
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight-driven Pickering interfacial catalysis for the oxidation of alkenes at near-room temperature†
Yaoyao
Feng
a,
Jean-François
Dechezelles
 a,
Quentin
D'Acremont
b,
Emmanuel
Courtade
b,
Vincent
De Waele
c,
Marc
Pera-Titus
*d and
Véronique
Nardello-Rataj
*a
a,
Quentin
D'Acremont
b,
Emmanuel
Courtade
b,
Vincent
De Waele
c,
Marc
Pera-Titus
*d and
Véronique
Nardello-Rataj
*a
aUniversité de Lille, CNRS, Centrale Lille, Université Artois, UMR 8181 – UCCS – Unité de Catalyse et Chimie du Solide, F-59000 Lille, France. E-mail: veronique.rataj-nardello@univ-lille.fr
bUniv. Lille, CNRS, UMR 8523 – PhLAM – Physique des Lasers Atomes et Molécules, F-59000 Lille, France
cUniv. Lille, CNRS, UMR 8516 – LASIRe – Laboratoire Avancé de Spectroscopie pour les Intéractions la Réactivité et l'Environnement, F-59000 Lille, France
dCardiff Catalysis Institute, School of Chemistry, Main Building, Cardiff University, Park Place, Cardiff CF10 3AT, UK. E-mail: peratitusm@cardiff.ac.uk
First published on 25th January 2023
Abstract
In this study, we have developed an emulsion system combining plasmonic Au-loaded amphiphilic silica nanoparticles (Au/SiO2–C3) and tri(dodecyltrimethylammonium) phosphotungstate ([C12]3[PW12O40]) nanoparticles acting as an on-site photoassisted heater/activator and a catalyst, respectively, at the water/oil interface. The system exhibits a 5-fold increase of activity compared to the thermal reaction for the near-room temperature oxidation of alkenes with H2O2. The nanoparticles show excellent recyclability and structural stability. This study opens an avenue to design multiphase photoreactors for oxidation reactions at mild temperature, with a potential energy saving of 74% compared to that of thermally heated reactors at isoconversion.
Introduction
Owing to localized surface plasmon resonance (LSPR), noble metal nanoparticles (NPs) can absorb light to generate hot electrons and cause local heating by hyperthermia.1 These properties allow efficient light-to-energy conversion and energy transfer to the local microenvironment around the NPs. Among noble metals, Au NPs are most commonly used as plasmonic materials and catalysts due to their low reactivity, easy fabrication and facile application in targeted therapies driven by local heating.2Depending on the decay pathways of plasmonic energy, the mechanisms of hot carriers in the chemical reaction are proposed as follows: (1) optically excited hot electrons can inject into the electron-accepting states of the nearby surface and can take part in the reaction, (2) photo-induced hot carriers play a negligible role in activating the adsorbed reactants, but convert energy into heat through energy exchange between electrons and phonons, and (3) both hot carrier-driven reactant activation and photothermal heating co-exist and can act synergistically to drive chemical reactions. The plasmonic properties of Au NPs are dictated by their size, shape (e.g., nanospheres, nanorods, nanoshells, etc.) and composition, as well as by the surrounding conditions such as pH, concentration and ionic environment.3
Based on their LSPR properties, Au NPs have been used as plasmonic/photothermal catalysts or catalyst components for a variety of reactions,4 including peroxide decomposition,5 oxidation,6 oxidation/amidation,7 reduction,8 and alkene/CO2 hydrogenation.9 Unlike conventional heating, plasmon-driven catalysis handles hot electrons and/or a temperature gradient on the catalyst surface under light irradiation to accelerate the activity and shift the selectivity of reactions by activating specific chemical bonds. Amphiphilic plasmonic NPs (e.g., HS-β-cyclodextrin modified with Ag NPs) can self-assemble at the water/oil interface giving improved signals for Surface-Enhanced Raman Spectroscopy (SERS) due to substrate activation by interfacial hot spots.10 All and all, these studies clearly demonstrate the possibility of using plasmonic metal NPs as energy converters to drive catalytic reactions at moderate bulk temperature.
Herein, we developed a light-driven catalytic system based on a Pickering emulsion affording the oxidation of olefins at mild temperature. The system makes use of the colloidal tectonics approach to prepare emulsions, encompassing the interfacial self-assembly of two surface-active particles with complementary hydrophilic/hydrophobic groups (tectons).11 The system combines silica NPs grafted with alkyl chains and Au NPs, acting as a heater/plasmon activator, and dodecyltrimethylammonium phosphotungstate NPs, [C12]3[PW12O40], acting as the catalyst (Fig. 1).12 The physicochemical properties, stability and interfacial plasmonic properties of the dual-NP stabilized emulsions were studied to assess the interaction between both NPs at the water/oil interface. The catalytic performance of the system was assessed for the oxidation of cyclooctene using H2O2 as the oxidant under UV light irradiation, and further extended to a panel of industrially relevant alkenes.
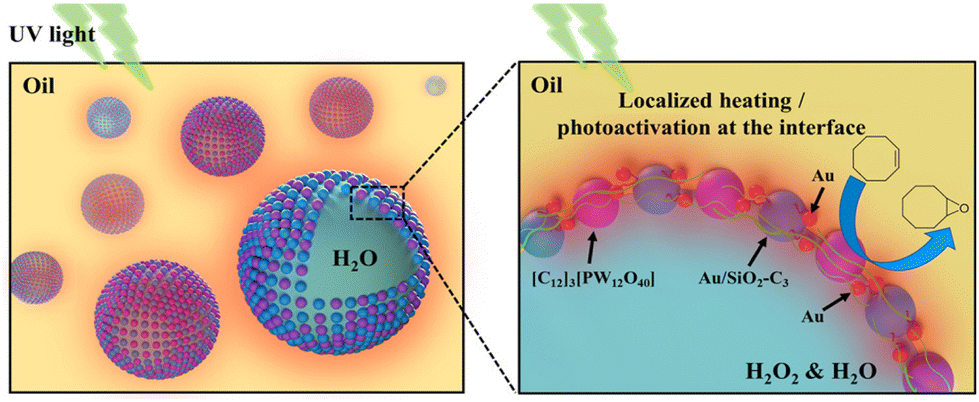 | ||
| Fig. 1 Schematic representation of the emulsion system designed in this study using self-assembled amphiphilic silica NPs loaded with Au NPs, acting as on-site heater/plasmon activators, and [C12]3[PW12O40] NPs, acting as the catalyst, under UV light irradiation. | ||
Results and discussion
Synthesis and characterization of particles
The hydrodynamic diameter and ζ-potential of the as-prepared NPs were measured by dynamic light scattering (DLS) (Table 1). In accordance with previous results,12a [C12]3[PW12O40] NPs have a hydrodynamic diameter of 48 nm with a ζ-potential of −31.7 mV at pH 7. Au NPs have a hydrodynamic diameter of 13 nm with a ζ-potential of −34.7 mV. The hydrodynamic diameter of bare SiO2 NPs is around 90 nm with a negative ζ-potential of −38 mV at pH 7. After surface modification, the ζ-potential of SiO2–C3 NPs becomes positive (+42.6 mV), which can be attributed to the protonation of amine groups on the silica surface. The hydrodynamic diameter of amphiphilic trimethoxy(propyl)- and aminopropyl-functionalized (SiO2–C3) NPs is about 200 nm, suggesting partial aggregation. The ζ-potential of SiO2–C3 NPs evolves from +59.9 mV to −49.4 mV on increasing the pH from 2 to 12 with the isoelectric point (IEP) at pH 10 (Fig. S1a†). After Au loading, Au/SiO2–C3 NPs exhibit a comparable hydrodynamic diameter (207 nm) and an analogous trend of the ζ-potential with pH, but the values are slightly lower than those of SiO2–C3 NPs (+40.3 vs. +42.6 mV at pH 7) (Fig. S1b†). This discrepancy suggests the presence of free amine groups after the decoration of SiO2–C3 with Au NPs. Increasing the Au loading in Au/SiO2–C3 from 8 to 24 μg (in 6 mL dispersion) results in a small decline of the ζ-potential. SiO2–C3 and Au/SiO2–C3 NP dispersions are stable below pH 8 and above 12, since the ζ-potential is outside the ±30 mV range.13The surface morphology of dispersed Au, SiO2–C3 and Au/SiO2–C3 NPs was characterized by HR-TEM (Fig. 2). Dispersed Au NPs show a uniform spherical shape with an average size of 4.5 nm. This value is ≈3 times lower than the hydrodynamic diameter measured by DLS (13 nm, Table 1) which can be explained by the aggregation of Au NPs. SiO2–C3 NPs are also spherical with an average size of 26 nm and large size polydispersity, which is also lower than the hydrodynamic diameter (190 nm), suggesting aggregation of SiO2–C3 NPs. Au NPs on Au/SiO2–C3 (highlighted with red circles) are well dispersed and show an average size of 4.5 nm. The Au loading in Au/SiO2–C3 measured by ICP-OES is ≈0.13 wt%. The binding energy of the Au (4f7/2) component for Au/SiO2–C3 measured by XPS is 83.9 eV (Fig. S2†), indicating that Au is mainly in its metallic state.14
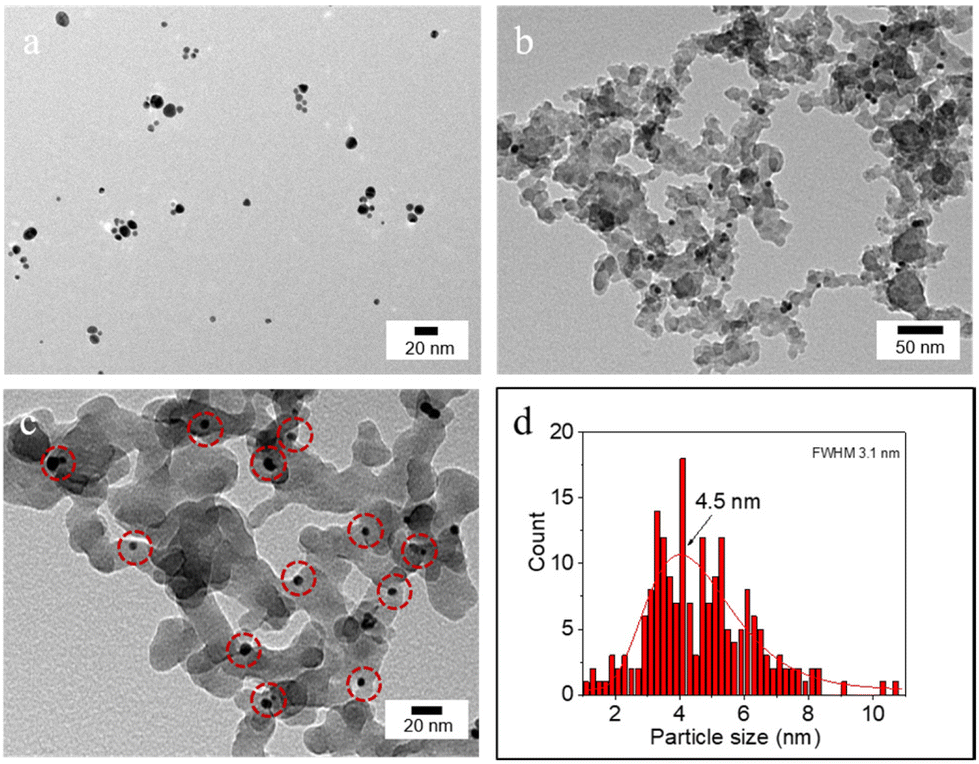 | ||
| Fig. 2 HR-TEM micrograph of (a) Au NPs and (b and c) Au/SiO2–C3 NPs and (d) particle size distribution of Au NPs. The red circles in (c) indicate Au NPs immobilized on Au/SiO2–C3. | ||
Au NPs exhibit a neat surface plasmon resonance band in the UV spectrum at 518 nm (Fig. S3†), which matches earlier observations.15 In the case of Au/SiO2–C3 NPs, the band slightly shifts to 528 nm, which can be explained by the shorter distance between Au NPs on Au/SiO2–C3 and by a change of the dielectric constant due to the silica environment.15 Pure [C12]3[PW12O40] NPs and a water solution of H3PW12O40 do not show any UV absorption band.
The stability of the particles was studied by TGA. SiO2 NPs show high thermal stability with a weight loss of 0.4% from 30 to 200 °C due to water desorption (Fig. S4†). A second weight loss is observed in the range of 200–900 °C which is attributed to the condensation of silanol groups. Both SiO2–C3 and Au/SiO2–C3 NPs show almost no weight loss (1.0 wt%) from 30 to 200 °C which is consistent with the hydrophobization of the samples. Both samples exhibit a weight loss of 15 wt% in the range of 400–600 °C which can be assigned to the decomposition of amine groups and C3 chains, but with no significant weight loss in the range of 600–900 °C. These profiles illustrate the excellent stability of SiO2–C3 and Au/SiO2–C3 NPs.
To confirm the presence of grafted alkyl chains and amine groups on the silica surface, we recorded the FT-IR spectra of SiO2, SiO2–C3 and Au/SiO2–C3 NPs (Fig. S5†). Two bands are visible at 1070 and 810 cm−1 for all samples which can be attributed to asymmetric and stretching vibrations of Si–O–Si bonds, respectively.16 Characteristic bands appear at 2993/2904 cm−1 and 1505 cm−1 for SiO2–C3 and Au/SiO2–C3 NPs which can be ascribed to –CH2 stretching vibrations in C3 chains and bending vibrations of N–H groups,17 respectively. Characteristic bands appearing at 930 cm−1 can be assigned to stretching modes of Si–C groups.17 These results prove the grafting of SiO2–C3 and Au/SiO2–C3 NPs with –C3 and –NH2 chains.
The surface wettability of SiO2, SiO2–C3 and Au/SiO2–C3 NPs was characterized by water contact angle analysis. SiO2 NPs show no measurable contact angle because of instantaneous water absorption. In contrast, the water contact angle of SiO2–C3 is 108°, indicating that C3 moieties successfully modify the silica surface (Fig. S6†). The contact angle for Au/SiO2–C3 NPs is slightly lower (98°), suggesting that the sample is slightly less hydrophobic. Finally, the contact angle of [C12]3[PW12O40] NPs is about 93°. According to the Finkle rule,18 emulsions stabilized with SiO2–C3 and Au/SiO2–C3 NPs are expected to be water-in-oil emulsions.
Physicochemical properties of Au/SiO2–C3/[C12]3[PW12O40] NP stabilized Pickering emulsions
We studied the emulsification properties of combined Au/SiO2–C3 NPs + [C12]3[PW12O40] NPs for a water/toluene (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 v/v) system as a function of the [C12]3[PW12O40] weight fraction (Fig. 3). Both [C12]3[PW12O40] and Au/SiO2–C3 NPs alone stabilize a water-in-toluene emulsion with an average droplet size of 18 μm and 22 μm, respectively. In the latter case, the emulsion becomes pink due to the presence of Au NPs. The combination of both NPs also generates a water-in-toluene emulsion, but the droplet size is much smaller (from 6 to 10 μm) than the values for both NPs alone. The combination of both NPs shows almost zero ζ-potential (−3 mV) at 0.2 wt%. Overall, these results confirm the remarkable synergy between both NPs at the water/oil interface.
:50 v/v) system as a function of the [C12]3[PW12O40] weight fraction (Fig. 3). Both [C12]3[PW12O40] and Au/SiO2–C3 NPs alone stabilize a water-in-toluene emulsion with an average droplet size of 18 μm and 22 μm, respectively. In the latter case, the emulsion becomes pink due to the presence of Au NPs. The combination of both NPs also generates a water-in-toluene emulsion, but the droplet size is much smaller (from 6 to 10 μm) than the values for both NPs alone. The combination of both NPs shows almost zero ζ-potential (−3 mV) at 0.2 wt%. Overall, these results confirm the remarkable synergy between both NPs at the water/oil interface.
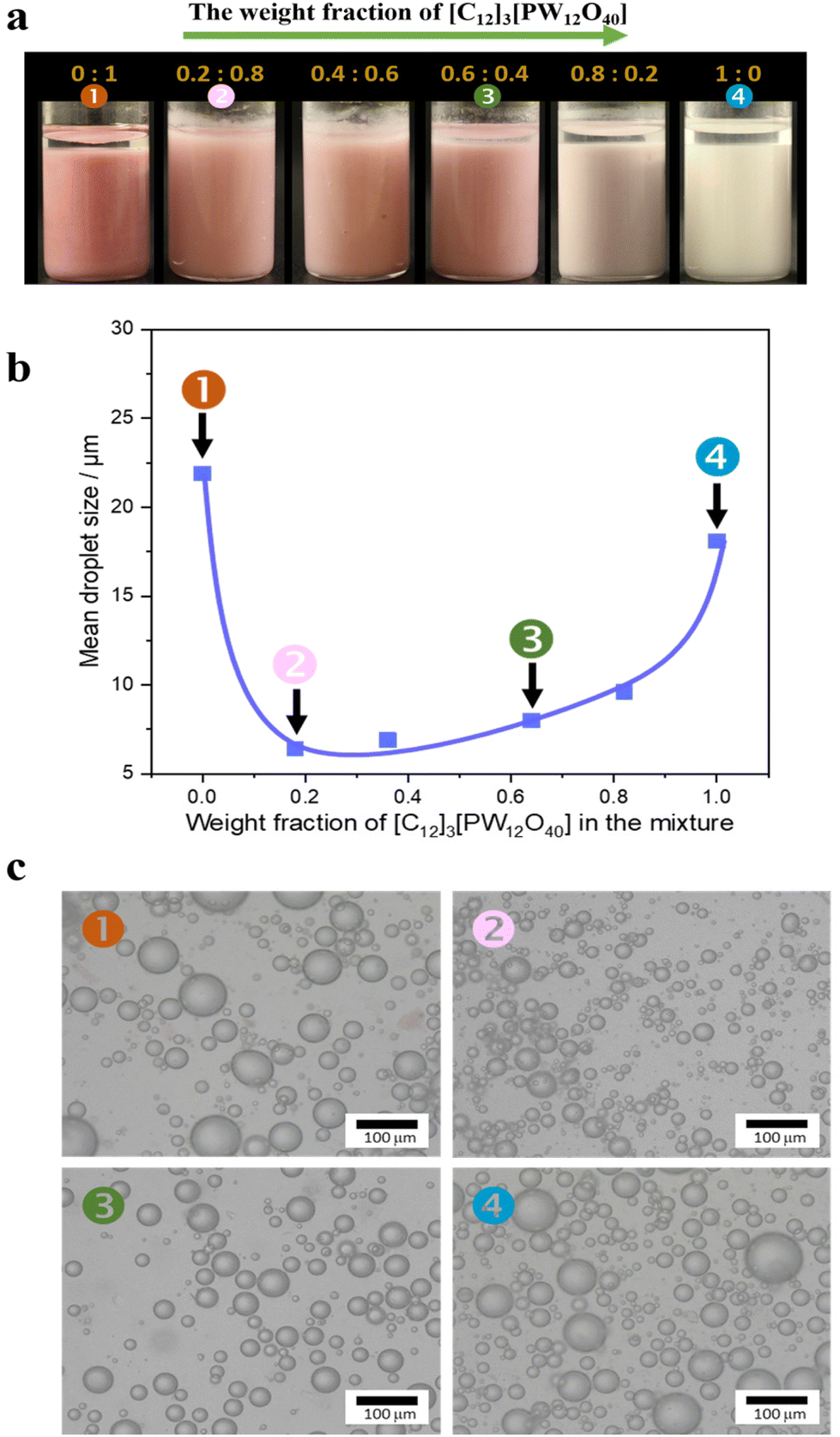 | ||
| Fig. 3 (a) Images of emulsions stabilized with Au/SiO2–C3 NPs + [C12]3[PW12O40] NPs at variable weight fractions; (b) evolution of the average droplet size against the [C12]3[PW12O40] weight fraction for emulsions stabilized with Au/SiO2–C3 NPs + [C12]3[PW12O40] NPs; (c) optical micrographs of water-in-oil emulsions. Emulsification conditions: 1.5 mL toluene, 1.5 mL water, 80 mg NPs (2.8 wt%), 11500 rpm for 2 min. | ||
We monitored the time-evolution of the emulsions stabilized with Au/SiO2–C3 NPs and [C12]3[PW12O40] NPs alone and with combinations of both NPs (Fig. S7†). The combinations afford much more stable emulsions with no obvious collapse after 6 h at 60 °C. To gain more insight into the emulsion stability, light scattering measurements were performed using Turbiscan to monitor the change of transmission (ΔT) and backscattering (ΔBS) light intensity along the emulsion height (Fig. S8†). In the presence of [C12]3[PW12O40] NPs, the emulsions exhibit a sharp decline of ΔBS in the top layer with a concomitant increase of the ΔT signal, which can be explained by clarification due to gravity-induced migration. Besides, ΔBS shows a slight decrease in the middle portion of the emulsion which can be interpreted as an increase in the droplet size due to either coalescence or flocculation. The ΔT signal remains almost unchanged over time, indicating no apparent stratification.
The emulsion is more stable when using combined Au/SiO2–C3 NPs + [C12]3[PW12O40] NPs, as shown by the changes in the backscattering and transmission results. The slight change of ΔBS suggests lower creaming. On the top of the emulsion, the lower ΔBS and higher ΔT are in agreement with a clarified layer due to sedimentation and/or aggregation of droplets. The emulsion stability can be explained by the formation of a compact interfacial layer of both NPs which likely protects the droplet surface and limits flocculation or coalescence of water droplets.19
With these results, we assessed the emulsion stability for Au/SiO2–C3 NPs + [C12]3[PW12O40] NPs in the presence of cyclooctene and cyclooctene oxide (2 mol L−1) (Fig. S9†). In both cases, the emulsion properties remain almost unchanged. However, in the latter case, the ΔBS signal is slightly lower within the tested height, but remains almost unchanged over time, suggesting neither creaming nor phase separation.
Study of plasmon-induced photothermal effects
We measured the photothermal curves of the Au NP dispersion and Au/SiO2–C3 NP-stabilized emulsion generated by light irradiation. The Au NP dispersion exhibits a marked increase of the bulk temperature (ΔT) of 24 °C vs. only 10 °C for pure water after 30 min (Fig. 4a). Increasing the amount of Au NPs from 4 to 40 μg (in 1 mL dispersion) leads to a ΔT increase from 16 to 24 °C with a maximal temperature of 48 °C at 40 μg Au. The Au/SiO2–C3 NP-stabilized emulsion displays a similar trend for 4 and 13 μg Au loading in the NPs (in 1 mL emulsion), with a ΔT of 20.6 °C and a maximal temperature of 47 °C at 13 μg Au after 30 min (Fig. 4b). However, no appreciable temperature change occurs upon further increasing the Au amount to 26 and 40 μg, encompassing a concomitant increase of the amount of Au/SiO2–C3 NPs in the system. This observation indicates a poorer heat transfer to the continuous phase at higher Au/SiO2–C3 NP loading, which can be attributed to two main reasons: (1) a high viscosity of the emulsion system that might hinder heat transfer from the water/toluene interface to the bulk phases20 and (2) a denser packing of Au/SiO2–C3 NPs at the inner and outer interface of the droplets. As a result, the local temperature of the interfacial shell is expected to be much higher than the bulk temperature, with photon absorption.21 The photothermal curves show a strong power-dependent effect for the Au NP dispersion and Au/SiO2–C3-stabilized emulsion upon increasing the power density from 260 to 1000 mW cm−2 (Fig. S10†).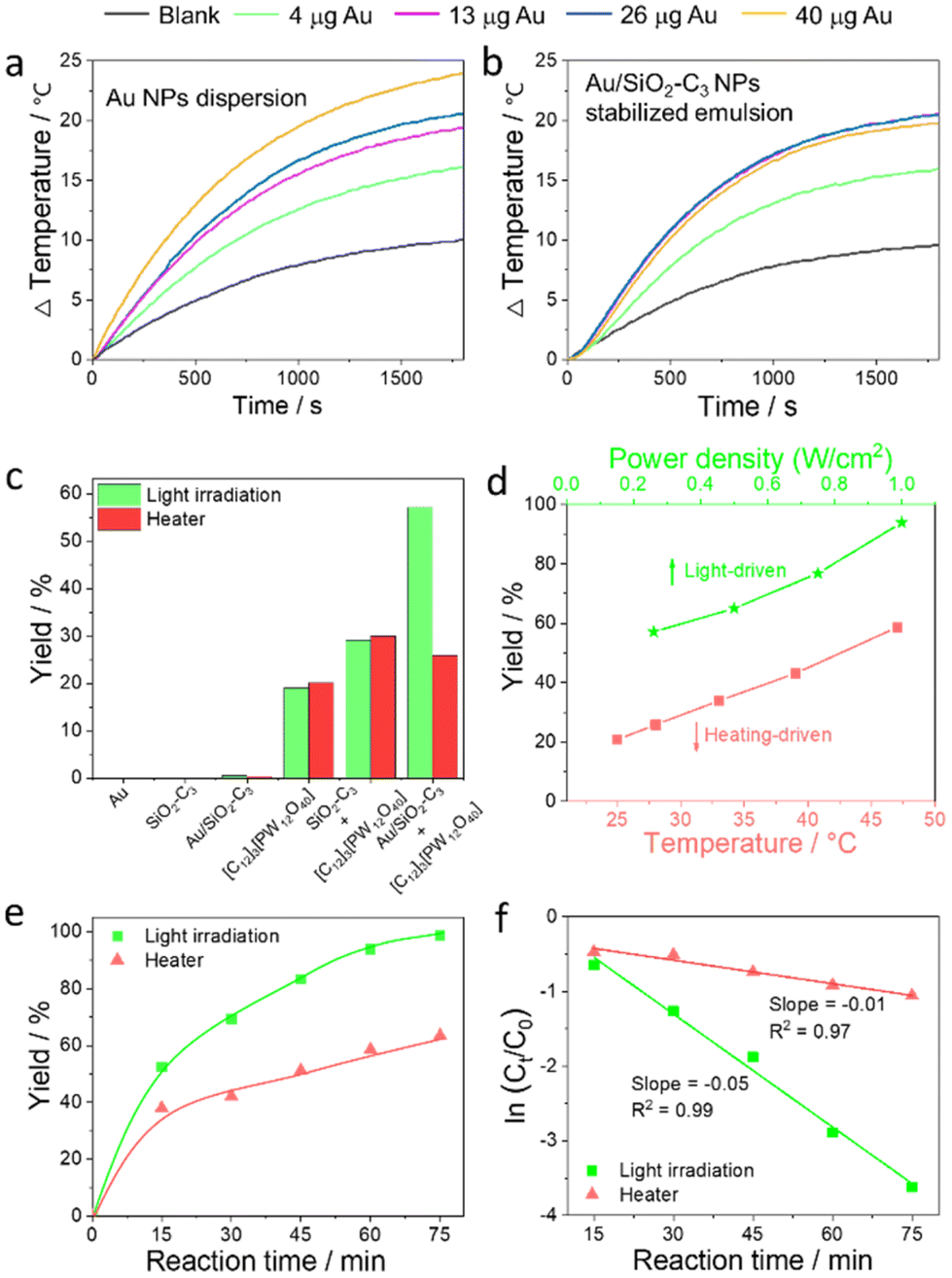 | ||
| Fig. 4 Photothermal profiles of (a) Au NP dispersion and (b) Au/SiO2–C3-stabilized emulsion against Au loading (in 1 mL dispersion) at 1000 mW cm−2 power density for 30 min. (c) Yield of cyclooctene oxide (60 min) in cyclooctene oxidation over various NPs (*). (d) Yield of cyclooctene oxide (60 min) over [C12]3[PW12O40] NPs + Au/SiO2–C3 NPs under light at variable power densities or after heating. (e and f) Time-evolution and kinetic plots of the yield of cyclooctene oxide over [C12]3[PW12O40] + Au/SiO2-C3 NPs under light and heating. Reaction conditions: 30 mg Au/SiO2–C3 NPs, 50 mg [C12]3[PW12O40] NPs, 1.5 mL toluene (2 mol L−1 cyclooctene), 1.5 mL water (containing 1.2 equiv. H2O2), emulsification at 11500 rpm for 2 min, 120 rpm. Results based on 1H NMR. (*) 40 μg Au NPs, 30 mg SiO2–C3 NPs, 30 mg Au/SiO2–C3 NPs, 50 mg [C12]3[PW12O40] NPs, 50 mg [C12]3[PW12O40] NPs + 30 mg SiO2–C3 NPs, 50 mg [C12]3[PW12O40, 260 mW cm−2. | ||
Catalytic performance
Given the photothermal properties of the Au/SiO2–C3-stabilized emulsion, we aimed to effectively utilize light-induced interfacial heating/hot electrons to enhance the catalytic performance of [C12]3[PW12O40] NPs. To this aim, we investigated the catalytic performance of combined Au/SiO2–C3 NPs + [C12]3[PW12O40] NPs in the oxidation of cyclooctene. For comparison, control experiments were carried out under the same reaction conditions, but the reactor was heated in a thermostated bath to the same temperature reached in the tests under light. For instance, the temperature increases from 25 to 28 °C under light (260 mW cm−2) for 60 min. Therefore, the bath temperature was set at 28 °C for the dark experiments. Preliminary tests were also conducted to optimize the reaction conditions (i.e. stirring rate, water/oil volume ratio, and weight of Au/SiO2–C3 and [C12]3[PW12O40] NPs) (Fig. S11†). It has been previously demonstrated that 1.0 equiv. of H2O2 is needed to get 99% yield for the epoxidation of cyclohexene and cyclooctene in the Pickering emulsion systems.12b Since some H2O2 molecules are decomposed into H2O and O2 under light irradiation, the decomposition rate of H2O2 in the Pickering emulsion system (without the addition of the substrate) under light irradiation was measured by KMnO4 titration.22 The decomposition rate of H2O2 was 16% higher under light irradiation for 1 h than that without light irradiation. Therefore, 1.2 equiv. H2O2 were added for the epoxidation of cyclooctene.No conversion is observed for blank experiments with Au/SiO2–C3 NPs under light and after heating to 28 °C (Fig. 4c). Using [C12]3[PW12O40] NPs, the yield of cyclooctene oxide is about 19% with/without light at 95–100% selectivity, suggesting that light does not impact the catalytic properties. Combined [C12]3[PW12O40] NPs and SiO2–C3 NPs exhibit a much higher yield with/without light (30%) compared to that achieved for [C12]3[PW12O40] NPs alone. This higher yield can be explained by the smaller droplet size and higher emulsion volume obtained when using combined SiO2–C3 NPs and [C12]3[PW12O40] NPs owing to their synergy at the water/toluene interface (Fig. S12†). A catalytic test carried out with Au/SiO2–C3 NPs, but replacing [C12]3[PW12O40] NPs with H3PW12O40, which has no interfacial properties, shows only 8% yield of cyclooctene oxide after 60 min under light (1000 W cm−2), whereas no conversion is observed for the tests performed either without light or in the absence of Au/SiO2–C3 NPs (Fig. S13†).
In contrast to these results, the reaction over combined Au/SiO2–C3 NPs and [C12]3[PW12O40] NPs gives 57% yield of cyclooctene oxide after 60 min under light, whereas it decreases to 26% at 28 °C. The yield increases linearly from 57% to 94% with the increase of power density (range 260–1000 mW cm−2) (Fig. 4d), but only an increase from 27% to 45% was achieved for the tests in a thermostated bath with the same temperature increase (from 28 to 47 °C after 60 min). The yield of cyclooctene oxide is almost 100% after 75 min under light (1000 mW cm−2), but it is only 63% at 47 °C (Fig. 4e).
Cyclooctene oxidation follows an apparent first-order kinetics (Fig. 4f) with a kinetic constant of k = 0.05 min−1 under light (1000 mW cm−2), which is much larger than k = 0.01 min−1 at 47 °C. In parallel, the turnover frequency (TOF) is 188 h−1 under light, whereas it is 88 h−1 when using [C12]3[PW12O40] NPs alone. Additional catalytic tests conducted using a 532 nm pulsed laser (100 mW cm−2), with higher photonic efficiency, deeper penetration and sharper spectrum than UV lamps, afforded 91% yield of cyclooctene oxide after 60 min despite the lower power density, while it is much lower (50%) at 43 °C corresponding to the temperature reached during the laser-driven catalytic reaction (Fig. S14†).
Overall, this body of results outline the enhanced catalytic efficiency of combined Au/SiO2–C3 NPs and [C12]3[PW12O40] NPs for cyclooctene oxidation. Such a promoting effect can be ascribed to three factors: (1) preferential location of catalytic sites (i.e. [C12]3[PW12O40]) at the water/toluene interface leading to greater contact with cyclooctene and favouring mass transfer; (2) synergy between both NPs allowing plasmon-driven heating/activation under light; and (3) coverage of the droplet surface by a thick layer of closely packed NPs allowing high emulsion stability and hindering heat transfer to the bulk phases.
Catalyst recyclability and reuse
The recyclability and reuse of Au/SiO2–C3 and [C12]3[PW12O40] NPs were studied for cyclooctene oxidation over five consecutive catalytic cycles. After each cycle, the reaction medium was centrifuged (5000 rpm, 30 min), and the particles were separated and dried at 80 °C for 12 h before use in the subsequent cycle. The particles can be conveniently recycled after each run without apparent loss of catalytic activity and emulsification properties (Fig. 5). This body of results evidence the absence of significant catalyst deactivation during the reaction. The HR-TEM micrographs of the recovered Au/SiO2–C3 NPs and [C12]3[PW12O40] NPs after the fifth cycle show no change in the size distribution of Au NPs over Au/SiO2–C3 compared to the fresh sample with an average size of 4.2 nm. However, partial agglomeration of Au NPs is observed (Fig. S15†). [C12]3[PW12O40] NPs are deposited on Au/SiO2–C3 NPs (not shown), confirming their interaction. On the other hand, it should also be mentioned that W-based catalysts can be prone to metal leaching. Indeed, while some studies have demonstrated that leached W species cannot act as a catalyst by themselves,23 Bisio et al. have shown that in the presence of hydrogen peroxide, they themselves can act as a catalyst for alkene epoxidation.23 So, a study of tungsten leaching in the system stabilized with Au/SiO2–C3 and [C12]3[PW12O40] NPs could be useful to confirm the impact of the nature of the polyoxometalates on different substrates.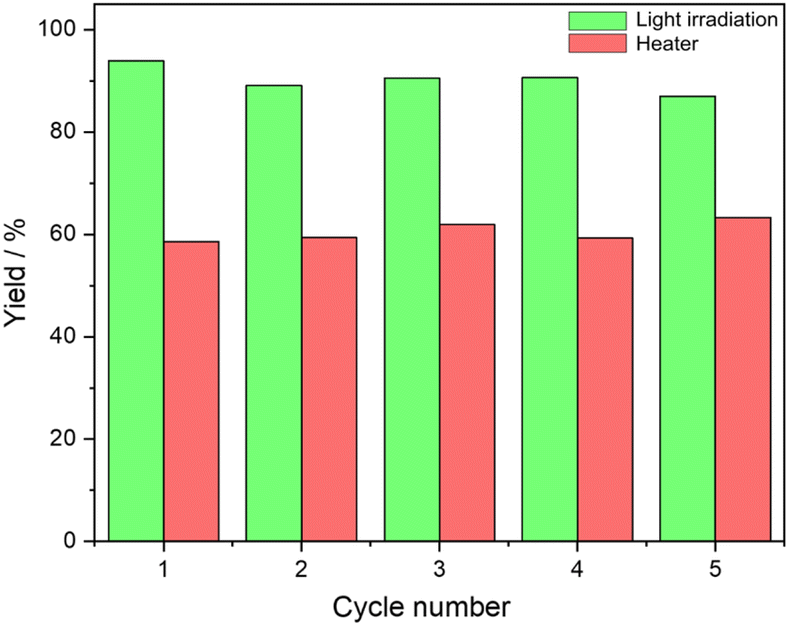 | ||
| Fig. 5 Recyclability and reuse of Au/SiO2–C3 and [C12]3[PW12O40] NPs (40/60 wt%) for cyclooctene oxidation over five consecutive cycles. Reaction conditions: 30 mg Au/SiO2–C3 NPs, 50 mg [C12]3[PW12O40] NPs, 1.5 mL toluene (2 mol L−1 cyclooctene), 1.5 mL water (containing 1.2 equiv. of H2O2), emulsification at 11500 rpm for 2 min, 120 rpm. Results based on 1H NMR. | ||
Scope of reactants
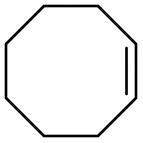
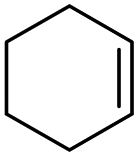
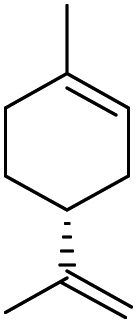
To assess the versatility of combined [C12]3[PW12O40] NPs and Au/SiO2–C3 NPs in emulsion for light-driven oxidation reactions, we tested cyclohexene, limonene and 1-octene, and compared the results obtained after heating to 47 °C which corresponds to the temperature reached under light (Table 2). Cyclohexene oxidation results mainly in 1,2-cyclohexanediol with 70% yield after 1 h under light irradiation (entry 2). In contrast, the yield is 57% at 47 °C. In both cases, no cyclohexene epoxide is observed due to fast ring opening in emulsion, matching the results of a previous study.11c In the case of limonene with two unsaturations, the epoxidation of the exocyclic unsaturation is achieved as a secondary reaction of the epoxidation of the endocyclic unsaturation. It is still challenging to get 8,9-limonene oxide or limonene dioxide, and 1,2-limonene oxide is obtained as the main product together with carvone as the minor product.24 Although limonene conversion is slightly lower under light than in the experiment at 47 °C (35% vs. 45% after 2 h), the yield of 1,2-limonene oxide is higher in the former case (29% vs. 11%) due to a much higher selectivity (83% vs. 24%). The oxidation of 1-octene shows 11% yield of 1,2-epoxyoctane after 2 h in the light-driven test, whereas only 3% yield is obtained at 47 °C. Overall, these results demonstrate the ability of combined Au/SiO2–C3 and [C12]3[PW12O40] NPs for the light-driven oxidation of olefins at the water/toluene interface.| Substrate | t/h | Light irradiation | Heater (47 °C) | ||
|---|---|---|---|---|---|
| Conv. (%) | Yield (%) | Conv. (%) | Yield (%) | ||
| Reaction conditions: 30 mg Au/SiO2–C3 NPs, 50 mg [C12]3[PW12O40] NPs, 1.5 mL toluene (containing 2 mol L−1 substrates) and 1.5 mL water (containing 1.2 equiv. of H2O2), emulsification at 11500 rpm for 2 min, 60 min, 120 rpm.a Results based on 1H NMR of cyclooctene oxide.b Results based on 1H NMR of 1,2-cyclohexanediol.c Results based on 1H NMR of 1,2-limonene oxide.d Results based on 1H NMR of carvone.e Results based on 1H NMR of 1,2-epoxyoctane. |
|||||
|
1 | 94 | 94a | 61 | 58a |
|
1 | 70 | 70b | 57 | 57b |
|
2 | 35 | 29c/8d | 45 | 11c/7d |
|
2 | 12 | 11e | 3 | 3e |
Photo- vs. thermal reactor: energy savings
Reduction of energy consumption is key to developing more eco-efficient processes. In this section, we aim to assess if plasmon-driven interfacial heating/activation under light irradiation could provide energy savings for biphasic reactions in emulsion compared to conventional thermal heating. In this view, by using the above body of results, we estimated the energy required in the light-driven and control experiments in our labscale reactor.The determination of the theoretical energy (QTH) that is required for a reaction mixture to reach a given temperature (heating + cooling) can be calculated using eqn (1), whereas the energy input supplied by the lamp (QL) can be computed using eqn (2) which depends on the power dissipation and time of exposure. From eqn (1) and (2), the overall energy saving factor (η) of light irradiation compared to the energy required in thermal experiments can be calculated using eqn (3)
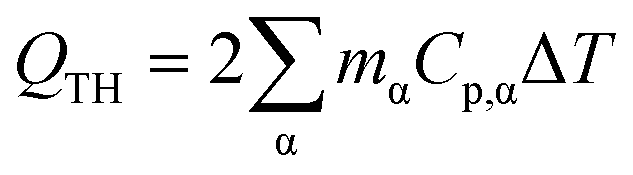 | (1) |
| QL = PavgΔT | (2) |
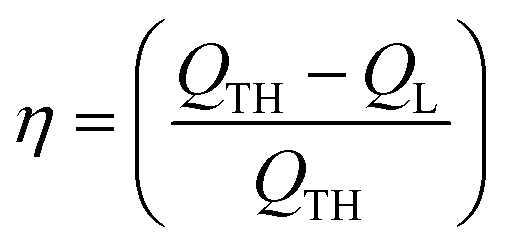 | (3) |
For a 10 mL water/toluene emulsion (50:50 v/v) in a lab scale batch reactor and cyclooctene oxidation for 1 h under light irradiation (260 mW cm−2 UV lamp) and thermal heating (47 °C), a similar cyclooctene yield (58%) is obtained, and the energy factor is about 74% (see detailed calculations in the ESI†). This result outlines the energy benefits of photoreactors based on particle-stabilised emulsions compared to standard thermal reactors for conducting oxidation reactions.
Conclusions
In summary, we have designed a novel emulsion system that combines interfacial heating/activation by plasmonic Au/SiO2–C3 NPs under light irradiation and the catalytic properties of [C12]3[PW12O40] NPs for near-room temperature oxidation of alkenes with H2O2. The dual-NP system exhibits a 5-fold increase of activity compared to the thermal reaction, and can be recycled and reused for at least five consecutive catalytic runs. The enhanced catalytic properties are ascribed to the intimate contact between both NPs at the water/toluene interface, discouraging heat transfer to the bulk phase. The concept presented in this study, although in its infancy, opens an avenue for engineering multiphase catalytic photoreactors with a potential energy saving of 74% at isoconversion compared to conventional thermal reactors. Further investigations will be conducted with a special emphasis on the effect of the wavelength of light irradiation on the system.Author contributions
YF: investigation, methodology, visualization, and writing – original draft; JFD: conceptualization, supervision, validation, and writing – review & editing; QA: investigation and writing – review & editing; EC: resources and writing – review & editing; VD: resources and writing – review & editing; MPT: conceptualization, validation, and writing – review & editing; VNR: conceptualization, acquisition, resources, supervision, validation, visualization, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Chevreul Institute (FR2638), Ministère de l'Enseignement et de la Recherche, Région Hauts de France and FEDER. Financial support from China Scholarship Council is also acknowledged. The authors would also like to thank Jean Pesez for his help in pulsed laser experiments and fruitful discussions.References
- (a) L. Wang, M. Hasanzadeh Kafshgari and M. Meunier, Adv. Funct. Mater., 2020, 30, 2005400 CrossRef CAS; (b) J. Liu, H. He, D. Xiao, S. Yin, W. Ji, S. Jiang, D. Luo, B. Wang and Y. Liu, Materials, 2018, 11, 1833 CrossRef PubMed; (c) A. Sanchot, G. Baffou, R. Marty, A. Arbouet, R. Quidant, C. Girard and E. Dujardin, ACS Nano, 2012, 6, 3434 CrossRef CAS PubMed.
- (a) M. Hu, J. Chen, Z.-Y. Li, L. Au, G. V. Hartland, X. Li, M. Marquez and Y. Xia, Chem. Soc. Rev., 2006, 35, 1084 RSC; (b) P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Plasmonics, 2007, 2, 107 CrossRef CAS; (c) X. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Lasers Med. Sci., 2008, 23, 217 CrossRef PubMed; (d) M. A. Mackey, M. R. K. Ali, L. A. Austin, R. D. Near and M. A. El-Sayed, J. Phys. Chem. B, 2014, 118, 1319 CrossRef CAS PubMed; (e) H. Liu, D. Chen, L. Li, T. Liu, L. Tan, X. Wu and F. Tang, Angew. Chem., Int. Ed., 2011, 50, 891 CrossRef CAS PubMed; (f) M. Sharifi, F. Attar, A. A. Saboury, K. Akhtari, N. Hooshmand, A. Hasan, M. A. El-Sayed and M. Falahati, J. Controlled Release, 2019, 311, 170 CrossRef PubMed.
- (a) K. Jiang, D. A. Smith and A. Pinchuk, J. Phys. Chem. C, 2013, 117, 27073 CrossRef CAS; (b) J. Qiu and W. D. Wei, J. Phys. Chem. C, 2014, 118, 20735 CrossRef CAS; (c) J. Zheng, X. Cheng, H. Zhang, X. Bai, R. Ai, L. Shao and J. Wang, Chem. Rev., 2021, 121, 13342 CrossRef CAS PubMed.
- (a) R. Ma, J. Sun, D. H. Li and J. J. Wei, Int. J. Hydrogen Energy, 2020, 45, 30288 CrossRef CAS; (b) S. Luo, X. Ren, H. Lin, H. Song and J. Ye, Chem. Sci., 2021, 12, 5701 RSC; (c) D. Mateo, J. L. Cerrillo, S. Durini and J. Gascon, Chem. Soc. Rev., 2021, 50, 2173 RSC.
- C. Fasciani, C. J. B. Alejo, M. Grenier, J. C. Netto-Ferreira and J. C. Scaiano, Org. Lett., 2011, 13, 204 CrossRef CAS PubMed.
- (a) X. Huang, Y. Li, Y. Chen, H. Zhou, X. Duan and Y. Huang, Angew. Chem., Int. Ed., 2013, 52, 6063 CrossRef CAS PubMed.
- A. Pineda, L. Gomez, A. M. Balu, V. Sebastian, M. Ojeda, M. Arruebo, A. A. Romero, J. Santamaria and R. Luque, Green Chem., 2013, 15, 2043 RSC.
- (a) G. Liu, Q. Xiong, Y. Xu, Q. Fang, K. C.-F. Leung, M. Sang, S. Xuan and L. Hao, Colloids Surf., A, 2022, 633, 127860 CrossRef CAS; (b) Y. Zhao, R. M. Sarhan, A. Eljarrat, Z. Kochovski, C. Koch, B. Schmidt, W. Koopman and Y. Lu, ACS Appl. Mater. Interfaces, 2022, 14, 17259 CrossRef CAS PubMed.
- (a) L. C. de la Garza, N. Brodusch, R. Gauvin and A. Moores, ACS Appl. Nano Mater., 2021, 4, 1596 CrossRef CAS; (b) Z. Tang, F. Zhu, J. Zhou, W. Chen, K. Wang, M. Liu, N. Wang and N. Li, Appl. Catal., B, 2022, 309, 121267 CrossRef CAS.
- L. Ouyang, D. Li, L. Zhu, W.-W. Yang and H. Tang, J. Mater. Chem. C, 2016, 4, 736 RSC.
- (a) L. Leclercq, Front. Chem., 2018, 6, 168 CrossRef PubMed; (b) B. Yang, L. Leclercq, V. Schmitt, M. Pera-Titus and V. Nardello-Rataj, Chem. Sci., 2019, 10, 501 RSC; (c) B. Yang, G. Douyère, L. Leclercq, V. Nardello-Rataj and M. Pera-Titus, Catal. Sci. Technol., 2020, 10, 6723 RSC.
- (a) L. Leclercq, A. Mouret, S. Renaudineau, V. Schmitt, A. Proust and V. Nardello-Rataj, J. Phys. Chem. B, 2015, 119, 6326 CrossRef CAS PubMed; (b) L. Leclercq, A. Mouret, A. Proust, V. Schmitt, P. Bauduin, J.-M. Aubry and V. Nardello-Rataj, Chem. – Eur. J., 2012, 18, 14352 CrossRef CAS PubMed.
- G. Douyère, L. Leclercq and V. Nardello-Rataj, Colloids Surf., A, 2021, 631, 127705 CrossRef.
- C. Oliveira, C. Ribeiro Chaves, P. Bargiela, M. da Graça Carneiro da Rocha, A. Ferreira da Silva, J. F. Diniz Chubaci, M. Boström, C. Persson and M. Malta, J. Mater. Res. Technol., 2021, 15, 768 CrossRef CAS.
- (a) S. M. Demers, C. R. Shirazinejad, J. L. A. Garcia, J. R. Matthews and J. H. Hafner, J. Phys. Chem. C, 2017, 121, 5201 CrossRef CAS; (b) S. S. Shankar, S. Bhargava and M. Sastry, J. Nanosci. Nanotechnol., 2005, 5, 1721 CrossRef CAS PubMed; (c) M. Santana Vega, F. Brisset and G. Laurent, Plasmonics, 2021, 16, 635 CrossRef.
- G. D. Chukin and V. I. Malevich, J. Appl. Spectrosc., 1977, 26, 223 CrossRef.
- (a) D. Lin-Vien, W. G. Fateley, N. B. Coltup and J. G. Grasselli, The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, Academic Press, 1991 Search PubMed; (b) B. Yang, L. Leclercq, J.-M. Clacens and V. Nardello-Rataj, Green Chem., 2017, 19, 4552 RSC.
- P. Finkle;, H. D. Draper; and J. H. Hildebrand, J. Am. Chem. Soc., 1923, 45, 2780 CrossRef.
- S. Arditty, C. P. Whitby, B. P. Binks, V. Schmitt and F. Leal-Calderon, Eur. Phys. J. E, 2003, 11, 273 CrossRef CAS PubMed.
- (a) F. Wang, W. Lin, Z. Ling and X. Fang, Sol. Energy Mater. Sol. Cells, 2019, 191, 218 CrossRef CAS; (b) R. Bielas, T. Hornowski, K. Paulovičová, M. Rajňák and A. Józefczak, J. Mol. Liq., 2020, 320, 114388 CrossRef CAS; (c) R. Bielas and A. Józefczak, Materials, 2020, 13, 4783 CrossRef CAS PubMed.
- (a) C.-H. Chou, C.-D. Chen and C. C. Wang, J. Phys. Chem. B, 2005, 109, 11135 CrossRef CAS PubMed; (b) L. François, M. Mostafavi, J. Belloni, J.-F. Delouis, J. Delaire and P. Feneyrou, J. Phys. Chem. B, 2000, 104, 6133 CrossRef; (c) M. Bryckaert, A. Kharchenko, O. Lebedev, B. Dong, I. De Waele, G. Buntinx, O. Poizat, S. Mintova and V. De Waele, J. Phys. Chem. C, 2017, 121, 26958 CrossRef CAS.
- C. E. Huckaba and F. G. Keyes, J. Am. Chem. Soc., 1948, 70, 1640 CrossRef CAS PubMed.
- (a) N. Mizuno, K. Yamaguchi and K. Kamata, Catal. Surv. Asia, 2011, 15, 68 CrossRef CAS; (b) N. Maksimchuk, I. Ivanchikova, O. Zalomaeva, Y. Chesalov, A. Shmakov, V. Zaikovskii and O. Kholdeeva, Catalysts, 2018, 8, 95 CrossRef; (c) C. Bisio, A. Gallo, R. Psaro, C. Tiozzo, M. Guidotti and F. Carniato, Appl. Catal., A, 2019, 581, 133 CrossRef CAS.
- (a) S. G. Casuscelli, M. E. Crivello, C. F. Perez, G. Ghione, E. R. Herrero, L. R. Pizzio, P. G. Vazquez, C. V. Caceres and M. N. Blanco, Appl. Catal., A, 2004, 274, 115 CrossRef CAS; (b) Y. M. Ahmat and S. Kaliaguine, Catal. Today, 2023, 407, 146 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc04591e |
| This journal is © The Royal Society of Chemistry 2023 |