
Nano-assemblies of phosphonium-functionalized diblock copolymers with fabulous antibacterial properties and relationships of structure-activity†
Peng
Cao
 a,
Xue
Bai
a,
Yufeng
He
a,
Pengfei
Song
a,
Rongmin
Wang
*a and
Junchao
Huang
*b
a,
Xue
Bai
a,
Yufeng
He
a,
Pengfei
Song
a,
Rongmin
Wang
*a and
Junchao
Huang
*b
aKey Lab. Eco-functional Polymer Materials of MOE, Institute of Polymers, College of Chemistry & Chemical Engineering, Northwest Normal University, Lanzhou 730070, China. E-mail: wangrm@nwnu.edu.cn
bSchool of Materials and Energy, Lanzhou University, Lanzhou 730000, China. E-mail: huangjunchaochitin@hotmail.com
First published on 18th October 2022
Abstract
As a novel antimicrobial material, quaternary phosphonium salts (QPSs) have been drawing close attention because of their excellent antimicrobial capacity with high activity and low bacterial survivability. Polymeric QPSs (PQPSs) also exhibit selectivity and long-term stability, however the polymerization of QPSs is severely challenged by low controllability and narrow selectivity of cation type. In this study, high-conversion RAFT polymerization is employed to prepare innovative phosphonium-functionalized diblock copolymers (PFDCs) with desired molecular weights and particle sizes. The excellent antibacterial activity of the PFDCs achieves lowest MIC values of 40 and 60 μg mL−1 (i.e., 1.4 and 2.2 μmol L−1) against E. coli and S. aureus, respectively. Mixing with an ink, dye, and latex coating does not weaken the antibacterial activity of the PFDCs, which inhibited 99.9% E. coli, showing broad applicability in different media. The effects of the cation type, synthesis medium, crosslinking content, and particle size on the morphology and antibacterial activity are studied. In summary, the RAFT polymerization of QPSs through the versatile design of ionic liquid monomers and the polymerization-induced self-assembly (PISA) method for constructing nano-assemblies with various micromorphology and particle size provides an exceedingly efficient way to build up multifunctional and multi-morphological polymeric nano-objects that open up vast possibilities in the fields of antibiotics, drug delivery, templated synthesis, and catalysis.
1. Introduction
Evolving and increasing numbers of superbugs (e.g., MRSA, MDRSP, VRE, MRAB, etc.), which have adapted to antibiotics owing to their abuse during infection treatments, are becoming one of the major risks that severely threaten human health and even the environment.1 The World Health Organization (WHO) reported that over 700 thousand people are killed by antibiotic-resistant bacteria every year, including infants and children.2 Fading disinfection or dysfunction of traditional antibiotics has significantly weakened the therapy of infections, especially against some vital superinfections.3 As a result, reinforcing the antimicrobial activity of antibiotics is now imperative to curb the havoc wreaked by superbugs. From the perspective of chemistry, creating specific cations with high polarity, e.g., quaternary ammonium ions, imidazole ions, and haloamides, is attractive and practicable due to the high antibacterial activity of these large cations.4–6 Polymeric antibiotics designed with the introduction of high polar cation ions are now gaining more and more attention due to their low toxicity, improved biodistribution, pharmacokinetics, biofilm penetration capacity, and bacterial selectivity, revealing great prospects as a new generation antibiotic.7,8 Compared to conventional quaternary cationic polymers, PQPSs have more effective antimicrobial activity thanks to the larger phosphor atom and lower electronegativity that induce stronger polarizability.9 Moreover, the high chemical and environmental stability of quaternary phosphonium salts allow them to be applied under various conditions and in different applications, such as the design of ionic liquids and nonviral gene delivery.10,11However, the synthesis of PQPSs is restricted by its low controllability, high price, and limited variety, causing their development to lag behind that of quaternary ammonium salts, imidazole salts, and pyrrolidine salts. The ionic liquids are novel organic solvents with a low melting point that have already been widely used in the preparation of functional polymers.12 High designability and variety of the chemical structure of both cations and anions can enable the synthesis of many polyelectrolytes, such as polycations, polyanions, polyzwitterions, and polyampholytes.2 Bringing quaternary phosphonium ions into an ionic liquid system significantly extends the variety of the cation type, which can be further designed as a highly selective antibiotic.13 On the other hand, employing reversible addition-fragmentation chain transfer (RAFT) polymerization in the preparation of poly(ionic liquids) has also been reported, as well as the advantages of high reactivity, high conversion rate, narrow molecular weight distribution, and good repeatability.14 RAFT polymerization-induced self-assembly (PISA), which realizes the precise manipulation of both molecular weight and particle morphology and size,15 is now commonly used to obtain multifunctional polymeric nanoparticles, e.g. micelles and vesicles,16 and it is also one of the optimal methods for the expandable design of PQPSs.
Our work aimed to prepare PQPSs through highly efficient RAFT polymerization with a tunable particle size and excellent antibacterial ability. To enhance the positivity of the quaternary phosphonium ion, the phenyl group with strong electron dispersion capacity was designed as the side group and introduced into the ionic liquids. Subsequently, the quaternary phosphonium ionic liquids were easily polymerized with a chain transfer agent to obtain a RAFT agent for further copolymerization with other monomers. Styrene (St) was used for the polymerization of the second block to control the molecular weight and particle size. Altering the hydrophilic quaternary phosphonium ion and hydrophobic styrene monomer provides high adaptability of the copolymers in various dispersions and application fields. The phosphonium-functionalized diblock copolymers (PFDCs) were further studied to evaluate their antibacterial activity, and the effects of the chemical structure of the cations and the molecular weight of the particles were studied to comprehend the possible antibacterial mechanism. The PFDCs with surprising antibacterial activity prepared through simple RAFT polymerization show great potential for future practical applications.
2. Experimental section
2.1 Materials and reagents
Azobisisobutyronitrile (AIBN, 98%, Shanghai Macklin Biochemical Co., Ltd, China) was recrystallized from methanol twice and stored in the fridge before use. Ionic liquid (IL) monomers of a quaternary phosphonium salt (QPS) (4-vinylbenzyl tributyl-phosphonium chloride (QPSBu3IL)) and 4-vinylbenzyl triphenyl-phosphonium chloride (QPSPh3IL) were prepared according to Scheme 1. Styrene (St, CP), ethylene glycol dimethacrylate (EGDMA, 98%), methyltrioctylammonium chloride (TOMAC, 97%), and 1-dodecanethiol (98%) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd, China. Carbon disulfide (CS2, 99%, Shanghai Saen Chemical Technology Co., Ltd) and Luria-Bertani (LB) broth medium (Qingdao Hope Bio-Technology Co., Ltd) were obtained commercially. Escherichia coli (E. coli, Gram-negative) and Staphylococcus aureus (S. aureus, Gram-positive) were kindly provided by Prof. Shiquan Niu (College of life sciences, Northwest Normal University, China) and incubated in nutrient agar (Qingdao Hi-Tech Industrial Park Haibo Biological Co., Ltd, biological-reagent grade). Deuterium oxide (D2O), Deuterated dimethyl sulfoxide (DMSO-d6), and deuterated chloroform (CDCl3) were of analytic grade and used as received without further purification. All other commercial reagents such as 2,6-di-tert-butyl-4-methylphenol (BHT), ether, acetone, 1,4-dioxane, methanol, isopropyl alcohol, n-hexane, trichloromethane, concentrated hydrochloric acid (HCl, 36–38%, w/w) and sodium hydroxide were of analytical grade and used as received unless otherwise noted. A dialysis bag (MWCO 8000–14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) was pretreated with 2% sodium bicarbonate solution and 1.0 mmol L−1 ethylenediamine tetraacetic acid (EDTA) solution, then washed with distilled water and stored in the fridge. All reactions were carried out under a nitrogen atmosphere with magnetic stirring unless otherwise noted. Distilled water was used throughout the experiment.
000 g mol−1) was pretreated with 2% sodium bicarbonate solution and 1.0 mmol L−1 ethylenediamine tetraacetic acid (EDTA) solution, then washed with distilled water and stored in the fridge. All reactions were carried out under a nitrogen atmosphere with magnetic stirring unless otherwise noted. Distilled water was used throughout the experiment.
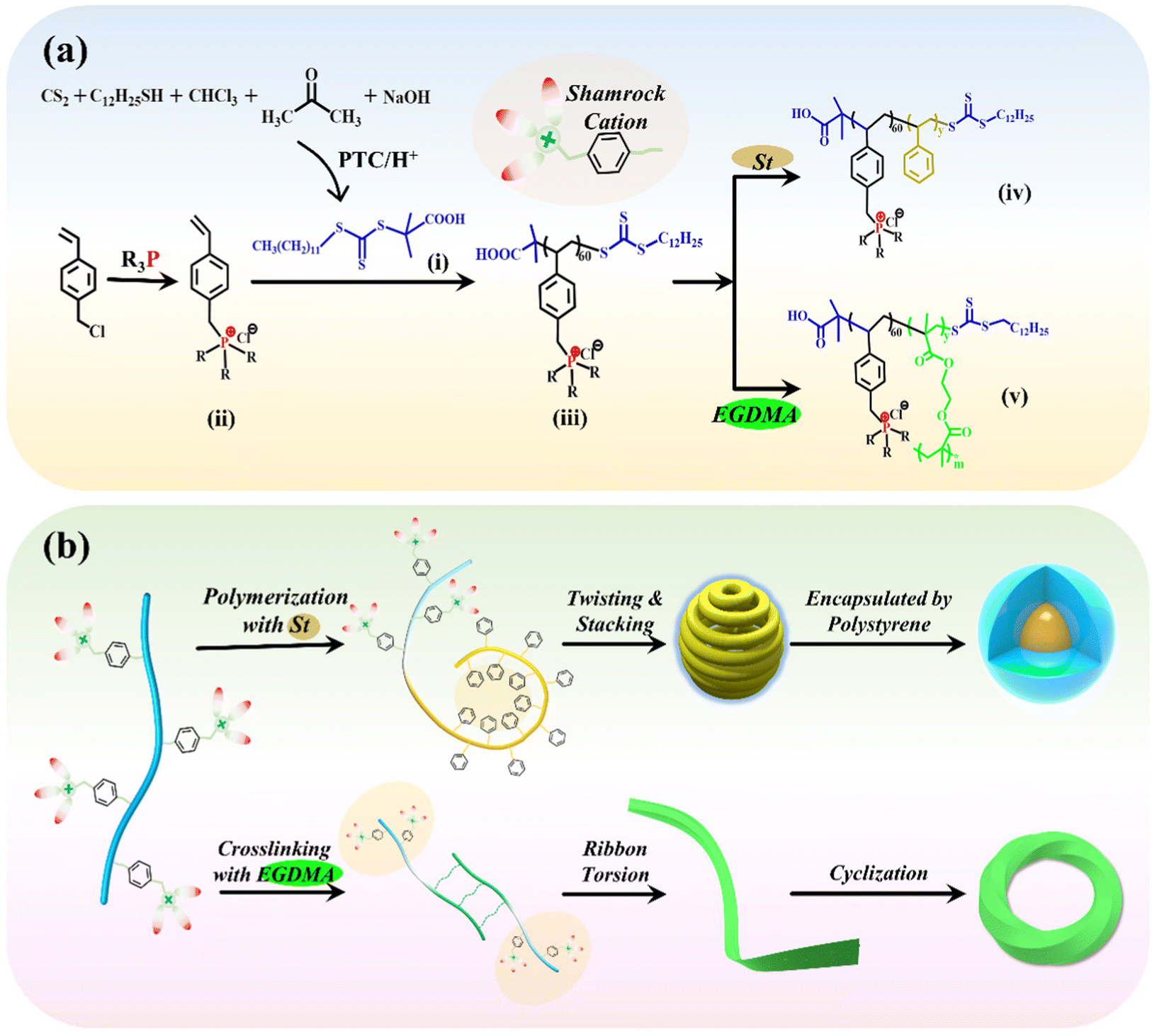 | ||
| Scheme 1 Schematic illustration of the synthesis of the CTA agent, IL monomers, and PFDCs (a), and the general assembly pattern for the micromorphology (b). (i) 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid chain transfer agent (DCMAT); (ii) quaternary phosphonium-based IL monomers (QPSPh3IL and QPSBu3IL); (iii) macromolecular chain transfer agent (macro-CTA, Ph60 and Bu60); (iv) phosphonium-functionalized diblock copolymers (PFDCs, Ph60Sty and Bu60Sty); (v) PEGDMA crosslinked Bu60/Ph60 diblock copolymers (BuEm and PhEm). | ||
2.2 Synthesis
QPSBu3IL was prepared according to a reported method,18 as shown in Scheme 1(a). 4-Vinylbenzyl chloride (8.06 g, 53 mol) and tributylphosphine (10.52 g, 53 mol) were added into a dried flask containing 100 mL of acetone, and the mixture was magnetically stirred at 60 °C for 48 h under a nitrogen atmosphere. After the reaction, the mixture cooled down to room temperature, then 150 mL of diethyl ether was added dropwise, which gradually generated a white precipitate. The precipitate was sequentially filtered, washed with diethyl ether, and vacuum-dried at room temperature for 24 h to obtain the pure product. The yield rate was 78.6% (m.p. 127.3 °C). The chemical shifts were measured by NMR as follows: 1H NMR (D2O, δ ppm from TMS, Fig. S3a, ESI†): 0.68 (t, 9H), 1.12–1.36 (m, 6H), 1.88 (s, 6H), 3.39 (t, 6H), 5.11 (d, 2H), 5.65 (d, 1H), 6.45–6.63 (m, 2H) and 7.0–7.4 (m, 4H). 13C NMR spectrum (D2O, δ ppm from TMS, Fig. S3b, ESI†): 12.8, 17.3, 17.8, 22.7, 23.5, 25.5, 26.1, 115.3, 127.2, 128.1, 130.3, 136.0 and 137.4. The molecular weight of QPSBu3IL (C21H36P) was characterized by ESI-MS (m/z calculated: 319.49, found: 319.31).
466 g mol−1 and 1.14, respectively (Table S1, ESI†). The chemical shifts of Bu60 were measured by NMR as follows: 1H NMR spectrum (D2O, broad peak δ ppm from TMS, Fig. 1): 7.09 (Ar–H, ortho to CH2–P), 6.39 (Ar–CH2–P), 3.63 (P–CH2–(CH2)2CH3), 2.00 (P–CH2–CH2–CH2–CH3 and backbone CH), 1.24 (P–(CH2)2–CH2–CH3 and backbone CH2), and 0.71 (P–(CH2)3–CH3 and backbone CH3). 31P{1H} NMR spectrum (D2O, δ ppm, Fig. S4, ESI†): 25.4.
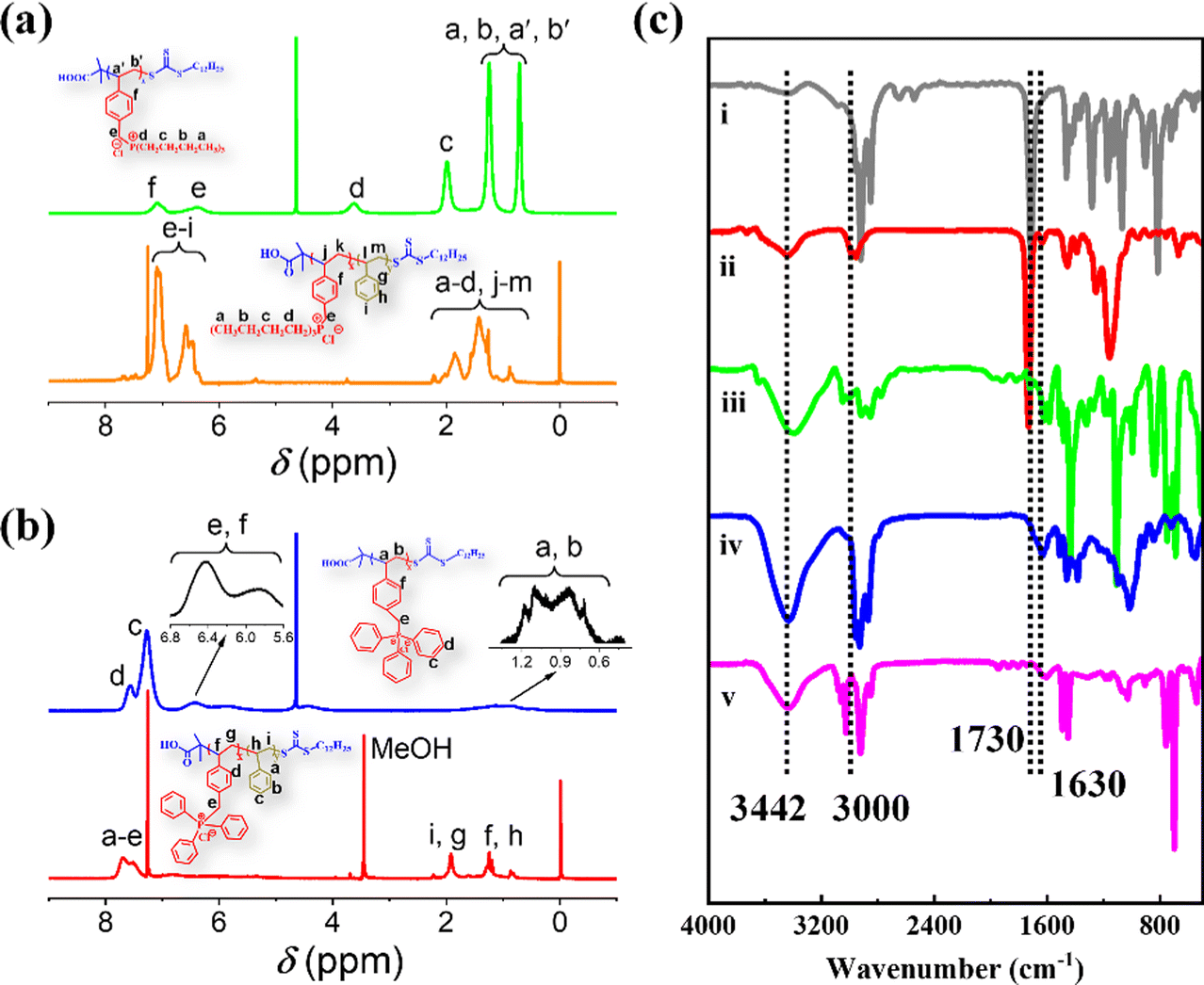 | ||
| Fig. 1 (a) and (b) 1H NMR spectra of Bu60 (green), Bu60St120-M (orange), Ph60 (blue) and Ph60St120-M (red). (c) FTIR spectra of DCMAT (i), Ph60 (ii), Ph60St120-M (iii), Bu60 (iv) and Bu60St120-M (v). | ||
Another macro-CTA, poly(triphenyl-(4-vinylbenzyl)-phosphonium chloride) chain transfer agent (Ph60), was also synthesized by RAFT solution polymerization. Briefly, DCMAT (91 mg, 2.5 × 10−4 mol), QPSPh3IL monomer (6.2337 g, 0.015 mol), and anhydrous methanol (20 g) were added into a 100 mL Schlenk flask to achieve a target degree of polymerization (DP) of 60. Subsequently, AIBN initiator (8.2 mg, 5 × 10−5 mol, nCTA/nAIBN = 5.0) was added to the mixture, followed by repeated vacuuming and filling with nitrogen for 30 min to eliminate dioxygen. The flask was then tightly sealed and immersed in an oil bath at 70 °C. The final monomer concentration was fixed at 24% (w/w). The reaction proceeded for 12 h and was quenched by exposing the mixture to air and plunging the tube into liquid nitrogen. After thawing, the polymer solution was transferred into a dialysis bag and dialyzed against abundant DI water for 24 hours. A light-yellow precipitate was generated during dialysis, then freeze-dried to obtain the macro-CTA agent (5.9452 g, 94% in conversion rate as determined by 1H NMR). The product was coded Ph60. Aqueous APC analysis (vs. poly (ethylene oxide) standards) indicated Mn and Mw/Mn values of 24667 g mol−1 and 1.13, respectively (Table S1, ESI†). The chemical shifts of Ph60 were measured by NMR as follows: 1H NMR spectrum (D2O, broad peak δ ppm from TMS, Fig. 1): 7.57 (P–Ar–H, para), 7.30 (P–Ar–H, ortho), 6.49 (H–Ar–CH2–P), 5.90 (H–Ar–CH2–P), 1.2 (P–(CH2)3–CH3 and backbone CH and CH2), and 0.6 (P–(CH2)3–CH3 and backbone CH3). 31P{1H} NMR (D2O, δ ppm, Fig. S4, ESI†): 32.8.
| PFDCs | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Synthesis and measured in methanol | Synthesis and measured in 1,4-dioxane | ||||||||
| Block copolymer a |
n
St:nmC:nI |
D h (nm) | PDIc | ξ-potential (mV) | Block copolymera |
n
St:nmC:nI |
D h (nm) | PDI | ξ-potential (mV) |
| a D: 1,4-dioxane; M: methanol; mC: macro-CTA; I: AIBN. b Average hydrodynamic diameter of the resultant nanoparticles. c Polydispersity index obtained from the accessory software. | |||||||||
| Bu60St30-M | 30:1:0.5 |
120 | 0.187 | 51.2 | Bu60St30-D | 30:1:0.5 |
126 | 0.154 | 49.8 |
| Bu60St120-M | 120:1:0.5 |
245 | 0.202 | 42.6 | Bu60St120-D | 120:1:0.5 |
244 | 0.150 | 43.2 |
| Bu60St200-M | 200:1:0.5 |
571 | 0.230 | 34.6 | Bu60St200-D | 200:1:0.5 |
464 | 0.121 | 33.3 |
| Bu60St700-M | 700:1:0.5 |
1255 | 0.208 | 28.7 | Bu60St700-D | 700:1:0.5 |
997 | 0.166 | 28.5 |
| Ph60St30-M | 30:1:0.5 |
40 | 0.289 | 55.8 | Ph60St30-D | 30:1:0.5 |
31 | 0.107 | 52.0 |
| Ph60St120-M | 120:1:0.5 |
185 | 0.275 | 48.2 | Ph60St120-D | 120:1:0.5 |
124 | 0.111 | 46.1 |
| Ph60St200-M | 200:1:0.5 |
427 | 0.202 | 44.2 | Ph60St200-D | 200:1:0.5 |
348 | 0.293 | 40.5 |
| Ph60St700-M | 700:1:0.5 |
1033 | 0.393 | 42.6 | Ph60St700-D | 700:1:0.5 |
1063 | 0.213 | 38.7 |
A series of poly[triphenyl-(4-vinylbenzyl)-phosphonium chloride-styrene] (Ph60Sty) diblock copolymers, was also synthesized by RAFT polymerization. For brevity, the example synthesis of Ph60St120 is given as follows. Briefly, AIBN (0.82 mg, 5.0 × 10−6 mol, nPh60/nAIBN = 2.0), Ph60 (202.1 mg, 1.0 × 10−5 mol) and methanol (1853.6 mg) were added into a Schlenk flask. After dissolution, styrene (St, 125 mg, 1.2 × 10−3 mol, nSt/nPh60 = 120) was added. The final content of St and Ph60 was fixed at 15% (w/w). The flask was deoxygenated five times by consecutive freezing-vacuuming-inflating with a nitrogen-thawing cycle. After the final cycle, the flask was thawed to room temperature and placed in an oil bath at 70 °C. The precursor solution was stirred for 16 h to ensure complete conversion of the St monomer. The reaction was then quenched by immersing the Schlenk flask in liquid nitrogen. After thawing, the polymer solution was mixed with diethyl ether, which generated a precipitate. The precipitate was washed with diethyl ether three times and freeze-dried to obtain the product. The product was coded Ph60St120-M, where M represents methanol solvent. Other diblock copolymers of Ph60Sty with different St monomer contents were prepared according to a similar protocol (Table 1). The conversion rate of St monomer for each Ph60Sty copolymer was determined by using the 1H NMR spectrum recorded in CDCl3 and the data are summarized in Table S1 (ESI†). For comparison, a copolymer prepared by using 1,4-dioxane as solvent was also synthesized, which was coded Ph60St120-D where D represents 1,4-dioxane solvent.
2.3 Characterization
1H, 13C, and 31P NMR spectra were recorded on a Brucker AM spectrometer (400 MHz) at 300 K with tetramethylsilane (TMS) as the internal standard. All samples were dissolved in CDCl3 to prepare a 2% (w/w) solution. Peak multiplicity was abbreviated as follows: singlet (s), doublet (d), triplet (t), quartet (q), and multiplet (m).Electrospray ionization mass spectrometry (ESI-MS, Agilent 7890/5975, USA) was used to identify the molecular weight of the monomer and CTA samples.
Fourier-Transform infrared (FTIR) spectra were measured on a DIGILAB FTS3000 spectrophotometer by using the KBr method with wavenumber ranging from 4000 cm−1 to 400 cm−1.
The average molecular weight of the PFDCs (1 mg mL−1 in HPLC THF solvent) was recorded using ACQUITY advanced polymer chromatography (APC) (Waters, USA) equipped with an ACQUITY APC XT 200 column (2.5 μm, 4.6 × 150 mm) and refractive index detector.
The surface compositions and chemical states of elements were determined by using ESCALAB Xi+ X-ray photoelectron spectroscopy (XPS) (Thermo Fisher Scientific Inc.) equipped with a monochromatic Al Kα X-ray source (1486.6 eV) with a spot size of 200–900 μm and an energy resolution of < 0.43 eV.
The morphology of the samples was observed using scanning electron microscopy (SEM, JEOL JSM 6701F) at an accelerating voltage of 5 kV.
The particle size and zeta potential (ξ) of the samples in the as-prepared solutions (1 mg mL−1 in methanol or 1,4-dioxane) were measured at 25 °C on a Malvern Panalytical Zetasizer Nano ZS90 instrument equipped with a 4 mW He–Ne laser operating at a wavelength of 633 nm and an avalanche photodiode (APD) detector. Each sample was measured three times, and the ξ values were calculated according to the Smolochowski–Helmholtz equation.
The contact angle (CA) of the samples was recorded on an SL200B goniometer (KINO Scientific Inc., USA). Briefly, dried sample powders were firstly ground and integrated by a tablet machine to obtain the membrane sample. The membrane sample was loaded on a slide and mounted on the platform. A drop of DI water (4 μL) was added from the top with an injection device. The images of the liquid on the sample were collected by a computer, and the contact angle was calculated automatically using the software.
2.4 Evaluation of antibacterial activity
The antibacterial activity of the PFDCs against Escherichia coli (E. coli, Grams-negative) and Staphylococcus aureus (S. aureus, Grams-positive) was evaluated in this work. Both strains were stored at 4 °C before the experiment. To activate the bacteria, a bacterial suspension (in LB broth) was incubated at 37 °C for 24 h. A bacterial suspension of 107 CFU mL−1 was prepared before the test. A nutrient agar hydrogel was used as the culture medium. The antibacterial activity of the PFDCs was assessed by using three different methods, colony formation assay, minimum inhibitory concentration (MIC), and SEM imaging.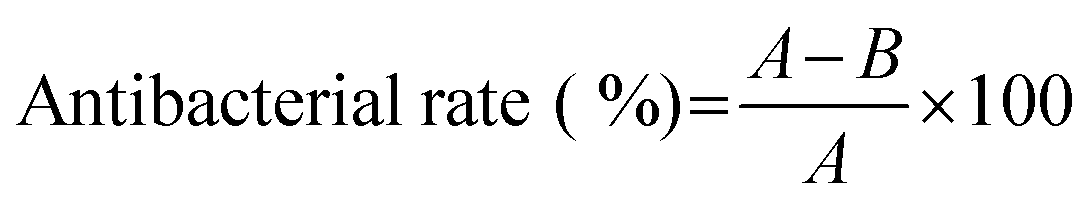 | (1) |
000 rpm for 10 min to harvest the bacteria. The bacteria were further dehydrated by 25%, 75%, 90%, and 100% (v/v) aqueous ethanol solutions every 15 min. The dehydrated bacteria were gently spread on conductive tape and then observed using SEM.
3. Results and discussion
3.1 Synthesis of phosphonium-functionalized diblock copolymers (PFDCs)
To prepare a series of PFDCs with long-acting and broad-spectrum antibacterial activity, RAFT polymerization-induced self-assembly (RAFT PISA) was employed to realize their efficient synthesis and micromorphology manipulations. As illustrated in Scheme 1(a), the requisite chain transfer agent (CTA) and quaternary phosphonium ionic liquid monomers were firstly synthesized.17,18 DCMAT (CTA) could be obtained through a designed nucleophilic reaction with phase transfer catalysis (PTC) according to R. Shea's work.20 Briefly, in the presence of a base, 1-dodecanethiol reacted with CS2, followed by nucleophilic reactions with chloroform and acetone, and yellow crystalline DCMAT was obtained after subsequent acidification. The characterization using NMR and FTIR spectra of DCMAT (Fig. S1, ESI†) demonstrated the successful synthesis of the desired product. On the other hand, the ionic liquid monomers of the QPS, 4-vinylbenzyl-tributylphosphonium chloride (QPSBu3IL), and 4-vinylbenzyl-triphenylphosphonium chloride (QPSPh3IL) were prepared using the substitution reaction between 4-vinylbenzyl chloride and tributylphosphine or triphenylphosphine. Fig. S2 and S3 (ESI†) show the NMR spectra with peak resolution to identify the chemical structure of QPSBu3IL and QPSPh3IL. Further, by mixing QPSBu3IL or QPSPh3IL, the DCMAT CTA, and AIBN (initiator) in the desired molar ratio, the active macro-CTA with a shamrock shape (regarding the electron cloud distribution), namely Bu60 or Ph60, was synthesized through RAFT solution polymerization.In the subsequent polymerization, the RAFT and PISA process was considered to regulate and control the micromorphology of the particles of PFDCs. Constructing an insoluble second block during RAFT polymerization from the reaction system is an effective method to realize PISA and produce nano-assemblies, which simplifies the synthesis process, shortens the equilibration time, and avoids extremely dilute conditions.21,22 Styrene is a typical rigid monomer that dissolves in methanol but precipitates after polymerization. Therefore, by mixing the Bu60 or Ph60 macro-CTA, styrene monomer, and AIBN (initiator) with methanol solvent, PFDC nano-assemblies could be achieved via the RAFT and PISA process at a relatively high polymer dosing concentration (15%, w/w), as shown in Scheme 1(b). Another solvent, 1,4-dioxane, was also employed to study the effect of solvent on the micromorphology of the nano-assemblies. The sample codes and their conversion rate and molecular weight are summarized in Table S1 (ESI†). A conversion rate beyond 96% revealed the high efficiency of the RAFT polymerization for PFDCs. Also, the experimental molecular weight was close to the theoretical calculation, indicating the that RAFT and PISA process in this work was a highly predictable polymerization method. The synthesis of PFDCs was also accompanied by the micromorphology formation by PISA.
A relatively high density of quaternary phosphonium cations allowed substantial intramolecular interactions that induced further self-assembly. Based on the principle of polymer physics, the shamrock cations are sequenced in the trans or gauche steric arrangement along the polymer chain due to steric hindrance and electrostatic repulsion and leading to a staggered distribution (Scheme 1(b)). The flexibility of the polymer chain and hydrophilic-hydrophobic interactions between polymer and solvent allowed the planar curling and twisting of PSt segments through the conjugations between adjacent phenyl groups on the inner side, and further lateral stacking on the outer side. Such a chain arrangement formed a spherical morphology with minimal configurational entropy and acted as the core encapsulated by Bu60 or Ph60 segments due to their higher affinity to solvent (methanol). A more interesting morphology was obtained through the altering of synthesis factors, which will be discussed in the next section.
Additional crosslinking along with RAFT polymerization by the introduction of EGDMA was also investigated to study the effect of crosslinking on the micromorphology and antibacterial activity of the nano-assemblies. After the styrene monomer was replaced by EGDMA for RAFT polymerization with Bu60 or Ph60, PEGDMA crosslinked PFDCs were obtained. Double vinyl groups allowed not only the polymerization of EGDMA itself but also copolymerization with different macro-CTAs, leading to the so-called “crosslinking polymerization”.23 The introduction of PEGDMA led to the formation of a crosslinked network between macro-CTA and a ribbon-like structure, which then twisted due to the chain flexibility and connected end-to-end through conjugation between quaternary phosphonium ions and the phenylene groups, giving a circle sheet or Mobius loop structure, which will be discussed further in the next section.
The PEGDMA crosslinked PFDCs coded REm are listed in Table S2 (ESI†), where R represents Bu or Ph, and m represents the initial dose in micromoles of EGDMA. The successful synthesis of PEGDMA crosslinked PFDCs manifested the favorable universality of the RAFT PISA method that might inspire more types and morphologies of PQPS in view of the required functionalities.
3.2 Chemical structure and micromorphology of PFDCs
Chemical structure characterization of the PFDCs was conducted on the representative copolymers Ph60St120-M and Bu60St120-M to exemplify each step during polymerization, and the corresponding macro-CTA (Bu60 and Ph60) was characterized by using 1H NMR and FTIR spectra, as shown in Fig. 1. The NMR peaks were also identified according to common knowledge of organic chemistry, as labeled in Fig. 1(a) and (b). Fig. S4 (ESI†) shows the 31P NMR spectra of Ph60, Bu60, Ph60St120-M, and Bu60St120-M, wherein the sharp signal for each sample confirmed the presence of phosphorus atoms in the polymer backbone. The higher chemical shift of Ph60 (32.83 ppm) than that of Bu60 (25.41 ppm) demonstrated that the triphenyl-phosphonium cation had lower electron density than that of the tributyl-phosphonium cation because of the stronger electron delocalization capacity provided by the phenyl groups. Copolymerization with St monomer did not sharply change the chemical shift in the 31P NMR spectra, which confirmed the diblock copolymerization.FTIR spectra were used to confirm the existence of the fundamental functional groups on the polymer molecules, as illustrated in Fig. 1(c). The characteristic peaks at 3442, 1730, 1180 and 1028 cm−1 appeared in the case of both macro-CTAs and the PFDCs, which were attributed to O–H in carboxyl groups, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in carboxyl groups, and thiocarbonyl (CS) groups, respectively, which belonged to DCMAT, one of the starting materials.24 The peaks at 1109 and 755 cm−1, 3000 and 3021 cm−1, and 1630 cm−1, which were assigned to the vibration of P–C, C–H aromatic stretching, and aromatic double bond stretching, respectively, were only observed in the spectra of the macro-CTAs and PFDCs, suggesting successful copolymerization.25 On the other hand, the chemical composition of the PEGDMA crosslinked PFDCs was characterized using XPS, as shown in Fig. S5 (ESI†). The survey spectrum clearly displayed the existence of C, O, P, S, and Cl elements for the PhE20, BuE25, and BuE30 samples as representatives. The peaks at 284.1, 531.7, 133.4, 163.64, and 196.9 eV were assigned to C1s, O1s, P2p, S2p, and Cl2p, respectively.26 This also confirmed that the PEGDMA crosslinked PFDCs were successfully synthesized.
O in carboxyl groups, and thiocarbonyl (CS) groups, respectively, which belonged to DCMAT, one of the starting materials.24 The peaks at 1109 and 755 cm−1, 3000 and 3021 cm−1, and 1630 cm−1, which were assigned to the vibration of P–C, C–H aromatic stretching, and aromatic double bond stretching, respectively, were only observed in the spectra of the macro-CTAs and PFDCs, suggesting successful copolymerization.25 On the other hand, the chemical composition of the PEGDMA crosslinked PFDCs was characterized using XPS, as shown in Fig. S5 (ESI†). The survey spectrum clearly displayed the existence of C, O, P, S, and Cl elements for the PhE20, BuE25, and BuE30 samples as representatives. The peaks at 284.1, 531.7, 133.4, 163.64, and 196.9 eV were assigned to C1s, O1s, P2p, S2p, and Cl2p, respectively.26 This also confirmed that the PEGDMA crosslinked PFDCs were successfully synthesized.
The molecular weights of the macro-CTAs and PFDCs were measured using advanced polymer chromatography (APC), and the trace spectra are shown in Fig. 2 and the conversion rate and molecular weight are summarized in Table S1 (ESI†). The macro-CTAs Bu60 and Ph60 with the theoretical molecular weight (Mn,th) of 21662 and 25259 g mol−1 showed a similar retention time at around 26 min, while the experimental molecular weight (Mn,APC) showed smaller values of 21466 and 24667 g mol−1, respectively, as shown in Fig. 2(a). All PFDCs were designed with a target degree of polymerization (DP) for the St monomer, and the corresponding theoretical molecular weight was calculated and is listed in Table S1 (ESI†). The retention time of the representative PFDCs ranged from 22 to 28 min (Fig. 2(b) and (c)), increasing in the order of descending molecular weight. The APC result showed that the experimental molecular weight of the obtained PFDCs was in good agreement with the theoretical value with an error less than 10%, suggesting the accurate controllability of the RAFT polymerization. Also, the high conversion rate of over 95% of the RAFT polymerization suggested the favorable possibility for industrial production of the PFDCs. Synthesis in different solvents with different relative polarities (methanol: 0.762; 1,4-dioxane: 0.164) had no significant influence on either molecular weight or conversion rate (Table S1, ESI†), suggesting a weak solvent dependence of the RAFT polymerization of PFDCs. The single peak of the APC curve and the polydispersity value measured by the APC method (Đ) ranged from 1.10 to 1.25, indicating a high uniformity of the synthesized PFDCs obtained from the living radical polymerization.27 The APC results confirmed that the RAFT polymerization designed in this work was highly applicable to industrial production.
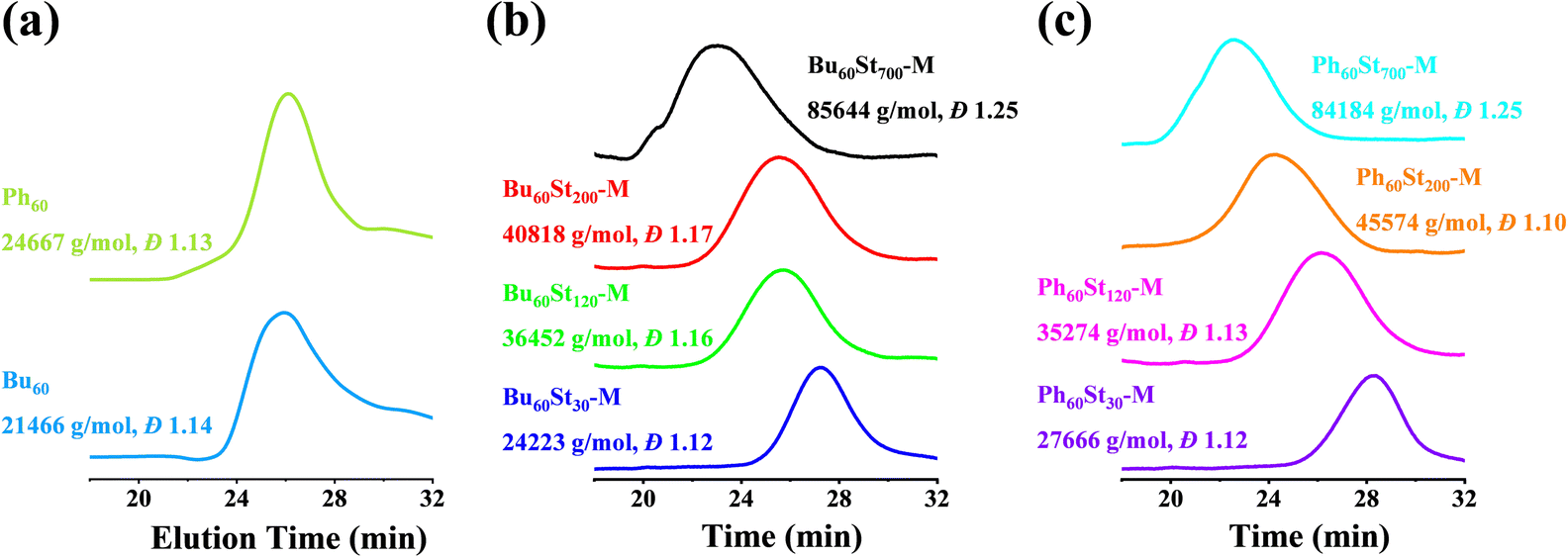 | ||
| Fig. 2 APC traces of the macro-CTAs and PFDCs. | ||
Contact angles (CAs) of the PFDC membranes were further measured to classify the hydrophilicity and hydrophobicity, as shown in Fig. 3. The macro-CTAs Bu60 and Ph60 were soluble in water possibly due to the strong polarization effect of the quaternary phosphonium ions. Undoubtedly, copolymerization with hydrophobic styrene strikingly enhanced the hydrophobicity of the PFDCs, which led to an increase in contact angle. Meanwhile, a bigger contact angle was observed with the increase of St content in both Bu60Sty-M and Ph60Sty-M groups. It was reported that the CA of PSt was around 85° in thin film form,28 which increased to 147° in foam form29 and 155° with a honeycomb microstructure,30 suggesting a nonnegligible effect of surface morphology on the hydrophobicity. The CA of the PFDCs with high St contents showed a bigger value (125–130°) than the case of the thin films; as a result, it was reasonable to deduce that a special morphology of the PFDCs was produced during polymerization. However, interestingly, the CA of Ph60Sty-M was similar to that of Bu60Sty-M even at a relatively low St content. The CA of several PEGDMA crosslinked PFDCs was also measured, as shown in Fig. S6 (ESI†). PEGDMA crosslinked PFDCs had a low CA, suggesting favorable hydrophilicity due to the low DP of the EGDMA monomer, while the CA increased with EGDMA content because of the insolubility of PEGDMA in water. It should be noted that the CA of PhE20 was bigger than that of BuE20, suggesting lower hydrophilicity.
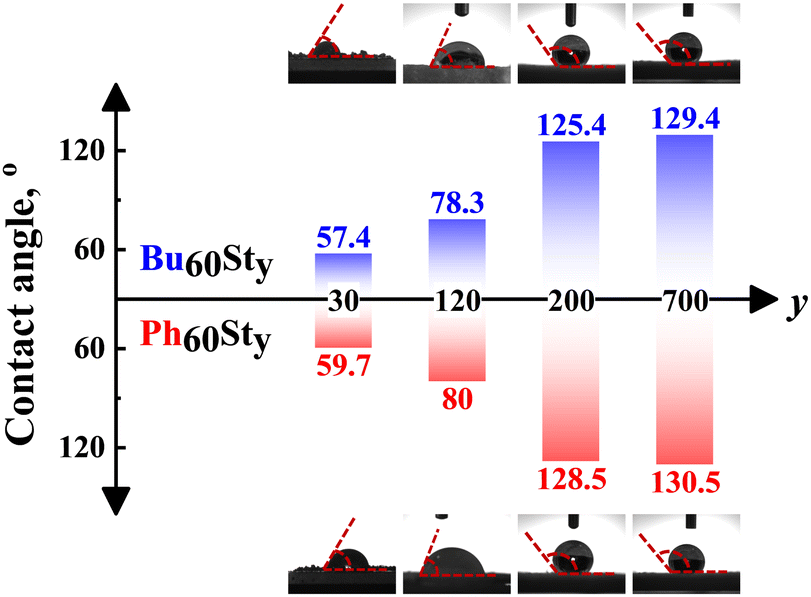 | ||
| Fig. 3 The contact angle (CA) of Bu60Sty-M and Ph60Sty-M in membrane form. The Bu60 and Ph60 macro-CTAs were soluble in water. | ||
As described above, when the copolymerized component was St, the CA tended to be the same, while it was markedly different in the case of EGDMA. The paradoxical phenomenon occurring between St and EGDMA monomers regarding the trends of contact angle possibly derived from the conjugation effect between the first macro-CTA block and the second block. In the case of PSt, an ultrawide conjugation effect along the whole polymer chain among phenyl groups averaged the charges and gave a similar surface with nearly equal electricity, leading to a similar contact angle regardless of the macro-CTA type. However, crosslinking polymerization by EGDMA only provided micromorphology tunability instead of averaging the polymer charges. The intermolecular conjugation effect of phenyl groups of the PhEm copolymer might induce a micellar shape with a hydrophilic inner (π–π stacking) but hydrophobic outer, resulting in a more hydrophobic surface and a higher CA compared to the BuEm copolymer, whose n-butyl side groups had weak interactions between each other and induced less special micromorphology or specific effects on the hydrophobicity.31 In summary, the hydrophilicity of the PFDCs could be controlled by quantitatively adjusting the ratio of two monomers, which will be useful for different practical applications.
Scanning electronic microscopy (SEM) was further conducted to examine the micromorphology of the PFDCs, as shown in Fig. 4. All PFDCs assembled into nanoparticles with a diameter ranging from 50 to 1600 nm related to the St content. With an increase in molar ratio of St monomer, the particle size of the nano-assemblies also increased. However, a dramatic decrease was observed from macro-CTA to PFDCs with relatively low contents of St (e.g. y = 30 and 120), which was induced by the π–π stacking between phenyl groups on cations and the PSt second block, which reduced the size of the particles. The large size of the macro-CTA might have originated from the adjacent quaternary phosphonium cations causing both strong electrostatic repulsion and steric hindrance that tremendously hampered the polymer chain assembly and expanded the size of the particles. The much smaller particle size of Ph60St30 synthesized in both methanol and 1,4-dioxane compared to that of Bu60St30 and Ph60 macro-CTA suggested that π–π stacking also occurred between phenyl groups on the cations and the PSt block, leading to a stronger intramolecular interaction that contracted the particle size of the nano-assemblies a lot more. Crosslinking polymerization with EGDMA increased the particle size of the Ph60-type PFDC but decreased it in the case of the Bu60-type PFDC, which might be due to the synergy of polymer chain elongation and intramolecular interactions (or repulsions) between PEGDMA and quaternary phosphonium cations.
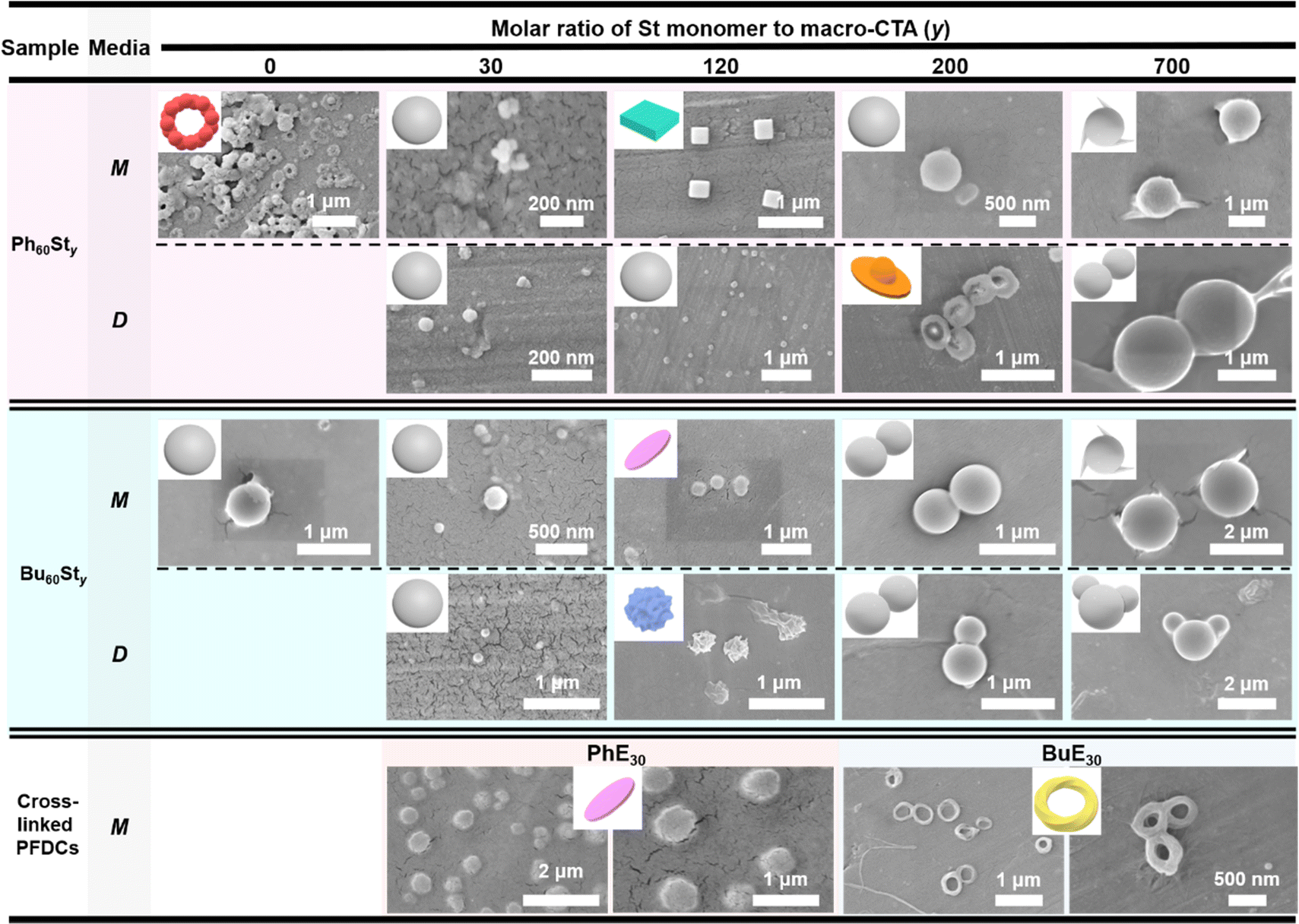 | ||
| Fig. 4 SEM images of PFDCs. Insets show the principal 3D schematic model for the nano-assemblies. M and D represent the methanol and 1,4-dioxane synthesis media, respectively. | ||
The major morphology of the nano-assemblies was a dispersed sphere, while in some cases, a special morphology in the shape of a ball-bearing ring, cuboid, disk-bowl (Chinese shoe-shape gold ingot), circle sheet, wrinkled sphere, or twisted strip loop (Mobius loop) was observed (Fig. 4). Such special morphologies appeared randomly regardless of either St content or crosslinking, while their size was mostly located in a narrow range from 300 to 450 nm, suggesting a size-related effect on the morphology. However, the changes in the special morphology did not follow a specific trend, making it difficult to identify the formation mechanism. One thing that could be certain is that the stacking and packing of macromolecules began with a positively charged nucleus and expanded from point to planar, and finally to a three-dimensional structure. Excess polymer segments are attached to the surface of the spherical particles and assembled as either cones or smaller spheres. A proper explanation for the special morphology is that the intramolecular interactions between phenyl groups and electrostatic repulsion among quaternary phosphonium cations induced certain polymer chain arrangements then various special morphologies,32–34 and even different contact angles as described above.
To obtain the particle information of the PFDC nano-assemblies, DLS was conducted to measure the hydrodynamic diameter (Dh), polydispersity index (PDI), and ξ-potential, as summarized in Table 1 and Table S2 (ESI†). Fig. 5 shows the tendency of Dh and PDI with the molar ratio of St monomer. The Dh value measured by DLS strongly agreed with the SEM results. A proximate linear increase of Dh with the molar ratio of St in both Bu60Sty and Ph60Sty PFDCs manifested the tunability of particle size through controlling DP (Fig. 5(a) and (c)). While in the case of the PEGDMA crosslinked PFDCs, the Dh value seemed to decrease in a logarithmic manner (Fig. S7a, ESI†), probably due to a different polymer chain rearrangement process during polymerization. The ξ-potential of all PFDCs was higher than +20 mV, indicating that they were positively charged particles. The ξ-potential decreased linearly with increasing molar ratio of St in the range from 0 to 200 and tended to be flat after that for the Bu60Sty and Ph60Sty PFDCs (Fig. 5(b)), suggesting lower dispersion stability. The ξ-potential slightly increased in the case of the PEGDMA crosslinked PFDCs (Fig. S7b, ESI†), which was attributed to a morphology stability increase through crosslinking. A higher ξ-potential of Ph60-type PFDCs than that of Bu60-type PFDCs suggested a stronger stabilization effect induced by the interactions between phenyl groups of quaternary phosphonium ions and the second block along the polymer chain, which was also consistent with the smaller particle size shown in Fig. 4. A sudden change in Dh and ξ-potential from macro-CTA to PFDCs was also observed from DLS results, demonstrating that copolymerization with St or EGDMA was able to stabilize the dispersion of PFDC nano-assemblies.
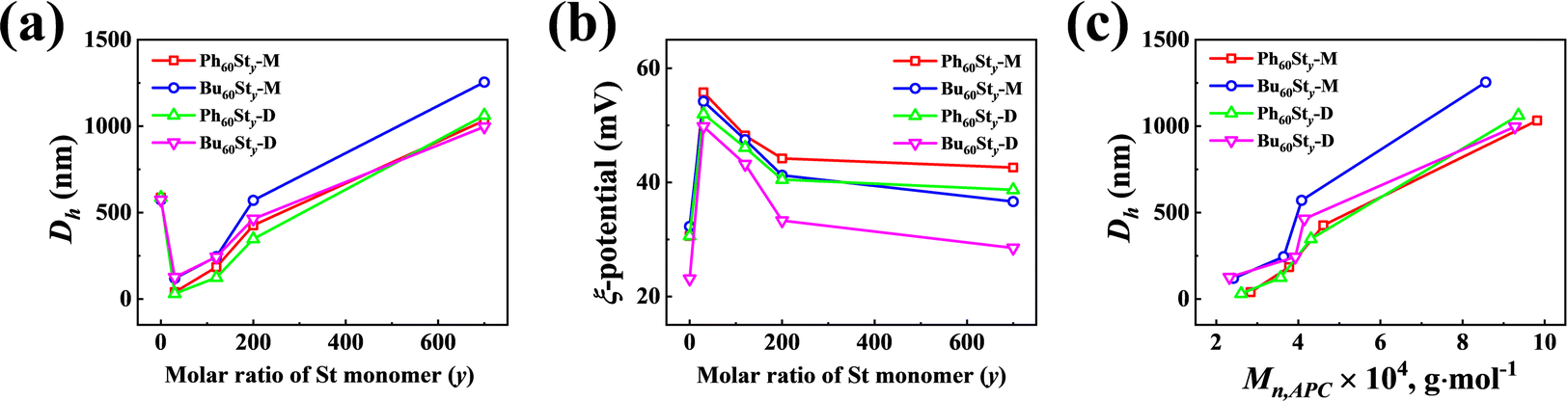 | ||
| Fig. 5 St content dependence of particle size (Dh) (a) and ξ-potential (b) for PFDCs measured by DLS. (c) Mn,APC dependence of particle size (Dh) for the PFDCs. | ||
The nanoparticle size and dispersion condition of the PFDCs could be directly visualized using the Tyndall effect in either methanol or 1,4-dioxane, as shown in Fig. S8 and S9 (ESI†). Homogenous suspensions of PFDCs with a distinct single light going through appeared as a white turbid emulsion with the increase of copolymerized St monomer (Fig. S8, ESI†) and crosslinking (Fig. S9, ESI†), also suggesting an increase of particle size but lower dispersion stability of the PFDCs. Selection of the type of suspension regarding the polymer molecular weight and particle size is essential in practical production. That means that there is a strong dependence of the molecular weight on either particle size or suspension condition. This could lead to the versatile production of phosphonium-functionalized diblock copolymers, and widen their application.
3.3 Antibacterial activities of PFDCs
The introduction of quaternary phosphonium cations brought about a high antibacterial activity for the PFDCs against E. coli and S. aureus, which was evaluated by using colony formation assay, as shown in Fig. 6 and Fig. S10 (ESI†). Semi-qualitative measurement by colony counting was used to characterize the antibacterial activities and confirm the effect of the molar ratio of macro-CTA and St monomer. The PFDCs showed significant antibacterial activities because of the quaternary phosphonium cations, with antibacterial rates beyond 95.7% and even 99.9% in most of the cases against E. coli. It is well known that lysis caused by cations originates from the electrostatic interactions between them and the negatively charged cell envelope of bacteria, which leads to a breakdown of cell envelope structure and leakage of organoids and ultimately death.35–37 In addition, with the increased ratio of St monomer, the A.R. value decreased significantly in both cases of E. coli and S. aureus, indicating a strong content dependence of antibacterial activity for the quaternary phosphonium cations (Fig. 6(b) and (c)). Notably, polymerization in either methanol or 1,4-dioxane did not alter the antibacterial activity of the PFDCs (Fig. 6(a)), making them highly adaptable in different production processes and applications. Interestingly, the PFDCs with different macro-CTAs (Bu60 or Ph60) showed similar antibacterial activity against the same bacteria, suggesting that the type of site groups on the phosphorus atom had no effect on the antibacterial activity. Some researchers have reported that the length of the site group can affect the antibacterial activity,38 but in our case, the geometrical length of the n-butyl and phenyl groups under ideal conditions were similar and had a very slight effect on antibacterial activity if this theory was applicable (Fig. 6(e)). Steric hindrance of the side groups might also affect the antibacterial activity,39 but this is not discussed here. The PEGDMA crosslinked PFDCs all showed excellent antibacterial activity against E. coli and S. aureus, as shown in Fig. S10 (ESI†), suggesting that the antibacterial capacity was endowed by the introduction of quaternary phosphonium cations instead of the nature of crosslinking.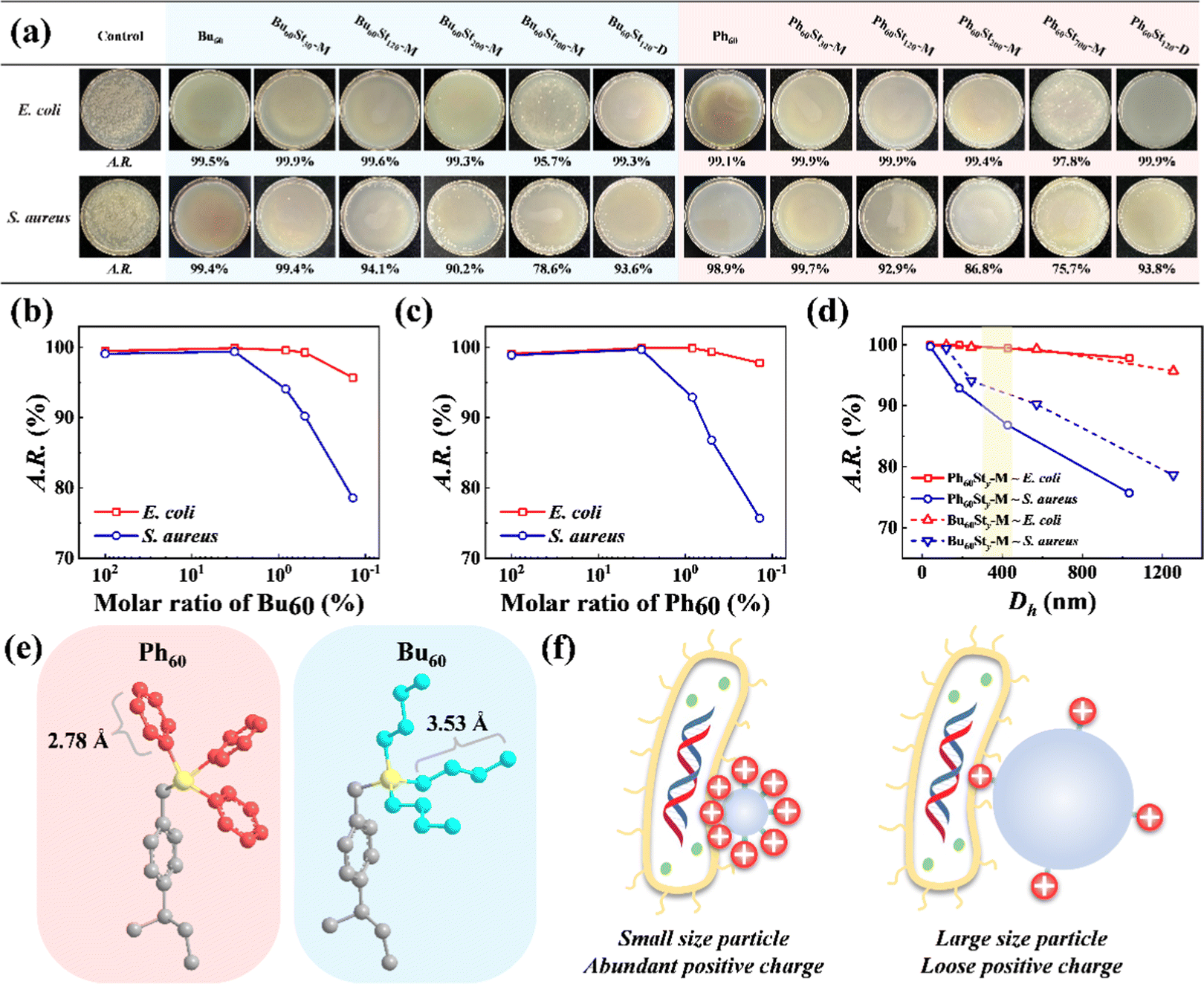 | ||
| Fig. 6 (a) Photographs of the culture plates of E. coli and S. aureus after exposure to different PFDCs at an initial dose of 40 μg mL−1. The A.R. value is the antibacterial rate according to the colony counting measurements. (b) and (c) The molar ratio of macro-CTA dependence of A.R. value for Bu60-type (b) and Ph60-type (c) PFDCs. (d) Particle size (Dh) dependence of A.R. value for Bu60Sty and Ph60Sty PFDCs. The light-yellow region is in the Dh range and highlights the special micromorphologies. (e) Calculation of the length of n-butyl and phenyl groups using a minimal energy model (calculated by ChemBio 3D Ultra). Red, blue, and yellow colors represent the phenyl group, n-butyl group, and phosphorus atom, respectively. (f) Schematic diagram of the particles with different sizes and positive charge density in contact with E. coli. The positive charge was supplied by the quaternary phosphonium cation, which had a trident shape according to the schematic diagram (e). | ||
As well as the content of macro-CTA, the particle size of the PFDCs showed a strong effect on antibacterial activity, as shown in Fig. 6(d). Generally, a bigger particle size led to a smaller A.R. value (i.e., antibacterial activity), and this tendency was more striking against S. aureus as the A.R. value decreased a lot more with the increase of Dh. A feasible explanation for this is that the total number of positive charges was constant on the surface of the PFDCs, while a large particle size led to a decrease in the positive charge density, leading to fewer cations in contact with the bacteria. This also explains why the A.R. value of the macro-CTA was a bit lower than that of either Ph60St30 or Bu60St30. On the other hand, E. coli has a long cylinder shape with a larger contact area and was able to contact more spherical PFDC particles compared to S. aureus, which had a circular shape. The two factors described here both affected the antibacterial activity, as illustrated in Fig. 6(f). Fig. 6(d) highlights the Dh region that produced the special morphologies observed using SEM, and it seems that the special morphologies had no specific antibacterial activity against E. coli, while in the case of S. aureus, the ball-bearing ring shaped PFDC had a better antibacterial activity, which might be due to its higher topological adaptability and contact area to contact with the spherical S. aureus, as shown in Fig. S11 (ESI†).
The results of the colony formation assay showed that the Ph60St30-M sample had the highest antibacterial activity against E. coli and S. aureus, whereby the A.R. values reached 99.9% and 99.7%, respectively. Therefore, the Ph60St30-M sample was further applied to evaluate the minimal inhibition concentration (MIC), as shown in Fig. 7. By measuring the optical density (OD) at 600 nm, the concentration dependence was examined to quantify the antibacterial activity against E. coli and S. aureus (107 CFU mL−1) for the Ph60St30-M sample. At a dose of 40.00 (μg·mL−1), E. coli growth was completely inhibited while in the case of S. aureus, a higher dose of 60 (μg mL−1) was required for full inhibition (Fig. 7), which was consistent with the colony formation assay results. In other words, the MIC for E. coli and S. aureus was 40 (1.4 μmol L−1) μg mL−1 and 60 μg mL−1 (2.2 μmol L−1), respectively, comparable to some commercial antibiotics and better than the reported polymeric antibiotics with a single active site (Table 2). Therefore, the synthesized copolymers showed excellent antibacterial activity.
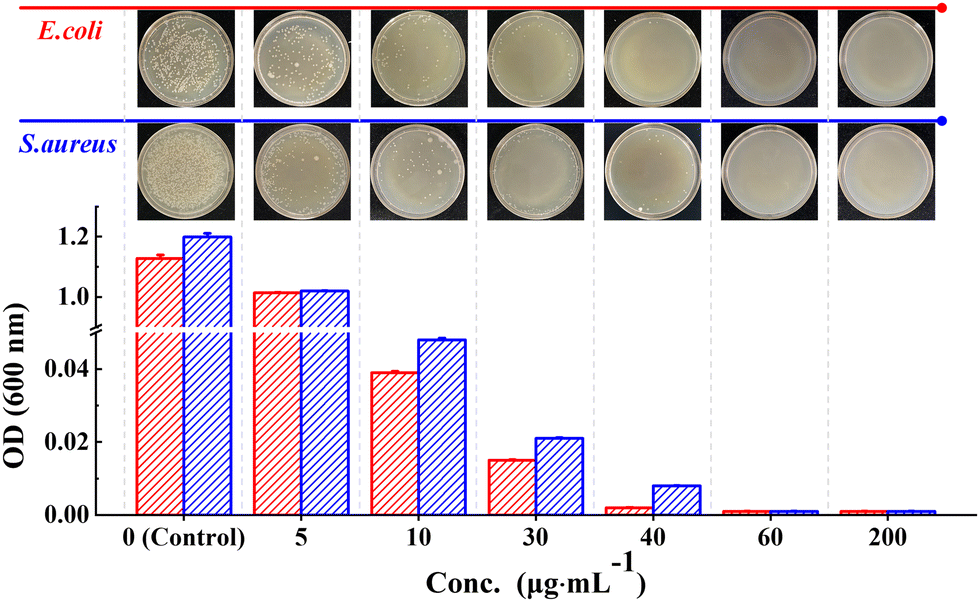 | ||
| Fig. 7 Concentration dependence of the OD value for the P60St30-M sample. The images show the visible inhibition effect at corresponding concentrations against E. coli and S. aureus. | ||
| Sample | MIC | Ref. | |
|---|---|---|---|
| E. Coli | S. aureus | ||
| Ampicillin | 2.4 μmol L−1 | 2 μg mL−1 | 40 and 41 |
| Tetracycline | 4.7 μmol L−1 | 64 μg mL−1 | 40 and 41 |
| Chitosan oligomer | 80 μg mL−1 | 80 μg mL−1 | 42 |
| Polyhexamethylene guanidine hydrochloride-based disinfectant | 50 μg mL−1 | 400 μg mL−1 | 43 |
| Cationic random methacrylamide polymers | 2.7 μmol L−1 | 3.8 μmol L−1 | 44 |
| N-Alkyl imidazolium-based poly(ionic liquids) | 1.2 μmol L−1 | 2.5 μmol L−1 | 38 |
| Imidazolium-type poly(ionic liquids) | 9 μmol L−1 | 12 μmol L−1 | 45 |
| Quaternary phosphonium salt carbon sphere | 150 μg mL−1 | 100 μg mL−1 | 46 |
| Quaternary ammonium salt-based crosslinked micelles | 298.4 μmol L−1 | 69.6 μmol L−1 | 47 |
| Phosphonium-based polymeric biocides | 2.7 μmol L−1 | 2.7 μmol L−1 | 13 |
| PFDCs | 1.4 μmol L−1 | 2.2 μmol L−1 | This work |
The time-related antibacterial effectiveness of the Ph60St30-M sample was also evaluated against E. coli to measure the long-acting antibacterial capacity, as shown in Fig. 8(a). A 107 CFU mL−1E. coli suspension mixed with 40.00 μg mL−1 Ph60St30-M and sufficient LB broth was incubated for 10 days to observe the growth condition of the bacteria. Unsurprisingly, only one small colony of bacteria grew within the whole experiment period compared to the control group, suggesting a long-acting antibacterial capacity of the PFDCs. SEM was also used to examine the cell membrane integrity of E. coli after Ph60St30-M treatment, as shown in Fig. 8(b) and (c). The E. coli maintained its rod-like shape in the absence of Ph60St30-M, while the outer membrane was damaged and cells exhibited a hollow morphology after being treated, indicating leakage of organoids that was responsible for the death of the bacteria.
 | ||
| Fig. 8 (a) Photographs of the culture plates of E. coli after exposure to Ph60St30-M for different numbers of days (107 CFU mL−1, 37 °C). (b) and (c) SEM images of E. coli before (b) and after (c) being treated with Ph60St30-M. (d) Photographs of the culture plates of E. coli after exposure to the modified ink, dye, and paint, respectively. | ||
Antibacterial materials in various forms and composites are crucial and frequently used in our daily lives. As demonstrated above, PFDCs in suspension forms like inks, dyes, or paints are one of the most common applications in practical usage. As illustrated in Fig. 8(d), Ph60St30-M samples mixed with blue ink or Rhodamine B (dye), or coated with commercial white latex paint were incubated with E. coli (107 CFU mL−1). Without the presence of the PFDC, E. coli grew very well with either the ink, dye, or paint. After the introduction of 40.00 μg mL−1 Ph60St30-M, an A.R. value of 99.9% for all mixing plates suggested that almost no bacteria grew in the plate compared to the control group, suggesting that the antibacterial activity was not affected after mixing with an ink, dye, or paint. The results suggested that the synthesized PFDCs had great potential as highly efficient antibacterial materials in many practical applications. They could also exhibit adaptability in different solvents by altering the ratio of hydrophilic and hydrophobic blocks in the copolymers.
4. Conclusion
Here, we demonstrated a RAFT polymerization method for the highly efficient synthesis of phosphonium-functionalized diblock copolymers with tunable particle size and excellent antibacterial activity. The conversion rate for the RAFT polymerization of PFDCs was beyond 95% synthesized in either methanol (strong polarity) or 1,4-dioxane (weak polarity) solvent, showing favorable producibility. In addition, polymerization-induced self-assembly (PISA) of PFDCs produced nano-assemblies of PFDCs whose particle size could be controlled by altering the initial dosing of St or EGDMA monomer. The quaternary phosphonium cations endowed the PFDCs with excellent antibacterial activity with a MIC of 40 and 60 μg mL−1 (1.4 and 2.2 μmol L−1) against E. coli and S. aureus, respectively, and the antibacterial mechanism was proposed regarding the size and charge density aspects. Interestingly, some special morphologies were generated during the synthesis of the PFDCs within a specific diameter range, which might have surprising antibacterial activity for specialized applications. Tuning the content of the monomer made it possible to modify the hydrophilicity and hydrophobicity of the PFDCs, which broadened their application. Combining the high practicability and functionality of PFDCs provides great prospects for their application as highly efficient and broad-spectrum antibiotics.Author contributions
Peng Cao: methodology, investigation, data curation, writing – original draft, formal analysis. Xue Bai: investigation, validation. Yufeng He: resources. Pengfei Song: resources. Rongmin Wang: resources, supervision, project administration, funding acquisition, conceptualization. Junchao Huang: writing – review & editing, data curation, visualization, formal analysis.Conflicts of interest
The authors declared no conflict of interest.Acknowledgements
This research was funded by the National Natural Science Foundation of China (21865030).References
- F. D. Lowy, Nat. Med., 2007, 13, 1418–1420 CrossRef CAS.
- N. Nikfarjam, M. Ghomi, T. Agarwal, M. Hassanpour, E. Sharifi, D. Khorsandi, M. Ali Khan, F. Rossi, A. Rossetti, E. Nazarzadeh Zare, N. Rabiee, D. Afshar, M. Vosough, T. Kumar Maiti, V. Mattoli, E. Lichtfouse, F. R. Tay and P. Makvandi, Adv. Funct. Mater., 2021, 31, 2104148 CrossRef CAS.
- J. N. Pendleton and B. F. Gilmore, Int. J. Antimicrob. Agents, 2015, 46, 131–139 CrossRef CAS.
- M. Wang, J. Shi, H. Mao, Z. Sun, S. Guo, J. Guo and F. Yan, Biomacromolecules, 2019, 20, 3161–3170 CrossRef CAS.
- M. D. T. Torres, S. Voskian, P. Brown, A. Liu, T. K. Lu, T. A. Hatton and C. de la Fuente-Nunez, ACS Nano, 2021, 15, 966–978 CrossRef CAS PubMed.
- B. Wang, T. Li, W. Guo, R. Wang, Y. Li, X. Zhu, P. Song and Y. He, Int. J. Biol. Macromol., 2021, 174, 198–206 CrossRef CAS.
- A. M. Carmona-Ribeiro and L. D. De Melo Carrasco, Int. J. Mol. Sci., 2013, 14, 9906–9946 CrossRef.
- M.-H. Xiong, Y. Bao, X.-Z. Yang, Y.-H. Zhu and J. Wang, Adv. Drug Delivery Rev., 2014, 78, 63–76 CrossRef CAS PubMed.
- H. Zhou, D. Tang, X. Kang, H. Yuan, Y. Yu, X. Xiong, N. Wu, F. Chen, X. Wang, H. Xiao and D. Zhou, Adv. Sci., 2022, 9, 2200732 CrossRef CAS PubMed.
- Y. Xiong, J. Liu, Y. Wang, H. Wang and R. Wang, Angew. Chem., Int. Ed., 2012, 51, 9114–9118 CrossRef CAS.
- V. Loczenski Rose, F. Mastrotto and G. Mantovani, Polym. Chem., 2017, 8, 353–360 RSC.
- W. Qian, J. Texter and F. Yan, Chem. Soc. Rev., 2017, 46, 1124–1159 RSC.
- T. J. Cuthbert, B. Hisey, T. D. Harrison, J. F. Trant, E. R. Gillies and P. J. Ragogna, Angew. Chem., Int. Ed., 2018, 57, 12707–12710 CrossRef CAS.
- B. Zhang, X. Yan, P. Alcouffe, A. Charlot, E. Fleury and J. Bernard, ACS Macro Lett., 2015, 4, 1008–1011 CrossRef CAS PubMed.
- Q. Zhang, R. Zeng, Y. Zhang, Y. Chen, L. Zhang and J. Tan, Macromolecules, 2020, 53, 8982–8991 CrossRef CAS.
- J. Cornel Erik, J. Jiang, S. Chen and J. Du, CCS Chem., 2020, 3, 2104–2125 CrossRef.
- Y. Xiong, H. Wang, R. Wang, Y. Yan, B. Zheng and Y. Wang, Chem. Commun., 2010, 46, 3399–3401 RSC.
- Y. Xiong, Y. Wang, H. Wang and R. Wang, Polym. Chem., 2011, 2, 2306–2315 RSC.
- S. Chen, S. Chen, S. Jiang, M. Xiong, J. Luo, J. Tang and Z. Ge, ACS Appl. Mater. Interfaces, 2011, 3, 1154–1162 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. P. Armes, Langmuir, 2013, 29, 7416–7424 CrossRef CAS.
- C. Gonzato, M. Semsarilar, E. R. Jones, F. Li, G. J. P. Krooshof, P. Wyman, O. O. Mykhaylyk, R. Tuinier and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 11100–11106 CrossRef CAS PubMed.
- M. Chen, J.-W. Li, W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, Macromolecules, 2019, 52, 1140–1149 CrossRef.
- A. J. D. Magenau, N. Martinez-Castro, D. A. Savin and R. F. Storey, Macromolecules, 2009, 42, 8044–8051 CrossRef CAS.
- B. Zeytuncu, E. Çakmakçı and M. V. Kahraman, Polym. Compos., 2015, 36, 946–954 CrossRef CAS.
- R. Bai, J. Kang, O. Simalou, W. Liu, H. Ren, T. Gao, Y. Gao, W. Chen, A. Dong and R. Jia, ACS Biomater. Sci. Eng., 2018, 4, 2193–2202 CrossRef CAS.
- C.-M. Liao, C.-C. Hsu, F.-S. Wang, B. B. Wayland and C.-H. Peng, Polym. Chem., 2013, 4, 3098–3104 RSC.
- Y. Li, J. Q. Pham, K. P. Johnston and P. F. Green, Langmuir, 2007, 23, 9785–9793 CrossRef CAS PubMed.
- N. Zhang, S. Zhong, T. Chen, Y. Zhou and W. Jiang, RSC Adv., 2017, 7, 22946–22953 RSC.
- P. Escalé, W. Van Camp, F. Du Prez, L. Rubatat, L. Billon and M. Save, Polym. Chem., 2013, 4, 4710–4717 Search PubMed.
- S. Hosseini, H. Savaloni and M. Gholipour Shahraki, Appl. Surf. Sci., 2019, 485, 536–546 CrossRef CAS.
- P. Xu, L. Gao, C. Cai, J. Lin, L. Wang and X. Tian, Angew. Chem., Int. Ed., 2020, 59, 14281–14285 CrossRef CAS PubMed.
- H. Zhu, X. Wang, Y. Cui, J. Cai, F. Tian, J. Wang and H. Qiu, Macromolecules, 2019, 52, 3479–3485 CrossRef CAS.
- H. Qiu, A. M. Oliver, J. Gwyther, J. Cai, R. L. Harniman, D. W. Hayward and I. Manners, Macromolecules, 2019, 52, 113–120 CrossRef CAS.
- A. Bhatnagar, W. Hogland, M. Marques and M. Sillanpää, Chem. Eng. J., 2013, 219, 499–511 CrossRef CAS.
- B. Wang, F. Wang, Y. Kong, Z. Wu, R.-M. Wang, P. Song and Y. He, Colloids Surf., A, 2018, 549, 122–129 CrossRef CAS.
- T. Zhang, J. Guo, Y. Ding, H. Mao and F. Yan, Sci. China: Chem., 2019, 62, 95–104 CrossRef CAS.
- C. Fang, L. Kong, Q. Ge, W. Zhang, X. Zhou, L. Zhang and X. Wang, Polym. Chem., 2019, 10, 209–218 RSC.
- T. R. Stratton, B. M. Applegate and J. P. Youngblood, Biomacromolecules, 2011, 12, 50–56 CrossRef CAS.
- C. L. Martini, C. C. Lange, M. A. V. P. Brito, J. B. Ribeiro, L. C. Mendonça and E. K. Vaz, J. Dairy Res., 2017, 84, 202–205 CrossRef CAS PubMed.
- V. Hoerr, G. E. Duggan, L. Zbytnuik, K. K. H. Poon, C. Große, U. Neugebauer, K. Methling, B. Löffler and H. J. Vogel, BMC Microbiol., 2016, 16, 82 CrossRef.
- A. Shanmugam, K. Kathiresan and L. Nayak, Biotechnol. Rep., 2016, 9, 25–30 CrossRef PubMed.
- M. K. Oulé, R. Azinwi, A.-M. Bernier, T. Kablan, A.-M. Maupertuis, S. Mauler, R. K. Nevry, K. Dembélé, L. Forbes and L. Diop, J. Med. Microbiol., 2008, 57, 1523–1528 CrossRef.
- A. Tyagi and A. Mishra, ACS Omega, 2021, 6, 34724–34735 CrossRef CAS PubMed.
- Z. Zheng, Q. Xu, J. Guo, J. Qin, H. Mao, B. Wang and F. Yan, ACS Appl. Mater. Interfaces, 2016, 8, 12684–12692 CrossRef CAS.
- Y. Yang, Q. Shi, J. Feng, X. Shu and J. Feng, RSC Adv., 2014, 4, 50708–50712 RSC.
- F. Liu, D. He, Y. Yu, L. Cheng and S. Zhang, Bioconjugate Chem., 2019, 30, 541–546 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tb01778d |
| This journal is © The Royal Society of Chemistry 2022 |