
Self-assembled halide perovskite quantum dots in polymer thin films showing temperature-controlled exciton recombination†
Most Farida
Khatun
ab,
Takuya
Okamoto
*ac and
Vasudevanpillai
Biju
 *ac
*ac
aGraduate School of Environmental Science, Hokkaido University, Sapporo, Hokkaido 060-0810, Japan. E-mail: biju@es.hokludai.ac.jp; okamotot@es.hokudai.ac.jp
bPharmacy Discipline, Khulna University, Khulna-9208, Bangladesh
cResearch Institute for Electronic Science, Hokkaido University, Sapporo, Hokkaido 001-0020, Japan
First published on 16th October 2023
Abstract
Rationally assembled supramolecular structures of organic chromophores or semiconductor nanomaterials show excitonic properties different from individual molecules or nanoparticles. We report polymer-assisted assembly formation and thermal modulation of excitonic recombination in self-assembled formamidinium lead bromide perovskite quantum dots.
Metal halide perovskites (MHPs) have gained considerable attention recently due to their excellent optoelectronic properties and low-cost synthesis.1 The size-, shape-, phase-, and halogen-dependent bandgap and excitonic properties make MHPs promising materials for lighting and energy harvesting applications.2 The bandgap of MHPs is tuned by halide exchange or size/shape controlling.3 MHPs are prepared in different sizes and shapes by solution processing. Hydrophobic ligand-capped low dimensional perovskite quantum dots (PQDs), nanocrystals (PNCs), and nanoplatelets (PNPs) self-assemble into supercrystals and superlattices. Like in supramolecular systems, dipole–dipole, hydrophobic, and van der Waals interactions drive assembly formation of low-dimensional MHPs, modifying optoelectronic and photophysical properties of individual PNCs, PQDs, or PNPs.4 Assembly formation weakens quantum confinement by increasing the local dielectric field.4a,b,5 As a result, the exciton binding energy decreases, photoluminescence (PL) spectra redshift, excitons split into free carriers, and the PL lifetime increases. Soetan et al. showed PL spectral redshift during solvent-assisted self-assembling of CsPbBr3 PNCs into superlattices.5b Liu et al. demonstrated light-induced self-assembling of cubic CsPbBr3 PNC into anisotropic nanowires.5a These nanowires showed a red-shifted PL spectrum and a longer PL lifetime than individual PNCs. Also, self-assembled PNPs show red-shifted PL bands and higher PL quantum yields than isolated particles in solutions.6 We previously reported the ligand-assisted assembly formation of FAPbBr34a or CsPbBr3 PQDs,4b and successfully dissociated hydrophobically-assembled FAPbBr3 by applying a lateral mechanical force.4a Conversely, stable CsPbBr3 PNC supercrystals prepared using bidentate ligands were free from mechanical deformation.4b Nevertheless, reversible changes to the optical properties of self-assembled PNCs are necessary for developing stimuli-responsive devices.
A polymer matrix is a technologically relevant host for dispersing and assembling optically active molecules and nanoparticles into stimuli-responsive devices. Williams et al. reported mechanically tuning optical properties of sticky polymer-grafted silica nanoparticle superlattices.7 Miyamoto et al. demonstrated mechanochromic perovskite nanosheet hydrogels.8 For thermochromism, Sussman et al. prepared a transparent film consisting of a polymer core and a composite shell, which becomes colored by heat-induced structural changes.9 So far, polymer hosts such as polyvinylpyrrolidone,10 polystyrene,11 polymethyl methacrylate,12 and polycarbonate13 have been applied to encapsulate nanoscale MHPs for protecting them from moisture and heat. Conversely, Gong et al. reported stretch-induced PL intensity enhancement for CsPbBr3 PQDs in a polymer composite.14 Also, Bade et al. demonstrated a stretchable perovskite LED using a MAPbBr3–polymer composite.15 Despite such emerging applications of MHP–polymer composites, stimuli-responsive excitonic recombination dynamics of perovskite assemblies in host materials remain largely unexplored.
We demonstrate reversible shifts in PL color and lifetime of FAPbBr3 PQD assemblies in polybutadiene (PBD) thin films as a function of temperature. FAPbBr3 PQD is selected due to the large differences in the PL spectral maxima and lifetimes between isolated and self-assembled PQDs.4a PBD is selected as the host matrix for making PQD assemblies, considering its elasticity, low glass transition temperature (Tg < 210 K), and optical transparency. The PL spectrum of isolated PQDs in toluene redshifts by >70 nm due to assembly formation during PQD–PBD film preparation. Also, the PL lifetime of a PQD assembly is much longer (345 ns) than isolated PQDs (1.3 ns). The green emission of a PQD assembly in a PBD film changes into cyan due to heat-induced (from 296 to 323 K) dissociation. This color change is due to a PL band ca. 480 nm corresponding to PQDs separated from assemblies. Correspondingly, the PL lifetime of a PQD assembly decreases from 345 to 74 ns. Interestingly, the PL color and lifetime are reversed by cooling the PQD–PBD film from 323 to 296 K. We discuss the PL color and lifetime changes of PQD assemblies from the viewpoint of exciton recombination in different dielectric environments.
We synthesized FAPbBr3 PQDs by the modified hot injection method.4a The synthesis procedure of PQDs is described in the ESI.†Fig. 1a shows the absorption and PL spectra of the as-synthesized PQD colloidal solution. The light-yellow-colored PQD solution shows blue emission under UV light (Fig. 1a inset). The sharp excitonic absorption peak (ca. 437 nm) and blue emission (ca. 441 nm) suggest strong quantum confinement and excitonic recombination of photo-excited PQDs, similar to low dimensional MHPs.4a,b,16 The excitonic absorption and PL bands are consistent with the quantum size (ca. 5 nm) of PQDs, as observed in the scanning transmission electron microscope (STEM) image (Fig. 1b). The as-synthesized PQD colloidal solution shows mono-exponential PL decay (Fig. 1c) with a short PL lifetime of 1.3 ns.
 | ||
| Fig. 1 (a) Absorption (i) and PL spectra (ii) of as-synthesized FAPbBr3 PQDs in toluene (λex = 400 nm). Inset: Photographs of a PQD colloidal solution under room light and a UV lamp. (b) A STEM image of as-synthesized PQDs (scale bar: 50 nm). (c) A PL decay curve of the as-synthesized PQD colloidal solution in toluene. (d) The SAXS pattern of a PQD-only film. The red line in d is the guide to the eyes. | ||
We prepared FAPbBr3 PQD-only and PQD–PBD films to verify PQD assembly formation in films. The films were prepared by drop-casting of the corresponding solutions in toluene. The PL color of a PQD-only film prepared from a 10 mg mL−1 solution in toluene is changed from blue to green during drying, consistent with our previous report.4a The small-angle x-ray scattering (SAXS) pattern (Fig. 1d) of the film showed an intense peak ca. 2.4 nm−1, supporting the quantum size of PQDs and a short interparticle distance of 2.6 nm. A PQD–PBD thin film was prepared by mixing a FAPbBr3 PQD colloidal solution (10 mg mL−1 in toluene) with a PBD solution (25 mg mL−1 in toluene), followed by drop-casting of 5 μL of the mixture on a glass coverslip. As the polymer host shrink during drying up, the polymer chains gathered isolated PQDs to form PQD assemblies in the film (Fig. 2a). STEM images of a PQD–PBD thin film (Fig. 2b and Fig. S1, ESI†) suggest the formation of polymer-assisted PQD assemblies.
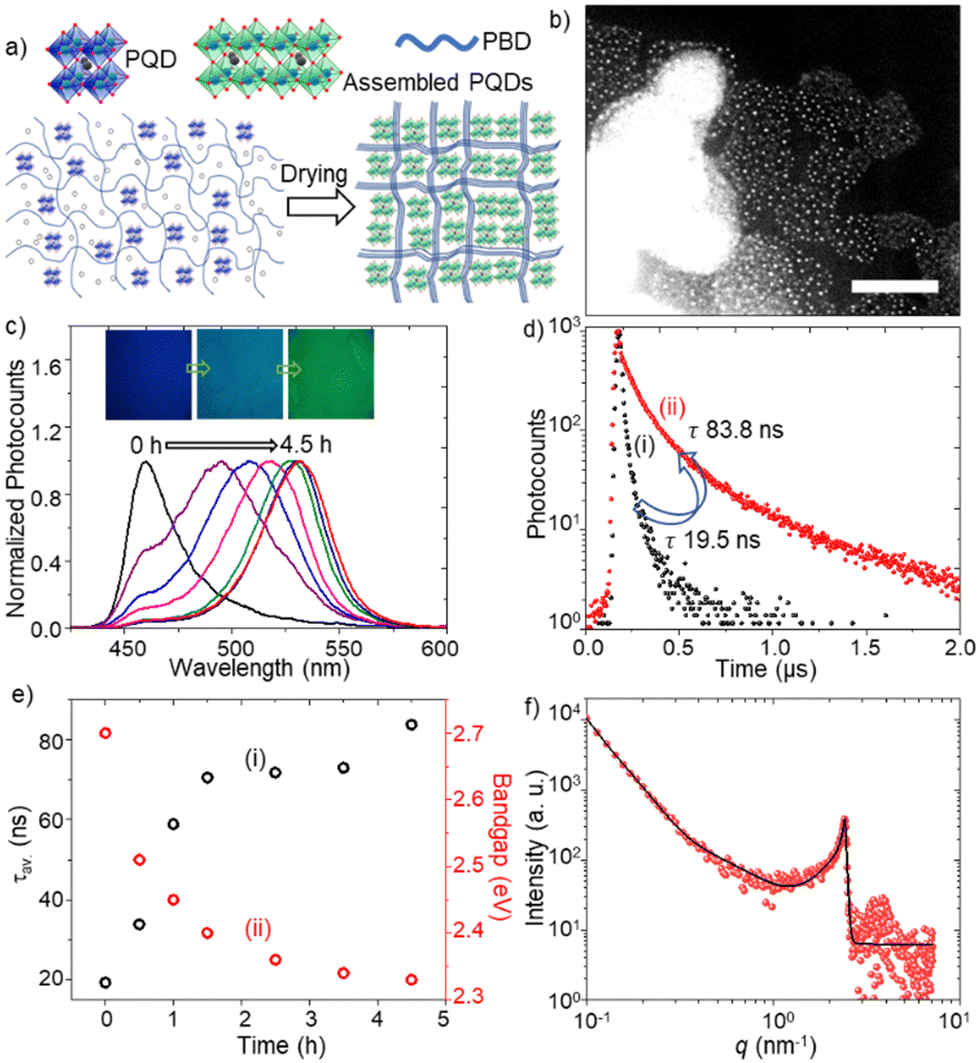 | ||
| Fig. 2 (a) A scheme of PQD assembly formation in PBD. (b) A STEM image (scale bar = 100 nm) of PQD assemblies in PBD. (c) PL spectra of a PQD–PBD mixture during drying (λex = 400 nm). Inset: PL images (1.2 mm × 1.2 mm) of a PQD–PBD film during drying. (d) PL decay curves of a PQD–PBD film at (i) 0 and (ii) 4.5 h (λex = 405 nm). (e) Plots of PL (i) lifetimes and (ii) optical bandgaps of a PQD–PBD film during drying. (f) The SAXS pattern of a PQD–PBD film. The black line is the guide to the eyes. | ||
To understand PL color and spectral changes of PQD samples during assembly formation, we examined the PL images and spectra of PQD-only and PQD–PBD films. Fig. 2c shows the corresponding PL images and spectra of a PQD–PBD film. The PL color of the film is changed from blue to green during assembly formation by solvent evaporation. Correspondingly, the PL spectrum is shifted from 460 to 532 nm. Low dimensional MHPs show strong quantum confinement, correspondingly blue-shifted absorption and PL bands, and short PL lifetimes. Conversely, due to strong dielectric screening, large PNCs and MHP microcrystals show red-shifted PL bands.17 Reabsorption-emission or inner filtering induces PL spectral redshift to thick MHP samples.17c,d Conversely, PNCs or PQDs in self-assembled films show red-shifted PL spectra and long PL lifetimes compared to isolated PNCs or PQDs due to increased interparticle-interaction-induced dielectric screening and exciton/carrier diffusion in films.4a,b,17b,d The PL lifetime of a PQD-only solution is increased from 1.3 (Fig. 1c) to 19.5 ns (Fig. 2d) in a PQD–PBD solution. As the PQD–PBD film was dried up in ambient air, the PL lifetime of PQDs increased to 83.8 ns (Fig. 2d). The PL lifetime was further increased to 345 ns after 4.5 h drying of the film. The PL spectral redshift and PL lifetime increase suggest PQD assembly formation in the film. Fig. 2e shows the plots of PL lifetime and optical bandgap of PQDs in a PQD–PBD film during drying. The correlation between PL lifetimes and optical bandgaps in Fig. 2e indicates increasing dielectric screening,4a,b weakening quantum confinement,4a,b increasing electronic coupling,4b,18 and decreasing surface trap density4a of PQDs during PQD assembly formation. The SAXS pattern of the PQD–PBD mixture shows a peak at the same position as the PQD film (Fig. 1d and 2f), suggesting PQD assembly formation in SAXS samples with or without PBD. In both cases, the interparticle spacing is determined by the capping ligands.
To understand the assembly formation mechanism and explore the thermal modulation of exciton dynamics in the assemblies, we investigated temperature-dependent PL properties of PQD–PBD films. A film was heated from 296 to 323 K on a micro heat plate (KITAZATO, KM-1), during which PL spectra, PL images, and PL decays of the film were recorded at different time intervals. Fig. 3a shows the PL spectra of a PQD–PBD film at different temperatures. Initially, the PL peak was at 544 nm [Fig. 3a(i)] for a completely dried PQD–PBD film at 296 K. As the temperature was increased to 323 K, the PL peak blue-shifted (Δλ = 14 nm) with the appearance of a shoulder band at 495 nm [Fig. 3a(ii)]. Correspondingly, the average PL lifetime of the film was decreased from 345 [Fig. 3b(i)] to 74 ns [Fig. 3b(ii) + (iii)]. Nevertheless, a PQD-only film does not show much PL spectral shift by heating from 296 to 323 K (Fig. S2a, ESI†). The largely blue-shifted PL spectrum (Δλ = 39 nm) and decreased PL lifetime of the film at 323 K are comparable to the mechanically dissociated FAPbBr3 PQD-only films (Fig S2b, ESI†).4a
 | ||
| Fig. 3 (a) PL spectra (λex = 400 nm) and (b) PL decays (λex = 405 nm) of a dried (4.5 h) PQD–PBD film. (a) The spectra at (i) 296 K before heating, (ii) 323 K, and (iii) 296 K after heating and cooling. (b) The decay (i) corresponds to the PL spectrum (i) in ‘a’, and the decay (ii) and (iii) correspond to the photons in the 440–515 nm range, and 515–570 nm range, respectively for the PL spectrum (ii) in ‘a’. (c) A scheme of PQD assembly formation and dissociation during drying and the heating–cooling cycle. (d) A scheme of energy states, photoexcitation, and relaxation in PQDs before and after assembly formation, heating, and cooling (knr: nonradiative recombination rate and kr: radiative recombination rate). | ||
The PL spectral blueshift of PQD assemblies with increasing temperature19 is the opposite of the Varshini model,20 which predicts PL redshift of semiconductors. Wang et al. reported the temperature-dependent (280 K → 320 K) PL spectral blueshift for a FAPbBr3 PNC film (Δλ ∼ 7 nm) due to optical phonon scattering.19a Yang et al. observed a PL spectral blueshift for FAPbBr3 PNCs with increasing temperature from 90 to 350 K due to phase transition (at 150 K) and thermal expansion (Δλ ∼ 3 nm for 290 K → 320 K).19b For other A-site cation MHPs, Woo et al. reported a PL spectral blueshift (Δλ < 5 nm) for MAPbBr3 with increasing temperature from 280 K to 300 K due to the thermal lattice expansion and electron–phonon interactions,19c and Li et al. reported a PL spectral blueshift (80 K to 280 K) and small redshift (from 280 K to 360 K) for a CsPbBr3 PNC film with increasing temperature.19d Although the small blueshift of the main PL band from 544 to 530 nm [from Fig. 3a(i) to Fig. 3a(ii)] can be attributed to the temperature-dependent exciton–phonon property of FAPbBr3 PQD assemblies, the largely blue-shifted (Δλ = 39 nm) shoulder band with a short PL lifetime suggests that the thermal expansion of the polymer chains partially dissociates PQD assemblies in the film, increasing inter-PQD distances and decreasing dielectric screening. The residual green emission after prolonged heating indicates reminiscent assemblies or fused PQDs. The PQD–PBD film was heated within the phase transition temperature of FAPbBr3.21
We repeatedly heated and cooled the PQD–PBD film to validate the reversibility of PQD assembly formation and dissociation. The main PL peak was red-shifted from 530 nm to 544 nm by cooling the film from 323 K to 296 K, with an appreciable decrease in the PL intensity of the blue-shifted shoulder band [Fig. 3a(ii)]. Also, the PL lifetime was increased from 74 to 128 ns by cooling down the film. The PL redshift from 530 to 544 nm is attributed to the temperature-dependent PL property of FAPbBr3 PQDs. However, the PL intensity decrease for the blue-shifted shoulder band and the PL lifetime increase during cooling are attributed to the reassembling of PQDs by the shrinking polymer host. Nevertheless, the complete disappearance of the shoulder band or complete recovery of the PL lifetime was not accomplished, suggesting PQD reassembling is partially hindered by the polymer chains occupying around a fraction of the thermally separated PQDs, which was confirmed by the PL spectral blueshift (Fig. S3a, ESI†) and lifetime decrease (Fig. S3b, ESI†) by increasing the polymer concentration in precursor solution of PQD–PBD film.
Finally, we rationalize the structural and PL property changes of the PQD assembly from the viewpoint of exciton/carrier recombination dynamics (Fig. 3c and d). A PL spectral redshift and long PL lifetime during PQD assembly formation are attributed to decreased inter-PQD distance. As the PQDs come closer, the electronic wavefunctions of neighboring PQDs overlap, leading to miniband formation.4b,18 Green-emitting PNCs with weak quantum confinement are also formed by ligand stripping of PQDs.4a The PL lifetime increase during assembly formation is attributed to decreased surface trap density and increased exciton dielectric screening.4a,b Consequently, the surface trap-related nonradiative recombination rate (knr) decreases, and the strong dielectric screening in the assemblies helps photogenerated excitons to split into carriers,22 increasing the PL lifetime. Conversely, the thermal expansion of the polymer dissociates PQDs from PQD assemblies, increasing the inter-PQD distance and lowering the strength of inter-PQD interactions. Therefore, when heated, a PQD–PBD film shows blue-shifted PL and a short PL lifetime. The incomplete recovery of the PL spectrum and lifetime is due to the incomplete separation of PQDs.
In summary, we demonstrate FAPbBr3 PQD assembly formation in a polymer and the heat-induced modulation of the PL color and lifetime of the assemblies. We concluded that the thermal expansion or shrinkage of the polymer host controls PQD assemblies and their excitonic properties, providing new dimensions to perovskite-based sensing devices.
We acknowledge the financial support under the MEXT JSPS grant-in-aid for scientific research (23H01781 and 21K14580). This work was performed under the Research Program of “Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials” in “Network Joint Research Center for Materials and Devices.” We acknowledge a JST DX doctoral scholarship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- L. Chouhan, S. Ghimire, C. Subrahmanyam, T. Miyasaka and V. Biju, Chem. Soc. Rev., 2020, 49, 2869 RSC.
- C.-Y. Chen, J.-H. Chang, K.-M. Chiang, H.-L. Lin, S.-Y. Hsiao and H.-W. Lin, Adv. Funct. Mater., 2015, 25, 7064 CrossRef CAS.
- (a) S. Ghimire, M. F. Khatun, B. M. Sachith, T. Okamoto, J. Sobhanan, C. Subrahmanyam and V. Biju, ACS Appl. Mater. Interfaces, 2023, 15, 41081 CrossRef CAS PubMed; (b) M. C. Weidman, A. J. Goodman and W. A. Tisdale, Chem. Mater., 2017, 29, 5019 CrossRef CAS.
- (a) Z. Zhang, S. Ghimire, T. Okamoto, B. M. Sachith, J. Sobhanan, C. Subrahmanyam and V. Biju, ACS Nano, 2022, 16, 160 CrossRef CAS PubMed; (b) T. Okamoto and V. Biju, Small, 2023, 19, 2303496 CrossRef CAS PubMed; (c) A. Jana, A. Meena, S. A. Patil, Y. Jo, S. Cho, Y. Park, V. G. Sree, H. Kim, H. Im and R. A. Taylor, Prog. Mater. Sci., 2022, 129, 100975 CrossRef CAS; (d) Y. Yang, J. T. Lee, T. Liyanage and R. Sardar, J. Am. Chem. Soc., 2019, 141, 1526 CrossRef CAS PubMed; (e) J. Liu, X. Zheng, O. F. Mohammed and O. M. Bakr, Acc. Chem. Res., 2022, 55, 262 CrossRef CAS PubMed.
- (a) J. Liu, K. Song, Y. Shin, X. Liu, J. Chen, K. X. Yao, J. Pan, C. Yang, J. Yin and L. J. Xu, et al. , Chem. Mater., 2019, 31, 6642 CrossRef CAS; (b) N. Soetan, W. R. Erwin, A. M. Tonigan, D. G. Walker and R. Bardhan, J. Phys. Chem. C, 2017, 121, 18186 CrossRef CAS.
- (a) J. Jagielski, et al. , Sci. Adv., 2017, 3, eaaq0208 CrossRef PubMed; (b) C. Bi, S. Wang, S. V. Kershaw, K. Zheng, T. Pullerits, S. Gaponenko, J. Tian and A. L. Rogach, Adv. Sci., 2019, 6, 1900462 CrossRef PubMed.
- G. A. Williams, R. Ishige, O. R. Cromwell, J. Chung, A. Takahara and Z. Guan, Adv. Mater., 2015, 27, 3934 CrossRef CAS PubMed.
- W. Yang, S. Yamamoto, K. Sueyoshi, T. Inadomi, R. Kato and N. Miyamoto, Angew. Chem., 2021, 133, 8547 CrossRef.
- J. Sussman, D. Snoswell, A. Kontogeorgos, J. J. Baumberg and P. Spahn, Appl. Phys. Lett., 2009, 95, 173116 CrossRef.
- M. J. Tan, et al. , Adv. Healthcare Mater., 2019, 8, 1900859 CrossRef CAS PubMed.
- Y. Wei, X. Deng, Z. Xie, X. Cai, S. Liang, P. A. Ma, Z. Hou, Z. Cheng and J. Lin, Adv. Funct. Mater., 2017, 27, 1703535 CrossRef.
- Y. Wang, L. Varadi, A. Trinchi, J. Shen, Y. Zhu, G. Wei and C. Li, Small, 2018, 14, 1803156 CrossRef PubMed.
- Z. Long, Y. Wang, Q. Fu, J. Ouyang, L. He and N. Na, Nanoscale, 2019, 11, 11093 RSC.
- Y. Gong, J. Shen, Y. Zhu, X. Yang, L. Zhang and C. Li, J. Mater. Chem. C, 2020, 8, 1413 RSC.
- S. G. R. Bade, X. Shan, P. T. Hoang, J. Li, T. Geske, L. Cai, Q. Pei, C. Wang and Z. Yu, Adv. Mater., 2017, 29, 1607053 CrossRef PubMed.
- (a) W. Tao, Q. Zhou and H. Zhu, Sci. Adv., 2020, 6, eabb7132 CrossRef PubMed; (b) M. C. Gélvez-Rueda, et al. , Nat. Commun., 2020, 11, 1901 CrossRef PubMed.
- (a) D. B. Mitzi, Chem. Mater., 1996, 8, 791 CrossRef CAS; (b) B. Sachith, T. Okamoto, S. Ghimire, T. Umeyama, Y. Takano, H. Imahori and V. Biju, J. Phys. Chem. Lett., 2021, 12, 8644 CrossRef CAS PubMed; (c) T. Okamoto, M. Shahjahan and V. Biju, Adv. Opt. Mater., 2021, 9, 2100355 CrossRef CAS; (d) S. Ghimire, K. Takahashi, Y. Takano, T. Nakamura and V. Biju, J. Phys. Chem. C, 2019, 123, 27752 CrossRef CAS.
- Y. Tong, E. P. Yao, A. Manzi, E. Bladt, K. Wang, M. Döblinger, S. Bals, P. Müller-Buschbaum, A. S. Urban and L. Polavarapu, Adv. Mater., 2018, 30, 1801117 CrossRef PubMed.
- (a) X. Wang, Q. Wang, Z. Chai and W. Wu, RSC Adv., 2020, 10, 44373 RSC; (b) L. Yang, K. Wei, Z. Xu, F. Li, R. Chen, X. Zheng, X. Cheng and T. Jiang, Opt. Lett., 2018, 43, 122 CrossRef CAS PubMed; (c) H. C. Woo, J. W. Choi, J. Shin, S. H. Chin, M. H. Ann and C. L. Lee, J. Phys. Chem. Lett., 2018, 9, 4066 CrossRef CAS PubMed; (d) J. Li, X. Yuan, P. Jing, J. Li, M. Wei, J. Hua, J. Zhao and L. Tian, RSC Adv., 2016, 6, 78311 RSC.
- Y. P. Varshni, Physica, 1967, 34, 149 CrossRef CAS.
- G. Mannino, I. Deretzis, E. Smecca, A. La Magna, A. Alberti, D. Ceratti and D. Cahen, J. Phys. Chem. Lett., 2020, 11, 2490 CrossRef CAS PubMed.
- S. Ghimire, L. Chouhan, Y. Takano, K. Takahashi, T. Nakamura, K. Yuyama and V. Biju, ACS Energy Lett., 2019, 4, 133 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, synthesis of PQDs, STEM image, PL spectra, PL lifetime plot, PL wavelength plot. See DOI: https://doi.org/10.1039/d3cc02621c |
| This journal is © The Royal Society of Chemistry 2023 |