
Enhancing the durability of Au clusters in CO2 photoreduction via encapsulation in Cu-based metal–organic frameworks†
Jun
Zhang
a,
Xiaofeng
Cui
*ac,
Yu
Zhou
c,
Tingting
Kong
*a,
Yixin
Wang
a,
Xianwen
Wei
 a and
Yujie
Xiong
*ab
a and
Yujie
Xiong
*ab
aAnhui Engineering Research Center of Carbon Neutrality, The Key Laboratory of Functional Molecular Solids, Ministry of Education, Anhui Laboratory of Molecular-Based Materials, College of Chemistry and Materials Science, Anhui Normal University, Wuhu, 241002, Anhui, P. R. China. E-mail: xfcui@ahnu.edu.cn; ktt@ahnu.edu.cn; yjxiong@ustc.edu.cn
bSchool of Chemistry and Materials Science, University of Science and Technology of China, Hefei, 230026, Anhui, P. R. China
cSchool of Chemistry and Chemical Engineering, Anqing Normal University, Anqing, 246011, Anhui, P. R. China
First published on 30th January 2023
Abstract
Here, we report the encapsulation of Au25 nanoclusters in a Cu3(BTC)2 metal–organic framework (Au25@Cu-BTC), which can achieve CO2 photoreduction for selective CO production in a gas–solid reaction system at low-concentration CO2 atmospheres (even to 0.1%), with remarkably enhanced durability up to at least 48 h.
Light-driven CO2 reduction has continuously received attention.1–3 To this end, various photocatalytic materials have been explored for achieving this promising reaction. Very recently, atomically precise metal nanoclusters (NCs) with highly tunable numbers of atoms have emerged as a new class of catalytic materials.4–7 In particular, their subnanometer sizes (∼1 nm), approaching the de Broglie wavelength, result in discrete electronic structures, which makes their light absorption readily tuned.8,9 Moreover, the ultrasmall sizes and high conductivity of metal NCs are beneficial for the transfer of photogenerated electrons to their surface.10–12 These characteristics endow the metal NCs with the capability of working for light-driven catalytic reactions. In practical applications, the light-driven catalysts based on metal nanoclusters still typically suffer from two limitations. First of all, metal nanoclusters more readily aggregate into nanoparticles under heating or light illumination due to their extremely high surface energy, leading to the decay of their specific photochemical properties.13 In the other limitation, the tightly capped ligands on the metal surface, as well as the lack of catalytically active sites, hinder the photogenerated electrons from participating in surface reactions, limiting the catalytic activity.6
To prevent aggregation, metal oxides,14 polymers15 and covalent organic frameworks16 can serve as surface coatings. However, these approaches often involve complicated processes and/or can hardly provide effective active sites. Ideally, a surface coating material should not only improve the stability of metal NCs but also offer catalytically active sites for enhancing their catalytic performance. Metal–organic frameworks (MOFs) are crystalline porous materials that can be prepared under mild conditions, and their abundant metal nodes may work as active sites for targeted catalytic applications.16,17 For this reason, the encapsulation of nanomaterials in MOFs has been intensively studied in recent years.18,19 However, it remains a great challenge to accomplish surface coating with MOFs because of the lattice mismatch between the guest materials and the MOFs.20 The abundant surface functional groups and ultrasmall sizes of metal NCs offer the possibility of packaging them in the cavities of a MOF without affecting the framework. Such promising features would make the MOF material an ideal candidate for encapsulating metal NCs to enhance CO2 photoreduction performance.
Herein, we report a facile process for encapsulating atomically precise Au25(p-MBA)18 (p-MBA = 4-mercaptobenzoic acid) clusters in MOFs to enhance the durability of the catalyst in the selective photoreduction of CO2 to CO. In our designed scheme, the MOFs are expected to introduce multiple advantages for CO2 photoreduction, i.e., confining Au NCs to prevent aggregation, offering active sites for CO2 reduction, and capturing CO2 to facilitate the conversion process. Inspired by these considerations, Cu3(BTC)2 (BTC = benzene-1,3,5-tricarboxylate) is selected as the MOF model, as it can provide a framework for CO2 capture and Cu sites for CO2 activation as demonstrated by our previous studies.21,22
Fig. 1a schematically illustrates the synthetic procedure of Cu3(BTC)2-encapsulated Au25(p-MBA)18 (denoted as Au25@Cu-BTC). Water-soluble Au25(p-MBA)18 was synthesized according to the method reported by Chen et al.23 The successful preparation of Au25(p-MBA)18 was confirmed by UV-vis absorption spectroscopy and ESI mass spectrum (Fig. S1, ESI†). Of particular note is that we selected Au25(p-MBA)18 as the model cluster on account of its good visible light response and facile synthesis. More importantly, the protected ligand of p-MBA has a similar structure to the BTC ligand of MOFs, and as such, its carboxyl group can directly be coordinated with the Cu nodes, which will facilitate electron transfer from the Au NCs to Cu sites in MOFs.
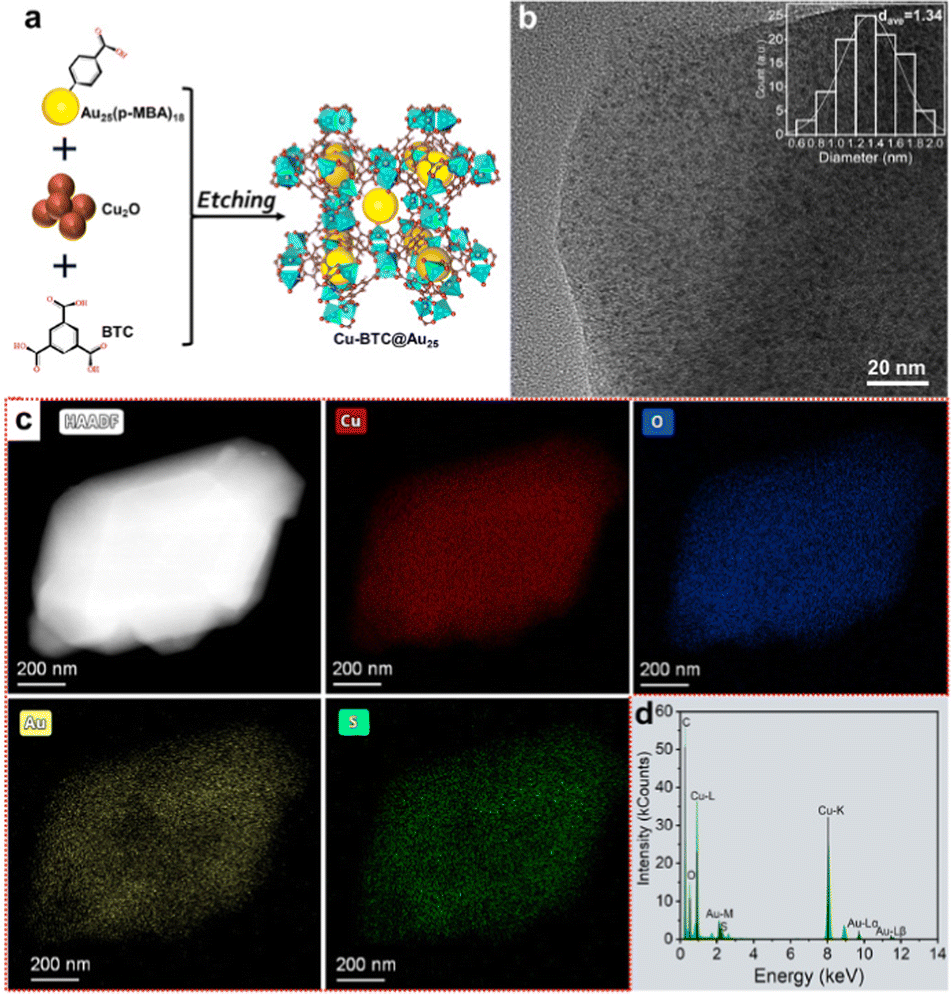 | ||
| Fig. 1 (a) Schematic illustration of the preparation process of the Au25@Cu-BTC composite. (b) HRTEM image, (c) STEM image and corresponding EDS elemental mapping images and (d) EDS spectrum of Au25@Cu-BTC. | ||
Unlike harsh synthesis conditions (i.e., solvo/hydrothermal methods), we assembled such a hierarchical composite through a mild one-pot solution-phase method to avoid altering the properties of the Au25 NCs. In our synthesis, Cu2O nanoparticles rather than Cu2+ ions were used as the Cu precursor for Cu3(BTC)2, as we found that directly adding Cu2+ inevitably induced Au NC aggregation. The Cu2O nanoparticles can be etched by the protons of carboxyl to slowly release Cu+ and then oxidized to Cu2+ by dissolved oxygen, which will coordinate with the carboxyl of BTC and p-MBA to in situ form the encapsulated Au25@Cu-BTC.22 As displayed in the powder X-ray diffraction (PXRD) pattern (Fig. S2, ESI†), the characteristic peak of Cu2O at 2θ = 36.6° (PDF#05-0667) decreased in intensity by prolonging the etching time. When it proceeded to 29 h, this characteristic peak faded thoroughly while the diffraction pattern is consistent with the simulated Cu3(BTC)2, indicating that Cu2O had been completely converted into Cu3(BTC)2. No characteristic peak of Au NCs was found in the PXRD pattern of the Au25@Cu-BTC composite, most likely due to the ultrasmall sizes of the Au NCs.
To investigate the structure, high-resolution transmission electron microscopy (HRTEM) was employed to examine Au25@Cu-BTC, as shown in Fig. 1b. Au NCs are clearly observed in the HRTEM image and uniformly dispersed in Cu3(BTC)2 with an average diameter of 1.34 nm (Fig. 1b inset). The size is consistent with that of the pristine Au NCs (Fig. S1b, ESI†), confirming that the Au NCs were well maintained during encapsulation. Cu3(BTC)2 was reported to have cavities with 1.33 nm size and 3D connected channels with 1.8 nm windows,24 so ultrasmall Au NCs can be confined in the cavities of Cu3(BTC)2 and/or implanted into their channels, preventing the aggregation of the Au NCs. The formation of Au25@Cu-BTC was further verified by scanning transmission electron microscopy (STEM) and corresponding energy-dispersive X-ray spectroscopy (EDS) elemental analysis (Fig. 1c and d). The EDS mapping images illustrate that Cu, Au, C, O and S are uniformly distributed, and the good coincidence relation between Cu and O as well as Au and S further confirms that the hierarchical structure has been successfully constructed. The amount of Au in the Au25@Cu-BTC composite is determined to be 11.6 wt% by ICP-AES, and the content of Au NCs in this composite is roughly calculated to be 18.1 wt% based on the molecular formula of Au25(p-MBA)18.
Upon forming the hierarchical structure, we further investigated the interaction between the embedded Au NCs and the coated Cu2(BTC)3 shell using X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared (FT-IR) spectroscopy. The high-resolution O1s XPS spectra (Fig. S4a, ESI†) reveal that Au NCs, Cu3(BTC)2 and Au25@Cu-BTC give the most distinct difference in both the existing forms and chemical shifts. The characteristic peaks around 532.5 eV and 531.7 eV can be assigned to hydroxide (H–C–O bonding) and carbonate (C–O bonding), respectively. H–C–O bonding is dominant in the Au NCs, which can be assigned to the uncoordinated carboxyl groups in p-MBA. When the Au NCs are incorporated into Cu3(BTC)2, C–O bonding becomes dominant in Au25@Cu-BTC, which can be attributed to the coordination of carboxyl groups of p-MBA with Cu nodes of Cu3(BTC)2 during in situ coating. This argument is confirmed by the shift of the O 1s (Fig. S4a, ESI†), S 2p (Fig. S4b, ESI†) and Au 4f (Fig. S4c, ESI†) characteristic peaks for Au25@Cu-BTC toward higher binding energy compared with the pristine Au NCs. This shift is caused by the coordination of carboxyl groups of Au NCs with Cu2+, reducing the electron densities of the p-MBA ligands. In the meantime, the characteristic peaks of Cu in the Cu LMM spectrum of Au25@Cu-BTC shift toward lower binding energy compared to bare Cu3(BTC)2 (Fig. S4d, ESI†), as the electron donation by O in the carboxyl of the Au NCs can increase the electron density of the Cu nodes in Cu3(BTC)2. These results demonstrate that the embedded Au NCs are directly connected with the Cu sites of coated Cu3(BTC)2 through coordination bonds, which is also confirmed by FT-IR (Fig. S3, ESI†), providing a good foundation for charge transfer between Au NCs and Cu3(BTC)2. No new Cu species are found on Au25@Cu-BTC (Fig. S4d, ESI† and Fig. S4, ESI†).
Upon acquiring the structural characteristics of Au25@Cu-BTC, we further examined its performance for CO2 capture and activation. Cu3(BTC)2 has been well demonstrated to have excellent selective sorption for CO2.25 The CO2 uptake capacity of Au25@Cu-BTC was evaluated by comparing the CO2 sorption isotherms with Cu3(BTC)2 under ambient conditions. As revealed by the adsorption and desorption isotherms (Fig. 2a), the CO2 uptake capacity of Cu3(BTC)2 and Au25@Cu-BTC was determined to be 100.63 and 62.17 cm3 g−1, respectively, based on the total weight. Given that Au NCs are not a characteristic material for CO2 adsorption, the CO2 uptake of the coated Cu3(BTC)2 component is calculated to be approximately 75.91 cm3 g−1, which is comparable to that of Cu3(BTC)2, indicating that the incorporation of Au NCs does not significantly block the channels of Cu3(BTC)2 for CO2 capture and mass transfer. This result is confirmed by N2 sorption measurements (Fig. S5, ESI†) The CO2 activation ability of the Cu3(BTC)2-based samples was examined by electrochemical linear sweep voltammetry (LSV) measurements in 0.1 M tetrabutylammonium hexafluorophosphate (TBAHFP) purged with CO2 or N2. As the reference sample, Cu3(BTC)2-loaded Au25(p-MBA)18 (denoted as Au25/Cu-BTC) was prepared via a mechanical mixing method. As shown in Fig. 2b, all the samples exhibit higher current density in the CO2 atmosphere than that in an inert atmosphere, indicating that all of them have the ability to activate CO2 under a reduction potential. Obviously, the Au25@Cu-BTC composite offers the largest increase of current density in the presence of CO2, which may be attributed to the improvement of electrical conductivity by embedding Au NCs into the framework of Cu3(BTC)2.
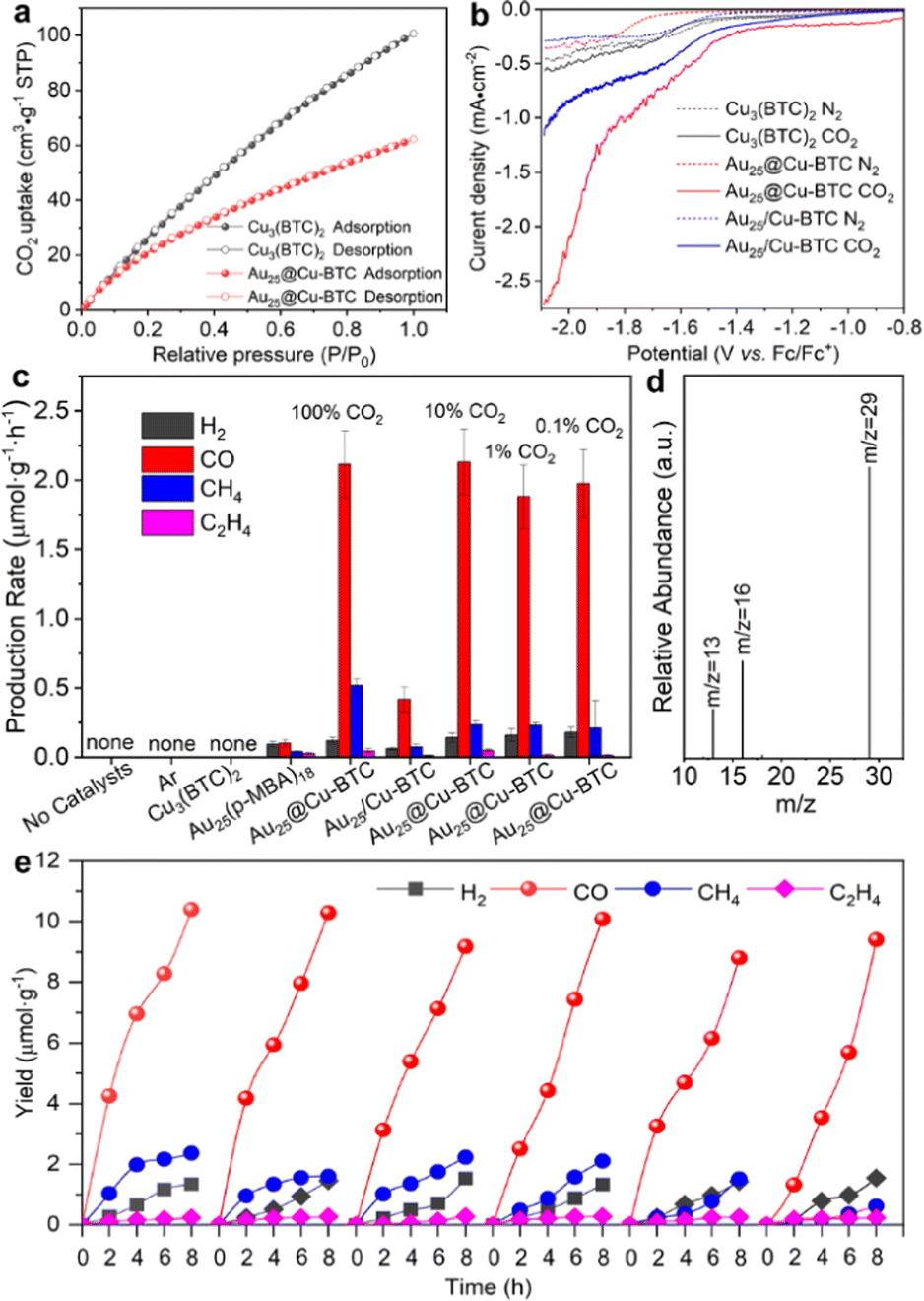 | ||
| Fig. 2 (a) CO2 adsorption behavior for Au25@Cu-BTC and bare Cu3(BTC)2. (b) LSV curves of pristine Cu3(BTC)2, Au25@Cu-BTC and mechanically mixed Au25/Cu-BTC in 0.1 M TBAHFP solution saturated with N2 or CO2. (c) Average production rates of H2, CO, CH4 and C2H4 in photoreduction CO2 by Au25@Cu-BTC in the first 2 h under visible-light (λ > 420 nm) irradiation, in comparison with those by Cu3(BTC)2, Au25(p-MBA)18, Au25/Cu-BTC and other control experiments under the same conditions. All measurements were performed on a gas–solid reactor in the presence of BIH and a trace amount of H2O. (d) GC–MS analysis of 13CO (m/z = 29) produced over Au25@Cu-BTC in light-driven reduction of 13CO2. (e) Light-driven catalytic durability over Au25@Cu-BTC. Each cycle takes 8 h. | ||
Given the excellent performance for CO2 capture and activation, we are now in a position to examine the performance of Au25@Cu-BTC as a light-driven catalyst for CO2 reduction. To fully take advantage of the outstanding CO2 uptake capability, the light-driven catalytic performance was assessed in a gas–solid reactor (Fig. S6, ESI†) under visible-light irradiation using a trace amount (100 μL) of H2O as a proton source and 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH) as a sacrificial agent to consume photogenerated holes. As shown in Fig. 2c, no reduction products were detected in the absence of catalysts and CO2, suggesting its strong dependence on both catalysts and CO2. Consistent with our previous study,18 pristine Cu3(BTC)2 did not exhibit catalytic activity for CO2 reduction because it lacks photoexcited electrons for the reduction reaction. Meanwhile, bare Au NCs also presented negligible photoreduction activity and lower selectivity for carbon products (84.1%), which should be due to their lack of active sites for catalyzing reactions.15 In sharp contrast, when Au NCs were embedded into the Cu3(BTC)2 matrix, the photoreduction activity of Au25@Cu-BTC was greatly improved (Fig. 2c and Fig. S7, ESI†). It turned out that the production rate of 2.13 μmol g−1 h−1 for CO is over 21 times that of bare Au NCs (0.102 μmol g−1 h−1), and the selectivity of the carbon products was increased up to 97.6% (Fig. 2c). This enhanced efficiency was achieved through the complementary roles of the two components, with Au NCs serving as a light-harvesting center and Cu3(BTC)2 providing catalytic sites. It has been well verified that Cu sites can suppress the side reaction of hydrogen evolution in photocatalytic and electrocatalytic CO2 reduction.21 However, it is difficult to accomplish such an effect by the simply mixed Au25/Cu-BTC, which yielded CO at a production rate of only 0.515 μmol g−1 h−1 (Fig. 2c). This indicates that the connection mode between them is the key for their efficient integration.
Considering the excellent CO2 capture capability of the Cu3(BTC)2 shell, we evaluated the photoreduction performance of Au25@Cu-BTC in a lower concentration CO2 (10%, 1%, and 0.1%) atmosphere (diluted with argon) under identical reaction conditions. Interestingly, our designed Au25@Cu-BTC gives roughly comparable photocatalytic performance in pure and diluted CO2 atmospheres (Fig. 2c), demonstrating that it has broad application potential in a low-concentration CO2 atmosphere. To determine the carbon source of the detected CO, isotopic 13CO2 was used as the reactant to carry out the light-driven catalytic reaction under the same conditions, and the product was analysed by gas chromatography–mass spectrometry (GC–MS) (Fig. 2d). The peak appearing at m/z = 29 in MS can be ascribed to 13CO, confirming that CO is indeed produced from the photoreduction of CO2.
To examine the durability of our catalysts, we performed a test in 6 successive cycles, each of which took 8 h. As shown in Fig. 2e, the catalytic performance of Au25@Cu-BTC remained steady for at least 48 h, indicating its excellent durability. HRTEM analysis confirms no distinct size change for the Au NCs confined in the Cu3(BTC)2 matrix (Fig. S8, ESI†). In contrast, bare Au NCs and physically mixed Au25/Cu-BTC can hardly maintain their catalytic activity for 2 successive cycles (Fig. S9, ESI†), as the Au NCs tend to be agglomerated into Au nanoparticles (ca. 4.5 nm) (Fig. S10, ESI†). These results fully prove that the good durability of Au25@Cu-BTC in CO2 photoreduction is attributed to the confinement effect of Cu3(BTC)2 encapsulation.
To gain a deeper understanding on the enhanced performance of Au25@Cu-BTC, we collected the photocurrent response on our catalysts, which has been widely used to reveal the charge separation efficiency. As shown in Fig. 3a, Au25@Cu-BTC exhibits about 4 times higher photocurrent density than Au25/Cu-BTC and 8 times that of bare Cu3(BTC)2, indicating the superiority of our designed composites for photogenerated charge separation. To find out the reason behind this improvement, Mott–Schottky measurements were applied to examine the charge transfer capability by comparing their resistance. As displayed in Fig. 3b, the charge transfer resistance R1 drops sharply from 7449 Ω of Cu3(BTC)2 to 2405 Ω of Au25/Cu-BTC and 1349 Ω of Au25@Cu-BTC, manifesting the smallest internal resistance of Au25@Cu-BTC for its efficient charge transfer. In combining the results of TEM, XPS and FT-IR with the preparation procedure of Au25@Cu-BTC, we can conclude that the efficient charge transfer and separation of Au25@Cu-BTC can be attributed to the following advantages compared with simple mixed Au25/Cu-BTC: (i) the high conductivity Au NCs (Fig. 3b), implanted in the Cu3(BTC)2 matrix, can improve the overall conductivity for fast transport of photogenerated charges; (ii) the in situ implanted Au NCs in Cu3(BTC)2 are directly connected with Cu sites through chemical bonds between the carboxyl of p-MBA and Cu nodes (Fig. S4, ESI†), offering more efficient transfer of photogenerated electrons than the Au25/Cu-BTC through physical contact; (iii) the photogenerated electrons on the embedded Au NCs have shorter migration distances to the Cu sites in the Cu3(BTC)2 channels than for Au25/Cu-BTC, which can greatly reduce the possibility of charge recombination during travelling to the Cu sites; (iv) more internal Cu sites can be utilized for efficient catalysis in the Au25@Cu-BTC system than in the counterpart of Au25/Cu-BTC.
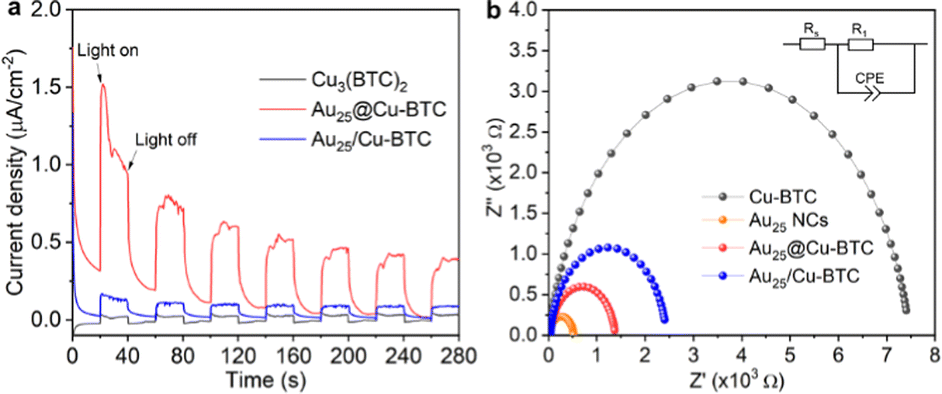 | ||
| Fig. 3 (a) Photocurrents and (b) Nyquist plots of Cu3(BTC)2, Au NCs, Au25@Cu-BTC and Au25/Cu-BTC. The inset in (b) is the equivalent circuit for fitting. | ||
In summary, we have developed a mild strategy for in situ encapsulating ultrasmall Au25 nanoclusters in a Cu-based MOF for CO2 photoreduction, in which the embedded Au NCs produce photoexcited charges while the coated microporous shells capture CO2 and provide active sites. Remarkably, the light-driven catalytic performance, particularly the durability, was enhanced by our designed Au25@Cu-BTC composite, in stark contrast to bare Au NCs and mechanically mixed Au25/Cu-BTC. The spectroscopic and morphological characterizations have proven that the performance enhancement is attributed to the direct chemical connection of Au NCs with the Cu sites of MOFs promoting charge transfer and the well-matched size between the Au NCs and MOF channels offering a strong confinement effect. This work highlights the importance of ligand and size matching in designing hierarchical photocatalytic materials based on molecular units.
We acknowledge the financial support in part by NSFC (91961106, 51902253, 21725102), Anhui Provincial Natural Science Foundation (Grant 2108085MB46), Key Project of Youth Elite Support Plan in Universities of Anhui Province (Grant gxyqZD2021121), and Shaanxi Provincial Natural Science Foundation (2020JQ-778).
Conflicts of interest
There are no conflicts to declare.Notes and references
- X. Wang, M. Sayed, O. Ruzimuradov, J. Zhang, Y. Fan, X. Li, X. Bai and J. Low, Appl. Mater. Today, 2022, 29, 101609 CrossRef.
- Y. Zhao, G. I. N. Waterhouse, G. Chen, X. Xiong, L. Z. Wu, C. H. Tung and T. Zhang, Chem. Soc. Rev., 2019, 48, 1972–2010 RSC.
- T. Kong, Y. Jiang and Y. Xiong, Chem. Soc. Rev., 2020, 49, 6579–6591 RSC.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443–6514 RSC.
- X. Cui, J. Wang, B. Liu, S. Ling, R. Long and Y. Xiong, J. Am. Chem. Soc., 2018, 140, 16514–16520 CrossRef CAS PubMed.
- M. Y. Gao, H. Bai, X. Cui, S. Liu, S. Ling, T. Kong, B. Bai, C. Hu, Y. Dai, Y. Zhao, L. Zhang, J. Zhang and Y. Xiong, Angew. Chem., Int. Ed., 2022, 61, e202215540 CAS.
- Q. Yao, X. Yuan, T. Chen, D. T. Leong and J. Xie, Adv. Mater., 2018, 30, 1802751–1802779 CrossRef PubMed.
- M. A. Abbas, T. Y. Kim, S. U. Lee, Y. S. Kang and J. H. Bang, J. Am. Chem. Soc., 2016, 138, 390–401 CrossRef CAS PubMed.
- Z. J. Guan, J. J. Li, F. Hu and Q. M. Wang, Angew. Chem., Int. Ed., 2022, 61, e202209725 CAS.
- R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Chem. Rev., 2021, 121, 567–648 CrossRef CAS PubMed.
- L. Sementa, G. Barcaro, A. Dass, M. Stener and A. Fortunelli, Chem. Commun., 2015, 51, 7935–7938 RSC.
- S. Liu and Y. J. Xu, Sci. Rep., 2016, 6, 22742 CrossRef CAS PubMed.
- B. Weng, K. Q. Lu, Z. Tang, H. M. Chen and Y. J. Xu, Nat. Commun., 2018, 9, 1543 CrossRef PubMed.
- Y. Deng, Z. Zhang, P. Du, X. Ning, Y. Wang, D. Zhang, J. Liu, S. Zhang and X. Lu, Angew. Chem., Int. Ed., 2020, 59, 6082–6089 CrossRef CAS PubMed.
- J. X. Gu, C. Y. Sun, X. L. Wang and Z. M. Su, Chem. Commun., 2022, 58, 10114–10126 RSC.
- M. L. Xu, X. J. Jiang, J. R. Li, F. J. Wang, K. Li and X. Cheng, ACS Appl. Mater. Interfaces, 2021, 13, 56171–56180 CrossRef CAS PubMed.
- Y. Luo, S. Fan, W. Yu, Z. Wu, D. A. Cullen, C. Liang, J. Shi and C. Su, Adv. Mater., 2017, 30, 1704576 CrossRef PubMed.
- M. L. Xu, M. Lu, G. Y. Qin, X. M. Wu, T. Yu, L. N. Zhang, K. Li, X. Cheng and Y. Q. Lan, Angew. Chem., Int. Ed., 2022, 61, e202210700 CAS.
- S. Dai, T. Kajiwara, M. Ikeda, I. R. Muñiz, G. Patriarche, A. E. Platero-Prats, A. Vimont, M. Daturi, A. Tissot, Q. Xu and C. Serre, Angew. Chem., Int. Ed., 2022, 61, e202211848 CAS.
- X. Deng, R. Li, S. Wu, L. Wang, J. Hu, J. Ma, W. Jiang, N. Zhang, X. Zheng, C. Gao, L. Wang, Q. Zhang, J. Zhu and Y. Xiong, J. Am. Chem. Soc., 2019, 141, 10924–10929 CrossRef CAS PubMed.
- R. Li, J. Hu, M. Deng, H. Wang, X. Wang, Y. Hu, H. L. Jiang, J. Jiang, Q. Zhang, Y. Xie and Y. Xiong, Adv. Mater., 2014, 26, 4783–4788 CrossRef CAS PubMed.
- T. Chen, V. Fung, Q. Yao, Z. Luo, D. Jiang and J. Xie, J. Am. Chem. Soc., 2018, 140, 11370–11377 CrossRef CAS PubMed.
- S. S.-Y. Chui, S. M.-F. Los, J. P. H. Charmant, A. G. Open and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- Z. Liang, M. Marshall and A. L. Chaffee, Energy Fuels, 2009, 23, 2785–2789 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc06719f |
| This journal is © The Royal Society of Chemistry 2023 |