
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembled systems for artificial photosynthesis†
Sebastiano
Campagna
 *,
Francesco
Nastasi
,
Giuseppina
La Ganga
,
Scolastica
Serroni
,
Antonio
Santoro
,
Antonino
Arrigo
and
Fausto
Puntoriero
*
*,
Francesco
Nastasi
,
Giuseppina
La Ganga
,
Scolastica
Serroni
,
Antonio
Santoro
,
Antonino
Arrigo
and
Fausto
Puntoriero
*
Department of Chemical, Biological, Pharmaceutical and Environmental Sciences, University of Messina, via F. Stagno d’Alcontres 31, 98166 Messina, Italy. E-mail: campagna@unime.it; fpuntoriero@unime.it
First published on 7th November 2022
Abstract
The last few decades have seen an impressive development in molecular-based artificial photosynthesis, thanks to the design of integrated light-harvesting antennae, charge separation systems, and catalysts for water oxidation or hydrogen production based on covalently linked subunits. However, in recent years, self-assembly and spontaneous aggregation of components emerged – sometimes also through serendipity – for the preparation of multicomponent systems aimed to perform the basic processes needed for artificial photosynthesis. Here we critically discuss some key articles that have recently shown the potential of self-assembly for artificial photosynthesis, ranging from self-assembly of antennae and charge separation systems to integrated antenna/catalyst assemblies, to planned co-localization of various components into restricted environments. It is evident that self-assembly can generate emerging properties with respect to the non-aggregated species, and such emerging properties can be quite convenient for designing efficient photocatalytic systems.
Introduction
Artificial photosynthesis, i.e. conversion of raw materials (like water and carbon dioxide) into high-energy content chemical species (ideally, hydrogen and reduced forms of CO2) by using solar light, is an important research field, on considering the expected increase of global energy demand and environmental problems which inevitably require the development of renewable energy sources.1 Within this framework, the long-standing research on the development of molecular-based components of artificial photosynthesis, essentially light-harvesting antenna systems and reaction centers featuring photoinduced charge separation, has mainly been centered on covalently linked systems. This has also produced important advancements of knowledge on the mechanism of photoinduced energy and electron transfer in multicomponent and supramolecular systems,2–4 strongly contributing to the development of many other areas of chemistry, such as luminescent sensors, bio-imaging devices, and molecular logics.5Indeed, covalently linked multicomponent systems allow having control on distances, to tune the energy gradient, and to investigate the role of spacers in the rate and efficiency of the intercomponent photoinduced processes,2–4 by avoiding the limit of diffusion, inherent to bimolecular reactions. However, in recent times, both planned and unexpected results have revealed that self-assembly or self-aggregation processes can give rise to organized assemblies capable of performing efficient artificial photosynthesis, in particular for photoinduced water oxidation (a needed process for water splitting) and photoinduced hydrogen production. Here, we review some examples of key reports which identified synthetic self-assembled or self-aggregated species are an emerging issue within the realm of artificial photosynthesis. Emerging features exclusively showed by the self-assembly or aggregate assemblies are also evidenced.
Light-harvesting antenna and charge separation systems
One of the first review-type reports discussing self-assembly strategies for preparing large light-harvesting systems was based on multicomponent building blocks made of arylene diimide species, including also some porphyrin subunits.6 Representative examples are shown in Fig. 1.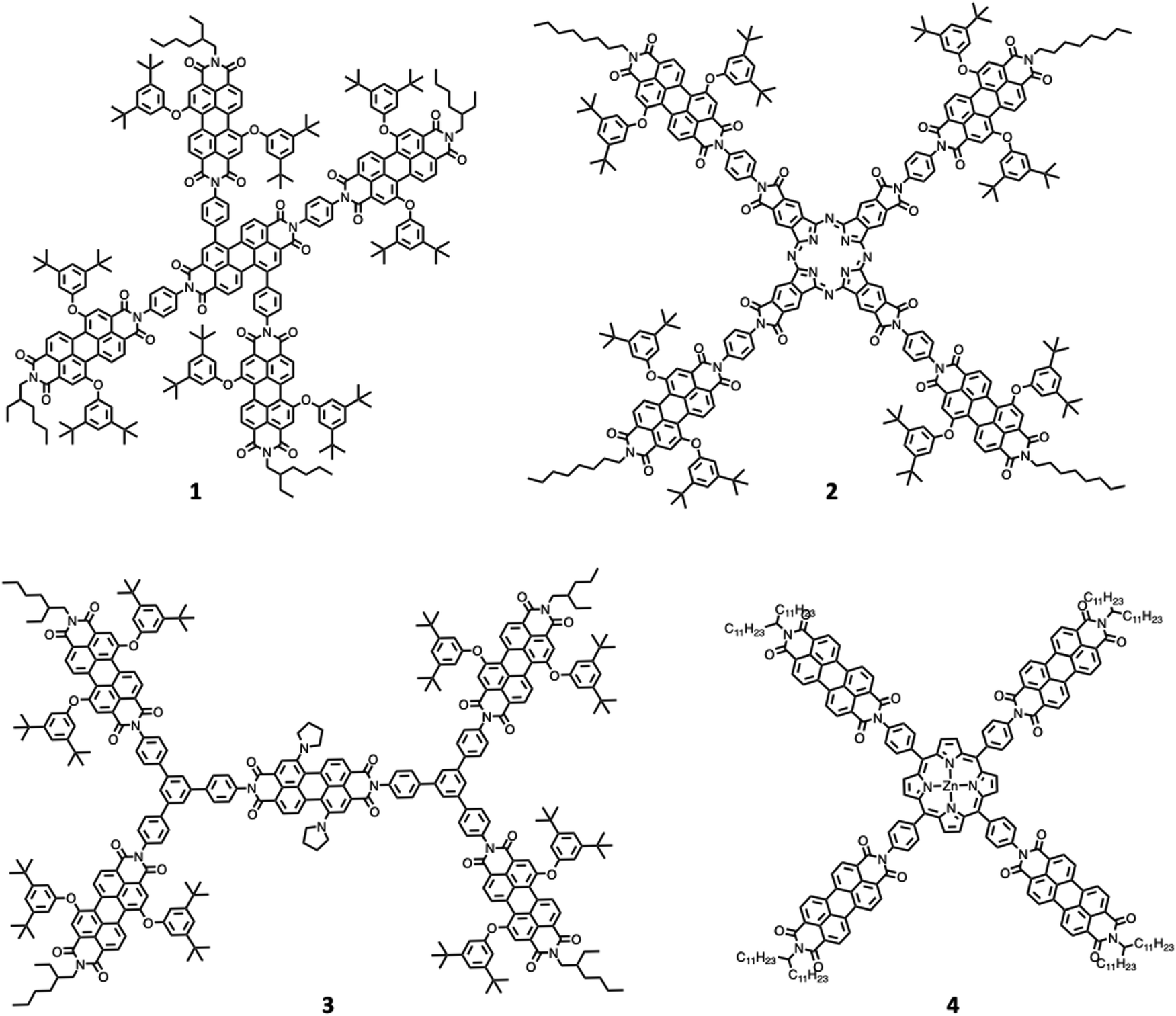 | ||
| Fig. 1 Chemical structures of representative arylene diimide 1–4 species. | ||
As indicated by small- and wide-angle X-ray scattering (SAXS/WAXS), these species, which can assume a somewhat flat conformation, tend to aggregate by π–π interactions, thus leading to monodisperse noncovalent assemblies. In particular, compound 3 forms π-stacked dimers in solution.7 This dimeric array exhibited an emergent behavior, only due to the aggregation: in fact, energy transfer from the excited states involving the peripheral perylene diimide units to the (lower-energy) excited states of the core occurs with a time constant of 21 ps and is followed by quantitative excited-state symmetry breaking of the core within the aggregated dimer, with a time constant of 7 ps. The so-formed ion pair recombines with a time constant of 420 ps.7Fig. 2 schematizes the processes. Therefore, electron transfer only occurs in the dimeric system, due to aggregation.
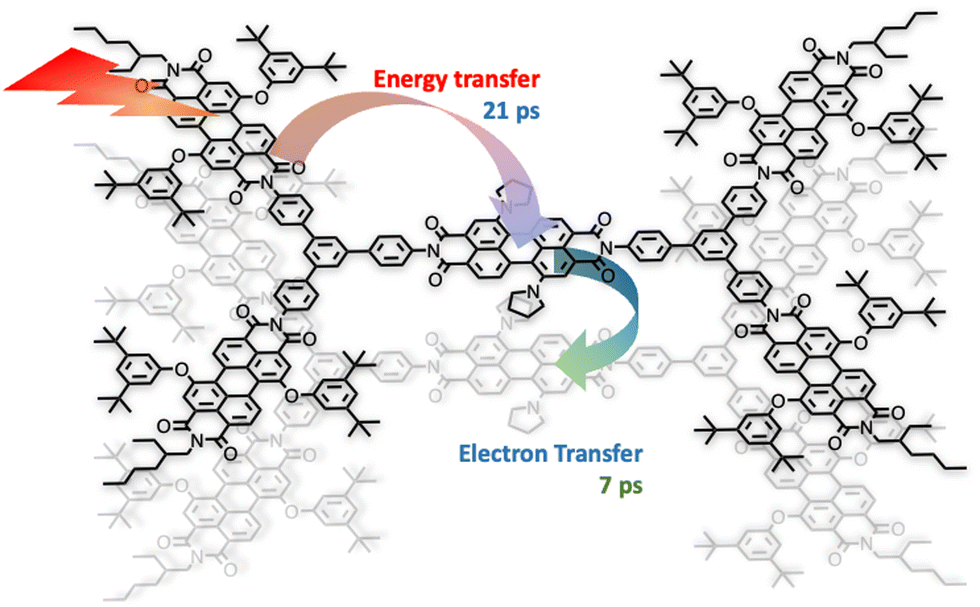 | ||
| Fig. 2 Schematization of the photoinduced processes in the dimeric array of 3. | ||
An intriguing behavior is also exhibited by compound 4, which self-assembles into nearly monodisperse assemblies comprising five molecules (pentamers) arranged in columnar stacks.8 In 4, excitation quantitatively produces a radical ion pair, with the radical cation on the Zn porphyrin core and the anionic radical on a perylene diimide subunit (ZnTPP+-PDI−), in a few picoseconds. Transient absorption spectroscopy indicates that in the aggregate pentamers, the PDI− radical anion strongly interacts with the adjacent PDI molecules leading to electron migration within the columnar stack. Charge recombination is consequently slowed down with respect to non-aggregated similar models (the time constant of charge recombination is 4.8 ns vs. 3.0 ns, for aggregate pentamers and the reference – non-aggregated – ZnTPP-PDI donor–acceptor bicomponent species, respectively). Also, in this case, this demonstrates that self-assembly can have a beneficial effect as far as important factors for artificial photosynthesis – in the specific case, charge recombination – are concerned. Further examples on aggregation of perylene bisimide species and the photophysical properties of the aggregates can be found in ref. 9.
Luminescent and redox-active metal dendrimers made of Ru(II) and Os(II) polypyridine building blocks have been extensively studied, mainly by our group, in the last thirty years. In most cases, the chelating ligands used were 2,2′-bipyridine (bpy) and 2,3-bis(2′-pyridyl)pyrazine (2,3-dpp) as terminal and bridging ligands, respectively.10 Due to the “complexes-as ligands/complexes-as metals” (cmpl) synthetic approaches coupled with iterative divergent or convergent strategies, dendrimers containing up to 22 metal-based chromophoric units were prepared, in which the large absorption cross-section with solar spectrum (a consequence of several, intense metal-to-ligand charge-transfer, MLCT, transitions) and quantitative electronic energy transfer occurring by Dexter mechanism on the fs timescale allows these dendrimers to behave as efficient light-harvesting antennae,10c–f also successfully employed for photoinduced water oxidation.11,12
However, recently it was shown that, despite the large charge of the multicomponent dendrimers, the second generation, decametallic dendrimers self-aggregate in solution, leading to improved energy transfer efficiency in some cases.13 Actually, SAXS experiments indicated that even at low concentrations (of the order of 10−5 M) in acetonitrile, the decanuclear dendrimers Ru10 and OsRu9 (see Fig. 3) aggregate to form larger structures. Whereas the consequence of the aggregation for Ru10 is relatively small (the energy emission shifts to slightly higher energy compared to the non-aggregated system, probably due to environmental effects), the effect on OsRu9 is much more relevant. In fact, energy transfer from MLCT excited states involving the peripheral Ru(II)-based chromophores to the lower-lying MLCT state involving the Os(II)-based core was negligible in the isolated OsRu9, due to the presence of intermediate Ru(II)-based chromophores, which have a highest-lying MLCT state(s) compared to peripheral and core chromophore units and behave as a barrier for the peripheral-to-core energy transfer.10a,c However, in the aggregated systems, femtosecond pump–probe spectroscopy indicates that quantitative peripheral-to-core energy transfer takes place, with a time constant of 18 ps.13 It is not totally clear whether the peripheral-to-core energy transfer involves different dendrimers within the aggregated assembly, taking advantage of possible inter–dendrimer interactions (so mimicking the energy transfer between LH2 and LH1 of natural systems;14 see Fig. 4) or the superexchange-assisted intercomponent energy transfer within a single dendrimer is accelerated in the aggregated systems: nevertheless, aggregation leads to an improved antenna effect. More recently, aggregation of dendrimers has also been proved by computation and visualized by scanning transmission electron microscopy, as shown in Fig. 5.15
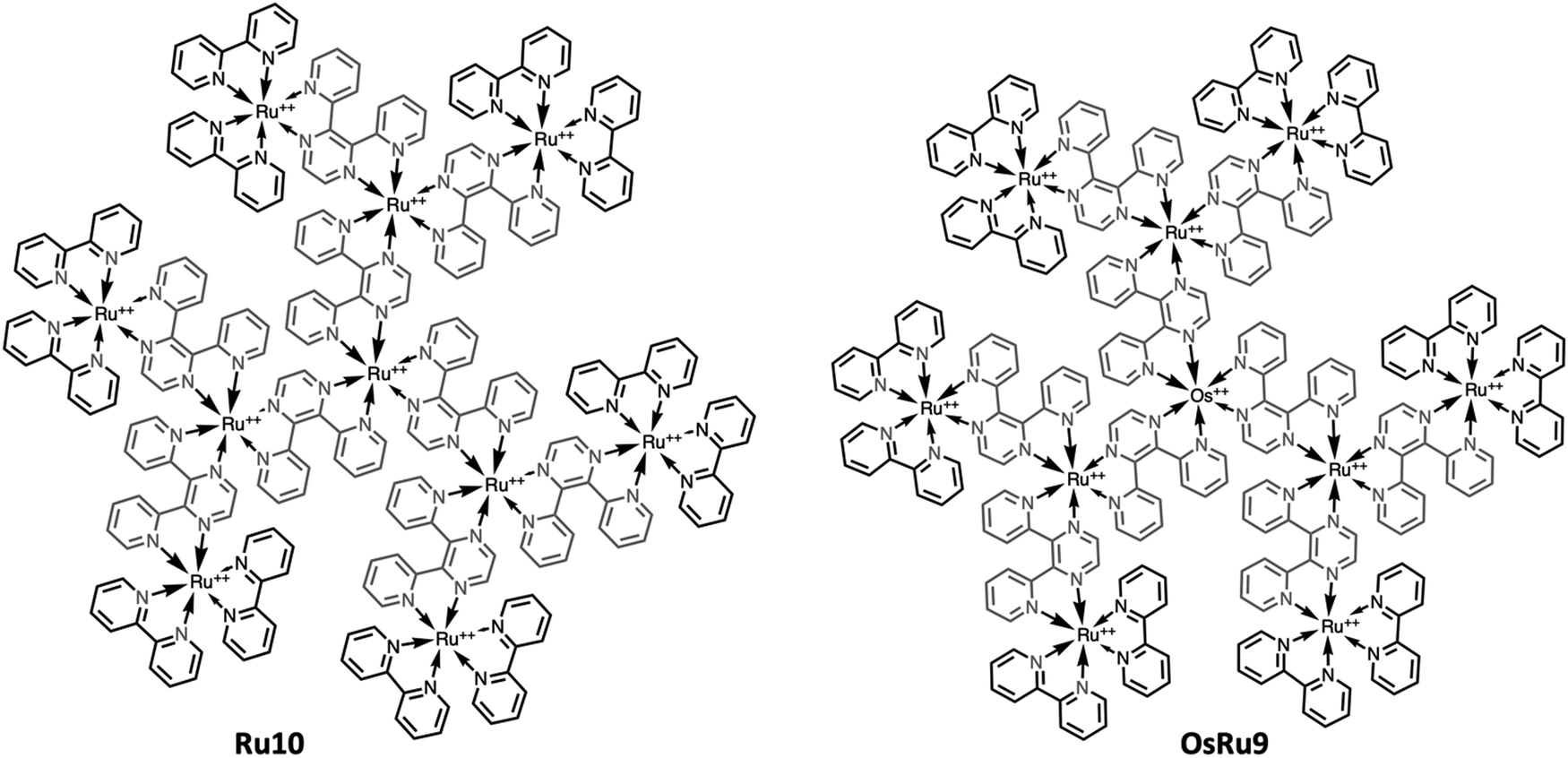 | ||
| Fig. 3 Chemical structures of light-harvesting metal dendrimers Ru10 (left) and OsRu9 (right). | ||
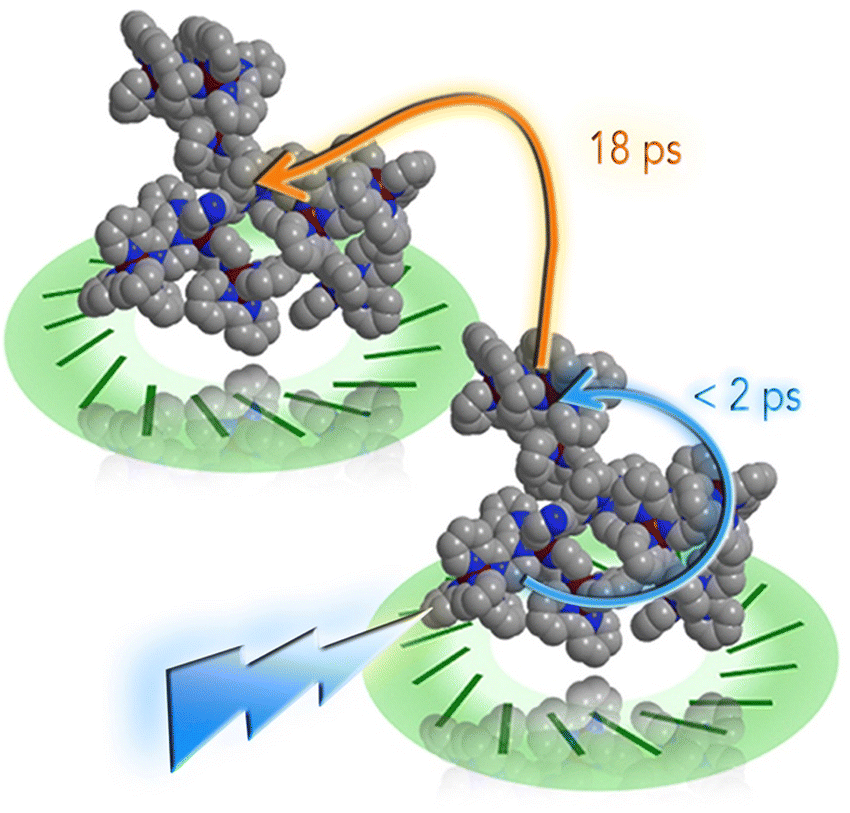 | ||
| Fig. 4 Cartoon of the photoinduced intra- and inter-molecular energy transfer in OsRu9 aggregates. Reprinted from ref. 15, Copyright (2017), with permission from Elsevier. | ||
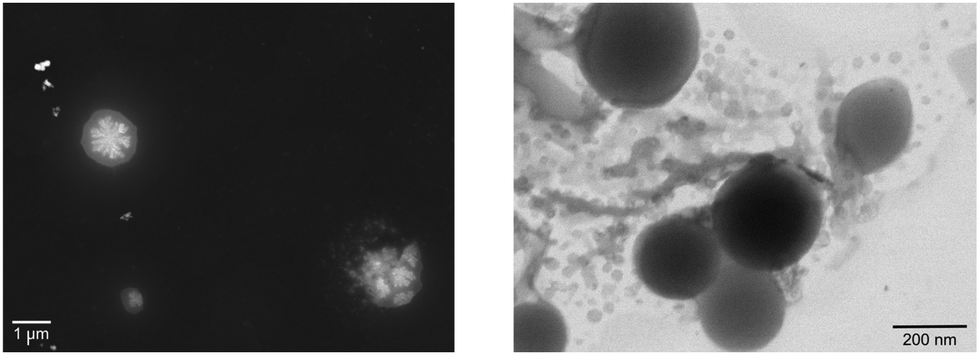 | ||
| Fig. 5 STEM image obtained from (left, registered in annular dark field-ADF) 5 × 10−6 M and from (right, registered in bright field-BF) 5 × 10−5 M acetonitrile solution of OsRu9. | ||
Organized aggregation of photosensitizers and catalysts
One of the most efficient systems for photoinduced water oxidation in the presence of a sacrificial acceptor was reported about 10 years ago,12 with the tetranuclear first-generation dendrimer [Ru{(μ-2,3-dpp)Ru(bpy)2}3]8+ (Ru4) used as the photosensitizer and the tetraruthenium polyoxometalate [Ru4(μ-O)4(μ-OH)2(H2O)4(γ-SiW10O36)2]10− (Ru4POM) as water oxidation photocatalyst (see Fig. 6). In the presence of persulfate salts as the sacrificial acceptor, the Ru4/Ru4POM system yields molecular oxygen with a quantum yield of 0.30 upon light excitation at 550 nm: considering that two photons were needed to produce an oxygen molecule, this meant that 60% of the absorbed photons were profitably used to perform the water oxidation process.11,12,16 Moreover, the absorption spectrum of Ru4, which extends over 700 nm, made it possible to take advantage of a large portion of visible light for driving the light-induced process. Quite interestingly, the photocatalytic water oxidation process was mainly limited by consumption of the sacrificial agent, so indicating the high photostability of the system.12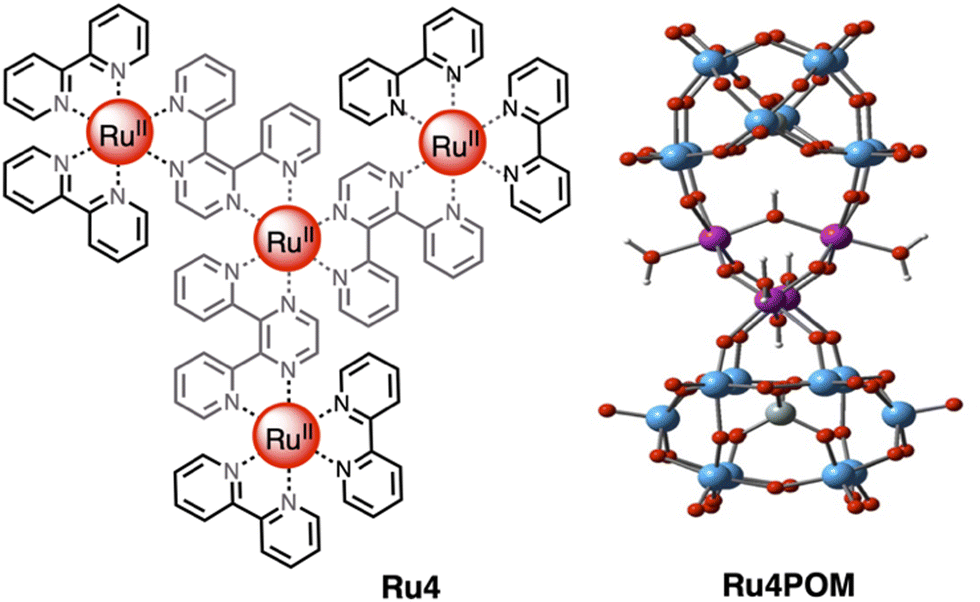 | ||
| Fig. 6 Chemical structures of the light-harvesting photosensitizer Ru4 (left) and of the water oxidation catalyst Ru4POM (right). Counterions omitted. | ||
Successive investigation of the machinery of the process, also taking advantage of ultrafast spectroscopy, revealed very interesting aspects:17 first of all, it was proved that persulfate anions, usually expected to be involved in the early event of the photoinduced electron transfer processes by oxidizing the excited photosensitizer, were not able to quench the MLCT state of Ru4, for thermodynamical reasons. The quenching process was indeed started by reductive electron transfer of the excited Ru4 by the Ru4POM catalyst. Successively, the reduced form of the photosensitizer transferred an electron to the sacrificial acceptor (the persulfate anion), a process that regenerated the photosensitizer ground state.
The sequence of electron transfer events occurring in the “electron acceptor/photosensitizer/catalyst” (SA/PS/Cat) system involving Ru4 and Ru4POM (and persulfate anion as an electron acceptor) was defined as an “anti-biomimetic scheme”, since it was reversed in comparison with the usual other SA/PS/Cat examples reported in the literature (and indeed also in comparison with the natural photosynthetic schemes18), for which the usual scheme assumes first (i) electron transfer from the excited photosensitizer to the electron acceptor, followed by (ii) regeneration of the photosensitizer ground state via the hole transfer from the oxidized form of the photosensitizer to the catalyst (Fig. 7). Noteworthy, the “anti-biomimetic” mechanism was later demonstrated to be efficient for charge injection into nanostructured TiO2 electrodes for dye-sensitized solar cells.19 However, the “anti biomimetic” mechanism, although representing an interesting novelty in the field, was not the unique novelty introduced by the Ru4/Ru4POM system: even more interestingly, the data indicated that self-assembling between Ru4 and Ru4POM (with the assembly also including the persulfate anion) was a needed requisite for photoinduced water oxidation.
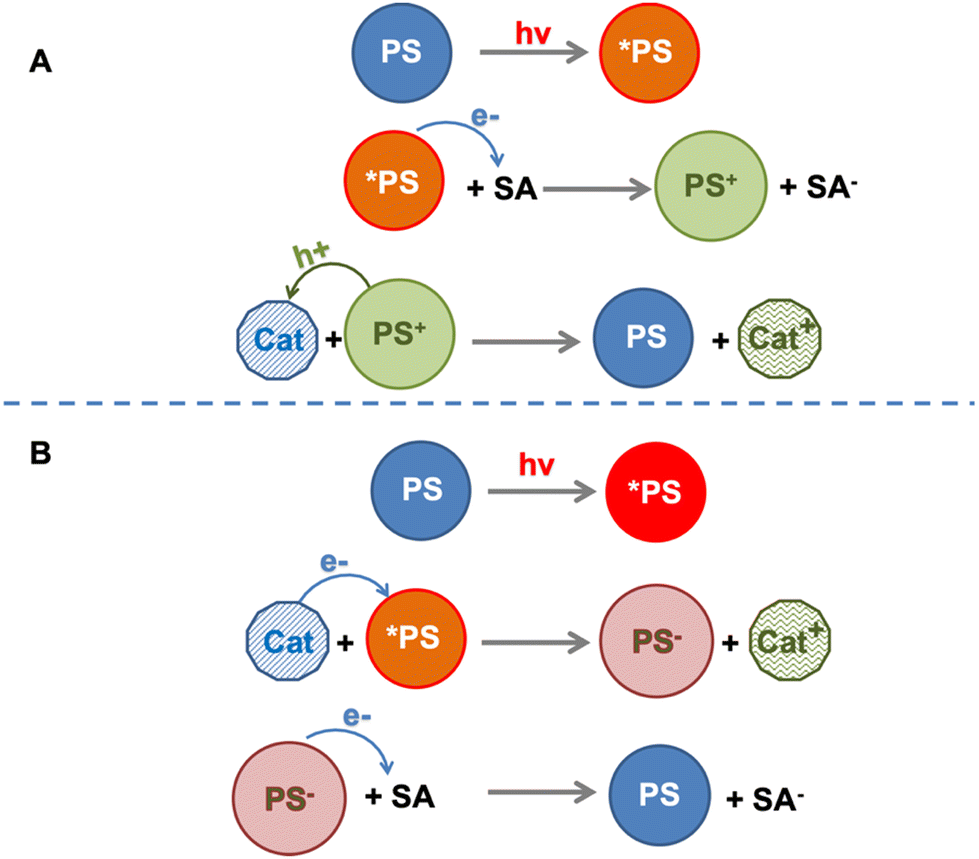 | ||
| Fig. 7 Schematization of the biomimetic (panel A) and anti-biomimetic (panel B) pathways for catalyst oxidation in a SA/PS/Cat system. SA = electron acceptor; PS = photosensitizer, Cat = catalyst. | ||
In fact, because of the short excited-state lifetime of the 3MLCT state of Ru4 in the experimental conditions, that is 18 ns in aqueous solution, and the micromolar concentration of Ru4POM that caused emission quenching, it was clear that such a quenching could not be dynamic, but it had to be static: in other words, the photosensitizer and the catalyst had to be associated in solution.17 Ultrafast experiments indeed showed that the excited-state lifetime of Ru4 in the water oxidation experimental conditions (Ru4: 50 μM; Ru4POM: 50 μM; Na2S2O8: 10 mM, in 10 mM phosphate buffer at pH 7) was 109 ps, indicating that photoinduced reductive electron transfer involving the MLCT state of Ru4 and Ru4POM has a rate constant of 9.2 × 109 s−1, so definitely confirming the association between Ru4 and Ru4POM. The opposite high charges of the photosensitizer (8+) and of the catalyst (10−) were proposed to be the mean driving force for the association, whose roughly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 nature was also confirmed by conductivity measurements.17 However, experiments performed on the system made by Ru4 and Ru4POM, in the absence of persulfate anions, showed that charge recombination within the associated Ru4-Ru4POM system was even faster than charge separation, since the reduced form of Ru4 and the oxidized form of Ru4POM did not accumulate. Since electron scavenging of the reduced Ru4 by persulfate was needed for the occurrence of the efficient water oxidation process as indicated by the photocatalytic data,12 such electron scavenging has to successfully compete with charge recombination, and therefore to be significantly faster than 9.2 × 109 s−1: the consequence is that persulfate anions must be part of the associated assembly, since diffusion would make the electron scavenging process not competitive with charge recombination, see Fig. 8.
:1 nature was also confirmed by conductivity measurements.17 However, experiments performed on the system made by Ru4 and Ru4POM, in the absence of persulfate anions, showed that charge recombination within the associated Ru4-Ru4POM system was even faster than charge separation, since the reduced form of Ru4 and the oxidized form of Ru4POM did not accumulate. Since electron scavenging of the reduced Ru4 by persulfate was needed for the occurrence of the efficient water oxidation process as indicated by the photocatalytic data,12 such electron scavenging has to successfully compete with charge recombination, and therefore to be significantly faster than 9.2 × 109 s−1: the consequence is that persulfate anions must be part of the associated assembly, since diffusion would make the electron scavenging process not competitive with charge recombination, see Fig. 8.
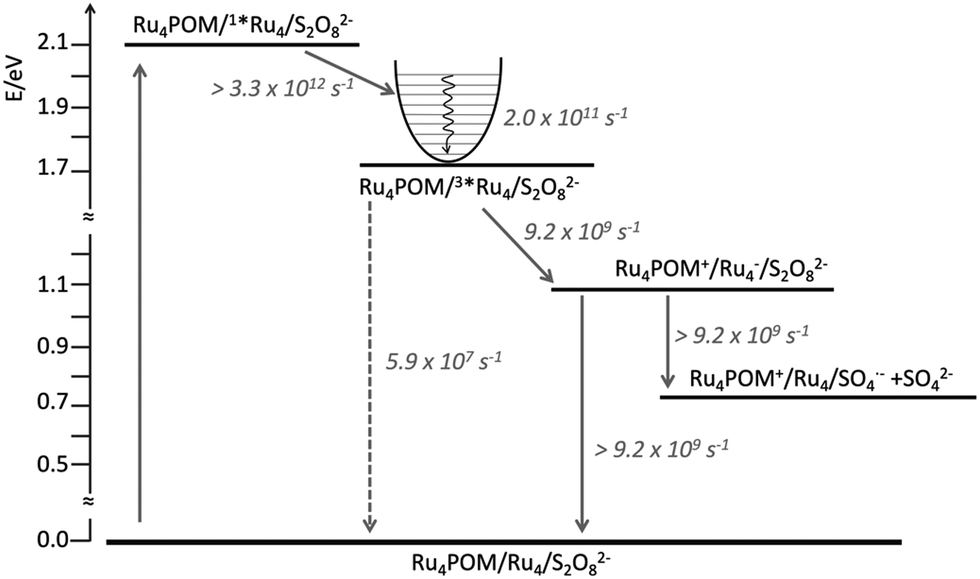 | ||
| Fig. 8 Energy level diagram and schematization of the processes (and rate constants) occurring upon excitation of Ru4 in the Ru4POM/Ru4/S2O82− system (solid and dashed lines refer to nonradiative and radiative processes, respectively). For further details, see ref. 17. | ||
The results obtained by Ru4 and Ru4POM represent a quite important breakthrough in the field: for the first time, it is demonstrated that aggregation between photosensitizers and catalysts (and acceptor sacrificial agents as well) can be obtained by taking advantage of intermolecular interaction essentially of electrostatic origin and that such an aggregation is fundamental to yield an efficient photochemical water oxidation. Such an aggregation was not planned, but undoubtedly indicated a new approach for organizing functional components for photoinduced water oxidation. It was clear that photosensitizers and catalysts could be assembled by playing on their opposite charges, so preparing efficient multicomponent systems for photoinduced processes in solution without necessarily synthesizing quite complicated covalently linked multicomponent structures.
The system Ru4/Ru4POM inspired successive works, still based on Ru4POM as the active oxygen-evolving catalyst. For example, perylene-bisimide chromophores (PBI) (see Fig. 9) were shaped to function by interaction with Ru4POM.20 The resulting ([PBI]5Ru4POM)n complex showed a robust amphiphilic structure and dynamic aggregation into large two-dimensional paracrystalline domains, a red-shifted light-harvesting efficiency of >40% and favorable exciton accumulation, with a peak quantum efficiency using ‘green’ photons (λ > 500 nm) when transferred onto a nanostructured tungsten oxide photoanode, and exhibiting an internal quantum efficiency (IQE, approximated to the absorbed photon-to-current conversion efficiency) of ca. 1.3%, in the wavelength range 470–540 nm.20 From the photochemical viewpoint, in the aggregated ([PBI]5Ru4POM)n system, the initially formed PBI-centered singlet excited state deactivates to a charge-separated state, in which the radical anion involves a PBI subunit and the radical cation involves a Ru4POM moiety, possibly via an intermediate charge-transfer excited state.20 A three-species model was indeed needed to fit the experimental data, indicating that the initially formed PBI-based excited state evolved with a rate constant of 7.3 × 1011 s−1 into the intermediate, charge-transfer state, which in its turn gave rise to the final charge-separated system with a rate constant of 4.0 × 1010 s−1. Charge recombination takes place with a rate constant of 9.5 × 108 s−1.20 Electron delocalization along the π-backbone of the ([PBI]5Ru4POM)n system after charge separation was proposed to explain the relatively slow (considering it occurs within an aggregated system) charge recombination, an essential element for the performance of the system.
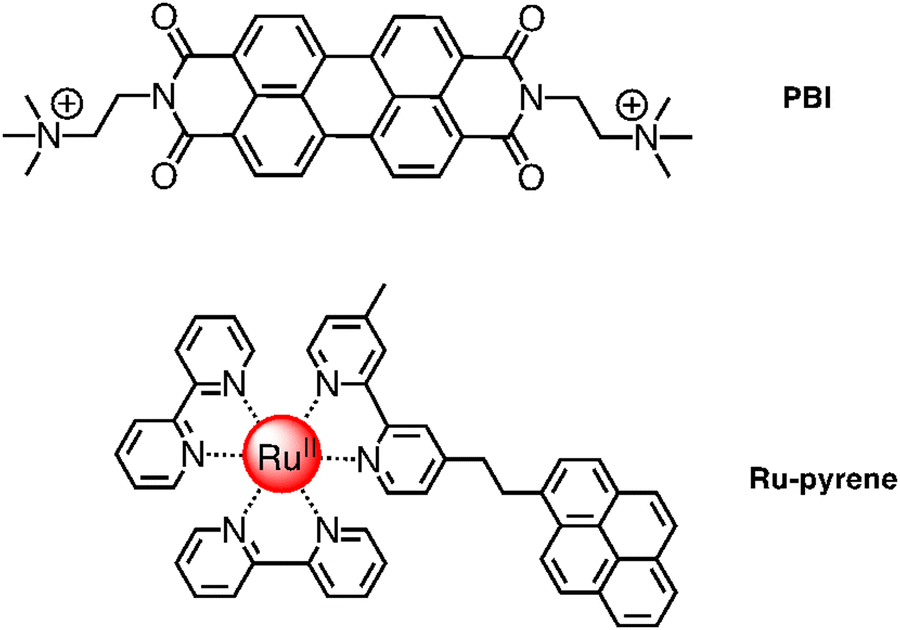 | ||
| Fig. 9 Chemical structures of PBI (top) and Ru-pyrene (bottom) species involved in self-assembled photosensitizer-catalyst arrays. | ||
A step forward in designing photosensitizers/catalysts assemblies was based on [Ru(bpy)2(bpy-pyrene)]2+ (Ru-pyrene; bpy-pyrene is 4-methyl-4′-[2-(1-pyrenyl)ethyl]-2,2′-bipyridine; see Fig. 9) as the photosensitizer and again Ru4POM as the catalyst. Here, the pyrene substituents have the function to promote intermolecular interactions between photosensitizer molecules, so favoring a further hierarchical organization of the Ru-pyrene/Ru4POM system. Indeed, self-organization takes place in solution, as shown by SAXS and transmission electron microscopy (TEM) and corroborated by molecular dynamics calculation.21 Pyrene–pyrene interactions are evidenced by modification of the absorption bands due to π–π* pyrene transitions with time in the aggregate system, clearly indicating that pyrene subunits are involved in a secondary self-assembly process: actually, characterization of the systems suggests that small, initially formed fibers organize themselves into larger (and probably more defined) structures, as inferred by SAXS analysis. Water molecules are an essential part of the aggregate, since they contribute to stabilize the systems, as inferred by molecular dynamics. The relatively rigid structure of the aggregate systems has an important role in accelerating specific electron transfer processes and delaying other electron transfer processes, probably by affecting their reorganization energies, so having a beneficial overall effect on the photocatalytic behavior. Indeed, the photochemical quantum yield of molecular oxygen production, in the presence of persulfate anions as sacrificial agents, reached the value of 0.30.21 The efficiency of the process strongly depends on the age of the solution, indicating that the excessive growth of the aggregated systems, increasing with time in dimension, makes the photochemical water oxidation process less effective.21
Forced co-localization of photosensitizers and catalysts
Although the former examples represent cases in which aggregation takes place spontaneously just dissolving the various components in solution, other efforts were aimed to force the organization of photosensitizers and catalysts: this concept is indeed inspired by the internal structure of organelles such as the plant chloroplasts, which co-localize the needed molecular components for chemical conversion of light energy. One interesting example was provided by the light-driven production of hydrogen inside a hydrogel scaffold obtained by the supramolecular self-assembly of perylene monoimide amphiphiles.22 In this work, the perylene monoimide chromophore amphiphile PMI (Fig. 10), in which the chromophore is decorated by a five-carbon linker bearing a carboxylic acid group, undergoes hydrophobic collapse of its aromatic units in water, so leading to self-assembly. Indeed, this was demonstrated by SAXS and cryo-TEM, which showed the formation of ribbons consisting of interdigitated bilayers of PMI. When salts were added to aqueous solutions of the ribbons, hydrogels were formed, as a consequence since the negatively charged supramolecular polymers are screened by electrolytes. In particular, organized gels are formed when poly(diallyldimethylammonium)chloride is employed as one of the added salts. Within the gels, strong electronic coupling among the PMI chromophore amphiphile molecules was demonstrated, so confirming that the organized structure of the chromophores is maintained in the hydrogel.21 Once the hydrogel formation was extensively investigated and characterized, a properly functionalized, nickel-based hydrogen catalyst (catalytic properties of the unfunctionalized nickel species were already well-established23) was added (see Ni-cat in Fig. 10). One night of aging let the nickel-based catalyst to permeate the hydrogel; in the presence of ascorbic acid as the sacrificial electron donor agent, irradiation of the hydrogel led to significant H2 production. Various relative concentrations of PMI and Ni catalyst were used: in all cases, the hydrogels showed significantly higher H2 formation compared to that obtained by using a solid precipitate of protonate (neutral) PMI. The authors proposed that these gels consist of porous networks of ribbons that allow diffusion of catalyst and ascorbic acid to enhance photocatalysis.22 However, it should be noted that, although the reported case is probably the first example of photocatalytic hydrogel based on supramolecular self-assembly of organic photosensitizers, H2 photoproduction from gel systems based on Ru(bpy)32+ and platinum catalysts was already reported.24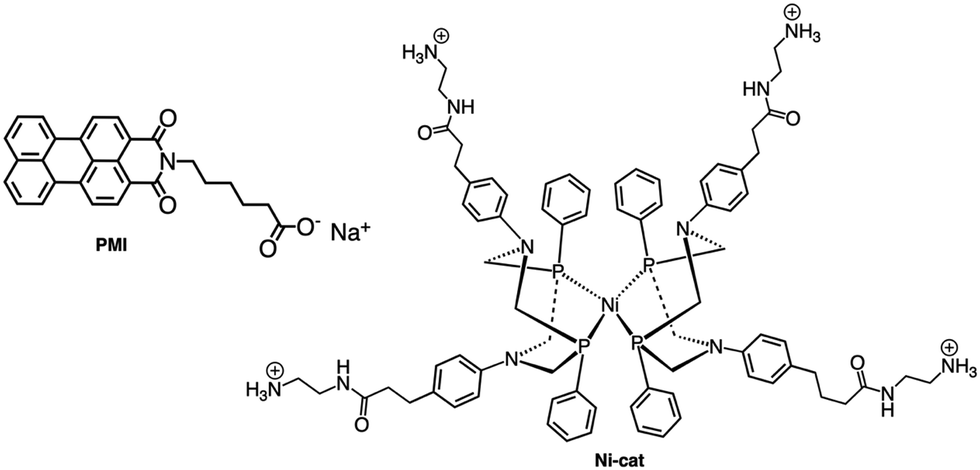 | ||
| Fig. 10 Chemical structures of PMI (left) and Ni-cat (right) species. | ||
The potential of hydrogels to provide an ideal environment for co-localizing photosensitizers and catalysts was recently shown also for photoinduced water oxidation. In particular, a hydrogel formed by polymerized chitosan, functionalized with bpy moieties serving to anchor Ru(bpy)32+-type chromophores on the chitosan structure, has been prepared in the presence of iridium oxide nanoparticles, the water oxidation catalysts.25 In these conditions, iridium oxide nanoparticles are incorporated within the hydrogel, forming a nanofiber-like structure, IrO2⊂NP-RuCh.25Fig. 11 shows the structural formula of the chitosan polymer containing the Ru(II) chromophore. Dynamic light scattering, scanning electronic microscopy (SEM), and energy-dispersive X-ray analysis (EDX) have been used to characterize the systems and indicated that in the prepared nanofiber-like hydrogel the ratio between Ru-based photosensitizer and the IrO2 nanoparticles was 2:1 (based on the Ru:Ir ratio). Photoinduced water oxidation with production of molecular oxygen takes place under visible irradiation in the presence of persulfate anions, with a quantum yield of 0.21.25 Interestingly, the quantum yield of an analogous solution containing the same amount of photosensitizers, catalysts, and sacrificial agents is 0.05. The better performance of the hydrogel, nano-fiber system is attributed to a kinetic effect, in particular to the faster hole scavenging process between the oxidized form of the photosensitizer (produced by photoinduced oxidative electron transfer from the excited Ru-based chromophore to the sacrificial electron acceptor) and the catalyst, which regenerate the ground state of the photosensitizer and contribute to producing the active form of the catalyst. Indeed, flash photolysis experiments showed that such a hole-scavenging process is faster in IrO2⊂NP-RuCh. For this latter species, the rate constant of the hole scavenging process is 7.4 × 104 s−1,25 a remarkable value when compared to the rate constant for the hole scavenging process in the [Ru(bpy)3]2+/IrO2/Na2S2O8 separated system, which is reported to be 8 × 102 s−1,26 that is two orders of magnitude slower. The relevant acceleration of the hole scavenging in IrO2⊂NP-RuCh is attributed to the increase in the proximity of both photosensitizer and catalyst subunits, thanks to the restricted environment promoted by the chitosan nanofibers, which play the role of concentrators. Such a relevant acceleration of the hole scavenging process is therefore held responsible for the improvement in the photochemical quantum yield of molecular oxygen production, that is in the water oxidation process, and has also a protective role in the stability of the photosensitizer, whose oxidized form is known to be subject to the nucleophilic attack leading to deactivation of the photocatalytic properties of the system.16,26
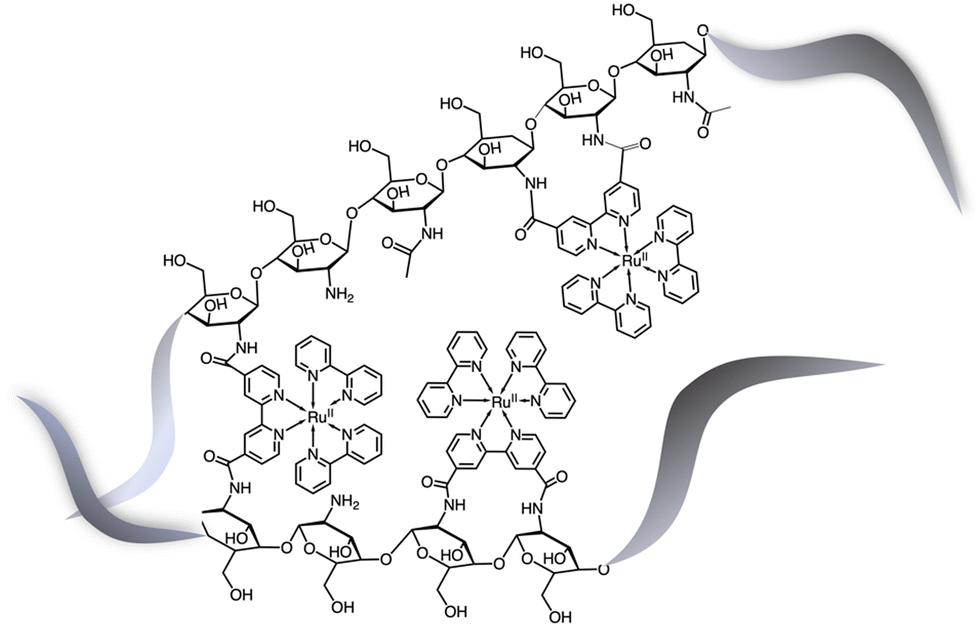 | ||
| Fig. 11 Schematization of the structural formula of the chitosan polymer containing the Ru(II) chromophores.25 The metal complexes are linked to the polysaccharide framework exclusively by chitosan–N(H)–C(O)–bipyridine connections. | ||
Noteworthily, nanofibers based on perylene bisimide subunits covalently linked to a ruthenium(II) water oxidation catalyst have also been prepared, and the chemically driven (using cerium(IV) ammonium nitrate) water oxidation was investigated, showing a good performance of the aggregate system, particularly as far as the stability of the catalytic activity is concerned.27 However, photocatalytic properties of these aggregates have not been studied, to the best of our knowledge. Moreover, for completeness, it should be mentioned that luminescent Ru(II) polypyridine complexes had already been incorporated in polysaccharide systems,28,29 although not for artificial photosynthesis purposes. For example, Ru(bpy)32+ has been incorporated in chitosan for developing luminescence temperature sensors.28
Concluding remarks
Although the previous decades have seen the impressive development of covalently linked light-harvesting antenna/reaction center/catalyst assemblies for artificial photosynthesis, the last decade has testified the potential of self-assembly and spontaneous aggregation of antennae, reaction centers and catalysts for obtaining improved systems capable of performing photo-induced charge separation and photocatalytic processes. In many cases, the aggregated systems also feature emerging properties, not accessible to non-aggregated components. Significant examples are the somewhat not expected formation of aggregated photosensitizer–catalyst assemblies made of light harvesting metal-based dendrimers and polyoxometalated catalysts, as well as the planned co-localization of photosensitizers and catalysts within hydrogel structures. These cases, as well as the other examples here discussed, identify self-assembly as one of the major breakthroughs for future developments of photo-active assemblies for artificial photosynthesis and open up the way for future avenues of research. Interfacing of such self-assembled hybrid structures on electrodes for regenerative solar cells also holds promise for further advancements in the field. It should be pointed out, moreover, that rationalizing the role of peripheral substituents of the monomeric subunits in the aggregation and self-assembling processes is impossible at present, owing to the absence of sufficient experimental data. This issue would require more specific investigation before general conclusions can be made.Finally, there is another point that is hidden or only partially discussed in the published literature concerning such self-assembled systems: the fact that a soft material that is formed by self-assembly, essentially made of photosensitizers (the antenna units) and catalysts, is permeable to water molecules as well as to sacrificial agents (in some cases, sacrificial agents are also strongly associated with the photosensitizer/catalyst assemblies). In fact, a fast exchange of sacrificial agents (and of their decomposition products) between bulk and aggregated systems is needed; otherwise, the relatively high efficiency of the systems would not be possible (for example, the Ru4/Ru4POM photoinduced water oxidation is only limited by the overall concentration of persulfate that is present in solution, thus indicating that all the concentrations of persulfate which are present in solution – initially associated or not – are used for the photoinduced processes). Therefore, although the reactions are probably optimized at the interfaces, the dynamic properties of the self-assembled systems most likely play an important role in the photocatalytic processes occurring in these systems.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank The Ministero degli Affari Esteri e della Cooperazione Internazionale (MAECI) for financial support (project title: Artificial photosynthesis. Light-driven hydrogen production and carbon dioxide reduction). The University of Messina is also acknowledged for funding and open access support.References
- See, for example: (a) N. Armaroli, V. Balzani and N. Serpone, Powering Planet Earth. Energy Solutions for the Future, Wiley-VCH, Weinheim, 2013 CrossRef; (b) L. Alibabaei, M. K. Brennaman, M. R. Norris, B. Kalanyan, W. J. Song, M. D. Losego, J. J. Concepcion, R. A. Binstead, G. N. Parsons and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20008 CrossRef CAS PubMed; (c) H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed; (d) T. Founce, S. Styring, M. R. Wasielewski, G. R. Brudvig, A. W. Rutherford, J. Messinger, A. F. Lee, C. L. Hill, H. deGroot, M. Fontecave, D. R. MacFarlane, B. Hankamer, D. Nocera, D. M. Tiede, H. Dau, W. Hillier, L. Wand and R. Amal, Energy Environ. Sci., 2013, 6, 1074 RSC; (e) T. J. Meyer, M. V. Sheridan and B. D. Sherman, Chem. Soc. Rev., 2017, 46, 6148 RSC; (f) D. D. Dogutan and D. G. Nocera, Acc. Chem. Res., 2019, 52, 3143 CrossRef CAS; (g) X. Fang, S. Kalathil and E. Reisner, Chem. Soc. Rev., 2020, 49, 4926 RSC; (h) D. Wang, L. Fei, Z. Huang and T. J. Meyer, Chem. Phys. Rev., 2022, 3, 011301 CrossRef CAS.
- This topic is too vast to be exhaustively quoted. For some example, see: (a) V. Balzani and F. Scandola, Supramolecular Photochemistry, Horwood, Chichester, 1991 Search PubMed; (b) M. N. Paddon-Row, Acc. Chem. Res., 1994, 27, 18 CrossRef CAS; (c) X.-H. Xu, X.-G. Fu, L.-Z. Wu, B. Chen, L.-P. Zhang, C.-H. Tung, H.-F. Ji, K. S. Schanze and R.-Q. Zhang, Chem. – Eur. J., 2006, 12, 5238 CrossRef CAS , and references therein; (d) S. Sato, K. Koike, H. Inoue and O. Ishitani, Photochem. Photobiol. Sci., 2007, 6, 454 CrossRef CAS PubMed; (e) M. J. Llansola-Portoles, D. Gust, T. A. Moore and A. L. Moore, C. R. Chim., 2017, 20, 296 CrossRef CAS , and references therein.
- (a) R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265 CrossRef CAS; (b) M. R. Wasielewski, Chem. Rev., 1992, 92, 435 CrossRef CAS; (c) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40 CrossRef CAS PubMed; (d) P. F. Barbara, T. J. Meyer and M. A. Ratner, J. Phys. Chem., 1996, 100, 13148 CrossRef CAS; (e) M. N. Paddon-Row, In Electron Transfer in Chemistry, ed. V. Balzani, Wiley-VCH, Weinheim, 2001, p. 179, vol. III, ch. 2.1 Search PubMed; (f) B. Albinsson and J. Martensson, J. Photochem. Photobiol., C, 2008, 9, 138 CrossRef CAS; (g) A. Arrigo, A. Santoro, M. T. Indelli, M. Natali, F. Scandola and S. Campagna, Phys. Chem. Chem. Phys., 2014, 16, 818 RSC; (h) S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455 CrossRef CAS PubMed; (i) M. Natali, S. Campagna and F. Scandola, Chem. Soc. Rev., 2014, 43, 4005 RSC; (j) I. Ma, G. Chen, C. Fave, L. Chen, R. Kuriki, K. Maeda, O. Ishitani, T.-C. Lau, J. Binin and M. Robert, J. Am. Chem. Soc., 2020, 142, 6188 CrossRef PubMed; (k) F. M. Toma, F. Puntoriero, T. V. Pho, M. La Rosa, Y. S. Jun, B. J. Tremolet de Villers, J. Pavlovich, G. D. Stucky, S. Campagna and F. Wudl, Angew. Chem., Int. Ed., 2015, 54, 6775 CrossRef CAS PubMed; (l) A. Arrigo, A. Santoro, F. Puntoriero, P. P. Lainé and S. Campagna, Coord. Chem. Rev., 2015, 304–305, 109 CrossRef CAS.
- M. Natali and F. Scandola in, Applied Photochemistry: When Light Meets Molecule, ed. G. Bergamini and S. Silvi, Springer, 2016, p. 1, and refs. therein Search PubMed.
- (a) V. Balzani, A. Credi and M. Venturi, Molecular Devices and Machines: Concepts and Perspectives for the Nanoworld, Wiley-VCH, 2nd edn, 2008 CrossRef; (b) A. P. De Silva, Nat. Mater., 2005, 4, 15 CrossRef CAS.
- M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910 CrossRef CAS PubMed.
- B. Rybtchinski, L. E. Sinks and M. R. Wasielewski, J. Am. Chem. Soc., 2004, 126, 12268 CrossRef CAS PubMed.
- M. J. Ahrens, R. F. Kelley, Z. E. X. Dance and M. R. Wasielewski, Phys. Chem. Chem. Phys., 2007, 9, 1469 RSC.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962 CrossRef.
- See, for example: (a) G. Denti, S. Campagna, S. Serroni, M. Ciano and V. Balzani, J. Am. Chem. Soc., 1992, 114, 2944 CrossRef CAS; (b) S. Campagna, G. Denti, S. Serroni, A. Juris, M. Venturi, V. Ricevuto and V. Balzani, Chem. – Eur. J., 1995, 1, 211 CrossRef CAS; (c) V. Balzani, S. Campagna, G. Denti, A. Juris, S. Serroni and M. Venturi, Acc. Chem. Res., 1998, 31, 26 CrossRef CAS; (d) S. Serroni, S. Campagna, F. Puntoriero, C. Di Pietro, F. Loiseau and N. D. McClenaghan, Chem. Soc. Rev., 2001, 30, 367 RSC; (e) A. Arrigo, G. La Ganga, F. Nastasi, S. Serroni, A. Santoro, M.-P. Santoni, M. Galletta, S. Campagna and F. Puntoriero, C. R. Chim., 2017, 20, 209 CrossRef CAS; (f) F. Puntoriero, S. Serroni, G. La Ganga, A. Santoro, M. Galletta, F. Nastasi, E. La Mazza, A. M. Cancelliere and S. Campagna, Eur. J. Inorg. Chem., 2018, 3887 CrossRef CAS.
- (a) G. La Ganga, F. Nastasi, S. Campagna and F. Puntoriero, Dalton Trans., 2009, 9997 RSC; (b) F. Puntoriero, A. Sartorel, M. Orlandi, G. La Ganga, S. Serroni, M. Bonchio, F. Scandola and S. Campagna, Coord. Chem. Rev., 2011, 255, 2594 CrossRef CAS.
- F. Puntoriero, G. La Ganga, A. Sartorel, M. Carraro, G. Scorrano, M. Bonchio and S. Campagna, Chem. Commun., 2010, 46, 4725 RSC.
- A. Arrigo, F. Puntoriero, G. La Ganga, S. Campagna, M. Burian, S. Bernstorff and H. Amenitsch, Chem, 2017, 3, 494 CAS.
- T. Pullerits and V. Sundström, Acc. Chem. Res., 1996, 29, 381 CrossRef CAS.
- G. M. G. Rogati, C. Capecci, E. Fazio, S. Serroni, F. Puntoriero, S. Campagna and L. Guidoni, Chem. – Eur. J., 2022, 28, e202103310 CrossRef CAS PubMed.
- (a) M. Hara, C. C. Waraksa, J. T. Lean, B. A. Lewis and T. E. Mallouk, J. Phys. Chem. A, 2000, 104, 5275 CrossRef CAS; (b) N. D. Morris, M. Suzuki and T. E. Mallouk, J. Phys. Chem. A, 2004, 108, 9115 CrossRef CAS.
- M. Natali, F. Puntoriero, C. Chiorboli, G. La Ganga, A. Sartorel, M. Bonchio, S. Campagna and F. Scandola, J. Phys. Chem. C, 2015, 119, 2371 CrossRef CAS.
- R. A. Blankenship, Molecular Mechanisms of Photosynthesis, Blackwell, Oxford, 2002 Search PubMed.
- F. Ronconi, M.-P. Santoni, F. Nastasi, G. Bruno, R. Argazzi, S. Berardi, S. Caramori, C. A. Bignozzi and S. Campagna, Dalton Trans., 2016, 45, 14109 RSC.
- M. Bonchio, Z. Syrgiannis, M. Burian, N. Marino, E. Pizzolato, K. Dirian, F. Rigodanza, G. A. Volpato, G. La Ganga, N. Demitri, S. Berardi, H. Amenitsch, D. M. Guldi, S. Caramori, C. A. Bignozzi, A. Sartorel and M. Prato, Nat. Chem., 2019, 11, 146 CrossRef CAS.
- S. Campagna, G. La Ganga, F. Puntoriero and M. Bonchio, M. Prato, work in progress.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, M. McClendon, A. R. Koltonow, A. P. S. Samuel, D. J. Kiebala, M. R. Wasielewski and S. I. Stupp, Nat. Chem., 2014, 6, 964 CrossRef CAS PubMed.
- A. D. Wilson, R. K. Shoemaker, A. Miedaner, J. T. Muckerman, D. L. DuBois and M. Rakowski DuBois, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6951 CrossRef CAS.
- K. Okeyoshi and R. Yoshida, Soft Matter, 2009, 5, 4118 RSC.
- G. La Ganga, F. Puntoriero, E. Fazio, M. Natali, F. Nastasi, A. Santoro, M. Galletta and S. Campagna, Chem. – Eur. J., 2021, 27, 16904 CrossRef CAS PubMed.
- P. G. Hoertz, Y.-I. Kim, W. J. Youngblood and T. E. Mallouk, J. Phys. Chem. B, 2007, 111, 6845–6856 CrossRef CAS PubMed.
- V. Kunz, V. Stepanenko and F. Würthner, Chem. Commun., 2015, 51, 290 RSC.
- (a) M. Tsvirko, S. Tkaczyk, M. Kozak and B. Kalota, Funct. Mater., 2013, 20, 127 CrossRef CAS; (b) K. Takato, N. Gokan and M. Kaneko, J. Photochem. Photobiol., A, 2005, 169, 109 CrossRef CAS.
- S. B. Marpu and E. N. Benton, Int. J. Mol. Sci., 2018, 19, 1795 CrossRef PubMed.
Footnote |
| † Dedicated to the memory of Prof. Franco Scandola, great scientist and friend. |
| This journal is © the Owner Societies 2023 |