
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-conventional low-temperature reverse water–gas shift reaction over highly dispersed Ru catalysts in an electric field†
Ryota
Yamano
a,
Shuhei
Ogo
 b,
Naoya
Nakano
a,
Takuma
Higo
a and
Yasushi
Sekine
*a
b,
Naoya
Nakano
a,
Takuma
Higo
a and
Yasushi
Sekine
*a
aDepartment of Applied Chemistry, Waseda University, 3-4-1, Okubo, Shinjuku, Tokyo, 169-8555, Japan. E-mail: ysekine@waseda.jp
bDepartment of Marine Resources Science, Faculty of Agriculture and Marine Science, Kochi University, Nankoku, 783-8502, Japan
First published on 29th November 2022
Abstract
The reverse water–gas shift (RWGS) reaction, a promising carbon-recycling reaction, was investigated by applying an electric field to promote the reaction at a temperature of 473 K or lower. The highly dispersed Ru/ZrTiO4 catalysts with an approximately 2 nm particle size of Ru showed high RWGS activity with a DC electric field below 473 K, whereas CO2 methanation proceeded predominantly over catalysts with larger Ru particles. The RWGS reaction in the electric field maintained high CO selectivity, suppressing CO hydrogenation into CH4 on the Ru surface by virtue of promoted hydrogen migration (surface protonics). The reaction mechanisms of the non-conventional low-temperature reverse water gas shift reaction were investigated and revealed using various characterization methods including in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements. With the DC electric field, the reaction proceeds via a redox reaction where the generated oxygen vacancies are involved in CO2 activation at low temperatures. As a result, the electric field promotes both hydrogen migration and redox reactions using lattice oxygen/vacancies, resulting in high RWGS activity and selectivity even at low temperatures.
Broader contextFor establishing carbon recycling processes, the reverse water gas shift reaction is a very important reaction. Due to the large endothermic reaction, the reaction does not proceed very well at low temperatures with conventional catalyst technology. In addition, at low temperatures, the side reaction, methanation, which is an exothermic reaction, occurs concurrently and dominates. We have discovered and established a new catalytic process that can selectively and rapidly proceed only with the reverse water gas shift on a fine Ru catalyst at temperatures as low as 473 K by means of a non-conventional catalytic process in which an electric field is applied. The reaction mechanisms of the reverse water gas shift on the non-conventional catalytic process were investigated and revealed by in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements and other characterizations. These revealed that CO hydrogenation into CH4 on the Ru surface was suppressed by virtue of promoted hydrogen migration (surface protonics). Our findings contribute to the establishment of efficient and selective CO2 conversion catalysis which can work at a low temperature of 473 K. |
Introduction
Recently, many countries have set a goal of reducing CO2 emissions, virtually to zero, during a half-century. Various efforts are being made to develop the necessary associated technologies.1 Catalytic conversion of CO2 with green hydrogen has drawn much attention from the perspective of CO2 capture and utilization (CCU).2,3 Numerous studies have revealed that CO2 can be converted into basic chemical building blocks such as methane (CH4),4 carbon monoxide (CO),5 methanol (CH3OH),6 and more complex chemical compounds.7–9Particularly, carbon monoxide, produced in the reverse water–gas shift (RWGS) reaction (eqn (1)), plays a central role in C1 chemistry, by which CO can be converted further to value-added chemicals such as hydrocarbons for liquid fuels via Fischer–Tropsch (FT) synthesis and oxygenated compounds through well-established industrial processes.5 However, the RWGS reaction is endothermic 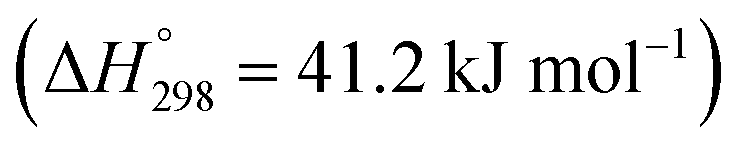 . Therefore, the activity is constrained heavily by thermodynamic equilibria. As a matter of fact, achieving 50% conversion requires extremely high temperatures of over 1000 K.
. Therefore, the activity is constrained heavily by thermodynamic equilibria. As a matter of fact, achieving 50% conversion requires extremely high temperatures of over 1000 K.
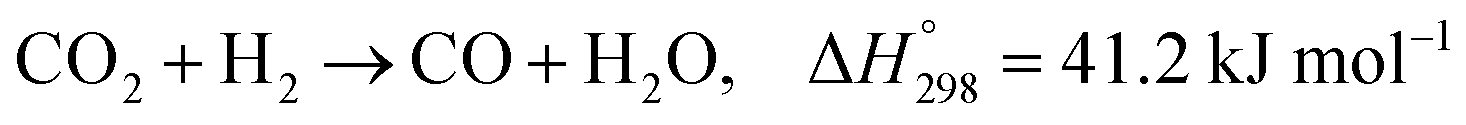 | (1) |
As earlier work, we have applied electric fields for several reactions including carbon dioxide activation (RWGS,17 dry reforming of methane,18,19 oxidative coupling of methane using carbon dioxide,20 and CO2 methanation21). Nevertheless, how CO2 is activated in catalytic reactions with an electric field remains unclear. Moreover, in our latest study of CO2 methanation in an electric field, kinetic analysis and spectroscopic measurements revealed that this reaction pathway included CO formation via RWGS, which is the rate-determining step. In addition, this reaction exhibited structural sensitivity in which Ru catalysts with a lower loading weight were favorable for the formation of CO, regarded as the by-product in this system. These findings suggest that the smaller Ru-particle-supported (highly dispersed) catalysts are rather suitable for the RWGS reaction. Tuning selectivity for CO2 hydrogenation has been examined in earlier studies, particularly addressing the necessity of sufficient sites for CO hydrogenation on the active metal surface and modification of the morphology or the electronic state of the metal associated with the stability of intermediates of the reaction.22–26
In this work, we aimed to use an electric field for the selective RWGS reaction to achieve high activity at lower temperatures and to shed light on the reaction mechanism of catalytic CO2 activation in the electric field. The catalysts appropriate for this reaction were prepared via the synthesis of colloidal Ru nanoparticles using the chemical reduction method and deposition on the supports because the conventional impregnation method produces large particle size distributions unless the Ru loading weight is decreased drastically. After confirming the structure of the synthesized Ru nanoparticles through characterizations, these catalysts were shown to exhibit high RWGS selectivity (>90%) in the low-temperature region of less than 423 K with the electric field. Furthermore, we evaluated the role of the electric field in the catalytic RWGS reaction through a comparative study of the activity behavior with conventional heated catalysis.
Experimental
Catalyst preparation
The catalyst supports were prepared using a complex polymerization method that includes the use of citric acid and ethylene glycol. Following the addition of citric acid and ethylene glycol to purified water, appropriate metal precursors were mixed with this solution. All reagents were obtained from Kanto Chemical Co. Inc. The precursors are presented in Table S1 (ESI†). The solution was stirred at 343 K for 18 h and then heated with a hot stirrer at 673 K to evaporate the water. The obtained resin was calcined at 673 K for 2 h with pre-calcination and then at 1073 K for 10 h to remove the residual carbons completely.In an attempt to prepare highly dispersed Ru catalysts, Ru nanoparticles were synthesized via colloidal synthesis using the liquid-phase reduction by ethylene glycol as described in several reports,27–29 but with slight modifications. First, tris(acetylacetonato)ruthenium(III) (Ru(acac)3, Tanaka Holdings Co., Ltd) was dissolved in 100 mL of ethylene glycol, which served as a reducing agent and solvent. After the addition of 10 mL of 1 M NaOH (Kanto Chemical Co. Inc.) aqueous solution to promote reduction, it was heated to 473 K and stirred for 2 h. The obtained colloidal solution of Ru nanoparticles was separated by centrifugation, washed thoroughly with ethanol, and dried at room temperature overnight. These colloidal nanoparticles were then re-dispersed in purified water. Subsequently, after this colloidal solution was mixed with supports and stirred for 5 h at room temperature, the suspension was filtered and dried at 393 K overnight. Finally, the resulting powder was treated under a reducing atmosphere (50% H2 flow) at 723 K for 2 h.
Catalyst preparation using a typical impregnation method was conducted to compare the activity among catalysts with different Ru dispersions. First, the Ru precursor (Ru(acac)3) was dissolved in acetone. Then the support was added to this solution. After being stirred at room temperature for 2 h, this slurry was heated to evaporate the acetone. It was then dried at 393 K overnight. The obtained powder was reduced in the gas flow of H2 (50 SCCM) and Ar (50 SCCM) for 2 h.
Activity tests
Activity tests were conducted using a fixed bed flow-type reactor equipped with a quartz tube (8.0 mm o.d., 6.0 mm i.d.), as shown in Fig. S1 (ESI†). The catalyst (100 mg) was pre-reduced at 723 K for 2 h under a flow of H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ar = 1:3 (100 SCCM). When the electric field was imposed, two stainless steel electrodes were attached to the top and bottom of the catalyst bed. Then direct current was applied using a DC power supply. The response voltage was monitored using an oscilloscope (TDS 3052B; Tektronix Inc.). It was stable (0.2–0.4 kV), not forming discharge/plasma during the reaction. The overall actual catalyst bed temperature was measured using a thermocouple inserted into the bottom of the catalyst bed, the local catalyst surface temperature was measured using an NIR camera, and the atomic temperature was measured by evaluating the Debye–Waller (DW) factor (coherent neutron scattering caused by thermal motion) using EXAFS measurements,16 and the effect of Joule heat caused by the electric field imposed was negligible in the promotion of the catalytic activity, selectivity to products, in this case. After removal of the produced H2O using a cold-trap, the outlet gases including CO, CH4, and CO2 were analyzed using a GC-FID (GC-14B; Shimadzu Corp.) equipped with a Porapak N packed column and a methanizer (Ru/Al2O3 catalyst). The respective calculation formulae for CO2 conversion, CO selectivity and CO2 consumption rate are shown below.
:Ar = 1:3 (100 SCCM). When the electric field was imposed, two stainless steel electrodes were attached to the top and bottom of the catalyst bed. Then direct current was applied using a DC power supply. The response voltage was monitored using an oscilloscope (TDS 3052B; Tektronix Inc.). It was stable (0.2–0.4 kV), not forming discharge/plasma during the reaction. The overall actual catalyst bed temperature was measured using a thermocouple inserted into the bottom of the catalyst bed, the local catalyst surface temperature was measured using an NIR camera, and the atomic temperature was measured by evaluating the Debye–Waller (DW) factor (coherent neutron scattering caused by thermal motion) using EXAFS measurements,16 and the effect of Joule heat caused by the electric field imposed was negligible in the promotion of the catalytic activity, selectivity to products, in this case. After removal of the produced H2O using a cold-trap, the outlet gases including CO, CH4, and CO2 were analyzed using a GC-FID (GC-14B; Shimadzu Corp.) equipped with a Porapak N packed column and a methanizer (Ru/Al2O3 catalyst). The respective calculation formulae for CO2 conversion, CO selectivity and CO2 consumption rate are shown below.| CO2 conversion (%) = (FCO,out + FCH4,out)/FCO2,in × 100 | (2) |
| CO selectivity (%) = FCO,out/(FCO,out + FCH4,out) × 100 | (3) |
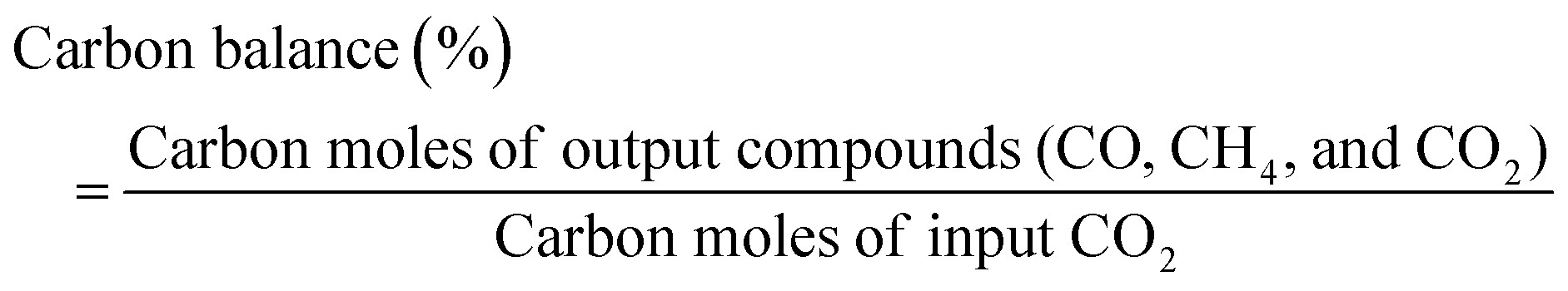 | (4) |
The reactant feed gas consisted of CO2, H2, and Ar (CO2:H2:Ar = 1:1:2, total flow rate: 100 SCCM) in the catalytic activity tests for comparison among catalysts synthesized with different methods or various supports, testing at various temperatures, and the evaluation of reaction stability with or without the electric field. In activity tests for evaluation of the H2/CO2 ratio in the feed gas on the RWGS activity, the reactant feed gases at various H2/CO2 ratios were arranged to the total flow rate of 50 SCCM and were diluted by Ar (50 SCCM). In activity tests for the evaluation of the contact time (W/F), the total flow rates changed to 20–200 SCCM with the CO2:H2:Ar ratio fixed at 1:1:2. The CO formation rate denoted as r is assumed according to the following equation using the partial pressures of CO2 and H2.
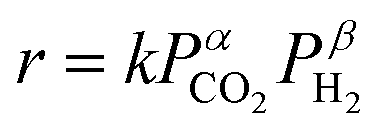 | (5) |
Characterization of catalysts
The actual Ru loading weight of the catalysts was quantified from inductively coupled plasma optical emission spectrometry (ICP-OES, 5100 ICP-OES; Agilent Technologies Inc.). Using about 50 mg of the catalyst, supported Ru was dissolved in sodium hypochlorite (Kanto Chemical Co. Inc.). The supports were then separated by filtration. The measurement was conducted using these solutions based on calibration curves recorded in Ru solutions ranging from approximately 0 to 5 ppm. Later, the residual supports were melted using lithium tetraborate (Kanto Chemical Co. Inc.) at 1273 K and dissolved in nitric acid. However, no Ru was detected in samples derived from residues.The catalyst surface morphology and the Ru particle size of the prepared catalysts were evaluated using a field emission transmission electron microscope (FE-TEM) equipped with an energy dispersive X-ray (EDX) spectrometer (JEM-2100F; JEOL Ltd). Each sample was dispersed ultrasonically in ethanol. Then the suspension was dropped onto a Cu micro-grid (NP-C15; Okenshoji Co. Ltd). The Ru particle size was found by calculating the mean values of more than 100 particles treated as spherical objects.
In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements were conducted using an FT-IR spectrophotometer (FT/IR-6200; Jasco Corp.) equipped with an MCT-M detector and a ZnSe window. For the application of the electric field, the DRIFTS cell was made of Teflon with pinholes to insert electrodes, as shown in Fig. S2 (ESI†). The sieved Ru/ZrTiO4 catalysts (about 120 mg) were used as a sample. Before measurements, the catalyst was reduced at 573 K in an H2 flow for 2 h. It was then purged in Ar flow for 1 h. First, background spectra were measured under Ar (40 SCCM) flow at 373 K (without the electric field) and 423 K (with the electric field). Then, after supplying the reactant gas (CO2:H2:Ar = 1:1:2, total flow rate: 40 SCCM), the measurement began at 373–673 K (without the electric field) and 423–573 K (with the electric field). When applying the electric field, 1 mA of direct current was imposed to minimize spectral distortion. All spectra from measurements were recorded with 4 cm−1 resolution and 100 scans.
To confirm the redox phenomena on the catalyst surface, H2-TPR (temperature programmed reduction) was performed in a dilute hydrogen atmosphere with and without the electric field. In the experiments, the catalyst was loaded into a reaction tube, then it was pre-treated at 523 K in a hydrogen atmosphere for 1 h. At this temperature, only Ru was thermally reduced. Then, after purging the gas, the temperature was controlled from 523 K to 773 K at a ramping rate of 5 K min−1 while observing the hydrogen consumption (m/z = 2) and the formation of water (m/z = 18) using a Q-Mass (OmniStar/ThermoStar GSD350; Pfeiffer Vacuum Co. Ltd) with/without the electric field. Also, the transient response of the electric field application was observed at a constant temperature of 423 K immediately after the field was applied.
Results and discussion
After initial screening tests, we have chosen 1.5 wt% Ru-supported catalysts for this purpose (see Table S2, ESI†). Also, we prepared two 1.5 wt% Ru-supported catalysts: one is prepared by the Ru-colloid supporting method and the other is an impregnation method. The comparison between these two catalysts in the electric field is presented in Table 1. The CO selectivity over the impregnated catalyst was only 24.7%, but the colloidal nanoparticle deposited catalyst showed high RWGS selectivity (95.9%) with a similar CO2 conversion in a kinetic region (11.2% for the colloidal Ru-catalyst and 9.7% for the impregnated Ru-catalyst, respectively). As a result of the TEM measurement (see Fig. S3, ESI†), the colloidal nanoparticle supported catalyst showed a fine dispersion of Ru, and the average Ru particle size was 2.37 nm; on the other hand, the impregnated catalyst showed a 7.97 nm particle size. The colloidal nanoparticle supported catalyst after the reaction with the electric field was also observed and the average Ru particle size was calculated to be 2.28 nm (Fig. S4, ESI†), which indicates that the Ru nanoparticles were not agglomerated in the reaction even with the application of the electric field. From these results, the highly dispersed Ru catalyst is appropriate for the selective RWGS reaction in the electric field. Therefore, for further investigation, we chose the colloidal nanoparticle supported Ru/ZrTiO4 catalyst (hereafter denoted as the Ru(col)/ZrTiO4 catalyst) as the best catalyst for RWGS in the electric field at a low temperature.| Catalysts | Average Ru particle size/nm | Catalyst-bed temperature/K | Response voltage/V | CO2 conversion/% | CO selectivity/% | CO formation rate per input power/mmol kJ−1 | TOF-sb/s−1 | TOF-pc/s−1 |
|---|---|---|---|---|---|---|---|---|
| CO2:H2:Ar = 1:1:2; 100 SCCM total flow rate; 100 mg catalyst weight; 5.0 mA imposed current.a The Ru loading weight was evaluated from the ICP-OES measurement.b TOF-s means the turnover frequency determined by the surface area of the metal particles.c TOF-p means the turnover frequency determined by the periphery of the metal particles. |
||||||||
| 1.5 wt% Ru/ZrTiO4a (Ru colloids supported) | 2.37 | 511 | 274 | 11.2 | 95.9 | 1.30 | 0.617 | 0.369 |
| 1.5 wt% Ru/ZrTiO4 (impregnation method) | 7.97 | 510 | 262 | 9.7 | 24.7 | 0.303 | 5.85 | 11.8 |
The electric field effects on the catalytic activity were investigated through activity tests conducted with and without the electric field at various temperatures (Fig. 1). Results indicated that the reaction proceeded even at a low temperature of 423 K, at which no activity was observed in the conventional RWGS reaction without the electric field, and that the selectivity was maintained higher than 90%. Furthermore, Arrhenius plots based on these tests (Fig. 2) showed that the apparent activation energy (6.74 kJ mol−1) with the electric field was much lower than that without the electric field (64.5 kJ mol−1), suggesting that the RWGS reaction with the electric field is promoted by a different mechanism from that of the conventional catalytic reaction without the electric field. The RWGS activity in the electric field was also evaluated by changing the imposed current. The results of the RWGS activity and the response voltage are shown in Fig. S5–S7, ESI.† In fact, the CO2 conversion increased in proportion to the imposed current (power), whereas the CO selectivity remained almost constant at more than 90%, irrespective of the imposed current.
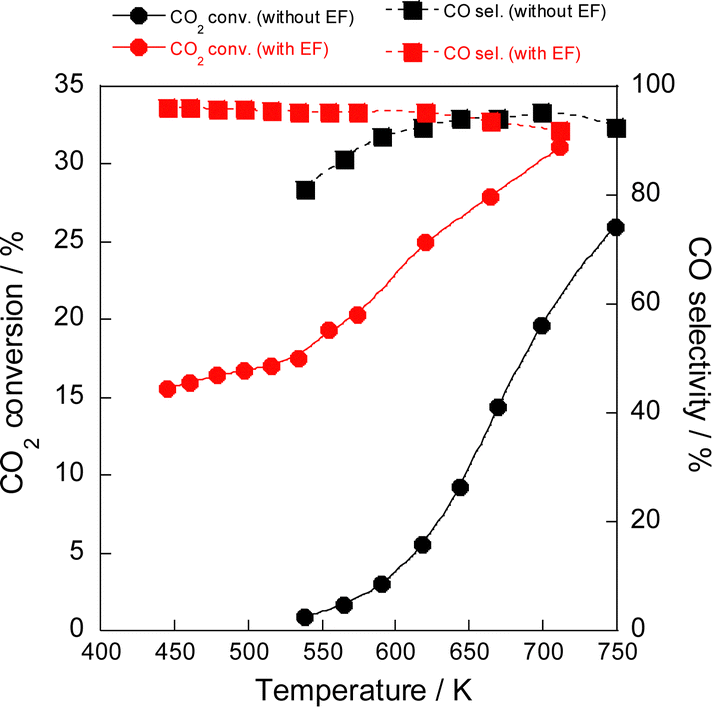 | ||
| Fig. 1 Temperature dependence of CO2 conversion and CO selectivity in the RWGS reaction with/without the electric field (EF) over the Ru(col)/ZrTiO4 catalyst; CO2:H2:Ar = 1:1:2; 100 SCCM total flow rate; 100 mg catalyst weight; 0 or 5.0 mA imposed current. | ||
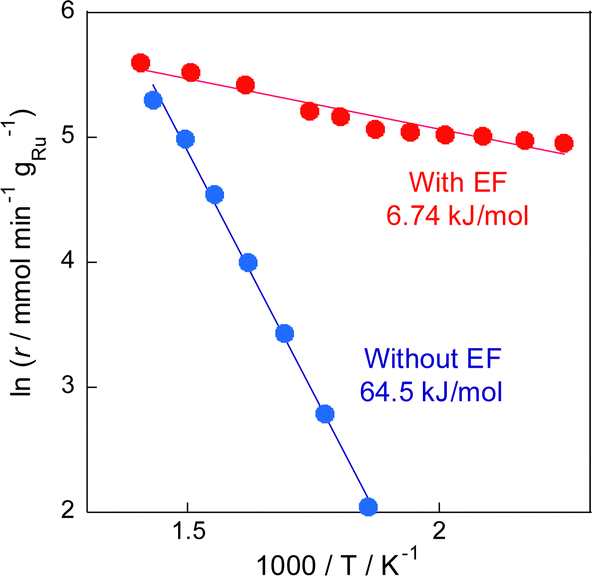 | ||
| Fig. 2 Arrhenius plots for the RWGS reaction over the Ru(col)/ZrTiO4 catalyst with and without the electric field; CO2:H2:Ar = 1:1:2; 100 SCCM total flow rate; 100 mg catalyst weight; 0 or 5.0 mA imposed current. | ||
Next, the reaction stability was evaluated with and without the electric field. The results are summarized in Fig. 3. The activity without the electric field decreased gradually over time: 56.6% decrease of the initial activity in 24 hours. On the other hand, the activity when subjected to an electric field was almost stable for the next 50 hours, although it dropped a little at the beginning. After reaction for 120 min in the case of the test with/without the electric field, the catalyst was purged with each component of the feed gas (Ar, H2, and CO2), then the activity was evaluated. As depicted in Fig. S8 – left in the ESI,† the RWGS activity without the electric field decreased and recovered by the greatest degree when purged with H2, which can be attributed to the removal of the CO2-derived adsorbates. On the other hand, it was found that the catalyst could easily recover from the initial slight decrease in activity when purged with any of the gases (Fig. S8 – right in the ESI†). We believe that this initial slight decrease in activity is due to weak adsorption, and that this weak adsorption of intermediates is the reason why the catalyst can maintain stable and high catalytic performance thereafter.
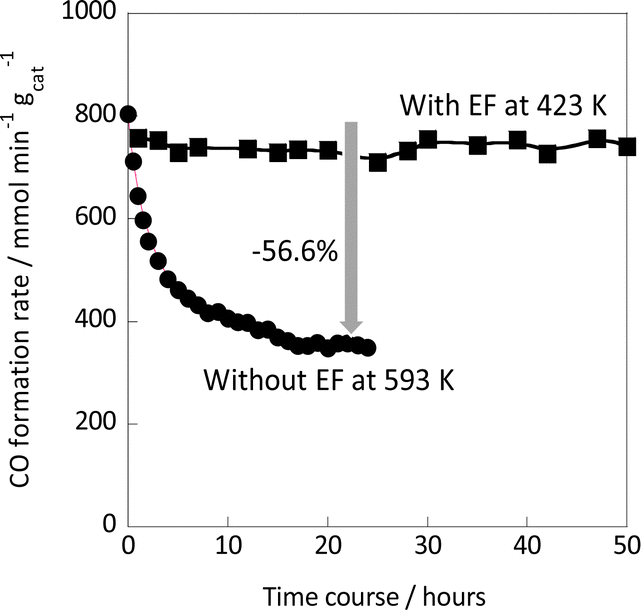 | ||
| Fig. 3 Catalytic stability during the RWGS reaction over the Ru(col)/ZrTiO4 catalyst with/without the electric field (5 mA); CO2:H2:Ar = 1:1:2; total flow rate: 100 SCCM; 100 mg catalyst weight. | ||
Next, the effects of the H2/CO2 ratio of the reactant gas on CO and CH4 formation rates were examined (Fig. 4). In the conventional RWGS reaction system, as the H2 concentration of the feedstock gas increased, CH4 formation proceeded dominantly and the CO2 consumption rate (CO formation rate + CH4 formation rate) decreased gradually with increasing CO2 concentration. However, in the system with the electric field, high RWGS selectivity was achieved even in the H2-rich condition. Moreover, the total CO2 consumption rate increased concomitantly in the CO2-rich condition, in contrast to the behavior observed without the electric field. These results revealed that the application of the electric field can suppress the side reaction (CO2 methanation).
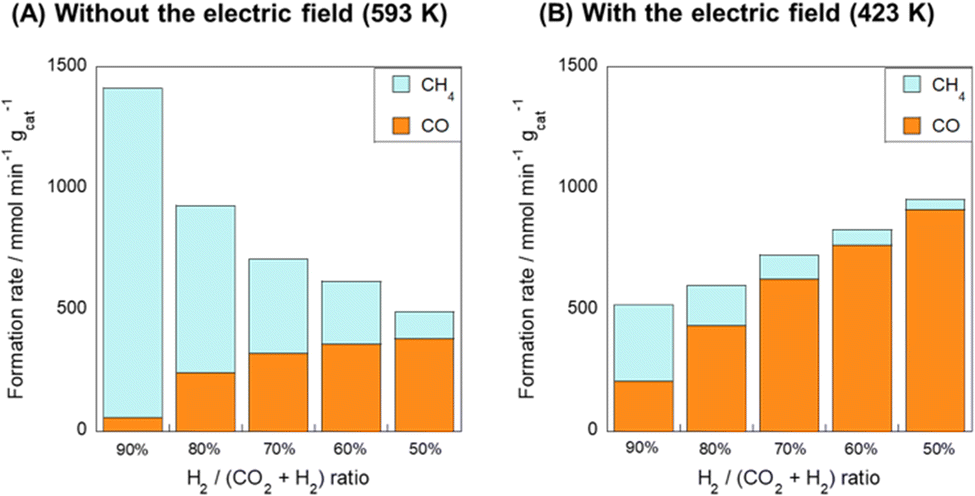 | ||
| Fig. 4 CO and CH4 formation rate over the Ru(col)/ZrTiO4 catalyst under various H2/CO2 ratios (A) without the electric field at 593 K and (B) with the electric field (5 mA) at 423 K; 100 SCCM total flow rate; 100 mg catalyst weight. | ||
To clarify the influence of CH4 formation, the influence of the contact time (W/F) on the catalytic activity was assessed (see Fig. S9, ESI†). In the thermal reaction system, the CO selectivity decreased considerably with increasing contact time. In the electric field, however, it is noteworthy that the CO selectivity remained higher than 90%, even in the high contact-time region. According to earlier reports,30,31 the dependence between the contact time and CO (or CH4) selectivity indicates that the reaction path of CO2 methanation goes through CO intermediates, i.e., the hydrogenation of CO on the Ru surface is the factor of the CH4 byproduct in RWGS.
The results presented above imply that the imposed electric field contributed to suppression of CO hydrogenation on the Ru surface. We can propose that this enhancement of selectivity is attributable to the surface protonics induced by the electric field, which facilitates hydrogen migration from the Ru surface to the support. A part of dissociatively adsorbed hydrogen is generally known to be transferred to the support. However, residual dissociated H atoms on the active metal can react with CO to form CH4. Considering another effect of the excess hydrogen on the Ru surface, it is claimed that electrons transfer from the hydrogen adsorbed onto Ru to the adsorbed CO via the metal. In this case, the Ru–C bond becomes stronger because of back-donation effects, making it more difficult to desorb.32 A recent study has revealed that CO2 hydrogenation using catalysts with high and low hydrogen-spillover ability dominates CO formation and CH4 formation, respectively.33 However, in the system with the electric field, as reported from experiments conducted with several systems of electric field promoted reactions, the induced surface proton conduction enables fast transfer of hydrogen on the catalyst surface.14–16 In our recent study, surface proton conduction over metal oxides has been observed even under an H2-dry atmosphere using electrochemical impedance spectroscopy.34 Indeed, it was also found that when applying the electric field to ammonia synthesis using Ru catalysts, hydrogen poisoning on the active metal surface, which is regarded as the main cause of the degradation of the activity in conventional thermal reactions, is eliminated.15 In general, hydrogen spillover accompanies electron transfer such as strong metal–support interaction (SMSI). Some room exists for the investigation of electric field effects, as discussed in the next section.
To elucidate the RWGS reaction mechanism in the electric field, in situ DRIFTS measurements were performed for comparison. First, DRIFTS spectra were measured without an electric field under the reaction atmosphere at 373–673 K. As shown in Fig. 5 and Fig. S10 in the ESI,† the IR feature developed after introduction of the reactant feed gas. Regarding carbonaceous species, CO adsorbed linearly (2012 cm−1) or bridged (1885 cm−1) on Ru was observed, whereas no band of gas-phase CO was detectable.35–37 Furthermore, the bands at 1357 cm−1 and 1601 cm−1 represent different vibrational modes of formates,38,39 which are most pronounced in the range of 1300–1700 cm−1 and which are found to be the main adsorbed species on the surface in this temperature region. The bands at 1669 cm−1 were assigned to bicarbonate species,33,40,41 but their intensity decreased considerably as the temperature increased from 373 K to 473 K. When the temperature exceeded 573 K, the formate band intensity decreased gradually, which corresponded to the increasing intensity of gaseous CO bands, indicating that the formate species are the main intermediates in the reaction.
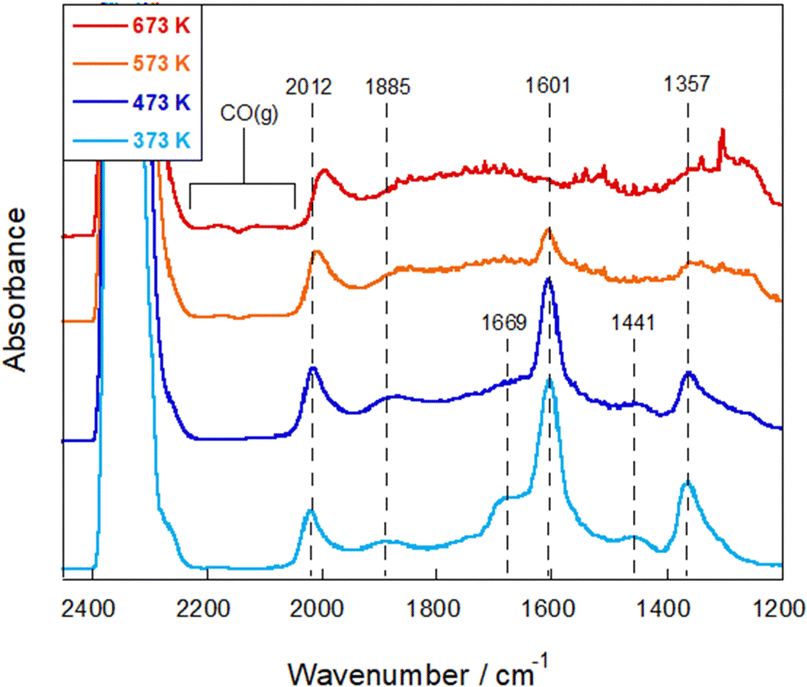 | ||
| Fig. 5
In situ DRIFTS spectra under the RWGS reaction atmosphere over the Ru(col)/ZrTiO4 catalyst without the electric field at 373–673 K; CO2:H2:Ar = 1:1:2; 40 SCCM total flow rate. | ||
In earlier reports on the conventional RWGS reaction, mainly two mechanisms have been proposed: the associative mechanism and the redox mechanism.42 In the associative mechanism, CO is produced via the decomposition of CO2-derived intermediates such as formate and carbonate species, which are formed in the reaction with the hydrogen species on the supports.43,44 In the redox mechanism, CO2 is dissociated into CO using oxygen vacancies in metal oxides. These vacancies are regenerated by H2 as a reducing agent.45,46 On a support with less reducibility, which makes it difficult for oxygen vacancies to be generated, formate species are known to form preferentially; the associative path becomes predominant.42 Moreover, formate species can stably present on the surface, which often accumulates on the surface and sometimes causes poisoning of the catalyst.47,48 Accordingly, one can infer that degradation of the activity over time without the electric field, as described above, can be attributed to the strong adsorption of formate species over the catalyst surface or CO species on the Ru surface. In earlier work, the activation energy barrier involving the formation and decomposition of formate species has been estimated to be about 50–70 kJ mol−1,42,49,50 which is close to our experimental value calculated with the Arrhenius plot.
In situ DRIFTS measurements were conducted by applying the electric field at various temperatures (Fig. 6). We were able to observe a marked change in adsorbed species after imposing the electric field: the formate peak intensity decreased drastically, whereas the gas CO band developed and some carbonaceous bands newly appeared. The bands at 1560 cm−1 and 1689 cm−1 were assigned, respectively, to bidentate carbonate51,52 and carboxylate species.53,54 Additionally, the DRIFTS spectrum when the imposed current was increased up to 10 mA, was measured (Fig. S11, ESI†) and the increase of gaseous CO species was observed corresponding to the evolution of the carboxylate and bidentate carbonate species. Considering the enhancement of the RWGS activity with the value of the imposed current (Fig. S5–S7, ESI†), this result suggests that carboxylate and carbonate species can be responsible for the CO formation in the electric field. These species were reported to form preferentially in the presence of oxygen vacancies and, especially, the carboxylate species was reported to be more reactive than formate species.22,42,55 In earlier studies of electrically assisted catalytic reactions, the electric field can activate surface lattice oxygens in metal oxides, which play an important role in the reaction with reactants at low temperatures.56 The findings obtained from this study also provide insight into the possibility that an electric field can promote the generation of oxygen vacancies and contribute to CO2 activation.
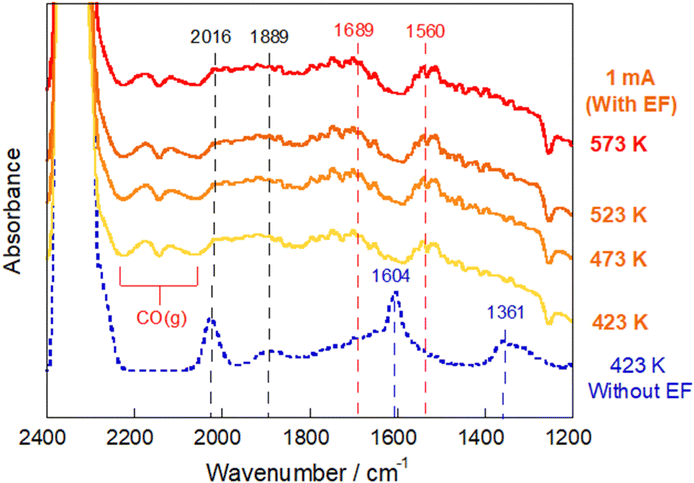 | ||
| Fig. 6
In situ DRIFTS spectra under the RWGS reaction atmosphere over the Ru(col)/ZrTiO4 catalyst recorded before/during application of the electric field (EF) at 423–573 K; CO2:H2:Ar = 1:1:2; 40 SCCM total flow rate. | ||
To ensure the reaction mechanism in the electric field, the ESR measurements were conducted using samples before and after applying the electric field (Fig. S12, ESI†). In both samples, sharp signals at g = 2.002 attributed to the surface-embedded Ti3+ sites57–60 were detected. In general, the formation of Vo (oxygen vacancy) requires the reduction of metal cations adjacent to the lattice oxygen (e.g. Ti4+ → Ti3+), so these sites are combined with the oxygen vacancy in the bulk. In addition, a broad signal assigned to surface-exposed Ti3+ sites was also observed only on catalyst treated in H2 flow with the electric field.56–61 According to a previous report,59 the presence of surface oxygen vacancies, which can play a role in the surface catalytic reaction, can be accompanied by the manifestation of this peak. Therefore, this result suggests that the electric field can efficiently introduce oxygen vacancies preferentially at the surface rather than the bulk under the reaction conditions through promoting the reduction of surface metal cations.
To confirm these phenomena, H2-TPR was performed in a dilute hydrogen atmosphere with and without the electric field. In the experiments, the catalyst was pre-reduced at 523 K in a hydrogen atmosphere, so only Ru was thermally reduced. Results are shown in Fig. S13 in the ESI;† for the TPR without the electric field, hydrogen was consumed and reacted with lattice oxygen to form water only above 623 K. On the other hand, as shown in Fig. S14 in the ESI,† for the TPR with the electric field, the results show that hydrogen is consumed and reacts with the lattice oxygen of the catalyst support to form water and lattice vacancies after the application of the electric field, even at a low temperature of 423 K. The surface oxygen consumption and vacancy formation corresponded to about five layers of surface oxygen in ZrTiO4. Thus, it was experimentally confirmed that the lattice oxygen reacts and forms lattice defects even at low temperatures in the electric field.
Discussion
Based on these results, we considered the reaction mechanism with and without an electric field in RWGS. Fig. 7 presents the proposed RWGS mechanism with/without the electric field over the Ru/ZrTiO4 catalyst. In the conventional thermal RWGS reaction (Fig. 7: left), CO2 initially reacts with the hydroxyl species on the ZrTiO4 surface, resulting in the formation of bicarbonate, which transforms into formate. This reaction path, however, proceeds quickly, considering the limited presence of bicarbonate species at low temperatures in DRIFTS spectra. Finally, these formate species decompose to CO at temperatures higher than 573 K. Generally, formate species at the metal–support interface can be involved in the series of reactions, whereas other species far from the support lack reactivity.49 On the other hand, in the system with the electric field (Fig. 7: right), CO2 adsorbs on the support in the form of carboxylate species using an oxygen vacancy. The vacancy could be formed mainly at the metal–support interface due to the modification of the electronic band structure (the Schottky junction).62 Subsequently, this intermediate dissociates into CO, followed by the release of water through the protonation of the surface lattice oxygen, leading to oxygen vacancy regeneration.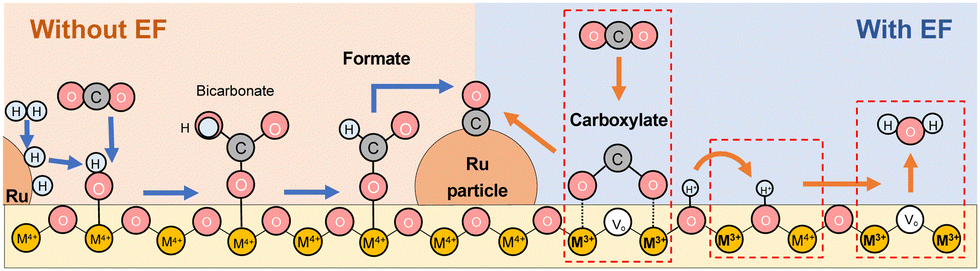 | ||
| Fig. 7 The schematic illustration of the proposed mechanism in the RWGS reaction with or without the electric field over the Ru(col)/ZrTiO4 catalyst. | ||
Specifically for the electron acceptance and donation in the redox system of this reaction, the proposed reaction pathway is found to comprise the following elementary reaction steps:
| H2 → 2H+ + 2e− | (6) |
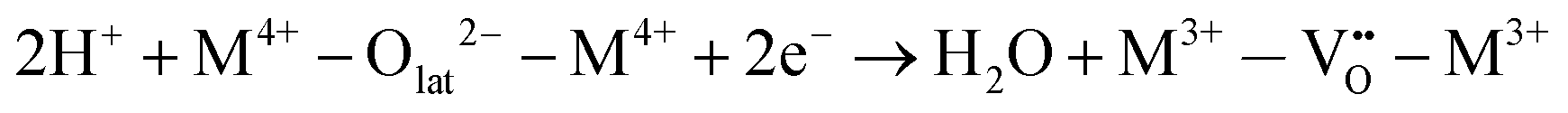 | (7) |
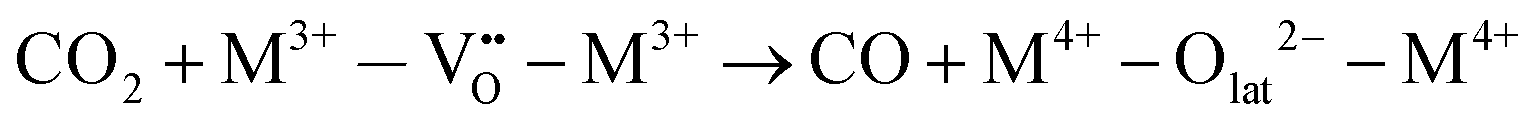 | (8) |
Many methods of applying an electric field to the reaction tube itself or to the catalyst layer have been reported recently,63–65 but these are promoted by heating by Joule heat, and the reaction mechanism is the same as that of an ordinary heated catalyst. On the other hand, in the case shown in this paper where a DC electric field is applied to a catalyst with a semiconductor as a support and a fine metal, the reaction mechanism is different from that of a catalyst heated as described above. Although Joule heat is generated to some extent by the application of the electric field, its contribution to the reaction activity, selectivity, and mechanism is very small. As for the effect of temperature, the overall catalyst layer temperature was measured using a thermocouple, the local catalyst surface temperature was measured using a NIR camera, and the atomic temperature was measured by evaluating the DW factor (coherent neutron scattering caused by thermal motion) using EXAFS measurements.
Therefore, we conclude that the electric field accelerates this charge transfer (6 & 7), which contributes to the achievement of high activity at low temperatures.
Conclusions
This study revealed that highly dispersed Ru catalysts exhibit high RWGS activity with the application of an electric field in the low-temperature region, where no activity was observed in the conventional heated catalytic RWGS reaction. In contrast to catalysts with larger Ru particles, the highly dispersed Ru catalysts showed high CO selectivity. Moreover, in the electric field, the catalyst maintained high CO selectivity under high H2 concentration or short contact time. This finding can be explained by surface protonics: fast hydrogen migration to the support restrains the side reaction of CO with residual hydrogen species on the Ru surface. Regarding the reaction mechanism, the RWGS reaction without the electric field proceeds through the formate-mediated path, followed by decomposition to CO. In the case of the reaction with the electric field, the main reaction path shifts to the redox mechanism, through which the generated oxygen vacancies can participate in CO2 activation at low temperatures. Consequently, the electric field promotes both the surface hydrogen migration and redox reaction using lattice oxygen vacancies, resulting in high RWGS activity and selectivity even at low temperatures and in a stoichiometric condition.Conflicts of interest
The authors have no conflict of interest.Acknowledgements
The authors thank Shinpei Enomoto (Kagami Memorial Research Institute for Materials Science and Technology, Waseda University) for great assistance in the FE-TEM operations and EDS measurements. The ESR measurement was conducted using the research equipment (Material Characterization Central Laboratory) shared in the MEXT Project for promoting public utilization of advanced research infrastructure (Program for supporting construction of core facilities) under Grant Number JPMXS0440500022. The TPR measurement (Fig. S13/S14) was conducted by Ms Ayaka Shigemoto, Waseda Univ. Some catalyst preparation and other experiments were assisted by Mr. Gao Yusuan, Waseda Univ.References
- International Energy Agency, Net Zero by 2050-A Roadmap for the Global Energy Sector, see https://www.iea.org/reports/net-zero-by-2050, accessed on 30 Oct/2022.
- P. Gabrielli, M. Gazzani and M. Mazzotti, Ind. Eng. Chem. Res., 2020, 59(15), 7033–7045 CrossRef CAS.
- W. Wang, S. Wang, X. Maa and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- P. Frontera, A. Macario, M. Ferraro and P. Antonucci, Catalyst, 2017, 7(2), 59 CrossRef.
- Y. A. Dazza and J. N. Kuhn, RSC Adv., 2016, 6, 49675–49691 RSC.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker and F. Abild-Pedersen, Science, 2012, 336(6038), 893–897 CrossRef CAS PubMed.
- S. Ba, Q. Shao, P. Wang, Q. Dai, X. Wang and X. Huang, J. Am. Chem. Soc., 2017, 139(20), 6827–6830 CrossRef PubMed.
- K. Zhao, X. Nie, H. Wang, S. Chen and X. Quan, Nat. Commun., 2020, 11, 2455 CrossRef CAS PubMed.
- R.-P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y.-G. Yao, Nat. Commun., 2019, 10, 5698 CrossRef CAS PubMed.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. R. Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- J. Ran, M. Jaroniec and S.-Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- J.-I. Makiura, T. Higo, Y. Kurosawa, K. Murakami, S. Ogo, H. Tsuneki, Y. Hashimoto, Y. Sato and Y. Sekine, Chem. Sci., 2021, 12, 2108–2113 RSC.
- R. Snoeckx and A. Bogaerts, Chem. Soc. Rev., 2017, 46, 5805–5863 RSC.
- R. Manabe, S. Okada, R. Inagaki, K. Oshima, S. Ogo and Y. Sekine, Sci. Rep., 2016, 6, 38007 CrossRef CAS PubMed.
- R. Manabe, H. Nakatsubo, A. Gondo, K. Murakami, S. Ogo, H. Tsuneki, M. Ikeda, A. Ishikawa, H. Nakai and Y. Sekine, Chem. Sci., 2017, 8, 5434–5439 RSC.
- Y. Hisai, Q. Ma, T. Qureishy, T. Watanabe, T. Higo, T. Norby and Y. Sekine, Chem. Commun., 2021, 57, 5737–5749 RSC.
- K. Oshima, T. Shinagawa, Y. Nogami, R. Manabe, S. Ogo and Y. Sekine, Catal. Today, 2014, 232, 27–32 CrossRef CAS.
- T. Yabe, K. Mitarai, K. Oshima, S. Ogo and Y. Sekine, Fuel Process. Technol., 2017, 158, 96–103 CrossRef CAS.
- T. Yabe, K. Yamada, K. Murakami, K. Toko, K. Ito, T. Higo, S. Ogo and Y. Sekine, ACS Sustainable Chem. Eng., 2019, 7(6), 5690–5697 CrossRef CAS.
- T. Yabe, K. Mitarai, K. Oshima, S. Ogo and Y. Sekine, J. CO2 Util., 2017, 20, 156–162 CrossRef CAS.
- K. Yamada, S. Ogo, R. Yamano, T. Higo and Y. Sekine, Chem. Lett., 2020, 49, 303–306 CrossRef CAS.
- S. Kattel, P. Liu and J. G. Chen, J. Am. Chem. Soc., 2017, 139(29), 9739–9754 CrossRef CAS PubMed.
- Y. Wang, L. R. Winter, J. G. Chen and B. Yan, Green Chem., 2021, 23, 249–267 RSC.
- J. C. Matusubu, V. N. Yang and P. Christopher, J. Am. Chem. Soc., 2015, 137(8), 3076–3084 CrossRef PubMed.
- J. H. Kwak, L. Kovarik and J. Szanyi, ACS Catal., 2013, 3(11), 2449–2455 CrossRef CAS.
- A. Aitbekova, L. Wu, C. J. Wrasman, A. Boubnov, A. S. Hoffman, E. D. Goodman, S. R. Bare and M. Cargnello, J. Am. Chem. Soc., 2018, 140(42), 13736–13745 CrossRef CAS PubMed.
- K. Kusada, H. Kobayashi, T. Yamamoto, S. Matsumura, N. Sumi, K. Sato, K. Nagaoka, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2013, 135, 5493–5496 CrossRef CAS PubMed.
- C. Bock, C. Paquet, M. Couillard, G. A. Botton and B. R. MacDougall, J. Am. Chem. Soc., 2004, 126, 8028–8037 CrossRef CAS PubMed.
- Y. Chen, K. Y. Liew and J. Li, Mater. Lett., 2008, 62, 1018–1021 CrossRef CAS.
- X. Chen, X. Su, H. Duan, B. Liang, Y. Huang and T. Zhang, Catal. Today, 2017, 281, 312–318 CrossRef CAS.
- D. J. Pettigrew and D. L. Trimm, Catal. Lett., 1994, 28, 313–319 CrossRef CAS.
- A. Erdöhelyi, M. Pásztor and F. Solymosi, J. Catal., 1986, 98, 166–177 CrossRef.
- X. Li, J. Lin, L. Li, Y. Huang, X. Pan, S. E. Collins, Y. Ren, Y. Su, L. Kang, X. Liu, Y. Zhou, H. Wang, A. Wang, B. Qiao, X. Wang and T. Zhang, Angew. Chem., Int. Ed., 2020, 59, 19983–19989 CrossRef CAS PubMed.
- Y. Hisai, K. Murakami, Y. Kamite, Q. Ma, E. Vøllestad, R. Manabe, T. Matsuda, S. Ogo, T. Norby and Y. Sekine, Chem. Commun., 2020, 56, 2699–2702 RSC.
- S. Ø. Stub, E. Vøllestad and T. Norby, J. Phys. Chem. C, 2017, 121(23), 12817–12825 CrossRef CAS.
- A. Takahashi, R. Inagaki, M. Torimoto, Y. Hisai, T. Matsuda, Q. Ma, J. G. Seo, T. Higo, H. Tsuneki, S. Ogo, T. Norby and Y. Sekine, RSC Adv., 2020, 10, 14487 RSC.
- S. Y. Chin, C. T. Williams and M. D. Amiridis, J. Phys. Chem. B, 2006, 110(2), 871–882 CrossRef CAS PubMed.
- Y. Wang, S. Kattel, W. Gao, K. Li, P. Liu, J. G. Chen and H. Wang, Nat. Commun., 2019, 10, 1166 CrossRef PubMed.
- N. H. M. D. Dostagir, R. Rattanawan, M. Gao, J. Ota, J. Hasegawa, K. Asakura, A. Fukouka and A. Shrotri, ACS Catal., 2021, 11, 9450–9461 CrossRef CAS.
- X. Wang, Y. Hong, H. Shi and J. Szanyi, J. Catal., 2016, 343, 185–195 CrossRef CAS.
- X. Wang, H. Shi, J. H. Kwak and J. Szanyi, ACS Catal., 2015, 5, 6337–6349 CrossRef CAS.
- L. F. Bobadilla, J. L. Santos, S. Ivanova, J. A. Odriozola and A. Urakawa, ACS Catal., 2018, 8(8), 7455–7467 CrossRef CAS.
- S. Choi, B.-I. Sang, J. Hong, K. J. Yoon, J.-W. Son, J.-H. Lee, B.-K. Kim and H. Kim, Sci. Rep., 2017, 7, 41207 CrossRef CAS PubMed.
- D. Heyl, U. Rodemerck and U. Bentrup, ACS Catal., 2016, 6(9), 6275–6284 CrossRef CAS.
- J. Wang, C.-Y. Liu, T. P. Senftle, J. Zhu, G. Zhang, X. Guo and C. Song, ACS Catal., 2020, 10(5), 3264–3273 CrossRef CAS.
- S. Mine, T. Yamaguchi, K. W. Ting, Z. Maeno, S. M. A. H. Siddiki, K. Oshima, S. Satokawa, K.-I. Shimizu and T. Toyao, Catal. Sci. Technol., 2021, 11, 4172–4180 RSC.
- A. Goguet, F. C. Meunier, D. Tibiletti, J. P. Breen and R. Burch, J. Phys. Chem. B, 2004, 108(52), 20240–20246 CrossRef CAS.
- E. T. Saw, U. Oemar, M. L. Ang, H. Kus and S. Kawi, Catal. Sci. Technol., 2016, 6, 5336–5349 RSC.
- F. C. Meunier, D. Reid, A. Goguet, S. Shekhtman, C. Hardacre, R. Burch, W. Deng and M. Flytzani-Stephanopoulos, J. Catal., 2007, 247, 277–287 CrossRef CAS.
- X. Wang, H. Shi and J. Szanyi, Nat. Commun., 2017, 8, 513 CrossRef PubMed.
- X. Jia, X. Zhang, N. Rui, X. Hu and C. Liu, Appl. Catal., B, 2019, 244, 159–169 CrossRef CAS.
- S. T. Korhonen, M. Calatayud and A. O. I. Krause, J. Phys. Chem. C, 2008, 112, 16096–16102 CrossRef CAS.
- L. Liu, C. Zhao and Y. Li, J. Phys. Chem. C, 2012, 116, 7904–7912 CrossRef CAS.
- H. He, P. Zapol and L. A. Curtiss, J. Phys. Chem. C, 2010, 114(49), 21474–21481 CrossRef CAS.
- J. A. Rodríguez, J. Evans, J. Graciani, J.-B. Park, P. Liu, J. Hrbek and J. F. Sanz, J. Phys. Chem. C, 2009, 113, 7364–7370 CrossRef.
- N. Nakano, M. Torimoto, H. Sampei, R. Yamashita, R. Yamano, K. Saegusa, A. Motomura, K. Nagakawa, H. Tsuneki, S. Ogo and Y. Sekine, RSC Adv., 2022, 12, 9036–9043 RSC.
- S. Mohajernia, P. Andryskova, G. Zoppellaro, S. Hejazi, S. Kment, R. Zboril, J. Schmidt and P. Schmuk, J. Mater. Chem. A, 2020, 8, 1432–1442 RSC.
- F. Amano, M. Nakata, A. Yamamoto and T. Tanaka, J. Phys. Chem. C, 2016, 120, 6467–6474 CrossRef CAS.
- Y. Yamazaki, K. Mori, Y. Kuwahara, H. Kobayashi and H. Yamashita, ACS Appl. Mater. Interfaces, 2021, 13, 48669–48678 CrossRef CAS PubMed.
- Y. Yamazaki, T. Toyonaga, N. Doshita, K. Mori, Y. Kuwahara, S. Yamazaki and H. Yamashita, ACS Appl. Mater. Interfaces, 2022, 14, 2291–2300 CrossRef CAS PubMed.
- E.-M. Köck, M. Kogler, T. Bielz, B. Klötzer and S. Penner, J. Phys. Chem. C, 2013, 117, 17666–17673 CrossRef PubMed.
- J. C. Frost, Nature, 1988, 334, 577–580 CrossRef CAS.
- S. T. Wismann, J. S. Engbæk, S. B. Vendelbo, F. B. Bendixen, W. L. Eriksen, K. Aasberg-Petersen, C. Frandsen, I. Chorkendorff and P. M. Mortensen, Science, 2019, 364(6442), 756–759 CrossRef CAS PubMed.
- Q. Dong, Y. Yao, S. Cheng, K. Alexopoulos, J. Gao, S. Srinivas, Y. Wang, Y. Pei, C. Zheng, A. H. Brozena, H. Zhao, X. Wang, H. E. Toraman, B. Yang, I. G. Kevrekidis, Y. Ju, D. G. Vlachos, D. Liu and L. Hu, Nature, 2022, 605, 470–476 CrossRef CAS PubMed.
- X. Mei, X. Zhu, Y. Zhang, Z. Zhang, Z. Zhong, Y. Xin and J. Zhang, Nat. Catal., 2021, 4, 1002–1011 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ey00004k |
| This journal is © The Royal Society of Chemistry 2023 |