DOI:
10.1039/D1TB02183D
(Paper)
J. Mater. Chem. B, 2022,
10, 2551-2560
Preparation of a thermo-responsive drug carrier consisting of a biocompatible triblock copolymer and fullerene†
Received
6th October 2021
, Accepted 19th November 2021
First published on 22nd November 2021
Abstract
A triblock copolymer (PEG-b-PUEM-b-PMPC; EUM) comprising poly(ethylene glycol) (PEG), thermo-responsive poly(2-ureidoethyl methacrylate) (PUEM), and poly(2-(methacryloyloxy)ethyl phosphorylcholine) (PMPC) blocks was synthesized via controlled radical polymerization. PEG and PMPC blocks exhibit hydrophilicity and biocompatibility. The PUEM block exhibits an upper critical solution temperature (UCST). PMPC can dissolve hydrophobic fullerenes in water to form a complex by grinding PMPC and fullerene powders. Fullerene-C70 (C70) and EUM were ground in a mortar and phosphate-buffered saline (PBS) was added to synthesize a water-soluble complex (C70/EUM). C70/EUM has a core–shell-corona structure, whose core is a complex of C70 and PMPC, the shell is PUEM, and corona is PEG. The maximum C70 concentration dissolved in PBS was 0.313 g L−1 at an EUM concentration of 2 g L−1. The C70/EUM hydrodynamic radius (Rh) was 34 nm in PBS at 10 °C, which increased due to the PUEM block's UCST phase transition with increasing temperature, and Rh attained a constant value of 38 nm above 36 °C. An anticancer drug, doxorubicin, was encapsulated in the PUEM shell by hydrophobic interactions in C70/EUM at room temperature, which can be released by heating. The generation of singlet oxygen (1O2) from C70/EUM upon visible-light irradiation was confirmed using the singlet oxygen sensor green indicator. Water-soluble C70/EUM may be used as a carrier that releases encapsulated drugs when heated and as a photosensitizer for photodynamic therapy.
Introduction
Fullerenes such as C60 and C70 and their derivatives have been reported to function as enzyme inhibitors,1–3 antivirals,4–6 DNA scission agents,7–9 and radical quenchers.10 Furthermore, fullerenes such as C60 and C70 are expected to be used as photosensitizers for photodynamic therapy (PDT) since they generate singlet oxygen with high efficiency upon visible-light irradiation.11–13 However, their biological applications are limited by their low water solubility.14 Various fullerene derivatives with improved water solubility have been reported.15–17 Fullerenes have been solubilized, forming a complex between fullerenes and host molecules such as cyclodextrins,18,19 calixarenes,20 micelles,21,22 liposomes,23,24 and poly(N-vinylpyrrolidone) (PVP).25–27 Particularly, PVP can solubilize fullerenes with high concentrations to form a water-soluble complex due to charge-transfer interactions. Yamakoshi et al.28 reported that C60 and C70 can be solubilized in water at 0.4 and 0.2 g L−1, respectively, using 50 g L−1 PVP.
Poly(2-(methacryloyloxy)ethyl phosphorylcholine) (PMPC) is water-soluble and biocompatible because the pendant phosphorylcholine group has the same chemical structure as the polar group in phospholipids from biological membranes.29–31 The phosphorylcholine group is an electrically neutral zwitterionic structure. PMPC shows no biological reactions with biomolecules because it does not interact with plasma proteins.32–34 We have reported that a water-soluble complex (C70/PMPC) can be synthesized by mixing C70 and PMPC powders and grinding them.35 0.49 g L−1 C70 can be solubilized in water using 1 g L−1 PMPC.
The photosensitizer should be accumulated around the affected region for effective PDT with low side effects. There are tens to hundreds of nanometer-sized pores that are not observed in normal tissue at the vascular endothelium of cancer tissue. Particles of several nanometers in size are immediately excreted from the kidneys, and those larger than 400 nm are rapidly eliminated by macrophages. However, tens to hundreds of nanometers of particles accumulate in cancer tissue due to enhanced permeation and retention (EPR) effects.36–38 The photosensitizer accumulated around the cancer tissue damages only cancer through the singlet oxygen generated upon light irradiation. The light should be irradiated to the photosensitizer inside the body for PDT. PDT uses ordinary visible or near-infrared light; however, it is difficult for it to penetrate deep inside the body.39 Thus, it should be combined with other PDT treatments for treating deep cancers inside the body. The photosensitizer C70 in a water-soluble C70/PMPC complex can generate singlet oxygen upon visible-light irradiation. Therefore, the controlled release function of anticancer drugs upon heating was added to the C70/PMPC complex. We focused on poly(2-ureidoethyl methacrylate) (PUEM), which exhibits an upper critical solution temperature (UCST).40 The PUEM's pendant ureido groups combine with interpolymer chains below the UCST due to hydrogen bonds, and PUEM cannot dissolve in water. Alternatively, PUEM can dissolve in water due to an increase in molecular motion to break the hydrogen bonds above the UCST. UCST type polymers are applied for controlled release for drug delivery systems.41 Deng et al.42 synthesized a diblock copolymer (PEG-b-P(NAGA-co-AN)) composed of a hydrophilic poly(ethylene glycol) (PEG) and a thermo-responsive random copolymer (P(NAGA-co-AN)) of N-acryloyl glycolinamide and acrylonitrile. P(NAGA-co-AN) exhibits UCST due to hydrogen bonding interactions. PEG-b-P(NAGA-co-AN) forms core–shell micelles to associate with the P(NAGA-co-AN) blocks below the UCST. Hydrophobic drugs can be encapsulated in the core and released by heating.
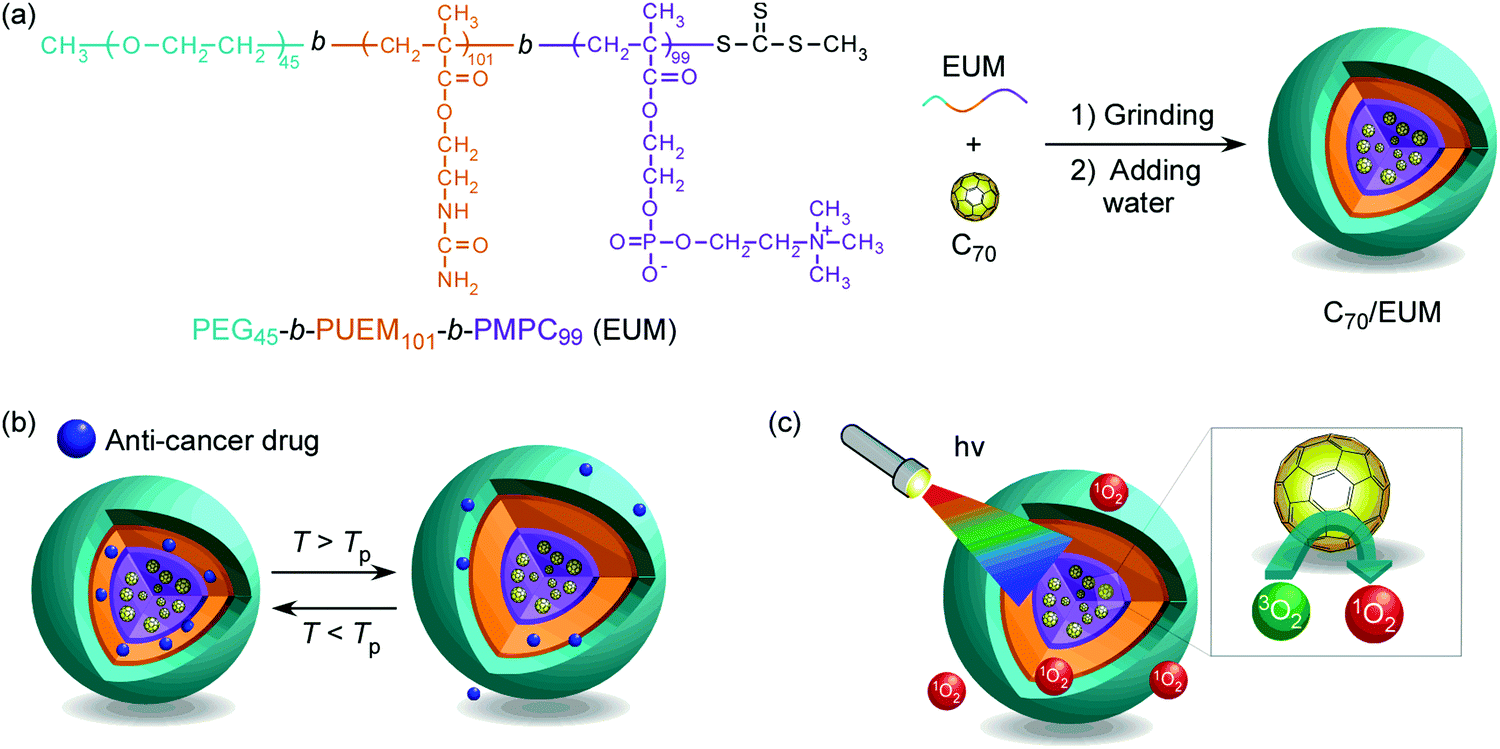
In this study, we have synthesized a triblock copolymer (EUM; PEG45-b-PUEM101-b-PMPC99) consisting of PEG for colloid stabilization in water, PUEM exhibiting UCST, and PMPC solubilizing C70 in water to synthesize a thermoresponsive drug carrier containing C70 photosensitizer. EUM was synthesized via reversible addition–fragmentation chain transfer (RAFT)-controlled radical polymerization using a PEG-macro chain transfer agent (CTA). The water-soluble complex (C70/EUM) was synthesized by crushing powders of EUM and C70 together in a mortar and then adding water. The C70/EUM complex formed a core–shell-corona micelle with a C70/PMPC core, thermoresponsive PUEM shell, and hydrophilic PEG corona (Fig. 1). C70/EUM can encapsulate guest molecules such as hydrophobic drugs in the PUEM shell because the PUEM block becomes hydrophobic below the UCST. Water-soluble PEG exhibits biocompatibility.43–45 C70/EUM covered with biocompatible PEG coronas is expected to accumulate in the affected region due to the EPR effect in the body. Heating above the UCST of the PUEM shell can release the encapsulated hydrophobic drugs in C70/EUM. C70/EUM is thought to have two functions: singlet oxygen generation upon visible-light irradiation and a drug carrier that can release the drug when heated.
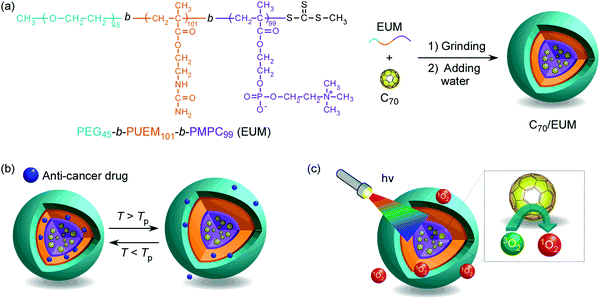 |
| | Fig. 1 (a) The chemical structure of PEG45-b-PUEM101-b-PMPC99 (EUM) and a conceptual illustration of C70/EUM micelle formation in water, (b) its thermo-responsive behavior, and (c) its light-responsive behavior upon visible-light irradiation. | |
Experimental
Materials
2-(Methacryloyloxy)ethyl phosphorylcholine (MPC) was purchased from NOF Corp. (Tokyo, Japan) and used after recrystallization from acetonitrile.31 Poly(2-(methacryloyloxy)ethyl phosphorylcholine) (PMPC, degree of polymerization (DP) = 100, number-average molecular weight (Mn) = 2.98 × 104 g mol−1, molecular weight distribution (Mw/Mn) = 1.16) was synthesized via RAFT polymerization using 4-cyanopentanoic acid dithiobenzoate as a CTA. The CTA, α-methyltrithiocarbonate-S-phenylacetic acid (MTPA), was synthesized according to the literature.46 4,4′-Azobis(4-cyanovaleric acid) dihydrochloride (V-501, 95%), N,N′-dicyclohexylcarbodiimide (DCC, 95%), imidazole (98%), potassium cyanate (KOCN, 95%), fullerene-C70 (C70, 99%), and doxorubicin (DOX) hydrochloride were purchased from Wako Pure Chemical (Osaka, Japan). 4-Dimethylaminopyridine (DMAP, 99%), poly(ethylene glycol) monomethyl ether (PEG45, Mn = 2000, DP = 45), 2-aminoethyl methacrylate hydrochloride (AEM, 90%), phosphate-buffered saline (PBS), and deuterium oxide (D2O) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Singlet oxygen sensor green (SOSG) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Ion-exchanged water was used as pure water.
Synthesis of PEG45-MTPA
PEG45 (22.4 g, 11.2 mmol), MTPA (3.47 g, 13.4 mmol), and DMAP (40.0 mg, 0.327 mmol) were dissolved in dichloromethane (100 mL). A dichloromethane solution (100 mL) of DCC (5.66 g, 27.4 mmol) was added dropwise to the solution for 30 min under an Ar atmosphere. After refluxing at 40 °C for 16 h, purification was performed on a silica-gel column using chloroform as the eluent. The solvent was evaporated. The residue was further dissolved in pure water, and PEG45-MTPA was obtained by a freeze-drying technique (6.48 g, 25.1%). According to 1H NMR, the number-average degree of polymerization (DP(NMR)) and number-average molecular weight (Mn(NMR)) determined from 1H NMR were 45 and 2.24 × 103 g mol−1, respectively (Fig. S1a, ESI†). According to gel-permeation chromatography (GPC) using tetrahydrofuran (THF) as an eluent, the number-average molecular weight Mn(GPC) and molecular weight distribution (Mw/Mn) were 4.40 × 103 g mol−1 and 1.03, respectively (Fig. S2a, ESI†).
Synthesis of PEG45-b-PAEM101-b-PMPC99 (EAM)
The triblock copolymer (EAM; PEG45-b-PAEM101-b-PMPC99) synthesis using PEG45-MTPA as a macro CTA was performed in one pot as follows: AEM (2.09 g, 12.6 mmol), PEG45-MTPA (274 mg, 0.121 mmol), and V-501 (13.9 mg, 0.0496 mmol) were dissolved in imidazole buffer ([imidazole] = 1.00 M, pH = 6.00, 12.1 mL) with a molar ratio of [AEM]/[PEG45-MTPA]/[V-501] = 104/1/0.4. The solution was heated at 70 °C for 6 h under an Ar atmosphere. The conversion (PEG45-b-PAEM101) after polymerization obtained using 1H NMR was 97.5%. DP(NMR) and Mn(NMR) obtained using 1H NMR were 101 and 1.90 × 104 g mol−1, respectively (Fig. S1b, ESI†). The Mn(GPC) and Mw/Mn determined by GPC measurement using acetate buffer as an eluent were 4.69 × 104 g mol−1 and 1.25, respectively (Fig. S2b, ESI†).
MPC (3.59 g, 12.2 mmol) and V-501 (14.1 mg, 0.0503 mmol) were dissolved in imidazole buffer (12.1 mL). The MPC solution was degassed with Ar and was added to the polymerization solution of PEG45-b-PAEM101 with a molar ratio of [MPC]/[PEG45-b-PAEM101]/[V-501] = 100/1/0.416. The solution was heated at 70 °C for 16 h under an Ar atmosphere. The polymer conversion after polymerization (PEG45-b-PAEM101-b-PMPC99) obtained from 1H NMR was 100%. Dialysis was performed overnight against pure water, and PEG45-b-PAEM101-b-PMPC99 (EAM) was obtained via freeze-drying (5.51 g, 92.6%). DP(NMR) and Mn(NMR) obtained from 1H NMR were 99 and 4.80 × 104 g mol−1, respectively (Fig. S1c, ESI†). The Mn(GPC) and Mw/Mn determined using GPC were 5.29 × 104 g mol−1 and 1.61, respectively (Fig. S2b, ESI†).
Synthesis of PEG45-b-PUEM101-b-PMPC99 (EUM)
Ureido groups were introduced in the pendant amino groups in the PAEM block in EAM as follows: EAM (4.00 g, 0.0828 mmol) and KOCN (808 mg, 9.94 mmol) were dissolved in imidazole buffer (16.0 mL) with molar ratio of [EAM]/[KOCN] = 1/120. The solution was heated at 50 °C for 24 h. PEG45-b-PUEM101-b-PMPC99 (EUM) was obtained via freeze-drying (3.30 g, 70.6%) after dialysis against pure water for three days. The conversion from the amine to ureido groups estimated using 1H NMR was 100% (Fig. S3, ESI†). The Mn(GPC) and Mw/Mn obtained using GPC were 8.91 × 104 g mol−1 and 1.64, respectively (Fig. S2b, ESI†).
Preparation of the C70/EUM Complex
C70 (6.0 mg, 7.14 × 10−3 mmol) and EUM (20 mg, 4.14 × 10−4 mmol, Mn(theo) = 4.83 × 104 g mol−1, Mw/Mn = 1.64) powders were ground in a mortar for 30 min. PBS (10 mL) was further added to adjust the polymer concentration (Cp) = 2 g L−1 to synthesize a water-soluble complex (C70/EUM). Undissolved C70 was eliminated by centrifugation at 6000 rpm for 30 min and then filtered through a 0.2 μm pore-sized filter. The C70/PMPC complex, which comprises C70 and PMPC, was synthesized in the same way as the C70/EUM complex for comparison.
Measurements
1H NMR spectra were obtained using a Bruker (Massachusetts, United States) DRX-500 and JEOL (Tokyo, Japan) ECZ400. GPC measurements using THF as the eluent were performed using a Shodex (Tokyo, Japan) DS-4 pump and a Shodex (Tokyo, Japan) RI-101 refractive index (RI) detector equipped with a Shodex (Tokyo, Japan) 10.0 μm bead size GF-7M column (exclusion limit ∼ 107) working at 40 °C, with a flow rate of 1.0 mL min−1. The Mn(GPC) and Mw/Mn were calibrated using standard poly(styrene) samples. GPC measurements using acetate buffer as the eluent were performed using a JASCO (Tokyo, Japan) PU-8020 pump and a JASCO (Tokyo, Japan) RI-2031 Plus RI detector equipped with a Shodex (Tokyo, Japan) 10.0 μm bead size Ohpak SB-804 HQ column (exclusion limit of approximately 107) working at 40 °C, with a flow rate of 0.6 mL min−1. A 0.5 M acetic acid aqueous solution containing 0.3 M sodium sulfate was used as the eluent. The Mn(GPC) and Mw/Mn were calibrated using standard poly(2-vinylpyridine) samples. Ultraviolet-visible (UV-vis) absorption spectra and percent transmittance (%T) at a wavelength of 700 nm were measured at varying temperatures using a JASCO (Tokyo, Japan) V-630BIO UV-vis spectrophotometer. Dynamic light scattering (DLS) measurements were performed with a Malvern (Worcestershire, UK) Zetasizer Nano ZS using a He–Ne laser (4 mW) with a wavelength of 632.8 nm as the light source. The hydrodynamic radius (Rh) and light scattering intensity (LSI) were obtained. Static light scattering (SLS) measurement was performed using an Otsuka Electronics Photal (Osaka, Japan) DLS-7000 HL light scattering spectrometer. A He–Ne laser (632.8 nm, 10.0 mW) was used as the light source. The weight-average molecular weight (Mw(SLS)) and radius of gyration (Rg) were estimated from the Zimm plots. The RI increment (dn/dCp) values at 633 nm were determined using an Otsuka Electronics Photal (Osaka, Japan) DRM-3000 differential refractometer. Transmission electron microscopy (TEM) observations were performed using a JEOL (Tokyo, Japan) JEM-2100 microscope at an acceleration voltage of 160 kV. The TEM sample was prepared by placing one drop of aqueous solution on a copper grid coated with Formvar thin films. Excess water was blotted using filter paper. The samples were stained with sodium phosphotungstate and dried under vacuum conditions. Fluorescence measurement was performed using a Hitachi F-2500 (Tokyo, Japan). Visible light at 590 nm was irradiated using OptCode (Tokyo, Japan) EX-590 with a power consumption of 3 W.
Characterization of C70/EUM
The complex solution was diluted 100-fold and the UV-vis absorption spectrum was measured at 25 °C. The complex solution's absorbance before dilution was calculated by multiplying the absorbance of the complex solution after dilution by 100. The concentration of C70 ([C70]s) was calculated from its molar extinction coefficient in water at 384 nm (40.4 L g−1 cm−1)28 and the absorbance.
Encapsulation and controlled release of DOX
DOX encapsulation in C70/EUM was confirmed via fluorescence measurements. DOX (0.02 g L−1) was dissolved in a C70/EUM (Cp = 2 g L−1, [C70]s = 0.313 g L−1) PBS solution and stirred at 25 °C for 3 days to synthesize DOX-encapsulating C70/EUM (DOX@C70/EUM). Dialysis was performed against a substantial excess of PBS at 25 °C for 3 days with a polycarbonate dialysis membrane (Harvard MFP, Massachusetts, USA) with a pore size of 10 nm to remove the DOX that could not be encapsulated. It was confirmed that the free DOX in the C70/EUM solution was eliminated by comparing the fluorescence intensity inside the dialysis membrane of the DOX@C70/EUM solution with the DOX-only blank solution. The concentration of DOX was calculated from the fluorescence intensity to estimate the amount of DOX in C70/EUM. The release of DOX from DOX@C70/EUM by heating was confirmed by fluorescence. The DOX@C70/EUM ([DOX] = 0.0099 g L−1) and DOX (0.01 g L−1) blank solutions were dialyzed against PBS using a polycarbonate dialysis membrane at 25 °C and 50 °C. The fluorescence intensity of DOX emitted to the outside of the dialysis membrane was used to estimate the release rate.
Generation of singlet oxygen from C70/EUM upon visible-light irradiation
SOSG was used to confirm 1O2 generation. The fluorescence intensity increases when SOSG is oxidized with 1O2 (Scheme S1, ESI†). The C70/EUM PBS solutions at Cp = 2 g L−1 and [C70]s = 0.313 g L−1 were diluted 10-fold with PBS, and SOSG was added to make it 2 μM. The solution was irradiated upon 590 nm visible light using an OptCode (Tokyo, Japan) EX-590 LED light, and the fluorescence spectra of SOSG were obtained. A quartz cell with a 1 cm optical path length was used. The SOSG fluorescence was measured at an excitation wavelength of 420 nm with excitation and emission slight widths at 20 nm.
Results and discussion
Preparation of PEG45-b-PAEM101-b-PMPC99 (EUM)
A macro CTA, PEG45-MTPA, was synthesized via condensation reaction of mono-methoxy PEG45 having an OH group at the chain end with MTPA having a carboxylic acid (Scheme S2, ESI†). DP(NMR) of PEG45-MTPA estimated from the integrated intensity ratio of the peaks attributed to PEG methylene protons at 3.4 ppm and the MTPA methyl proton at 2.7 ppm was 45 (Fig. S1a, ESI†). AEM and MPC were continuously polymerized in one pot via RAFT polymerization using PEG45-MTPA. The PAEM block's DP(NMR) estimated from the integrated intensity ratio of the peaks attributed to PEG methylene proton at 3.6 ppm to the pendant methylene protons in the PAEM block at 3.3 ppm was 101 (Fig. S1b, ESI†). The PMPC block's DP(NMR) estimated from the integrated intensity ratio of the peaks of the pendant methylene protons in the PAEM block at 3.3 ppm to the pendant methyl protons in the PMPC block at 3.1 ppm was 99 (Fig. S1c, ESI†). GPC measurements were performed for PEG45-MTPA using THF as the eluent (Fig. S2a, ESI†), and for PEG45-b-PAEM101, EAM, and EUM using aqueous acetic acid solution (Fig. S2b, ESI†). Mw/Mn of PEG45-MTPA, PEG45-b-PAEM101, EAM, and EUM were 1.03, 1.25, 1.61, and 1.64, respectively. Theoretical DP (DP(theo)) and number-average molecular weight (Mn(theo)) values were calculated from the following equations| | 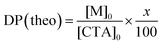 | (1) |
| | | Mn(theo) = DP(theo) × Mm + MCTA | (2) |
where x is the monomer's percent conversion, [M]0 and [CTA]0 are the initial concentrations of the monomer and CTA, respectively, and Mm and MCTA are the molecular weights of the monomer and CTA, respectively. Table 1 summarizes the results.
Table 1 Molecular characteristics of the synthesized polymers
| Polymers |
M
n (theoretical) × 10−4 (g mol−1) |
DP (theoretical) |
M
n (NMR) × 10−4 (g mol−1) |
DP (NMR) |
M
n (GPC) × 10−4 (g mol−1) |
M
w/Mn |
|
DP of PEG.
DP of PAEM.
DP of PMPC.
DP of PUEM.
The signals were overlapped.
|
| PEG45-MTPA |
— |
— |
0.224 |
45a |
0.44 |
1.03 |
| PEG45-b-PAEM101 |
1.90 |
101b |
1.90 |
101b |
4.69 |
1.25 |
| PEG45-b-PAEM101-b-PMPC99 |
4.82 |
100c |
4.80 |
99c |
5.29 |
1.61 |
| EUM |
5.24 |
101d |
—e |
—e |
8.91 |
1.64 |
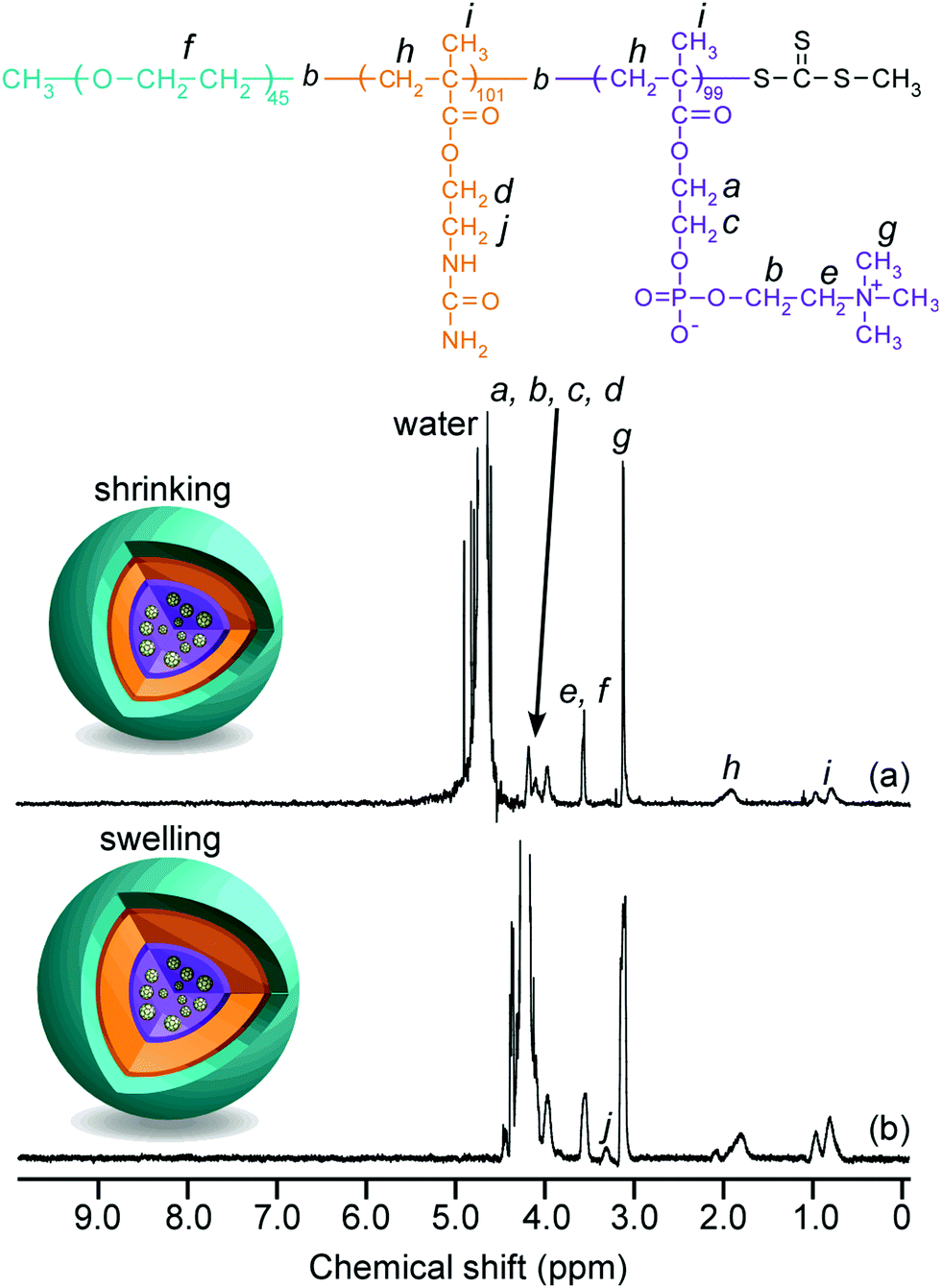
The 1H NMR spectra for EAM and EUM were measured in D2O at 25 °C and 80 °C (Fig. S3, ESI†). The peak of pendant methylene protons adjacent to the primary amine in the PAEM block at 3.3 ppm (g) disappeared in the 1H NMR for EUM. This suggests that the pendant primary amine groups in the PAEM block were replaced with 100% ureido groups in the PUEM block. Although the peak intensity of pendant methylene protons adjacent to the ureido in the PUEM block in EUM at 3.4 ppm (i) was barely observed at 25 °C (Fig. S3b, ESI†), the peak intensity at 3.4 ppm (i) increased at 80 °C (Fig. S3c, ESI†). The NMR peak becomes broad when the proton mobility decreases due to a decrease in the spin–spin relaxation time.47 Therefore, this result suggests that the PUEM block's motion in EUM was restricted in D2O at 25 °C.
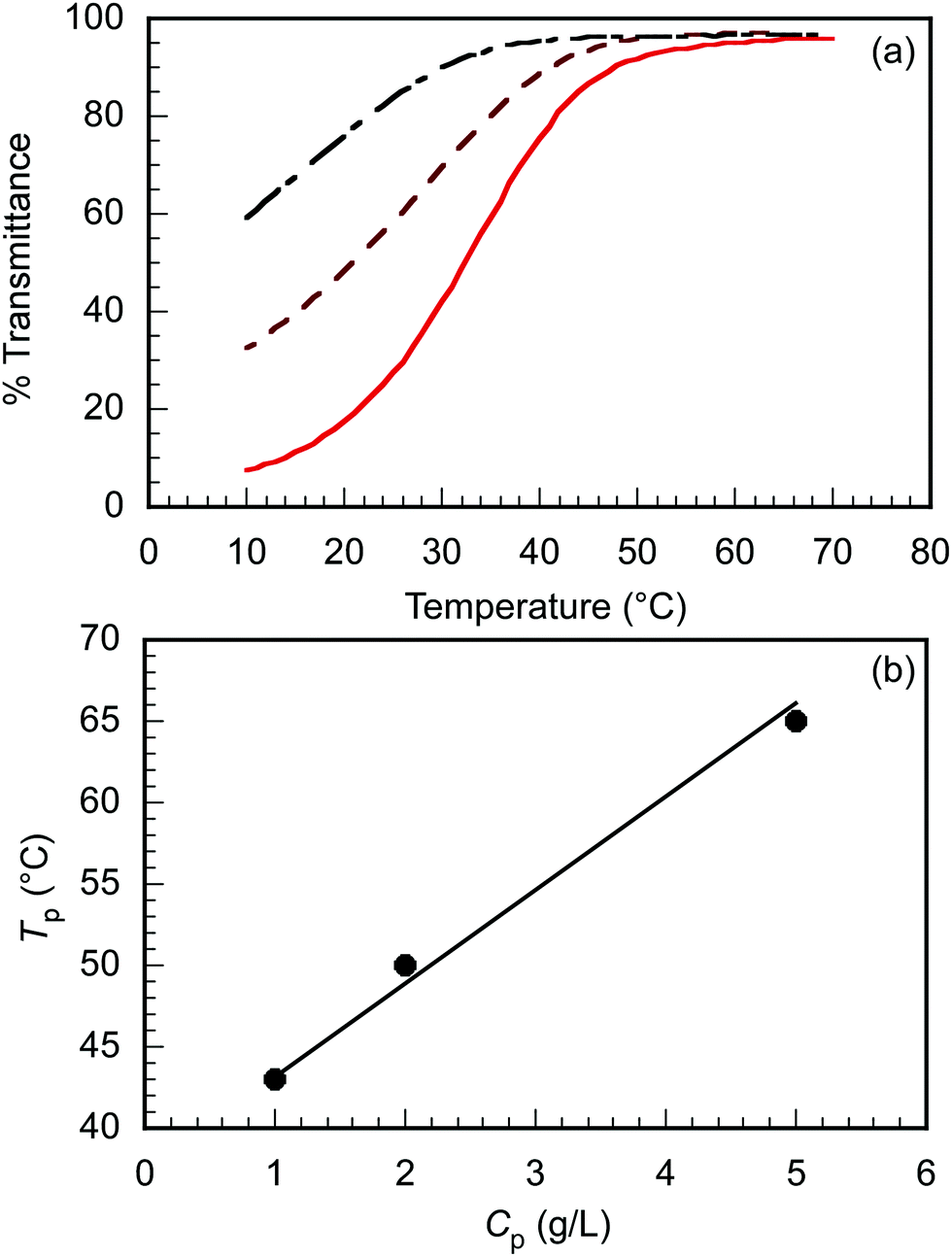
The triblock copolymer, EUM, which contains the thermo-responsive PUEM block bearing pendant ureido groups exhibits UCST behavior in water. The polymer concentration (Cp) dependence on the thermo-responsive behavior of EUM was investigated. EUM was dissolved in PBS at various Cp values. The percent transmittance (%T) at 700 nm was measured as a function of temperature at the cooling process (Fig. 2a). The phase transition temperatures (Tp) at Cp = 1.0, 2.0, and 5.0 g L−1 were 43 °C, 50 °C, and 65 °C, respectively. Tp increased linearly with Cp (Fig. 2b). This phenomenon suggests that the frequency of collisions between polymer chains increased with increasing Cp to easily form hydrogen bonds between the pendant ureido groups in the PUEM blocks.
 |
| | Fig. 2 (a) Percent transmittance (%T) of EUM in PBS at various Cp = 1.0 ( ), 2.0 ( ), 2.0 ( ), and 5.0 g L−1 ( ), and 5.0 g L−1 ( ) as a function of temperature with the cooling process, and (b) phase transition temperature (Tp) of EUM in PBS as a function of Cp. ) as a function of temperature with the cooling process, and (b) phase transition temperature (Tp) of EUM in PBS as a function of Cp. | |
Preparation of C70/EUM
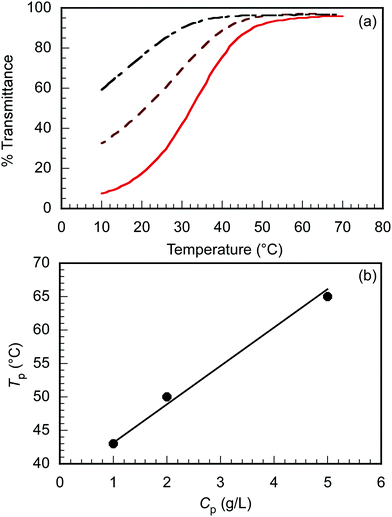
The maximum C70 concentration ([C70]s) to be solubilized by the formation of a water-soluble C70/EUM complex was determined. EUM and various amounts of C70 ([C70]f) powders were ground and mixed in a mortar for 30 min. PBS was added to the mixture to synthesize C70/EUM aqueous solution with Cp = 2.0 g L−1. A water-soluble C70/PMPC complex was prepared using C70 and PMPC powders by the same method for comparison. From the UV-vis absorption spectra of C70/EUM (Fig. 3) and C70/PMPC (Fig. S4, ESI†), maximum absorptions are observed at 250 and 384 nm for C70, suggesting that C70 could be solubilized in PBS. The absorbance of C70 increased for C70/EUM and C70/PMPC with an increase in [C70]f. The [C70]s values were calculated using the molar extinction coefficient of C70 in water at 384 nm (40.4 L g−1 cm−1)28 and the absorbance of the complex aqueous solution. The maximum [C70]s values using EUM and PMPC were 0.31 g L−1 (Fig. 3b) and 0.83 g L−1 (Fig. S4b, ESI†), respectively. The maximum EUM [C70]s was lower than that of PMPC at Cp = 2 g L−1 because the EUM's PEG and PUEM blocks cannot contribute to solubilizing C70. C70 powder was mixed with PEG45 or PUEM100 powders at [C70]f = 0.10 g L−1; however, C70 could not be solubilized in PBS (Fig. S5, ESI†). This observation suggests that the C70/EUM core was formed from the C70 and PMPC block. The maximum [C70]s (0.313 g L−1) obtained using EUM with Cp = 2 g L−1 was higher than that ([C70]s = 0.2 g L−1) obtained using PVP (MW = 4.00 × 104 g mol−1) with Cp = 50 g L−1.28 All subsequent experiments were conducted using C70/EUM with [C70]s = 0.313 g L−1.
 |
| | Fig. 3 (a) UV-vis absorption spectra of C70/EUM at Cp = 2 g L−1 and (b) the amount of solubilized C70 in PBS ([C70]s) as a function of the amount of C70 ([C70]f) used. | |
Characterization of C70/EUM
1H NMR spectra for C70/EUM were obtained in D2O at 25 °C and 80 °C (Fig. 4). Although the NMR signal of the pendant methylene protons adjacent to the ureido group in the PUEM block was observed at 3.4 ppm at 80 °C, the signal disappeared at 25 °C. This observation suggests that 80 °C was higher than the phase transition temperature (Tp) of the PUEM block in C70/EUM. Alternatively, the NMR signal for the PUEM shells in C70/EUM became broad at 25 °C due to the decreasing mobility caused by the hydrophobic interactions of the PUEM shells below Tp.
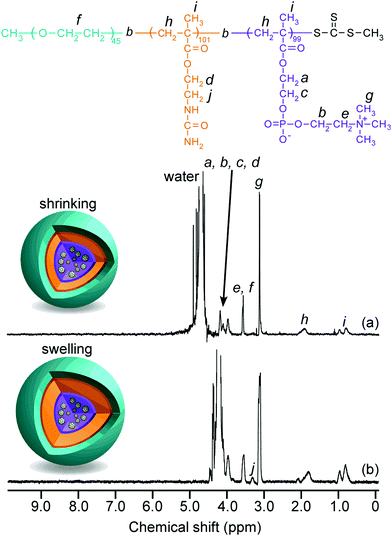 |
| | Fig. 4
1H NMR spectra for C70/EUM in D2O at (a) 25 °C and (b) at 80 °C. | |
SLS measurements were performed in PBS to characterize the association state of the complex (Fig. S6 and Table 2, ESI†). The theoretical expanded polymer chain lengths calculated from the DP values of EUM (245) and PMPC (100) were 61.3 and 25.0 nm, respectively, based on the C–C bond chain length. The Rg values of C70/EUM and C70/PMPC were 58.4 and 40.0 nm, respectively. The Rg value of C70/EUM was similar to the theoretical expanded polymer chain length of EUM. This result suggests that C70/EUM formed the core–shell-corona micelle structure without intermicellar aggregation. The Rg value of C70/PMPC was larger than the theoretical chain length of 25.0 nm. This suggests that C70/PMPC cannot form a clear core–shell structure. The aggregate's shape is a rigid sphere when the Rg/Rh value is 0.8, a random coil when it is 1, and a rod when it is larger than 2.48 The Rg/Rh values of C70/EUM and C70/PMPC were 1.60 and 0.885, respectively. Therefore, C70/EUM's shape was almost spherical, and C70/PMPC formed aggregates similar to a rigid sphere. The inside of the C70/PMPC complex was tightly packed because it does not have the shell and corona layers. Therefore, the Rg/Rh value of C70/PMPC was similar to that of a rigid sphere.
Table 2 DLS and SLS data for the C70/EUM and C70/PMPC complexes in PBS at 25 °C
| Samples |
r
|
M
w(SLS)b × 10−6 (g mol−1) |
R
g
(nm) |
R
h
(nm) |
R
g/Rh |
d
(g cm−3) |
N
agg
|
N
C70
|
dn/dCph (mL g−1) |
|
Molar ratio of C70 and polymer ([C70]/[polymer]).
Weight-average molecular weight estimated from SLS.
Radius of gyration estimated from SLS.
Hydrodynamic radius estimated from DLS.
Density of complex calculated from eqn (4).
Aggregation number of the polymer chains calculated from eqn (3).
Number of C70 in one complex calculated from r and Nagg.
Refractive index increment.
|
| C70/EUM |
9.8 |
6.67 |
58.4 |
36.3 |
1.60 |
0.0133 |
70.9 |
691 |
0.068 |
| C70/PMPC |
15 |
2.32 |
40.0 |
45.2 |
0.885 |
0.0144 |
46.7 |
686 |
0.780 |
The aggregation number (Nagg), which is the number of polymer chains forming one aggregate, can be calculated from the following equation
| | 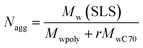 | (3) |
where
Mwpoly and
MwC70 are the weight-average molecular weights of the polymer and C
70, respectively, and
r is the molar ratio of C
70 to the polymer.
Nagg of C
70/EUM was calculated to be 70.9. The number of C
70 (
NC70) contained in one C
70/EUM complex was calculated to be 691 from
r and
Nagg. A single EUM polymer chain solubilized about 10 C
70 molecules based on the
Nagg and
NC70 of C
70/EUM. Similarly, a single PMPC chain can solubilize 15 C
70 molecules in the case of C
70/PMPC. PMPC can solubilize a greater amount of C
70 than EUM in water because the PEG and PUEM blocks in EUM cannot contribute to C
70 solubilization. The density (
d) of the complex was calculated from the following formula
| | 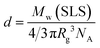 | (4) |
where
NA represents Avogadro's number. The
d values of C
70/EUM and C
70/PMPC were 0.0133 and 0.0144 g/cm
3, respectively. The
d value of C
70/EUM was lower than that of C
70/PMPC, because C
70/EUM contains the PUEM shell and PEG corona layers, which are well-hydrated.
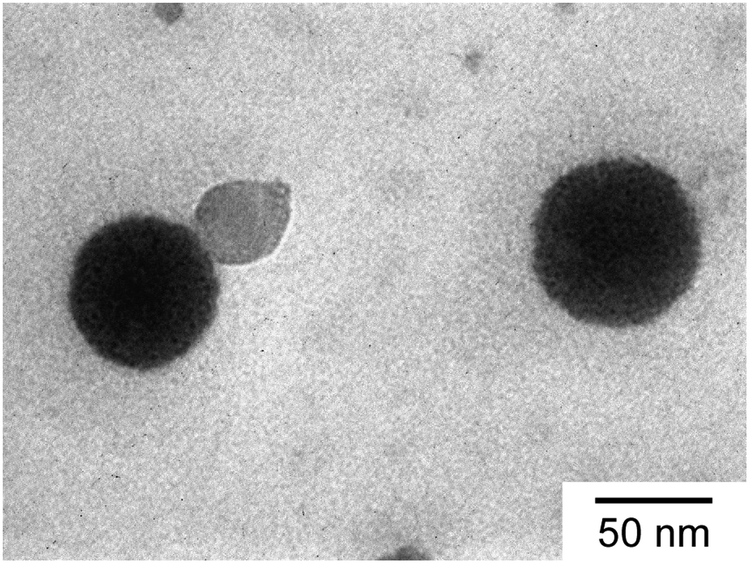
TEM observation confirmed the spherical shape of the C70/EUM complex (Fig. 5). The average RTEM obtained from TEM was 33.5 nm, which was lower than the Rh = 36.3 nm calculated by DLS at 25 °C due to shrinkage of the TEM sample during the drying process.
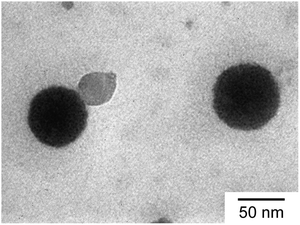 |
| | Fig. 5 TEM image of C70/EUM. | |
Thermo responsive behavior of C70/EUM
R
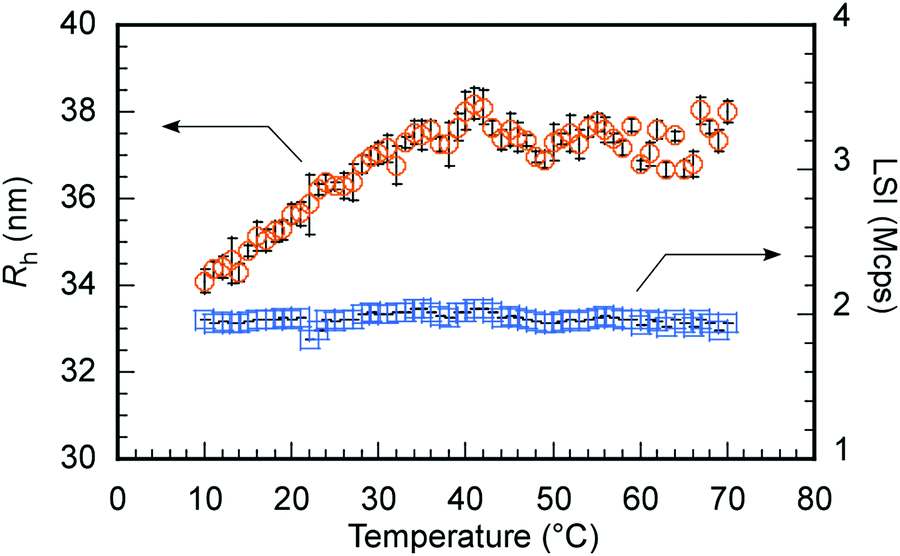
h and LSI were investigated as a function of temperature to confirm the swelling of C70/EUM in PBS due to the PUEM shell's UCST behavior upon heating (Fig. 6). Tp was defined as the temperature at which Rh became constant during a heating process. Below 36 °C, Rh increased with temperature. However, Rh became constant above 36 °C at about 38 nm. Therefore, C70/EUM's Tp was determined to be 36 °C. The Tp of EUM without C70 was 50 °C (Fig. 2); however, the Tp of C70/EUM decreased to 36 °C. The PUEM block in C70/EUM is anchored to the hydrophobic core containing C70. The Tp of C70/EUM was decreased because the PUEM polymer chain was affected by the core's hydrophobicity. LSI is sensitive to the formation of large aggregates due to intermicellar aggregations. No intermicellar aggregation of C70/EUM occurred because the LSI was constant regardless of temperature. The Rh values were repeatedly measured at 25 °C and 70 °C to confirm the reversibility of swelling and shrinking of C70/EUM by temperature (Fig. S7, ESI†). The Rh values at 25 °C and 70 °C were ca. 36.5 and ca. 38.2 nm, respectively, for five temperature cycles. Thus, the C70/EUM complex phase transition is reversible.
 |
| | Fig. 6 Hydrodynamic radius (Rh,  ) and light scattering intensity (LSI, ) and light scattering intensity (LSI,  ) of C70/EUM at Cp = 2 g L−1 and [C70]s = 0.313 g L−1 in PBS as a function of temperature. ) of C70/EUM at Cp = 2 g L−1 and [C70]s = 0.313 g L−1 in PBS as a function of temperature. | |
Generation of singlet oxygen from C70/EUM upon visible-light irradiation
The C70/EUM solution with Cp = 2 g L−1 and [C70]s = 0.313 g L−1 was diluted 10-fold with PBS, and SOSG was added to make it 2 μM. The solution was irradiated with light at 590 nm and SOSG's fluorescence spectra were measured for the irradiation time, t (Fig. S8, ESI†). The ratios (I/I0) of SOSG's maximum fluorescence intensity (I) at t to that before irradiation (I0) were plotted against t (Fig. 7). SOSG is oxidized by 1O2 and the fluorescence intensity at 500–550 nm increased. The I/I0 ratios were almost constant regardless of light irradiation when the light was irradiated towards SOSG PBS solution and SOSG PBS solution with EUM. Alternatively, when the SOSG PBS solution with the C70/EUM complex containing C70 was irradiated, I/I0 of SOSG increased with the irradiation time. This observation indicates that C70 in C70/EUM's core can generate 1O2 under light irradiation. There is almost no error for the samples that cannot generate 1O2. The plots of C70/EUM contains up to 1.5% error.
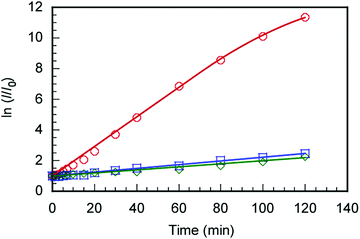 |
| | Fig. 7 Changes in SOSG fluorescence intensity (2 μM) with C70/EUM at [C70]s = 0.03 g L−1 ( ), without C70 ( ), without C70 ( ), and without EUM and C70 ( ), and without EUM and C70 ( ) as a function of irradiation time at 25 °C; I and I0 were the fluorescence intensities at the maximum wavelength (525 nm) after and before 590 nm light irradiation. ) as a function of irradiation time at 25 °C; I and I0 were the fluorescence intensities at the maximum wavelength (525 nm) after and before 590 nm light irradiation. | |
Encapsulation and controlled release of doxorubicin (DOX)
The following method was used to encapsulate a guest molecule, DOX, in C70/EUM: C70/EUM and DOX were stirred in PBS for 3 days at room temperature, and any DOX that could not be encapsulated was removed by dialysis. The removal of free DOX from DOX@C70/EUM solution was confirmed by comparing the fluorescence intensity inside the dialysis membrane of the DOX@C70/EUM solution with that of the DOX-only solution (Fig. S9, ESI†). The fluorescence intensity of DOX@C70/EUM was almost constant after 24 h dialysis, whereas that of the DOX-only solution decreased. These results show that DOX can be encapsulated in C70/EUM. From eqn (5) and (6), DOX encapsulation efficiency (EE) and DOX encapsulation content (EC) were estimated after 72 h dialysis. EE is the ratio of the encapsulated DOX concentration ([DOX]) to the used DOX concentration ([DOX]0). [DOX] can be estimated from the fluorescence intensity. EC is the ratio of [DOX] to Cp. EE(%) and EC(%) were calculated using the following equations:| | 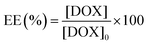 | (5) |
| | 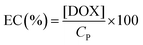 | (6) |
EE(%) and EC(%) for DOX@C70/EUM were 49.5% and 0.495%, respectively.
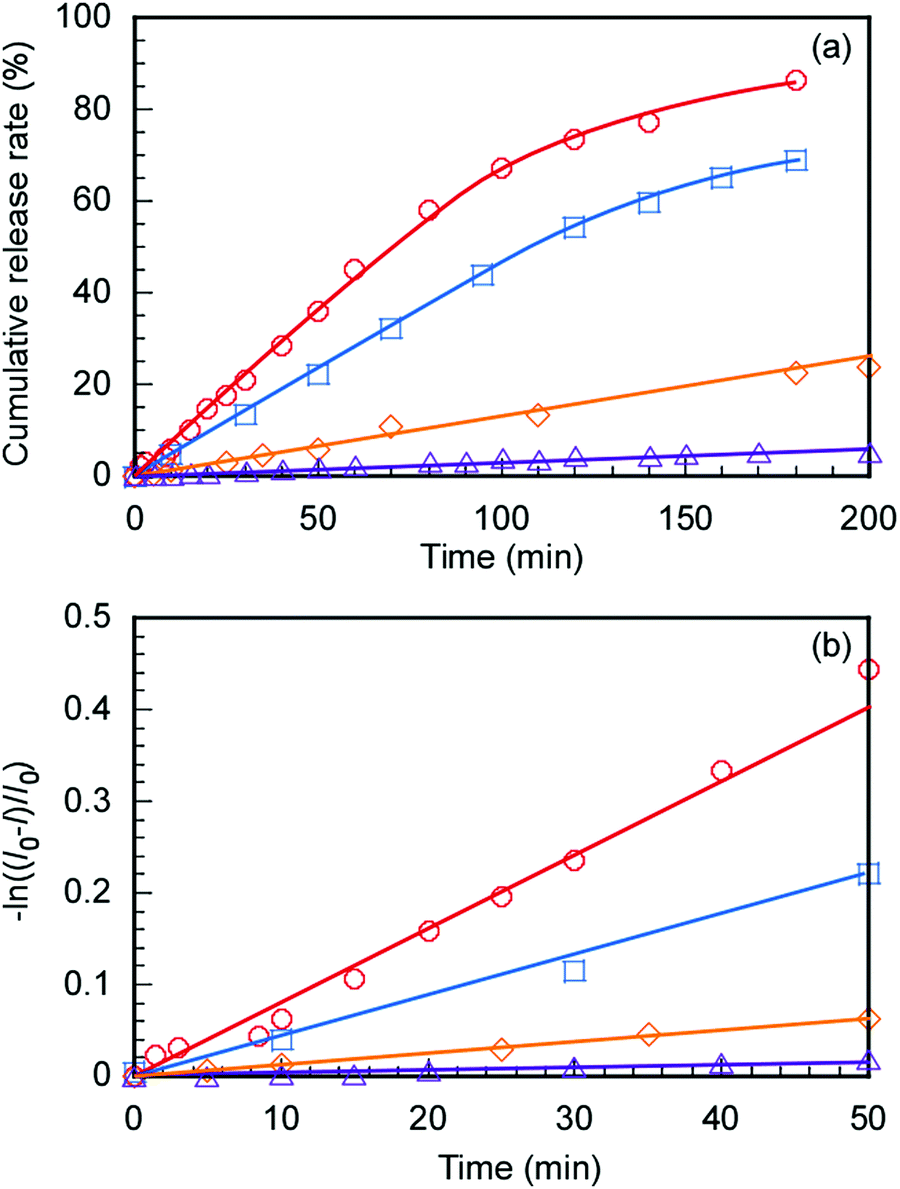
The controlled release of DOX from DOX@C70/EUM with increasing temperature was evaluated. The DOX@C70/EUM solutions were dialyzed against PBS at 25 °C and 50 °C. The dialysis membrane with a 10 nm pore size was used. DOX can permeate the membrane, whereas large-sized C70/EUM cannot. DOX fluorescence intensity released to the outside of the membrane was investigated with respect to the dialysis time (Fig. 8a). The DOX-only solution was studied using the same method for comparison. DOX release amounts at 50 °C and 25 °C after 200 min were 90% and 70%, respectively, for the DOX-only solution. The significant increase at 50 °C was attributed to an increase in DOX thermal motion due to high temperature. The amounts of DOX released from DOX@C70/EUM at 50 °C and 25 °C after 200 min were 20% and 5.0%, respectively. The plots contain up to 1.2% error. At 50 °C the release amount of DOX@C70/EUM solution was lower than that of the DOX-only solution. This suggests that not only is DOX encapsulated in the PUEM shell in C70/EUM due to hydrophobic interactions but it also interacts with the hydrophobic core containing hydrophobic C70. Assuming that DOX release follows the first-order mechanism,49,50 the release rate constant (k) can be calculated from the following equation:
where
I0 indicates the initial fluorescence intensity of DOX before dialysis,
I indicates the fluorescence intensity outside of the membrane at
t, and
t is the dialysis time. The slope of the plots in
Fig. 8b shows that
k can be calculated. The
k values for the comparative DOX-only solution at 25 °C and 50 °C were 0.264 s
−1 and 0.481 s
−1, respectively. Therefore, the release rate increased by 1.82 times at 50 °C compared to 25 °C due to the increase in DOX thermal motion. The
k values for the DOX@C
70/EUM solution at 25 °C and 50 °C were 0.0180 s
−1 and 0.0792 s
−1, respectively. The rate of release rose by 4.40 times when the temperature was raised to 50 °C from 25 °C. The increase in
k due to heating in the presence of C
70/EUM was larger than in the absence of C
70/EUM. This suggests that DOX release could be controlled by increasing the temperature rather than just a simple change in thermal motion. The affinity between DOX and the PUEM block of C
70/EUM was changed drastically at 25 °C and 50 °C due to the UCST behavior of the PUEM block. At 50 °C the affinity of DOX with the PUEM block was low. Therefore, DOX can be released from C
70/EUM at 50 °C.
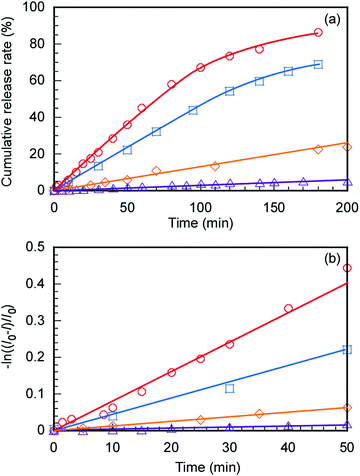 |
| | Fig. 8 (a) DOX release from DOX@C70/EUM at 25 °C ( ) and 50 °C ( ) and 50 °C ( ) and a blank solution without C70/EUM at 25 °C ( ) and a blank solution without C70/EUM at 25 °C ( ) and 50 °C ( ) and 50 °C ( ) in PBS. (b) −ln((I0 − I)/I0) was plotted against dialysis time; I is the fluorescence intensity and I0 is the initial intensity. ) in PBS. (b) −ln((I0 − I)/I0) was plotted against dialysis time; I is the fluorescence intensity and I0 is the initial intensity. | |
The effect of DOX encapsulation and release on C70/EUM size was evaluated using DLS (Fig. S10, ESI†). The Rh value of C70/EUM without DOX was 36.3 nm. The Rh value of DOX@C70/EUM was 39.8 nm. The Rh value of C70/EUM decreased to 32.7 nm after DOX release. The LSIs of DOX@C70/EUM before and after the release of DOX were 2160 and 1830 kcps, respectively. From these results, the influence of DOX encapsulation and release on the size and structure of the complex was minimal.
Conclusions
A thermo-responsive triblock copolymer, EUM, was synthesized via controlled radical polymerization. EUM can dissolve C70 in PBS to form a C70/EUM complex. The maximum solubilized C70 concentration in PBS by EUM was 0.313 g L−1, which was superior to a previously reported solubilization method using PVP. The Tp value of C70/EUM in PBS was 36 °C according to DLS measurements. C70/EUM can generate 1O2 by visible-light irradiation at 590 nm in PBS. C70/EUM can encapsulate the anticancer agent, DOX, at 25 °C by hydrophobic interactions between the PUEM block in EUM and DOX. The encapsulated DOX can be released when heated. C70/EUM may be expected to function as both a thermo-responsive drug carrier and a photosensitizer for PDT.
Author contributions
Conceptualization, S. Y.; data curation, K. K. and S. Y.; formal analysis, K. K. and S. Y.; investigation, K. K., K. I., and S. Y.; methodology, K. I. and S. Y.; project administration, S. Y.; supervision, S. Y.; visualization, K. K.; writing – original draft, K. K. and S. Y.; writing – review and editing, K. K. K. I. and S. Y. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This research was supported by KAKENHI grants (21H02005, 21K19931, 21H05027, 21H05535) from the Japan Society for the Promotion of Science (JSPS), JSPS Bilateral Joint Research Projects (JPJSBP120203509). and the Cooperative Research Program of “Network Joint Research Center for Materials and Devices (20214044).” The author would like to thank Enago for the English language editing.
References
- S. Durdagi, T. Mavromoustakos, N. Chronakis and M. G. Papadopoulos, Bioorg. Med. Chem., 2008, 16, 9957–9974 CrossRef CAS.
- G. Pastorin, S. Marchesan, J. Hoebeke, T. Da Ros, L. Ehret-Sabatier, J. P. Briand, M. Prato and A. Bianco, Org. Biomol. Chem., 2006, 4, 2556–2562 RSC.
- A. Innocenti, S. Durdagi, N. Doostdar, T. Amanda Strom, A. R. Barron and C. T. Supuran, Bioorg. Med. Chem., 2010, 18, 2822–2828 CrossRef CAS PubMed.
- Á. Serrano-Aroca, K. Takayama, A. Tuñón-Molina, M. Seyran, S. S. Hassan, P. Pal Choudhury, V. N. Uversky, K. Lundstrom, P. Adadi, G. Palù, A. A. A. Aljabali, G. Chauhan, R. Kandimalla, M. M. Tambuwala, A. Lal, T. M. Abd El-Aziz, S. Sherchan, D. Barh, E. M. Redwan, N. G. Bazan, Y. K. Mishra, B. D. Uhal and A. Brufsky, ACS Nano, 2021, 15, 8069–8086 CrossRef PubMed.
- A. B. Kornev, A. S. Peregudov, V. M. Martynenko, J. Balzarini, B. Hoorelbeke and P. A. Troshin, Chem. Commun., 2011, 47, 8298–8300 RSC.
- E. Nakamura and H. Isobe, Acc. Chem. Res., 2003, 36, 807–815 CrossRef CAS.
- S. Samal and K. E. Geckeler, Macromol. Biosci., 2001, 1, 329–331 CrossRef CAS.
- A. Kumar, M. V. Rao and S. K. Menon, Tetrahedron Lett., 2009, 50, 6526–6530 CrossRef CAS.
- F. Paquin, J. Rivnay, A. Salleo, N. Stingelin and C. Silva, J. Mater. Chem. C, 2015, 3, 10715–10722 RSC.
- Z. Markovic and V. Trajkovic, Biomaterials, 2008, 29, 3561–3573 CrossRef CAS PubMed.
- M. Wang, S. Maragani, L. Huang, S. Jeon, T. Canteenwala, M. R. Hamblin and L. Y. Chiang, Eur. J. Med. Chem., 2013, 63, 170–184 CrossRef CAS PubMed.
- S. K. Sharma, L. Y. Chiang and M. R. Hamblin, Nanomedicine, 2011, 6, 1813–1825 CrossRef CAS.
- P. Mroz, A. Pawlak, M. Satti, H. Lee, T. Wharton, H. Gali, T. Sarna and M. R. Hamblin, Free Radical Biol. Med., 2007, 43, 711–719 CrossRef CAS.
- V. Milic, M. Posa, B. Srdjenovic and A. Luísa, Colloids Surf., B, 2011, 82, 46–53 CrossRef.
- C. F. Richardson, D. I. Schuster and S. R. Wilson, Org. Lett., 2000, 2, 1011–1014 CrossRef CAS PubMed.
- Z. Yao and K. C. Tam, Macromol. Rapid Commun., 2011, 32, 1863–1885 CrossRef CAS PubMed.
- S. Bosi, L. Feruglio, T. Da Ros, G. Spalluto, B. Gregoretti, M. Terdoslavich, G. Decorti, S. Passamonti, S. Moro and M. Prato, J. Med. Chem., 2004, 47, 6711–6715 CrossRef CAS.
- H. M. Wang and G. Wenz, Beilstein J. Org. Chem., 2012, 8, 1644–1651 CrossRef CAS.
- K. Komatsu, K. Fujiwara and T. Braun, J. Chem. Soc., Perkin Trans. 1, 1999, 2963–2966 RSC.
- S. Kunsági-Máté, G. Vasapollo, K. Szabó, I. Bitter, G. Mele, L. Longo and L. Kollár, J. Incl. Phenom. Macrocycl. Chem., 2008, 60, 71–78 CrossRef.
- T. Metanawin, T. Tang, R. Chen, D. Vernon and X. Wang, Nanotechnology, 2011, 22, 235604–235613 CrossRef PubMed.
- C. C. Cheng, W. L. Lin, Z. S. Liao, C. W. Chu, J. J. Huang, S. Y. Huang, W. L. Fan and D. J. Lee, Polym. Chem., 2017, 8, 7469–7474 RSC.
- Y. Doi, A. Ikeda, M. Akiyama, M. Nagano, T. Shigematsu, T. Ogawa, T. Takeya and T. Nagasaki, Chem. – Eur. J., 2008, 14, 8892–8897 CrossRef CAS PubMed.
- A. Ikeda, T. Sato, K. Kitamura, K. Nishiguchi, Y. Sasaki, J. Kikuchi, T. Ogawa, K. Yogo and T. Takeya, Org. Biomol. Chem., 2005, 3, 2907–2909 RSC.
- L. Xiao, K. Matsubayashi and N. Miwa, Arch. Dermatol. Res., 2007, 299, 245–257 CrossRef CAS.
- H. Takada, K. Kokubo, K. Matsubayashi and T. Oshima, Biosci., Biotechnol., Biochem., 2006, 70, 3088–3093 CrossRef CAS.
- H. Aoshima, S. Yamana, S. Nakamura and T. Mashino, J. Toxicol. Sci., 2010, 35, 401–409 CrossRef CAS PubMed.
- Y. Yamakoshi, T. Yagami, K. Fukuhara, S. Sueyoshi and N. Miyata, Chem. Commun., 1994, 517–518 RSC.
- L. Chrit, P. Bastien, B. Biatry, J. T. Simonnet, A. Potter and A. M. Minondo, Biopolymers, 2007, 85, 359–369 CrossRef CAS PubMed.
- O. Tairy, N. Kampf, M. J. Driver, S. P. Armes and J. Klein, Macromolecules, 2015, 48, 140–151 CrossRef CAS.
- K. Ishihara, T. Ueda and N. Nakabayashi, Polym. J., 1990, 22, 355–360 CrossRef CAS.
- A. Lewis, Y. Tang, S. Brocchini, J. W. Choi and A. Godwin, Bioconjugate Chem., 2008, 19, 2144–2155 CrossRef CAS.
- Y. Iwasaki and K. Ishihara, Sci. Technol. Adv. Mater., 2012, 13, 064101–064115 CrossRef CAS PubMed.
- T. Konno, J. Watanabe and K. Ishihara, J. Biomed. Mater. Res., Part A, 2003, 65, 209–214 CrossRef PubMed.
- T. Ohata, K. Ishihara, Y. Iwasaki, A. Sangsuwan, S. Fujii, K. Sakurai, Y. Ohara and S. Yusa, Polym. J., 2016, 48, 999–1005 CrossRef CAS.
- H. Hatakeyama, H. Akita and H. Harashima, Adv. Drug Delivery Rev., 2011, 63, 152–160 CrossRef CAS PubMed.
- H. Maeda, J. Controlled Release, 2012, 164, 138–144 CrossRef CAS PubMed.
- V. Torchilin, Adv. Drug Delivery Rev., 2011, 63, 131–135 CrossRef CAS PubMed.
- M. Ethirajan, Y. Chen, P. Joshi and R. K. Pandey, Chem. Soc. Rev., 2011, 40, 340–362 RSC.
- N. Shimada, H. Ino, K. Maie, M. Nakayama, A. Kano and A. Maruyama, Biomacrimolecules, 2011, 12, 3418–3422 CrossRef CAS PubMed.
- G. Huang, H. Li, S. T. Feng, X. Li, G. Tong, J. Liu, C. Quan, Q. Jiang, C. Zhang and Z. Li, Macromol. Chem. Phys., 2015, 216, 1014–1023 CrossRef CAS.
- Y. Deng, F. Käfer, T. Chen, Q. Jin, J. Ji and S. Agarwal, Small, 2018, 14, 1802420 CrossRef.
- L. Douziech-Eyrolles, H. Marchais, K. Hervé, E. Munnier, M. Soucé, C. Linassier, P. Dubois and I. Chourpa, Int. J. Nanomed., 2007, 2, 541–550 Search PubMed.
- T. Il Kim, H. J. Seo, J. S. Choi, H. S. Jang, J. U. Baek, K. Kim and J. S. Park, Biomacromolecules, 2004, 5, 2487–2492 CrossRef PubMed.
- G. N. Grover, J. Lam, T. H. Nguyen, T. Segura and H. D. Maynard, Biomacromolecules, 2012, 13, 3013–3017 CrossRef CAS.
- S. Yusa, T. Endo and M. Ito, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6827–6838 CrossRef CAS.
- S. Yusa, K. Fukuda, T. Yamamoto, K. Ishihara and Y. Morishima, Biomacromolecules, 2005, 6, 663–670 CrossRef CAS PubMed.
- A. Z. Akcasu and C. C. Han, Macromolecule, 1979, 12, 276–280 CrossRef CAS.
- F. Meng, G. H. M. Engbers and J. Feijen, J. Controlled Release, 2005, 101, 187–198 CrossRef CAS.
- S. Dash, P. N. Murthy, L. Nath and P. Chowdhury, Acta Pol. Pharm., 2010, 67, 217–223 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tb02183d |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Kazuhiko
Ishihara
a,
Kazuhiko
Ishihara
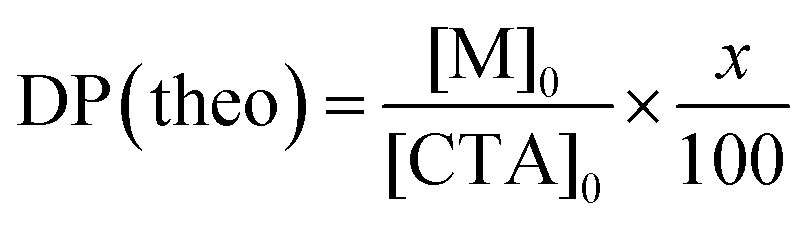
 ), 2.0 (
), 2.0 ( ), and 5.0 g L−1 (
), and 5.0 g L−1 ( ) as a function of temperature with the cooling process, and (b) phase transition temperature (Tp) of EUM in PBS as a function of Cp.
) as a function of temperature with the cooling process, and (b) phase transition temperature (Tp) of EUM in PBS as a function of Cp.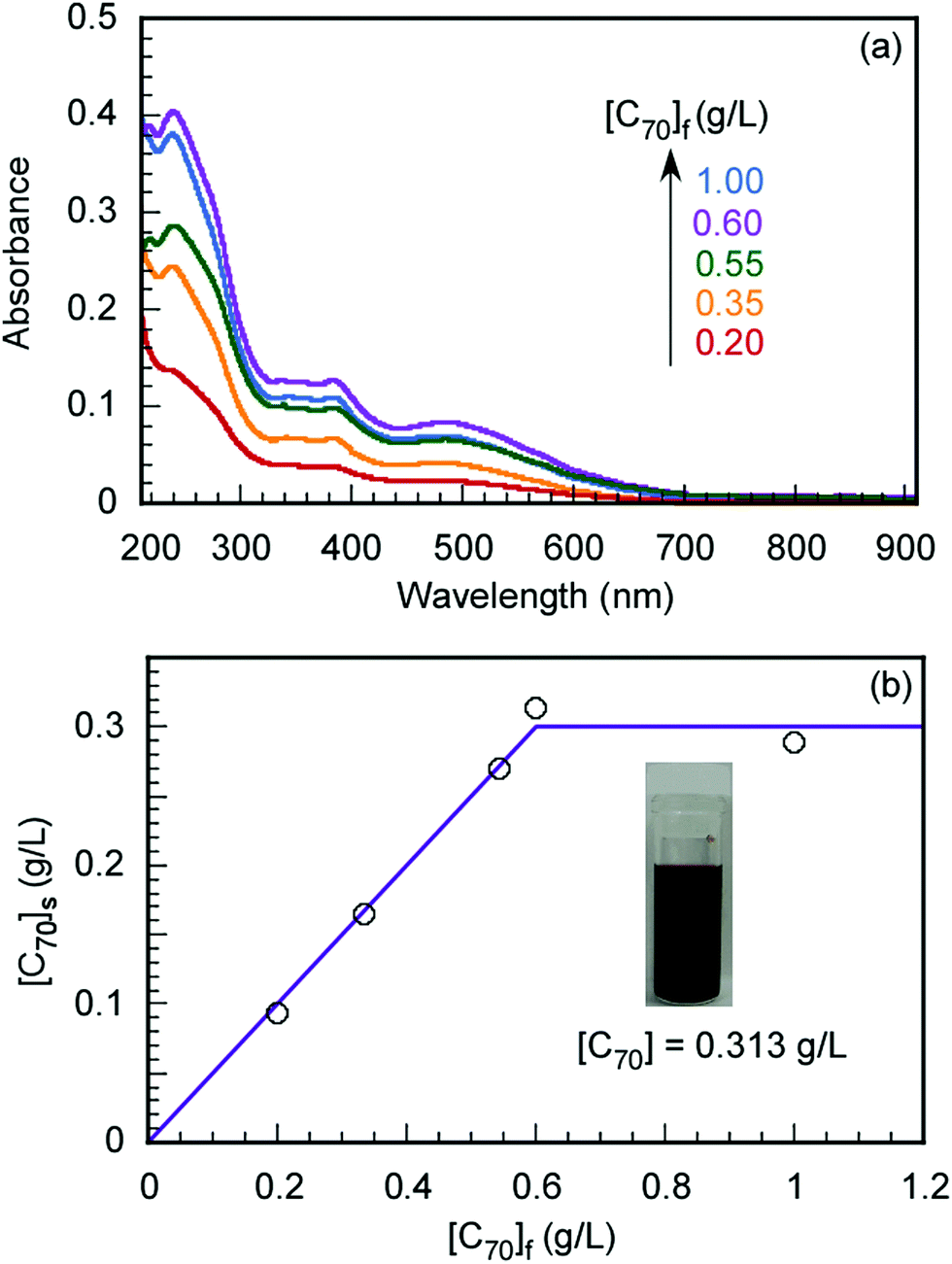
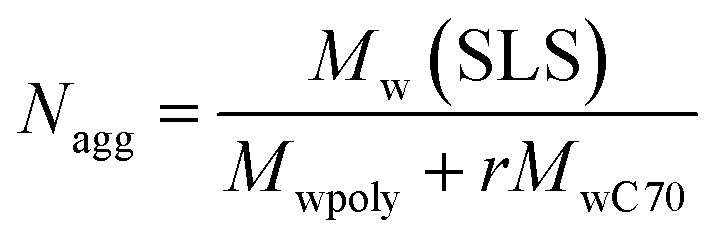
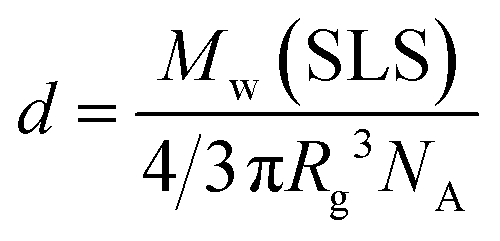
 ) and light scattering intensity (LSI,
) and light scattering intensity (LSI,  ) of C70/EUM at Cp = 2 g L−1 and [C70]s = 0.313 g L−1 in PBS as a function of temperature.
) of C70/EUM at Cp = 2 g L−1 and [C70]s = 0.313 g L−1 in PBS as a function of temperature.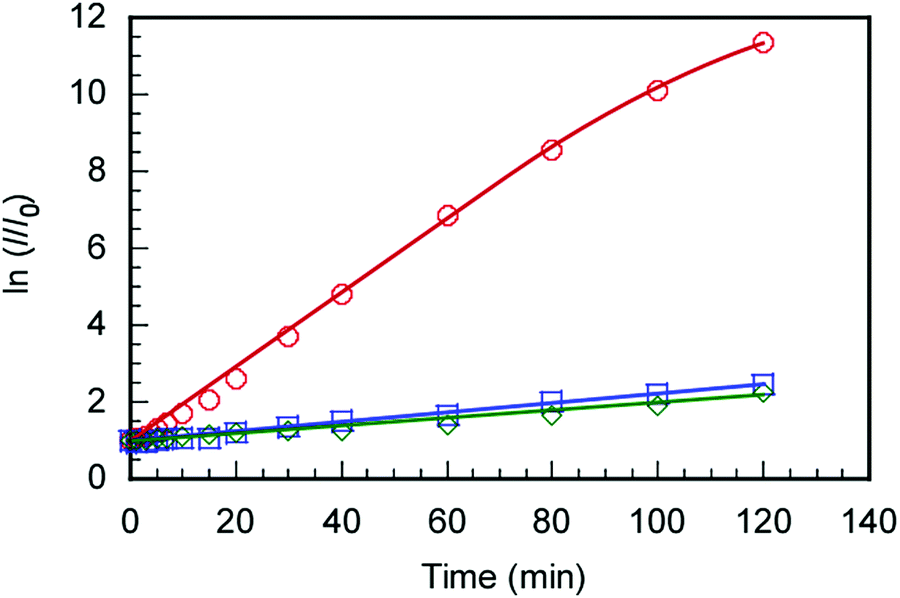
 ), without C70 (
), without C70 ( ), and without EUM and C70 (
), and without EUM and C70 ( ) as a function of irradiation time at 25 °C; I and I0 were the fluorescence intensities at the maximum wavelength (525 nm) after and before 590 nm light irradiation.
) as a function of irradiation time at 25 °C; I and I0 were the fluorescence intensities at the maximum wavelength (525 nm) after and before 590 nm light irradiation.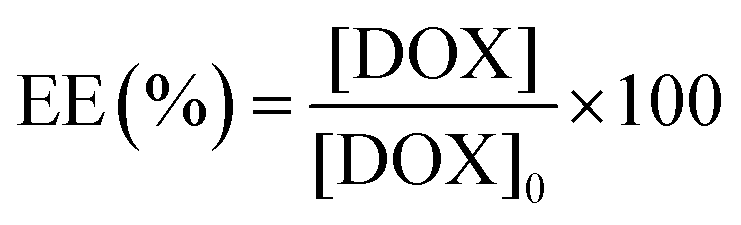
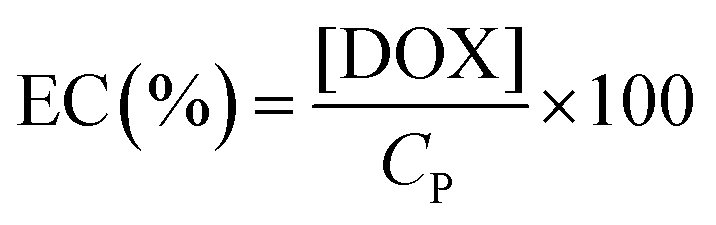
 ) and 50 °C (
) and 50 °C ( ) and a blank solution without C70/EUM at 25 °C (
) and a blank solution without C70/EUM at 25 °C ( ) and 50 °C (
) and 50 °C ( ) in PBS. (b) −ln((I0 − I)/I0) was plotted against dialysis time; I is the fluorescence intensity and I0 is the initial intensity.
) in PBS. (b) −ln((I0 − I)/I0) was plotted against dialysis time; I is the fluorescence intensity and I0 is the initial intensity.