
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral supramolecular polymers
Fátima
García
 ,
Rafael
Gómez
and
Luis
Sánchez
*
,
Rafael
Gómez
and
Luis
Sánchez
*
Departamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, Ciudad Universitaria s/n, 28040-Madrid, Spain. E-mail: lusamar@ucm.es
First published on 11th October 2023
Abstract
As an active branch within the field of supramolecular polymers, chiral supramolecular polymers (SPs) are an excellent benchmark to generate helical structures that can clarify the origin of homochirality in Nature or help determine new exciting functionalities of organic materials. Herein, we highlight the most utilized strategies to build up chiral SPs by using chiral monomeric units or external stimuli. Selected examples of transfer of asymmetry, in which the point or axial chirality contained by the monomeric units is efficiently transferred to the supramolecular scaffold yielding enantioenriched helical structures, will be presented. The importance of the thermodynamics and kinetics associated with those processes is stressed, especially the influence that parameters such as the helix reversal and mismatch penalties exert on the achievement of amplification of asymmetry in co-assembled systems will also be considered. Remarkable examples of breaking symmetry, in which chiral supramolecular polymers can be attained from achiral self-assembling units by applying external stimuli like stirring, solvent or light, are highlighted. Finally, the specific and promising applications of chiral supramolecular polymers are presented with recent relevant examples.
 From left to right: Rafael Gómez, Fátima García and Luis Sánchez | Fátima García received her PhD degree in 2013 from the Universidad Complutense de Madrid (ES) for research in the group of Prof. Luis Sánchez. After postdoctoral positions at the Wageningen University (NL) and CiQUS-Universidad de Santiago (ES) de Compostela, in 2021 she took up a position as an assistant professor in the Organic Chemistry Department at the Universidad Complutense de Madrid (ES). Her current research interests lie in supramolecular chemistry and catalysis. The research of Prof. Rafael Gómez Aspe is focused on the design, synthesis and characterization of organic molecules that self-assemble into supramolecular structures. Although helixes are the most intriguing to him, he is also interested in opto- and electroactive materials and in the chemistry of natural products. He has worked in different universities and in industry and has been part of the group “Amphiphilic molecules and supramolecular polymers” for many years now at UCM, where he combines research and educational responsibilities. Prof. Luis Sánchez is a Full Professor of Organic Chemistry at the Universidad Complutense de Madrid (UCM), Spain. In 2002, he was appointed as an associate professor at UCM and he was promoted to Full Professor in October, 2017. He is currently the principal investigator of the group “Amphiphilic molecules and supramolecular polymers” at UCM. He was awarded with the Prize to Novel Researchers of the RSEQ in 2003. He has directed and co-directed several Doctoral Theses, three of which have been awarded as the best theses in Chemistry at the Community of Madrid (2009, 2013 and 2018). In 2020, he was awarded with the Ignacio Ribas Medal by the Specialized Group on Organic Chemistry of the Spanish Royal Society of Chemistry (RSEQ). He is currently the vice Dean of research and doctorate at the Faculty of Chemical Sciences at UCM. |
Introduction
Chirality, from the Greek word kheir which means hand, is the geometric property of an object of not being superimposable on its mirror image (Fig. 1(a)). Chirality, and some related terms such as asymmetry, handedness, symmetry breaking, parity violation, or enantiorecognition are present in the natural world, which ranges from the nanoscale, involving particles like neutrinos or molecules, to the macroscale, including living systems and galaxies (Fig. 1(b)–(d)). The presence of chirality in such a vast range of scales makes this property an object of study for many disciplines in life sciences, like maths, physics, biology and, very especially, chemistry. Since the discovery of chirality and the first chiral resolution made by Pasteur by separating the enantiomeric crystals of the double sodium–ammonium salt of tartaric acid,1 an enormous number of studies about chirality have been developed. From a chemical point of view, the effects of thalidomide on children which were detected in the 1950s, prompted an investigation of chirality in the context of new enantiomeric and diastereomeric synthetic protocols,2 new chiral resolution processes3 or in the emergence of new technologies.4 However, one of the most challenging goals related to chirality is unravelling the origin of homochirality, that is, how does Nature chose D-sugars and L-amino acids endowed with a specific handedness as the chiral constitutive units of functional biomolecules like DNA, RNA, proteins or carbohydrates.5 It is generally accepted that in the prebiotic world, a minute enantiomeric excess (ee) emerged to initiate an initial asymmetry. Then, the propagation of this initial chiral differentiation should have been accompanied by an autocatalytic process that finally yielded families of compounds with high optical purity.6 The extensive work developed by Soai and co-workers on the chiral autocatalytic alkylation of pyrimidyl aldehydes with diisopropylzinc to afford chiral alcohols brought to light the concept of amplification of asymmetry.7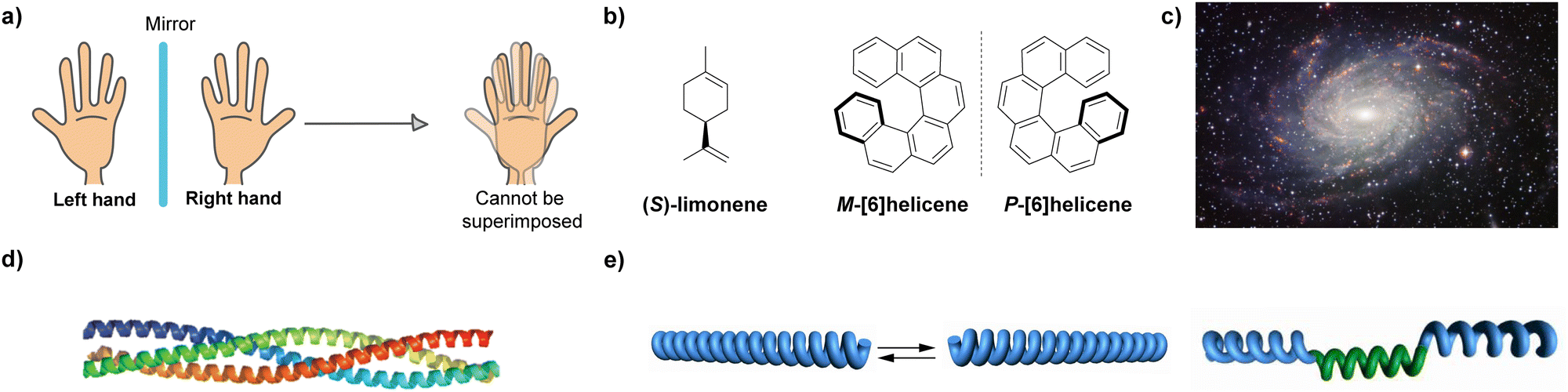 | ||
| Fig. 1 Paradigms of chiral entities: (a) Representation of the non-superimposable mirror images of right and left hands. (b) Chiral molecules endowed with point (limonene) and axial ([6]helicene) chirality. (c) Spiral arrangement of the galaxy NGD 6744. (d) Cartoon representation of a trimeric coiled coil. (Reproduced from Mol. BioSyst., 2015, 11, 1794 with permission from the Royal Society of Chemistry, copyright 2015.) (e) Schematic representation of helical covalent polymers as a mixture of right- and left-handed species (left) and a helical polymer with fragments of opposite turns (right). | ||
To unveil the origin of homochirality, and taking the helical nature of DNA or a number of proteins as examples, it is clear that the helix is a paradigm among the chiral entities called to play a crucial role in this area.8 The clockwise or anticlockwise twist of helical structures yields an intrinsic chiral entity whose asymmetry is propagated along the helical axis. On the other hand, it is well established that L-amino acids usually form diastereomeric, right-handed (P-type) α-helices. Building up the diastereomeric, left-handed (M-type) helices from such L-amino acids would be possible, but the energetic difference between these two diastereomeric structures is so high (around 21 kJ mol−1 in the simulated neutral α-helical Ala6) that interconversion is not feasible.9
The helix motif has been demonstrated to be of paramount relevance in the field of polymeric materials since the properties of the isotactic, atactic and syndiotactic versions of a certain polymer are drastically different.10 Many efforts have been made to achieve helical polymers in a non-racemic way,11 prompted by the applicability of helical polymers in a number of scientific and technological applications like ferroelectric liquid crystals, enantiomer separation or asymmetric catalysis.12 However, many helical polymers are still prepared in an achiral way that involves helical polymers with fragments of opposite turn or racemic mixtures of M- and P-type helical structures (Fig. 1(e)).13 Especially relevant were the seminal studies carried out by Green and co-workers on the amplification of asymmetry on polyisocyanides. Thus, copolymerization with minute amounts of chiral monomers in the synthesis of poly(n-hexylisocyanate) afforded homochiral polyisocyanides.14 Furthermore, these authors demonstrated that mixing unequal amounts of two enantiomeric polyisocyanides generated homochiral mixtures depending on the enantiomeric excess. The former effect was termed “sergeants-and-soldiers” (SaS) and the latter phenomenon was termed “majority-rules” (MR).15 Both experiments opened the door to the studies on amplification of asymmetry in some other covalent polymers like poly(phenylacetylene)s and polysilanes.16
In recent decades, and taking into account that the screw-sense of helical structures is strongly determined by non-covalent interactions among chiral entities, a new concept for helical polymers emerged from supramolecular chemistry. Thus, the seminal works by Nolte on phthalocyanines17 and Aida and co-workers on self-assembled porphyrins,18 complemented by later works by Fujiki revealing the importance of a substitution pattern with multiple hydrogen-bonding pendants at peripheral positions to attain a favourable supramolecular aggregation of porphirines,19 and the posterior report by Lehn and co-workers on the complementary H-bonding interactions between ditopic uracils and 2,6-diacetylamidopyridines,20 triggered the area of supramolecular polymers (SPs).21 In good analogy with a covalent polymer, a SP can be defined as a macromolecule in which the monomeric units are held together by non-covalent interactions. In the last few years, SPs have found applicability in a number of scientific areas like elastomers,22 biomedicine,23 optoelectronic devices,24 adhesives,25 enantioselective catalysis,26 photocatalysis27 or just to create beautiful and amazing objects.28 Despite the different approaches to classify SPs depending on the nature of the self-assembling units or on the non-covalent forces operating to build up the aggregated species, the most utilized feature is the supramolecular polymerization mechanism. The classification of SP by the mechanism of formation involves the evaluation of the Gibbs free energy of the polymer as a function of conversion (thermodynamical classification) and is based on the classification made by Carothers for covalent polymers in 1931.29 Two main supramolecular polymerization mechanisms have been described for SPs. The first of them is the isodesmic mechanism, comparable to the step-growth polymerization in covalent polymers, and the cooperative or nucleation-elongation mechanism that is comparable to the chain-growth covalent polymerization. In the former, the supramolecular polymerization is well-described by a single binding constant while in the latter, the supramolecular polymerization involves two different regimes: the nucleation, a thermodynamically unfavourable initial step that yields nuclei, and the elongation regime in which these nuclei grow up to form the final aggregated species. Therefore, a cooperative mechanism is defined by two different binding constants, the nucleation Kn and the elongation Ke, and the degree of cooperativity (σ) described as the quotient between Kn and Ke. Thus, the lower the value of σ is, the higher the cooperativity of the process.30 In cooperative SPs, generally multiple but strongly directional Pauling type C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/H–N hydrogen bonding is combined with co-facial π–π stacking,31 but several non-directional ‘attractive’ weak forces, including C–H/π, C–H/O–C, C–H/OC, C–H/N and other interactions also play a key role.32
O/H–N hydrogen bonding is combined with co-facial π–π stacking,31 but several non-directional ‘attractive’ weak forces, including C–H/π, C–H/O–C, C–H/OC, C–H/N and other interactions also play a key role.32
Importantly, the presence of elements of asymmetry in the monomeric units that upon self-assembly afford SPs has contributed to the development of this field generating chiral, aggregated species. In many examples of chiral SPs, the chiral monomeric species form helical structures in a controlled fashion. To the best of our knowledge, and despite the relevance of these aggregated species in different areas, the formation of chiral SPs has been reviewed in very few articles.33 We will outline herein the most relevant processes generally used to yield chiral SPs: transfer of asymmetry; amplification of asymmetry and symmetry breaking. We will highlight the main achievements in the field that are related not only to the structural requirements of the monomeric units to generate these chiral supramolecular entities but also to unravel relevant thermodynamic and kinetic insights useful to establish structure–function relationships. Furthermore, recent examples of chiral SPs in research areas like light emitting chiral materials or spin-filtering due to chirality-induced spin selectivity properties demonstrate the applicability of these supramolecular structures to develop stimuli-responsive circularly polarized light (CPL) and spintronic materials.34
Chiral supramolecular polymers
Transfer of asymmetry under thermodynamic control
Hierarchical self-assembly can be defined as an organizational process in which elementary building blocks are ordered into more complex secondary structures via non-covalent interactions. Therefore, the presence of asymmetry elements in molecules endowed with suitable functional groups can efficiently generate chiral SPs. The three sources of structural chirality, point, axial or planar chirality, either independently or in combination, can influence the formation of these chiral SPs by the so-called transfer of asymmetry process: the asymmetry outcome at the self-assembled level is influenced and/or determined, completely or partially, by the chirality of the corresponding building blocks. Thus, in the non-covalent interaction of two chiral molecules to form a supramolecular dimer, the spatial restrictions imposed by the inherent three-dimensional orientation of their constitutive parts can favour only one of the possible stereochemical configurations. When more molecules come together to form oligomers first, and then SPs, the preferential orientation of the dimer is transferred to the growing aggregate, thus resulting in the propagation of the preferred stereochemical outcome.35 These space-dependent interactions can also take place when non-identical molecules overcome the sterical restrictions imposed in the supramolecular aggregate, as in the SaS and MR experiments that will be discussed in the following sections.As stated in the Introduction section, the stereogenic centres in D-carbohydrates or L-amino acids are responsible for a vast majority of the examples of transfer of asymmetry found in natural supramolecular systems.5 Inspired by these natural supramolecular systems, synthetic chemists have used point chirality as a convenient source of chirality in the monomeric species to afford helical supramolecular structures.36 Among all the building blocks available, peptides and their derivatives emerge as excellent building blocks for the construction of chiral SPs;37 although in these review only some of them will be covered in which the asymmetry present in the final SPs is extensively studied. These structures are often investigated by circular dichroism (CD), a sensitive spectroscopic technique for detecting helical assemblies since they show preferential absorption of left or right-handed CPL.38
When molecules, oligomers, polymers, etc. self-assemble into supramolecules with a preferred-handedness helical conformation, these assemblies are optically active, as a result of the inherent asymmetry of the helix topology, even if the constitutive units lack asymmetric carbons or other stereogenic centres. However, in this case, an equal mixture of the two possible helical configurations M and P is obtained, and it makes no sense to talk about transfer of asymmetry. Apart from symmetry breaking, which will be also discussed in this review, for a real transfer of asymmetry to occur in helical SPs, it is necessary to use chiral monomeric species or a chiral external stimulus. The transfer of asymmetry from a chiral centre to a helical aggregate is a common process in self-assembly, either under thermodynamic or kinetic control, and depends on various factors such as the distance between the chiral centre and the assembly site, the strength of the non-covalent forces, the competition between chiral and achiral interactions or even the chiral nature of the solvent. Another factor that greatly determines the possibility of transfer of asymmetry is the type of supramolecular polymerization mechanism governing the self-assembly process. Most examples described in the literature occur in cooperative SPs, in which, very often, the participation of two or more different types of directional non-covalent forces determines the stability of the final helical polymer. In fact, for a long time it was considered that those SPs formed from chiral monomeric units following an isodesmic supramolecular polymerization mechanism were not able to experience transfer of asymmetry. Furthermore, most of the examples of transfer of asymmetry in SPs were investigated for thermodynamically controlled processes, i.e. one monomeric species generates a single helical aggregated species.
The paradigm of the formation of helical SP under thermodynamic control is probably the self-assembly of benzene-1,3,5-tricarboxamides (BTAs, compounds 1 and 2 in Fig. 2(a)). These C3-symmetric self-assembling units can form helical SPs by the intermolecular H-bonding of the three amide groups, rotated 40° out of the plane, and the π-stacking of the aromatic cores, separated by 3.5 Å, following a highly cooperative supramolecular polymerization mechanism (Fig. 2(b) and (c)).39 The rotation of the adjacent monomeric units within the columnar stack is ∼60°. The presence of stereocenters in the aliphatic side chains dictates the preferential P or M helicity of the resulting supramolecular aggregate, which is confirmed by the appearance of a strong Cotton effect in the CD spectrum (Fig. 2(d)).39 For a typical BTA substituted with chiral alkyl chains, such as (S)-1 and (R)-1 (Fig. 2(a)), the formation of the SP takes place in nonpolar solvents, usually methylcyclohexane (MCH), to yield helical aggregates with opposite helicity, as a direct consequence of the transfer of asymmetry from the peripheral chiral centres to the supramolecular level (Fig. 2(d)).40,41
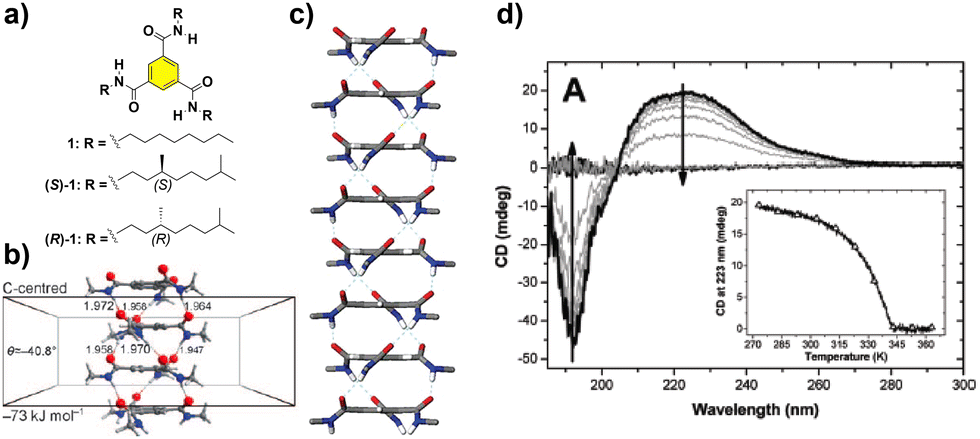 | ||
| Fig. 2 (a) Chemical structure of BTAs 1. (b) Optimized structure of a tetramer of the BTA showing the rotation angle of the amide functional groups and the distance between the aromatic cores in the ensemble (reproduced from ref. 39b with permission from the American Chemical Society, copyright 2017). (c) Right-handed helical structure proposed for the stacking of the BTAs, based on the X-ray structure reported by Lightfoot41 (reproduced from ref. 73 with permission from the American Chemical Society, copyright 1986). (d) CD spectra of (S)-1 (heptane, cT = 14 μM). The inset represents the molar ellipticity at λ = 223 nm as a function of temperature for the same solution (reproduced from ref. 44 with permission from Wiley-VCH, copyright 2022). | ||
Despite the structural simplicity of BTAs, the diversity of the different peripheral substituents utilized has allowed the preparation and investigation of a myriad of these derivatives. The extensive studies carried out by Prof. Meijer's research group with BTAs since the 90s have provided an increased understanding about the supramolecular behaviour of these structures and the subtle effects exerted by the interaction with solvents,42 traces of water43 or impurities44 which can provoke the appearance of complex polymerization pathways;45 the development of different synthetic approaches46 or determine their potential applications.47 All the progress on BTAs has paved the way for a better understanding of many practical and theoretical aspects related to supramolecular polymerization, which have been further extended to other supramolecular motifs.
Our research group has investigated the self-assembling features of C3-symmetric tricarboxamides closely related to BTAs. One of these C3-symmetric tricarboxamides are 1,3,5-triphenylbenzenetricarboxamides (TPBAs, compounds 2 in Fig. 3(a)).48 Similarly to BTAs, the self-assembly of these derivatives proceeds through the formation of a triple array of H-bonds between the amide groups and π-stacking of the aromatic cores and generally follows a cooperative mechanism. In this case, and to maximize the H-bonding of the amide functional groups and the π-stacking interactions between the adjacent aromatic moieties, BTAs are rotated out of plane ca. 31° yielding enantioenriched helical stacks when the asymmetry of the chiral chains is transferred to the aggregate. In helical aggregates formed by TPBAs, the rotation of the adjacent monomeric units is 26° (Fig. 3(c)). Indeed, CD spectra evidenced the formation of right-handed helices from (S)-2 and left-handed helices from (R)-2 (Fig. 3(e)). Surprisingly, the non-aggregated monomers also showed mirror-like CD spectra, what are explained in terms of a restricted rotation of the benzamide groups that afforded a propeller-like configuration and the generation of M- and P-type atropisomers (Fig. 3(b)). This atropisomerism is cancelled upon the formation of the helical, columnar SPs since the supramolecular polymerization of compound 2 imposes the planarization of the triphenylbenzene moiety (Fig. 3(c)).
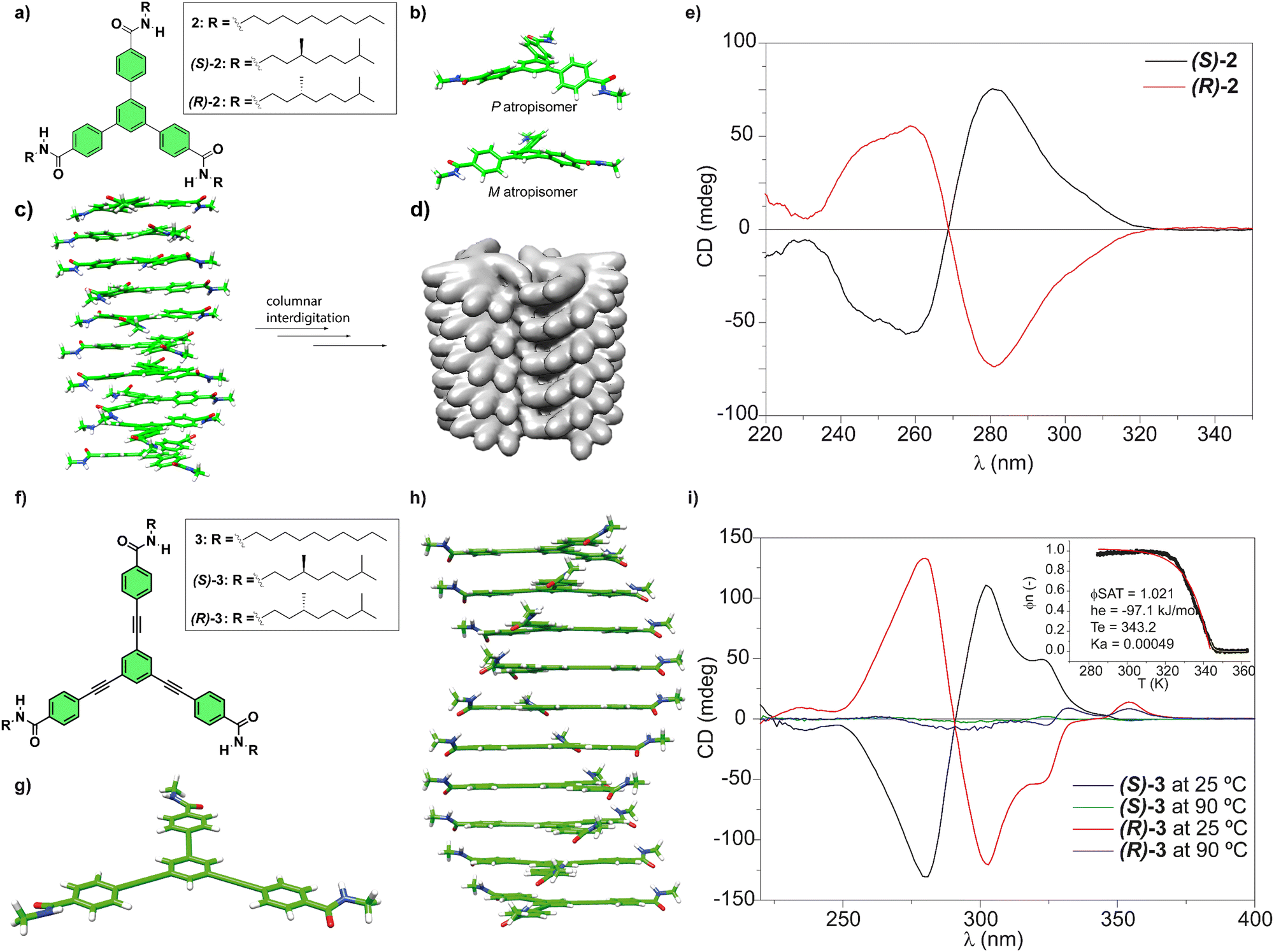 | ||
Fig. 3 (a) Chemical structure of TPBAs 2. (b) Optimized propeller-shaped structure of the monomeric units of 2. (c) Optimized structure of a P-type helical, columnar decamer formed from TPBAs 2. (d) Illustration of a bundle of two helices of achiral 2 formed by the interdigitation of the achiral side chains (reproduced from ref. 48 with permission from Wiley-VCH, copyright 2018). (e) Experimental CD spectra of chiral (S)-2 and (R)-2 (MCH/dichloroethane 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, 10 μM, 298 K) (reproduced from ref. 48 with permission from Wiley-VCH, copyright 2018). (f) Chemical structure of OPETAs 3. (g) Optimized structure of the planar monomeric OPETAs. (h) Optimized structure of a P-type helical decamer formed from OPETAs 3. (i) Experimental CD spectra of (S)-3 and (R)-3 in MCH at different temperatures. The inset shows the non-sigmoidal variation of the degree of aggregation (α) versus the temperature, which demonstrates the cooperative character of the supramolecular polymerization of OPETAs 3 with the corresponding fitting (reproduced from ref. 49a with permission from Wiley-VCH, copyright 2011). :5, 10 μM, 298 K) (reproduced from ref. 48 with permission from Wiley-VCH, copyright 2018). (f) Chemical structure of OPETAs 3. (g) Optimized structure of the planar monomeric OPETAs. (h) Optimized structure of a P-type helical decamer formed from OPETAs 3. (i) Experimental CD spectra of (S)-3 and (R)-3 in MCH at different temperatures. The inset shows the non-sigmoidal variation of the degree of aggregation (α) versus the temperature, which demonstrates the cooperative character of the supramolecular polymerization of OPETAs 3 with the corresponding fitting (reproduced from ref. 49a with permission from Wiley-VCH, copyright 2011). | ||
Interestingly, the branched chiral nature of the peripheral chain prompted not only the transfer of asymmetry from the molecular to the supramolecular level but also determines higher levels of organization. Thus, whereas the linear chains of the achiral TPBA 2 allowed the columnar stacks to efficiently interdigitate to form bundles of fibres (Fig. 3(e)), ramification of the chains in compounds (S)-2 and (R)-2 prevents such helix-to-helix interdigitation and no tertiary level of organization was attained, evidencing how subtle variations alters the supramolecular polymerization.48
To further investigate the effect of increasing the aromatic core size on the self-assembly of discotic chiral tricarboxamides, oligo(phenylene-ethynylene)tricarboxamides (OPETAs) 3 have also been synthesized (Fig. 3(f)).49 OPETAs exhibit a larger, planar conjugated central core compared to BTAs 1 or TPBAs 2 and also lack the atropisomerism found in the latter (Fig. 3(g)). As is common in discotic C3-symmetry tricarboxamides, supramolecular polymerization in OPETAs takes place in a cooperative fashion by the formation of a triple array of H-bonds between the amide functional groups concomitant to the π-stacking of the aromatic moieties. In these C3-symmetric scaffolds, the rotation angle of the amides, to maximize the H-bonding interactions between them and the π-stacking of the aromatic cores, is 34.9°. Furthermore, the adjacent monomers in the columnar stack are rotated 18° (Fig. 3(h)). Similarly to chiral TPBAs 2, the (S) peripheral chiral chains in (S)-3 yielded enantioenriched P-type helices and the (R) configuration of the chiral centres in the peripheral chains affords helical structures with an M-type handedness in (R)-3 (Fig. 3(i)). Our research group also synthesized a series of TPBAs and OPETAs in which a variable number of chiral side chains are attached to the central aromatic core. In all cases, the presence of only one stereogenic centre per monomeric unit is sufficient to bias the right- or left-handedness of the resulting helical aggregated species.50
A vast number of chiral SPs are obtained by the efficient transfer of asymmetry from peripheral stereogenic centres (point chirality) to the central aromatic backbones. However, axial chirality, and more specifically, atropisomers in which a contorted aromatic backbone is responsible for the source of asymmetry have also been utilized as self-assembling units to construct chiral SPs. In 2012, Würther and co-workers reported on the formation of J-type helical aggregates formed by the head-to-tail supramolecular interaction of a contorted perylenediimide (PDI) in its enantioenriched form. The cooperative supramolecular polymerization of this contorted enantioenriched PDI-based atropisomer yielded rope-like fibrillar structures whose helicity was visualized by atomic force microscopy (AFM).51
However, an intriguing question arose from this study: in the formation of a chiral SP, which element of asymmetry would predominate, point or axial chirality? To shed light in this challenging issue, we prepared a series of bis(N-annulated perylenecarboxamides) endowed with achiral and chiral side chains (compounds 4 in Fig. 4(a)).
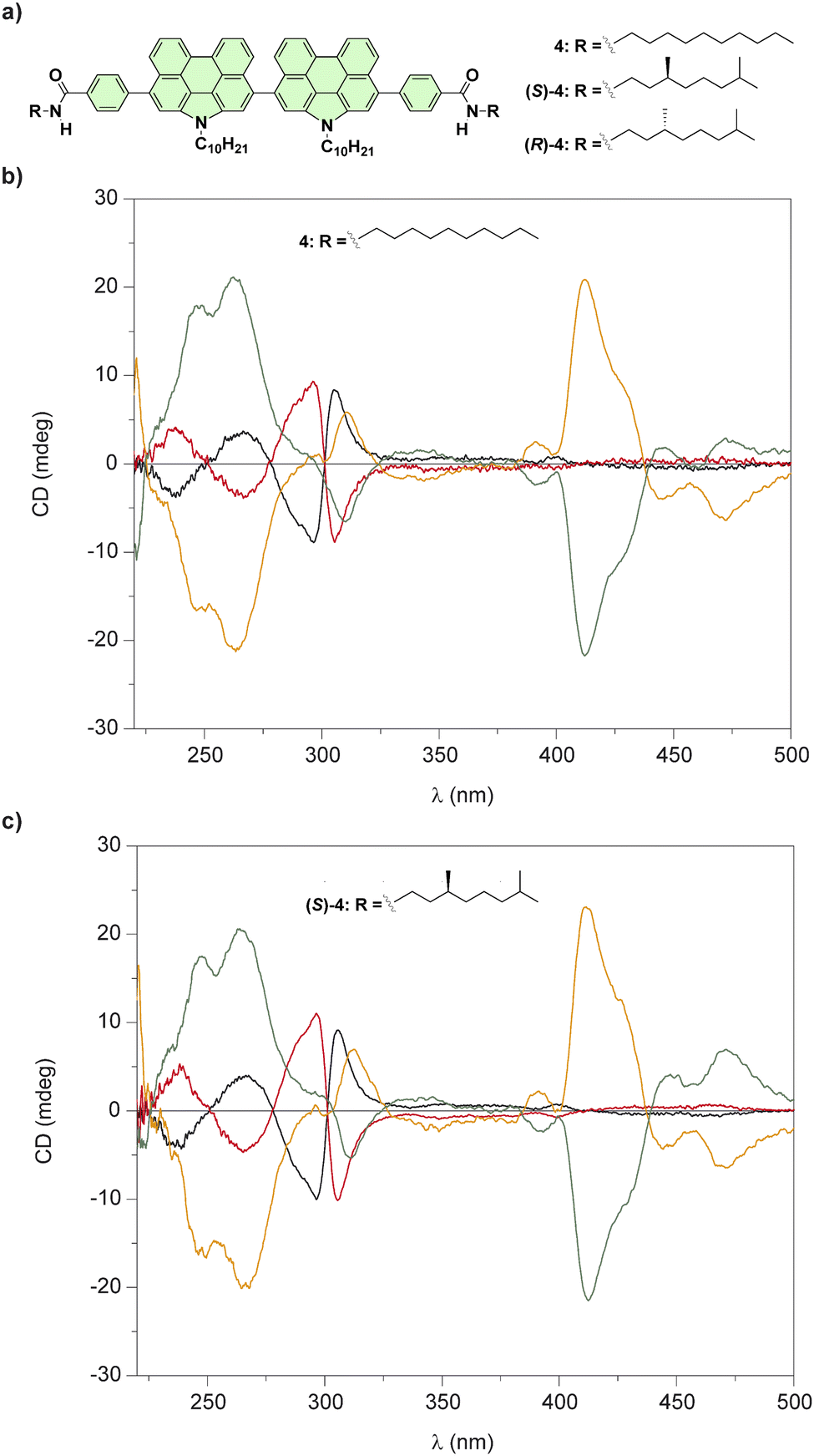 | ||
| Fig. 4 (a) Chemical structures of bis(N-annulated) perylenecarboxamides 4. CD spectra of all stereoisomers of 4 (b) and (S)-4 (c) in a molecularly dissolved state (black and red lines; CH2Cl2, cT = 10 μM, 298 K) and in the aggregated state (green and orange lines); 95/5 cyclohexane/CH2Cl2, cT = 10 μm, 298 K (reproduced from ref. 52 with permission from the American Chemical Society, copyright 2015). | ||
The restricted rotation of the perylenecarboxamide moieties around the single bond that separate both aromatic units generates atropisomers that can be separated by chiral HPLC. The enantioenriched samples of compounds 4 in solution of CH2Cl2, a good solvent that favours their disassembly, yields the atropisomers in the molecularly dissolved state (Fig. 4(b)). The addition of MCH, a bad solvent that induces their cooperative supramolecular polymerization, results in noticeable changes in the CD spectra in comparison to those registered in CH2Cl2 (Fig. 4(c)). However, the CD spectra of any of the atropisomers, regardless of the lack or the presence of point chirality in the peripheral side chains, displayed the same dichroic pattern which demonstrates that axial chirality predominates over point chirality in the formation of these chiral SPs.52
These selected examples illustrate the efficient transfer of the point chirality embedded in the peripheral side chains of the monomeric units to the supramolecular level in cooperative supramolecular polymerizations. However, only few examples of transfer of asymmetry in SPs governed by isodesmic supramolecular polymerization mechanisms have been reported to date. Isodesmic supramolecular polymerizations, usually detected in scaffolds that are not able to form highly directional non-covalent forces, produce a polydisperse mixture of oligomers of different lengths in which no helical preference is achieved or, at least, detected. However, there are a selected number of examples of helical aggregates formed following an isodesmic mechanism. To the best of our knowledge, the first example of a chiral SP formed through an isodesmic mechanism was reported by Meijer and co-workers for the BTAs endowed with three bipyridine moieties 5. In these BTAs, the formation of a twofold H-bonding array between the amide of the BTA with one of the pyridine rings and the amide of the peripheral benzamide fragments with the other pyridine ring of the bipyridine unit. These intramolecular H-bonding interactions planarize the whole monomeric unit and prevent the formation of intermolecular H-bonding interactions (Fig. 5(a)).53 Closely related to this example are the BTAs endowed with a fluorinated L-phenylalanine and an aminobenzoate spacer connected to peripheral hydrophilic M(III) complexes reported by Meijer and co-workers (compounds 6 in Fig. 4(a)),54 and the BTAs decorated with three naphthalenediimides reported by George and co-workers (compounds 7 in Fig. 5(a)).55 In these three examples, the bulkiness of the peripheral side chains of the BTA core and the intramolecular H-bonding interactions could be responsible for the isodesmic character of the supramolecular polymerization mechanism but, at the same time, the large size of the oligomers formed from these monomeric units could justify the appearance of the transfer of asymmetry.
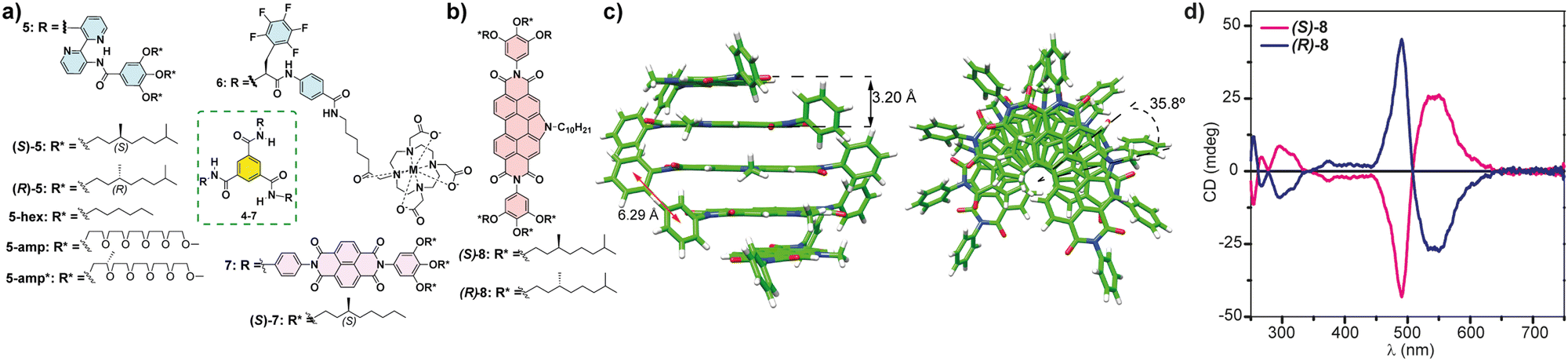 | ||
| Fig. 5 (a) Chemical structure of the BTAs 5–7 that form chiral SPs following an isodesmic mechanism. (b) Chemical structure of chiral NPDIs 8. (c) Minimum-energy structures of a pentamer of 8 displaying the distances and the rotation angle between the monomeric units (reproduced from ref. 56 with permission from the American Chemical Society, copyright 2021). (d) CD spectra of chiral NPDIs 8 in MCH (reproduced from ref. 56 with permission from the American Chemical Society, copyright 2021). | ||
Very recently, our research group has reported the efficient transfer of asymmetry in the N-annulated perylenediimide (NPDI) 8 decorated with lateral trialkoxyphenyl groups that form chiral SPs by following an isodesmic supramolecular polymerization mechanism (Fig. 5(b)). In this case, no H-bonding interactions are possible. However, theoretical calculations performed on this NPDI revealed that the more stable aggregated species are formed by the π-stacking of the aromatic backbones separated by distances of 3.20 Å. In addition, the attractive π-stacking of the lateral trialkoxyphenyl groups results in the formation of columnar stacks in which the monomeric units are rotated 35.8° (Fig. 5(c)). In fact, the experimental CD spectra of NPDIs 8 display an intense bisignated Cotton effect diagnostic of the formation of helical aggregated species (Fig. 5(d)).56
Transfer of asymmetry under kinetic control
Contrarily to the SPs formed under thermodynamic control, kinetically controlled supramolecular polymerization can provide more complex situations in which on-pathway and off-pathway intermediate aggregated species are formed in a competitive or consecutive way.57 This is generally a consequence of the formation of metastable species that retard the supramolecular polymerization process as they remain kinetically trapped on their evolution to a thermodynamically stable equilibrium.19 Therefore, in later years, some supramolecular systems have been described that show complex energy landscapes until they reach thermodynamic equilibrium. In this section, we will focus mainly on systems showing complex supramolecular polymerization that finally afford chiral SPs. On the other hand, some examples of differentiation, i.e. the process in which a single monomeric species forms two or more aggregated species and, at least one of them is a metastable one not accessible through a spontaneous thermodynamic process, will also be presented.For many years, the development of novel π-conjugated materials has been an area of active interest, as they offer many technological advantages over their inorganic counterparts. However, some issues related to the morphological aspects of the active layers must still be addressed before their realistic applicability. In this regard, supramolecular chemistry can play an important role considering the possibility to construct well-ordered supramolecular structures that can become part of the electroactive layer in devices.58 The group of Prof. Meijer was pioneering in the study of complex supramolecular polymerizations of π-conjugated systems and described, for the first time, a differentiation process in a chiral oligo-p-phenylene (OPV) substituted with branched chiral chains able to form helical aggregates. The polymerization process of the (S)-OPV 9 (Fig. 6) was promoted by the formation of H-bonded dimers between the ureidotriazine blocks, that later elongated by aromatic stacking to yield M-type helical SPs under thermodynamic control. However, when a solution of 9 was quickly cooled in an ice bath, a mixture of M- and P-type aggregates was obtained, and conversion of the P into the M-aggregate at room temperature was also observed after some time. This observation evidenced the coexistence of two polymerization pathways leading to different aggregates from the same monomeric species. On the one hand, the on-pathway route, leading to M-type thermodynamically stable helical aggregates and, on the other, the off-pathway that yielded kinetically trapped, metastable, P-type helical aggregates (Fig. 6(c)). Interestingly, when the P-type aggregates were reacted with dibenzoyl D-tartaric acid (DTA), their time-dependent evolution was prevented, remaining stable. However, when DTA was removed by extraction with an aqueous solution of ethylenediamine the conversion of the kinetically trapped P-type aggregates into the thermodynamically stable M-OPV was again favoured, evidencing the possibility to control these types of complex supramolecular polymerizations through the presence of additives.59 This seminal example, in which the temperature quenching was the strategy utilized to achieve the differentiation, was, at the same time, the starting point for the development of kinetically controlled SPs. Further investigation of kinetically controlled SPs was elegantly complemented by the development of living supramolecular polymerization (LSP) and seeded supramolecular polymerization (SSP).57 The first example of a living supramolecular polymerization was accomplished by Sugiyasu, Takeuchi and co-workers by utilizing porphyrins 10 that, initially, form J-type aggregated species as nanoparticles that evolve with time to the thermodynamically controlled fibrillar H-type aggregates (Fig. 6(d)). This kinetic evolution can be accelerated by the addition of seeds of the thermodynamically H-type aggregated species, achieved by the sonication of a solution of these H-type fibrillar structures, and repeated for several cycles yielding low polydisperse SPs.60 An outstanding achievement in this field was the LSP of corannulenes 11–13 reported by Aida and co-workers in 2015 (Fig. 6(e)). In these corannulenes, the formation of a pentafold intramolecular H-bonding interaction between the amide functional groups of compounds 11a–13a completely cancels the ability of these monomeric units to self-assemble. On the other hand, the N-methylated species 11b–13b are not able to form either intra- or intermolecular H-bonding interactions between the amide groups. However, these methylated species can act as initiators in the supramolecular polymerization of the non-methylated corannulenes. Thus, the addition of minute amounts of these methylated species 11b–13b in a solution of non-methylated 11a–13a provokes the rapid growth of SPs, a process that can be repeated by the addition of subsequent aliquots of the non-methylated species in a living manner.61 This seminal work brought to light the relevance to produce metastable monomeric units as dormant species able to experience kinetically controlled supramolecular polymerizations.57,62
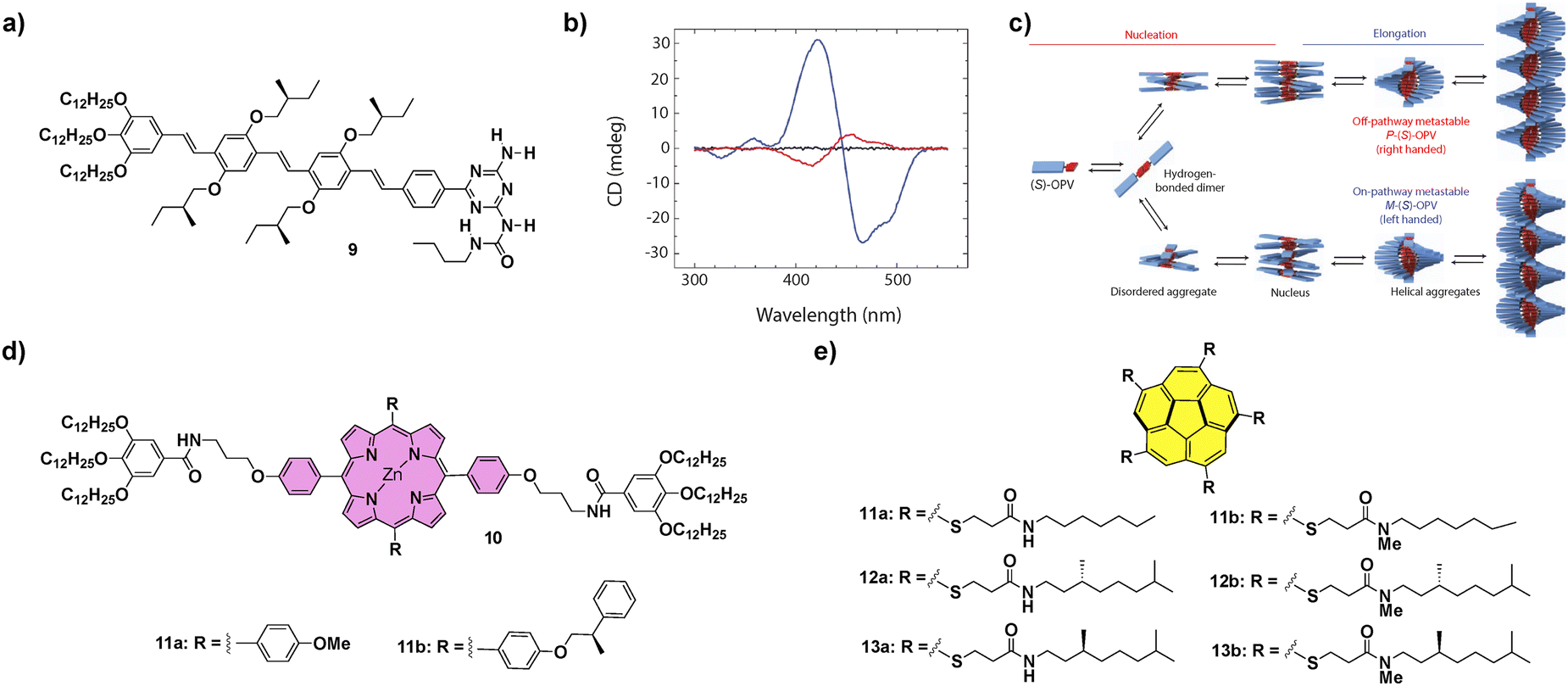 | ||
| Fig. 6 (a) Chemical structure of (S)-OPV 9. (b) CD spectra of (S)-OPV 9 in MCH solution: disassembled state (343K, black line), thermodynamically stable M-OPV (273K, blue line) and a mixture of M-OPV and metastable P-OPV (273K, red line); (reproduced from ref. 33c with permission from Wiley-VCH, copyright 2019). (c) Schematic representation of the aggregation pathways of (S)-OPV 9, including the growth of two competing assemblies (reproduced from ref. 33c with permission from Wiley-VCH, copyright 2019). Chemical structures of porphyrins 10 (d) and corannulene derivatives 11–13 (e) that have been reported to experience LSP. | ||
A very useful strategy to generate metastable monomeric species that retard supramolecular polymerization was reported by Würthner and co-workers on achiral PDIs. In these PDIs the two peripheral ethylen-3,4,5-trialkoxybenzamide moieties were able to form two seven-membered pseudocycles by the intramolecular H-bonding interaction of the NH of the benzamide and one of the carbonyls of the central imides. These pseudocycles, acting as monomeric metastable species that evolve with time, retard the supramolecular polymerization of the PDIs that finally yield the corresponding H-type SPs.63 Interestingly, Würthner and coworkers also reported on the seeded polymerization kinetics of this PBI, that were derived in values of 6 × 10−3 s−1.63 Taking advantage of the formation of such intramolecularly H-bonded pseudocycles, a variety of kinetically controlled supramolecular polymerizations have been reported to date.64 Nevertheless, only a reduced number of them deals with the formation of kinetically controlled chiral SPs. One of the first examples of the relevance of these metastable monomers to trigger pathway complexity in chiral SPs was reported for the solvent-dependent supramolecular polymerization of carbonyl-bridged triarylamines (CBTs) 14 (Fig. 7(a)).65
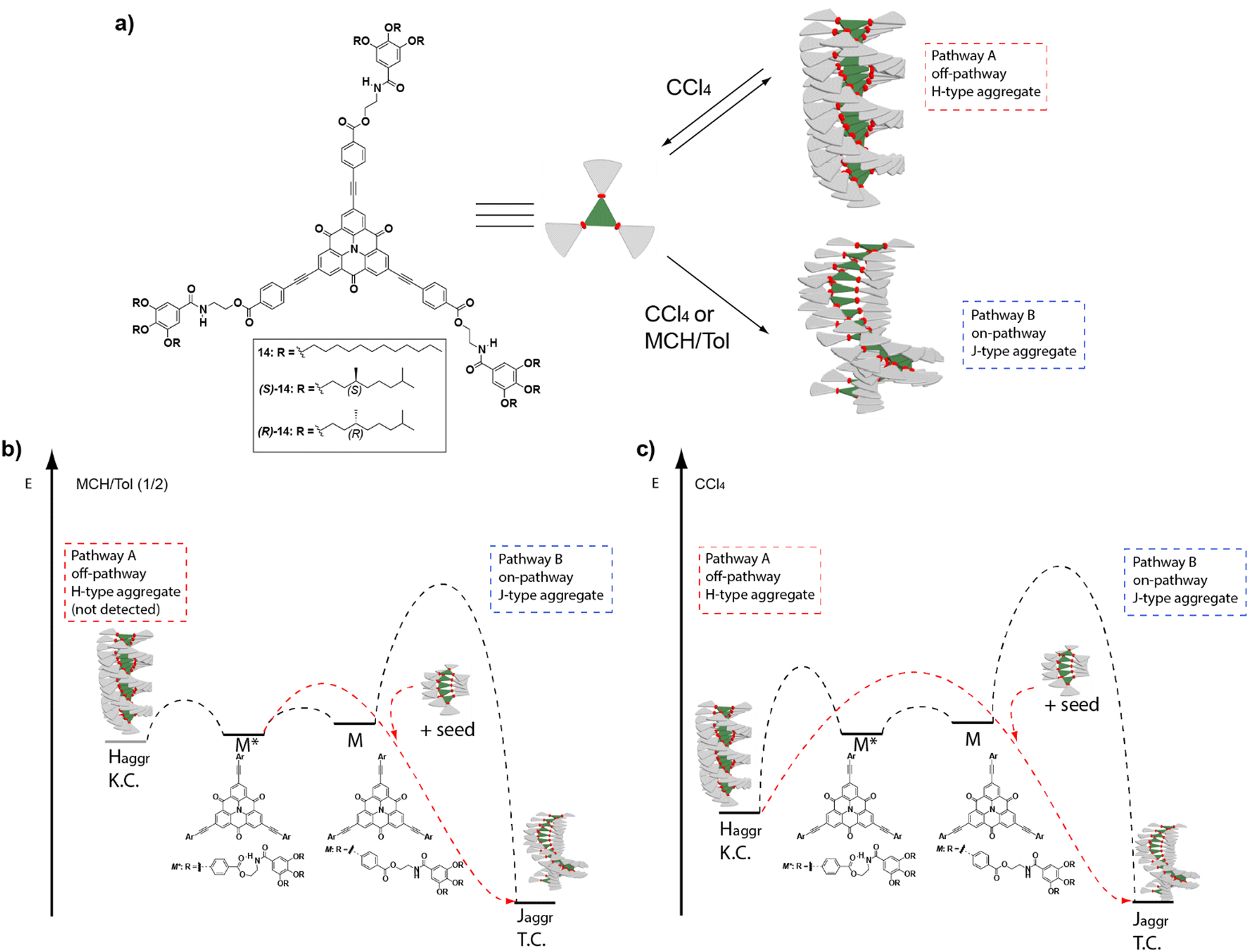 | ||
| Fig. 7 (a) Chemical structure of the investigated CBTs 14. Energy landscapes of the supramolecular polymerization coupled with a competing kinetic trap in (b) an MCH/Tol (1/2) mixture and (c) CCl4. In MCH/Tol (1/2), the complexity pathway could not be detected most probably because the kinetic trap is too shallow in energy and only the on-pathway J-type supramolecular polymerization is spectroscopically detected (Pathway B). In pure CCl4, the energy level of the kinetic trap is lower, and it is possible to detect both the off-pathway H-type aggregates (Pathway A) and the on-pathway J-type aggregates (Pathway B). In both solvents, the addition of J-type seeds accelerates the conversion (reproduced from ref. 65 with permission from Wiley-VCH, copyright 2017). | ||
The strong tendency of these compounds to self-assemble in MCH yielded J-type helical aggregates with the CBT disks slipped with staggered orientations. However, when these systems were investigated in toluene solution, the large hysteresis observed between the disassembly and self-assembly processes evidenced a complex mechanism. Indeed, if carbon tetrachloride was the solvent of choice, the formation of H-type, face-to-face, kinetically trapped aggregates could be detected. These species originated from the operation of a triple array of H-bonding interactions between the amide groups and the π-stacking between the CBT cores, and eventually evolved into a J-type helical aggregate. Whereas in AFM the metastable H-type aggregates appeared as disk-like micelles, the thermodynamically stable J-type aggregates appeared as rope-like fibrils. The increasing rate of conversion of the kinetically trapped aggregate into the thermodynamic one with temperature, and the sigmoidal profile obtained for this conversion, are characteristic of an autocatalytic process in which the thermodynamically stable species accelerates the transformation of the kinetic aggregate. This observation is in agreement with the operation of a complex parallel pathway mechanism. SSP were carried out by adding seeds of the thermodynamically stable J-type aggregate, prepared by sonication, to the kinetically trapped H-type aggregate, which acted as a monomer reservoir, increasing the rate of transformation. On the other hand, LSP experiments were also carried out. In these experiments, the thermodynamically controlled J-aggregates acted as the active seeds to trigger supramolecular polymerization upon the addition of successive aliquots of the kinetically controlled H-type aggregates. A reduction of the evolution time of the supramolecular polymerization was observed in all the cases. Additionally, it is important to note the retardation of all the kinetically mediated transformations in the presence of the chiral chains, in comparison with the analogue achiral derivatives, due to the steric demands imposed by the branched side chains. This constitutes another example of how subtle chemical modifications can enormously influence the pathway complexity (Fig. 7(b) and (c)).
Another example of π-conjugated systems in which a complex supramolecular polymerization was observed are the p-phenylene-cyanovinylene luminogens 15 and 16 (Fig. 8).66
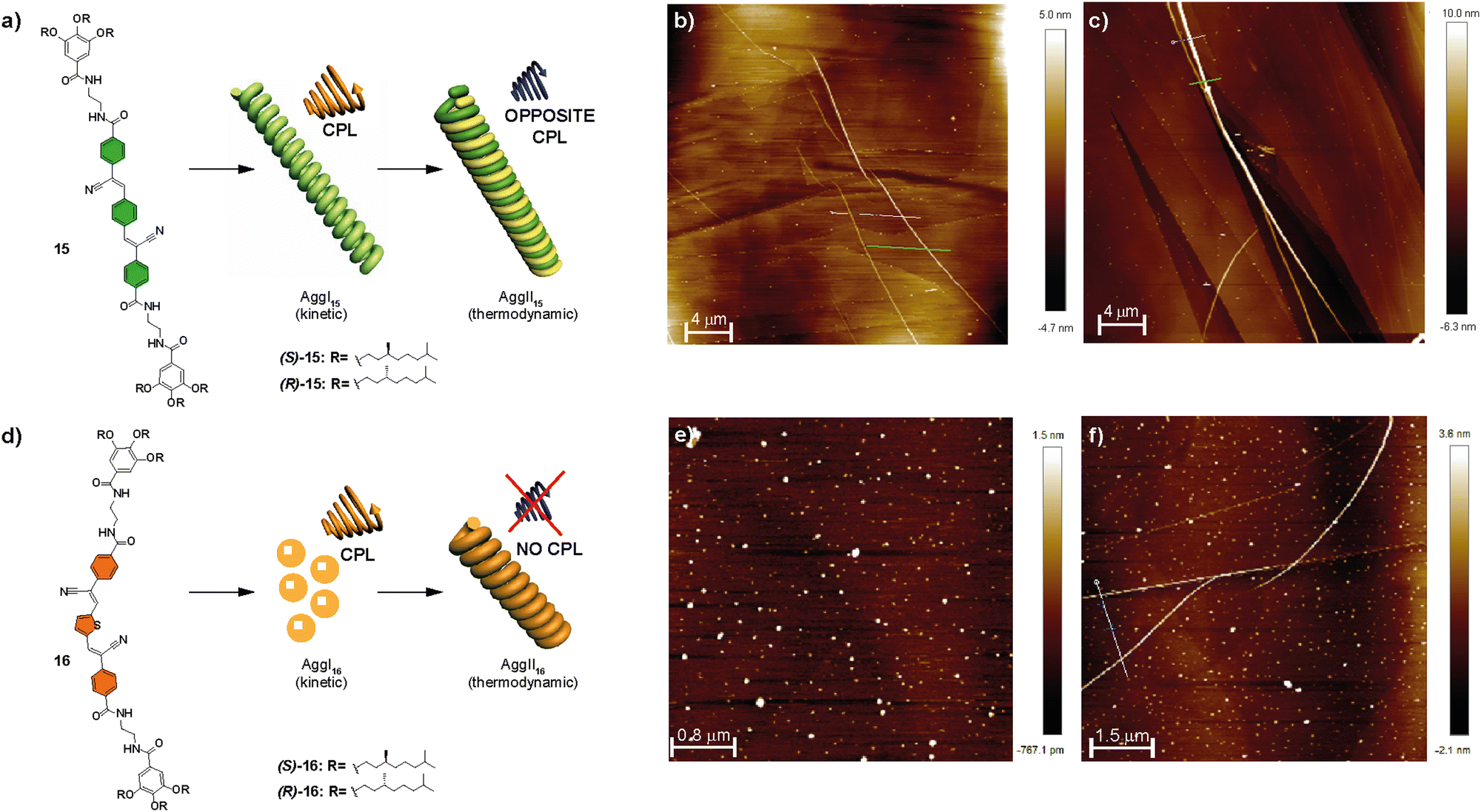 | ||
| Fig. 8 Chemical structures of compounds 15 (s) and 16 (d) and schematic representation showing the CPL-emitting kinetic and thermodynamically controlled aggregated structures formed by 15 and 16. AFM images of the kinetically controlled AggI15 (b), thermodynamically controlled AggII15, (c) kinetically controlled AggI16 (e) and thermodynamically controlled AggII16 (f) (MCH/CHCl3 9/1 as the solvent; HOPG as the surface; cT = 10 mM) (reproduced from ref. 66 with permission from the Royal Society of Chemistry, copyright 2022). | ||
These compounds were capable of forming helical H-type aggregates by the aromatic stacking between the self-assembling conjugated oligomeric units and by the formation of a four-fold array of H-bonding interactions. The handedness of these helical aggregates was determined by the chirality of the peripheral chains due to an efficient transfer of asymmetry from the peripheral chains to the aggregate level. FT-IR spectroscopy revealed the formation of intramolecularly H-bonded pseudocycles. These metastable species determine the final chiroptical outcome of the corresponding chiral SPs. Thus, for both compounds 15 and 16, two different H-type helical aggregates, namely AggI and AggII, were differentiated. The former evolved into AggII after time, indicating that it is a kinetically trapped species; whereas AggII was the thermodynamically most stable state. AFM images showed that, while AggI15 was constituted by isolated fibrillary structures, AggII15 appeared as thicker fibres formed by the bundling experienced by the AggI15 fibres into superhelices (Fig. 8(a)–(c)). On the other hand, AggI16 consisted of nanoparticles that evolved over time into the fibrillar aggregates that could be observed with time for AggII16 (Fig. 8(d)–(f)).
Investigation of the fluorescent and CPL emission properties of the chiral congeners of 15, (R)-15 and (S)-15, revealed aggregation-induced enhanced emission and CPL activity for both aggregates, AggI15 and AggII15, of each enantiomer and showed opposite signs depending on the aggregation state (and also, depending on the (R) or (S) nature of the chiral chain). This inversion was explained by considering that the CPL signal of the kinetically controlled AggI15 followed the helicity dictated by the stereogenic centres, whereas the intertwining responsible for the formation of the AggII15 was responsible for the inversion of the CPL signal in this state. A similar investigation for 16 disclosed an aggregation induced enhancement of emission with CPL activity for AggI16. However, the type of aggregation in AggII16 causes a strong quenching of the emissive properties, responsible for a lack of CPL activity. In any case, the evolution of the kinetically trapped AggI species into AggII depends on the evolution of the intramolecular H-bonds in the pseudocyclic metastable kinetically trapped species into the more stable intermolecular four-fold H-bonding array in the thermodynamically stable AggII.66
Besides helical aggregates, that have been extensively revised in this section, other supramolecular structures can benefit from the transfer of asymmetry from chiral monomers, for instance nanotubes. Thus, compound (S)-17 (Fig. 9) was capable of self-assembling into nanotoroids that further stacked into nanotubes, through H-bonding between the amide groups and π-stacking of two azobenzene units, producing a spontaneous curvature responsible for this initial aggregation into toroidal structures.67 CD experiments evidenced the formation of chiral nanotubes from chiral 17 and an enhancement of the dichroic signal as the temperature decreased.68 The replacement of the peripheral alkyl chains in 17 by the more sterically demanding trialkoxybenzene units, together with the introduction of a benzamidoethyl trialkoxy-benzamide spacer in 18 had a deep impact not only on the supramolecular aggregation mechanism but also in the hierarchy of the self-assembly.69
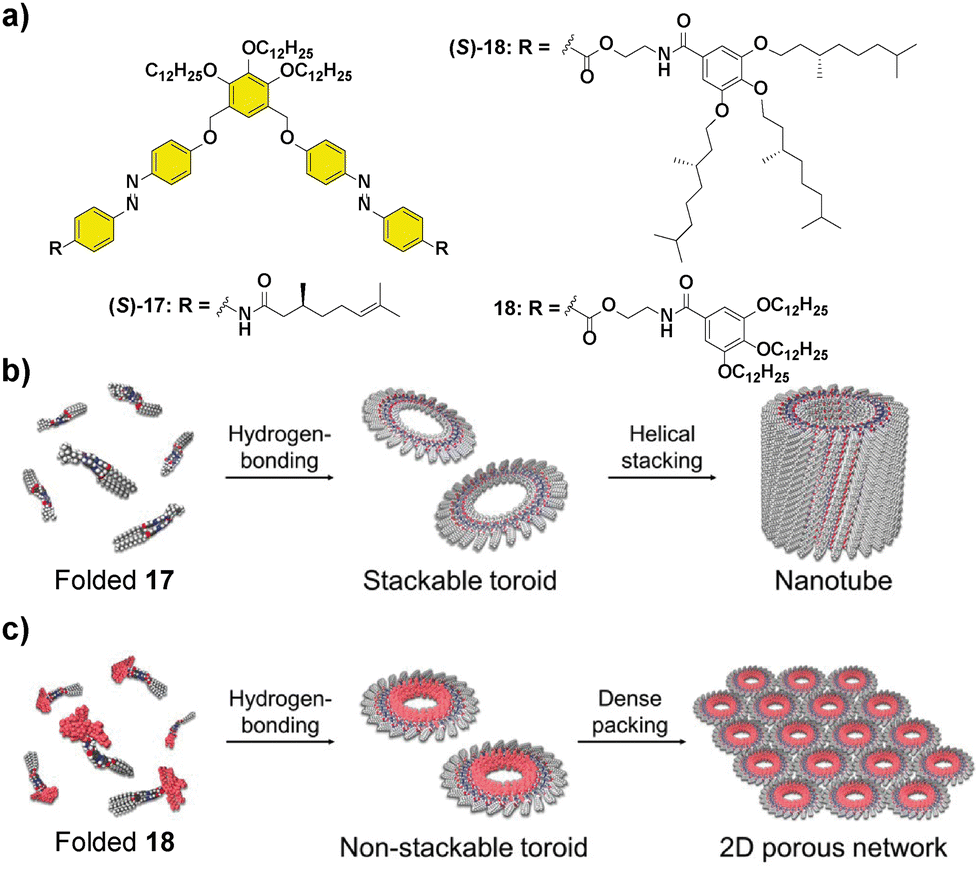 | ||
| Fig. 9 Molecular structures of scissor-shaped azobenzene dyads 17 and 18 (a) and illustrations of the self-assembly of 17 into nanotubes (b) and 18 into 2D porous networks (c) (reproduced from ref. 69 with permission from Wiley-VCH, copyright 2022). | ||
As in the case of 17, compound 18 aggregated into toroids that, in contrast, formed a highly ordered 2D network instead of the tubular structures due to the steric demands imposed by the trialkoxybenzylbenzamide units. Besides, the two-step process observed when the degree of aggregation was plotted vs. temperature evidenced a pre-nucleation phase that was controlled by a conformational change in the monomer. The hysteresis between the cooling and heating cycles was in this case attributed to the formation of an inactive kinetically trapped species, involving an open conformation with intramolecularly H-bonded pseudocycles that later evolved into a folded conformation with inter- and intramolecular H-bonds (Fig. 9). The photoresponsive behaviour, arising from the trans–cis isomerization of the azobenzene units and the ratio between the three possible trans,trans; cis,cis and trans,cis isomers were investigated. Photoisomerization of the azobenzene units is a suitable input to reversibly modulate the formation of the nanotoroids, which is interesting for the preparation of photoresponsive porous nanosheets.
It is worth mentioning that, in literature, there are also relevant examples of kinetically controlled supramolecular polymerizations in which no metastable pseudocycles are involved but pathway complexity is experimentally observed. Interestingly, some of these reports rely on the supramolecular polymerization of chiral CBTs in which the formation of the intramolecularly H-bonded pseudocycles by the peripheral side chains is not feasible.70 These examples open new avenues to be explored in the field.
To summarize this section, systematic investigations of the effect of structural variations on the supramolecular polymerization, even if becoming more common, are still somewhat scarce, thus greatly limiting the knowledge about clear structure–property relationships. However, from the remarkable studies of supramolecular aggregates highlighted in this section, showing asymmetry that has been transferred from different elements of chirality and non-classical polymerization pathways, one clear conclusion can be extracted: even very subtle structural modifications on the same motif can trigger differential polymerization pathways with different degrees of complexity to afford materials with a variety of morphologies and, consequently, different properties and potential applications.
Amplification of asymmetry: moving forward with controlled supramolecular copolymers
The previous section collected some representative examples of transfer of asymmetry from chiral monomeric units to the supramolecular level. However, both natural events and man-made processes require multicomponent assembly to achieve complexity and functionality. In the area of SPs and, of course, to achieve chiral SPs, the utilization of such multicomponent systems is a useful strategy. Thus, the term supramolecular copolymer or co-assembly is gaining more and more attention.71 The above-mentioned SaS and MR experiments are clear examples of supramolecular copolymerization in which two closely related, but sufficiently different, monomeric species are mixed to achieve a homochiral co-assembled system.How homochirality appeared in biomolecules has fascinated researchers for decades and it will continue to because it can lead to a complete understanding of the origin of life as mentioned before.5 When referring to catalysis, asymmetric amplification is a phenomenon in which the ee of a product is higher than that of a chiral auxiliary for a catalyst,72 and was first noted by Kagan et al. in 1986 in the Sharpless epoxidation of geraniol.73 Discovery of asymmetric autocatalysis in the alkylation of pyrimidine-5-carbaldehyde with diisopropylzinc is considered to be a chiral amplification process.7 In polymers, asymmetric amplification is a process in which the ee of the polymer formed is higher than the ee of the monomer counterparts. It was observed first in covalent polymers, especially for polyisocyanates. In these pioneering investigations, Green and co-workers discovered that only small amounts of the chiral monomer were able to bias the helicity of the polymer.14 Pioneering and exhaustive work by Palmans and Meijer in the 1990s demonstrated that this phenomenon could also take place in SPs.53a As in the case of covalent polymers, the experiments to investigate amplification of asymmetry in SPs are called SaS and MR.13a,16 In a SaS experiment, increasing amounts of a chiral monomer are mixed with an achiral one and, if amplification of asymmetry takes place, a few of the chiral monomers, the “sergeants”, will command the helicity of a large number of the achiral monomers, the “soldiers”. In a MR experiment, monomers with an opposite element of asymmetry at different ratios are mixed and, if amplification of asymmetry takes place, the most abundant monomer imposes its preferred helicity to the others. In both experiments, a non-linear increase in the ee is distinctive of the existence of chiral amplification phenomena. This ee is monitored mainly through CD spectroscopy. A modified and less utilized version of these experiments is the so-called diluted majority-rules (DMR) experiment, in which an ee is introduced in the sergeant fraction of a SaS experiment.74
In any case, to attain chiral amplification in SPs, co-assembly between the different monomers involved, is needed. This is favoured when the different monomers involved in the polymerization process present geometrical complementarity but also, are energetically favoured to give rise to a chiral enriched SP. If the monomers are not energetically favoured to co-assemble, both monomers will give rise to independent SPs, and self-sorted homopolymers will be obtained. In this situation, the ee of the mixture will be the algebraic sum of the ee of both self-sorted SPs and therefore, chiral amplification will not take place. On the other hand, if both monomers co-assemble, a supramolecular copolymer will be formed and depending on the degree of mixing, the distribution of each of the monomers along the polymer chain will be different ranging from block, blocky, periodic, statistic or alternating and due to the interaction of both monomers within the polymer chain, and amplification of asymmetry will be plausible (Fig. 10).71
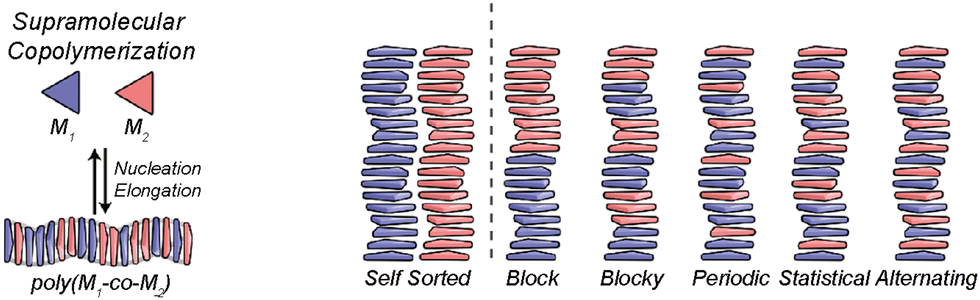 | ||
| Fig. 10 Possible microstructures for each type of supramolecular copolymerization (reproduced from ref. 71 with permission from the American Chemical Society, copyright 2019). | ||
To explain the amplification of asymmetry phenomena, van Gestel et al. described a model where two different energy penalties were considered: the helix reversal penalty (HRP) and the mismatch penalty (MMP).75 The HRP is the energy paid when there is a helix reversal within the stack of the SP. The MMP is the energy paid when a chiral monomer is introduced in a stack of its unpreferred helicity.76–78 Depending on the relative values of these energy penalties, the supramolecular copolymer formed will prefer to incorporate a wrong chiral monomer into the unpreferred helicity stack (HRP > MMP), or the copolymer formed will have different sections of opposite helicity within the same stack (HRP ≈ MMP). In the former, amplification of the asymmetry process will be strengthened whereas in the latter, the different helicity sections within the stack will decrease the ee. Another parameter used in the amplification of asymmetry experiments is the net helicity, which is defined as the difference between the fractions of right- and left-handed chiral monomers.75 Despite the utility provided by this model to achieve accurate thermodynamic data, it has not been used systematically for other systems than BTAs and OPETAs showing amplification of asymmetry. This lack of data impedes direct comparison of chiral amplification phenomena in different SPs.
The first report of a SP showing amplification of asymmetry was reported by Meijer in 1997 in which disc-shaped bipyridine compounds self-assembled through an isodesmic mechanism in alkane solution (BTAs 5 in Fig. 5).53a,c As stated before, these discotics aimed to be planar by the formation of intramolecular H-bonds between the N atoms of the bipyridines and the amide functional groups to show liquid crystallinity, which indeed they showed.53b The formation of intramolecular H-bonds prevents the formation of intermolecular directional H-bonds favouring an isodesmic supramolecular polymerization. Moreover, curiosity regarding the ability to yield chiral amplification led to the discovery that upon adding (S)-5 to 5 solutions through a SaS experiment, small amounts of the chiral sergeant (S)-5 (∼10%) were enough to achieve the maximum dichroic signal (Fig. 11(a)).33a The MR experiment upon mixing (S)-5 and (R)-5 showed a pronounced non-linear response (Fig. 11(b)).76 The MMP (0.94 kJ mol−1) calculated for this experiment is much lower than the HRP (7.8 kJ mol−1), therefore, the energy gained by the formation of the stack is much higher than the energy lost by the mismatch of the “wrong” enantiomer incorporated into the unpreferred helical stack at low ratios.
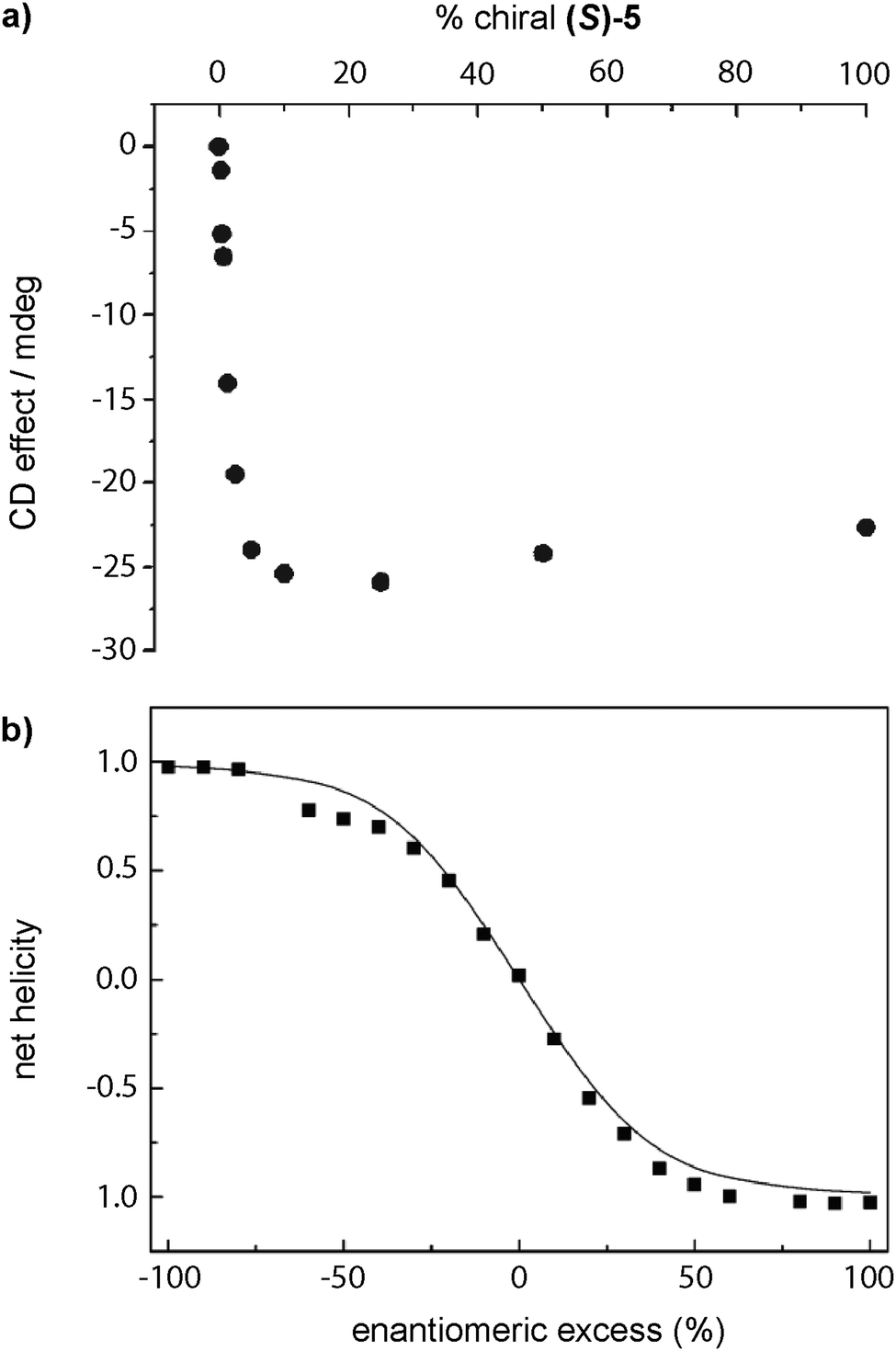 | ||
| Fig. 11 (a) SaS experiment by mixing solutions of (S)-5 and 5 in hexane. (b) Net helicity as a function of ee measured by CD spectroscopy in n-octane in a MR experiment between (S)-5 and (R)-5 (20 °C, cT = 79 μM) (reproduced from ref. 33a with permission from Wiley-VCH, copyright 2007). | ||
The introduction of hydrophilic chains of oligoethyleneglycol (compounds 5-amp and 5-amp* in Fig. 5) resulted in successful amplification of the asymmetry between intrinsic chiral 5-amp and achiral 5-amp* in a polar solvent such as n-butanol.78 But the behaviour of this system is quite different from 1a–c with aliphatic chains, in hexane. Here, the kinetics is much slower, playing an essential role in the aggregation and amplification of asymmetry phenomenon. Moreover, this system is able to self-assemble in water and the SaS experiment evidences amplification of asymmetry, showing a maximum when 25–30% of 5-amp is mixed with 5-amp*.79,80
In 2000, Meijer and co-workers described the supramolecular polymerization of N,N′,N′′-alkyl substituted BTAs 1 in heptane (Fig. 2).81 Their self-assembling features were studied by IR and CD spectroscopy and confirmed the formation of SPs in which the monomers are joined together by intermolecular H-bonds, which results in helices with a preferred handedness for the chiral congener (S)-1. SaS experiments in heptane demonstrated that only 2.5% of sergeant (S)-1 was enough to acquire the same helicity as the stacks composed only by sergeants.
Replacing the amide functional groups of BTAs 1 with urea functionalities (compounds 19 in Fig. 12(a)) resulted in a decrease of the amplification of the asymmetry ability of the system in comparison with previously reported bipyridine-based discs 1 and 5 and large hysteresis effects.82 Additionally, co-assembly between chiral units was observed in a SaS experiment at low loadings of chiral (S)-1 and achiral 19 (Fig. 12(b)). However, mixing 15% of chiral amide (S)-1 with 85% of achiral 19 firstly resulted in the formation of a supramolecular copolymer that upon 1 hour evolved into the self-sorting homopolymers (Fig. 12(c)). This experiment is representative of the necessity of an efficient co-assembly between the monomers for yielding successful amplification of asymmetry.
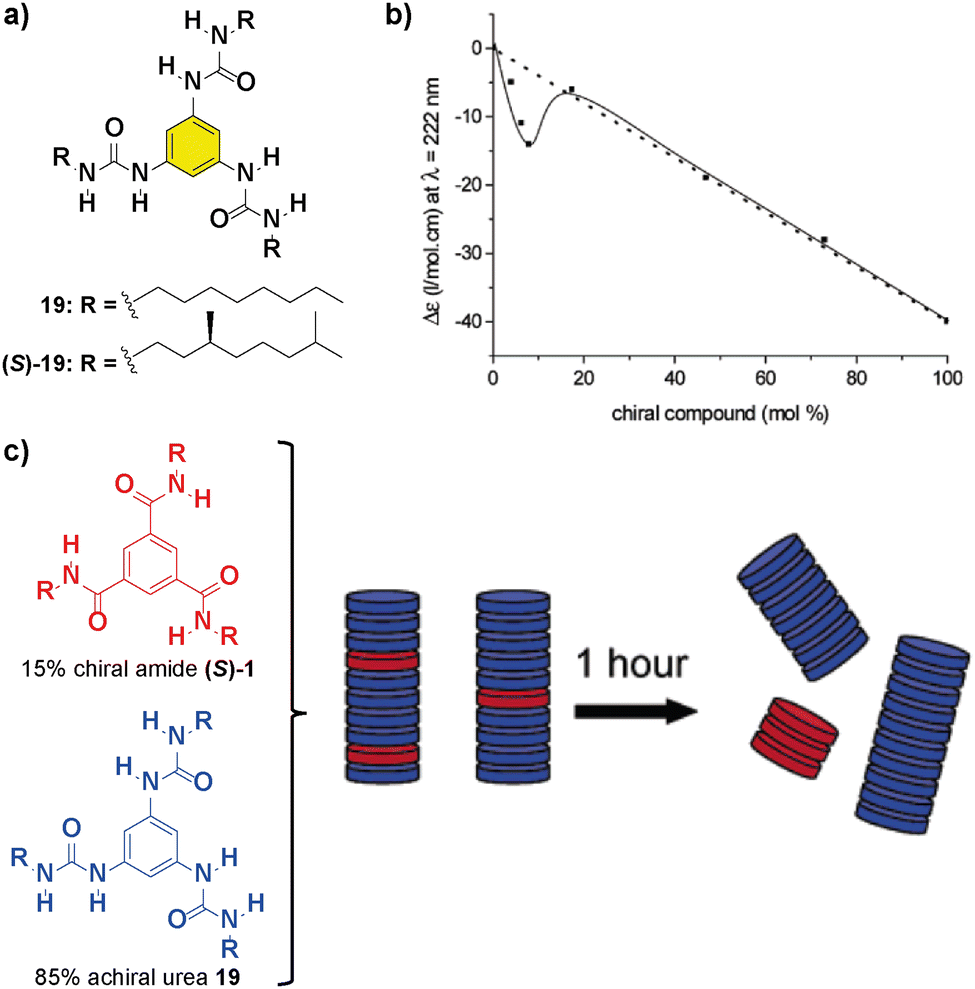 | ||
| Fig. 12 (a) Chemical structure of benzene trisureas 19. (b) SaS experiment between chiral (S)-1 and achiral 19 (cT = 50 μM in heptane). (c) Schematic representation of the aggregates formed during the mixing of 15% of (S)-1 and 85% of 19 (reproduced from ref. 82 with permission from the American Chemical Society, copyright 2022). | ||
The influence of temperature in the amplification of asymmetry was studied for the first time in 2010 by Smulders et al. on BTAs 1.77 They hypothesized that the amplification of asymmetry should be affected by temperature, since the degree of aggregation and the average stack length are also affected. They found that the HRP was little affected by temperature. But in contrast, the MMP decreased when the temperature was increased. This inverse dependence of the MMP with temperature produces opposite effects in the amount of chiral amplification in the SaS and the MR experiments: while the degree of chiral amplification is decreased with temperature in the SaS, it is increased in the MR (Fig. 13(a) and (b)). In a different report by Smulders et al., they investigated the systematic variation on the number and position of the stereogenic centres present on the chemical structure of BTAs (Fig. 13(c)).78 In these studies, they observed high and constant HRP values for all the BTAs studied (∼10–15 kJ mol−1), which is explained by the fact that the same number of intermolecular H-bonds are present in all studied BTAs derivatives. On the other hand, they observed that the ee necessary to reach the maximum CD signal in the MR experiments was minimum when only one stereogenic centre was present at the BTA (Fig. 13(d)). This observation was explained by the increase in the MMP upon increasing the number of stereogenic centres. The increase in the MMP was also supported by the so-called mixed MR experiments in which two monomers with opposite chirality and different numbers of stereogenic centres were mixed. Another important conclusion was extracted varying the position of the stereogenic centre with respect to the amide group: it was found that the helical sense was inverted within the same type of stereogenic centre depending on the relative position within the amido group in what is known as the “odd–even effect”. Moreover, in this work, simulation curves given a certain HRP, revealed that there is an optimum to which the MMP can be reduced while also enhancing the degree of amplification of asymmetry.
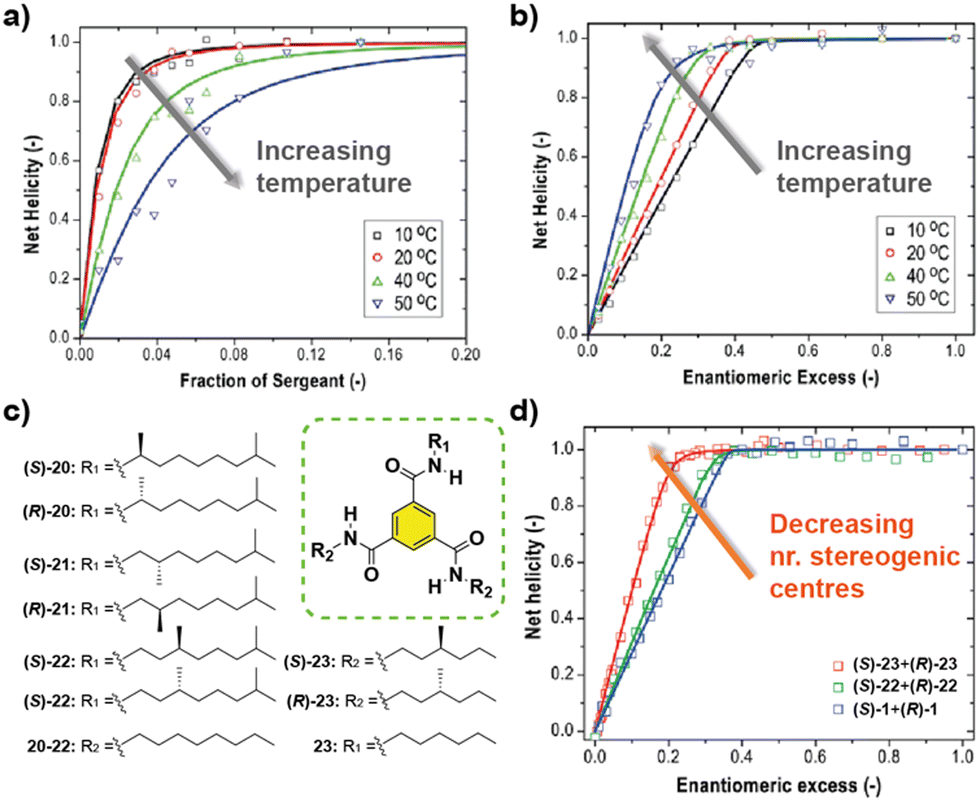 | ||
| Fig. 13 SaS (a) and MR (b) experiments of (S)-1 and (R)-1 at different temperatures (cT = 20 μM in MCH). (c) Chemical structure of asymmetric BTAs. (d) MR experiments with BTAs containing a different number of stereogenic centres (cT = 30 μM in MCH) (reproduced from ref. 77 and 78 with permission from the American Chemical Society, copyright 2010). | ||
Another C3-symmetric family of compounds where amplification of asymmetry has been widely studied is OPETAs 3. This system consists of a C3-symmetric extended aromatic central disc in comparison to BTAs surrounded by 3 amido groups. In 2011, our group described the SaS experiments in achiral OPETA 3 upon additions of chiral sergeant in MCH solution finding a complete chiral amplification upon the addition of 20% of the sergeant (Fig. 14 bottom).49a In order to study the influence on the number of stereogenic centres in chiral amplification, OPETAs, with a variable number of stereogenic centres, 25 and 26 were synthesized (Fig. 14).49c,50b HRPs calculated from the SaS experiments using the reported model from Smulders et al.78 revealed a decrease of this penalty (from 13 to 8 kJ mol−1) upon decreasing the number of stereogenic centres from 3 to 1, which can justify the gradual decrease in the chiral amplification (Fig. 14). When 3 stereogenic centres are present, the HRP is rather high, impeding the formation of inverse helical domains within the same stack. This HRP values makes the presence of homochiral stacks easier even at low concentration of the chiral sergeant. Upon decreasing the number of stereogenic centres, the decrease in the HRP favours the apparition of domains of inverse helicity within the same stack impeding the chiral amplification process in the SaS experiments. Therefore, for OPETAs, the decrease in the number of stereogenic centres decreases its amplification of asymmetry ability, in contrast to that observed for BTAs as described above.77
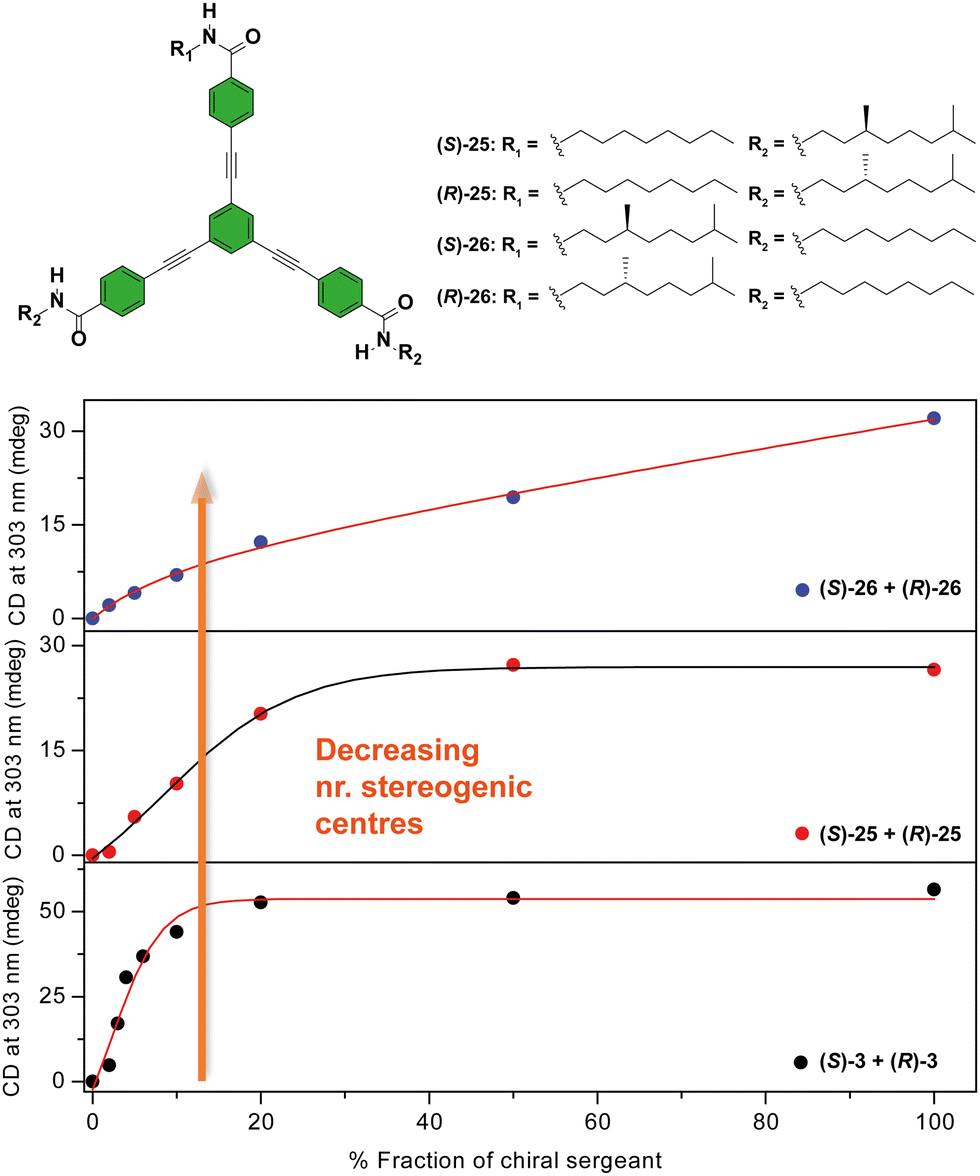 | ||
| Fig. 14 Chemical structure of asymmetric OPETAs (top). SaS experiments for OPTAs (298 K, cT = 10 μM in MCH) (bottom) (reproduced from ref. 50b with permission from the American Chemical Society, copyright 2012). | ||
In accordance with that reported for BTAs, temperature decreases the MMP and, therefore, decreases the chiral amplification in MR experiments (Fig. 15).49c The MR experiments revealed that a 48% of ee is necessary to attain a complete amplified state in the OPETAs bearing 3 stereogenic centres.49b Decreasing the number of stereogenic centres present in the OPETAs, increases the amount of ee that is necessary to obtain a complete chiral response (with ee of 57% and >80% for 2 and 1 stereogenic centres, respectively).49c This observation was explained in terms of the values of MMP obtained from the MRs experiment fitting to the two component model.83,84 In this case, and in agreement with the values obtained from the SaS experiments, it is assumed that the HRP would be high and this effect can be ruled out. The values of calculated MMP showed that this energy penalty increases upon decreasing the number of stereogenic centres. This value is minimum for MR experiments between OPETAs (S)-26 and (R)-26, containing only one stereogenic centre (Fig. 15), where the MMP value (5.4 kJ mol−1) is comparable to the calculated HRP from the SaS experiments. Therefore, in these tricarboxamides, it is more probable to have helical reversals within the stacks impeding the chiral amplification. As a conclusion, increasing the number of stereogenic centres per monomeric unit results in a lower MMP, enhancing the chiral amplification phenomenon in OPETAs, in sharp contrast with BTAs.78
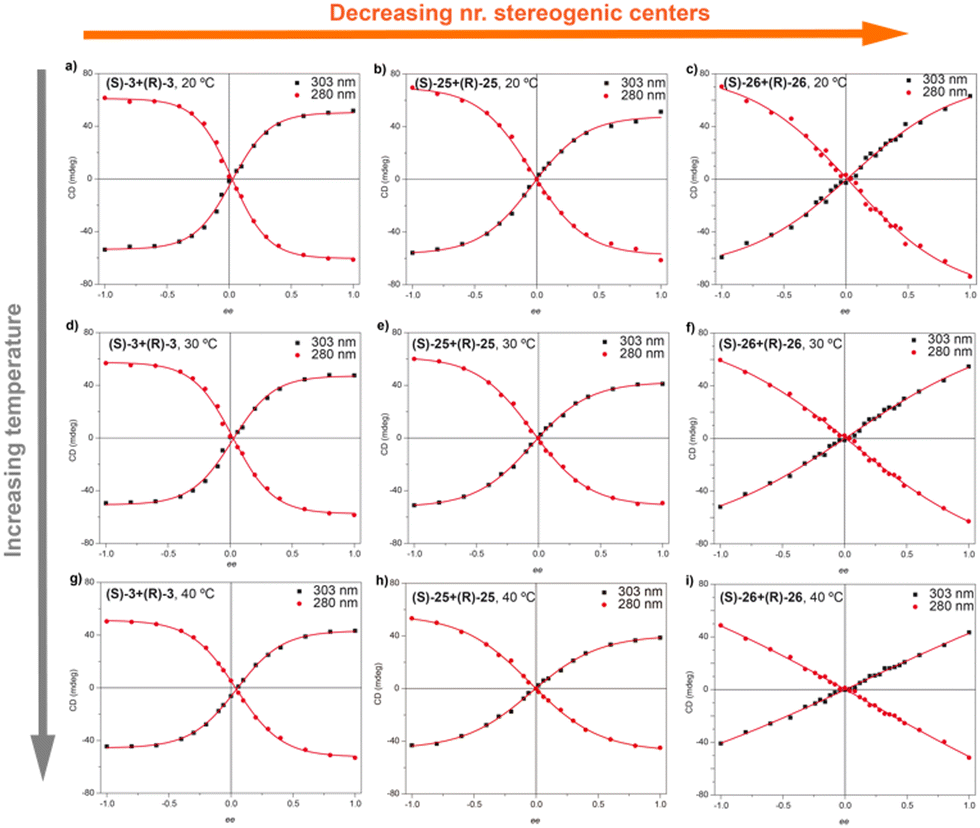 | ||
| Fig. 15 MR experiments for mixtures of OPTAs 3, 25 and 26 at different temperatures: 293 K (top), 303 K (middle) and 313 K (top) (cT = 10 μM in MCH) (reproduced from ref. 49c with permission from the Royal Society of Chemistry, copyright 2019). | ||
Theoretical calculations by using the GFN2-xTB approach permitted comparison of the temperature dependence behaviour of chiral amplification in BTAs with OPETAs.49c These calculations showed the rotation between neighbouring OPETA units is ca. 18°, whereas for BTA units it is ca. 65° within the stack (Fig. 16). The smaller angle present in OPETAs increases the steric hindrance and, therefore, upon increasing the number of stereogenic centres, this angle will increase to reduce the steric hindrance which decreases the MMP. The bigger angle in stacked BTAs gives rise to weak interactions between the side chains and remains mostly unchanged within the number of stereogenic centres. Therefore, increasing the number of chiral side chains in BTAs increases the steric hindrance, which results in larger MMP values. Chiral amplification experiments were also performed by our research group in related C3-symmetric TPBAs with a propeller shape.50a MR experiments performed between (S)-2 and (R)-2 revealed that 22% of ee excess was enough to bias the helicity of the mixture to yield homochiral helixes. Moreover, a MMP of 1.00 kJ mol−1 was calculated, a slightly lower value in comparison with related C3-symmetric BTAs (S)-1 and (R)-1, thus decreasing the amount of ee needed to yield homochiral helices.77 SaS experiments performed in these TPBAs were not conclusive due to the contribution of linear dichroism. In this case, theoretical calculations reveal that the rotation angle between adjacent TPBAs is 26°. This rotation angle is optimum to favour the van der Waals interactions between the aliphatic side chains which results in a very efficient chiral co-assembly (Fig. 16).
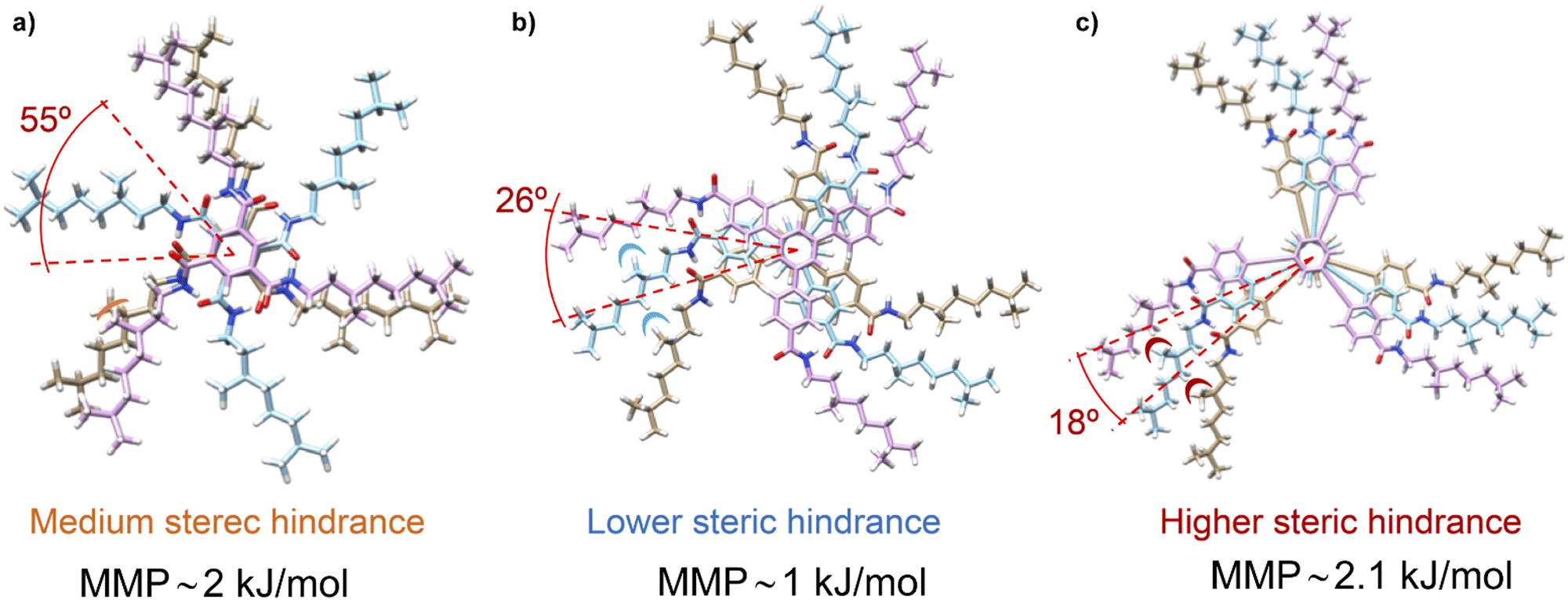 | ||
| Fig. 16 (a) Schematic illustration of the steric hindrance effect exerted by the chiral aliphatic chains of vicinal units in BTAs 1 (a), TPBAs 2 (b) and OPETAs 3 (c). | ||
OPETAs have also been used to co-assemble with oligophenylene based discotics bearing a variable number of amide groups.50b Oligophenylene ethynylene (OPE) with a variable number of amido and ether functional groups are not able to form helical SPs. Remarkably, performing a mixed SaS experiment between achiral OPETA 3 and chiral (S)-27, the copolymer poly-3-co-(S)-27 with a preferred helical sense was formed. This copolymerization takes place by incorporation of (S)-27 within the helical stacks formed by 3 together with a successful amplification of asymmetry between (S)-27 and 3, with this effect reaching the maximum when a 1:1 mixture of each monomer is present (Fig. 17(a)).
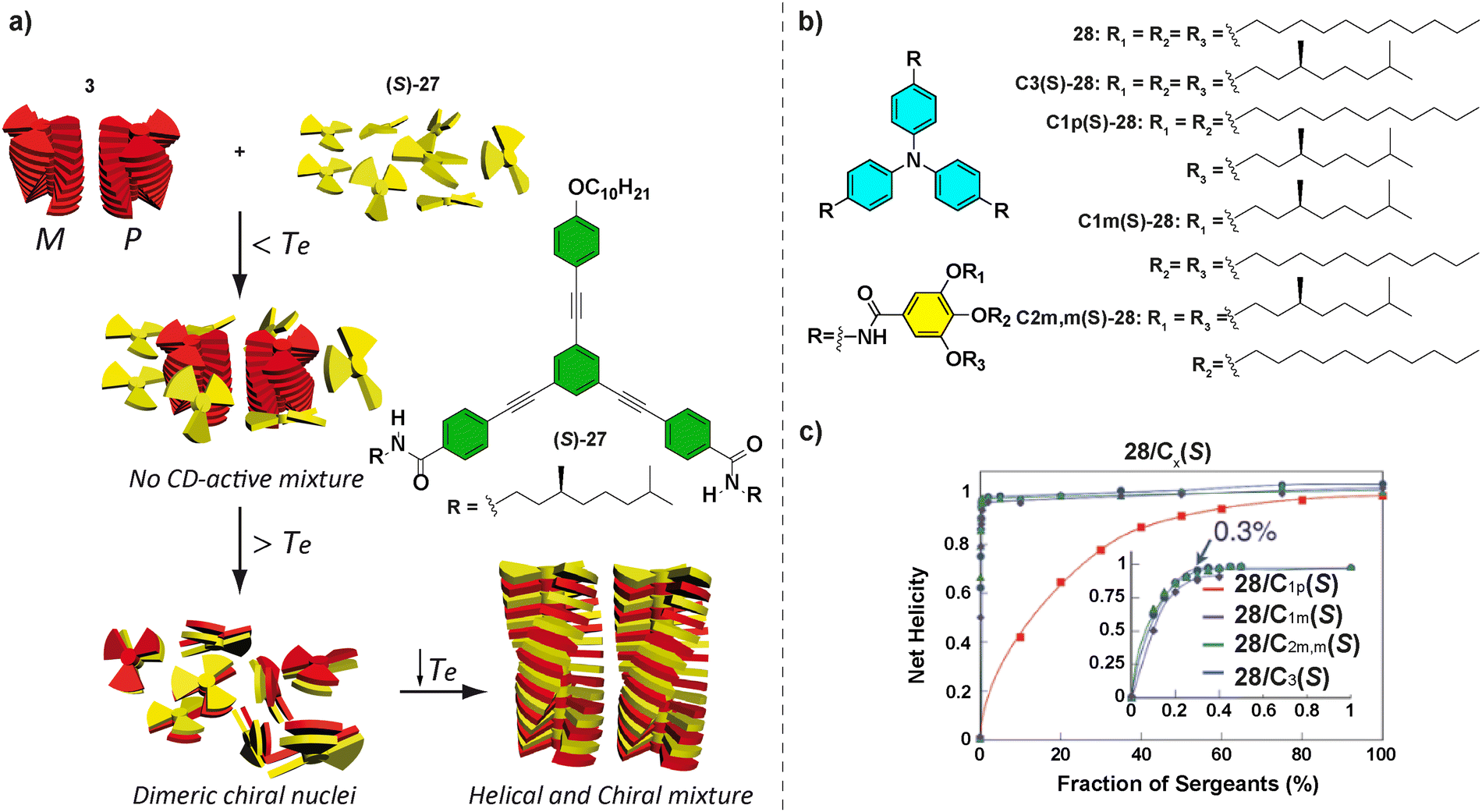 | ||
| Fig. 17 (a) Chemical structure of OPE (S)-27 and schematic representation of the formation of poly-5-co-(S)-27 (reproduced from ref. 50b with permission from the American Chemical Society, copyright 2012). (b) Chemical structures of TPATAs 28. (c) SaS experiments for mixtures of 28/C1p(S)-28, 28/C1m(S)-28, 28/C2m,m(S)-28, and 28/C3(S)-28 (298 K, cT = 30 μM in cyclohexane) (reproduced from ref. 85 with permission from the Royal Society of Chemistry, copyright 2016). | ||
Other systems, in which a very efficient amplification of asymmetry has been demonstrated, are the propeller-shaped triphenylamine tricarboxamides (TPATAs) 28 reported by Aida and co-workers in 2016 (Fig. 17(b)).85 The SaS experiments evidenced that only 0.3% of the sergeant was enough to yield complete homochiral helices.
The importance of completely understanding the amplification phenomena is exemplified by the works described by our group.86,87 In particular, MR experiments performed in TPATAs 30 (Fig. 18(a)) show different behaviour if they are recorded immediately after heating and cooling the samples or after aging them for 24 hours (Fig. 18(b)). The samples measured immediately after their mixing, yielded amplification of asymmetry only at large ees (∼65%) while the aged samples showed complete homochiral helical sense at ee values of ∼30%. This behaviour is explained by the formation of an intramolecular H-bond yielding a seven-membered metastable kinetic species which acts as a chain capper in the supramolecular polymerization of 30 (Fig. 18(a)) and thus, retarding the amplification of the asymmetry process. It is worth mentioning that substitution of the phenyl into a biphenyl (compounds 30 in Fig. 18(a)) in the aromatic scaffold in TPATAs results in the absence of amplification of asymmetry in the SaS experiments.88 This behaviour is explained by the lack of co-assembly between achiral 30 and chiral (S)-30 as evidenced by the apparition of two transitions in the UV-vis cooling curves of the 1/1 mixture. These transitions are the same as those of the two components separately, indicating a narcissistic self-sorting phenomenon. The self-sorting homopolymerization was previously shown by Meijer and co-workers in the copolymerization of TPATAs and pyridyl triamides.89
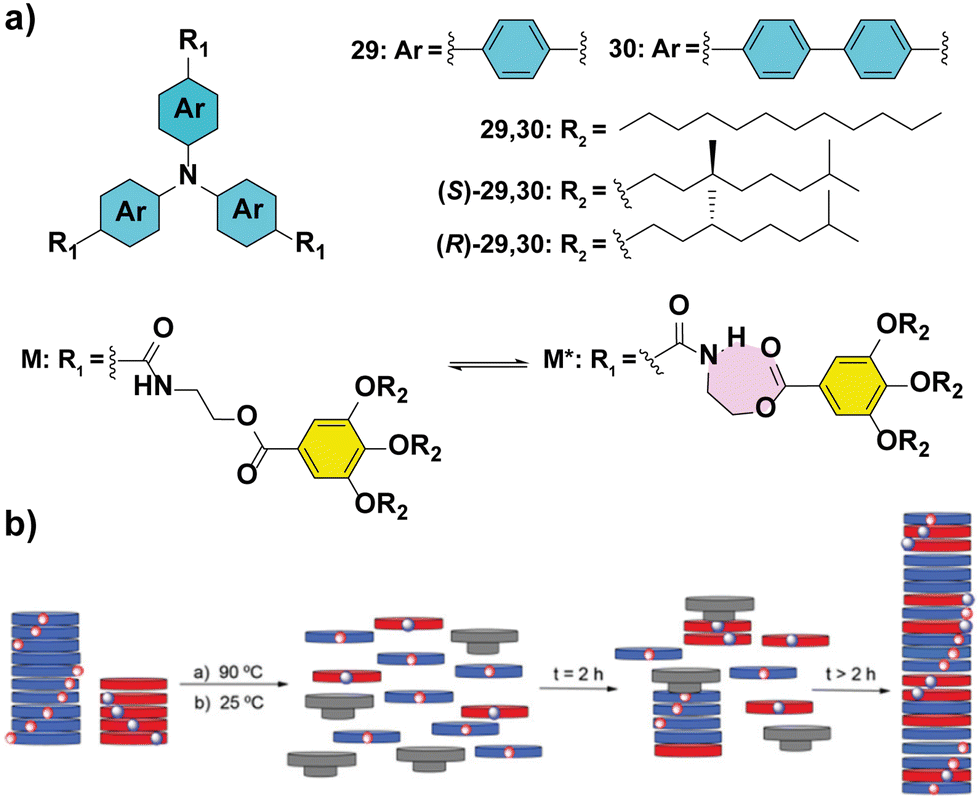 | ||
| Fig. 18 (a) Chemical structures of triarylamine tricarboxamides 29 and 30 indicating the formation of the intramolecular H-bond. (b) Schematic illustration of the chain-capper effect exerted by the intramolecular H-bond acting as a chain capper in the MR experiments (reproduced from ref. 87 with permission from the Royal Society of Chemistry, copyright 2021). | ||
Co-assembly of different enantiomers can lead to a change in the type of the aggregates formed. For example, Nakashima and co-workers described the supramolecular polymerization of chiral binaphthalenes 31.90 The temperature dependent absorption experiments suggested an isodesmic mechanism. When the MR experiments on (S)-31 and (R)-31 were performed, a hypsochromic shift in the CD was observed when decreasing the ee suggesting a change in the molecular packing (Fig. 19(a)–(c)). Below an ee of 0.6 the aggregates formed were short fibers in contrast with the long fibers formed for enantioenriched copolymers as visualized by transmission electron microscopy (TEM).
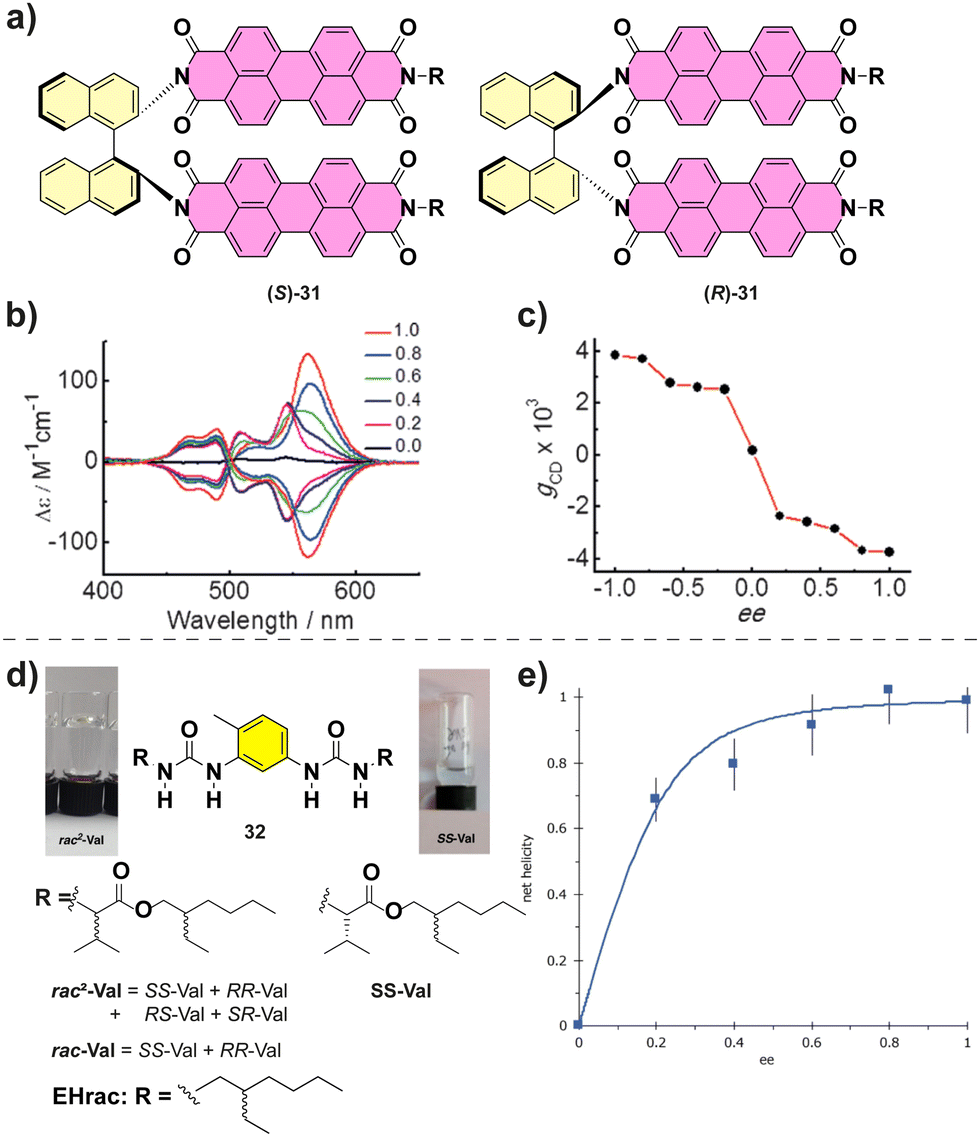 | ||
| Fig. 19 (a) Chemical structure of (S)-31 and (R)-31. (b) CD spectra of the co-assembly of (S)-31 and (R)-31. (c) Plot of the maximum gcdversus ee value (reproduced from ref. 90 with permission from Wiley-VCH, copyright 2015). (d) Structure of bisureas 32 and the solution and gel formed. (e) Net helicity of the low temperature SP measured at −10 °C (103 μM in MCH, λ = 233 nm) (reproduced from ref. 91 with permission from Wiley-VCH, copyright 2021). | ||
Recently, Boutellier and co-workers, studied the supramolecular polymerization of bisureas 32 (Fig. 19(d) and (e)).91 In this work, they found the existence of two competing helical assemblies, a double helical and a single helical one depending on the temperature. The discrimination between both types of assemblies was assessed by CD spectroscopy, small angle neutron scattering (SANS) and nanodifferential scanning calorimetry (nDSC). For the heterochiral assemblies (SS-Val + RR-Val) the double helix was favoured while for the homochiral (SS-Val or RR-Val) assemblies the single one predominated. MR experiments between SS-Val and RR-Val at different temperatures in which the double helical and the single helical assemblies are predominant (80 °C and −10 °C respectively) affording the corresponding HRP and MMP data. Therefore, this investigation reveals how subtle differences in the energetic values ended in the formation of different aggregation modes.
Amplification of asymmetry has been also observed for PDIs. Würthner et al. studied the influence of the peripheral side chains in the supramolecular polymerization of a series of PDIs.92 They found that for these PDIs when a methyl substituent was present in the peripheral side chains (PDIs 33–39 in Fig. 20(a)), J-aggregates were formed to avoid the steric repulsion between them, whereas for the less crowded PDIs 33 and 34, H-aggregates were formed. Co-assembly of achiral PDI 33, which forms H-aggregates, and chiral PDI 35, which forms J-aggregates, was studied through a SaS experiment (Fig. 20(b)) which showed the appearance of a CD signal upon increasing additions of chiral PDI 33 to achiral PDI-35 resulting in a H-type aggregate. A further increase in the ratio of PDI 33 resulted in the appearance of a separated band at 623 due to the formation of J-aggregates and a significant decrease in the CD signal due to a decrease in the concentration of H-aggregates upon increasing the steric repulsion (Fig. 20(c)). This change in the aggregation mode highlights that a geometrical complementarity between the monomers should be present for an effective amplification of asymmetry.
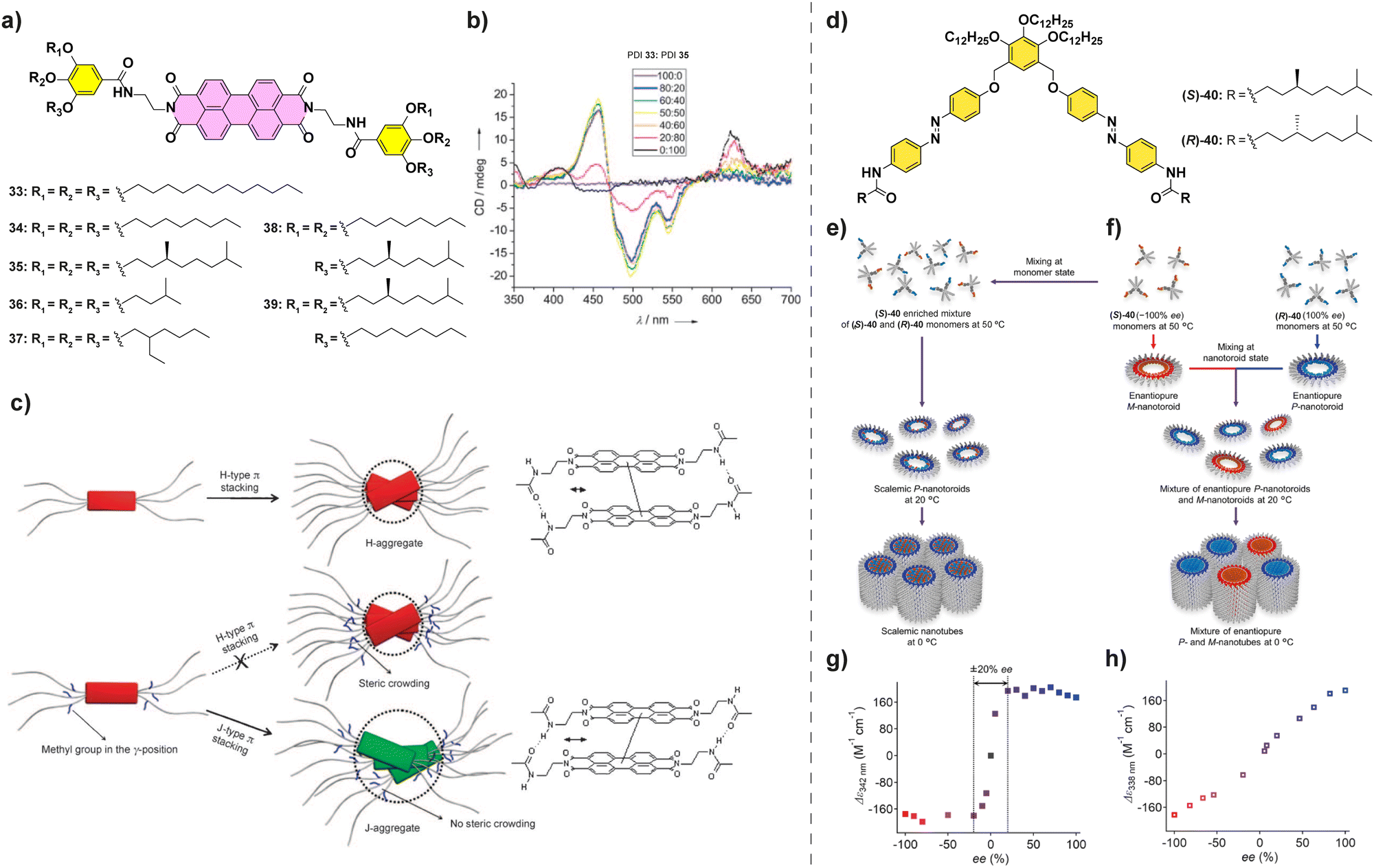 | ||
| Fig. 20 (a) Chemical structure of PDIs 33–39. (b) CD spectra of mixtures of PDIs 33 and 35 at different ratios (cT = 10 μM in MCH/CHCl3 80/20). (c) Schematic representation of the polymerization of PBIs 33–39 into H- or J-aggregates (reproduced from ref. 92 with permission from Wiley-VCH, copyright 2008). (d) Chemical structure of (S)- and (R)-40. (e) and (f) Schematic representations of the distinct hierarchical organization of (S)-40 and (R)-40. (g) MR experiment performed in scalemic nanotoroids as prepared in (e). (h) MR experiment performed in mixtures of enantiopure nanotubes as prepared in (f) (cT = 300 μM in MCH) (reproduced from ref. 96 with permission from the American Chemical Society, copyright 2023). | ||
In all the examples of amplification of asymmetry described above, the MR experiments show a positive nonlinear response in which a small ee of the chiral building blocks leads to unproportionally large chiroptical signals near zero ee. In contrast, there is also an effect shown by some systems known as the “racemate rules effect” (RRE), where a linear response of the CD is observed near zero ee while giving large nonlinear chiroptical responses at high values of ee. Only few published systems exhibit this phenomenon described by Jiang and co-workers.93,94 These systems were formed by dicationic and anionic hosts such as malate and amino acids. An interesting application for this RRE effect is the determination of the ee of an enantioenriched mixture because of its non-linear behaviour at high ee.93 A plausible explanation is that a particular polymer cooperatively binds opposite enantiomers (a racemic mix) until the solution is so depleted in enantiomers that the polymer can only bind those of the same handedness, which would occur with solutions of higher ee values. The interest of this system relies on the origin of this behaviour because the systems by themselves cannot yield high amplification of asymmetry unless there are high loadings of enantioenriched mixtures.
Very recently, Yagai and co-workers described the supramolecular polymerization of scissor-shape azobenzene dyads 40 functionalized with chiral side chains (Fig. 20(d)).95,96 These dyads give rise to two levels of organized aggregates: firstly, toroids are formed when cooling hot MCH solutions to 20 °C and, secondly, these toroids grow into nanotubes when cooled further down to 0 °C. When the chiral monomers are mixed in a MR experiment and allowed to cool to 20 °C, enriched nanotoroids are formed by the copolymerization of both enantiomers (Fig. 20(e) and (f)). These asymmetrically enriched nanotoroids, when cooled down to 0 °C, give rise to nanotubes of preferred helicity with a strong majority-rules effect (Fig. 20(g)). However, if the solutions are mixed at 20 °C the nanotoroids give rise via a narcissistic polymerization to homochiral nanotubes showing the absence of amplification of asymmetry (Fig. 20(e)). This exemplifies the importance of understanding the aggregation modes of the system of choice to successfully allow the correct mixing and copolymerization of the constituting monomers.
It is important to remark that not all helical SP give rise to amplification of asymmetry, as demonstrate the NPBIs 8 described in our research group, which self-assemble through an isodesmic mechanism into helical aggregates with a preferred helicity depending on the chiral nature of the peripheral chains (Fig. 5(b)–(d)).56 Co-assembly between chiral (S)-8 and (R)-8 did not result in any MR effect. The lack of a MR effect could be justified by considering the isodesmic character of the supramolecular polymerization of 8. In fact, only few examples have been described where amplification of asymmetry was found for isodesmic or small cooperative mechanisms.53c,97 It has already been mentioned that isodesmic mechanisms decrease the degree of polymerization and increase the polydispersity of the SP formed in comparison to cooperative mechanisms. Therefore, a decrease in the amplification of asymmetry phenomenon could be explained by the similar energy associated to the formation of homopolymers, formed between pristine monomeric units (achiral or chiral), and the heteropolymers formed by the mixture of both species.
Therefore, and according to the literature, the co-assemblies must fulfil some requisites to execute and efficient amplification of asymmetry: (i) the congeners must exhibit a geometry complementary in such a way that they efficiently co-assemble, instead of having a narcissistic polymerization; (ii) the preferred supramolecular polymerization mechanism should be cooperative, and (iii) there should be a favourable energetic balance described by the MMP and the HRP.
Symmetry breaking in supramolecular polymers: chiral structures from achiral units
A common feature of all the examples collected in the previous sections is the presence of a chiral monomeric unit to achieve a helical SP. The self-assembly or the co-assembly of such monomeric units finally affords such helical species in a process involving transfer or amplification of asymmetry. It is worth mentioning that a less common strategy to achieve homochiral entities involves achiral monomeric units and the application of an external, chiral stimulus that finally yields the helical aggregate in a process termed symmetry breaking. Symmetry breaking implies the spontaneous generation of enantiomeric excess from an equimolecular mixture of two enantiomers through the application of an external stimulus, usually stirring, solvent, a magnetic field or light.98 Symmetry breaking is often observed in the field of crystallization upon the application of controlled stirring. Following this strategy, Ribó and co-workers were able to generate chiral aggregates from diluted solutions of 4-sulfonatephenylporphyrins by biasing the stirring direction. The helical aggregation of this porphyrin derivative is governed by the spinning direction of the vortex created. Importantly, once formed, these aggregates always exhibit the same helicity regardless of the spinning sense which implies the generation of chiral entities in a static situation.99 Despite the examples of kinetically controlled supramolecular polymerization yielding chiral (or achiral) supramolecular polymers, the correlation between the different methodologies used to initiate the aggregation and the rate constants of the aggregation process that finally generates optically active aggregated species is less studied. In this regard, the studies reported by Monsù Scolaro and coworkers on the effect of acidification of meso-tetrakis(4-sulfonatophenyl)porphyrin (TPPS) shed relevant light on the achievement of chiral entities upon a symmetry breaking event. Thus, the addition of acid traces to a dilute solution of the TPPS results in a sigmoidal profile featuring an induction period at the beginning of the process that resembles an autocatalytic process. However, inverting the sequence, that is adding a solution of the TPPS into an acidic solution, results in an exponential growth of the dichroic response without any initial induction time. These studies indicate the strong influence of the protocol utilized to generate the nucleation regime. Thus, adding TPPS to the acidic solution affords a large number of nuclei and, consequently, a large number of CD-inactive nuclei. On the other hand, the addition of acid into the TPPS solution shortens the nucleation regime thus resulting in a more sensitive chiral response.100 Interestingly, controlling the stirring direction can also provoke the appearance of dynamic symmetry breaking effects in SPs. Stirring forces to generate chiral SPs have also been utilized to afford chiral structures with other porphyrins,101a or BTA derivatives able to form organogels.101b Thus, the flexibility of SPs formed from achiral dendronized porphyrins,102 an oligo(p-phenylene vinylene) derivative103 or N-annulated perylenedicarboxamides104 favours the switch of the macroscopical helical alignment with the spinning sense. This dynamic effect is an outstanding probe to sense the macroscopic chiral character of local fluidic flows.The solvent–solute interactions are crucial in SPs to control the stability and, consequently, the final properties of the aggregated species.42 The relevance of such solvent–solute interactions has been also harnessed to bias solvent-controlled symmetry breaking. In this area, the pioneering reports by Green et al. demonstrating that optically active solvents generate minute solvation energy differences between the enantiomeric P and M helices of achiral polyisocyanates,105 prompted the use of chiral solvents to achieve helical SPs. In good agreement with that reported by Green et al. on polyisocyanates, a linear correlation between the CD response and the excess of a chiral solvent like (S)-2,6-dimethyloctane or enantiomerically enriched limonene, that corroborates the formation of helical SPs from achiral monomeric units, was observed for achiral BTA 5-hex,53a achiral urea 32EHrac,106 or achiral PBI 33.107 However, more challenging is deciphering the influence exerted by a chiral solvent in the formation of helical SPs built up from chiral self-assembling units. Thus, Palmans, Meijer and co-workers reported in 2021 the diastereomeric differences in the supramolecular polymerization of enantiomers of chiral triphenylene-2,6,10-tricarboxamides TTAs (compounds 41 in Fig. 21(a)) by using chiral chlorinated solvents (Fig. 21(b)).108
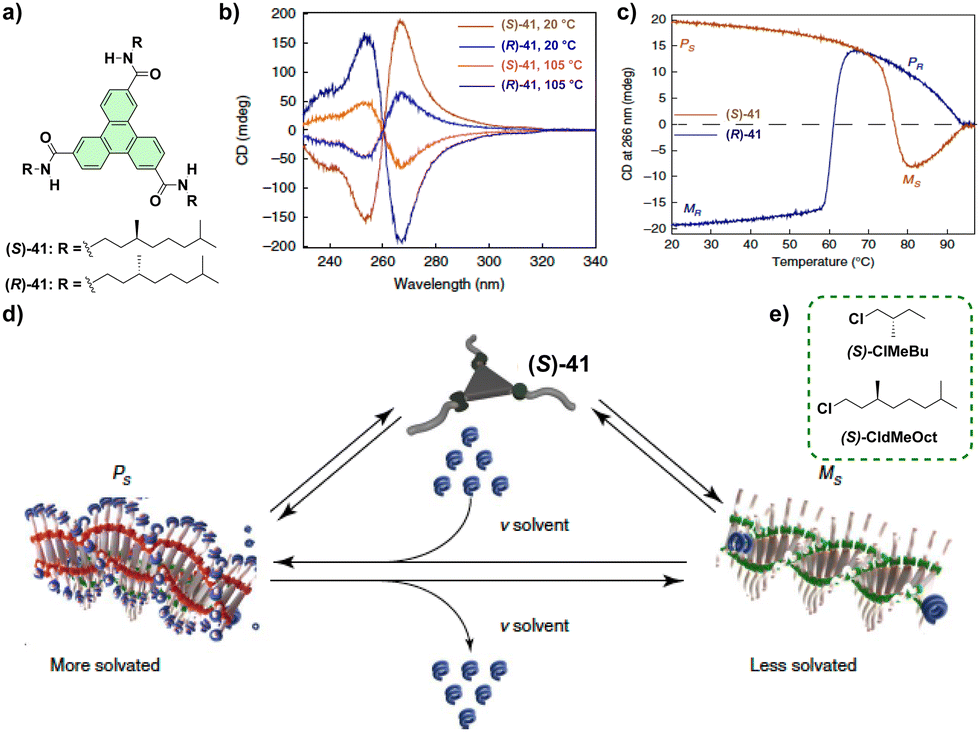 | ||
| Fig. 21 Chemical structure of chiral TTAs 41 (a) and the chiral solvents (S)-ClMeBu and (S)-CldMeOct (b). (c) Schematic illustration of the supramolecular polymerization of TTAs 41 depicting the role of solvent in the thermodynamic equilibrium between two competing helical aggregates for a single enantiomer (HT and LT high and low temperature, respectively; reproduced from ref. 106 with permission from Wiley-VCH, copyright 2010). | ||
Importantly, the results reported for these systems indicate that the energetic difference between the helical SPs formed by the self-assembly of compounds 41 is similar to the interaction energy of these helical SPs with the chiral solvent. The CD spectra of both enantiomers of TTAs 41 are mirror images and, interestingly, increasing the temperature yields CD spectra for each enantiomer that are mirror images to those registered at 20 °C.
Variable temperature CD experiments show a thermoresponsive stereomutation in the formation of helical SPs. The temperature at which the stereomutation is produced depends on the point chirality of the side chains of the monomeric unit and the point chirality of the chiral solvent. Therefore, the dissimilar helix inversion temperatures indicate that the chiral solvent breaks the mirror symmetry between the enantiomers, inducing a diastereomeric relationship based on additive solvation effects by the chiral solvent (Fig. 21(c)). The results presented in this recent report indicate that the helical preference induced by chiral monomers and chiral solvents is similar in energy, but becomes visible only in cooperative systems since minute energy differences accumulate along the sufficiently long supramolecular polymer structure.108
The interaction of light, and more specifically, CPL is a very intriguing way to achieve symmetry breaking in supramolecular polymerization thus yielding helical SPs. Le Bel and van’t Hoff proposed that CPL could be responsible for the origin of natural homochirality. The irradiation of natural samples with CPL could trigger the achievement of an enantioenriched sample from a racemic mixture.109 Inspired by this theory, some reports, still very scarce, collect the formation of chiral SPs from achiral monomeric units upon the irradiation with CPL. To the best of our knowledge, the first example describing the formation of helical SPs from achiral monomeric units upon the irradiation with CPL was reported by Kim et al. for C3-symmetric triarylamines endowed with three amide functional groups and diacetylene moieties (compound 42 in Fig. 22(a)). Whilst a diluted solution of 42 in dichloroethane is CD silent, the irradiation of this solution with l-CPL or r-CPL gives rise to rich CD spectra with opposite Cotton effects diagnostic of the formation of helical aggregated species due to the generation of radical cations. The subsequent photopolymerization of the diacetylene moieties by irradiating the samples with UV light generates covalent polymers that lock the handedness achieved by irradiating the monomeric units with CPL (Fig. 22(b)).110 A similar strategy has been followed by Zou and co-workers by using an achiral porphyrin scaffold also endowed with diacetylene groups.111 Triarylamine 42, and its chiral congeners (S)-42 and (R)-42, have been utilized to perform SaS experiments in which the samples are irradiated with CPL. These experiments demonstrated that point chirality and CPL irradiation compete to generate helical aggregates. Thus, for the CPL with mismatched handedness, above a critical molar fraction of the sergeant, the handedness of the co-assembled system is dictated by this chiral component. However, below this critical molar fraction, CPL overrides the point chirality with increasing irradiation time which results in helical aggregated species of opposite handedness.110b
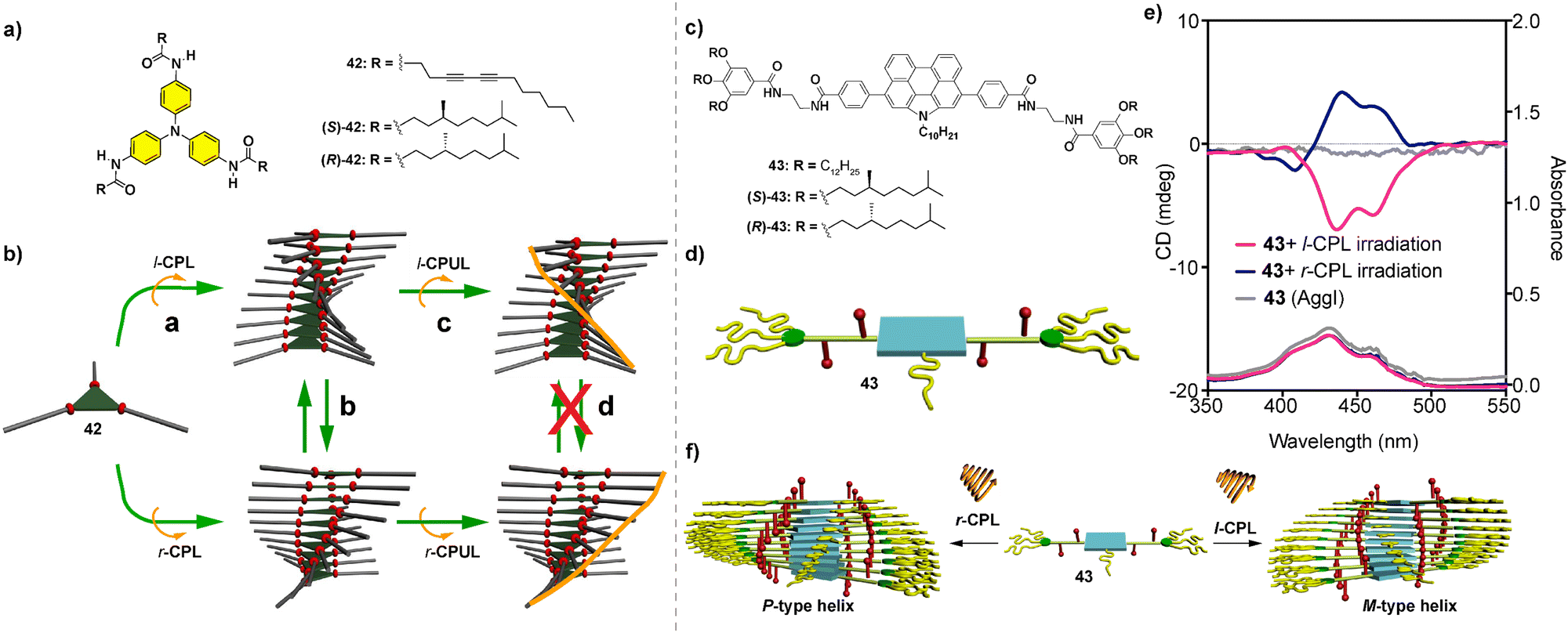 | ||
| Fig. 22 (a) Chemical structure of triarylamines 42. (b) Schematic representation of the formation of helical SPs and covalent polymers from 43 upon CPL irradiation. (c) and (d) Chemical structure and schematic illustration of the N-annulated perylenes 43. (e) UV-Vis (bottom) and CD (top) spectra of the aggregated species formed from 43 upon CPL irradiation. (f) Schematic illustration of the formation of M- or P-type helical SPs upon irradiation of 43 with l-CPL or r-CPL, respectively (reproduced from ref. 110b with permission from the American Chemical Society, copyright 2010). | ||
Our research group has recently reported the formation of helical SPs from the achiral N-annulated perylenetetracarboxamide 43 by irradiating with l-CPL or r-CPL (Fig. 22(c) and (d)). Previously, we observed that the chiral congeners (S)-43 and (R)-43 form enantioenriched helical SPs in nonpolar solvents like MCH that are able to generate CPL of opposite sign depending on the point chirality of the side chains. The irradiation of a diluted solution of achiral 43 in MCH with l-CPL or r-CPL results in a null dichroic response diagnostic of the presence of a racemic mixture of both M and P helices and without experiencing any symmetry breaking effect. However, if this diluted MCH solution of 43 is heated up to 90 °C, the temperature at which a complete disassembly of the aggregates is produced, and irradiated with l-CPL or r-CPL, the formation of enantioenriched helical SPs is observed, as demonstrated by the appearance of a clear CD response (Fig. 22(e) and (f)).112
Conclusions and outlook
The research field of SPs is a very active one, in which systems with increasing complexity are constantly emerging. The mathematical models elaborated to investigate the formation of SPs have contributed to explore new ways and find new functionalities for these self- or co-assembled supramolecular species. As an active branch within the ample field of SPs, chiral SPs are an excellent benchmark to generate helical structures that can model the origin of homochirality in Nature or determine other exciting functionalities. In this review, we highlight the most utilized strategies to build up chiral SPs. The most common strategy implies the self-assembly of chiral monomeric units endowed with any element of asymmetry, point or axial chirality, and functional groups able to establish stable non-covalent interactions. The supramolecular polymerization of a single, chiral component into helical aggregates is termed transfer of asymmetry. On the other hand, chiral SPs can be efficiently attained by the co-assembly of two structurally close species. Two different experiments are mainly used to afford chiral SPs from mixtures of two components: (i) the SaS experiments, in which achiral soldiers and chiral sergeants are mixed together to yield a homochiral mixture (Fig. 23(a)) and (ii) the MR experiments, in which a mixture of two enantiomers of the same scaffold at different ratios is mixed together to give rise to enantioenriched heteropolymers (Fig. 23(b)). In these experiments, the thermodynamic penalties HRP and MMP play a crucial role in controlling the ability of the system to experience amplification of asymmetry. Finally, we highlight some examples of symmetry breaking in which physical stimuli (stirring, solvent or light) generate an unbalanced mixture of enantiomers to produce a final enantioenriched sample. SPs can present enhanced mechanical, biomedical or optoelectronic features in comparison to covalent polymers or isolated molecules which results in outstanding applications.113 In addition to these applications, chiral SPs can also be applied in specific fields for chiral entities. Thus, chiral SPs are being elegantly utilized in materials science as CPL-emitters with remarkable emissive properties (Fig. 23(c)).66,114 Chiral SPs have been also reported to act as efficient electron-spin filtering materials upon deposition on conductive surfaces due to their chirality-induced spin selectivity properties (Fig. 23(d)). These spin selectivity properties make them candidates for various spintronic applications.34,114d,115 The helical aggregated species formed upon supramolecular polymerization of chiral monomeric units have also been utilized to induce chirality in catalytic processes. This induction can be achieved by functionalizing the monomeric units with elements amply utilized in catalytic reactions like phosphines or proline residues.116 A second strategy for chiral SPs to induce chirality in organic reactions involves the incorporation of chiral self-assembling units as pendants in copolymers that also contain these elements utilized in catalytic processes.117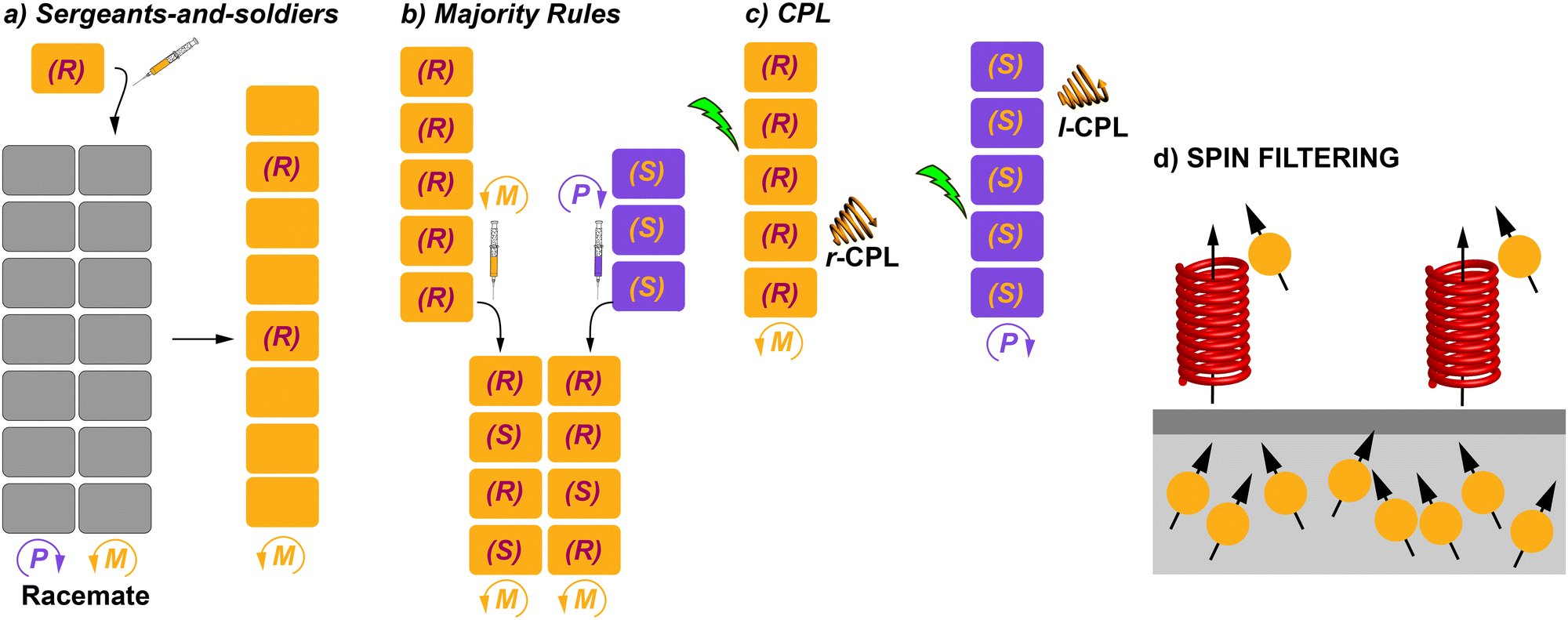 | ||
| Fig. 23 Schematic illustration of the function that chiral supramolecular polymers can exert in the field of amplification of asymmetry by developing sergeants-and-soldiers (a) and majority rules (b) experiments; the generation of efficient CPL-emitters (c) and the application of these helical structures as spin filters (d). | ||
Finally, this review contains a section devoted to amplification of asymmetry in which several co-assembly structures have been highlighted. A common feature of all those systems collected in this section is the structural similarity between the molecular components that finally generate the helical SPs. However, recent literature collected exciting examples of co-assemblies constituted by very dissimilar structures (covalent polymers, organogels, nanoparticles, etc.) able to form chiral aggregated species able to exert thrilling functions.118
In summary, this review highlights a collection of relevant examples of the techniques utilized to build up chiral SPs, highlighting relevant thermodynamic issues associated with the generation of such helical assemblies. In addition, and in a succinct manner, the outlook regarding the specific applications of chiral SPs is presented to display the interest of these supramolecular structures in the quest for new functionalities.
Abbreviations
| AFM | Atomic force microscopy |
| Agg | Aggregated species |
| BTA | 1,3,5-Benzene tricarboxamide |
| CBT | Carbonyl-bridged triarylamine |
| CD | Circular dichroism |
| CldMeOct | Chlorodimethyloctane |
| CPL | Circularly polarized luminescence |
| DMR | Diluted majority-rules |
| DTA | Dibenzoyl D-tartaric acid |
| ee | Enantiomeric excess |
| HRP | Helix reversal penalty |
| K e | Elongation binding constant |
| K n | Nucleation binding constant |
| LS | Living supramolecular polymerization |
| MCH | Methylcyclohexane |
| MMP | Mismatch penalty |
| MR | Majority rules |
| nDSC | Nanodifferential scanning calorimetry |
| NPDI | N-Annulated perylenediimides |
| OPETA | Oligo(phenylene ethynylene)tricarboxamide |
| OPV | Oligo-p-phenylene |
| PDI | Perylenediimide |
| RRE | Racemate rules effect |
| SANS | Small angle neutron scattering |
| SaS | Sergeants and soldiers |
| SP | Supramolecular polymer |
| SSP | Seeded supramolecular polymerization |
| TPBA | 1,3,5-Triphenylbenzenetricarboxamide |
| TPTA | Triphenylamine tricarboxamide |
| TTA | Triphenylene-2,6,10-tricarboxamide |
Author contributions
All authors have contributed to the preparation of the manuscript and agreed with the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was given by the MCIN/AEI of Spain (PID2020-113512GB-I00 and TED2021-130285B-I00). We sincerely acknowledge all our collaborators working in the field of chiral supramolecular polymers and, in particular, to Prof. Enrique Ortí for the continuous and helpful discussions.Notes and references
- L. Pasteur, C. R. Acad. Sci., 1848, 26, 535 Search PubMed.
- (a) S. P. Jacqueline, Chiral Auxiliaries and Ligands in Asymmetric Synthesis, Wiley, New York, NY, 1995 Search PubMed; (b) E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Comprehensive Asymmetric Catalysis, Springer, Berlin, 1999 Search PubMed; (c) R. Noyori, Asymmetric Catalysis in Organic Synthesis, Wiley, New York, NY, 1994 Search PubMed.
- (a) P. Peluso and B. Chankvetadze, Chem. Rev., 2022, 122, 13235 CrossRef CAS PubMed; (b) T. Buhse, J.-M. Cruz, M. E. Noble-Terán, D. Hochberg, J. M. Ribó, J. Crusats and J.-C. Micheau, Chem. Rev., 2021, 121, 2147 CrossRef CAS PubMed.
- (a) D. Amabilino, Chirality at the Nanoscale, Wiley-VCH, Weinheim, 2009 CrossRef; (b) W. H. Brooks, W. C. Guida and K. G. Daniel, Curr. Top. Med. Chem., 2011, 11, 760 CrossRef CAS PubMed; (c) G. Albano, G. Pescitelli and L. Di Bari, Chem. Rev., 2020, 120, 10145 CrossRef CAS PubMed.
- (a) F. C. Frank, Biochim. Biophys. Acta, 1953, 11, 459 CrossRef CAS PubMed; (b) M. M. Green, J.-W. Park, T. Sato, A. Teramoto, S. Lifson, R. L. B. Selinger and J. V. Selinger, Angew. Chem., Int. Ed., 1999, 38, 3138 CrossRef; (c) B. Lovejoy, S. Choe, D. Cascio, D. K. McRorie, W. F. DeGrado and D. Eisenberg, Science, 1993, 259, 1288 CrossRef CAS PubMed; (d) D. P. L. Luisi, The Emergence of Life: From Chemical Origins to Synthetic Biology, Cambridge University Press, Cambridge, 2006 CrossRef; (e) Q. Sallembien, L. Bouteiller, J. Crassous and M. Raynal, Chem. Soc. Rev., 2022, 51, 3436 RSC.
- (a) M. H. Todd, Chem. Soc. Rev., 2002, 31, 211 RSC; (b) D. G. Blackmond, Chem. Rev., 2020, 120(11), 4831 CrossRef CAS PubMed.
- (a) K. Soai, T. Shibata, H. Morioka and K. Choji, Nature, 1995, 378, 767 CrossRef CAS; (b) K. Soai and T. Kawasaki, Top. Curr. Chem., 2008, 284, 1 CrossRef CAS.
- D. T. J. Morris and J. Clayden, Chem. Soc. Rev., 2023, 52, 2480 RSC.
- (a) M. Novotny and G. J. Kleywegt, J. Mol. Biol., 2005, 347, 231 CrossRef CAS PubMed; (b) A. Banerjee, S. Datta, A. Pramanik, N. Shamala and P. Balaram, J. Am. Chem. Soc., 1996, 138, 9477 CrossRef.
- J. Bicerano, Prediction of Polymer Properties, CRC Press, Boca Raton, Fl, 3rd edn, 2002 Search PubMed.
- (a) Q. Wang, Y.-Q. Liu, R.-T. Gao and Z.-Q. Wu, J. Polym. Sci., 2023, 61, 189 CrossRef CAS; (b) M. J.-L. Tschan, R. M. Gauvin and C. M. Thomas, Chem. Soc. Rev., 2021, 50, 13587 RSC.
- (a) B. Zhao, J.-P. Deng and W.-T. Yang, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1679 CrossRef CAS; (b) M. Gingras, G. Félix and R. Peresuttia, Chem. Soc. Rev., 2013, 42, 1007 RSC; (c) N. Liu, L. Zhou and Z.-Q. Wu, Acc. Chem. Res., 2021, 54, 3953 CrossRef CAS PubMed.
- (a) T. Nakano and Y. Okamoto, Chem. Rev., 2001, 101, 4013 CrossRef CAS PubMed; (b) E. Yashima, Polym. J., 2010, 42, 3 CrossRef CAS; (c) E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102 CrossRef CAS PubMed; (d) F. Freire, E. Quiñoá and R. Riguera, Chem. Rev., 2016, 116, 1242 CrossRef CAS PubMed.
- (a) H. Gu, Y. Nakamura, T. Sato, A. Teramoto, M. M. Green, S. K. Jha, C. Andreola and M. P. Reidy, Macromolecules, 1998, 31, 6362 CrossRef CAS; (b) M. M. Green, M. P. Reidy, R. D. Johnson, G. Darling, D. J. O’Leary and G. Willson, J. Am. Chem. Soc., 1989, 111, 6452 CrossRef.
- M. M. Green, B. A. Garetz, B. Muñoz, H. Chang, S. Hoke and R. G. Cooks, J. Am. Chem. Soc., 1995, 117, 4181 CrossRef CAS.
- (a) M. Fujuki, Macromol. Rapid Commun., 2001, 22, 539 CrossRef; (b) M. Fujiki, J. R. Koe, K. Terao, T. Sato, A. Teramoto and J. Watanabe, Polym. J., 2003, 35, 297 CrossRef CAS.
- H. Engelkamp, S. Middelbeek and R. J. M. Nolte, Science, 1999, 284, 785 CrossRef CAS PubMed.
- T. Aida, A. Takemura, M. Fuse and S. Inoue, J. Chem. Soc., Chem. Commun., 1988, 391 RSC.
- (a) M. Fujiki, H. Tabei and T. Kurihara, J. Phys. Chem., 1988, 92, 1281 CrossRef CAS; (b) W. Zhang, M. Fujiki and X. Zhu, Chem. – Eur. J., 2011, 17, 10628 CrossRef CAS PubMed.
- C. Fouquey, J.-M. Lehn and A.-M. Levelut, Adv. Mater., 1990, 2, 254 CrossRef CAS.
- (a) L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071 CrossRef CAS PubMed; (b) M. Wehner and F. Würthner, Nat. Rev., 2019, 4, 38 Search PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, K. J. H. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601 CrossRef CAS PubMed.
- Z. Álvarez, A. N. Kolberg-Edelbrock, I. R. Sasselli, J. A. Ortega, R. Qiu, Z. Syrgiannis, P. A. Mirau, F. Chen, S. M. Chin, S. Weigand, E. Kiskinis and S. I. Stupp, Science, 2021, 374, 848 CrossRef PubMed.
- Y. Yamamoto, T. Fukushima, Y. Suna, N. Ishii, A. Saeki, S. Seki, S. Tagawa, M. Taniguchi, T. Kawai and T. Aida, Science, 2006, 314, 1761 CrossRef CAS PubMed.
- W. Zhao, J. Tropp, B. Qiao, M. Pink, J. D. Azoulay and A. H. Flood, J. Am. Chem. Soc., 2020, 142, 2579 CrossRef CAS PubMed.
- M. A. Martínez-Aguirre, Y. Li, N. Vanthuyne, L. Bouteiller and M. Raynal, Angew. Chem., Int. Ed., 2021, 133, 4229 CrossRef.
- O. Dumele, L. Đorđević, H. Sai, T. J. Cotey, M. H. Sangji, K. Sato, A. J. Dannenhoffer and S. I. Stupp, J. Am. Chem. Soc., 2022, 144, 3127 CrossRef CAS PubMed.
- S. Datta, Y. Kato, S. Higashiharaguchi, K. Aratsu, A. Isobe, T. Saito, D. D. Prabhu, Y. Kitamoto, M. J. Hollamby, A. J. Smith, R. Dalgliesh, N. Mahmoudi, L. Pesce, C. Perego, G. M. Pavan and S. Yagai, Nature, 2020, 583, 400 CrossRef CAS PubMed.
- W. H. Carothers, Chem. Rev., 1931, 8, 353 CrossRef CAS.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687 CrossRef CAS PubMed.
- C. R. Martínez and B. L. Iverson, Chem. Sci., 2012, 3, 2191 RSC.
- (a) O. Takahashi, Y. Kohno and M. Nishio, Chem. Rev., 2010, 110, 6049 CrossRef CAS PubMed; (b) G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565 CrossRef CAS PubMed; (c) T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef CAS.
- (a) A. R. A. Palmans, E. W. Meijer, A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8948 CrossRef CAS PubMed; (b) E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752 CrossRef CAS PubMed; (c) Y. Dorca, E. E. Greciano, J. S. Valera, R. Gómez and L. Sánchez, Chem. – Eur. J., 2019, 25, 5848 CrossRef CAS PubMed.
- (a) J. Han, S. Guo, H. Lu, S. Liu, Q. Zhao and W. Huang, Adv. Opt. Mater., 2018, 6, 1800538 CrossRef; (b) R. Naaman, Y. Paltiel and D. H. Waldeck, Nat. Rev. Chem., 2019, 3, 250 CrossRef CAS.
- in Fundamentals of Supramolecular Chirality, ed. R. Purrello and A. D’Urso, World Scientific, 2021 Search PubMed.
- (a) M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304 CrossRef CAS PubMed; (b) Y. Dorca, J. Matern, G. Fernández and L. Sánchez, Isr. J. Chem., 2019, 59, 869 CrossRef CAS; (c) S. Huang, H. Yu and Q. Li, Adv. Sci., 2021, 8, 2002132 CrossRef PubMed.
- (a) H. Cui, M. J. Webber and S. I. Stupp, Pept. Sci., 2010, 94, 1 CrossRef CAS PubMed; (b) Y. Shen, Y. Wang, I. W. Hamley, W. Qi, R. Su and Z. He, Prog. Polym. Sci., 2021, 123, 101469 CrossRef CAS; (c) Q. Li, Y. Wang, G. Zhang, R. Su and W. Qi, Chem. Soc. Rev., 2023, 52, 1549 RSC; (d) G. Zhang, L. Zhang, H. Rao, Y. Wang, Q. Li, W. Qi, X. Yang, R. Su and Z. He, J. Colloid Interface Sci., 2020, 577, 388 CrossRef CAS PubMed.
- C. C. Lee, C. Grenier, E. W. Meijer and A. P. H. J. Schenning, Chem. Soc. Rev., 2009, 38, 671 RSC.
- (a) S. Cantekin, T. F. A. de Greef and A. R. A. Palmans, Chem. Soc. Rev., 2012, 41, 6125 RSC; (b) C. Kulkarni, E. W. Meijer and A. R. A. Palmans, Acc. Chem. Res., 2017, 50, 1928 CrossRef CAS PubMed.
- M. M. J. Smulders, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2008, 130, 606 CrossRef CAS PubMed.
- M. P. Lightfoot, F. S. Mair, R. G. Pritchard and J. E. Warren, Chem. Commun., 1999, 1945 RSC.
- (a) S. A. H. Jansen, E. Weyandt, T. Aoki, T. Akiyama, Y. Itoh, G. Vantomme, T. Aida and E. W. Meijer, J. Am. Chem. Soc., 2023, 145, 4231 CrossRef CAS PubMed; (b) M. F. J. Mabesoone, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2020, 142, 19781 CrossRef CAS PubMed.
- R. P. M. Lafleur, S. M. C. Schoenmakers, P. Madhikar, D. Bochicchio, B. Baumeier, A. R. A. Palmans, G. M. Pavan and E. W. Meijer, Macromolecules, 2019, 52, 3049 CrossRef CAS PubMed.
- T. Schnitzer, M. D. Preuss, J. van Basten, S. M. C. Schoenmakers, A. J. H. Spiering, G. Vantomme and E. W. Meijer, Angew. Chem., Int. Ed., 2022, 61, e202206738 CrossRef CAS PubMed.
- (a) A. Das, G. Vantomme, A. J. Markvoort, H. M. M. ten Eikelder, M. García-Iglesias, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2017, 139, 7036 CrossRef CAS PubMed; (b) L. Su, J. Mosquera, M. F. J. Mabesoone, S. M. C. Schoenmakers, C. Muller, M. E. J. Vleugels, S. Dhiman, S. Wijker, A. R. A. Palmans and E. W. Meijer, Science, 2022, 377, 213 CrossRef CAS PubMed.
- K. M. Vonk, E. W. Meijer and G. Vantomme, Chem. Sci., 2021, 12, 13572 RSC.
- (a) S. Varela-Aramburu, G. Morgese, L. Su, S. M. C. Schoenmakers, M. Perrone, L. Leanza, C. Perego, G. M. Pavan, A. R. A. Palmans and E. W. Meijer, Biomacromolecules, 2020, 21, 4105 CrossRef CAS PubMed; (b) G. T. Williams, C. J. E. Haynes, M. Fares, C. Caltagirone, J. R. Hiscock and P. A. Gale, Chem. Soc. Rev., 2021, 50, 2737 RSC; (c) P. K. Hashim, J. Bergueiro, E. W. Meijer and T. Aida, Prog. Polym. Sci., 2020, 105, 101250 CrossRef CAS; (d) T. Aida and E. W. Meijer, Isr. J. Chem., 2020, 60, 33 CrossRef CAS.
- S. Díaz-Cabrera, Y. Dorca, J. Calbo, J. Aragó, R. Gómez, E. Ortí and L. Sánchez, Chem. – Eur. J., 2018, 24, 2826 CrossRef PubMed.
- (a) F. García, P. M. Viruela, E. Matesanz, E. Ortí and L. Sánchez, Chem. – Eur. J., 2011, 17, 7755 CrossRef PubMed; (b) F. García, P. A. Korevaar, A. Verlee, E. W. Meijer, A. R. A. Palmans and L. Sánchez, Chem. Commun., 2013, 49, 8674 RSC; (c) E. E. Greciano, J. Calbo, J. Buendía, J. Cerdá, J. Aragó, E. Ortí and L. Sánchez, J. Am. Chem. Soc., 2019, 141, 7463 CrossRef CAS PubMed.
- (a) Y. Dorca, R. Sánchez-Naya, J. Cerdá, J. Calbo, J. Aragó, R. Gómez, E. Ortí and L. Sánchez, Chem. – Eur. J., 2020, 26, 14700 CrossRef CAS PubMed; (b) F. García and L. Sánchez, J. Am. Chem. Soc., 2012, 134, 734 CrossRef PubMed.
- Z. Xie, V. Stepanenko, K. Radacki and F. Würthner, Chem. – Eur. J., 2012, 18, 7060 CrossRef CAS PubMed.
- J. Buendía, E. E. Greciano and L. Sánchez, J. Org. Chem., 2015, 80, 12444 CrossRef PubMed.
- (a) A. R. A. Palmans, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Angew. Chem., Int. Ed. Engl., 1997, 36, 2648 CrossRef CAS; (b) A. R. A. Palmans, J. A. J. M. Vekemans, R. A. Hikmet, H. Fischer and E. W. Meijer, Adv. Mater., 1998, 10, 873 CrossRef CAS; (c) T. Metzroth, A. Hoffmann, R. Martín-Rapún, M. M. J. Smulders, K. Pieterse, A. R. A. Palmans, J. A. J. M. Vekemans, E. W. Meijer, H. W. Spiess and J. Gauss, Chem. Sci., 2011, 2, 69 RSC.
- P. Besenius, G. Portale, P. H. H. Bomans, H. M. Janssen, A. R. A. Palmans and E. W. Meijer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17888 CrossRef CAS PubMed.
- B. Narayan, C. Kulkarni and S. J. George, J. Mater. Chem. C, 2013, 1, 626 RSC.
- M. A. Martínez, A. Doncel-Giménez, J. Cerdá, J. Calbo, R. Rodríguez, J. Aragó, J. Crassous, E. Ortí and L. Sánchez, J. Am. Chem. Soc., 2021, 143, 13281 CrossRef PubMed.
- J. Matern, Y. Dorca, L. Sánchez and G. Fernández, Angew. Chem., Int. Ed., 2019, 58, 16730 CrossRef CAS PubMed.
- (a) S.-S. Sun and L. R. Dalton, Introduction to Organic Electronic and Optoelectronic Materials and Devices, CRC Press, New York, 2008 Search PubMed; (b) S. R. Forrest, Nature, 2004, 428, 911 CrossRef CAS PubMed; (c) G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153 CrossRef CAS.
- P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. De Greef and E. W. Meijer, Nature, 2012, 481, 492 CrossRef CAS PubMed.
- (a) S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188 CrossRef CAS PubMed; (b) S. Ogi, T. Fukui, M. L. Jue, M. Takeuchi and K. Sugiyasu, Angew. Chem., Int. Ed., 2014, 53, 14363 CrossRef CAS PubMed.
- J. Kang, D. Mijayima, T. Mori, Y. Inoue, Y. Itoh and T. Aida, Science, 2015, 347, 646 CrossRef CAS PubMed.
- S. Dhiman and S. J. George, Bull. Chem. Soc. Jpn., 2018, 91, 687 CrossRef CAS.
- S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, J. Am. Chem. Soc., 2015, 137, 3300 CrossRef CAS PubMed.
- (a) W. Wagner, M. Wehner, V. Stepanenko and F. Würthner, J. Am. Chem. Soc., 2019, 141, 12044 CrossRef CAS PubMed; (b) A. Chakraborty, G. Ghosh, D. S. Pal, S. Varghese and S. Ghosh, Chem. Sci., 2019, 10, 7345 RSC; (c) E. E. Greciano, J. Calbo, E. Ortí and L. Sánchez, Angew. Chem., Int. Ed., 2020, 59, 17517 CrossRef CAS PubMed; (d) J. Matern, Z. Fernández, N. Bäumer and G. Fernández, Angew. Chem., Int. Ed., 2022, 61, e202203783 CrossRef CAS PubMed; (e) C. Naranjo, S. Adalid, R. Gómez and L. Sánchez, Angew. Chem., Int. Ed., 2023, 62, e202218572 CrossRef CAS PubMed.
- J. S. Varela, R. Gómez and L. Sánchez, Small, 2017, 1702437 Search PubMed.
- L. López-Gandúl, C. Naranjo, C. Sánchez, R. Rodríguez, R. Gómez, J. Crassous and L. Sánchez, Chem. Sci., 2022, 13, 11577 RSC.
- K. Tashiro, T. Saito, H. Arima, N. Suda, B. Vedhanarayanan and S. Yagai, Chem. Rec., 2022, 22, e202100252 CrossRef CAS PubMed.
- S. Yagai, M. Yamauchi, A. Kobayashi, T. Karatsu, A. Kitamura, T. Ohba and Y. Kikkawa, J. Am. Chem. Soc., 2012, 134, 18205 CrossRef CAS PubMed.
- J. S. Varela, H. Arima, C. Naranjo, T. Saito, N. Suda, R. Gómez, S. Yagai and L. Sánchez, Angew. Chem., Int. Ed., 2022, 134, e202114290 CrossRef.
- (a) A. T. Haedler, S. C. J. Meskers, R. H. Zha, M. Kivala, H.-W. Schmidt and E. W. Meijer, J. Am. Chem. Soc., 2016, 138, 10539 CrossRef CAS PubMed; (b) Y. Dorca, J. Cerdá, J. Aragó, E. Ortí and L. Sánchez, Chem. Mater., 2019, 31, 7024 CrossRef CAS.
- B. Adelizzi, N. J. Van Zee, L. N. J. de Windt, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2019, 141, 6110 CrossRef CAS PubMed.
- D. R. Fenwick and H. B. Kagan, Top. Stereochem., 1999, 257 CAS.
- C. Puchot, O. Samuel, E. Dunach, S. Zhao, C. Agami and H. B. Kagan, J. Am. Chem. Soc., 1986, 108, 2353 CrossRef CAS PubMed.
- (a) J. V. Selinger and R. L. B. Selinger, Macromolecules, 1998, 31, 2488 CrossRef CAS; (b) S. K. Jha, K. S. Cheon, M. M. Green and J. V. Selinger, J. Am. Chem. Soc., 1999, 121, 1665 CrossRef CAS; (c) A. J. Wilson, J. Van Gestel, R. P. Sijbesma and E. W. Meijer, Chem. Commun., 2006, 4404 RSC.
- J. van Gestel, Macromolecules, 2004, 37, 3894 CrossRef CAS.
- J. van Gestel, A. R. A. Palmans, B. Titulaer, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 5490 CrossRef CAS PubMed.
- M. M. J. Smulders, I. A. W. Filot, J. M. A. Leenders, P. van der Schoot, A. R. A. Palmans, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2010, 132, 611 CrossRef CAS PubMed.
- M. M. J. Smulders, P. J. M. Stals, T. Mes, T. F. E. Paffen, A. P. H. J. Schenning, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2010, 132, 620 CrossRef CAS PubMed.
- L. Brunsfeld, B. G. G. Lohmeijer, J. A. J. M. Vekemans and E. W. Meijer, J. Incl. Phenom. Macrocycl. Chem., 2001, 41, 61 CrossRef CAS.
- L. Brunsveld, B. G. G. Lohmeijer, J. A. J. M. Vekemans and E. W. Meijer, Chem. Commun., 2000, 2305 RSC.
- L. Brunsveld, A. P. H. J. Schenning, M. A. C. Broeren, H. M. Janssen, J. A. J. M. Vekemans and E. W. Meijer, Chem. Lett., 2000, 292 CrossRef CAS.
- J. J. van Gorp, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2002, 124, 14759 CrossRef CAS PubMed.
- H. M. M. ten Eikelder, B. Adelizzi, A. R. A. Palmans and A. J. Markvoort, J. Phys. Chem. B, 2019, 123, 6627 CrossRef CAS PubMed.
- A. J. Markvoort, H. M. M. ten Eikelder, P. A. J. Hilbers, T. F. A. de Greef and E. W. Meijer, Nat. Commun., 2011, 2, 509 CrossRef PubMed.
- T. Kim, T. Mori, T. Aida and D. Miyajima, Chem. Sci., 2016, 7, 6689 RSC.
- J. S. Valera, R. Gómez and L. Sánchez, Angew. Chem., Int. Ed., 2019, 58, 510 CrossRef CAS PubMed.
- C. Naranjo, Y. Dorca, G. Ghosh, R. Gómez, G. Fernández and L. Sánchez, Chem. Commun., 2021, 57, 4500 RSC.
- Y. Dorca, C. Naranjo, G. Ghosh, R. Gómez, G. Fernández and L. Sánchez, Org. Mater., 2020, 02, 41 CrossRef CAS.
- B. Adelizzi, A. Aloi, A. J. Markvoort, H. M. M. Ten Eikelder, I. K. Voets, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2018, 140, 7168 CrossRef CAS PubMed.
- J. Kumar, H. Tsumatori, J. Yuasa, T. Kawai and T. Nakashima, Angew. Chem., Int. Ed., 2015, 54, 5943 CrossRef CAS PubMed.
- V. Ayzac, M. Dirany, M. Raynal, B. Isare and L. Bouteiller, Chem. – Eur. J., 2021, 27, 9627 CrossRef CAS PubMed.
- S. Ghosh, X.-Q. Li, V. Stepanenko and F. Würthner, Chem. – Eur. J., 2008, 14, 11343 CrossRef CAS PubMed.
- X.-X. Chen, Y.-B. Jiang and E. V. Anslyn, Chem. Commun., 2016, 52, 12669 RSC.
- X.-X. Chen, X.-Y. Lin, X. Wu, P. A. Gale, E. V. Anslyn and Y.-B. Jiang, J. Org. Chem., 2019, 84, 14587 CrossRef CAS PubMed.
- N. Suda, T. Saito, H. Arima and S. Yagai, Chem. Sci., 2022, 13, 3249 RSC.
- T. Saito, T. Kajitani and S. Yagai, J. Am. Chem. Soc., 2023, 145, 443 CrossRef CAS PubMed.
- J. Buendía, F. García, B. Yelamos and L. Sánchez, Chem. Commun., 2016, 52, 8830 RSC.
- A. Arango-Restrepo, O. Arteaga, D. Barragán and J. M. Rubi, Phys. Chem. Chem. Phys., 2023, 25, 9238 RSC.
- (a) J. M. Ribó, J. Crusats, F. Sagués, J. Claret and R. Rubires, Science, 2001, 292, 2063 CrossRef PubMed; (b) N. Micali, H. Engelkamp, P. G. van Rhee, P. C. M. Christianen, L. Monsù Scolaro and J. C. Maan, Nat. Chem., 2012, 4, 201 CrossRef CAS PubMed; (c) M. Gaeta, R. Randazzo, C. Costa, R. Purrello and A. D’Urso, Chem. – Eur. J., 2023,(29), e202202337 CrossRef CAS PubMed.
- (a) A. Romeo, M. A. Castriciano, I. G. Occhiuto, R. Zagami, R. F. Pasternack and L. Monsù Scolaro, J. Am. Chem. Soc., 2014, 136, 40 CrossRef CAS PubMed; (b) I. G. Occhiuto, M. A. Castriciano, M. Trapani, R. Zagami, A. Romeo, R. F. Pasternack and L. Monsù Scolaro, Int. J. Mol. Sci., 2020, 21, 4001 CrossRef CAS PubMed.
- (a) A. D'Urso, R. Randazzo, L. Lo Faro and R. Purrello, Angew. Chem., Int. Ed., 2010, 49, 108 CrossRef PubMed; (b) J. Sun, Y. Li, F. Yan, C. Liu, Y. Sang, F. Tian, Q. Feng, P. Duan, L. Zhang, X. Shi, B. Ding and M. Liu, Nat. Commun., 2018, 9, 2599 CrossRef PubMed.
- A. Tsuda, M. D. Alam, T. Harada, T. Yamaguchi, N. Ishii and T. Aida, Angew. Chem., Int. Ed., 2007, 46, 8198 CrossRef CAS PubMed.
- M. Wolffs, S. J. George, Z. Tomovic, S. C. J. Meskers, A. P. H. J. Schenning and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8203 CrossRef CAS PubMed.
- J. Buendía, J. Calbo, E. Ortí and L. Sánchez, Small, 2017, 13, 1603880 CrossRef PubMed.
- M. M. Green, C. Khatri and N. C. A. Peterson, J. Am. Chem. Soc., 1993, 115, 4941 CrossRef CAS.
- B. Isare, M. Linares, L. Zargarian, S. Fermandjian, M. Miura, S. Motohashi, N. Vanthuyne, R. Lazzaroni and L. Bouteiller, Chem. – Eur. J., 2010, 16, 173 CrossRef CAS PubMed.
- V. Stepanenko, X. Q. Li, J. Gershberg and F. Würthner, Chem. – Eur. J., 2013, 19, 4176 CrossRef CAS PubMed.
- (a) M. L. Ślęczkowski, M. F. J. Mabesoone, P. Ślęczkowski, A. R. A. Palmans and E. W. Meijer, Nat. Chem., 2021, 13, 200 CrossRef PubMed; (b) M. D. Preuss, S. A. H. Jansen, G. Vantomme and E. W. Meijer, Isr. J. Chem., 2021, 61, 622 CrossRef CAS.
- (a) J. A. Le Bel, Bull. Soc. Chim. Fr., 1874, 22, 337 Search PubMed; (b) J. H. Van’t Hoff, Pamphlet, Utrecht, 1874.
- (a) J. Kim, J. Lee, W. Y. Kim, H. Kim, S. Lee, H. C. Lee, Y. S. Lee, M. Seo and S. Y. Kim, Nat. Commun., 2015, 6, 6959 CrossRef CAS PubMed; (b) J. S. Kang, S. Kang, J.-M. Suh, S. M. Park, D. K. Yoon, M. H. Lim, W. Y. Kim and M. Seo, J. Am. Chem. Soc., 2022, 144, 2657 CrossRef CAS PubMed.
- J. Hu, J. Xie, H. Zhang, C. He, Q. Zhang and G. Zou, Chem. Commun., 2019, 55, 4953 RSC.
- E. E. Greciano, R. Rodríguez, K. Maeda and L. Sánchez, Chem. Commun., 2020, 56, 2244 RSC.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813 CrossRef CAS PubMed.
- (a) Y. Wang, Y. Jiang, X. Zhu and M. Liu, J. Phys. Chem. Lett., 2019, 10, 5861 CrossRef CAS PubMed; (b) S. Lee, K. Y. Kim, S. H. Jung, J. H. Lee, M. Yamada, R. Sethy, T. Kawai and J. H. Jung, Angew. Chem., Int. Ed., 2019, 58, 18878 CrossRef CAS PubMed; (c) Y. Wang, K. Wan, F. Pan, X. Zhu, Y. Jiang, H. Wang, Y. Chen, X. Shi and M. Liu, Angew. Chem., Int. Ed., 2021, 60, 16615 CrossRef CAS PubMed; (d) R. Rodríguez, C. Naranjo, A. Kumar, P. Matozzo, T. Kumar Das, Q. Zhu, N. Vanthuyne, R. Gómez, R. Naaman, L. Sánchez and J. Crassous, J. Am. Chem. Soc., 2022, 144, 7709 CrossRef PubMed.
- (a) C. Kulkarni, A. K. Mondal, T. K. Das, G. Grinbom, F. Tassinari, M. F. J. Mabesoone, E. W. Meijer and R. Naaman, Adv. Mater., 2020, 32, 1904965 CrossRef CAS PubMed; (b) A. K. Mondal, M. D. Preuss, M. L. Ślęczkowski, T. K. Das, G. Vantomme, E. W. Meijer and R. Naaman, J. Am. Chem. Soc., 2021, 143, 7189 CrossRef CAS PubMed.
- (a) M. Raynal, F. Portier, P. W. N. M. van Leeuwen and L. Bouteiller, J. Am. Chem. Soc., 2013, 135, 17687 CrossRef CAS PubMed; (b) A. Desmarchelier, X. Caumes, M. Raynal, A. Vidal-Ferran, P. W. N. M. van Leeuwen and L. Bouteiller, J. Am. Chem. Soc., 2016, 138, 4908 CrossRef CAS PubMed; (c) M. A. Martínez-Aguirre, Y. Li, N. Vanthuyne, L. Bouteiller and M. Raynal, Angew. Chem., Int. Ed., 2021, 60, 4183 CrossRef PubMed; (d) K. S. Lee and J. R. Parquette, Chem. Commun., 2015, 51, 15653 RSC.
- E. Huerta, P. J. M. Stals, E. W. Meijer and A. R. A. Palmans, Angew. Chem., Int. Ed., 2013, 52, 2906 CrossRef CAS PubMed.
- (a) T. Zhao, J. Han, P. Duan and M. Liu, Acc. Chem. Res., 2020, 53, 1279 CrossRef CAS PubMed; (b) S. Maniappan, C. Dutta, D. M. Solís, J. M. Taboada and J. Kumar, Angew. Chem., Int. Ed., 2023, 62, e202300461 CrossRef CAS PubMed; (c) S. K. Sen, R. D. Mukhopadhyay, S. Choi, I. Hwang and K. Kim, Chem., 2023, 9, 624 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |