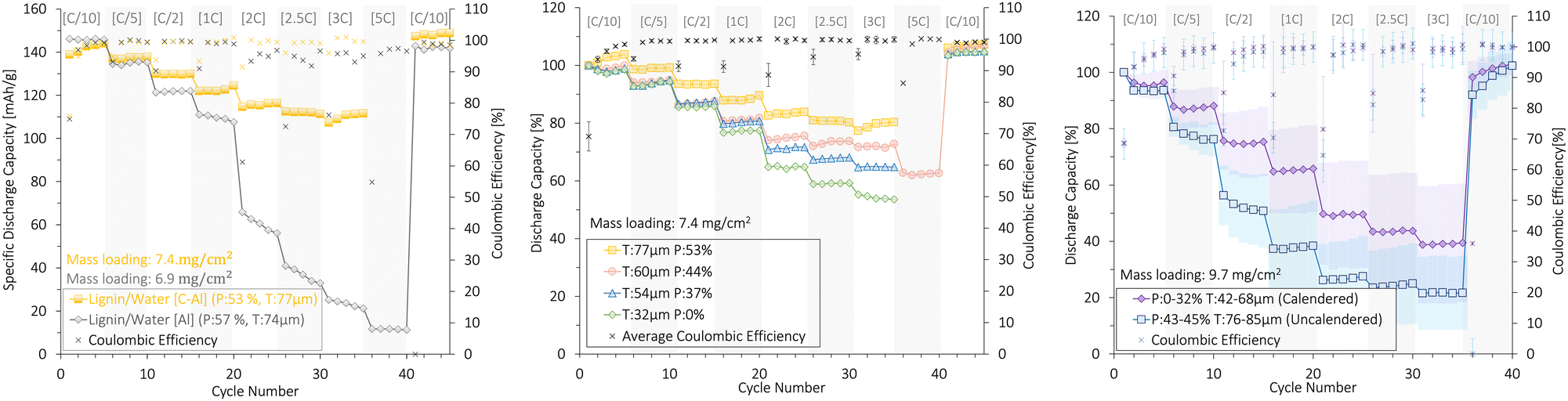 | ||
| Fig. 10 The rate capability (%) and coulombic efficiency (%) of uncalendered and calendered lignin/water-based NMC111 cathodes with (a), (b) low and (c) high mass loading, resulting in different thicknesses (T) and porosities (P). An SEM analysis of the cathode surfaces in (b) is presented in Fig. 7. The cathodes were cycled at different [C-rates] (1C = 160 mA g−1) in coin cells at 22 °C between 3.0 and 4.3 V (vs. Li+/Li) using a Li-metal anode. The plots with deviation bars show the average and of three cells deviation of three cells. | ||
First, Fig. 10a shows the rate capability of the uncalendered lignin/water cathodes coated onto a C-Al-foil and an Al-foil with similar thickness and porosities. The carbon-coated Al-foil (C-Al) current collector presented high mechanical strength compared with the plain Al-foil (Subsection 3.4). This created a higher electronic conductivity to the Al-foil which was crucial for rate capability. Due to the superior rate performance and mechanical stability, all other lignin/water-based cathodes in this section were coated onto a carbon-coated Al-foil.
The capacity decrease for the Al-foil cathode could also partly be caused by the pit corrosion of the Al (Fig. S7, ESI†). However, the CMC/lignin in Fig. 9 showed no particular difference in rate capability when comparing C-Al and Al, although corrosion also occurred for these cathodes. Thus, we believe the Al corrosion was not the main issue for a low rate performance at elevated C-rates, but rather during the long-term cycling. It is noteworthy that the lower rate capability of the lignin/water-cathodes (7.4 mg cm−2) compared to the CMC/lignin (5.1–5.5 mg cm−2) cathodes was likely caused by differences in the cathode coating's density and mass loading.
When increasing the mass loading from 7.4 shown in Fig. 10b to 9.7 mg cm−2 shown in Fig. 10c, the rate capability decreased despite the degree of porosity. For the high mass loading cathodes, the activation time for the NMC111 particles close to the current collector increased, and the Li+ diffusion pathways within the cathodes were generally elongated despite having a high porosity.36 At a thickness of >76 μm and a porosity of >43%, the capacity retention at high C-rates (3C) was therefore limited to 20%.
The large standard deviations for the rate capability within each plot were likely caused by the variation in the thickness and porosity of these cathodes. Generally, the lignin-cathodes with a lower mass loading were less sensitive to these variations than those of high mass loading. This gives an insight into the difficulties associated with reproducing cathodes with a high mass loading.
By calendering the lignin/water-cathodes with low mass loading (7.4 mg cm−2), the porosity dropped with high calendaring pressures and the rate capability of lignin/water-cathodes decreased accordingly, as illustrated in Fig. 10b. Despite having a relatively high porosity (44–53%), the uncalendered lignin-cathodes with low mass loading obtained a higher rate capability at 3C (84%) compared with the calendered cathodes (54%).
This indicated that even at a high porosity of 53%, the C-Al-foil enabled proper connections between the coating and the Al and that the large carbon/lignin-matrix in these 80:11:9 NMC:CB:lignin cathodes provided proper cohesion forces between the coated particles (i.e., a high electronic conductivity). The calendering, therefore, seemed to limit the ionic conduction pathways for the Li+ by removing pores, without improving the electrical conductivity. We conclude that the large CB/lignin-matrix already created a sufficient electronic conductivity for the uncalendered low mass loading (7.4 mg cm−2) cathodes and that the low thickness (<76 μm) provided an effective Li+-diffusion path. Thus, the calendering sacrificed the Li+-diffusion network by decreasing the porosity without improving the electrical conductivity, and consequently, the rate capability decreased.
However, the opposite trend was detected for the lignin/water cathodes with higher mass loading (9.7 mg cm−2). When increasing the mass loading, and thus the cathode thickness (>76 μm), the calendering likely improved the ionic conductivity by decreasing the thickness.
Thus, calendering lignin cathodes with a certain high thickness (>76 μm) improved the ionic and electronic conductivity, whereas calendering cathodes with a low thickness (<76 μm) reduced the ionic pathways without improving the electronic network.
Interestingly, the PVDF/NMP-fabricated cathodes of similar mass loading (6.6 ± 0.6 (Al) and 6.1 ± 0.8 (C-Al) mg cm−2) shown in Fig. S9a (ESI†) also showed the opposite trend to the 7.4 mg cm−2 lignin/water-based cathodes. The rate capability increased with lower porosities (30%). This was in agreement with other studies on PVDF/NMP-based cathodes, which reported that calendering down to a porosity of 30–40% was necessary to provide an optimal electronic conductivity.53 The need for a lower porosity indicated that the CB/binder-network within the 85:10:5 PVDF-based coating provided poor electrical conductivity compared with the 80:11:9 lignin-based cathodes.
It was, however, difficult to distinguish to what extent the improved electrical conductivity for the lignin-based cathodes was attributed to (1) the higher electrical conductivity of lignin relative to PVDF or (2) the higher amount of inactive material (CB and lignin) which provided more connection points to the active NMC111 particles in the 80:11:9 lignin/water-cathodes compared with the 85:10:5 PVDF/NMP-cathodes.51
An important factor to contemplate when comparing the rate capability of lignin/water-based cathodes to PVDF/NMP-based cathodes is the additional LiOH and Li2CO3 surface layer formed when the NMC111 particles are exposed to water. This phenomena will be further discussed in Subsubsection 3.7.2.
To conclude, the relationship between ionic and electronic conductivity within the different cathode coatings explained why the capacity decreased under high calendering pressures for lignin/water-based cathodes with a large carbon matrix (20%) shown in Fig. 10b, while the opposite trend was observed for the lignin/water-based cathodes of high mass loading shown in Fig. 10c and the PVDF-based cathodes with similar mass loading (6.1–7.2 mg cm−2) but lower carbon content (10 wt%) shown in Fig. S9a (ESI†).
Additionally, the low rate capability at high calendering pressure (Phigh: 40–45 μm gap) for the lignin/water-cathodes of low mass loading (7.4 mg cm−2) may to some extent be explained by the NMC111 particle crushing found in cathodes with ∼0% porosities (Fig. 7), or a Li+-transport pore-blocking effect formed when calendering cathodes with a large carbon-based matrix (80 wt% NMC111).
Kang et al.70 found that crushed NMC111 particles decreased the rate capability as many loosely connected NMC111 particles became inactive at high C-rates.
Zheng et al.53 and Rollag et al.9 reported that NMC111 cathodes with extremely low porosities (∼0%) suffered from a high activation barrier for Li+-transfer at the electrode/electrolyte interphase which reduced the cathode's rate capability. Haselrieder et al.71 reported that such an activation barrier on the surface of anodes may introduce poor electrolyte penetration and problems associated with electrolyte wetting. However, also reported that the Li+-transfer was closely associated with the fraction of inactive material (carbon black and lignin) rather than with the degree of the calendering pressure (i.e., porosity).53
In the next section, the lignin/water-based cathodes with large (80:11:9) and small (90:5:5) carbon/lignin matrices are compared to understand how the carbon/lignin-matrix influenced the electrolyte wettability. The porosity was kept constant (∼45–50%).
:11:9 cathode and the Li-metal anode and is presented in Fig. 11. The potential plateau indicating Li+-(de)intercalation was initially short but extended over multiple cycles. This feature was assigned to the cathode since the cathode's capacity curve featured the same response as the total cell (Ecell). The poor wetting of the cathode prevented the pores in the cathode coating from being filled with the electrolyte which hindered Li+-(de)intercalation and activation of the NMC111 close to the Al-foil. This resulted in a low cell capacity during the first cycles.
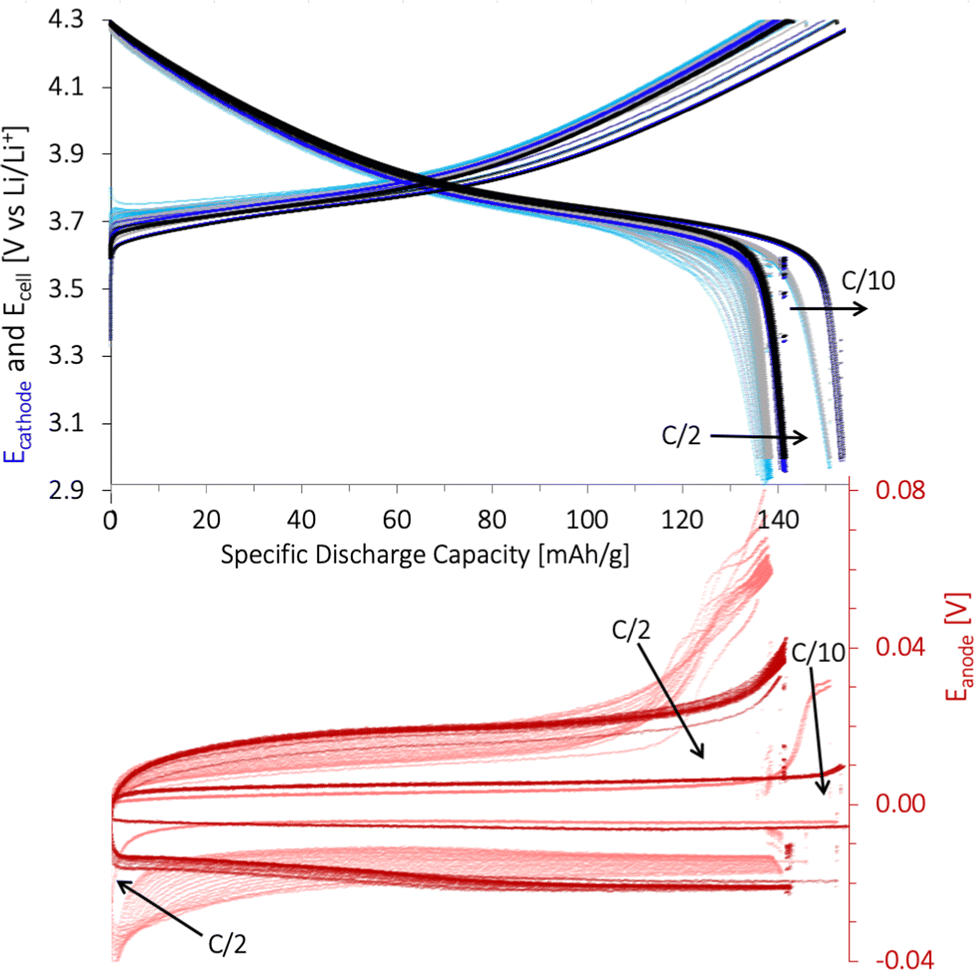 | ||
| Fig. 11 The galvanostatic cycling performance of a PAT cell using Li metal as the anode and the reference electrode. The response from the Li-metal anode (red Eanode), NMC111 working cathode (blue Ecathode), and both (black ECell) was measured. The lignin/water-cathode (80:11:9 wt% NMC111:CB:lignin) was coated on C-Al-foil, with 7.4 mg cm−2 mass loading and calendered at Pmedium. The cell was cycled after a 8 days wetting period for 5 cycles at C/10, 21 cycles at C/2 (light colour), 5 cycles at C/10, and 21 cycles at C/2 (dark colour) between 3.0 and 4.3 V vs. Li+/Li (1C = 160 mA g−1) at 22 °C. Arrows represent an increasing cycle number. The cathode surface area used for PAT cells was larger (2.54 cm2) compared to the coin cells (1.78 cm2) and the separator was glass fiber. | ||
The poor wetting behavior became apparent when comparing the soaking time of a 10 μL drop of electrolyte on a lignin-based 80:11:9 and PVDF-based 90:5:5 cathode. The former showed >12 times slower wetting (Fig. S1b, ESI†).
To understand the influence of the wetting and CB/lignin-matrix on the long-term cyclability, lignin/water-cathodes with 80:11:9 and 90:5:5 wt% NMC111:CB:lignin were soaked in the electrolyte for 1, 3, 6, or 35 day(s) before being cycled. The corresponding discharge capacities and coulombic efficiencies are shown in Fig. 12.
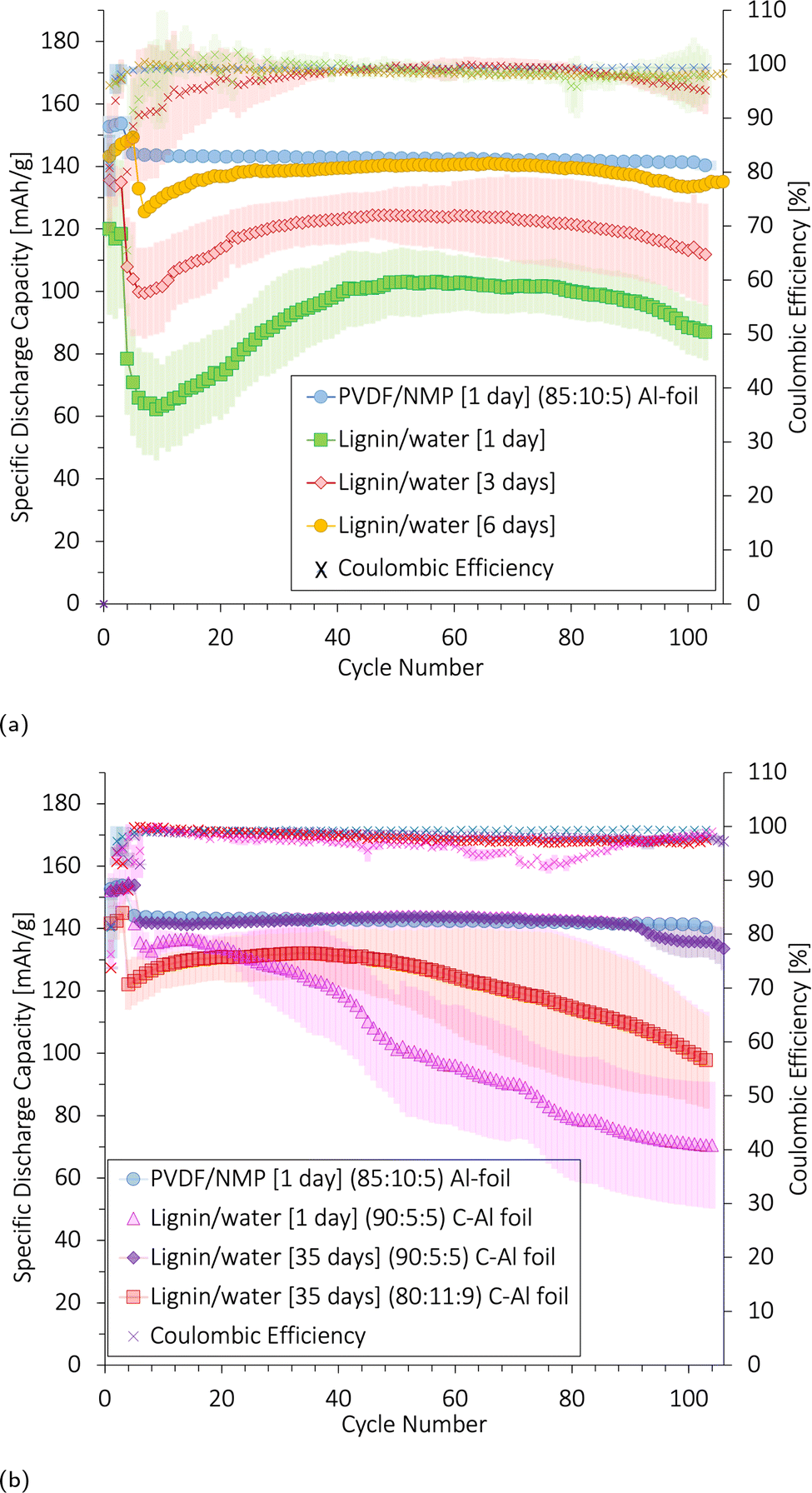 | ||
| Fig. 12 The average specific discharge capacity (mA h g−1) and coulombic efficiency (%) of three coin cells and their standard deviation. The lignin/water cathodes were coated onto a C-Al current collector with 80:11:9 wt% NMC111:CB:binder unless specified. The days refer to the mass loading for all cathodes was 7.4 ± 0.3 mg cm−2. An 85:10:5 PVDF/NMP-based cathode with comparable mass loading (6.6 ± 0.6 mg cm−2) coated onto a plain Al-foil and wetted for 1 day (blue circles) was used as the baseline. The cathodes were cycled in coin cells between 3.0 and 4.3 V (vs. Li+/Li) using a Li-metal anode. The C-rate was C/10 for the first 3 or 5 cycles and C/2 for the following 100 cycles. | ||
Both Fig. 12a and b show a decreasing initial discharge capacity (at C/10) and capacity retention (after 100 cycles at C/2) with shorter electrolyte wetting times for the lignin/water-cathodes. When the electrolyte exposure time was short, the cathodes with an 80:11:9 ratio in Fig. 12a showed a decreasing capacity in the first ∼25 cycles. The capacity drop gradually became less prominent by increasing the electrolyte wetting time from 1 to 6 days before cycling the cell. This phenomenon is also often recognized in silica-based anodes as poor electrolyte wetting.37 Thus, we believe that a poor electrolyte penetration into the cathode explained the declining capacity detected for lignin/water-based 80:11:9 cathodes during the first ∼25 cycles.
When reducing the CB and lignin wt% to about half (90:5:5 wt% NMC111:CB:lignin), the wetting process improved as shown in Fig. 12b. A 90:5:5 cathode soaked for 35 days in the electrolyte before cycling (red) obtained similar electrochemical performance over 100 cycles as the PVDF/NMP-based baseline cathode (blue).
On the other hand, when cycling such lignin/water-based 90:5:5 cathodes after being soaked in electrolyte for 1 day (pink), the capacity decrease caused by wetting was less apparent than for those with a high matrix, but the capacity decreased rapidly over the next 100 cycles. The CE was also unstable, suggesting that cracking and loss of active material occurred.
Although wetting was improved by reducing the CB/lignin matrix to 10%, the insufficient soaking of the cathode may cause local differences in the CEI layer formation and non-uniform stress accumulation in the cathode material, as volume expansion and contraction take place in the NMC111 particles during cycling. A large amount of NMC111 (90%) in combination with a small CB/lignin matrix did not seem to provide sufficient mechanical strength to sustain these stresses during cycling. This provoked crack propagation, and loss of active material, and eventuated in a rapid capacity fade for the 90:5:5 cathodes wetted for 1 day.
To conclude, the short voltage plateau during the first cycles in Fig. 11 was attributed to poor cathode wetting. Lu et al.22 reported the same reduction of the voltage plateau and a lower discharge capacity when applying lignin as a binder material. They did not detect such a trend for the uncalendered lignin-cathodes, and wettability is known to decrease with pore removal by calendering.37
This phenomenon explains why there is a dip during the first 5 formation cycles for the calendered lignin/water-based cathodes in the rate capability plot in Fig. 10. In contrast, no such dip was seen for the uncalendered cathodes with proper wetting. Thus, the poor electrolyte wettability of the 80:11:9 lignin/water cathodes can be improved by either increasing the porosity or reducing the CB/lignin-matrix from 20 to 10 wt%.
Problems related to wetting are common for other electrode chemistries, and other methods have been assessed to increase the wetting rate.37,72 Al-Shroofy et al.46 reported that wetting abilities decreased for calendered PVDF/NMP-cathodes as pores usually increase the electrolyte uptake. Sheng et al.15 utilized additives and different electrolytes to improve the electrode's electrolyte-wettability. Pfeging and Pröll36 observed a dramatic acceleration of electrode wetting by utilizing a set of laser-induced micro-channels with widths of 40–55 μm (NMC111 cathode) and 70 μm (LMO cathode).
3.7 Water exposed NMC111
No significant difference was detected in the initial capacity at low C-rates (C/10) when comparing the aqueous processed NMC111 cathodes to those fabricated with PVDF/NMP (Table 2). However, there was a larger capacity loss at higher C-rates (C/2-5C) and after long-term cycling (100 cycles) at C/2 for the lignin/water cathodes compared to the NMP/PVDF cathodes. This was not necessarily caused by the lignin binder, but rather the water solvent. In this section, we discuss the formation of restive Li2CO3 and LiOH surface layers on NMC111 during the aqueous slurry production, HF-formation during cycling, and the corrosion of the Al-foil.XRD profiles in Fig. S15 (ESI†) confirmed that no major change in the bulk crystallinity nor the appearance of secondary phases were observed for the water- and humidity-exposed NMC111 particles. A Rietveld refinement of the XRD spectra (Table S3, ESI†) showed a slight reduction in the unit cell volume for the NMC111 after being exposed to water (−0.011%) and air (−0.015%). Such a trend was reported earlier for NMC111 particles exposed to acid to be a sign of Li+ leaching from the surface of NMC111,73 although those values were >40 times higher.
Furthermore, XRD spectra may be used to determine the degree of Ni2+/Li+ cation mixing in the NMC111 particles. Cation mixing occurs when Ni2+ of similar radius as Li+ migrate from the octahedral 3b transition metal-(TM)-sites to the octahedral 3a Li+-sites. If the charge compensation of the lost Li+ during water exposure promotes the reduction of Ni3+ to Ni2+ and the migration of surface Ni2+ into the Li+-depleted layer, the XRD Rietveld refinement is expected to show a higher Li+/Ni2+ cation mixing in water- and humidity-exposed NMC111 particles.29,43,74
No such increase was observed from the Rietveld refinement (Table S3, ESI†) and the degree of cation mixing remained low for NMC111 particles regardless of the storage conditions or solvent exposure (<1.99% Ni2+ in the Li+-layers). In fact, the cation mixing decreased relative to the pristine NMC111.74 Thus, few migrating Ni2+ species were likely present, and there was no sign of Ni3+ reduction upon water exposure. These findings supported the conclusion drawn from the XPS measurements in Fig. S17 (ESI†), which showed no change in the Ni 2p peak for humidity- or water-exposed particles. Thus, we conclude that the charge compensation of Li+ during Li+ leaching of the water- and humidity-exposed NMC111 particles was mainly H+-exchange rather than the reduction of Ni3+ to Ni2+.29
It should be mentioned that the cation mixing is generally more prominent in Ni-rich NMC materials (NMC811), as the ratio between Ni3+/Ni2+ is higher.74 There are more Ni3+ available for reduction, which explains why an increase in cation mixing was detected in the XRD scans for water-exposed Ni-rich NMC in earlier reports,74 and not for NMC111 particles.29
First, the electrodes were dried at 120 °C under vacuum to remove all moisture before cell assembly. The voltage profiles of the water-based NMC111 cathodes assembled in half-cells showed a delithiation voltage peak at the beginning of the first charge Fig. 13. This initial potential peak completely disappeared from the profile in the subsequent cycles (Fig. S11, ESI†) and was not present for the NMP/PVDF-based cathodes. It is generally attributed to the dissolution of the insulating species formed at a humidity-exposed NMC particle surface, and usually increases with thicker LiOH and Li2CO3-layers for Ni-rich electrodes.43 This surface impurity acts as a resistive film around the NMC111 particles, and must be partially decomposed before delithiation can occur.43 Since it decreases for 90:5:5 cells, it indicates that the higher lignin content promoted this peak. We believe that there is a surface layer on the NMC particles in the lignin cathodes that dissolve upon delithiation, which may be a combination of LiOH or Li2CO3 from the water–NMC111 reaction and NaOH or N2S residuals deposited from the lignin-binder.
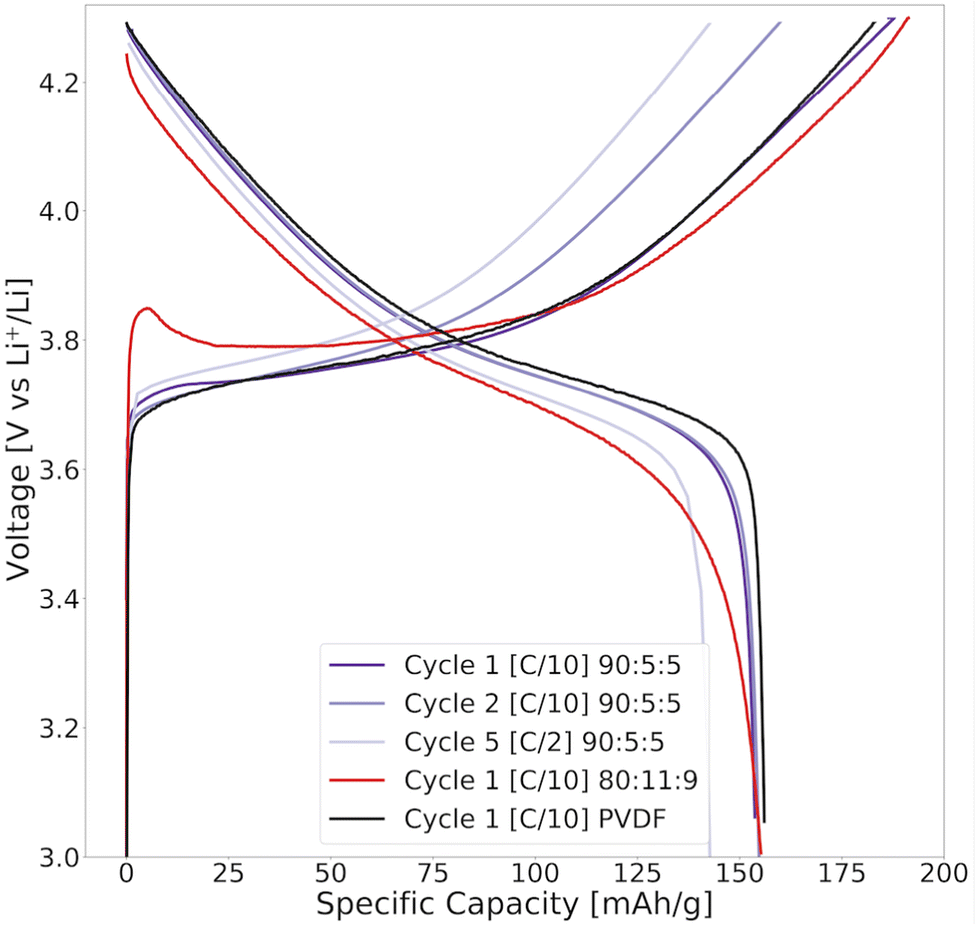 | ||
| Fig. 13 The voltage profile of the first and second cycles at C/10 and the fifth cycle at C/2 for NMC111 cathodes fabricated using 80:11:9 (red) or 90:5:5 (purple) lignin/water (7.4 mg cm−2), and 85:10:5 PVDF/NMP-fabricated cathodes (black) (6.9 mg cm−2). The cathodes were cycled in coin cells between 3.0 and 4.3 V (vs. Li+/Li) at different [C-rates] at 22 °C using a Li metal anode. 1C = 160 mA g−1. | ||
Studies report an absence of such a peak during the first charge of full cells,29,43 implying that such decomposition reaction occurs between the insulating surface compounds and the Li metal anode, rather than at the cathode. However, it should be mentioned that in the case of a graphite anode, the reactive surface area increases, and the compounds formed on the water-based cathode can be diluted at the graphite surface.29
At a low rate of C/10, the lignin/water-based cathodes did not face any significant loss in capacity due to the water treatment of NMC111, Fig. 13. Most studies report little to no difference in the capacity at low C-rates (<C/2) when comparing water-exposed NMC111 cathodes with those exposed to NMP.41,64,74 According to Memm et al.64 only 0.63% of Li+ per formula unit Li1.034Co0.33Mn0.33Ni0.33O2 were lost from the NMC111 surface due to Li+/H+ exchange, and were considered negligible for the performance.41,64,74,75
Such small Li+-depletion in the NMC111 surface was negligible for the capacity and was likely compensated for already in the first charge cycle as the cells were cycled in excess Li+ using a Li metal anode. However, since we assume that the main charge compensation for the Li+ leaching in the surface layer of the NMC111 lattice was H+/Li+-exchange, the H+ may react with fluorine (F) in the electrolyte and form HF during cycling. This reaction directly dissolves the metal ions in the cathode lattice leading to gas evolution, and phase transformation from the layered to spinel at the charged state.34 This reaction is therefore known to be detrimental to the battery capacity upon subsequent cycles and is likely causing more damage to these half-cells rather than the loss of Li+.76
It should, however, be mentioned that the charge capacity is highest for the lignin/water cathodes relative to the PVDF/NMP cathodes. The additional charge capacity in the first cycle (at high voltages >4.2 V) could potentially be attributed to the large oxidation current peak provided by lignin in the CV scan (Fig. 4b). Both features disappeared upon cycles indicating that these were irreversible depletion reactions.
When increasing the C-rate to C/2 in Fig. 13, there was a more prominent capacity loss for the lignin/water-based NMC111 cathodes compared with the PVDF/NMP-based cathodes with similar mass loading. There was also a higher polarization for the water-exposed NMC111 cathode indicating a higher resistance, which can be caused by the poor wetting of the lignin binder, the oxidation of the binder, or the presence of alkali residuals. This was in agreement with other studies looking at cathodes with water- or humidity-exposed NMC111 particles.16,41,45,74 Looking at the subsequent cycles with the same C-rate (Cycle 10 and 15 in Fig. S10b, ESI†), the polarization decreased.
Most studies agree that the Li+-diffusion limitation restricts the (de)intercalation of Li+ and is the main reason for a larger capacity loss at high C-rates when comparing water- and NMP-exposed NMC111 particles. However, the mechanism behind the Li+-diffusion limitation varies across scientific articles.
Azhari et al.74 reported that LiOH and Li2CO3 surface layers increased the surface resistance on the NMC111 particles, which kinetically limited the intercalation. Meanwhile, Wood et al.41 reported that the lower rate capability for water-exposed NMC811 particles was caused by the different slurry fabrication procedure, as SEM images revealed more binder coverage on water-processed NMC particles compared with NMP-fabricated NMC particles. A higher coverage ultimately increased the Li+-diffusion resistance.
In this article, no additional Li2CO3 and LiOH residuals were found. The different binder compositions, slurry mixture procedures, and coverage of NMC111 particles made it hard to distinguish whether the capacity loss at high C-rates for aqueous processed cathodes was caused by the Na2S and NaOH residuals formed upon water exposure or other fabrication parameters.41
When looking at the capacity loss over multiple cycles, studies generally conclude that cathodes made of water-exposed NMC111 particles deteriorate more rapidly during cycling compared to pristine particles. For example, Azhari et al.74 reported a capacity fade of 69% over 100 cycles at C/2 for water exposed NMC111 cathodes.
Even the best performing lignin/water-cathodes (lignin/water, 90:5:5, 35 days) obtained a slightly worse cyclability (100 cycles) than the PVDF/NMP-cathodes. This capacity deterioration over multiple cycles is likely explained by the water exposure of NMC111 particles and is caused by a series of phenomena; including the partial isolation of active NMC111 material,45,74 growth of a restive surface layer,74 and reaction between the electrolyte and H+ residuals to form hydrofluoric acid (HF).69,76 These degradation reactions usually get progressively larger for Ni-rich cathodes NMC811 cathodes with a more prominent LiOH and Li2CO3 surface layers.41,43
:11:9):1.7 (NMC111:CB:lignin):solvent ratio. The measurements are presented in Fig. 14, and at the initial pH, no NMC111 particles were present. The CB and lignin were already mixed with the water for the slurry mixtures. No pH increase was detected when adding the CB and the NMC111 was the main species contributing to the measured pH increase presented in Fig. 14.
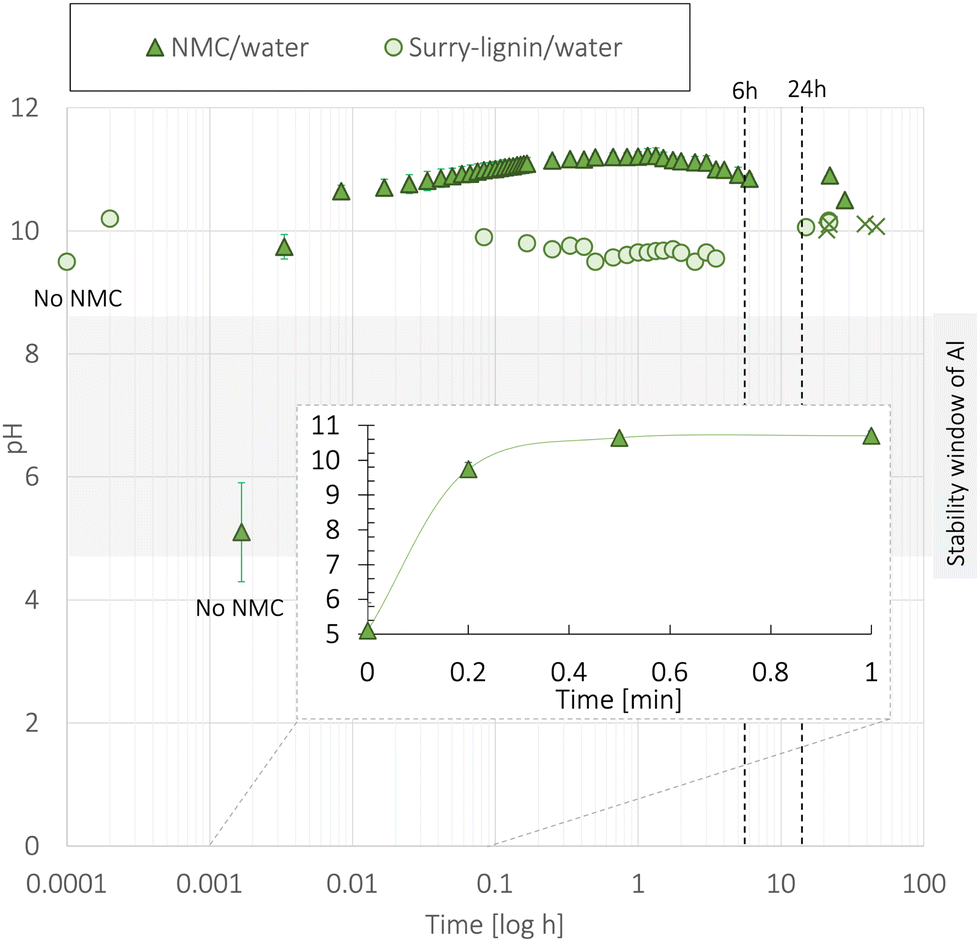 | ||
| Fig. 14 The pH measurement during the mixture of water with a low concentration of NMC111 powder (11.45 wt%) or as a slurry ((NMC111:CB:lignin):water) with a high NMC111 concentration ((80:11:9):1.7) at 22 °C. The point measurements display the average of two measurements. The points denoted “x” are measurements taken after adding extra water to the lignin/water slurry. | ||
Fig. 14 shows that when exposing NMC111 particles to water, the pH drastically changed during the first minute. Wood et al.41 reported a similar increase in the initial pH and long-term pH values for the NMC111 particles as shown in Fig. 14. The increase was caused by Li+ leaching from the NMC111 surface and originated from a combination of dissolution of LiOH and Li2CO3 surface species from NMC111,41 following the mechanism presented in Fig. 2.
The lignin/water-based slurry showed a high initial pH of 9.5. This was caused when dissolving lignin in water, as the pH increased from ∼6 to 9.5. The pre-mixed lignin/water (1:10 wt%) solution used for slurry fabrication had a high initial pH (9.4). Such a high initial pH for the lignin/water-slurry shown in Fig. 14 was caused by the dissolution of kraft lignin in water. Kraft lignin is extracted from the oil using an alkali process, and alkali residuals (such as NaOH or Na2S) are often left on the lignin if a neutralization process does not take place after the extraction. During the dissolution of lignin in water, these alkali residuals increased the pH from 5.8 to 9.4. Lignin is usually insoluble in water but becomes soluble if dissociation of its phenolic hydroxy group in Fig. 1 occurs.17 At pH 9.4, the phenolic hydroxy group was activated by the removal of its H+ which enabled the dissolution of lignin in water. The alkali species were, therefore, necessary to dissolve the lignin and make a homogeneous dispersion, and eventually a homogeneous cast. The phenolic hydroxy groups have a pKa around 10, indicating that at a pH of 9.4, nearly all phenolic hydroxy groups were activated.
After the initial increase in pH to 9.4, the pH increased to 10.4 after the first minute, before stabilizing at 10.5 after a day. In Fig. 14, such a small increase may indicate that the initial increase in pH caused by dissolving lignin in water suppressed the Li+/H+-exchange that occurs when NMC111 is introduced into water. However, the characteristic overpotential increase at the beginning of the first charge curve in Fig. S10b (ESI†) indicated that the Li residuals were still present on the NMC111 particle surface. Wood et al.41 also reported that the Li+ leaching took place in basic solutions (pH 12) as well as acidic solutions (pH 2). The high initial pH caused by adding lignin likely camouflaged the pH rise associated with Li+ leaching from NMC111. After 24 hours, twice the amount of water was added to the lignin/water-slurry mix (points marked X in Fig. 14). The pH remained stable, indicating that the Li+ leaching had reached a saturation point.41
Since the maximum content of lignin in any lignin/water-cathode was 9 wt%, only a trace amount of the alkali species was assumed to be present in the lignin/water-based 80:11:9 cathodes. EDS scans in Table S2 (ESI†) reveal that sodium (Na) (2.72 ± 0.84 wt%) and sulphur (S) residuals (0.35 ± 0.18 wt%) were present in the uncycled lignin/water-cathodes, and their presence decreased to 0.28 ± 0.02 and 0.25 ± 0.04 after 54 cycles, respectively. These species were not found in the PVDF/NMP-based cathode's EDS and XRD spectra in Fig. S6 and S16a (ESI†), respectively. An additional XRD scan of the lignin raw material confirmed their origin (Fig. S16b, ESI†).
The cycling curve generally obtained the typical NMC111 shape, and no apparent signs of the S and Na species were observed in the cycling curves. However, the impact of these species on the electrochemical performance should be further investigated before the commercialization of kraft lignin as a binder material. Interestingly, Na is often used as an additive in Na-CMC binders,3,16,77–79 and its presence is not considered harmful to the battery. S was present in such a small amount and was neglected in this study.
The corrosion of the Al can be avoided by using acids such as phosphoric acid (PA) as an additive; however, a high pH was needed for lignin to dissolve in water. Furthermore, the NaOH in the slurry during lignin/water-slurry fabrication may undergo neutralization reactions with the PA additive to form Na3PO4, and a protective carbon-coated Al-foil (C-Al-foil) was, therefore, the focus of this paper. An additional study on the use of PA as a pH controller during slurry fabrication and the use of a PA coating to eliminate the surface residuals (LiOH and Li2CO3) can be found in Subsection 5.4.
4 Conclusions
Cyclic voltammetry (CV) revealed that lignin undergoes an oxidation reaction during the first cycle with the electrolyte between 2.8 and 4.5 V, but stabilized over multiple cycles. Irreversible oxidation reactions contributed to an overall higher charge capacity and lower average coulombic efficiency over 100 cycles for lignin/water-cathodes (98.2%) compared with PVDF/NMP cathodes (99.2%). Differential scanning calorimetry (DSC) reveals that lignin was thermally stable at temperatures up to 152 °C. When replacing the PVDF/NMP with lignin/water during slurry fabrication, rheology measurements revealed that a lower liquid content was needed to obtain the same optimal viscosity for tape casting.When gradually replacing the PVDF/NMP with lignin/NMP, the lignin accumulated at the surface at high evaporation rates (90 °C), the adhesion to Al decreased, and the initial specific discharge capacity dropped. By decreasing the drying temperature to 50 °C, a more controlled NMP evaporation occurred, the accumulation of lignin at the cathode surface diminished, and its mechanical strength and specific capacity increased accordingly. By replacing the NMP solvent with water, the accumulated surface layer of lignin diminished and the capacity increased. The high surface tension of water relative to NMP caused a higher capillary pressure in the vacuum drying processes of aqueous slurries, which led to extensive cracking in the coating at 90 °C. Electrode drying at 50 °C and calendering reduced surface cracks. The calendering magnified the nature of the initial mechanical strength, intensifying a poor adhesion of the coating to the Al-foil and strong cohesion between particles. Pen and scratch tests confirmed that the lignin binder provided strong cohesion forces to carbon black. By replacing the Al current collector with a carbon-coated Al current collector (C-Al), the aqueous processed lignin-based NMC111 cathodes obtained mechanical stability compared to the PVDF/NMP baseline cathodes.
Calendering cathodes with a large binder matrix decreased the thickness and porosity, induced pore blocking, and created a barrier for Li+-transport, which eventually lowered the rate capability. While calendering and pore removal improved the rate capability for PVDF/NMP-based cathodes, the rate capability of lignin/water-based cathodes decreased with lower porosity and higher mass loading (9.7 mg cm−2).
The NMC111 cathode using pure lignin as a binder, coated onto a carbon-coated Al (C-Al) current collector, dried at 50 °C, with a mass loading of 7.4 mg cm−2 obtained 63% capacity retention at 5C. The cathodes using a CMC/lignin binder and a mass loading of 5.1–5.6 mg cm−2 obtained 60% capacity retention at 5C, independent of the current collector (Al or C-Al).
The aqueous-produced NMC111 cathodes with lignin binder obtained poor electrolyte wetting compatibility with the organic 1 M LiPF6 in the EC/DMC/DEC electrolyte. The cycling performance of the lignin-based cathodes improved with longer electrolyte wetting exposure before cycling, additional formation cycles at C/10, higher porosity, and a smaller CB/lignin matrix. Consequently, the 90:5:5 wt% lignin/water-cathodes wetted for 35 days with 5 formation cycles at C/10 showed similar initial discharge capacity and capacity retention (154 mA h g−1 and 89%) as to 85:10:5 PVDF/NMP-cathodes (153 mA h g−1 and 93%) after 100 cycles at C/2.
During the first charge cycle, lignin underwent an oxidation reaction that diminished over subsequent cycling. This caused a large charge capacity detected in the voltage curve during the first galvanostatic cycle. This current contribution was almost undetectable in the second cycle and diminished over multiple cycles.
The electrochemical, thermal, and mechanical stabilities of lignin suggest high applicability to other high Ni-containing NMC cathodes. Since the aqueous production of CMC-based NMC811 cathodes with minimal Li+ leaching has been reported,41 the electrolyte wettability of lignin needs to be improved before its commercialization as a binder.
The pH measurements and XRD confirmed the presence of Li+ leaching from the NMC111 particle surface during water exposure and slurry fabrication. The aqueous processing of NMC111 cathodes did not decrease the initial capacity of the cells at a low rate (C/10) but decreased the rate capability and long-term cyclability at higher C-rates.
Author contributions
O. S. B.: writing – review and editing, supervision, conceptualization, project administration, and funding acquisition. A. M. S.: writing – review and editing, supervision, and validation. J. J. L.: writing – review and editing, supervision, and validation. P. R. S.: writing – review and editing, supervision, and validation. I. T.: data curation, and formal analysis. S. N. B: methodology, investigation, data curation, formal analysis, conceptualization, and writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge financial support from FREYR (Project number: 90492502), ENERSENCE (Project number: 68024013), and InnoEnergy. Our gratitude is extended to the Department of Materials Science and Engineering, NTNU, as the XPS measurement and data analysis were conducted by Dr Verner Håkonsen and Dr Mahsa Ebadi, and to PhD candidate Harald Pollen, Dr Julian Richard Tolchard, and Prof. Nils Peter Wagner for conducting the Rietvelt analysis of the XRD spectra. Furthermore, we thank BSc. Nora Kvalsvik for assistance with the pH measurements and the graphic illustration of Li+ leaching.References
- S. D. Kurland, Environ. Res. Commun., 2020, 2, 012001 CrossRef
.
- L. A.-W. Ellingsen, B. Singh and A. H. Strømman, Environ. Res. Lett., 2016, 11, 1–9 CrossRef
- S. N. Bryntesen, A. H. Strømman, I. Tolstorebrov, P. R. Shearing, J. J. Lamb and O. Stokke Burheim, Energies, 2021, 14, 1406 CrossRef CAS
- D. L. Wood, J. D. Quass, J. Li, S. Ahmed, D. Ventola and C. Daniel, Drying Technol., 2018, 36, 234–244 CrossRef CAS
- D. L. Wood, J. Li and C. Daniel, J. Power Sources, 2015, 234–242 CrossRef CAS
- J. Li, Y. Lu, T. Yang, D. Ge, D. L. Wood and Z. Li, iScience, 2020, 23, 101081 CrossRef CAS PubMed
- Y. Li, IOP Conf. Ser.: Mater. Sci. Eng., 2020, 514, 042019 CrossRef
- I. Doberdò, N. Löffler, N. Laszczynski, D. Cericola, N. Penazzi, S. Bodoardo, G. T. Kim and S. Passerini, J. Power Sources, 2014, 248, 1000–1006 CrossRef
- K. Rollag, D. Juarez-Robles, Z. Du, D. L. Wood and P. P. Mukherjee, ACS Appl. Energy Mater., 2019, 4464–4476 CrossRef CAS
-
T. C. Nirmale, B. B. Kale and A. J. Varma, A review on cellulose and lignin based binders and electrodes: Small steps towards a sustainable lithium ion battery, 2017 Search PubMed
- R. Sahore, M. Wood, A. Kukay, Z. Du, K. M. Livingston, D. L. Wood and J. Li, J. Electrochem. Soc., 2022, 169, 040567 CrossRef
- S.-L. Chou, Y. Pan, J.-Z. Wang, H.-K. Liu and S.-X. Dou, Phys. Chem. Chem. Phys., 2014, 16, 20347 RSC
- D. Bresser, D. Buchholz, A. Moretti, A. Varzi and S. Passerini, Energy Environ. Sci., 2018, 11, 3096–3127 RSC
- Y. Ma, J. Ma and G. Cui, Energy Storage Mater., 2019, 20, 146 CrossRef
- Y. Sheng, Theses Dissertations, 2015, 1–146 CAS
- F. A. Çetinel and W. Bauer, Bull. Mater. Sci., 2014, 37, 1685–1690 CrossRef
- M. Ragnar, C. T. Lindgren and N.-O. Nilvebrant, J. Wood Chem. Technol., 2000, 20, 277–305 CrossRef CAS
- F. S. Chakar and A. J. Ragauskas, Ind. Crops Prod., 2004, 20, 131–141 CrossRef CAS
- V. K. Thakur, M. K. Thakur, P. Raghavan and M. R. Kessler, ACS Sustainable Chem. Eng., 2014, 2, 1072–1092 CrossRef CAS
- W. Li, Y. Zhang, L. Das, Y. Wang, M. Li, N. Wanninayake, Y. Pu, D. Y. Kim, Y. T. Cheng, A. J. Ragauskas and J. Shi, RSC Adv., 2018, 8, 38721–38732 RSC
- S. D. Gong, Y. Huang, H. J. Cao, Y. H. Lin, Y. Li, S. H. Tang, M. S. Wang and X. Li, J. Power Sources, 2016, 307, 624–633 CrossRef CAS
- H. Lu, A. Cornell, F. Alvarado, M. Behm, S. Leijonmarck, J. Li, P. Tomani and G. Lindbergh, Materials, 2016, 9, 127 CrossRef PubMed
- V. Ponnuchamy and E. S. Esakkimuthu, Appl. Surf. Sci., 2022, 573, 151461 CrossRef CAS
- W. E. Tenhaeff, O. Rios, K. More and M. A. McGuire, Adv. Funct. Mater., 2014, 24, 86–94 CrossRef CAS
- D. Jeong, J. Shim, H. Shin and J. C. Lee, ChemSusChem, 2020, 13, 2642–2649 CrossRef CAS PubMed
- D. Kai, M. J. Tan, P. L. Chee, Y. K. Chua, Y. L. Yap and X. J. Loh, Green Chem., 2016, 18, 1175–1200 RSC
- W. O. Doherty, P. Mousavioun and C. M. Fellows, Ind. Crops Prod., 2011, 33, 259–276 CrossRef CAS
- Z. Du, K. M. Rollag, J. Li, S. J. An, M. Wood, Y. Sheng, P. P. Mukherjee, C. Daniel and D. L. Wood, J. Power Sources, 2017, 200–206 CrossRef CAS
- M. Bichon, D. Sotta, N. Dupré, E. De Vito, A. Boulineau, W. Porcher and B. Lestriez, ACS Appl. Mater. Interfaces, 2019, 11, 18331–18341 CrossRef CAS PubMed
- W. Man and W. B. Russel, Phys. Rev. Lett., 2008, 100, 198302 CrossRef PubMed
- H. Zheng, J. Li, X. Song, G. Liu and V. S. Battaglia, Electrochim. Acta, 2012, 71, 258–265 CrossRef CAS
- F. Buss, C. C. Roberts, K. S. Crawford, K. Peters and L. F. Francis, J. Colloid Interface Sci., 2011, 359, 112–120 CrossRef CAS PubMed
- M. Stein, A. Mistry and P. P. Mukherjee, J. Electrochem. Soc., 2017, 164, A1616–A1627 CrossRef CAS
- C. Busà, M. Belekoukia and M. J. Loveridge, Electrochim. Acta, 2021, 366, 137358 CrossRef
- P. Li, Y. Zhao, Y. Shen and S.-H. Bo, JPhys: Energy, 2020, 2, 022002 CAS
- W. Pfleging and J. Pröll, J. Mater. Chem. A, 2014, 2, 14918–14926 RSC
- A. Davoodabadi, J. Li, H. Zhou, D. L. Wood, T. J. Singler and C. Jin, J. Energy Storage, 2019, 26, 101034 CrossRef
-
H. Berg, Batteries for electric vehicles: Materials and Electrochemistry, Cambridge University Press, 2015, pp. 1–250 Search PubMed
- J. Xiao, Q. Li, Y. Bi, M. Cai, B. Dunn, T. Glossmann, J. Liu, T. Osaka, R. Sugiura, B. Wu, J. Yang, J. G. Zhang and M. S. Whittingham, Nat. Energy, 2020, 5, 561–568 CrossRef CAS
- A. Tornheim and D. C. O'Hanlon, J. Electrochem. Soc., 2020, 167, 110520 CrossRef CAS
- M. Wood, J. Li, R. E. Ruther, Z. Du, E. C. Self, H. M. Meyer, C. Daniel, I. Belharouak and D. L. Wood, Energy Storage Mater., 2020, 24, 188–197 CrossRef
- N. V. Faenza, L. Bruce, Z. W. Lebens-Higgins, I. Plitz, N. Pereira, L. F. J. Piper and G. G. Amatucci, J. Electrochem. Soc., 2017, 164, A3727 CrossRef CAS
- R. Jung, R. Morasch, P. Karayaylali, K. Phillips, F. Maglia, C. Stinner, Y. Shao-Horn and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, A132 CrossRef CAS
- I. A. Shkrob, J. A. Gilbert, P. J. Phillips, R. Klie, R. T. Haasch, J. Bareño and D. P. Abraham, J. Electrochem. Soc., 2017, 164, A1489–A1498 CrossRef CAS
- X. Zhang, W. J. Jiang, X. P. Zhu, A. Mauger, Qilu and C. M. Julien, J. Power Sources, 2011, 196, 5102–5108 CrossRef CAS
-
M. N. Al-Shroofy, PhD thesis, University of Kentucky, 2017
- L. C. Thomas, Why Modulated DSC®?; An Overview and Summary of Advantages and Disadvantages Relativeto Traditional DSC, 2005, p. 8.
- S. J. Cho, C. C. Chung, S. Podowitz-Thomas and J. L. Jones, Mater. Chem. Phys., 2019, 228, 32–36 CrossRef CAS
- N. Loeffler, J. Von Zamory, N. Laszczynski, I. Doberdo, G. T. Kim and S. Passerini, J. Power Sources, 2014, 248, 915–922 CrossRef CAS
- J. Liu, X. Lu and C. Wu, Membranes, 2013, 3, 389–405 CrossRef PubMed
- B. G. Westphal and A. Kwade, J. Energy Storage, 2018, 18, 509–517 CrossRef
- B. Westphal, H. Bockholt, T. Gunther, W. Haselrieder and A. Kwade, ECS Trans., 2015, 57–68 CrossRef CAS
- H. Zheng, L. Tan, G. Liu, X. Song and V. S. Battaglia, J. Power Sources, 2012, 208, 52–57 CrossRef CAS
- H. Bockholt, W. Haselrieder and A. Kwade, Powder Technol., 2016, 297, 266–274 CrossRef CAS
- J. Liu, B. Ludwig, Y. Liu, Z. Zheng, F. Wang, M. Tang, J. Wang, J. Wang, H. Pan and Y. Wang, Adv. Mater. Technol., 2017, 1700106 CrossRef
- W. B. Hawley, H. M. Meyer and J. Li, Electrochim. Acta, 2021, 380, 138203 CrossRef CAS
- N. Susarla, S. Ahmed and D. W. Dees, J. Power Sources, 2018, 378, 660–670 CrossRef CAS
- F. Font, B. Protas, G. Richardson and J. M. Foster, J. Power Sources, 2018, 393, 177–185 CrossRef CAS
- M. Malamatari, A. Charisi, S. Malamataris, K. Kachrimanis and I. Nikolakakis, Processes, 2020, 8, 788 CrossRef CAS
- M. Baunach, S. Jaiser, S. Schmelzle, H. Nirschl, P. Scharfer and W. Schabel, Drying Technol., 2016, 34, 462–473 CrossRef CAS
- R. Vanholme, B. Demedts, K. Morreel, J. Ralph and W. Boerjan, Plant Physiol., 2010, 153, 895–905 CrossRef CAS PubMed
- J. Zhu, C. Yan, X. Zhang, C. Yang, M. Jiang and X. Zhang, Prog. Energy Combust. Sci., 2020, 76, 100788 CrossRef
-
J. Li, Z. Du, R. E. Ruther, S. J. An, L. A. David, K. Hays, M. Wood, N. D. Phillip, Y. Sheng, C. Mao, S. Kalnaus, C. Daniel and D. L. Wood, Toward Low-Cost, High-Energy Density, and High-Power Density Lithium-Ion Batteries, 2017 Search PubMed
- M. Memm, A. Hoffmann and M. Wohlfahrt-Mehrens, Electrochim. Acta, 2018, 260, 664–673 CrossRef CAS
- Z. Du, C. J. Janke, J. Li, C. Daniel and D. L. Wood, J. Electrochem. Soc., 2016, 163, A2776–A2780 CrossRef CAS
- H. Zheng, G. Liu, X. Song, P. Ridgway, S. Xun and V. S. Battaglia, J. Electrochem. Soc., 2010, 157, A1060 CrossRef CAS
- MTI Conductive Carbon Coated Aluminum Foil for Battery Cathode Substrate, https://www.mtixtl.com/ConductiveCarbonCoatedAluminumFoil.
- L. Qiao, U. Oteo, M. Martinez-Ibañez, A. Santiago, R. Cid, E. Sanchez-Diez, E. Lobato, L. Meabe, M. Armand and H. Zhang, Nat. Mater., 2022, 455–462 CrossRef CAS PubMed
- C. Pastor-Fernandez, W. Dhammika Widanage, J. Marco, M.-A. Gama-Valdez and G. H. Chouchelamane, 2016 IEEE Transportation Electrification Conference and Expo (ITEC), 2016, pp. 1-6.
- H. Kang, C. Lim, T. Li, Y. Fu, B. Yan, N. Houston, V. De Andrade, F. De Carlo and L. Zhu, Electrochim. Acta, 2017, 232, 431–438 CrossRef CAS
- W. Haselrieder, S. Ivanov, D. K. Christen, H. Bockholt and A. Kwade, ECS Trans., 2013, 50–59 Search PubMed
- M. Li, J. Wei, L. Ren, Y. Zhao, Z. Shang, D. Zhou, W. Liu, L. Luo and X. Sun, Cell Rep. Phys. Sci., 2021, 2, 100374 CrossRef CAS
- E. Billy, M. Joulié, R. Laucournet, A. Boulineau, E. De Vito and D. Meyer, ACS Appl. Mater. Interfaces, 2018, 10, 16424–16435 CrossRef CAS PubMed
- L. Azhari, X. Zhou, B. Sousa, Z. Yang, G. Gao and Y. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 57963–57974 CrossRef CAS PubMed
- K. Sahni, M. Ashuri, Q. He, R. Sahore, I. D. Bloom, Y. Liu, J. A. Kaduk and L. L. Shaw, Electrochim. Acta, 2019, 301, 8–22 CrossRef CAS
- C. H. Jo, D. H. Cho, H. J. Noh, H. Yashiro, Y. K. Sun and S. T. Myung, Nano Res., 2015, 8, 1464–1479 CrossRef CAS
- H. Buqa, M. Holzapfel, F. Krumeich, C. Veit and P. Novák, J. Power Sources, 2006, 161, 617–622 CrossRef CAS
- C. C. Li and Y. W. Wang, J. Electrochem. Soc., 2011, 158, A1361 CrossRef CAS
- N. Loeffler, G. T. Kim, F. Mueller, T. Diemant, J. K. Kim, R. J. Behm and S. Passerini, ChemSusChem, 2016, 9, 1112–1117 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ma00850e |
| This journal is © The Royal Society of Chemistry 2023 |