
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Benzylic C–H arylation with dicyanoarenes via convergent paired electrolysis†
Shanyu
Tang
and
Guillaume
Vincent
 *
*
Institut de Chimie Moléculaire et des Matériaux d'Orsay (ICMMO), Université Paris-Saclay, CNRS, Bâtiment Henri Moissan, 17 Avenue des Sciences, 91400 Orsay, France. E-mail: guillaume.vincent@universite-paris-saclay.fr
First published on 21st June 2023
Abstract
We describe the convergent paired electrolysis of methylarene derivatives and 1,4-dicyanoarenes to perform the arylative functionalization of a benzylic C(sp3)–H bond to form 1,1-biarylmethane derivatives that are found in several drugs and biologically active compounds. This electrochemical process proceeds via the coupling of a benzylic radical or a benzylic carbocation with a 1,4-dicyanoarene radical anion to form the desired C–C bond followed by elimination of the cyanide anion which could be trapped as a cyanhydrin by an aldehyde. These reactive species are produced, respectively, by the oxidation of the benzylic substrate at the anode and the reduction of the dicyanoarene at the cathode. One of the key challenges that we have overcome is avoiding the formation of overoxidized coupling products at the bisbenzylic position of the biarylmethane products obtained.
Introduction
In recent years, several electrochemical methods for the functionalization of benzylic C(sp3)–H bonds have been reported that form C–N, C–O, C–halogen or even C–C bonds.1,2 These processes usually involve the oxidation of the benzylic position of 1 directly at the anode via single electron transfers (SET) or indirectly with an electro mediator. The resulting benzylic radical A could react with a radical acceptor or be further oxidized into a carbocation that could be intercepted by a nucleophile already present in the reaction media to yield 2 (Scheme 1).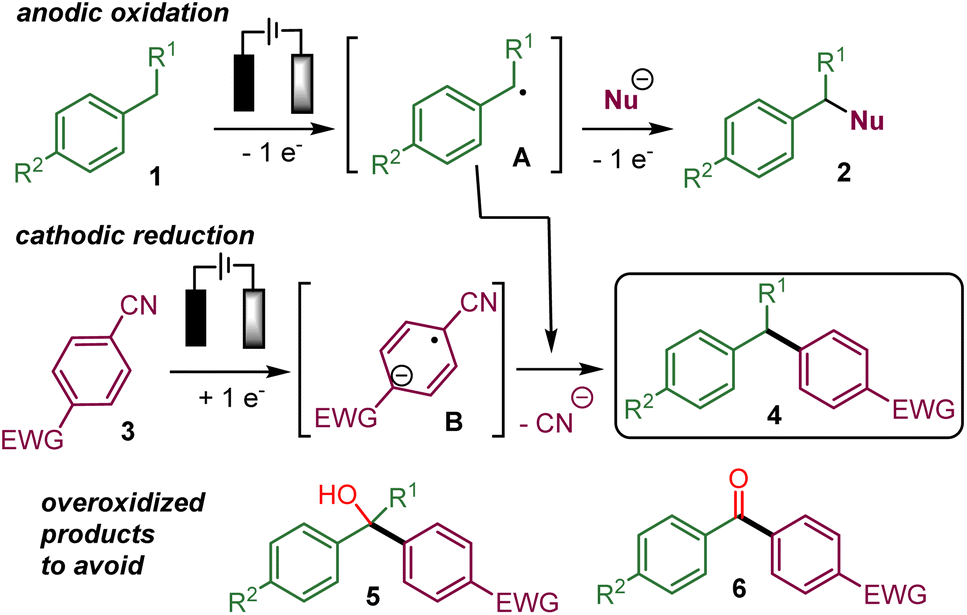 | ||
| Scheme 1 The proposed electrochemical benzylic arylation via convergent paired electrolysis. | ||
For example, we have performed the addition of an isocyanide to the electrogenerated benzylic carbocation.3 Most of the time, the cathodic counter-reaction, such as the reduction of protons into hydrogen, does not produce species reacting with the benzylic substrate. An attractive refinement of the electrochemical benzylic C(sp3)–H functionalization would be to take advantage of the cathodic reduction to generate a nucleophilic intermediate that would add to the anodically oxidized benzylic substrate in a convergent paired-electrolysis process.4
In this context, we turned our attention to developing an electrochemical benzylic arylation using this concept to form the 1,1-biarylmethane motif,5 which can be found in several drugs or biologically active compounds such as diisopromine, fenpiprane, prozapine, fendilline, milverine, indatraline, sertraline and elvitegravir among others (Fig. 1).5,6
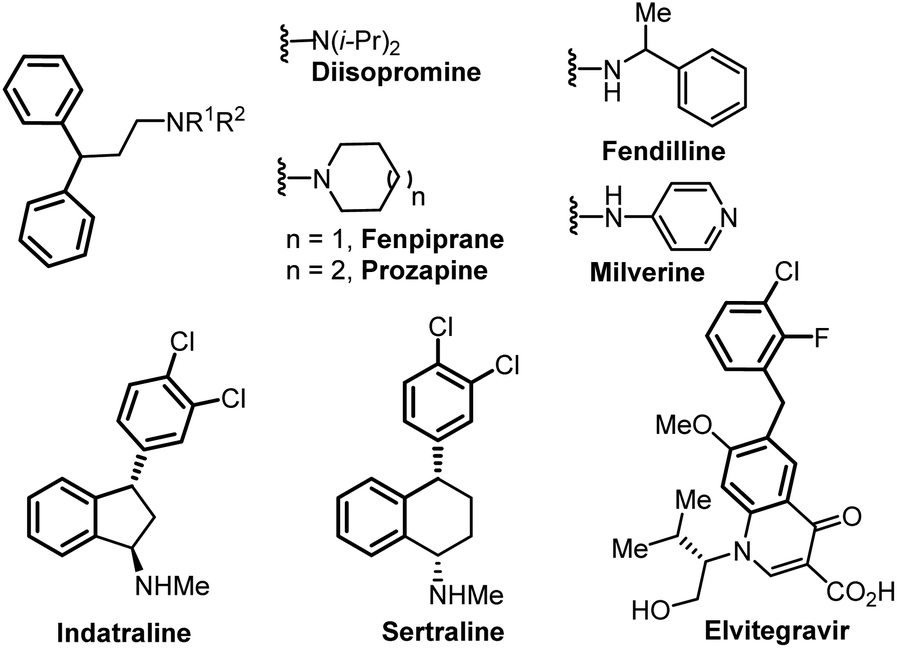 | ||
| Fig. 1 Biologically active compounds containing a 1,1-diarylmethane motif. | ||
Indeed, the anodic oxidation of biarylmethane derivatives delivered a particularly stabilized carbocation in which an electron-rich arene could be added during the electrolysis in a few cases7 or in the absence of electric current (cation-pool method)8 to deliver triarylmethane derivatives. Anodically generated less stable benzylic carbocations from monoarylmethane derivatives could be trapped in situ by a stabilizing agent during the electrolysis and then an electron-rich arene could be added to deliver biarylmethanes (stabilized cation pool method).9 Electrogenerated benzylic radicals could also react in a Minisci-type process with heteroarenes to achieve a C–H bond functionalization of the latter.10
On the other hand, the electrochemical nickel-catalysed arylation of electron-rich primary benzylic C(sp3)–H bonds with electron-deficient arylbromides were deployed in a paired-electrolysis process via the reductive regeneration of the nickel catalyst at the cathode.11
Electron-deficient aryl nitrile derivatives appeared to be pertinent partners to develop a benzylic arylation via convergent paired electrolysis4a and related transformations have been achieved under photochemical irradiation.12
The cathodic reduction of an electron-deficient aryl nitrile 3, such as 1,4-dicyanobenzene, is known to produce a persistent radical-anion B, which could add to a radical or an electrophilic species.13,14 Indeed, such a concept has been recently employed in convergent paired electrolysis involving the arylation of electron-deficient intermediates produced by anodic oxidation,15 such as the α-arylation of amines,15a,b alkenes,15b benzylic alcohols15c and benzylic ethers,15d the decarboxylative arylation of carboxylic acid15b and the deboronative arylation of trifluoroborate salts.15b In addition, the α-hydroxylation-α-arylation of benzylic C(sp3)–H substrates to afford 5 has also been very recently reported via preliminary oxidation into a benzylic alcohol.16
Therefore, it appears that the present challenge would be to favour the coupling of the persistent aryl nitrile radical anion with the transient benzylic radical over the evolution of the latter into the corresponding benzylic alcohol that could lead to overoxidized products 5 or 6.16,17 Indeed, conducting the reaction in the absence of water or oxygen should prevent the evolution of A into a benzylic alcohol or ketone leading to 5 or 6. However, the risk is that the highly reactive benzylic radical A and the corresponding carbocation resulting from further anodic oxidation would be involved in undesired reaction processes instead of reacting with the radical anion B.
Results and discussion
To test the feasibility of this approach, we selected ethylbiphenyl 1a and 1,4-dicyanobenzene 3a as substrates to generate 4a (Table 1).|
|
||
|---|---|---|
| Entry | Deviation from standard conditionsa | Yield 4a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
a Undivided cell, graphite-SK50 anode (1.5 cm × 0.8 cm × 0.2 cm submerged), nickel cathode (1.5 cm × 0.8 cm × 0.2 cm submerged), 7.5 mA, 6 F mol−1, 1a (0.2 mmol), 2a (0.2 mmol), n-Bu4NOTs (0.2 mmol), 3 mL of CH3CN/THF (1:1), room temperature, under an argon atmosphere.
b Isolated yield of 4a.
c
7a and 8a were obtained in 14% and 4% yields.
d
5a was obtained in 13% yield. 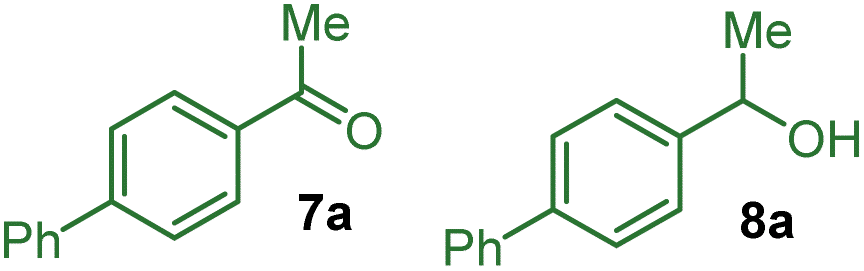 . .
|
||
| 1 | None | 58% |
| 2 | 2 equiv. of 1a | 60% |
| 3 | Under air instead of argon | 12%c |
| 4 | No t-BuCHO | 48% |
| 5 | CH3CN instead of THF/CH3CN (1:1), no t-BuCHO |
27% |
| 6 | THF instead of THF/CH3CN (1:1), no t-BuCHO |
8% |
| 7 | 2Me-THF instead of THF, no t-BuCHO | 22%d |
| 8 | THF/CH3CN (2:1) instead of (1:1), no t-BuCHO |
44% |
| 9 | THF/CH3CN (1:2) instead of (1:1), no t-BuCHO |
46% |
| 10 | DMF instead of THF/CH3CN (1:1), no t-BuCHO |
39% |
| 11 | n-Bu4NClO4 instead of n-Bu4NOTs, no t-BuCHO | Traces |
| 12 | n-Bu4NBF4 instead of n-Bu4NOTs, no t-BuCHO | Traces |
| 13 | n-Bu4NOTf instead of n-Bu4NOTs, no t-BuCHO | 0% |
| 14 | Et4NOTs instead of n-Bu4NOTs, no t-BuCHO | 37% |
| 15 | C(+)/C(−) instead of C(+)/Ni(−), no t-BuCHO | 37% |
| 16 | C(+)/Pt(−) instead of C(+)/Ni(−), no t-BuCHO | 53% |
| 17 | Pt(+)/Ni(−) instead of C(+)/Ni(−), no t-BuCHO | Traces |
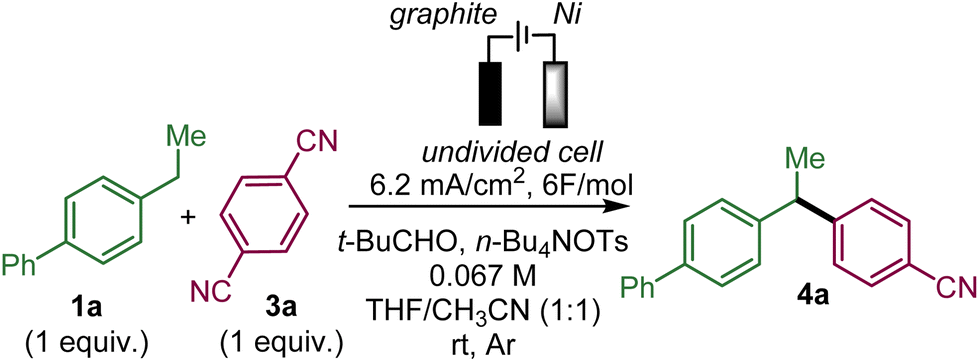
Upon evaluation of the parameters of the reaction, the optimal conditions appear to be those of the electrolysis in an undivided cell at a constant current of 6.2 mA cm−2 with a graphite anode and a nickel cathode in a 1:1 mixture of acetonitrile and THF with tetrabutylammonium tosylate as the electrolyte and pivaldehyde as a trap for the released cyanide ion at room temperature; a 58% yield of 4a was obtained from equimolar amounts of 1a and 3a (entry 1 with 19% recovery of 1a) while 60% yield was obtained with an excess of 1a (entry 2). Performing the reaction under argon is critical since under air, only 12% of 4a was obtained along with 47% of unreacted 1a with 14% and 4% ketone 7a and benzyl alcohol 8a, respectively, which are supposedly the products of the reaction of benzyl radical A with oxygen (entry 3). In the absence of pivaldehyde as a scavenger for the toxic cyanide ion, the yield diminished to 48% (entry 4). The screening of solvents indicated that the association of THF and acetonitrile indeed played a crucial role in this electrochemical arylation protocol. Lower yields were obtained when pure acetonitrile (47% of 1a recovered) or THF (85% recovery of 1a) were used (entries 5 and 6). Interestingly, replacing THF by 2-MeTHF resulted in a decrease in the yield and formation of undesired bisbenzylic alcohol 5a (entry 7). The ratio of THF and acetonitrile also had an influence on the yield (entries 8 and 9). DMF is also a suitable solvent and can promote the reaction but with a moderate yield of 39% (entry 10), while other solvents, such as DMA, NMP or DMSO, are ineffective (see the ESI†). The nature of the electrolyte was critical, particularly that of the anion, since replacing the tosylate anion of n-BuNOTs by a perchlorate, a tetrafluoroborate or a triflate led to almost no formation of the desired product, which is explained by the non-reactivity of 1a (entries 11–13). On the other hand, the tetrabutylammonium cation could be substituted by a tetraethylammonium cation with a moderately decreased yield (entry 14). The effect of the material of the electrodes was also investigated. Replacing nickel with graphite at the cathode was less effective affording a 37% yield, while a more expensive platinum cathode afforded a similar yield of 53% (entries 15 and 16). On the other hand at the anode, switching graphite to platinum led only to trace amounts of 4a due to very poor conversion of 1a (entry 17). It should be noted that the use of an electrochemical mediator to generate the benzyl radical A was ineffective (see the ESI†).
Overall, we were able to attain our goal, i.e. the arylation of benzylic C(sp3)–H bonds without overoxidation or other undesired reactions.
We then studied the scope of this benzylic arylation with para-dicyanobenzene (Scheme 2).
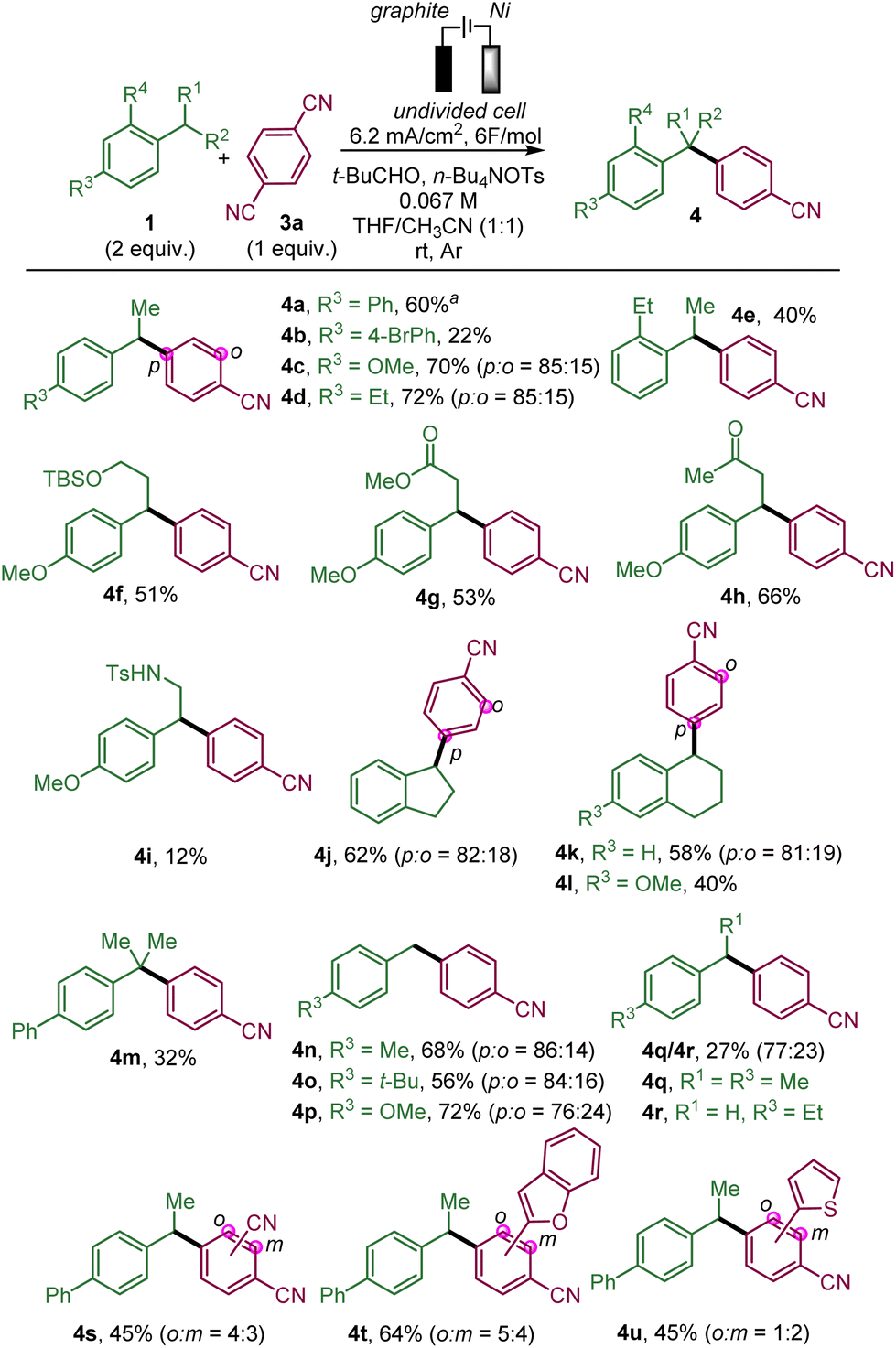 | ||
| Scheme 2 Electrochemical benzylic arylation with dicyanoarenes via convergent paired electrolysis. Reaction conditions: an undivided cell, a graphite-SK50 anode (1.5 cm × 0.8 cm × 0.2 cm submerged), a nickel cathode (1.5 cm × 0.8 cm × 0.2 cm submerged), 7.5 mA, 6 F mol−1, 1a (0.4 mmol), 2a (0.2 mmol), n-Bu4NOTs (0.2 mmol), 3 mL of CH3CN/THF (1:1), room temperature, under an argon atmosphere; isolated yield of 4; a55% yield on a 1 mmol scale. | ||
We began by arylating secondary benzylic positions. In addition to para-ethylbiphenyl 1a, para-substituted ethyl benzene derivatives were able to deliver the desired targets in modest yields for the para-bromo bipenyl 4b but with more success with electron-donating groups, such as para-methoxy 4c or para-ethyl 4d. Symmetrical ortho-ethyl benzene also led to the arylated product 4e. We further showed the compatibility of this benzylic electrochemical arylation with para-methoxy alkyl benzenes bearing a functional group on the alkyl chain, such as a protected alcohol (4f), an ester (4g) or a ketone (4h). The reaction proceeded affording a modest yield in the presence of a tosyl amide on the ethyl chain (4i). Cyclic substrates fused with the aromatic ring proved to be amenable for producing the desired arylation products, such as indane 4j, and tetrahydronaphthalene derivatives 4k and 4l. The tertiary benzylic position of iso-propyl-biphenyl could also be arylated (4m) albeit in moderate yield probably due to steric hindrance. The primary benzylic position of para-substituted toluene derivatives is also prone to react with the formation of para-methyl, t-butyl and methoxy biarylmethane products 4n–p. para-Ethyl toluene reacts mainly to afford the ethyl-substituted compound (4q) over the methyl-substituted compound (4r), which could be explained by the stabilisation of the secondary radical being favoured over the primary one. It should be noted that in some cases (4c,d,j,k and 4n–p), a minor amount of the ortho-substituted arylation product relative to the cyanobenzene part has been formed, as it has been observed previously.14e Subsequently, 1,4-dicyanobenzene derivatives, which have an additional substituent such as a cyano group (4s), and heteroaromatic substituents (benzofurane and thiophene) (4t and 4u), could also deliver the arylation products as a mixture of two regioisomers from para-ethylbiphenyl.
This protocol could be scaled-up to provide almost 1 g of 4a in 67% yield, which proved the synthetic utility of this protocol (Scheme 3). In order to shorten the reaction time, the current density and concentration were increased compared to the standard conditions.
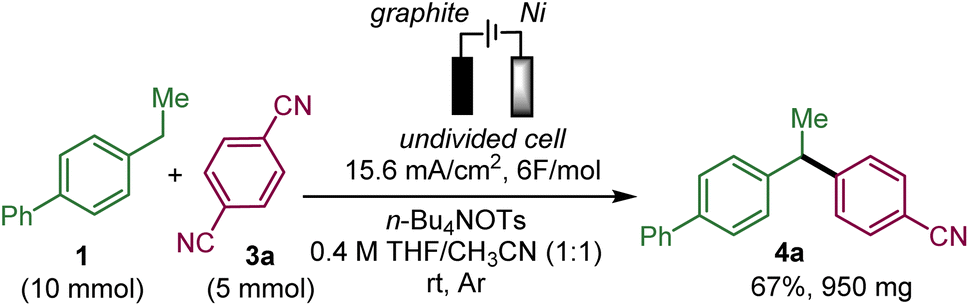 | ||
| Scheme 3 Scale-up experiment. Reaction conditions: an undivided cell, a graphite-SK50 anode (3 cm × 0.8 cm × 0.2 cm submerged), a nickel cathode (3 cm × 0.8 cm × 0.2 cm submerged), 37.5 mA, 6 F mol−1, 1a (10 mmol), 2a (5 mmol), n-Bu4NOTs (5 mmol), 12 mL of CH3CN/THF (1:1), room temperature, under an argon atmosphere. | ||
From a mechanistic point of view, cyclic voltammetry results show that the oxidation potential of 4-ethylbiphenyl 1a in a mixture of THF and acetonitrile is sharply lower than that of 1,4-dicyanobenzene (Fig. 2), which indeed implies that the benzylic substrate is preferentially oxidized directly at the anode. In the cathodic process, 1,4-dicyanobenzene displays a reduction peak, which is reversible and corresponds to the formation of the persistent radical anion B, while the benzylic substrate is not reduced in the range studied as well as pivaldehyde.
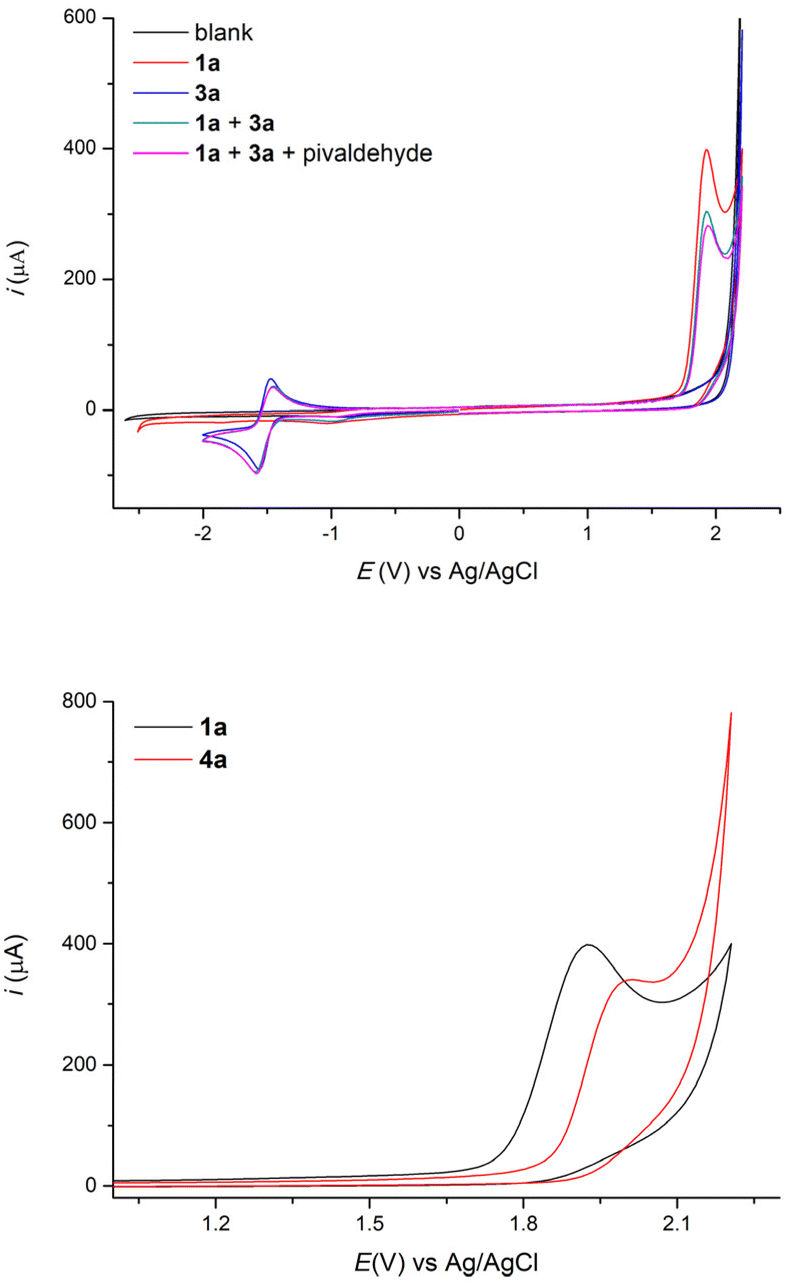 | ||
| Fig. 2 Cyclic voltammetry studies of the reactants of the electrochemical benzylic arylation. CV scans were performed using a glassy carbon disk (d = 3 mm) as the working electrode, a platinum wire as the counter electrode and an Ag/AgCl electrode as the reference electrode at a scan rate of 0.1 V s−1 at 20 °C in 5 mL of a 2 mM solution of each substrate and 0.1 M of n-Bu4N·BF4 in a 1:1 mixture of THF and acetonitrile. | ||
In addition, we demonstrated that the benzylic substrate 1a is oxidized at a lower potential than the obtained 1,1-biarylethane product 4a (Fig. 2) and this explains why we do not observe any noticeable amount of 5.
We performed a competitive experiment between 1a and its bisbenzylic deuterated analogue 1a-d2 (Scheme 4), which resulted in a 2.8/1 ratio of 4a and 4a-d corresponding to a primary KIE similar to a previously reported anodic benzylic functionalization.1e
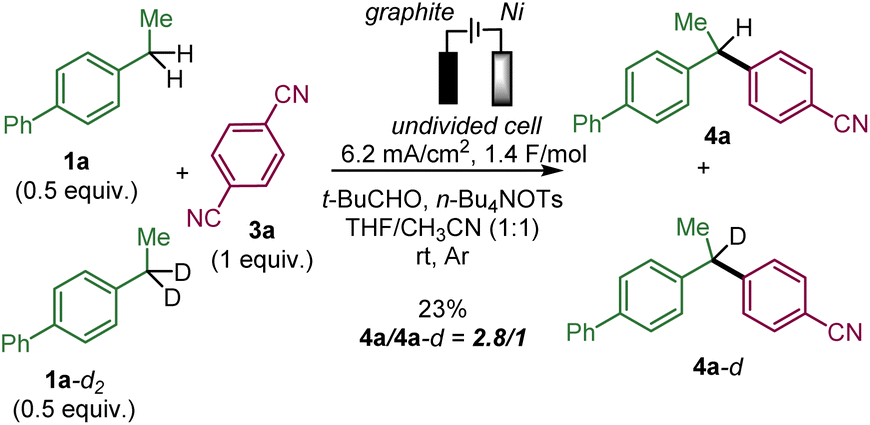 | ||
| Scheme 4 The competitive experiment between 1a and 1a-d2. | ||
Based on this study and previous works, the following mechanisms are proposed involving a convergent paired-electrolysis process (Scheme 5).15,16 The benzylic substrate 1 is presumably oxidized directly at the anode to the radical cation C.1e Upon the elimination of a proton, the transient benzylic radical A could be generated. On the other hand, the cathodic reduction of 3a produces a persistent radical anion B. According to previously proposed mechanisms of related transformations,15,16 the radical anion B could react with the transient benzyl radical Avia radical–radical recombination to form the desired C–C bond and the anionic intermediate D (path a). The desired arylated benzylic product could be obtained via aromatization through the elimination of a cyanide ion, which could be trapped by pivaldehyde to probably yield cyanohydrin 9.
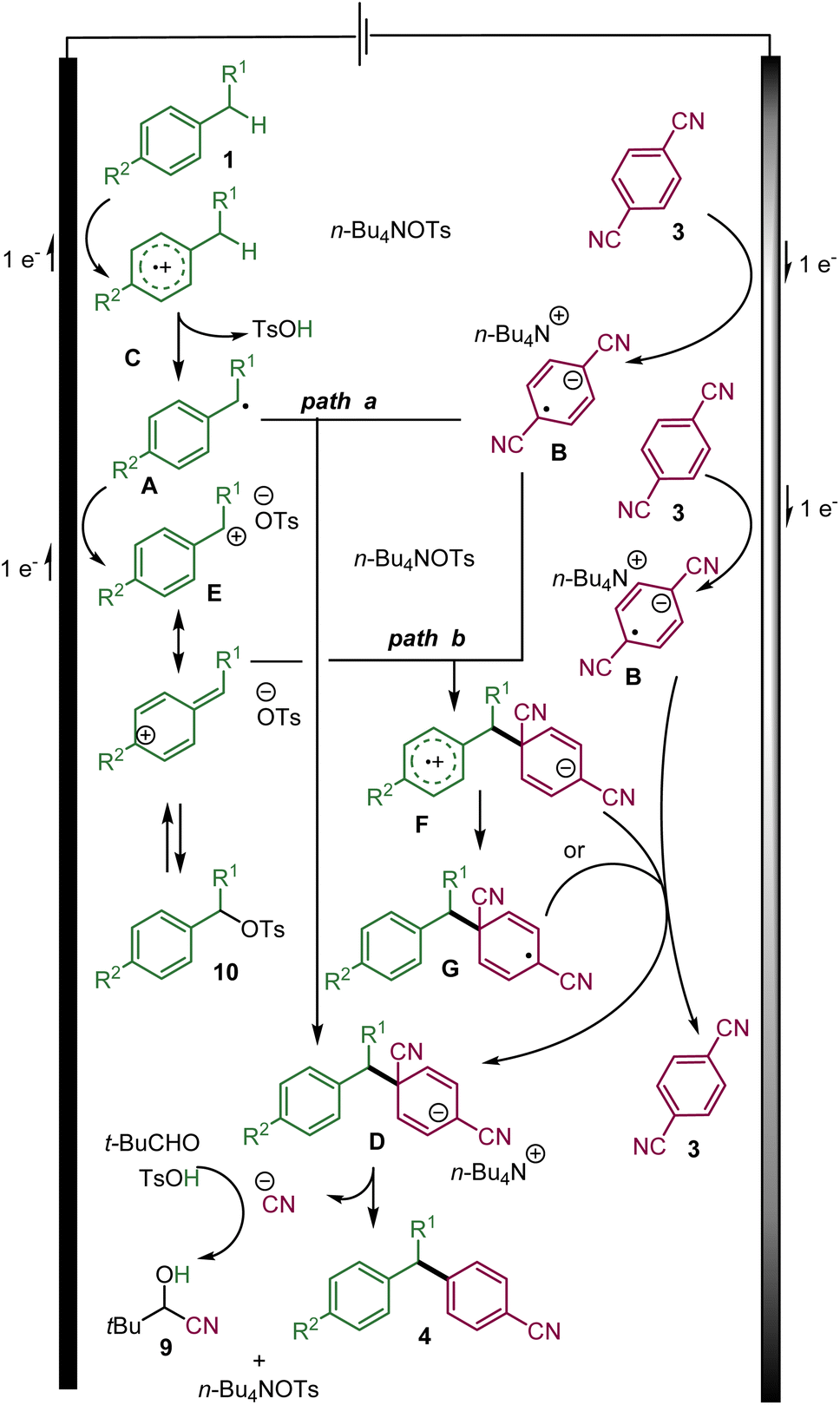 | ||
| Scheme 5 The proposed mechanism of the convergent paired electrolysis of benzylic substrates and dicyanoarenes. | ||
Nevertheless, it is more plausible that before leaving the double layer and reacting with a radical anion B, benzylic radical A could rapidly be oxidized at the anode into the corresponding benzylic carbocation E.1e,3
Upon its diffusion into the solution, the carbocation E could react with the radical anion B leading to the zwitterionic radical F that could evolve into radical Gvia a fast intramolecular electron transfer. Either radical F or G could be reduced by the radical anion B into anion D with the regeneration of 1,4-dicyanoarene 3 (path b).
The role of a tosylate as the counter-anion of the tetrabutylammonium electrolyte is the key to the success of the reaction. It might be explained by its basicity, which helps in eliminating a proton from the radical cation C. In addition, the benzylic carbocation E (in the case of path b) might also be trapped by the tosylate anion to form the rather stable benzylic tosylate 10.
Conclusions
In conclusion, we report the direct electrochemical arylation of benzylic C(sp3)–H bonds via a convergent paired electrolysis. We merged the anodic benzylic C–H bond oxidation and the cathodic reduction of dicyanobenzenes to produce two reactive intermediates that can merge to form the desired biarylmethane derivatives 4.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are very grateful to Dr Maxime Vitale and Dr Laurence Grimaud (Ecole Normale Supérieure, Paris) for very helpful discussions and their help in the cyclic voltammetry studies. ST thanks the China Scholarship Council (CSC) for her PhD fellowship. We also gratefully acknowledge the ANR (ANR-20-CE07-0020; “ELMER”), the Université Paris-Saclay and the CNRS for financial support.References
- For a review, see: (a) M. Oliva, G. A. Coppola, E. V. V. der Eycken and U. K. Sharma, Adv. Synth. Catal., 2021, 363, 1810–1834 CrossRef CAS. For selected examples of C–N bonds, see: (b) T. Morofuji, A. Shimizu and J. Yoshida, J. Am. Chem. Soc., 2014, 136, 4496–4499 CrossRef CAS; (c) R. Hayashi, A. Shimizu, Y. Song, Y. Ashikari, T. Nokami and J. Yoshida, Chem. – Eur. J., 2017, 23, 61–64 CrossRef CAS PubMed; (d) L. Niu, C. Jiang, Y. Liang, D. Liu, F. Bu, R. Shi, H. Chen, A. D. Chowdhury and A. Lei, J. Am. Chem. Soc., 2020, 142, 17693–17702 CrossRef CAS PubMed; (e) Z.-W. Hou, D.-J. Liu, P. Xiong, X.-L. Lai, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2021, 60, 2943–2947 CrossRef CAS; (f) T. Shen and T. H. Lambert, J. Am. Chem. Soc., 2021, 143, 8597–8602 CrossRef CAS PubMed; (g) Y. Yu, X.-B. Zhu, Y. Yuan and K.-Y. Ye, Chem. Sci., 2022, 13, 13851–13856 RSC. C–O bonds: (h) Y. Ashikari, T. Nokami and J. Yoshida, Org. Lett., 2012, 14, 938–941 CrossRef CAS; (i) H. Wang, K. Liang, W. Xiong, S. Samanta, W. Li and A. Lei, Sci. Adv., 2020, 6, eaaz0590 CrossRef CAS PubMed. C–Halogen bonds: (j) T. Raju, K. Kulangiappar, M. A. Kulandainathan and A. Muthukumaran, Tetrahedron Lett., 2005, 46, 7047–7050 CrossRef CAS; (k) M. Rafiee, F. Wang, D. P. Hruszkewycz and S. S. Stahl, J. Am. Chem. Soc., 2018, 140, 22–25 CrossRef CAS PubMed. C–C bonds: (l) X.-H. Ho, S. Mho, H. Kang and H.-Y. Jang, Eur. J. Org. Chem., 2010, 4436–4441 CAS; (m) N. Fu, L. Li, Q. Yang and S. Luo, Org. Lett., 2017, 19, 2122–2125 CrossRef CAS; (n) Y.-Z. Yang, Y.-C. Wu, R.-J. Song and J.-H. Li, Chem. Commun., 2020, 56, 7585–7588 RSC; (o) Q. Zhang, X. Chang, L. Peng and C. Guo, Angew. Chem., Int. Ed., 2019, 58, 6999–7003 CrossRef CAS PubMed.
- For general reviews on benzylic C(sp3)–H functionalization, see: (a) R. Yazaki and T. Ohshima, Tetrahedron Lett., 2019, 60, 151225 CrossRef; (b) R. Vanjari and K. N. Singh, Chem. Soc. Rev., 2015, 44, 8062–8096 RSC.
- S. Tang, R. Guillot, L. Grimaud, M. R. Vitale and G. Vincent, Org. Lett., 2022, 24, 2125–2130 CrossRef CAS.
- For selected reviews, see: (a) A. Claraz and G. Masson, ACS Org. Inorg. Au, 2022, 2, 126–147 CrossRef CAS PubMed; (b) S. Zhang and M. Findlater, Chem. – Eur. J., 2022, 28, e202201152 CAS; (c) N. Sbei, T. Hardwick and N. Ahmed, ACS Sustainable Chem. Eng., 2021, 9, 6148–6169 Search PubMed; (d) W. Zhang, N. Hong, L. Song and N. Fu, Chem. Rec., 2021, 21, 2574–2584 CrossRef CAS PubMed; (e) F. Marken, A. J. Cresswell and S. D. Bull, Chem. Rec., 2021, 21, 2585–2600 CrossRef CAS PubMed.
- (a) Md. Belal, Z. Li, X. Lu and G. Yin, Sci. China: Chem., 2021, 64, 513–533 CrossRef CAS; (b) T. Jia, P. Cao and J. Liao, Chem. Sci., 2018, 9, 546–559 RSC; (c) S. Mondal and G. Panda, RSC Adv., 2014, 4, 28317–28358 RSC.
- D. Ameen and T. J. Snape, MedChemComm, 2013, 4, 893–907 RSC.
- (a) B. Wei, J.-H. Qin, Y.-Z. Yang, Y.-X. Xie, X.-H. Ouyang and R.-J. Song, Org. Chem. Front., 2022, 9, 816–821 RSC; (b) X. Chen, H. Liu, H. Gao, P. Li, T. Miao and H. Li, J. Org. Chem., 2022, 87, 1056–1064 CrossRef CAS PubMed; (c) H. Gao, X. Chen, P.-L. Wang, M.-M. Shi, L.-L. Shang, H.-Y. Guo, H. Li and P. Li, Org. Chem. Front., 2022, 9, 1911–1916 RSC.
- (a) M. Okajima, K. Soga, T. Nokami, S. Suga and J. Yoshida, Org. Lett., 2006, 8, 5005–5007 CrossRef CAS PubMed; (b) T. Nokami, K. Ohata, M. Inoue, H. Tsuyama, A. Shibuya, K. Soga, M. Okajima, S. Suga and J. Yoshida, J. Am. Chem. Soc., 2008, 130, 10864–10865 CrossRef CAS.
- (a) R. Hayashi, A. Shimizu and J. Yoshida, J. Am. Chem. Soc., 2016, 138, 8400–8403 CrossRef CAS PubMed; (b) Y. Imada, J. L. Röckl, A. Wiebe, T. Gieshoff, D. Schollmeyer, K. Chiba, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 12136–12140 CrossRef CAS.
- P. Xu, P.-Y. Chen and H.-C. Xu, Angew. Chem., Int. Ed., 2020, 59, 14275–14280 CrossRef CAS.
- (a) L. Zhang and X. Hu, Chem. Sci., 2020, 11, 10786–10791 RSC; (b) K.-J. Jiao, D. Liu, H.-X. Ma, H. Qiu, P. Fang and T.-S. Mei, Angew. Chem., Int. Ed., 2020, 59, 6520–6524 CrossRef CAS.
- (a) R. Bernardi, T. Caronna, S. Morrocchi, P. Traldi and B. M. Vittimberga, J. Chem. Soc., Perkin Trans. 1, 1981, 1607–1609 RSC; (b) T. Hoshikawa and M. Inoue, Chem. Sci., 2013, 4, 3118–3123 RSC; (c) K. Qvortrup, D. A. Rankic and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 626–629 CrossRef CAS PubMed; (d) J. D. Cuthbertson and D. W. C. MacMillan, Nature, 2015, 519, 74–77 CrossRef CAS PubMed; (e) H. Tanaka, K. Sakai, A. Kawamura, K. Oisaki and M. Kanai, Chem. Commun., 2018, 54, 3215–3218 RSC.
- For a review on the reductive and decyanative functionalization of cyanoarenes, see: S. Tong, K. Li, X. Ouyang, R. Song and J. Li, Green Synth. Catal., 2021, 2, 145–155 CrossRef.
- (a) D. Lehnherr, Y. Lam, M. C. Nicastri, J. Liu, J. A. Newman, E. L. Regalado, D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2020, 142, 468–478 CrossRef CAS PubMed; (b) S. Zhang, L. Li, X. Li, J. Zhang, K. Xu, G. Li and M. Findlater, Org. Lett., 2020, 22, 3570–3575 CrossRef CAS; (c) X. Zhang, C. Yang, H. Gao, L. Wang, L. Guo and W. Xia, Org. Lett., 2021, 23, 3472–3476 CrossRef CAS PubMed; (d) H. Xu, J. Liu, F. Nie, X. Zhao and Z. Jiang, J. Org. Chem., 2021, 86, 16204–16212 CrossRef CAS PubMed; (e) S. Zhang, W. Gao, J. Shi, J. Li, F. Li, Y. Liang, X. Zhan and M.-B. Li, Org. Chem. Front., 2022, 9, 1261–1266 RSC; (f) H.-J. Zhou and J.-M. Huang, J. Org. Chem., 2022, 87, 5328–5338 CrossRef CAS; (g) W.-M. Zeng, Y.-H. He and Z. Guan, Org. Lett., 2022, 24, 7178–7182 CrossRef CAS PubMed; (h) L. Yang, M. Sun, L. Cao, C. Liang, J. Yang, J. Yi, R. Cheng, Y. Ma and J. Ye, Chem. Commun., 2022, 58, 13345–13348 RSC; (i) W. Yu, S. Wang, M. He, Z. Jiang, Y. Yu, J. Lan, J. Luo, P. Wang, X. Qi, T. Wang and A. Lei, Angew. Chem., Int. Ed., 2023, 62, e202219166 CrossRef CAS PubMed.
- (a) Y. Ma, X. Yao, L. Zhang, P. Ni, R. Cheng and J. Ye, Angew. Chem., Int. Ed., 2019, 58, 16548–16552 CrossRef CAS PubMed; (b) Y. Mo, Z. Lu, G. Rughoobur, P. Patil, N. Gershenfeld, A. I. Akinwande, S. L. Buchwald and K. F. Jensen, Science, 2020, 368, 1352–1357 CrossRef CAS PubMed; (c) S. Zhang, L. Li, J. Li, J. Shi, K. Xu, W. Gao, L. Zong, G. Li and M. Findlater, Angew. Chem., Int. Ed., 2021, 60, 7275–7282 CrossRef CAS PubMed; (d) A. F. Zahrt, Y. Mo, K. Y. Nandiwale, R. Shprints, E. Heid and K. F. Jensen, J. Am. Chem. Soc., 2022, 144, 22599–22610 CrossRef CAS PubMed.
- X.-W. Wang, R.-X. Li, Y. Deng, M.-Q.-H. Fu, Y.-N. Zhao, Z. Guan and Y.-H. He, J. Org. Chem., 2023, 88, 329–340 CrossRef CAS.
- C. Yu, R. Huang and F. W. Patureau, Angew. Chem., Int. Ed., 2022, 61, e202201142 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc00866e |
| This journal is © The Royal Society of Chemistry 2023 |