
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly selective hydroxylation of gaseous alkanes at the terminal position by wild-type CYP153A33†
Yusaku
Kodama
a,
Shinya
Ariyasu
 *a,
Masayuki
Karasawa
a,
Yuichiro
Aiba
a and
Osami
Shoji
*ab
*a,
Masayuki
Karasawa
a,
Yuichiro
Aiba
a and
Osami
Shoji
*ab
aDepartment of Chemistry, Graduate School of Science, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8602, Japan. E-mail: ariyasu.shinya.v4@f.mail.nagoya-u.ac.jp; shoji.osami.w3@f.mail.nagoya-u.ac.jp
bCore Research for Evolutional Science and Technology (Japan), Science and Technology Agency, 5 Sanbancho, Chiyoda-ku, Tokyo, 102-0075, Japan
First published on 13th September 2023
Abstract
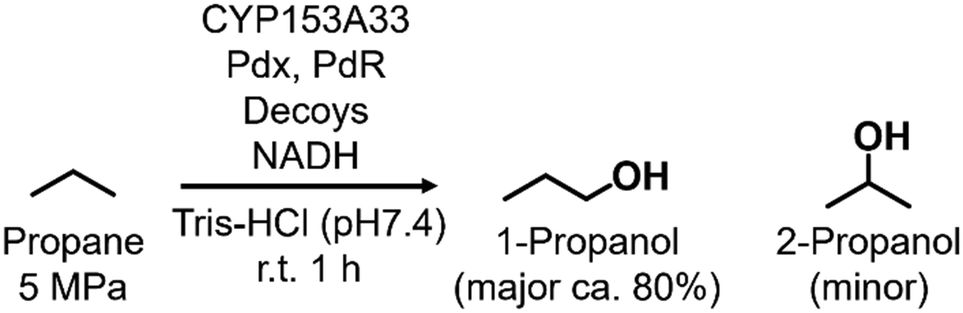
In the direct hydroxylation of C–H bonds by cytochrome P450s, their regioselectivities between primary and secondary carbons depend strongly on their C–H bond-dissociation energies. Thus, selective hydroxylation at the terminal position is a challenging task because of its low reactivity compared with internal positions. On the other hand, CYP153A33 can hydroxylate long fatty acids with high regioselectivity at the terminal position. For highly selective hydroxylation of gaseous alkanes at the terminal position, we herein combined CYP153A33 with substrate analog decoy molecules to alter the substrate specificity of the enzyme. Perfluoroacyl amino acids can effectively activate CYP153A33 and facilitate the regioselective hydroxylation of propane at the terminal position to afford 1-propanol (1-propanol/2-propanol = 80/20). In addition, we found that the use of CYP153A33 with decoy molecules also enables the hydroxylation of ethane and methane.
Introduction
Enabling unreactive C–H bonds to undergo transformations is a crucial chemical process, allowing for the creation of a wide range of functional molecules. Among these, the direct addition of hydroxyl groups to alkanes has garnered significant attention in both synthetic catalyst research and natural enzyme research due to the versatility of hydroxyl groups in various chemical conversions.1 The primary challenge encountered in the direct hydroxylation of hydrocarbons through C–H bond abstraction lies in the manipulation of regioselectivity. This is attributed to the fact that the effectiveness of hydroxylation at different C–H positions within substrates is greatly influenced by the bond-dissociation energy (BDE) of the respective C–H bonds. Taking for example propane, the BDE of primary C–H bonds (101 kcal mol−1) surpasses that of secondary C–H bonds (98 kcal mol−1). This discrepancy leads to elevated hydroxylation efficiencies at secondary carbon sites, giving rise to the formation of 2-propanol.2 Achieving selective hydroxylation of terminal C–H bonds in hydrocarbons remains a considerable hurdle in the realms of both synthetic catalysis and enzymatic chemistry.3In nature, cytochrome P450s (P450s), which contain heme as a reaction center, catalyze the direct hydroxylation of inert C–H bonds. This enzyme is essential for many different functions in living organisms, such as making important molecules and breaking down drugs.4 P450s generate a highly reactive oxidative species, known as ‘Compound I’, during their catalytic cycle through the reductive activation of molecular oxygen.5,6 Because they possess potent hydroxylation abilities and can effectively use molecular oxygen, P450s are gaining attention for their possible use as biocatalysts in alkane hydroxylation reactions.7,8 Among the abundant series of P450s, P450BM3 (CYP102A1), which is isolated from Priestia megaterium, can be regarded as a promising candidate for biocatalysts because P450BM3 exhibits one of the highest catalytic efficiencies in subterminal hydroxylation of long fatty acids (Fig. 1a).9 To apply P450BM3 for the hydroxylation of non-natural substrates, including small alkanes, a multitude of modified versions of P450BM3 have been engineered.10–20 These variants have been generated with the aim of modifying the pockets in which the substrates bind. Deviating from conventional mutagenesis methods, we have previously pioneered a distinctive approach to modify the substrate preferences of P450BM3. This innovative technique involves the utilization of synthetic substrate analogs, decoy molecules with lengths that are shorter than that of natural substrates, to create an artificial reaction space around the heme in P450BM3 (Fig. 1b).21–24 Within this reaction setup, referred to as a substrate-misrecognition system, the molecular configurations of decoy molecules wield significant influence over substrate preferences, stereoselective behaviors, and catalytic effectiveness.25 We have achieved extremely efficient hydroxylation of propane (2200 min−1) and ethane (83 min−1) by using optimal decoy molecules with the support of an original high-pressure reactor.26,27 While the hydroxylation of alkanes by P450BM3 has been subjected to extensive investigation, achieving proficient terminal hydroxylation of these compounds has remained elusive. For instance, our reaction system in wild-type P450BM3 shows high selectivity to the internal position of propane to afford 2-propanol (1-propanol/2-propanol = ca. 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :95) even in the presence of a wide variety of decoy molecules,26 possibly due to the larger size of the reaction space around the heme formed by decoy molecules than those by small alkanes such as propane.
:95) even in the presence of a wide variety of decoy molecules,26 possibly due to the larger size of the reaction space around the heme formed by decoy molecules than those by small alkanes such as propane.
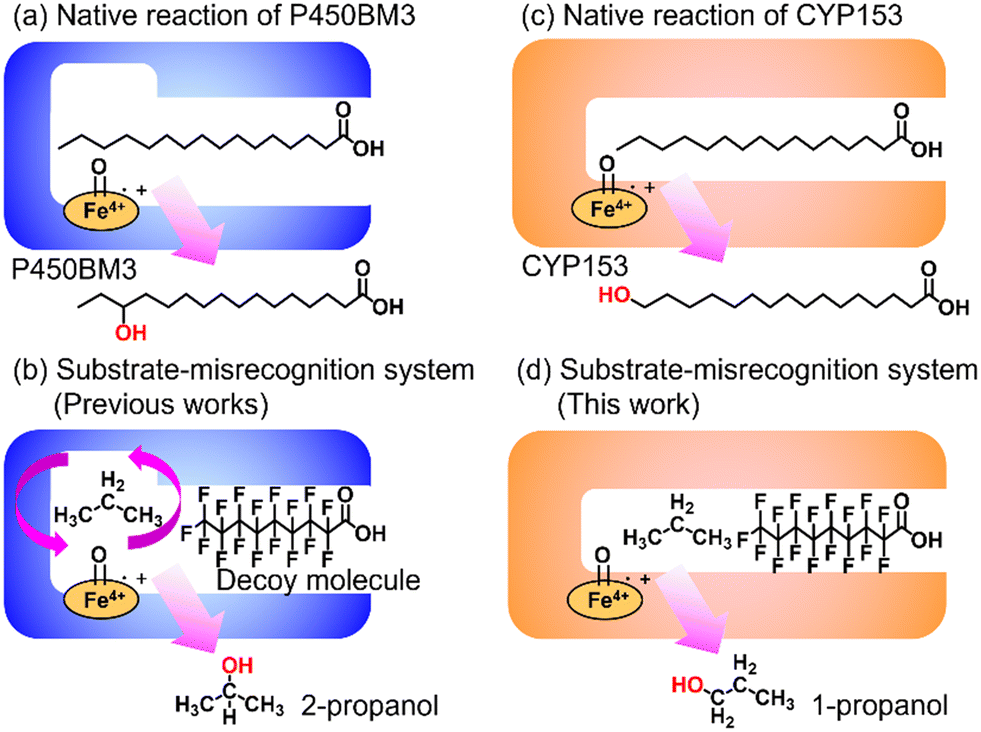 | ||
| Fig. 1 Schematic images of native reactions and substrate-misrecognition systems with P450BM3 and CYP153. (a) Native reaction of P450BM3. (b) Substrate-misrecognition system of P450BM3 for propane hydroxylation to afford 2-propanol. (c) Native reaction of CYP153. (d) Substrate-misrecognition system of CYP153 for propane hydroxylation to afford 1-propanol. | ||
In pursuit of achieving effective terminal hydroxylation in short alkanes such as propane, our attention turned to CYP153A33 derived from Marinobacter aquaeolei. This enzyme has the capacity to facilitate hydroxylation in fatty acids spanning a wide spectrum of chain lengths (C9–C20). Moreover, it demonstrates a pronounced preference for terminal C–H bonds, showcasing significant regioselectivity. CYP153A33 belongs to the CYP153 family, which is classified under bacterial class I P450 enzymes. These enzymes necessitate three key components: the P450 enzyme itself, along with two supplementary reductases. These components work collaboratively to achieve a sequential intermolecular electron transfer from NADH to the heme group.28 Founded on the crystal structures of CYP153A33, it harbors a tunnel-like, slender substrate pocket that is directed toward the heme region.29 Owing to this constrained pocket, the end portion of extended fatty acids can be precisely positioned near the heme moiety. This positional control substantially contributes to the remarkable preference of the enzyme for terminal sites in its catalytic reactions. While CYP153A33 has been documented for its ability to execute the terminal hydroxylation of n-butane, it struggles to engage proficiently with smaller, non-native substrates. This limitation is attributed to the specialized shape of its pocket, which is tailored primarily for fatty acids.30 Accomplishing the target hydroxylation of propane at the terminal site to produce 1-propanol remains an ongoing challenge.
In this study, we endeavored to overcome the regioselectivity influenced by BDE during the hydroxylation of hydrocarbons, with a specific focus on gaseous alkanes. To address this challenge utilizing P450 enzymes, it becomes imperative to resolve two issues simultaneously: enhancing the substrate specificity of P450s to accommodate small alkanes, and refining their terminal regioselectivity. We identified CYP153A33 as a promising hydroxylase for the terminal hydroxylation of gaseous alkanes, including propane. Given that CYP153A33 potentially possesses an optimal structure around the heme for terminal hydroxylation, our remaining task was to alter its substrate specificity to accommodate small alkanes (Fig. 1d). We implemented a substrate-misrecognition approach for CYP153A33 to accomplish the terminal hydroxylation of gaseous alkanes such as propane. CYP153A33 can efficiently respond to second-generation decoy molecules, especially PFC9-Gly, and induce hydroxylation from propane to 1-propanol with high regioselectivity to the terminal position.
Experimental
Hydroxylation of propane, n-butane, or ethane by CYP153A33 using decoy molecules: general procedure
A gas-saturated buffer solution (propane/oxygen: 80:20, v:v) containing CYP153A33, PdR, Pdx (final 1 μM, 5 μM, 10 μM) and a DMSO solution of decoy molecules (final 100 μM) with 20 mM Tris-HCl (pH 7.4) and 100 mM KCl was injected into a high-pressure reaction vessel. The reaction vessel was connected to an HPLC-based high-pressure reactor.26 After exchanging the gas in the system with N2, the substrate gas (propane, n-butane, or ethane; 0.5 MPa) was introduced into both the reaction vessel and 1 mL loop. Pressurization buffer (20 mM Tris-HCl pH 7.4 and 100 mM KCl) was then flowed to the reaction vessel by using an HPLC pump to pressurize the substrate gas in the reaction vessel. During pressurization, a buffer solution containing NADH (final 5 mM) was injected into the reaction vessel from the NADH loop. When the internal pressure reached the desired pressure (5 MPa), the valve of the reaction vessel was closed, and the reaction vessel was vortexed at room temperature for 1 h. After the enzymatic reaction, a sample of the reaction mixture (15 μL) was taken to determine the consumption of NADH by measuring the absorption at 340 nm. To the reaction mixture (200 μL) was added a solution of 3-pentanol in DMSO as an internal standard, and the mixture was extracted with CH3Cl (200 μL). The extracted organic phase was directly analyzed using a GCMS 2010SE (Shimazu, DI 2010) equipped with an Rtx-1 column (Restek Corp.).
Hydroxylation of 13C-labeled methane (13CH4) in a high-pressure reactor at 10 MPa
13C-labeled methane (13CH4) hydroxylation by CYP153A33 was performed based on a procedure for methane hydroxylation by P450BM3.31 A 13CH4-saturated buffer solution containing CYP153A33, PdR, Pdx (final 1 μM, 5 μM, 10 μM, respectively) and a DMSO solution of decoy molecules (final 100 μM) in 50 mM KPi buffer (pH 7.4) was injected into a high-pressure reaction vessel. Initially, the fluid lines of the reactor were purged with N2 gas, and then ethane gas was injected into both the reaction vessel and a line connected to the reaction vessel. Thereafter, to pressurize 13CH4 gas in the reaction vessel, a buffer solution (50 mM KPi buffer (pH 7.4)) was flowed into the line using an HPLC pump. Simultaneously, a buffer solution containing NADH (final 10 mM) was also added to the reaction vessel to start the reaction. When the pressure in the reaction vessel reached 10 MPa, the pressurization line was closed, and the reaction vessel was vortexed at room temperature for 1 h. The NADH consumption was estimated as follows: 20 μL of the reaction mixture following a 1 h reaction was diluted by a factor of 50 and the absorbance of NADH at 340 nm was measured. The concentration of NADH was calculated by using the reported molar extinction coefficient ε340 of 6220 M−1 cm−1. The amount of 13CH3OH produced was estimated with a GCMS QP2020 (Shimadzu) equipped with an Rtx-1 column (Restek Corp.) through concentrating 13CH3OH by SPME (PAL RTC system (CTC)). The SPME method was constructed through optimization based on a reported method for concentrating ethanol from water.32 The SPME method utilized an SPME Arrow Carbon WR/PDMS-120 μm fiber (CTC) that was cleaned at 250 °C before sample extraction and after sample desorption. The reaction mixture of 13CH4 hydroxylation (200 μL) in a 20 mL vial was heated at 50 °C for 30 min prior to a 10 min extraction by SPME performed by inserting the fiber into the headspace above the reaction solution in the vial. Samples were agitated during extraction with a Heatex stirrer (CTC). The adsorbed 13CH3OH on the fiber was desorbed after extraction for 1 min in the injection port at 240 °C.Results and discussion
CYP153A33 was expressed by following established protocols.29 A plasmid containing a chemically synthesized gene coding for the CYP153A33 sequence was purchased, and then amplified using polymerase chain reaction. The extracted gene was inserted into a template plasmid (pET28a(+)) and then transformed into Escherichia coli JM109 for the amplification of plasmids coding for CYP153A33. After purification of the plasmids and transformation into competent cells of E. coli Arctic Express (DE3), the overexpression of CYP153A33 was induced by the addition of isopropyl-β-D(−)-thiogalactopyranoside, and the desired CYP153A33 was purified by using anion-exchange and gel permeation columns. Because we mainly obtained CYP153A33 as inclusion bodies by expression using conventional E. coli strains such as BL21(DE3), E. coli Arctic Express (DE3) was used to improve the expression level of CYP153A33 in soluble fractions through slow expression at low temperatures. In contrast to self-sufficient P450s such as P450BM3, CYP153A33, which is classified as a class I P450, requires appropriate redox partners. Although the native partners of CYP153A33 are ferredoxin (Fdx) and ferredoxin reductase (FdR) from M. aquaeolei, it has been reported that putidaredoxin (Pdx) and putidaredoxin reductase (PdR) from Pseudomonas putida can also activate the CYP153A family.33 Pdx and PdR were separately prepared by conventional E. coli expression systems. Because their catalytic performances in class I P450s strongly depend on the efficiencies of electron transfers between P450s and two redox partners, we carried out the optimization of the ratio of these three components through hydroxylation of lauric acid without decoy molecules.34 Comparison of two series of CYP153A33/Pdx/PdR ratios, 1:10:5 and 1:60:1, respectively, shows that the former ratio with 3 μM CYP153A33 exhibited the highest hydroxylation efficiencies (Fig. S1†). Therefore, we used this ratio and concentration as optimal conditions for further investigation of the adaptation of decoy molecules.
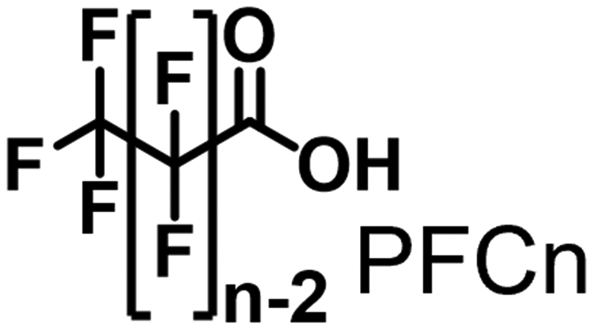
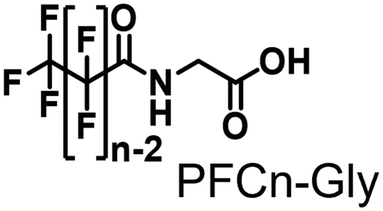
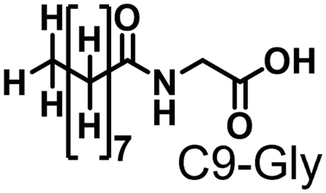
Propane hydroxylation was conducted in the presence of decoy molecules to confirm the applicability of CYP153A33 to a misrecognition system for alteration of its substrate specificity. In the case of P450BM3, third-generation decoy molecules that consist of amino acids or dipeptides modified with various N-substituents without fluorine atoms can serve as the most potent decoys in the hydroxylation of non-native substrates such as benzene.24 However, the unique shape of the substrate pocket of CYP153A33 prompted us to test its responsiveness toward various types of decoy molecules, including first- (simple perfluorocarboxylic acids, PFC9-12),21,22 second- (perfluorocarboxylic acid-modified amino acids, PFCn-Gly (n = 6–11) and PFC9-AA (AA = Ala, Val, Leu, Phe, or Trp)),23 and third-generation decoys (non-fluorinated carboxylic acid-modified amino acids such as C9-Gly)24 (Table 1). As expected, high terminal selectivity (ca. 80%) in propane hydroxylation by CYP153A33 was observed in the case of all tested decoy molecules, indicating that the unique structure of CYP153A33 plays a dominant role in achieving such a high terminal selectivity. The third-generation decoy molecules, such as C9-Gly, were not effective for the activation of CYP153A33, which is clearly different from P450BM3. CYP153A33 preferentially recognized decoy molecules bearing long perfluoroalkyl chains, which seems to be essential for the induction of its catalytic properties. Similar to P450BM3, second-generation decoy molecules can activate CYP153A33 more efficiently than first-generation decoy molecules.23 Although P450BM3 can be effectively activated by second-generation decoy molecules containing bulky amino acids such as Phe and Trp, the efficiency of propane hydroxylation by CYP153A33 was observed to increase linearly as the size of the amino acid side chain in the second-generation decoy molecules decreased (Fig. S2a and b†). In addition, unlike P450BM3, which can utilize decoy molecules of a wide range of lengths, CYP153A33 strongly prefers those with PFC9 and PFC10 chain lengths of the PFCn-Gly type (Fig. S2c and d†).26 Among the tested decoys, PFC9-Gly demonstrated the highest propane hydroxylation efficiency, achieving a turnover number (TTN) of 1163 in an enzymatic reaction over 1 h while maintaining high terminal selectivity.
|
|
||||||
|---|---|---|---|---|---|---|
| Decoy molecule | Propanol concentration (mM) | TTN (1 h) | 1-Propanol/2-propanol | NADH consumption (mM) | Coupling efficiency (%) | |
| No decoy | 0.246 ± 0.009 | 246 ± 9 | 77/23 | 1.2 ± 0.1 | 20 ± 1 | |
| PFC9 | 0.311 ± 0.039 | 311 ± 39 | 79/21 | 1.2 ± 0.2 | 25 ± 5 |
|
| PFC10 | 0.237 ± 0.058 | 237 ± 58 | 78/22 | 1.3 ± 0.2 | 18 ± 2 | |
| PFC11 | 0.173 ± 0.027 | 173 ± 27 | 77/23 | 1.1 ± 0.3 | 16 ± 4 | |
| PFC12 | 0.457 ± 0.042 | 457 ± 42 | 79/21 | 1.6 ± 0.1 | 28 ± 1 | |
| PFC6-Gly | 0.159 ± 0.013 | 159 ± 13 | 89/11 | 1.0 ± 0.1 | 16 ± 5 |
|
| PFC7-Gly | 0.276 ± 0.005 | 276 ± 5 | 79/21 | 1.1 ± 0.1 | 26 ± 2 | |
| PFC8-Gly | 0.380 ± 0.039 | 380 ± 39 | 79/21 | 1.6 ± 0.3 | 24 ± 3 | |
| PFC9-Gly | 1.163 ± 0.087 | 1163 ± 87 | 81/19 | 3.0 ± 0.3 | 39 ± 2 | |
| PFC10-Gly | 1.095 ± 0.047 | 1095 ± 47 | 81/19 | 2.3 ± 0.1 | 47 ± 4 | |
| PFC11-Gly | 0.491 ± 0.069 | 491 ± 69 | 80/20 | 1.6 ± 0.1 | 32 ± 3 | |
| C9-Gly | 0.286 | 286 | 80/20 | 1.1 | 25 |
|
| PFC9-Ala | 0.844 ± 0.112 | 844 ± 112 | 80/20 | 2.2 ± 0.1 | 38 ± 5 |
|
| PFC9-Val | 0.763 ± 0.032 | 763 ± 32 | 81/19 | 1.9 ± 0.1 | 39 ± 2 | |
| PFC9-Leu | 0.630 ± 0.069 | 630 ± 69 | 81/19 | 1.6 ± 0.2 | 39 ± 3 | |
| PFC9-Phe | 0.304 ± 0.052 | 304 ± 52 | 79/21 | 1.2 ± 0.1 | 25 ± 3 | |
| PFC9-Trp | 0.237 ± 0.150 | 237 ± 15 | 78/22 | 1.8 ± 0.2 | 14 ± 2 | |
To obtain insights into the binding modes of decoy molecules with CYP153A33, docking simulations using the crystal structure of CYP153A33 with hydroxylated lauric acid (PDB:5FYG) were performed (Fig. 2).35 The distances of the termini of PFC9-Gly or PFC9-Ala to heme were about 7 Å, which is longer than that of hydroxylated lauric acid, supporting the conclusion that these decoy molecules can form a vacant space around heme to bind non-native substrates such as propane. The docking structures around amino acid moieties of decoy molecules show that only small spaces might remain around the methyl group of the side chain of PFC9-Ala, which are consistent with the high availabilities of second-generation decoys having small side chains toward CYP153A33.
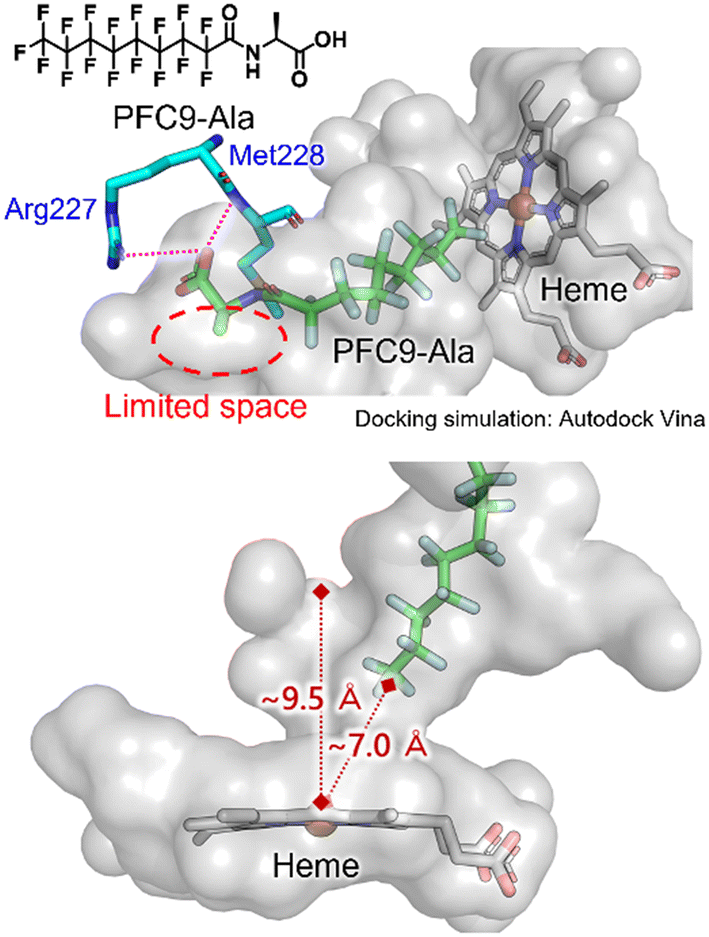 | ||
| Fig. 2 Docking simulation of CYP153A33 with PFC9-Ala calculated with AutoDock Vina. The crystal structure of the lauric acid-bound form of CYP153A33 (PDB: 5FYG) was used as a rigid receptor for the docking of PFC9-Ala. | ||
In time-course studies of propane hydroxylation with CYP153A33, both the production of propanol and the consumption of NADH slowed down after 1 h, potentially because of the inactivation of its redox partners (Fig. S3†). When comparing the regioselectivities and efficiencies of CYP153A33 and P450BM3 in propane hydroxylation for 1 h under the same reaction conditions, the regioselectivities for 1-propanol/2-propanol were the opposite. This indicates that the desired propanol isomers can be separately obtained by the choice of appropriate P450 enzymes (Fig. 3). Compared with those of P450BM3, which consumed almost all NADPH molecules (5 mM) within 10 min and showed extremely high turnover frequencies,26 the reaction rates of CYP153A33 were significantly slower. However, CYP153A33 displayed approximately half the total turnover number for 1 h, with around 60% consumption of NADH, indicating that CYP153A33 may be a promising counterpart to P450BM3 for regioselective propane hydroxylation.
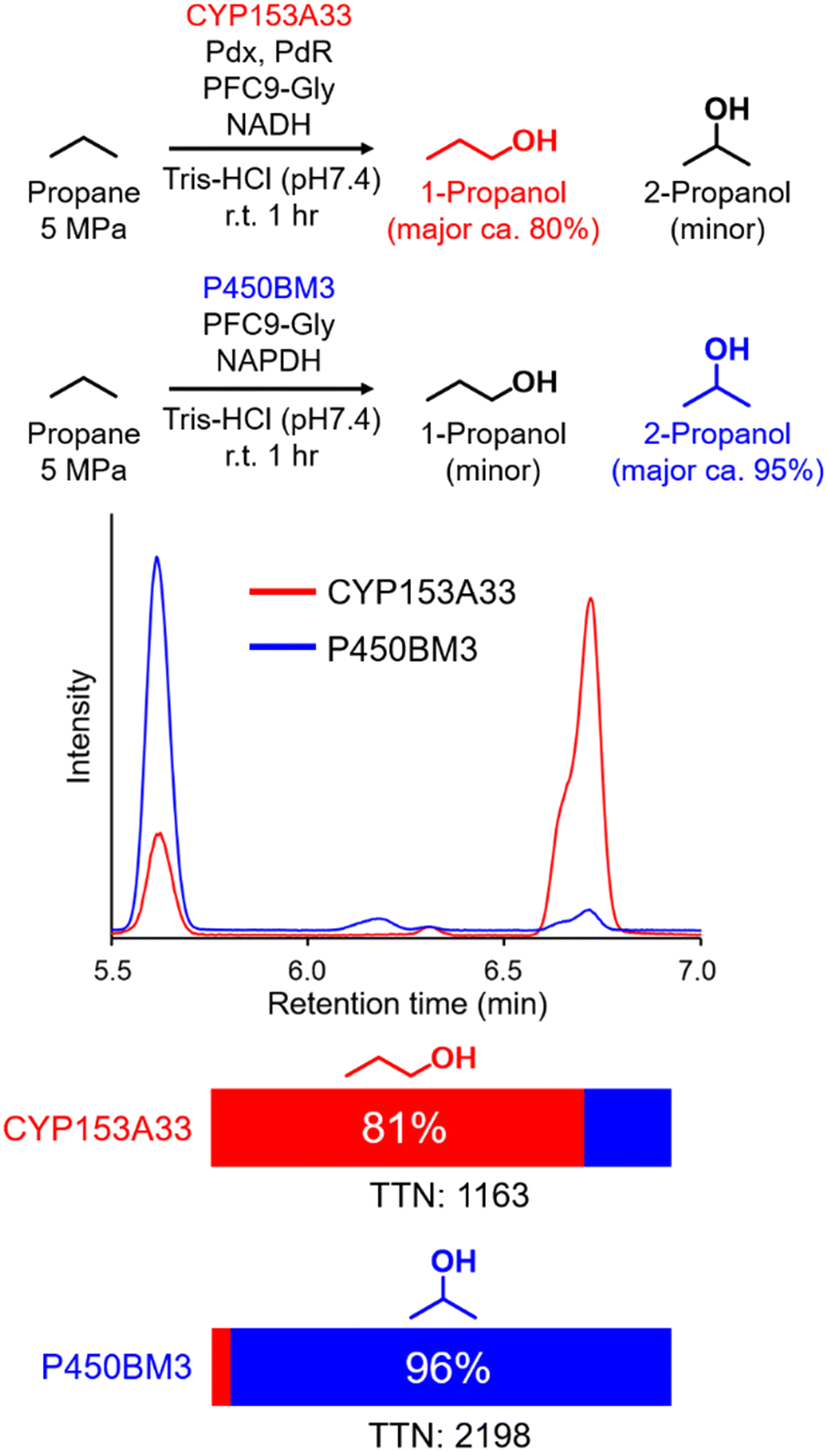 | ||
| Fig. 3 Propane hydroxylation with CYP153A33 or P450BM3 in the presence of PFC9-Gly under similar conditions. Reaction conditions for CYP153A33:CYP153A33 (1 μM), Pdx (10 μM), PdR (5 μM), propane (5 MPa), PFC9-Gly (100 μM), NADH (5 mM), Tris-HCl buffer (20 mM Tris-HCl, 100 mM KCl, pH 7.4), at room temperature for 1 h. Reaction conditions for P450BM3:P450BM3 (1 μM), propane (5 MPa), PFC9-Gly (100 μM), NADPH (5 mM), Tris-HCl buffer (20 mM Tris-HCl, 100 mM KCl, pH 7.4), at room temperature for 1 h. | ||
Finally, the applicability of the reaction system of CYP153A33 with decoy molecules was tested through the hydroxylation of n-butane, ethane, and methane (Fig. 4). As expected, high terminal selectivity was also observed in the hydroxylation of n-butane. As might be expected due to the low maximum pressure of n-butane for phase transition to a liquid, the TTN values in n-butane hydroxylation were lower than those in propane hydroxylation. In addition, CYP153A33 can also hydroxylate ethane in the presence of suitable decoy molecules, with a TTN of 301 for a 1 h reaction; the TTN values were comparable to those observed with P450BM3 (TTN of 280 for 10 min in the presence of PFC9-Phe). Finally, we tested direct methane hydroxylation with CYP153A33 with its redox partners in the presence of PFC9-Gly under 10 MPa 13CH4. After reaction for 1 h at room temperature, a peak corresponding to 13CH3OH was detected by GCMS analysis, and its TTN reached 0.82, which is lower than that by P450BM3 under similar conditions (TTN = 4.0 for 1 h).31 Given that the catalytic properties of CYP153A33 differ from those of P450BM3, there may be potential for further improvement of methane hydroxylation efficiencies through the precise optimization of reaction conditions.
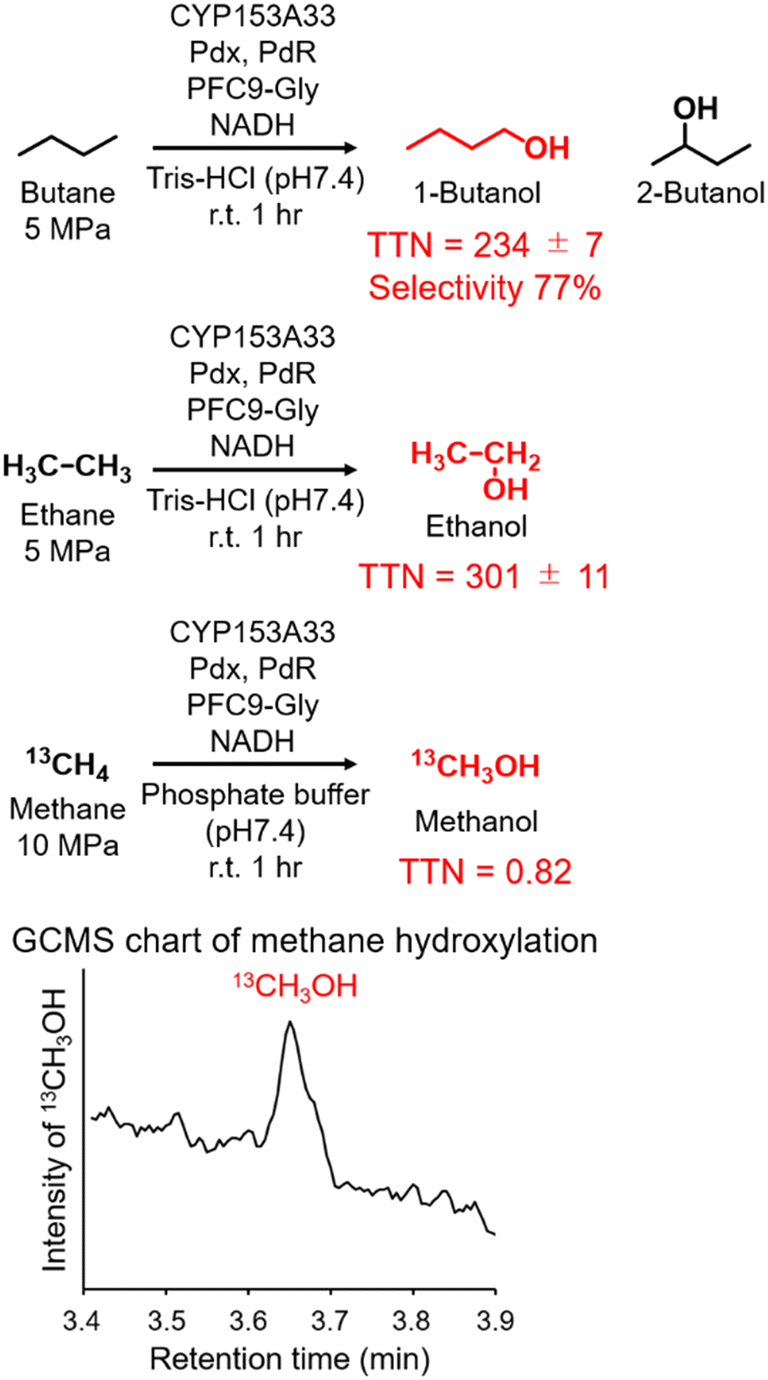 | ||
| Fig. 4 Hydroxylation of n-butane, ethane, and methane with CYP153A33 in the presence of PFC9-Gly. Reaction conditions: CYP153A33 (1 μM), Pdx (10 μM), PdR (5 μM), substrate gas (n-butane and ethane: 5 MPa, methane (13CH4): 10 MPa), PFC9-Gly (100 μM), NADH (5 mM), Tris-HCl buffer (20 mM Tris-HCl, 100 mM KCl, pH 7.4), at room temperature for 1 h. | ||
Conclusions
We have succeeded in the regioselective hydroxylation of propane and n-butane at their terminal positions by using a combination of CYP153A33, which originally has terminal selectivity in the hydroxylation of long fatty acids, and appropriate decoy molecules. Because P450BM3 can efficiently convert propane into 2-propanol, we have the option to choose the most suitable P450 enzyme: either CYP153A33 for 1-propanol or P450BM3 for 2-propanol. We have demonstrated that the combined system of CYP153A33 with decoy molecules exhibits hydroxylation reactivities with ethane and methane. While our research focused solely on the hydroxylation of small alkanes, it is expected that CYP153A33 may exhibit different selectivities for other substrates, such as small aromatic compounds. Furthermore, the terminal-selective reaction system of CYP153 could potentially be applied to whole-cell transformation through the development of cell-membrane-permeable decoy molecules. Research along these lines is ongoing in our lab.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JST CREST Grant Number JPMJCR15P3, Japan to O. S. This work was also supported by JSPS KAKENHI (JP22H05129 and JP21H04704 to O. S., JP21K05271 to S. A.), Japan. Gratitude goes to Dr. Joshua Kyle Stanfield for checking the English of this manuscript.Notes and references
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- M. Yuan, S. Abdellaoui, H. Chen, M. J. Kummer, C. A. Malapit, C. You and S. D. Minteer, Angew. Chem., Int. Ed., 2020, 59, 8969–8973 CrossRef CAS.
- W. Huttel and M. Muller, Nat. Prod. Rep., 2021, 38, 1011–1043 RSC.
- I. Schlichting, J. Berendzen, K. Chu, A. M. Stock, S. A. Maves, D. E. Benson, R. M. Sweet, D. Ringe, G. A. Petsko and S. G. Sliger, Science, 2000, 287, 1615–1622 CrossRef CAS.
- J. Rittle and M. T. Green, Science, 2010, 330, 933–937 CrossRef CAS.
- R. Fasan, ACS Catal., 2012, 2, 647–666 CrossRef CAS.
- S. Ariyasu, J. K. Stanfield, Y. Aiba and O. Shoji, Curr. Opin. Chem. Biol., 2020, 59, 155–163 CrossRef CAS PubMed.
- H. M. Girvan, K. R. Marshall, R. J. Lawson, D. Leys, M. G. Joyce, J. Clarkson, W. E. Smith, M. R. Cheesman and A. W. Munro, J. Biol. Chem., 2004, 279, 23274–23286 CrossRef CAS PubMed.
- R. Fasan, M. M. Chen, N. C. Crook and F. H. Arnold, Angew. Chem., Int. Ed., 2007, 46, 8414–8418 CrossRef CAS.
- Y. Li and L. L. Wong, Angew. Chem., Int. Ed., 2019, 58, 9551–9555 CrossRef CAS.
- H. Zhou, B. Wang, F. Wang, X. Yu, L. Ma, A. Li and M. T. Reetz, Angew. Chem., Int. Ed., 2019, 58, 764–768 CrossRef CAS PubMed.
- A. J. Ruff, A. Dennig, G. Wirtz, M. Blanusa and U. Schwaneberg, ACS Catal., 2012, 2, 2724–2728 CrossRef CAS.
- S. G. Bell, J. A. Stevenson, H. D. Boyd, S. Campbell, A. D. Riddle, E. L. Orton and L. L. Wong, Chem. Commun., 2002, 490–491 RSC.
- F. Xu, S. G. Bell, J. Lednik, A. Insley, Z. Rao and L. L. Wong, Angew. Chem., Int. Ed., 2005, 44, 4029–4032 CrossRef CAS.
- R. Fasan, Y. T. Meharenna, C. D. Snow, T. L. Poulos and F. H. Arnold, J. Mol. Biol., 2008, 383, 1069–1080 CrossRef CAS PubMed.
- D. J. Koch, M. M. Chen, J. B. van Beilen and F. H. Arnold, Appl. Environ. Microbiol., 2009, 75, 337–344 CrossRef CAS PubMed.
- C. J. Whitehouse, W. Yang, J. A. Yorke, B. C. Rowlatt, A. J. Strong, C. F. Blanford, S. G. Bell, M. Bartlam, L. L. Wong and Z. Rao, ChemBioChem, 2010, 11, 2549–2556 CrossRef CAS PubMed.
- F. E. Zilly, J. P. Acevedo, W. Augustyniak, A. Deege, U. W. Hausig and M. T. Reetz, Angew. Chem., Int. Ed., 2011, 50, 2720–2724 CrossRef CAS PubMed.
- M. M. Chen, C. D. Snow, C. L. Vizcarra, S. L. Mayo and F. H. Arnold, Protein Eng., Des. Sel., 2012, 25, 171–178 CrossRef CAS PubMed.
- N. Kawakami, O. Shoji and Y. Watanabe, Angew. Chem., Int. Ed., 2011, 50, 5315–5318 CrossRef CAS.
- O. Shoji, T. Kunimatsu, N. Kawakami and Y. Watanabe, Angew. Chem., Int. Ed., 2013, 52, 6606–6610 CrossRef CAS.
- Z. Cong, O. Shoji, C. Kasai, N. Kawakami, H. Sugimoto, Y. Shiro and Y. Watanabe, ACS Catal., 2014, 5, 150–156 CrossRef.
- O. Shoji, S. Yanagisawa, J. K. Stanfield, K. Suzuki, Z. Cong, H. Sugimoto, Y. Shiro and Y. Watanabe, Angew. Chem., Int. Ed., 2017, 56, 10324–10329 CrossRef CAS PubMed.
- K. Suzuki, J. K. Stanfield, O. Shoji, S. Yanagisawa, H. Sugimoto, Y. Shiro and Y. Watanabe, Catal. Sci. Technol., 2017, 7, 3332–3338 RSC.
- S. Ariyasu, Y. Kodama, C. Kasai, Z. Cong, J. K. Stanfield, Y. Aiba, Y. Watanabe and O. Shoji, ChemCatChem, 2019, 11, 4709–4714 CrossRef CAS.
- K. Yonemura, S. Ariyasu, J. K. Stanfield, K. Suzuki, H. Onoda, C. Kasai, H. Sugimoto, Y. Aiba, Y. Watanabe and O. Shoji, ACS Catal., 2020, 10, 9136–9144 CrossRef CAS.
- D. Scheps, S. H. Malca, H. Hoffmann, B. M. Nestl and B. Hauer, Org. Biomol. Chem., 2011, 9, 6727–6733 RSC.
- S. Honda Malca, D. Scheps, L. Kuhnel, E. Venegas-Venegas, A. Seifert, B. M. Nestl and B. Hauer, Chem. Commun., 2012, 48, 5115–5117 RSC.
- B. A. Nebel, D. Scheps, S. Honda Malca, B. M. Nestl, M. Breuer, H. G. Wagner, B. Breitscheidel, D. Kratz and B. Hauer, J. Biotechnol., 2014, 191, 86–92 CrossRef CAS PubMed.
- S. Ariyasu, K. Yonemura, C. Kasai, Y. Aiba, H. Onoda, Y. Shisaka, H. Sugimoto, T. Tosha, M. Kubo, T. Kamachi, K. Yoshizawa and O. Shoji, ACS Catal., 2023, 13, 8613–8623 CrossRef CAS.
- R. J. Kieber, A. L. Guy, J. A. Roebuck, A. L. Carroll, R. N. Mead, S. B. Jones, F. F. Giubbina, M. L. Campos, J. D. Willey and G. B. Avery, Anal. Chem., 2013, 85, 6095–6099 CrossRef CAS.
- E. Jung, B. G. Park, M. M. Ahsan, J. Kim, H. Yun, K. Y. Choi and B. G. Kim, Appl. Microbiol. Biotechnol., 2016, 100, 10375–10384 CrossRef CAS.
- S. Kochius, J. van Marwijk, A. Ebrecht, D. Opperman and M. Smit, Catalysts, 2018, 8, 531 CrossRef.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00752a |
| This journal is © The Royal Society of Chemistry 2023 |