
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic strategies to enhance the long-term stability of polymer brush coatings
Zhichao
Ding†
a,
Changyou
Chen†
a,
Yunlong
Yu
 *a and
Sissi
de Beer
*b
*a and
Sissi
de Beer
*b
aInstitute of Burn Research, Southwest Hospital & State Key Lab of Trauma, Burn and Combined Injury, Third Military Medical University (Army Medical University), Chongqing, 400038, China. E-mail: yuyunlong@scu.edu.cn
bSustainable Polymer Chemistry, Department of Molecules & Materials, MESA+ Institute for Nanotechnology, University of Twente, P.O. Box 217, 7500 AE Enschede, The Netherlands. E-mail: s.j.a.debeer@utwente.nl
First published on 10th March 2022
Abstract
High-density, end-anchored macromolecules that form so-called polymer brushes are popular components of bio-inspired surface coatings. In a bio-mimetic approach, they have been utilized to reduce friction, repel contamination and control wetting, in particular in the development of biomedical materials. For reliable application of these coatings, it is critical that the performance of these coatings does not degrade in time. Yet, it is well-known that polymer brushes can deteriorate and degraft when exposed to water(–vapor) and this strongly limits the durability of these coatings. In this article, we provide an overview of the current status of research on the stability of polymer brushes. Moreover, we review different synthetic strategies, some of which are bio-inspired by itself, to enhance the long-term stability of these brushes. Based on this overview, we identify open question and issues to be resolved for brushes to be applied as durable bio-inspired surface coatings.
 Zhichao Ding | Zhichao Ding obtained his bachelor degree in polymer science and engineering from the College of Polymer Materials and Engineering at Sichuan University in 2020. In his final year project, he focuses on the preparation of stable polymer brushes for biomedical applications. Now he is a PhD candidate at National University of Singapore. |
 Changyou Chen | Changyou Chen obtained his bachelor degree in Pharmaceutical Engineering from Xihua University in 2015 and a Master degree in Medicinal Chemistry from Southwest Medical University in 2018. He is currently working as an assistant researcher in Prof. Yunlong Yu's group. His research interests include biomaterial design and biomedical applications. |
 Yunlong Yu | Prof. Yunlong Yu obtained his master degree at the Sichuan University and his PhD at the University of Twente. Now he is working at Third Military Medical University (Army Medical University) Southwest Hospital. His research areas include design and fabrication of biomaterials, antibacterial research, hemostasis and wound healing. |
 Sissi de Beer | Dr Sissi de Beer obtained her PhD degree at the University of Twente. After postdoctoral fellowships at the Forschungszentrum in Jülich and the University of Toronto, she is now an associate professor at the University of Twente. In her research, Sissi combines molecular modeling with laboratory experiments to design functional polymer surfaces for application in lubrication, sensing and molecular separations. |
1. Introduction
Polymer brushes consist of long macromolecules that are end-tethered to surfaces at sufficiently high densities such that the polymers stretch out, away from the substrate.1,2 The advantage of end-anchoring of the polymers is that the coating can be utilized in good solvents, without being dissolved in the solvent. In the last decades, these brushes have been utilized in a multitude of bioinspired materials. For example, the strong swelling of brushes in good solvents can be utilized to mimic key elements of biological lubricants.3–5 This has resulted in the development of highly effective synthetic lubricants.6–8 Moreover, this strong brush swelling can be employed in the design of anti-fouling surfaces as well.9–13 Other biomimetic materials, where polymer brushes have been incorporated, have been designed to display environmentally responsive14–16 and structural17–19 colours, as found in e.g. chameleons.20 Next to these bioinspired approaches, brushes can also be applied in smart adhesives,21,22 energy devices,23,24 bactericidal25–27 and blood compatible28,29 coatings, membranes,30,31 nano-theranostics,32 sensing33–35 and many more applications.36–39End-anchored polymers can adopt different conformations. When the distance between the surface bonds is larger than two times the radius of gyration, the polymers do not interact and form mushrooms in good solvents. When these low-density polymers are in a poor solvent and/or strongly interacting with the substrate, pancakes instead of mushrooms are formed.40 Only when the grafting density is sufficiently high and the distances between anchors is much smaller than the radius of gyration,2 polymer brushes are formed. At these grafting densities, the chains interact and stretch away from the substrate.41 Besides the grafting density, the degree of stretching is also determined by the solvent quality. In poor solvents, the polymers form a dense film on the substrate, while in good solvents, the polymers absorb the solvent and stretch more strongly away from the substrate.42,43 When polymers are in the brush regime, their change in swelling in response to adjustments in the solvent environments acts in the direction perpendicular to the grafting substrate alone. This enhances their responsivity and can be utilized in the development of switchable or adaptive coatings.44,45 Those high-density stretched surface grafts are the focus material of this review.
For the preparation of polymer grafted surfaces, three strategies can be followed46 (see Fig. 1). Polymers can be physisorbed to substrates e.g., via diblock copolymers, where the shorter block has an affinity for the substrate (see Fig. 1a). This method is straightforward to apply. However, it is difficult to obtain sufficiently high grafting densities due to steric hindrance. Therefore, often mushrooms, or pancakes are formed instead of brushes. Moreover, the surface bonds are relatively weak such that the polymers can easily detach by thermal of solvation effects.47 Polymers that are covalently bonded to the substrate will be more stable. This can be achieved by “grafting to”,48 as depicted in Fig. 1b. In this method, reactive end-groups on the polymers can bond via complementary reactive groups on the substrate. Even though these coatings are more stable, it remains challenging to obtain grafting densities that are high enough to reach the brush regime.
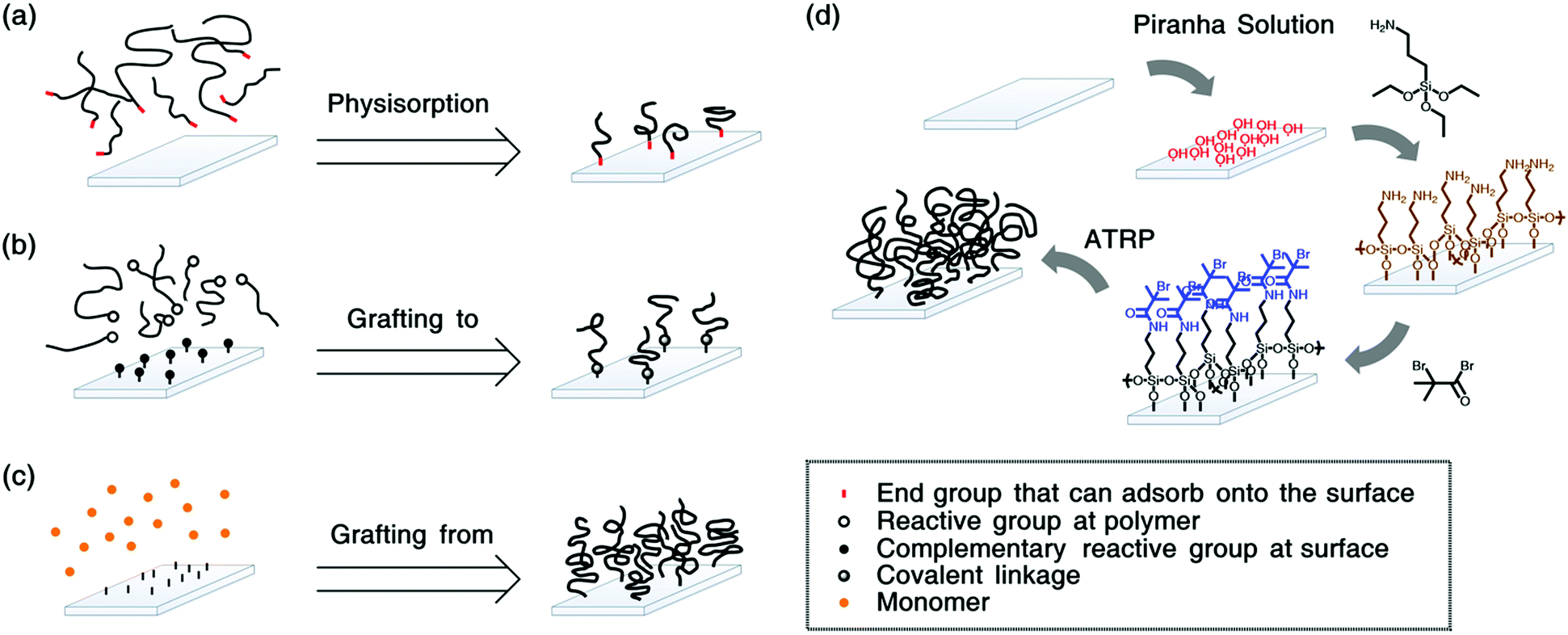 | ||
| Fig. 1 Synthesis techniques for polymer brushes (a) physisorption of polymers by surface attractive groups or blocks, (b) grafting of polymers with reactive groups to substrates with complementary reactive groups, (c) grafting of polymers from the substrate. In (d) a typical “grafting from” synthesis scheme is shown, where first silicon wafers are activated via immersion in piranha solution. Next, they are functionalised with aminoalkylsilanes to which the initiators (here 2-bromo-2-methylpropionyl bromide (BIBB)) are coupled. Finally, the brushes are polymerized from the initiators, e.g. via atom transfer radical polymerization (ATRP). These brushes are typically not stable. | ||
To obtain polymer brushes with high grafting densities, the “grafting from” synthesis-method is typically employed.49,50 (see Fig. 1c) In this method, the macromolecules are polymerized from initiators on the substrate. For these surface-initiated (SI) approaches, controlled radical polymerization techniques, such as atom transfer radical polymerization (ATRP),51–53 nitroxide mediated polymerization (NMP)54 or reversible addition–fragmentation chain transfer (RAFT),55–57 are utilized to ensure a narrow dispersity of the polymers. Fig. 1d shows a typical synthesis procedure to grow brushes via SI-ATRP.
Polymer brushes are typically grown from well-defined surfaces such as silicon58–60 or gold.61–63 For silicon wafers, initiator coupling is often achieved by silane chemistry,64,65 while gold surfaces are frequently decorated with initiators using disulphides or thiols.66–68 We note that the surface initiated synthesis is not limited to these substrates and it has been shown that brushes can be grown from more complex surfaces, such as cellulose nanocrystals69 or porous membranes,70 carbon nanotubes,71 mica,72,73 silk fibrion,74 or cotton fibres.75
Most applications of polymer brushes rely on the coatings being stable for long times. However, it was realized early on that the brushes can degrade or degraft in time.76,77 In Fig. 2, two examples of degrafted brushes are given. There can be different reasons for brush deterioration. For example, the polymers themselves can oxidize and degrade. This has been observed for e.g. poly(ethylene glycol) polymers,78 poly(lactic acid) brushes79 and poly(phosphoester)s80 and it can be prevented by choosing more stable monomers/polymers81 in the preparation of the brushes.82 Yet, in most dense brush systems, the polymers are reported to degraft at their substrate anchors, where, depending on the type of anchor, bonds can be weaker83 and tension is the highest.84 This degrafting can be intentional85–87 (e.g. to characterize the molecular weight of the polymers) or unintentional. In the following, we focus on the latter.
 | ||
| Fig. 2 (a) Scanning electron microscopy pictures displaying the detachment of poly(poly(ethylene glycol) methacrylate) (PPEGMA) brushes grafted from SiOx substrates using silane chemistry, after 7 days of immersion in a cell culture medium at 37 °C (reprinted with permission from ref. 76. Copyright 2008 American Chemical Society), (b) photos displaying the detachment of poly(sulfobetaine methacrylamide) (SBMAm) brushes grafted from silicon wafers via silane linkers (left), after immersion for 3 months in seawater and the prevention of this detachment by utilisation of diblock copolymer brushes (see also Section 3.2) (reprinted with permission from ref. 141. Copyright 2013 American Chemical Society). | ||
In this review, we provide an overview of synthetic strategies that are aimed at anchoring polymer brushes more strongly to the substrate. Though strategies for stable brushes have been developed for “grafting to” as well,88,89 we will focus here on brushes prepared by “grafting from”, because they lead typically to high grafting density brushes. We start by discussing the proposed mechanisms for degrafting of polymer brushes and describe the different bonds that can break depending on the substrate and the type of substrate anchor. Next, we will provide an overview of the different synthetic strategies that have been developed to reduce the probability of degrafting such that the brushes will be stable for longer times. We finalize this review with an outlook and open questions and challenges in the field.
2. Proposed mechanisms for degrafting
When polymers are end-tethered to a substrate, they stretch perpendicular to the surface to form a brush. This stretching results in an enhanced tension, which is highest at the anchor-points to the substrate. The tension is enhanced when polymers stretch more. Therefore, it increases for higher grafting densities or when the brushes are swollen in good solvents. Taking these effects into account, the tension at the anchors can be estimated to be around 1–10 pN.84 These forces are too low to break covalent bonds. To break C–C bonds, forces on the order of nN are needed, which can only be achieved by dendrimer structures84 or bottlebrushes on substrates.90 However, the lower tensions in brushes can still facilitate degrafting, since they can “mechanochemically” reduce activation energies,91 in particular at the weaker or hydrolysis sensitive surface bonds.83Experimental observations are consistent with the hypothesis that polymer brushes detach by tension-enhanced breakage of the surface bonds. Wang and Klok have shown in a recent publication92 that the degrafting rate for brushes increases upon increasing the solvent quality and thus swelling of these brushes in different (water-miscible) organic solvents, which shows that a higher tension results in faster degrafting, in agreement with earlier observations.93 Another confirmation that tension enhances the degrafting has been provided by Chen et al.94 These authors observed that the detachment rate of polymers in nanopatterned brushes decreased for decreasing pattern sizes. The reason for this is that in smaller patches the polymers are stretched less, because they can relax in the lateral directions.94 All these results are also consistent with the general observation that the degrafting rate increases with increasing grafting density and, thus, tension at the anchors.95–97 Moreover, it is often observed that degrafting proceeds until a finite film thickness is reached,98,99 which indicates that the mechanochemical effect reduces when the tension reduces, until it is too low to facilitate degrafting over the timescale of the experiment.
To ensure that polymers indeed detach and not degrade via side chain or backbone breakage, Du et al.100 used X-ray photoelectron spectroscopy (XPS) to show that the composition of the material on the substrate does not change before and after degrafting. Moreover, they checked with proton nuclear magnetic resonance (H-NMR) and gel permeation chromatography (GPC) that the polymers in bulk do not change in chemical composition or molecular weight.100 This has been confirmed by Wang and Klok,92 who analyzed the molecular weight of polymers that are intentionally cleaved off at the start of the experiment and after partial degrafting. They observed a small decrease in the molecular weight (∼6%), which indicates that longer polymers detach first,95 but that they stay intact. In addition, Wang and Klok showed in the same article that their hydrophobic brushes did not degraft when being swollen in organic solvents alone. Their polymers only detach when sufficient water is present. These results are a strong indication that the brushes break at their anchors because the brushes were grafted from hydrolysis-sensitive anchors and water is needed for the hydrolysis reaction to occur (see also Section 2.1).
Degrafting of brushes is mainly observed in liquid media. Yet, it was recently shown that brushes101 or even initiator layers102 can degraft in air as well. When brushes are exposed to vapors that are good solvents, they swell and absorb the vapor.103–106 Vapor-solvated swelling ratios are smaller than the swelling ratios in liquid.60 Nevertheless, hydrophilic brushes such as poly(3-sulfopropyl methacrylate potassium) (PSPMA) and poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC) can absorb sufficient amounts of water vapor from the air to allow for the detachment of ∼10–50% of the polymers over 8 weeks.101
In the discussions above, it has become clear that the experimental evidence supports the hypothesis that brushes degraft by tension-enhanced breakage of surface bonds. What surface bonds are broken exactly, depends on the grafting substrate and the type of anchor that is used. Since brushes are grafted most often from silicon wafers, we will discuss potential mechanisms for debonding on those substrates first.
2.1 Hydrolysis of siloxane, amide or ester bonds
For brushes that are grafted from silicon wafers, the initiators are often coupled to the substrates via silanes.49 This can be achieved via a single-step or a two-step process. Fig. 1d gives an example of a two-step process. There, first, (3-aminopropyl) triethoxysilane (APTES) is coupled to hydroxyl groups on the substrate. Next, the initiator 2-bromo-2-methylpropionyl bromide (BIBB) is linked to the silanes, from where the brushes are polymerized.107,108 Brushes grown from these anchors are well known to degraft,101,109,110 even though they are more stable than monosilanes97,111,112 and can be utilized with minimal degrafting for over an hour.113 The higher stability of APTES and other trifunctional silanes can be attributed to their ability to potentially form three bonds with the substrate and/or neighbouring silanes.65,114 In addition, it has been observed that the silane stability depends on the pre-treatment of the SiO2 as well as temperature.115 Nevertheless, the anchors can hydrolyse via their siloxane or amide bonds. Thus, eventually, they will degraft.Polymer brushes grafted from silicon wafers have been proposed to detach via hydrolysis of three bonds:98 siloxane, amide and ester bonds. The hydrolysis of siloxanes has an activation energy of approximately 100 kJ mol−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 116–118 and is commonly observed.119 However, amide and ester bonds are not expected to be cleaved at neutral pH. Nevertheless, some polymers (such as poly(acrylic acid) (PAA)120) might change the local acidity or basicity in the brush, such that cleavage can occur. Moreover, it has been suggested that tension can lower the activation barrier for hydrolysis and thereby allow for amide and ester bond breakage at neutral pH.121 Indeed, Galvin et al. have shown that brushes grown from surface anchors that contain amide groups detach faster than brushes grafted from anchors that are similar, but have ester instead of amide groups.96 Moreover, Ataman and Klok observed that amide- or ester-bearing brush-anchors without siloxane bonds degraft at rates that are comparable to anchors with siloxane bonds.98 This indicates that siloxanes, amides and esters can all contribute to the degrafting process.
116–118 and is commonly observed.119 However, amide and ester bonds are not expected to be cleaved at neutral pH. Nevertheless, some polymers (such as poly(acrylic acid) (PAA)120) might change the local acidity or basicity in the brush, such that cleavage can occur. Moreover, it has been suggested that tension can lower the activation barrier for hydrolysis and thereby allow for amide and ester bond breakage at neutral pH.121 Indeed, Galvin et al. have shown that brushes grown from surface anchors that contain amide groups detach faster than brushes grafted from anchors that are similar, but have ester instead of amide groups.96 Moreover, Ataman and Klok observed that amide- or ester-bearing brush-anchors without siloxane bonds degraft at rates that are comparable to anchors with siloxane bonds.98 This indicates that siloxanes, amides and esters can all contribute to the degrafting process.
2.2 Cleavage of Au–S
Gold is another substrate material from which brushes are commonly grown, since it allows for surface plasmon resonance measurements122 or to exploit the electro-responsiveness of charged polyelectrolyte brushes.123 On such Au substrates, the initiators are often coupled via thiols or disulphides. However, the Au–S binding energy is only 120 kJ mol−1,124,125 which makes the bond thermally unstable for temperatures above 60 °C.126 This troubled early synthesis of brushes on gold, in particular for polymerization temperatures above room temperature.127 Yet, with ATRP at room temperature61 or UV photo-polymerizations,128 it is possible to grow brushes from gold. Nevertheless, even when the grafting of brushes from gold or other metals using thiol linkers is successful, the brush polymers can degraft when immersed in good solvents.129–131 For example, Zhang et al.129 observed the degrafting rate of carboxylated poly(oligo(ethylene glycol) methacrylate-random-2-hydroxyethylmethacrylate) (carboxylated poly(OEGMA-r-HEMA)) is mainly determined by the swelling ratio, which has been attributed to the increased tension at the surface bonds for more highly swollen brushes.In summary, brushes can degraft from different commonly employed substrates (e.g. SiOx, gold or other metals). Therefore, multiple strategies have been developed to prevent this degrafting. In the following section, we will provide an overview of these strategies and discuss the advantages and disadvantages of the proposed synthesis routes.
3. Synthetic strategies against degrafting
To counteract the degrafting of polymer brushes, many different synthesis routes have been developed. We have grouped these routes into seven strategies (see Fig. 3). Each of these strategies has different advantages and disadvantages, which we will discuss in more detail below, and it will depend on the application, substrate material and synthesis skills of the user, which strategy is the best solution.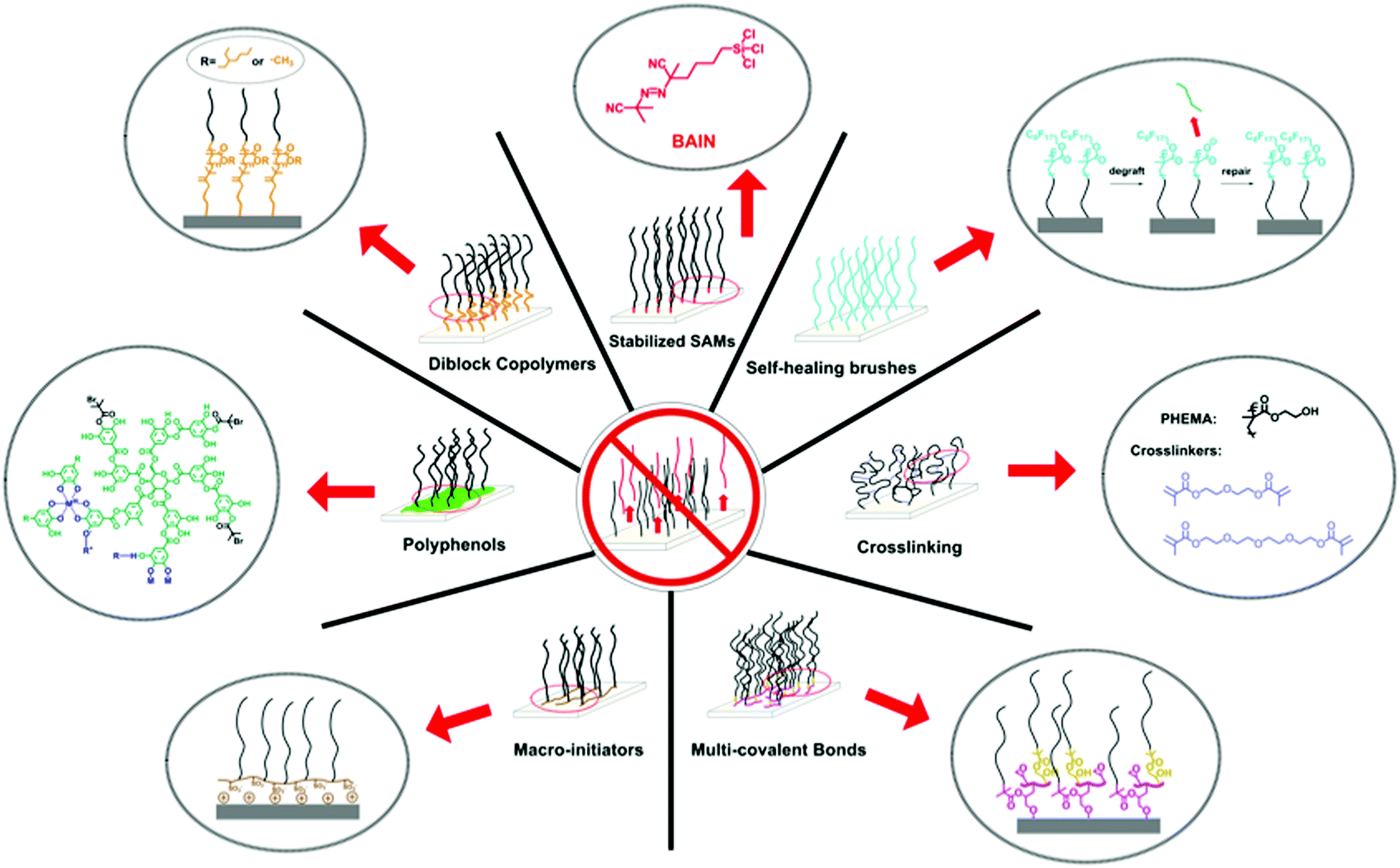 | ||
| Fig. 3 Overview of the different synthetic strategies that have been developed to prepare long-term stable polymer brushes and that are being reviewed in this article. | ||
3.1 Stabilized SAMs
Because the hydrolysis of siloxanes is one of the main mechanisms for degrafting, linker molecules have been designed that do not form Si–O–Si bonds. For example, Borozenko et al.132 showed that poly(acrylic acid) (PAA) brushes grafted from phosphonic acid-based initiators resist degrafting more than brushes grown from organosiloxanes. Using density functional theory (DFT), they show that the enhanced stability is a result of the surface bond strength for organophosphonic acid initiators in water being 120 kJ mol−1 higher than for organosiloxanes.132 Also Nguyen et al.133 designed a method to attach initiators without having to form hydrolyzable Si–O–C and Si–O–Si bonds. They utilized a three-step reaction to couple initiators that are anchored by stable Si–C bonds and showed that low-fouling hydrophilic zwitterionic brushes grafted from these initiators are stable for >1 week.Besides circumventing siloxane bonds, the long-term stability of brushes can be improved by preventing the usage of ester-bonds as well. Bain et al. synthesized ester-free initiator molecules called BAIN (named after the first author). They showed that quaternized poly(2-(dimethylamino)ethyl methacrylate) (qPDMAEMA) brushes degraft by less than 10% over 5 days (see Fig. 4a).134 For comparison (Fig. 4b), when the same brushes are grafted from [11-(2-bromo-2-methyl)propionyloxy]undecyltrichlorosilane (BMPUS), which is an initiator with an ester bond, the brushes degraft by up to 80% over 5 days in solution (pH = 9).
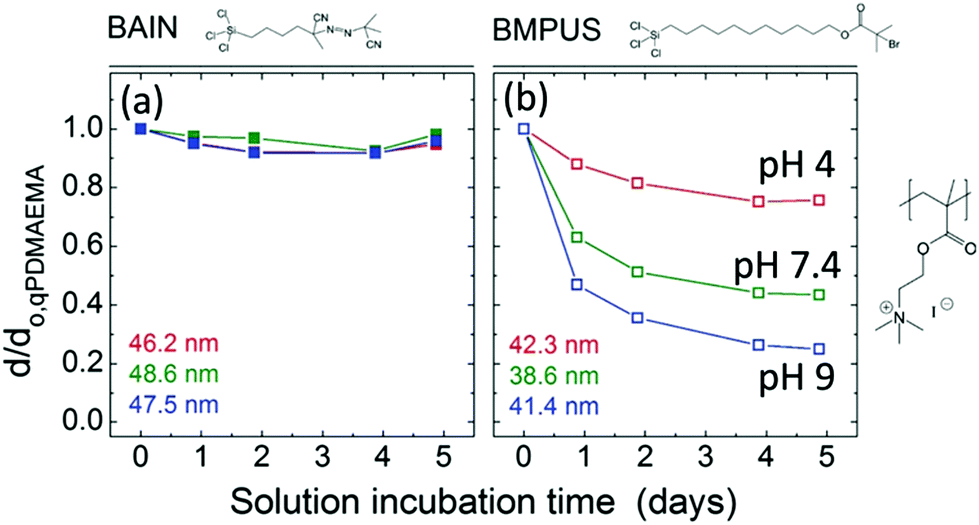 | ||
| Fig. 4 The brush height d normalised by the initial brush height d0,qPDMAEMA for brushes (a) grafted from ester-free BAIN initiator molecules and (b) BMPUS initiator molecules. The heights given in colour in the bottom left of the graphs are the initial heights of the brushes in the experiments given in the respective colour (adapted with permission from ref. 134. Copyright 2012 American Chemical Society). | ||
For gold substrates, synthetic routes that result in more stable polymer brushes have been developed as well. Park et al.135 showed that brushes can be grafted from gold at elevated temperatures (120 °C) using bromoisobutyrate-terminated alkanethiols, 16-(3,5-bis(mercaptomethyl)phenoxy)hexadecyl 2-bromo-2-methylpropanoate (BMTBM) as initiators. The enhanced stability of these molecules was attributed to the chelate effect: the molecule has an aromatic ring with two thiol groups that can chelate readily to the gold substrate. An alternative, more stable synthesis route is the silanisation of thiol based initiators,136 which links the initiators to each other. This allows for stable grafting of the brushes from the initiators. Moreover, it makes the grafted brushes stable for 3 h at temperature of 150 °C. However, due to the presence of siloxane and amide bonds, these brushes can still degraft. A more stable route for gold (or metal) attachment can be achieved via aryl diazonium salts.137,138 For example, Matrab et al. electrografted initiators based on aryl diazonium salts.137 Optimization of this process by Iruthayaraj et al.,138 resulted in stable high density brushes via covalently bonded initiators without any hydrolyzable bonds.
The advantage of the linker molecules described above is that they form dense self-assembled monolayers (SAMs), such that the initiator density at the substrate is high as well. Therefore, polymer brushes can be grown with grafting densities that are comparable to the grafting densities obtained with the more common silanes used for silicon and mica or thiols used for gold. A disadvantage of most of the described linkers is that the molecules are not commercially available, and the synthesis involves several steps. This makes it more challenging to employ these techniques on large scales. Moreover, the proposed initiator linkers might be less effective for initiation of the polymerization.49,51
3.2 Diblock copolymers
Several groups have proposed diblock copolymers as promising systems for enhancing the stability of polymer brushes.139–143 They are prepared in a two-step SI-ATRP reaction. First, hydrophobic anchor blocks are grafted from the surface. Their function is to protect the sensitive bonds near the surface. Next, the second block is polymerized from the chain ends of the first block. This block will be hydrophilic. When immersed in water, the diblock copolymer brushes will consist of a dense, collapsed layer near the substrate (anchor blocks) and a swollen brush on top of them (hydrophilic block, see also Fig. 3).An overview of different proposed diblock copolymer brush systems and their performance is given in Table 1. From these performances several conclusions can be drawn. First of all, more hydrophobic anchor blocks appear to result in more stable brushes. Quintana et al.141 compared poly(styrene) (PS) and poly(methyl methacrylate) (PMMA) anchor blocks and found that the hydrophilic poly(sulfobetaine methacrylamide) (PSBMAm) brushes grafted from the more hydrophobic PS are more stable. However, not only the hydrophobicity is important to consider. Divandari et al.142 observed that PMMA anchors are more stable than poly(lauryl methacrylate) (PLMA) anchors, even though PLMA is more hydrophobic. They attribute this to the PMMA being glassy, while PLMA has a glass transition temperature below the experimental temperature of 37 °C. Consequently, PMMA will be mechanically more stable than PLMA.
| Anchor | d a [nm] | Brush | d b [nm] | medium | pH | T [°C] | Performance | Ref. |
|---|---|---|---|---|---|---|---|---|
| PMMA | 40 | PMAA | −1 | NaOH solution | 12 | 37 | 100% after 80 h | 140 |
| PEHMA | 48 | PMAA | 42 | NaOH solution | 12 | 37 | 65% after 10 h | 140 |
| PMMA | 20 | PSBMAm | 8 | Sea water | 8.2 | 37 | 0% after 4 weeks | 141 |
| PS | 19 | PSBMAm | 9 | Sea water | 8.2 | 37 | 33% after 14 weeks | 141 |
| PMMA | 23 | POEGMA | 21 | DMEM solution | 7.4 | 37 | No observable changes after 7 days | 142 |
| PLMA | 21 | POEGMA | 27 | DMEM solution | 7.4 | 37 | Degrafting after 6 days | 142 |
| PBMA | 27 | POEGMA | 36 | DMEM solution | 7.4 | 37 | Degrafting after 4 days | 142 |
| PMMA | 11 | PAA | 70 | Ethanolamine solution | 9 | RT | 88% after 120 h | 143 |
| PMMA | 2.8 | PAA | 70 | Ethanolamine solution | 9 | RT | 78% after 120 h | 143 |
| PPEGMA | 10 | PAA | 70 | Ethanolamine solution | 9 | RT | 77% after 120 h | 143 |
| PPEGMA | 3.5 | PAA | 70 | Ethanolamine solution | 9 | RT | 69% after 120 h | 143 |
Besides the hydrophobicity and the glassiness of the anchor blocks, also the thickness of these blocks is important. Li et al. observed that the height of PMMA-b-PAA polymer brushes with a PMMA anchor thickness of 11 nm decreased to 88%, while the same brushes with a PMMA anchor height of 2.8 nm decreased to 78% (see also Table 1). From all these observations we can conclude that the anchor block of diblock copolymer brushes needs to be hydrophobic, glassy and of sufficient thickness (>20 nm) to prepare long-term stable polymer brushes.
A major advantage of diblock copolymers as a synthetic strategy for stable brushes is that truly long-term stability can be achieved (even after several months of immersion in seawater141). Nevertheless, there are several disadvantages as well. The grafting density of the second block is typically lower than the first block due to termination of the polymerization during the grafting of the anchor block, and limited availability of chain ends during second polymerization. Moreover, the synthesis relies on multiple complex steps, which limits its applicability.
3.3 Bio-inspired polyphenols
Since the first report of mussel-inspired, strongly and (almost) universally adhering polydopamine films,144 these coatings have been utilized as an initiator primer for surface initiated polymerizations as well.145–147 Brushes grafted from these mussel-adhesive inspired films on noble metals, metal oxides, and inert polymers have been reported to be stable against degrafting.148,149 For example, Kuang et al. synthesized bifunctional tripeptide bromide (BrYKY),148 which bears both a initiating and surface attachment functionality. High-density pSBMAm brushes grafted from BrYKY kept their low-fouling performance for at least 4 months. This is much longer than observed for anchors based on SAMs of individual catechols,150 for which the performance declined after 1 month already. This has been attributed to the multiple bonds that BrYKY can form with the substrates.In a similar bio-inspired approach, initiator primers can be formed by tea stains.151–153 By bromination151,153 or direct UV activation152 of tannic acid based anchoring layers, long term stable brushes can be prepared by consecutive surface initiated polymerization. For example, Pranantyo et al.151 showed several cationic and zwitterion brushes grafted from brominated tannic acid attached to stainless steel (see Fig. 5 for the synthesis scheme) are stable against degrafting for 14 days in a stream of sea water.
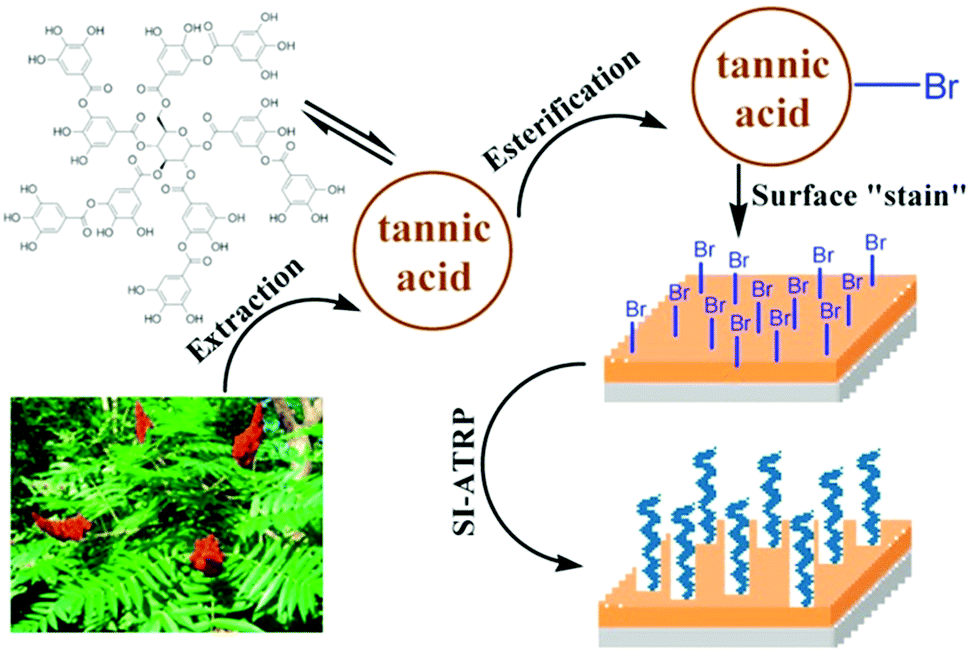 | ||
| Fig. 5 Schematic representation of the synthesis of long-term stable polymer brushes from brominated, tea-stain inspired tannic acid films. Adapted with permission from ref. 151. Copyright 2015 American Chemical Society. | ||
The synthetic strategy based on bio-inspired polyphenols has the advantage that the initiator primers are easy to apply (e.g. by dip coating or drop casting). Moreover, true long term stability of several months148 can be achieved. Nevertheless, the primer cannot be strictly universally applied. Though the polyphenol layers bind strongly to metal(-oxide) substrates154 and carbon-based materials,155 they not adhere well to SiO2 substrates. In fact, this weak adherence to silicon has been utilized to prepare brush-coated polydopamine-based nanosheets.156,157 This makes this method less suitable for several applications, for example in micro-electromechanical systems (MEMS)158 and (nano-)photonic devices159 where often SiOx's are the primary material.
3.4 Macro-initiators
In the descriptions of the previous sections, we stated several times that initiator molecules that have multiple bonds to the substrate exhibit an enhanced stability compared to molecules, with single surface bonds (e.g. in silane,111 thiol135 and catechol148 binding). Macro-initiators function by the same mechanism. Macro-initiators are composed of long macromolecules with both initiating sites and moieties that can bind to the substrate.160 This binding is often physical (electro-static). Therefore, the binding strength of a single moiety with the substrate is not strong. Yet, the total binding strength can be very strong, since it is amplified by the inherent cooperativity via the many binding sites between the macromolecules and the substrate.The macro-initiators are typically bonded to the substrate via hydrogen bonding or electrostatic interactions. An example of the former is the binding of diazo bearing poly(ε-caprolactone)s to hydroxyl groups on silicon.161 To exploit electrostatic interactions, cationic or anionic polyelectrolytes can be used as macroinitiators.162,163 For example, Chen et al. synthesised cationic random copolymers of 2-(dimethylamino)ethyl methacrylate (DMA) and 2-hydroxy-ethyl methacrylate (HEMA), in which the hydroxy groups of the HEMA monomers were esterified using 2-bromoisobutyryl bromide to form the initiator groups and the DMA was quaternized to introduce the positive charges. These macroinitiators can be bound to negatively charged substrates such as silica.
From macro-initiators one can graft stable polymer brushes, as was shown by Rodda et al.164 They grafted poly(oligo(ethylene glycol) methacrylate) (pOEGMA) brushes from poly(styrene-co-vinylbenzyl chloride) macroinitiators and observed that these brushes were stable for at least 24 h in cell culture media.164 This is comparable to the performance of pOEGMA brushes grafted from silane-based initiators.165 Nevertheless, using a different technique, the same group showed that pOEGMA brushes grafted from brominated polycaprolactone (PCL) macroinitiators resulted in them being stable for >21 days.166 Alternatively, brushes can be grafted from crosslinked physisorbed polyvinylpyrrolidone (PVP) on the substrate. Sun et al. showed that PVP brushes grafted from such crosslinked PVP films are stable for 4 weeks.167
A major advantage of the utilization of macro-initiators is that these initiator coatings are easy to apply as a primer layer on the substrate. Nevertheless, the synthesis of the macro-initiators often involves several synthesis steps. Moreover, the initiator surface coverage is often smaller than for SAM-based initiator layers. Though higher density brushes can be obtained by layer-by-layer deposition of the anchor.168 Nevertheless, the bonding is based on physical interactions, which can change if the environment (e.g. pH or salt concentration) is varied. Therefore, the brushes can degraft under unfavorable conditions.
3.5 Multi-covalent bonds
The synthetic strategy that utilizes multi covalent bonds is closely related to the macro-initiators described in Section 3.4. Yet, the strategy described here eliminates an important disadvantage of macro-initiators, which is that debonding of the physisorbed macromolecules can occur by e.g. pH changes. This debonding can be circumvented by covalently bonding the macromolecules to the substrate.Multi-covalent bonding has been obtained by poly(glycidyl methacrylate) (PGMA) polymers. The epoxy groups of PGMA can react with for example hydroxyl groups on different types of substrates. Next, initiators can be attached to the PGMA, from which brushes can be grafted109,169 (see Fig. 6a). These brushes are very stable, even under harsh oxidative environments.109 The enhanced stability can be attributed to different reasons. First, the PGMA can only detach if all bonds are broken, which will slow down degrafting. Moreover, the hydrophobicity of PGMA can prevent water from reaching the hydrolysis sensitive PGMA-substrate bonds. In fact, when we make the PGMA anchor more hydrophilic by coupling polyethylenimine (PEI) before polymerization, the brushes do degraft.170
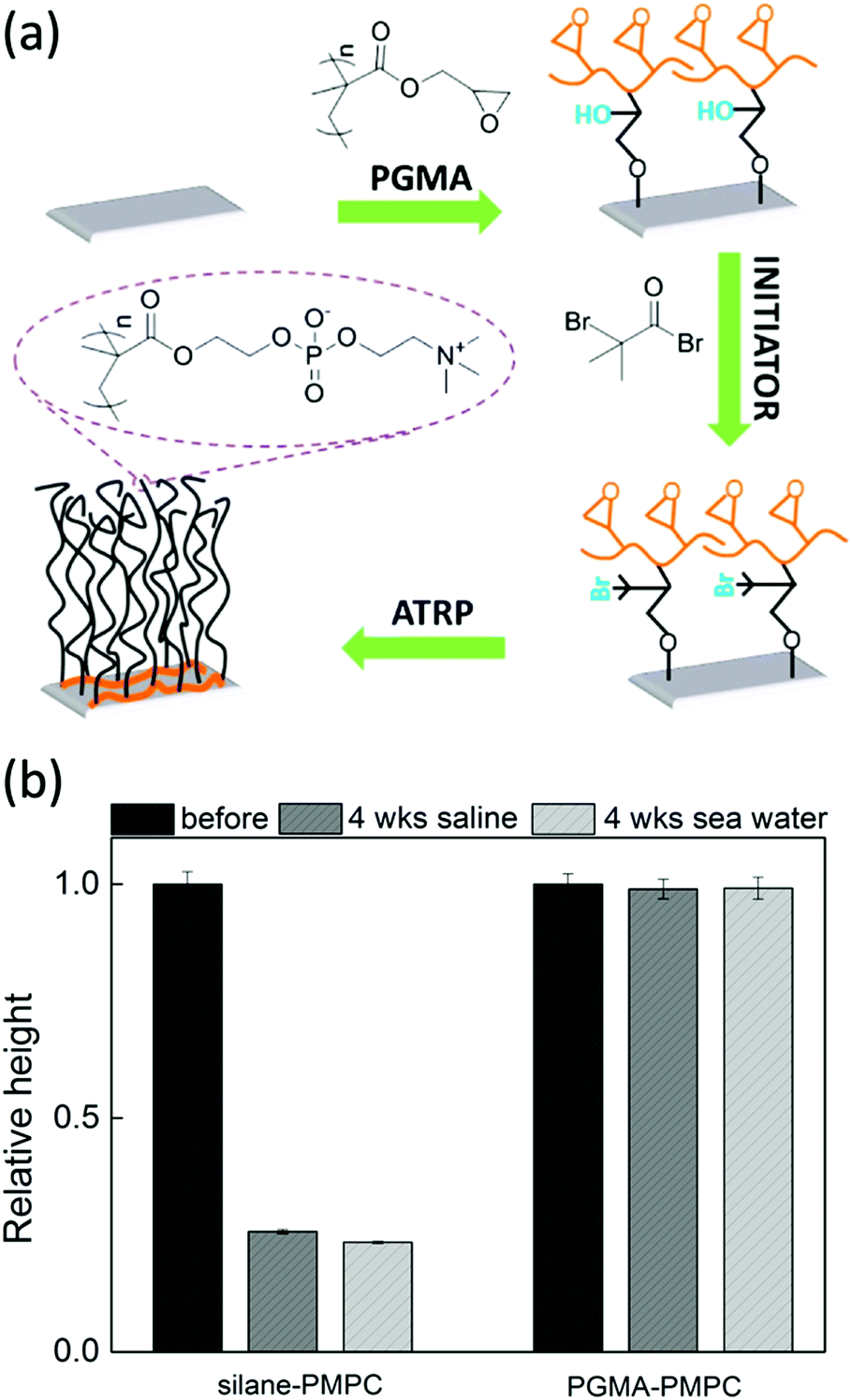 | ||
| Fig. 6 (a) Synthesis scheme for the preparation of poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC) brushes on silicon surfaces by grafting the brushes from initiators attached to PGMA. The PGMA is attached to the substrate via multiple covalent bonds (adapted from ref. 109). (b) The relative height of PMPC brushes grafted from unstable silanes (left) and from PGMA attached initiators (right) after immersion for 4 weeks in saline solution (dark gray) or artificial seawater (light gray). | ||
For example, in our research group, we have shown that poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC) brushes grafted from multi-covalently bonded PGMA are stable for more than 4 weeks while being immersed in saline solution or artificial seawater. Moreover, the brushes keep their hydrophilic properties even after being kept in a highly oxidative sodium hypochlorite solution for 100000 ppm hours.109 In a different study, we showed that PGMA can be utilised as a stable anchor-layer to graft anti-fouling poly(3-sulfopropyl methacrylate potassium) (PSPMAK) brushes on artificial implants.110 The brushes displayed less than 10% degrafting after being immersed in aqueous solutions for more than 32 days.
An alternative route to prepare multi-covalently bonded initiator layers was presented by Kang et al.171 They prepared crosslinked P(S-r-GMA) anchor films to graft brushes that were stable upon repetitive washing and during multiple thermal annealing cycles. Moreover, the grafting density could be controlled by varying the GMA/S ratio.
A major advantage of this synthetic strategy is that the initiator film is strongly bonded to the substrate via multiple covalent bonds, giving rise to long-term stable brushes even under harsh oxidative conditions. Nevertheless, there is also a clear disadvantage: the initiator density in the film is low compared to that for SAMs. Moreover, the initiator moieties are ‘hidden’ under the protective PGMA film. Therefore, one typically obtains lower grafting densities of 0.1–0.2 chains per nm2, especially for bulky monomers such as MPC and SPMAK.
3.6 Crosslinking
In parallel with the development of strong substrate anchors, researchers have designed complementary strategies to increase the stability of polymer brushes. Crosslinking of the brush can strongly enhance the long-term stability of the brushes.76 In a recent report, Chen et al.172 showed that the crosslinked edges of patterned brushes do not degraft, while the non-crosslinked centers of the patterned brushes do degraft. To effectively increase the stability of brushes, one needs to keep in mind that enough links are formed such that one is beyond the gelation threshold for crosslinked brushes.173 Then long-term stable brushes can be formed: Wu et al. showed that crosslinked brushes can keep their antibacterial properties for more than 4 weeks of immersion in water.174 Moreover, crosslinked brushes can resist high shear stresses, while maintaining their lubricating properties.175An alternative method to introduce extra bonds can be achieved via a ladder-structure, as recently presented by the lab of Zapotoczny.176 Słowikowska et al. observed that the degrafting of ladder-structured poly(3-trimethylsilyl-2-propynylmethacrylate) (PTPM) brushes in tetra-butyl-ammonium fluoride (TBAF) solution is significantly slowed down compared to the linear counter parts. While the linear brushes degraft completely in 2 h, the ladder brushes still have 33% of their brush height after 25 h.177 The increased stability can be attributed to the additional intra- and intermolecular bridges as well as the reduced swelling.
Though the synthesis of ladder-brushes is highly specialized, a primary advantage of crosslinked brushes is that this crosslinking is often easy to achieve. Nevertheless, there are several disadvantages as well. The brushes of these alternative topologies will swell less and have different mechanical properties than traditional brushes. Moreover, the degrafting is only delayed. Since water can still reach hydrolysis-sensitive bonds, degrafting will eventually occur.76
3.7 Self-healing brushes
An out-of-the-box solution that does not prevent degrafting but will, nevertheless, give rise to a longer performance time of brush-based coatings, is the development of self-healing brushes. Instead of initiator-engineering or crosslinking, as described in the previous strategies, the polymer of the brushes are now replenished e.g. via 3D grafting or surface re-organisation.178 This brings the advantage of regeneration of the coating. Kuroki et al. designed polymer networks in which polymers were grafted both at the surface of the network as inside the network material.179 After the surface polymers had been detached, polymers in the bulk network can be brought to the surface. With this network the authors could achieve that the longevity of the antifouling behavior of these 3D grafted brushes was 4 times as long as for 2D brushes.While improved stability has been observed for these self-healing brushes, there are several disadvantages to this technique. The synthesis procedures for the routes developed so far are complex. Moreover, the replenishing of the coating is limited and are after a few healing cycles the healing process no longer works.
The different synthetic strategies discussed above (and summarized in Fig. 3) each have specific advantages and disadvantages. We summarize these advantages and disadvantages in Table 2. Which synthetic strategy is the best for the preparation of long-term stable polymer, depends on the demands on the coating by the application as well as the substrate materials. With the overview provided in the text and Table 2, the readers can make a more informed decision on the best solution for their specific application.
| Strategy | Chemistry | Advantages | Disadvantages |
|---|---|---|---|
| (1) Stabilized SAMs | (a) Phosphonic acid based initiators on SiO2132 | High density brushes | Multiple-step synthesis |
| (b) Direct Si−C linkage on SixN4133 | |||
| (c) Ester-free initiators on SiO2134 | |||
| (d) Chelate effect on gold135 | |||
| (e) Silanisation thiols on gold136 | |||
| (f) Aryl diazonium salts on gold137,138 | |||
| (2) Diblock copolymers | Consecetive polymerizations via SI-ATRP140–143 | Strongly enhanced stability (up to months) | Multiple synthesis steps and low grafting densities |
| (3) Polyphenols | (a) Polydopamine148,149 | Synthesis is easy and long-term stability | Cannot be applied to all substrates (not for SiOx) |
| (b) Tannic acid151–153 | |||
| (4) Macro-initiators | (a) Brominated polycaprolactone166 | Synthesis is easy | Often low grafting density and not stable under all solvent conditions |
| (b) Polyvinylpyrrolidone167 | |||
| (c) Multi-layer bonding for high grafting density168 | |||
| (5) Multi-covalent bonds | (a) PGMA109,110 | Synthesis is easy and long-term stability even under harsh conditions | Low grafting density, especially for bulky monomers |
| (b) p(S-r-GMA)171 | |||
| (6) Cross-linking | (a) Random crosslinking76,174 | Synthesis is easy (crosslinking) | Brush characteristics can disappear |
| (b) Ladder structure177 | |||
| (7) Self-healing | 3D grafted brushes178,179 | Possibility for regeneration | Complex synthesis, limited self-healing events |
4. Outlook and open questions in the field
Though many synthetic strategies have been developed, there are still open questions that need answering. As is clear from the overview provided in Table 2, the perfect synthetic strategy to prepare long-term stable brushes does not yet exist. In particular for SiOx substrates, most strategies (e.g. diblock copolymers, macro-initiators and multi covalent bonds) result in brushes with lower grafting densities than can be obtained via SAM-based initiators. How to obtain high grafting density, stable brushes, is currently carefully examined. Similar to layer-by-layer macro-initiators,168 layer-by-layer multi-covalent bonds could be prepared, which might result in higher grafting densities. Additionally, the anchors could be functionalized with groups that allow for a high density initiator coupling (such as tris(hydroxymethyl)aminomethane).Additionally, to translate research on polymer brushes to applications, the synthesis route for grafting brushes from substrates needs to be simplified. Recently, several synthesis techniques have been designed that allow grafting under ambient conditions, for example by large scale Cu0-mediated surface-initiated ATRP,180 filter paper assisted Cu0-mediated surface-initiated controlled polymerizations181 or oxygen tolerant photoinduced electron transfer (PET) controlled polymerization techniques.57,182 This will make it possible to scale up the synthesis of these brushes. Nevertheless, the synthesis for initiator primer films that give rise to long-term stable brushes still must be scaled up.
How to prepare large scale initiator primer layers is not yet widely explored. Nevertheless, the first techniques have been reported.21,183 For example, Sato et al. reported on the large-scale (∼40 m2) production of initiator layers by spin-, wire-bar-, or roll-to-roll-coating of a simple sol–gel solution of (p-chloromethyl)phenyl trimethoxysilane (CMPTMS) and tetraethoxysilane (TEOS)183 (see also Fig. 7). Developing routes like this will be key in the translation of polymer brush research to the large-scale application of these coatings and they need to be developed for the synthetic strategies reviewed in this article as well.
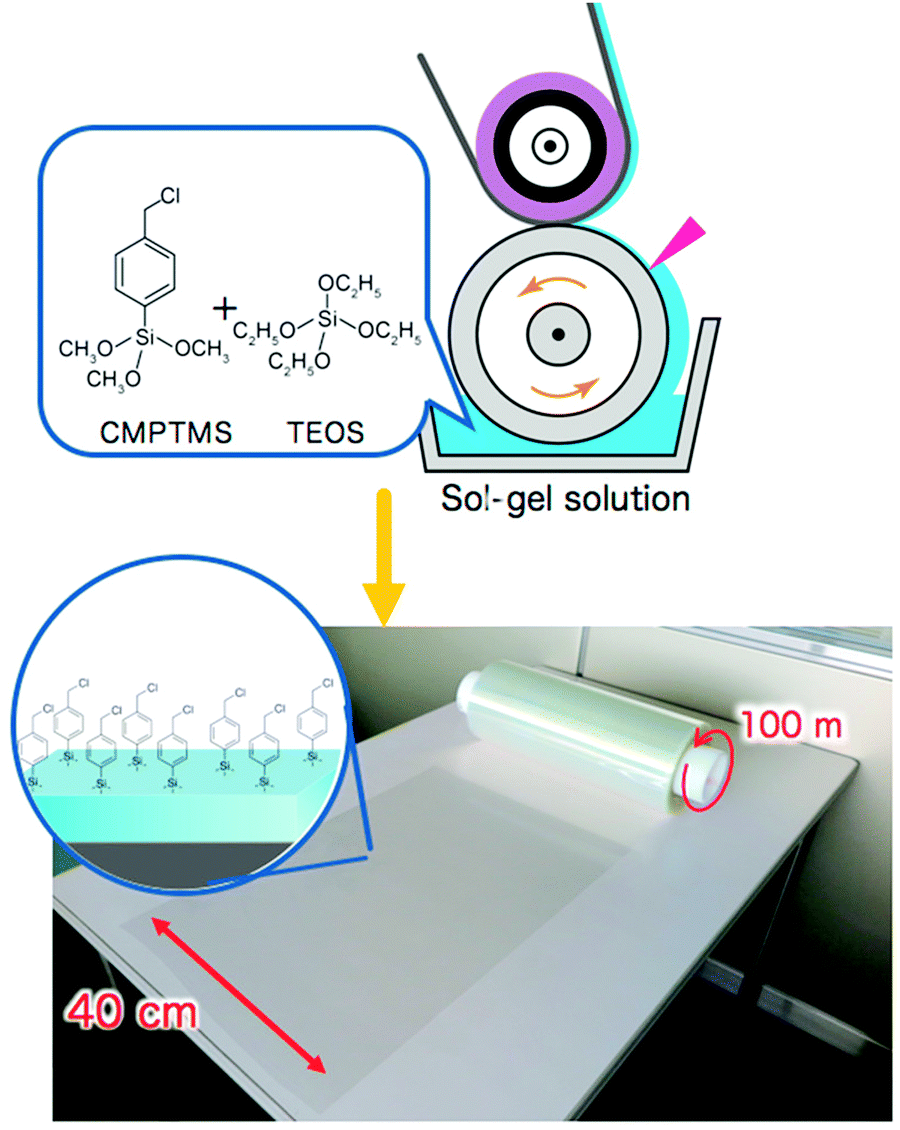 | ||
| Fig. 7 Large-scale production of initiator layers by casting of a sol–gel solution of (p-chloromethyl)phenyltrimethoxysilane (CMPTMS) and tetraethoxysilane (TEOS), which will allow for scale up in polymer brush synthesis. Adapted with permission from ref. 183. Copyright 2018 American Chemical Society. | ||
Finally, we would like to discuss the relevance of the different synthetic strategies reviewed in this article for preparing patterned brushes. Recently, there has been a renewed interest in such patterned brushes50 because they allow for more precise control of for example adhesion184 and membrane transport.185 Patterned brushes can be prepared by micro-contact127 or inkjet printing186 of thiols or silanes. Alternatively, photo-lithography can employed to spatially control brush growth.187–189 However, when anchors are utilized that easily degraft, the patterned brushes will also degraft. Several strategies presented in this article allow for printing stable surface anchors. Macro-initiators,190 anchors based on multi-covalent bonding and polyphenol-based anchors, can be printed straightforwardly, since they do not immediately wet the substrate and, therefore, these strategies are recommended for preparing patterned brushes by printing. Brushes prepared by photo-lithography can utilize all presented strategies.
5. Summary
In this review article, we have presented an overview of proposed mechanisms and causes for the deterioration and degrafting of polymer brushes. Moreover, we have reviewed the status on synthetic strategies that have been developed to prevent degrafting and we identified open questions in the field. We hope that with this review we will help the reader in making an informed decision on the best strategy in synthesizing more stable polymer brushes.Author contributions
Zhichao Ding: data curation, investigation, resources, writing – original draft. Changyou Chen: investigation, visualisation, writing – original draft. Yunlong Yu: conceptualization, funding acquisition, project administration, supervision, writing – original draft. Sissi de Beer: conceptualization, formal analysis, visualization, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank G. J. Vancso for fruitful discussions during the start of this research. This work has been financially supported by the National Natural Science Foundation of China (51703145), China Postdoctoral Science Foundation (2017M620426 and 2019T120844), and Natural Science Foundation of Chongqing (cstc2021jcyj-msxmX0513).Notes and references
- S. T. Milner, Science, 1991, 251, 905–914 CrossRef CAS PubMed.
- W. J. Brittain and S. Minko, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3505–3512 CrossRef CAS.
- S. Lee and N. D. Spencer, Science, 2008, 319, 575 LP–576 LP CrossRef.
- J. Klein, Science, 2009, 323, 47 LP–48 LP CrossRef PubMed.
- T. Kreer, Soft Matter, 2016, 12, 3479–3501 RSC.
- J. Klein, E. Kumacheva, D. Mahalu, D. Perahia and L. J. Fetters, Nature, 1994, 370, 634–636 CrossRef CAS.
- S. de Beer, E. Kutnyanszky, P. M. Schön, G. J. Vancso and M. H. Müser, Nat. Commun., 2014, 5, 3781 CrossRef PubMed.
- W. Yan, S. N. Ramakrishna, N. D. Spencer and E. M. Benetti, Langmuir, 2019, 35, 11255–11264 CrossRef CAS PubMed.
- Y. Higaki, M. Kobayashi, D. Murakami and A. Takahara, Polym. J., 2016, 48, 325–331 CrossRef CAS.
- B. Li, P. Jain, J. Ma, J. K. Smith, Z. Yuan, H.-C. Hung, Y. He, X. Lin, K. Wu, J. Pfaendtner and S. Jiang, Sci. Adv., 2019, 5, eaaw9562 CrossRef CAS PubMed.
- X. Su, D. Hao, Z. Li, X. Guo and L. Jiang, J. Mater. Chem. B, 2019, 7, 1322–1332 RSC.
- J. Baggerman, M. M. J. Smulders and H. Zuilhof, Langmuir, 2019, 35, 1072–1084 CrossRef CAS PubMed.
- Y. Liu, D. Zhang, B. Ren, X. Gong, L. Xu, Z.-Q. Feng, Y. Chang, Y. He and J. Zheng, J. Mater. Chem. B, 2020, 8, 3814–3828 RSC.
- M. Wei, Y. Gao and M. J. Serpe, J. Mater. Chem. B, 2015, 3, 744–747 RSC.
- T. Wang, Y. Yu, D. Chen, S. Wang, X. Zhang, Y. Li, J. Zhang and Y. Fu, Nanoscale, 2017, 9, 1925–1933 RSC.
- D. Chen, T. Wang, G. Song, Y. Du, J. Lv, X. Zhang, Y. Li, L. Zhang, J. Hu, Y. Fu and R. Jordan, ACS Appl. Mater. Interfaces, 2019, 11, 41668–41675 CrossRef CAS PubMed.
- W. Chen, K. J. Shea, M. Xue, L. Qiu, Y. Lan and Z. Meng, Anal. Bioanal. Chem., 2017, 409, 5319–5326 CrossRef CAS PubMed.
- K. Ohno, M. Sakaue and C. Mori, Langmuir, 2018, 34, 9532–9539 CrossRef CAS PubMed.
- K. Ohno and Y. Mizuta, ACS Appl. Polym. Mater., 2020, 2, 368–375 CrossRef CAS.
- M. Qin, M. Sun, M. Hua and X. He, Curr. Opin. Solid State Mater. Sci., 2019, 23, 13–27 CrossRef CAS.
- K. B. Buhl, A. H. Agergaard, M. Lillethorup, J. P. Nikolajsen, S. U. Pedersen and K. Daasbjerg, Polymers, 2020, 12, 1475 CrossRef CAS PubMed.
- Y. Yu, M. Brió Pérez, C. Cao and S. de Beer, Eur. Polym. J., 2021, 147, 110298 CrossRef CAS.
- J. M. Giussi, M. L. Cortez, W. A. Marmisollé and O. Azzaroni, Chem. Soc. Rev., 2019, 48, 814–849 RSC.
- S. Wang, Z. Wang, J. Li, L. Li and W. Hu, Mater. Chem. Front., 2020, 4, 692–714 RSC.
- N. Hadjesfandiari, K. Yu, Y. Mei and J. N. Kizhakkedathu, J. Mater. Chem. B, 2014, 2, 4968–4978 RSC.
- P. Li, Z. Ding, Y. Yin, X. Yu, Y. Yuan, M. Brió Pérez, S. de Beer, G. J. Vancso, Y. Yu and S. Zhang, Eur. Polym. J., 2020, 134, 109845 CrossRef CAS.
- H. Choi, A. Schulte, M. Müller, M. Park, S. Jo and H. Schönherr, Adv. Healthcare Mater., 2021, 10, 2100069 CrossRef CAS PubMed.
- K. Ishihara, Langmuir, 2019, 35, 1778–1787 CrossRef CAS PubMed.
- C. Yoshikawa, S. Hattori, C.-F. Huang, H. Kobayashi and M. Tanaka, J. Mater. Chem. B, 2021, 9, 5794–5804 RSC.
- J. J. Keating, J. Imbrogno and G. Belfort, ACS Appl. Mater. Interfaces, 2016, 8, 28383–28399 CrossRef CAS PubMed.
- E. N. Durmaz, S. Sahin, E. Virga, S. de Beer, L. C. P. M. de Smet and W. M. de Vos, ACS Appl. Polym. Mater., 2021, 3, 4347–4374 CrossRef CAS PubMed.
- D. Li, L. Xu, J. Wang and J. E. Gautrot, Adv. Healthcare Mater., 2021, 10, 2000953 CrossRef CAS PubMed.
- H. Liu, Y. Li, K. Sun, J. Fan, P. Zhang, J. Meng, S. Wang and L. Jiang, J. Am. Chem. Soc., 2013, 135, 7603–7609 CrossRef CAS PubMed.
- N. Fortin and H.-A. Klok, ACS Appl. Mater. Interfaces, 2015, 7, 4631–4640 CrossRef CAS PubMed.
- L. A. Smook, G. C. Ritsema van Eck and S. de Beer, ACS Appl. Polym. Mater., 2021, 3, 2336–2340 CrossRef CAS PubMed.
- O. Azzaroni, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3225–3258 CrossRef CAS.
- S. Ma, X. Zhang, B. Yu and F. Zhou, NPG Asia Mater., 2019, 11, 1–39 CrossRef.
- G. Cavallaro, S. Micciulla, L. Chiappisi and G. Lazzara, J. Mater. Chem. B, 2021, 9, 594–611 RSC.
- G. C. Ritsema van Eck, L. Chiappisi and S. de Beer, ACS Appl. Polym. Mater., 2022 DOI:10.1021/acsapm.1c01615.
- X. Sui, S. Zapotoczny, E. M. Benetti, P. Schön and G. J. Vancso, J. Mater. Chem., 2010, 20, 4981–4993 RSC.
- T. Wu, K. Efimenko and J. Genzer, J. Am. Chem. Soc., 2002, 124, 9394–9395 CrossRef CAS PubMed.
- G. S. Grest and M. Murat, Macromolecules, 1993, 26, 3108–3117 CrossRef CAS.
- D. I. Dimitrov, A. Milchev and K. Binder, J. Chem. Phys., 2007, 127, 84905 CrossRef CAS PubMed.
- S. Minko, J. Macromol. Sci., Part C, 2006, 46, 397–420 CrossRef CAS.
- T. Chen, R. Ferris, J. Zhang, R. Ducker and S. Zauscher, Prog. Polym. Sci., 2010, 35, 94–112 CrossRef CAS.
- R. C. Advincula, W. J. Brittain, K. C. Caster and J. C. Rühe, Polymer Brushes, Wiley, 2004 Search PubMed.
- G. J. Fleer, M. A. C. Stuart, J. M. H. M. Scheutjens, T. Cosgrove and B. Vincent, Polymers at Interfaces, Springer Netherlands, 1998 Search PubMed.
- B. Zdyrko and I. Luzinov, Macromol. Rapid Commun., 2011, 32, 859–869 CrossRef CAS PubMed.
- J. O. Zoppe, N. C. Ataman, P. Mocny, J. Wang, J. Moraes and H. A. Klok, Chem. Rev., 2017, 117, 1105–1318 CrossRef CAS PubMed.
- M. Fromel, M. Li and C. W. Pester, Macromol. Rapid Commun., 2020, 41, 2000177 CrossRef CAS PubMed.
- S. Edmondson, V. L. Osborne and W. T. S. Huck, Chem. Soc. Rev., 2004, 33, 14–22 RSC.
- J. Pyun, T. Kowalewski and K. Matyjaszewski, Macromol. Rapid Commun., 2003, 24, 1043–1059 CrossRef CAS.
- A. Khabibullin, E. Mastan, K. Matyjaszewski and S. Zhu, Surface-Initiated Atom Transfer Radical Polymerization, in Controlled Radical Polymerization at and from Solid Surfaces, ed. P. Vana, Springer International Publishing, Cham, 2016, pp. 29–76 Search PubMed.
- M. Husseman, E. E. Malmström, M. McNamara, M. Mate, D. Mecerreyes, D. G. Benoit, J. L. Hedrick, P. Mansky, E. Huang, T. P. Russell and C. J. Hawker, Macromolecules, 1999, 32, 1424–1431 CrossRef CAS.
- M. Baum and W. J. Brittain, Macromolecules, 2002, 35, 610–615 CrossRef CAS.
- K. Ohno, Y. Ma, Y. Huang, C. Mori, Y. Yahata, Y. Tsujii, T. Maschmeyer, J. Moraes and S. Perrier, Macromolecules, 2011, 44, 8944–8953 CrossRef CAS.
- A. R. Kuzmyn, A. T. Nguyen, L. W. Teunissen, H. Zuilhof and J. Baggerman, Langmuir, 2020, 36, 4439–4446 CrossRef CAS PubMed.
- H. Sakata, M. Kobayashi, H. Otsuka and A. Takahara, Polym. J., 2005, 37, 767–775 CrossRef CAS.
- Y. Yu, B. D. Kieviet, E. Kutnyanszky, G. J. Vancso and S. De Beer, ACS Macro Lett., 2015, 4, 75–79 CrossRef CAS.
- R. J. Horst, M. Brió Pérez, R. Cohen, M. Cirelli, P. S. Dueñas Robles, M. G. Elshof, A. Andreski, M. A. Hempenius, N. E. Benes, C. Damen and S. de Beer, Langmuir, 2020, 36, 12053–12060 CrossRef CAS PubMed.
- J.-B. Kim, M. L. Bruening and G. L. Baker, J. Am. Chem. Soc., 2000, 122, 7616–7617 CrossRef CAS.
- K. Ohno, K. Koh, Y. Tsujii and T. Fukuda, Macromolecules, 2002, 35, 8989–8993 CrossRef CAS.
- S. de Beer, E. Kutnyanszky, M. H. Müser and G. J. Vancso, J. Vis. Exp., 2014, 94, e52285 Search PubMed.
- F. Zhang, K. Sautter, A. M. Larsen, D. A. Findley, R. C. Davis, H. Samha and M. R. Linford, Langmuir, 2010, 26, 14648–14654 CrossRef CAS PubMed.
- M. Zhu, M. Z. Lerum and W. Chen, Langmuir, 2012, 28, 416–423 CrossRef CAS PubMed.
- L. Bertilsson and B. Liedberg, Langmuir, 1993, 9, 141–149 CrossRef CAS.
- M. Jaschke, H. Schönherr, H. Wolf, H.-J. Butt, E. Bamberg, M. K. Besocke and H. Ringsdorf, J. Phys. Chem., 1996, 100, 2290–2301 CrossRef CAS.
- H. Schönherr, F. J. B. Kremer, S. Kumar, J. A. Rego, H. Wolf, H. Ringsdorf, M. Jaschke, H.-J. Butt and E. Bamberg, J. Am. Chem. Soc., 1996, 118, 13051–13057 CrossRef.
- J. Majoinen, A. Walther, J. R. McKee, E. Kontturi, V. Aseyev, J. M. Malho, J. Ruokolainen and O. Ikkala, Biomacromolecules, 2011, 12, 2997–3006 CrossRef CAS PubMed.
- C. J. Porter, J. R. Werber, C. L. Ritt, Y.-F. Guan, M. Zhong and M. Elimelech, J. Membr. Sci., 2020, 596, 117719 CrossRef CAS.
- M. U. Khan, K. R. Reddy, T. Snguanwongchai, E. Haque and V. G. Gomes, Colloid Polym. Sci., 2016, 294, 1599–1610 CrossRef CAS.
- M. Chen, W. H. Briscoe, S. P. Armes, H. Cohen and J. Klein, ChemPhysChem, 2007, 8, 1303–1306 CrossRef CAS PubMed.
- B. Lego, M. François, W. G. Skene and S. Giasson, Langmuir, 2009, 25, 5313–5321 CrossRef CAS PubMed.
- D. L. Heichel, N. C. H. Vy, S. P. Ward, D. H. Adamson and K. A. Burke, J. Mater. Chem. B, 2020, 8, 10392–10406 RSC.
- J. Jia, C. Liu, L. Wang, X. Liang and X. Chai, Chem. Eng. J., 2018, 347, 631–639 CrossRef CAS.
- S. Tugulu and H. A. Klok, Biomacromolecules, 2008, 9, 906–912 CrossRef CAS PubMed.
- B. Liberelle and S. Giasson, Langmuir, 2007, 23, 9263–9270 CrossRef CAS PubMed.
- S. Han, C. Kim and D. Kwon, Polymer, 1997, 38, 317–323 CrossRef CAS.
- L. Xu, K. Crawford and C. B. Gorman, Macromolecules, 2011, 44, 4777–4782 CrossRef CAS.
- T. Steinbach and F. R. Wurm, Angew. Chem., Int. Ed., 2015, 54, 6098–6108 CrossRef CAS PubMed.
- E. Schönemann, A. Laschewsky and A. Rosenhahn, Polymers, 2018, 10, 639 CrossRef PubMed.
- G. Morgese, B. Verbraeken, S. N. Ramakrishna, Y. Gombert, E. Cavalli, J.-G. Rosenboom, M. Zenobi-Wong, N. D. Spencer, R. Hoogenboom and E. M. Benetti, Angew. Chem., Int. Ed., 2018, 57, 11667–11672 CrossRef CAS PubMed.
- H. A. Klok and J. Genzer, ACS Macro Lett., 2015, 4, 636–639 CrossRef CAS.
- S. S. Sheiko, S. Panyukov and M. Rubinstein, Macromolecules, 2011, 44, 4520–4529 CrossRef CAS PubMed.
- C. Kang, R. M. Crockett and N. D. Spencer, Macromolecules, 2014, 47, 269–275 CrossRef CAS.
- R. Patil, J. Miles, Y. Ko, P. Datta, B. M. Rao, D. Kiserow and J. Genzer, Macromolecules, 2018, 51, 10237–10245 CrossRef CAS.
- A. H. Agergaard, S. U. Pedersen, H. Birkedal and K. Daasbjerg, Polym. Chem., 2020, 11, 5572–5577 RSC.
- W. Yan, M. Divandari, J. G. Rosenboom, S. N. Ramakrishna, L. Trachsel, N. D. Spencer, G. Morgese and E. M. Benetti, Polym. Chem., 2018, 9, 2580–2589 RSC.
- N. Kyriakou, M.-A. Pizzoccaro-Zilamy, A. Nijmeijer, M. Luiten-Olieman and L. Winnubst, Microporous Mesoporous Mater., 2020, 307, 110516 CrossRef CAS.
- S. S. Sheiko, F. C. Sun, A. Randall, D. Shirvanyants, M. Rubinstein, H. Lee and K. Matyjaszewski, Nature, 2006, 440, 191–194 CrossRef CAS PubMed.
- B. Lyu, W. Cha, T. Mao, Y. Wu, H. Qian, Y. Zhou, X. Chen, S. Zhang, L. Liu, G. Yang, Z. Lu, Q. Zhu and H. Ma, ACS Appl. Mater. Interfaces, 2015, 7, 6254–6259 CrossRef CAS PubMed.
- J. Wang and H. A. Klok, Angew. Chem., Int. Ed., 2019, 58, 9989–9993 CrossRef CAS PubMed.
- K. Enomoto, S. Takahashi, T. Iwase, T. Yamashita and Y. Maekawa, J. Mater. Chem., 2011, 21, 9343–9349 RSC.
- W.-L. Chen, M. Menzel, T. Watanabe, O. Prucker, J. Rühe and C. K. Ober, Langmuir, 2017, 33, 3296–3303 CrossRef CAS PubMed.
- K. A. Melzak, K. Yu, D. Bo, J. N. Kizhakkedathu and J. L. Toca-Herrera, Langmuir, 2015, 31, 6463–6470 CrossRef CAS PubMed.
- C. J. Galvin, E. D. Bain, A. Henke and J. Genzer, Macromolecules, 2015, 48, 5677–5687 CrossRef CAS.
- Y. Ko and J. Genzer, Macromolecules, 2019, 52, 6192–6200 CrossRef CAS.
- N. C. Ataman and H. A. Klok, Macromolecules, 2016, 49, 9035–9047 CrossRef CAS.
- M. Menzel, W.-L. Chen, K. Simancas, H. Xu, O. Prucker, C. K. Ober and J. Rühe, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1283–1295 CrossRef CAS.
- Y. Du, J. Gao, T. Chen, C. Zhang, J. Ji and Z.-K. Xu, Langmuir, 2017, 33, 7298–7304 CrossRef CAS PubMed.
- M. Brió Pérez, M. Cirelli and S. de Beer, ACS Appl. Polym. Mater., 2020, 2, 3039–3043 CrossRef PubMed.
- M. Li, M. Fromel, D. Ranaweera and C. W. Pester, Macromol. Rapid Commun., 2020, 41, 2000337 CrossRef CAS PubMed.
- M. Biesalski and J. Rühe, Langmuir, 2000, 16, 1943–1950 CrossRef CAS.
- C. J. Galvin and J. Genzer, Macromolecules, 2016, 49, 4316–4329 CrossRef CAS.
- G. C. Ritsema Van Eck, L. B. Veldscholte, J. H. W. H. Nijkamp and S. de Beer, Macromolecules, 2020, 53, 8428–8437 CrossRef CAS PubMed.
- L. A. Smook, G. C. Ritsema van Eck and S. de Beer, Macromolecules, 2020, 53, 10898–10906 CrossRef CAS PubMed.
- Y. Yu, B. D. Kieviet, F. Liu, I. Siretanu, E. Kutnyánszky, G. J. Vancso and S. de Beer, Soft Matter, 2015, 26–28 Search PubMed.
- J. D. Willott, T. J. Murdoch, G. B. Webber and E. J. Wanless, Macromolecules, 2016, 49, 2327–2338 CrossRef CAS.
- Y. Yu, G. J. Vancso and S. de Beer, Eur. Polym. J., 2017, 89, 221–229 CrossRef CAS.
- Y. Yu, M. Cirelli, P. Li, Z. Ding, Y. Yin, Y. Yuan, S. De Beer, G. J. Vancso and S. Zhang, Ind. Eng. Chem. Res., 2019, 58, 21459–21465 CrossRef CAS.
- X. Sui, A. Di Luca, M. K. Gunnewiek, E. S. Kooij, C. A. Van Blitterswijk, L. Moroni, M. A. Hempenius and G. J. Vancso, Aust. J. Chem., 2011, 64, 1259–1266 CrossRef CAS.
- G. Panzarasa, S. Aghion, G. Marra, A. Wagner, M. O. Liedke, M. Elsayed, R. Krause-Rehberg, R. Ferragut and G. Consolati, Macromolecules, 2017, 50, 5574–5581 CrossRef CAS.
- Z. Dong, J. Mao, M. Yang, D. Wang, S. Bo and X. Ji, Langmuir, 2011, 27, 15282–15291 CrossRef CAS PubMed.
- S. R. Wasserman, Y. T. Tao and G. M. Whitesides, Langmuir, 1989, 5, 1074–1087 CrossRef CAS.
- N. Aissaoui, L. Bergaoui, J. Landoulsi, J.-F. Lambert and S. Boujday, Langmuir, 2012, 28, 656–665 CrossRef CAS PubMed.
- J. D. Rimstidt and H. L. Barnes, Geochim. Cosmochim. Acta, 1980, 44, 1683–1699 CrossRef CAS.
- Y. Xiao and A. C. Lasaga, Geochim. Cosmochim. Acta, 1996, 60, 2283–2295 CrossRef CAS.
- J. E. Del Bene, K. Runge and R. J. Bartlett, Comput. Mater. Sci., 2003, 27, 102–108 CrossRef CAS.
- N. S. Bhairamadgi, S. P. Pujari, F. G. Trovela, A. Debrassi, A. A. Khamis, J. M. Alonso, A. A. Al Zahrani, T. Wennekes, H. A. Al-Turaif, C. van Rijn, Y. A. Alhamed and H. Zuilhof, Langmuir, 2014, 30, 5829–5839 CrossRef CAS PubMed.
- O. Borozenko, R. Godin, K. L. Lau, W. Mah, G. Cosa, W. G. Skene and S. Giasson, Macromolecules, 2011, 44, 8177–8184 CrossRef CAS.
- J. Ribas-Arino and D. Marx, Chem. Rev., 2012, 112, 5412–5487 CrossRef CAS PubMed.
- E. Wassel, S. Jiang, Q. Song, S. Vogt, G. Nöll, S. I. Druzhinin and H. Schönherr, Langmuir, 2016, 32, 9360–9370 CrossRef CAS PubMed.
- F. Zhou, P. M. Biesheuvel, E.-Y. Choi, W. Shu, R. Poetes, U. Steiner and W. T. S. Huck, Nano Lett., 2008, 8, 725–730 CrossRef PubMed.
- R. G. Nuzzo, B. R. Zegarski and L. H. Dubois, J. Am. Chem. Soc., 1987, 109, 733–740 CrossRef CAS.
- R. G. Nuzzo, F. A. Fusco and D. L. Allara, J. Am. Chem. Soc., 1987, 109, 2358–2368 CrossRef CAS.
- C. D. Bain, E. B. Troughton, Y. T. Tao, J. Evall, G. M. Whitesides and R. G. Nuzzo, J. Am. Chem. Soc., 1989, 111, 321–335 CrossRef CAS.
- R. R. Shah, D. Merreceyes, M. Husemann, I. Rees, N. L. Abbott, C. J. Hawker and J. L. Hedrick, Macromolecules, 2000, 33, 597–605 CrossRef CAS.
- E. M. Benetti, S. Zapotoczny and G. J. Vancso, Adv. Mater., 2007, 19, 268–271 CrossRef CAS.
- Y. Zhang, B. Lv, Z. Lu, J. He, S. Zhang, H. Chen and H. Ma, Soft Matter, 2011, 7, 11496–11500 RSC.
- Y. Zhang, J. He, Y. Zhu, H. Chen and H. Ma, Chem. Commun., 2011, 47, 1190–1192 RSC.
- B. Lv, Y. Zhou, W. Cha, Y. Wu, J. Hu, L. Li, L. Chi and H. Ma, ACS Appl. Mater. Interfaces, 2014, 6, 8313–8319 CrossRef CAS PubMed.
- O. Borozenko, V. Machado, W. G. Skene and S. Giasson, Polym. Chem., 2014, 5, 5740–5750 RSC.
- A. T. Nguyen, J. Baggerman, J. M. J. Paulusse, C. J. M. Van Rijn and H. Zuilhof, Langmuir, 2011, 27, 2587–2594 CrossRef CAS PubMed.
- E. D. Bain, K. Dawes, A. E. Özçam, X. Hu, C. B. Gorman, J. Šogl and J. Genzer, Macromolecules, 2012, 45, 3802–3815 CrossRef CAS.
- C. S. Park, H. J. Lee, A. C. Jamison and T. R. Lee, ACS Appl. Mater. Interfaces, 2016, 8, 5586–5594 CrossRef CAS PubMed.
- X. Liu, K. Sun, Z. Wu, J. Lu, B. Song, W. Tong, X. Shi and H. Chen, Langmuir, 2012, 28, 9451–9459 CrossRef CAS PubMed.
- T. Matrab, M. M. Chehimi, C. Perruchot, A. Adenier, A. Guillez, M. Save, B. Charleux, E. Cabet-Deliry and J. Pinson, Langmuir, 2005, 21, 4686–4694 CrossRef CAS PubMed.
- J. Iruthayaraj, S. Chernyy, M. Lillethorup, M. Ceccato, T. Røn, M. Hinge, P. Kingshott, F. Besenbacher, S. U. Pedersen and K. Daasbjerg, Langmuir, 2011, 27, 1070–1078 CrossRef CAS PubMed.
- K. Nagase, J. Kobayashi, A. Kikuchi, Y. Akiyama, H. Kanazawa and T. Okano, ACS Appl. Mater. Interfaces, 2012, 4, 1998–2008 CrossRef CAS PubMed.
- D. Paripovic and H. A. Klok, Macromol. Chem. Phys., 2011, 212, 950–958 CrossRef CAS.
- R. Quintana, M. Gosa, D. Jańczewski, E. Kutnyanszky and G. J. Vancso, Langmuir, 2013, 29, 10859–10867 CrossRef CAS PubMed.
- M. Divandari, E. S. Dehghani, N. D. Spencer, S. N. Ramakrishna and E. M. Benetti, Polymer, 2016, 98, 470–480 CrossRef CAS.
- Y. Li, Y. Ko, Y. Lin, D. Kiserow and J. Genzer, Macromolecules, 2017, 50, 8580–8587 CrossRef CAS.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- W. Sheng, B. Li, X. Wang, B. Dai, B. Yu, X. Jia and F. Zhou, Chem. Sci., 2015, 6, 2068–2073 RSC.
- S. Wang, J. Song, Y. Li, X. Zhao, L. Chen, G. Li, L. Wang, Z. Jia and X. Ge, React. Funct. Polym., 2019, 140, 48–55 CrossRef CAS.
- N. Li, T. Li, X.-Y. Qiao, R. Li, Y. Yao and Y.-K. Gong, ACS Appl. Mater. Interfaces, 2020, 12, 12337–12344 CrossRef CAS PubMed.
- J. Kuang and P. B. Messersmith, Langmuir, 2012, 28, 7258–7266 CrossRef CAS PubMed.
- H. Watanabe, A. Fujimoto, R. Yamamoto, J. Nishida, M. Kobayashi and A. Takahara, ACS Appl. Mater. Interfaces, 2014, 6, 3648–3653 CrossRef CAS PubMed.
- X. Fan, L. Lin and P. B. Messersmith, Biomacromolecules, 2006, 7, 2443–2448 CrossRef CAS PubMed.
- D. Pranantyo, L. Q. Xu, K.-G. Neoh, E.-T. Kang, Y. X. Ng and S. L.-M. Teo, Biomacromolecules, 2015, 16, 723–732 CrossRef CAS PubMed.
- G. Bai, S. Ma, R. Qie, Z. Liu, Y. Shi, C. Li, R. Wang, X. Guo, F. Zhou and X. Jia, Macromol. Rapid Commun., 2016, 37, 1256–1261 CrossRef CAS PubMed.
- W. Jeong, H. Kang, E. Kim, J. Jeong and D. Hong, Langmuir, 2019, 35, 13268–13274 CrossRef CAS PubMed.
- G. Kafkopoulos, C. J. Padberg, J. Duvigneau and G. J. Vancso, ACS Appl. Mater. Interfaces, 2021, 13, 19244–19253 CrossRef CAS PubMed.
- Q. Wei, X. Pei, J. Hao, M. Cai, F. Zhou and W. Liu, Adv. Mater. Interfaces, 2014, 1, 1400035 CrossRef.
- M. Kohri, Y. Shinoda, H. Kohma, Y. Nannichi, M. Yamauchi, S. Yagai, T. Kojima, T. Taniguchi and K. Kishikawa, Macromol. Rapid Commun., 2013, 34, 1220–1224 CrossRef CAS PubMed.
- D. Hafner, L. Ziegler, M. Ichwan, T. Zhang, M. Schneider, M. Schiffmann, C. Thomas, K. Hinrichs, R. Jordan and I. Amin, Adv. Mater., 2016, 28, 1489–1494 CrossRef CAS PubMed.
- N. I. Abu-Lail, M. Kaholek, B. LaMattina, R. L. Clark and S. Zauscher, Sens. Actuators, B, 2006, 114, 371–378 CrossRef CAS.
- S. P. Wetzler, K. A. Miller, L. Kisley, A. L. D. Stanton, P. V. Braun and R. C. Bailey, Langmuir, 2020, 36, 10351–10360 CrossRef CAS PubMed.
- S. Edmondson and S. P. Armes, Polym. Int., 2009, 58, 307–316 CrossRef CAS.
- T. Stöhr and J. Rühe, Macromolecules, 2000, 33, 4501–4511 CrossRef.
- M. Chen, W. H. Briscoe, S. P. Armes and J. Klein, Science, 2009, 323, 1698–1701 CrossRef CAS PubMed.
- E. Roeven, A. R. Kuzmyn, L. Scheres, J. Baggerman, M. M. J. Smulders and H. Zuilhof, Langmuir, 2020, 36, 10187–10199 CrossRef CAS PubMed.
- A. E. Rodda, F. Ercole, D. R. Nisbet, J. S. Forsythe and L. Meagher, Macromol. Biosci., 2015, 15, 799–811 CrossRef CAS PubMed.
- A. Hucknall, A. J. Simnick, R. T. Hill, A. Chilkoti, A. Garcia, M. S. Johannes, R. L. Clark, S. Zauscher and B. D. Ratner, Biointerphases, 2009, 4, FA50–FA57 CrossRef CAS PubMed.
- A. E. Rodda, F. Ercole, V. Glattauer, D. R. Nisbet, K. E. Healy, A. P. Dove, L. Meagher and J. S. Forsythe, J. Mater. Chem. B, 2016, 4, 7314–7322 RSC.
- M. Sun, H. Qiu, C. Su, X. Shi, Z. Wang, Y. Ye and Y. Zhu, ACS Appl. Bio Mater., 2019, 2, 3983–3991 CrossRef CAS PubMed.
- D. Li, L. Wu, F. Qu, M. C. Ribadeneyra, G. Tu and J. Gautrot, Chem. Commun., 2019, 55, 14166–14169 RSC.
- Y. Yu, Y. Yao, S. van Lin and S. de Beer, Eur. Polym. J., 2019, 112, 222–227 CrossRef CAS.
- Y. Yu, Switchable adhesion and friction by stimulus responsive polymer brushes, University of Twente, 2017 Search PubMed.
- H. Kang, S. An, W. J. Lee, G. R. Kang, S. Kim, S.-M. Hur, K. Paeng and M. Kim, RSC Adv., 2018, 8, 24166–24174 RSC.
- W.-L. Chen, M. Menzel, O. Prucker, E. Wang, C. K. Ober and J. Rühe, Macromolecules, 2017, 50, 4715–4724 CrossRef CAS.
- M. Hoffmann, M. Lang and J.-U. Sommer, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 83, 21803 CrossRef PubMed.
- J. Wu, D. Zhang, L. Zhang, B. Wu, S. Xiao, F. Chen, P. Fan, M. Zhong, J. Tan, Y. Chu and J. Yang, Prog. Org. Coat., 2019, 134, 153–161 CrossRef CAS.
- H. Nakano, Y. Noguchi, S. Kakinoki, M. Yamakawa, I. Osaka and Y. Iwasaki, ACS Appl. Bio Mater., 2020, 1071–1078 CrossRef CAS PubMed.
- A. J. Wójcik, K. Wolski and S. Zapotoczny, Eur. Polym. J., 2021, 155, 110577 CrossRef.
- M. Słowikowska, K. Wolski, A. J. Wójcik, D. Wesner, H. Schönherr and S. Zapotoczny, Polym. Chem., 2020, 11, 7050–7062 RSC.
- Z. Wang and H. Zuilhof, Langmuir, 2016, 32, 6310–6318 CrossRef CAS PubMed.
- H. Kuroki, I. Tokarev, D. Nykypanchuk, E. Zhulina and S. Minko, Adv. Funct. Mater., 2013, 23, 4593–4600 CrossRef CAS.
- W. Yan, M. Fantin, S. Ramakrishna, N. D. Spencer, K. Matyjaszewski and E. M. Benetti, ACS Appl. Mater. Interfaces, 2019, 11, 27470–27477 CrossRef CAS PubMed.
- W. Li, W. Sheng, B. Li and R. Jordan, Angew. Chem., Int. Ed., 2021, 60, 13621–13625 CrossRef CAS PubMed.
- S. E. Seo, E. H. Discekici, Y. Zhang, C. M. Bates and C. J. Hawker, J. Polym. Sci., 2020, 58, 70–76 CrossRef CAS.
- T. Sato, G. J. Dunderdale, C. Urata and A. Hozumi, Macromolecules, 2018, 51, 10065–10073 CrossRef CAS.
- J. Hou, R. Chen, J. Liu, H. Wang, Q. Shi, Z. Xin, S.-C. Wong and J. Yin, J. Mater. Chem. B, 2018, 6, 4792–4798 RSC.
- J. Madsen, R. E. Ducker, O. Al Jaf, M. L. Cartron, A. M. Alswieleh, C. H. Smith, C. N. Hunter, S. P. Armes and G. J. Leggett, Chem. Sci., 2018, 9, 2238–2251 RSC.
- M. Kopeć, S. Tas, M. Cirelli, R. van der Pol, I. de Vries, G. J. Vancso and S. de Beer, ACS Appl. Polym. Mater., 2019, 1, 136–140 CrossRef PubMed.
- H. Zhao, J. Sha, X. Wang, Y. Jiang, T. Chen, T. Wu, X. Chen, H. Ji, Y. Gao, L. Xie and Y. Ma, Lab Chip, 2019, 19, 2651–2662 RSC.
- C. Carbonell, D. Valles, A. M. Wong, A. S. Carlini, M. A. Touve, J. Korpanty, N. C. Gianneschi and A. B. Braunschweig, Nat. Commun., 2020, 11, 1244 CrossRef CAS PubMed.
- M. Fromel, R. L. Crisci, C. S. Sankhe, D. Reifsnyder Hickey, T. B. Tighe, E. W. Gomez and C. W. Pester, Eur. Polym. J., 2021, 158, 110652 CrossRef CAS.
- J. E. Gautrot, B. Trappmann, F. Oceguera-Yanez, J. Connelly, X. He, F. M. Watt and W. T. S. Huck, Biomaterials, 2010, 31, 5030–5041 CrossRef CAS PubMed.
Footnote |
| † Joint first authorship. These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |