
Remarkable COx tolerance of Ni3+ active species in a Ni2O3 catalyst for sustained electrochemical urea oxidation†
Muhammed
Safeer N. K.
ab,
Chandraraj
Alex
 a,
Rajkumar
Jana
c,
Ayan
Datta
c and
Neena S.
John
*a
a,
Rajkumar
Jana
c,
Ayan
Datta
c and
Neena S.
John
*a
aCentre for Nano and Soft Matter Sciences (CeNS), Shivanapura, Bengaluru 562162, India. E-mail: jsneena@cens.res.in
bManipal Academy of Higher Education, Manipal 576104, India
cSchool of Chemical Sciences, Indian Association for the Cultivation of Science (IACS), Kolkata 700032, India
First published on 5th January 2022
Abstract
The electrochemical urea oxidation reaction (UOR) provides a cost-effective way of generating hydrogen owing to its low thermodynamic energy barrier. Although the UOR is an effective way to generate hydrogen, sustained activity and long-term catalyst usage are retarded by COx poisoning. In nickel oxide-based UOR electrocatalysts, enhancing Ni3+ active species (NiO(OH)) is one of the effective ways to improve the activity. In this study, we show that Ni2O3 is a promising UOR catalyst with Ni3+ ions being highly active and the UOR activity is seen to be almost six times higher than that of NiO. Ni2O3 shows retention of 70% UOR performance even after 25 hours at an average current density of 25 mA cm−2, whereas the NiO system loses 50% of its activity within 10 h even at a low current density of 5.1 mA cm−2. The efficient and sustained UOR activity of Ni2O3 can be correlated with the highly tolerant Ni3+ ions in the Ni2O3 system towards COx poisoning compared to the tolerance of NiO, as proved by impedance studies. The impedance spectra acquired at various applied potentials in the UOR regime show a highly significant reverse loop (real impedance is negative) for NiO indicative of impeding COx desorption as a rate-determining step in the 0.48 to 0.52 V potential window, whereas for Ni2O3, it is less influential under the same conditions indicating the better tolerance of Ni2O3 towards COx poisoning. Furthermore, this is reflected in the fast electro-oxidation kinetics of Ni2O3 with a lower Tafel slope value (21 mV dec−1) than NiO (104 mV dec−1). The higher COx tolerance of the Ni2O3 catalyst is the main reason behind the remarkable stability of the catalyst. Theoretical modeling supports the high activity of Ni2O3 through effective adsorption of reactants such as urea and hydroxide with feasible CO2 removal on the Ni2O3 surface.
Introduction
Hydrogen production from water electrolysis holds a clear advantage of using the earth-abundant source of water resources; however, it suffers from the requirement of high potential for the counter water oxidation reaction (1.23 V vs. RHE).1 The higher overpotential for water oxidation or the oxygen evolution reaction (OER) seriously affects the efficiency of hydrogen production and has a huge bearing on commercialization.2 The requirement of high applied potential for hydrogen production can be dramatically reduced by replacing the OER with the urea electro-oxidation reaction (UOR). Urea is a waste product, abundantly available in human and animal urine and it is also present in sewage. In the year 2010, approximately 2000 billion tons of wastewater was generated worldwide and most of it has flowed into other water bodies.3 The human body generates on average 20–30 mg of urea in a day.4 UOR and urea-based fuel cells have gained a lot of importance recently, facilitating waste water purification combined with energy generation. Nickel-based catalysts are extensively used for the UOR in alkaline medium as nickel oxyhydroxide (NiO(OH)) is shown to be the active intermediate. Vedharathinam et al. have proven mechanisms for urea electro-oxidation using in situ Raman spectroscopy.5,6 The overall reaction scheme and cell potential involved in hydrogen production are given below.Anode:
| CO(NH2)2 + 6OH− ↔ N2(g) + 5H2O + CO2(g) + 6e−, (E0ox. = −0.46 V vs. SHE) | (1) |
Cathode:
| 6H2O + 6e− ↔ 6OH− + 3H2(g), (E0red = −0.828 V vs. SHE) | (2) |
Overall:
| CO(NH2)2 + H2O → N2(g) + 3H2(g) + CO2(g), (E0cell = −0.37 V vs. SHE) | (3) |
Urea electrolysis requires only a potential of −0.37 V vs. SHE for the generation of H2. Nickel oxide and hydroxide-based nanomaterials have been employed for the electrochemical UOR in previous studies. Most of the earlier studies have been focused on improving the catalyst activity. The sustained activity of the catalyst for a longer duration is challenging.7 Guo et al. synthesized Ni nanowires by electrodeposition. They measured the stability in 5 M KOH containing 0.33 M urea, and a rapid current density decrease from 140 mA cm−2 to 50 mA cm−2 within 0.5 h is noticed due to the occupation of gaseous intermediates on the active site of the catalyst.8 Hameed et al. have studied the stability of nickel oxide deposited on various substrates, and NiO deposited on a graphite substrate9 has shown stability for 0.5 h at a current density of 6.67 mA cm−2. In the case of carbon fiber incorporated NiO, UOR activity shows a current degradation from 30 to 19.99 mA cm−2 in a concise duration of 800 seconds,10 indicating the possibility of catalyst poisoning. Very recently, in 2021, Lu et al.11 have studied the UOR activity of nano-nickel oxide in eggshell membrane-derived carbon, which shows an initial current density of 0.05 mA cm−2 that degraded to 0.015 mA cm−2 in 0.5 h. A nickel oxide–nickel hybrid nanoarray on nickel foam has been studied for the electrochemical UOR by Yue et al.12 and has shown 16 h stable catalytic activity at a current density of 10 mA cm−2. Zhang et al.13 synthesized Ni0 rich Ni/NiO by acid etching followed by (H2/Ar) annealing. The above catalyst has shown a superior activity towards the UOR with minimal degradation in 50 mA cm−2 (current density) up to 20 h at a potential of 1.62 V. In many reports, higher current density for the UOR is obtained only when the catalyst is loaded on nickel foam, which itself is UOR active.
The challenge in sustained catalyst activity and stability is explained mainly due to catalyst surface poisoning by the adsorption of reaction intermediates such as NCO−, CO2, and others. Among these intermediates, the potential poison that causes severe degradation in the activity is CO2.14 The catalyst poison leads to the unavailability of active catalytic sites for further reaction and shortens the catalyst life span. Very recently, from our group, Alex et al.15 reported the importance of surface defects and effective removal of adsorbed catalyst poisons for improving electrochemical urea oxidation. They have observed that strong urea adsorption on defective NiO leads to higher catalytic surface poisons favoring the direct UOR and can be removed by adjusting the urea and KOH ratio. In search of a better nickel oxide-based catalyst with a higher number of active species (NiO(OH)) that can tolerate COx poisons, we investigated the Ni2O3 system for the UOR and compared it with NiO. To the best of our knowledge, Ni(III)oxide has not been reported for the UOR application previously. Both the catalysts have Ni3+ active species (NiO(OH)) on their surface with Ni2O3 possessing higher Ni3+ active species providing effective interaction with urea and hydroxyl. A combination of voltammetry and impedance studies in various UOR potential regimes and at different urea–KOH electrolyte concentrations is performed to have a better understanding of the influence of COx poisons in both cases. The impedance study reveals the presence of less significant surface oscillatory phenomena with negative real impedance values for Ni2O3 indicative of COx poisoning to a lesser extent. The excellent COx removal comes from effective OH− ion adsorption and its surface saturation on Ni2O3 samples, whereas NiO has poor OH− adsorption. The high COx tolerance of the active species on the Ni2O3 surface is correlated with the superior stability of UOR activity up to 25 h at an average current density of 25 mA cm−2 in dilute urea solution (0.150 M urea in 1 M KOH). The effective interaction of urea and hydroxyl with the Ni2O3 surface and weak CO2 interaction are supported by density functional theory (DFT) calculations.
Experimental details
Synthesis of NiO and Ni2O3
Nickel hydroxide was precipitated by adding 5 mL of 0.92 g of KOH (Merck) to a solution containing 2.48 g of NiNO3·6H2O (SDFCL) in 25 mL of ultra-pure (type 1) water. The solution is heated to 90 °C for 1 h. The obtained green color precipitate was washed with DI water and ethanol several times to remove unreacted nickel nitrate impurities, and finally, the green Ni(OH)2 powder was dried at 90 °C.16 The obtained Ni(OH)2 powder was annealed at 400 °C in air for 1 h at a heating rate of 5 °C per minute to obtain black NiO powder.17Nickel nitrate was oxidized directly to obtain Ni2O3. We have synthesized Ni(III)oxide using a precipitation method with slight modification to the synthesis reported by Dey et al.18 2.48 g of NiNO3·6H2O was dissolved in 25 mL of water (type 1) and NaClO (4% w/v available chlorine, Merck) is added dropwise to this solution under magnetic stirring at 500 rpm under ambient conditions until the solution completely turns black. The reaction was kept for 1 h and the obtained precipitate was washed with DI water followed by drying in an oven at 90 °C for 1 h.
Characterization techniques
X-ray diffraction (XRD) analyses were performed using a Rigaku SmartLab diffractometer with CuKα incident radiation (40 kV and 30 mA) from nickel oxide powder. Raman spectra were acquired using a HORIBA XploRA PLUS confocal Raman microscope using a 532 nm excitation laser (10% laser power) with an acquisition time of 30 seconds. A field emission scanning electron microscope (FESEM) (TESCAN MIRA3 LM, Brno, Czech Republic) operated at 10 kV with an attached Bruker Nano XFlash 6|30 was used to study the surface morphology and perform energy dispersive spectroscopy (EDS). XPS measurements were carried out with an Omicron ESCA+ integrated with dual-beam charge neutralization. A high-resolution transmission electron microscope (HRTEM, TALOS F200S G2, Thermo Fisher Scientific) operated at 200 kV was used to perform further structural and morphological characterization and electron diffraction.Electrochemical measurements
All electrochemical measurements were performed using a CHI 760 E electrochemical workstation with a three-electrode setup. 2 mg of the catalyst (NiO or Ni2O3) was dispersed in a solution containing a 200 μL isopropyl alcohol–water (type 1) mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio) along with 50 μL (1 wt%) of Nafion solution by sonication for 2 h to obtain a homogeneous catalyst ink. 5 μL of the above solution was drop-cast onto a previously cleaned glassy carbon electrode (GCE) of diameter 0.07 cm2 and used as the working electrode. A Pt spring and Ag/AgCl(sat.KCl) are used as the counter and reference electrodes, respectively. Cyclic voltammetry (CV) was performed at various scan rates in the potential window of −0.2 V to +0.65 V vs. Ag/AgCl(sat.KCl) in mixtures of urea–KOH solutions as the electrolyte. The concentrations of KOH (Merck) and urea (Merck) are varied for each catalyst to obtain the best solution for electrochemical urea oxidation. Electrochemical impedance spectroscopy (EIS) was performed in a frequency range of 105 to 10−2 Hz. Before capturing the impedance data, an equilibration time of 180 s was allowed for the system at each polarization potential. The amplitude of the sinusoidal wave was set to 5 mV. Amperometric measurements were done in a 6 M KOH electrolyte solution containing 0.150 M urea at a potential of 0.40 V vs. Ag/AgCl(sat.KCl) for the NiO catalyst. The stability of the Ni2O3 catalyst was monitored at a potential of 0.58 V vs. Ag/AgCl(sat.KCl) in a 1 M KOH electrolyte solution containing 0.150 M urea. The initial current density during chronoamperometry was verified to match the current density obtained for the linear sweep voltammogram (LSV) at that particular potential and pH. The electrochemical stability of the catalyst was performed using an L-shaped GCE (surface area 0.07 cm2) for feasible removal of the gaseous products of the UOR during a 25 h run.
:3 ratio) along with 50 μL (1 wt%) of Nafion solution by sonication for 2 h to obtain a homogeneous catalyst ink. 5 μL of the above solution was drop-cast onto a previously cleaned glassy carbon electrode (GCE) of diameter 0.07 cm2 and used as the working electrode. A Pt spring and Ag/AgCl(sat.KCl) are used as the counter and reference electrodes, respectively. Cyclic voltammetry (CV) was performed at various scan rates in the potential window of −0.2 V to +0.65 V vs. Ag/AgCl(sat.KCl) in mixtures of urea–KOH solutions as the electrolyte. The concentrations of KOH (Merck) and urea (Merck) are varied for each catalyst to obtain the best solution for electrochemical urea oxidation. Electrochemical impedance spectroscopy (EIS) was performed in a frequency range of 105 to 10−2 Hz. Before capturing the impedance data, an equilibration time of 180 s was allowed for the system at each polarization potential. The amplitude of the sinusoidal wave was set to 5 mV. Amperometric measurements were done in a 6 M KOH electrolyte solution containing 0.150 M urea at a potential of 0.40 V vs. Ag/AgCl(sat.KCl) for the NiO catalyst. The stability of the Ni2O3 catalyst was monitored at a potential of 0.58 V vs. Ag/AgCl(sat.KCl) in a 1 M KOH electrolyte solution containing 0.150 M urea. The initial current density during chronoamperometry was verified to match the current density obtained for the linear sweep voltammogram (LSV) at that particular potential and pH. The electrochemical stability of the catalyst was performed using an L-shaped GCE (surface area 0.07 cm2) for feasible removal of the gaseous products of the UOR during a 25 h run.
Results and discussion
The crystal structures of the synthesized systems NiO and Ni2O3 have been verified by XRD. The XRD peaks obtained at 37.2°, 43.3°, 62.9°, 75.4° and 79.4° correspond to the planes (111), (200), (220), (311) and (222), respectively, of NiO as shown in Fig. 1(a). These diffraction peaks are in perfect agreement with the face-centered cubic (FCC) structure of NiO (JCPDS no. 075-0269). The XRD pattern of the as-obtained black shiny powder sample of Ni2O3 is presented in Fig. 1(b). Diffraction peaks observed at 27.5°, 31.8°, 45.6°, 56.6° and 66.3° could be indexed to (101), (002), (111), (202) and (004) planes corresponding to hexagonal Ni2O3 (JCPDS no. 014-0481). Additionally, broad peaks corresponding to the (001) and (101) planes of Ni(OH)2 are also observed in the case of the synthesized Ni2O3. It has been reported by Fingerle et al.19 that the high oxidation state of Ni3+ ions is feasible to undergo surface reduction to a lower oxidation state of Ni2+ ions. This unstable nature of Ni3+ ions leads to the formation of Ni(OH)2. The simulated XRD patterns of the corresponding compounds from the JCPDS files are shown just below the obtained diffraction patterns (Fig. 1(a) and (b)). The Raman spectra of the synthesized systems, NiO and Ni2O3, are shown in Fig. S1.† The two broad peaks observed around 515 and 1075 cm−1 belong to the longitudinal one phonon (1P) and two phonon (2P) modes due to Ni–O vibrations.20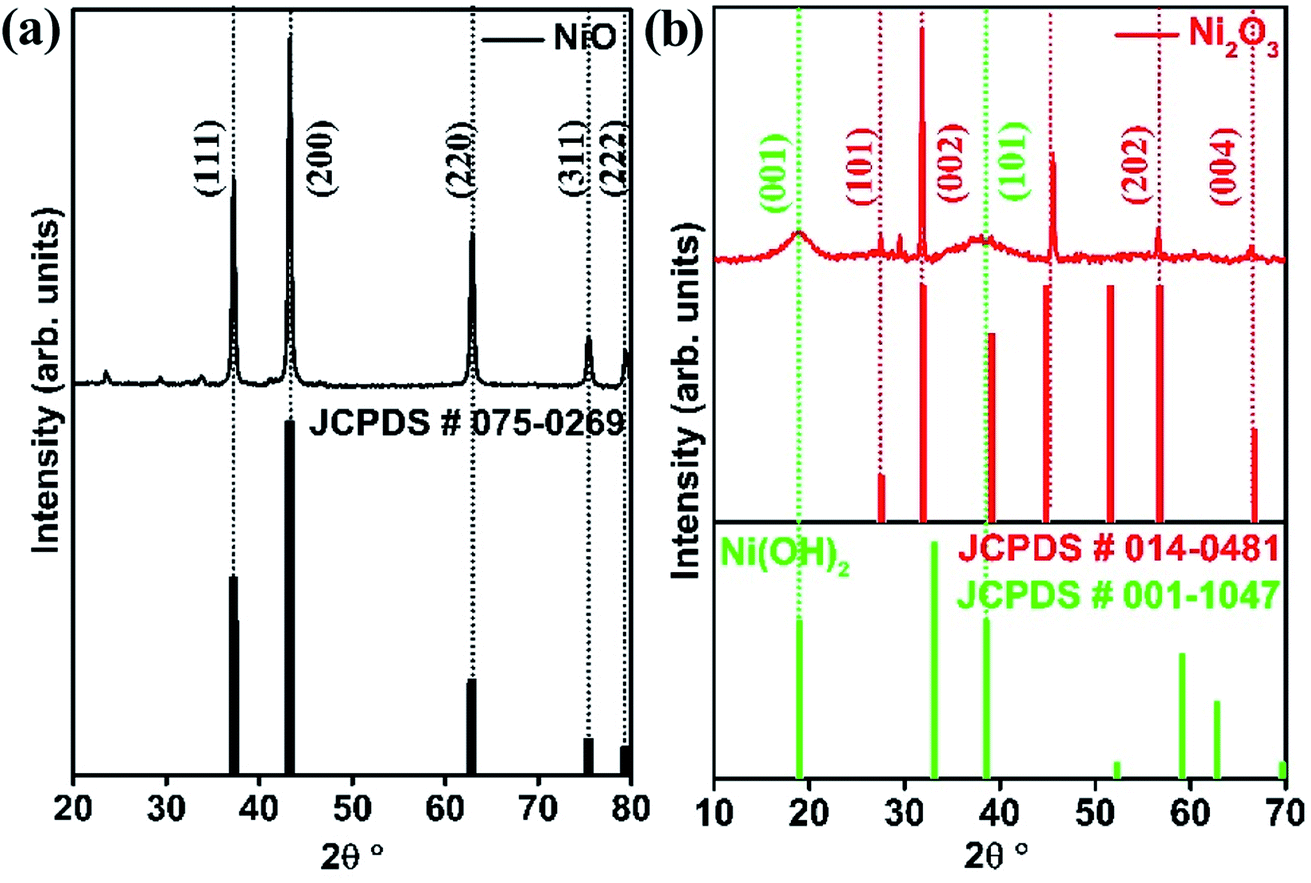 | ||
| Fig. 1 XRD pattern of (a) NiO and (b) Ni2O3. | ||
The morphology of the nickel oxides is examined using FESEM and TEM. Fig. S2† shows the SEM image of NiO crystallites and a magnified image of the same shows that NiO crystallites have an approximate diameter in the range 10–70 nm. In comparison, Ni2O3 has an agglomerated sheet-like morphology (Fig. 2(c)). A similar but clear image of the NiO crystallites and Ni2O3 sheets is observed under TEM (Fig. 2(a) and (d)) as well. Fig. 2(b) shows the high-resolution TEM (HRTEM) image of a NiO crystallite with well-defined lattice fringes analogous to the (111) plane with an interplanar spacing distance of 0.241 nm. Lattice fringes are visible for Ni2O3 and are shown in Fig. 2(e) with an interlayer spacing of 0.280 nm corresponding to the (002) plane. The lattice fringes of the (101) plane belonging to Ni(OH)2 are identified by TEM and support the presence of Ni(OH)2 in accordance with the observation from XRD. The selected area electron diffraction (SAED) pattern of NiO shows planes such as (111), (200) and (220) corresponding to NiO (Fig. 2(c)), whereas Ni2O3 gives the (002) and (202) planes of Ni2O3, along with the (101) plane of Ni(OH)2 (Fig. 2(f)). A uniform distribution of the constituents, nickel and oxygen for both the prepared samples is confirmed by elemental mapping in TEM (Fig. S3†).
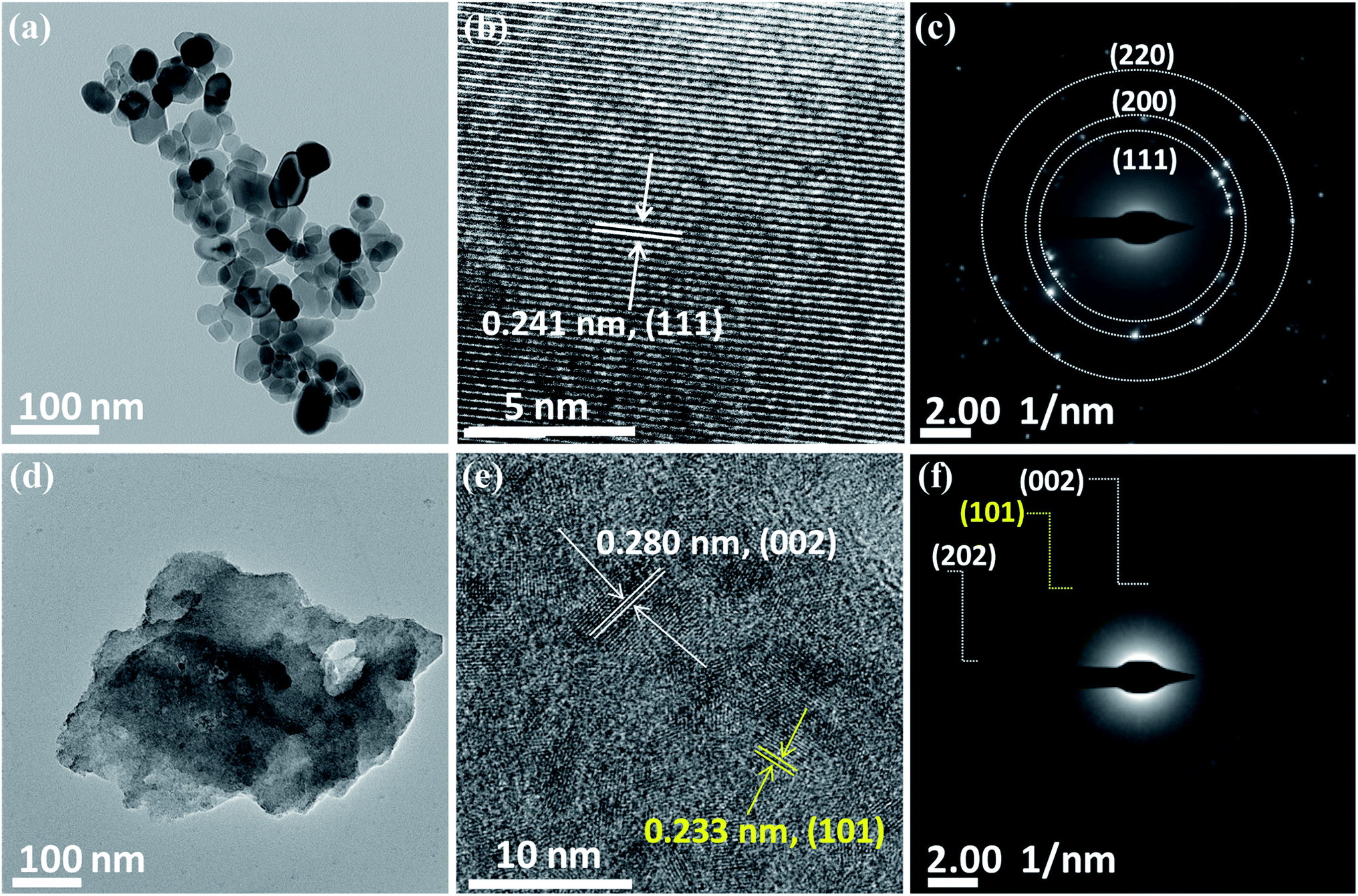 | ||
| Fig. 2 TEM images of NiO (a–c) and Ni2O3 (d–f). (a) NiO crystallites, (b) HRTEM of NiO, (c) SAED pattern of NiO and (d) Ni2O3 sheets, (e) HRTEM of Ni2O3 sheets and (f) SAED pattern of Ni2O3 sheets (HRTEM and SAED of Ni(OH)2 are highlighted in yellow color). | ||
XPS measurements are carried out to examine the surface chemical composition of the as-synthesized nickel oxides. The XPS survey spectra of NiO and Ni2O3 are given in Fig. S4.†Fig. 3(a) shows the Ni 2p3/2 and Ni 2p1/2 spin–orbit splitting of NiO and Ni2O3 samples. The Ni 2p3/2 component of NiO can be deconvoluted into peaks contributed by Ni2+ and Ni3+ states, which are located at 853.4 and 855.5 eV.21 Similarly, peaks due to Ni2+ and Ni3+ states can be discerned from Ni 2p1/2 with a binding energy of 871.1 and 873.2 eV, respectively. The satellite peaks of NiO are represented as Sat1 and Sat2 with a binding energy of 860.9 and 879.0 eV. Similarly, the Ni 2p3/2 and Ni 2p1/2 peaks of the Ni2O3 sample are deconvoluted into peaks due to Ni2+ and Ni3+ located at binding energies of 854.1 and 855.8 eV (2p3/2) and 871.9 and 873.7 eV (2p1/2), respectively. The satellite peaks from Ni2O3 are observed at 861.4 and 879.6 eV. The presence of Ni2+ peaks in Ni2O3 further indicates the formation of NiO or hydroxide arising from surface reduction.
 | ||
| Fig. 3 High-resolution XPS of NiO and Ni2O3: (a) Ni 2p spectra and (b) O 1s spectra. | ||
We have also calculated the ratio of Ni3+/Ni2+ from the deconvoluted spectra of NiO and Ni2O3. A higher Ni3+/Ni2+ intensity ratio of 2.07 was noticed for Ni2O3 as compared to NiO (Ni3+/Ni2+ = 1.36). The O 1 s spectra, along with their deconvoluted peaks, are displayed in Fig. 3(b). The O 1s peak from NiO is deconvoluted into two peaks: O1 at 529.3 eV and O2 situated at 531.3 eV. The intense peak O1 is attributed to O2− from the Ni–O lattice.22 The O2 peak at higher binding energy indicates surface adsorbed oxygen (O2), which belongs to defective sites.23,24 Similarly, the O 1s peak of Ni2O3 samples can be deconvoluted into three peaks: O1 (lattice oxygen) at a lower binding energy value of 529.4 eV, O2 (defect state) observed as a broader peak at 531.3 eV and the O3 peak at 531.5 eV (hydroxide from Ni(OH)2).25,26 The percentage of lattice oxygen (O1), adsorbed oxygen (O2) and hydroxide (O3) are given in Table S2.† The EDS study shows that Ni2O3 has a high O/Ni ratio of 3.48, which is 1.48 in the case of NiO and supports the presence of a high oxygen content in Ni2O3 (Table S1†).
Electrochemical studies
GCEs modified with NiO and Ni2O3 are studied for electrochemical urea oxidation employing cyclic voltammetry, linear sweep voltammetry, impedance analysis, and chronoamperometry. Fig. 4(a) shows the cyclic voltammograms of NiO and Ni2O3 in 1 M KOH containing 0.150 M urea acquired at a scan rate of 10 mV s−1. Despite having an equal onset oxidation potential value of the UOR at 0.34 V, the peak current density for Ni2O3 (36.32 mA cm−2) is almost six times higher than that of NiO (5.94 mA cm−2), revealing the higher activity of the Ni2O3 catalyst towards the UOR. When the scan polarity is reversed, we observe an anodic current arising from the oxidation of reactive intermediates adsorbed on the catalyst surface (intermediates generated in the forward scan get adsorbed on the catalyst surface). The oxidation of intermediates during the reverse scan continues till 0.32 V but the electrochemical UOR starts at a higher potential of 0.34 V vs. Ag/AgCl(sat.KCl), resulting in a CV crossover. A similar CV crossover has been observed by Vedharathinam et al. for the UOR in alkaline electrolytes.5 The Tafel slope was taken by plotting log(current density) vs. applied UOR potential. The obtained Tafel slope for NiO and Ni2O3 is 104 and 21 mV dec−1. The significantly less value obtained for Ni2O3 indicates faster kinetics exhibited by Ni2O3 than NiO and further confirms the superior activity of Ni2O3 (Fig. 4(b)). The Tafel slope obtained for Ni2O3 is relatively less than the values reported for the UOR in recent literature.26 A Tafel slope of 31 mV dec−1 is shown for a MOF-derived nickel nitride catalyst,27 while 49 mV dec−1 is reported for a nickel phosphide nanoflake array.28 Ding et al.29 compared the activity of atomically thin Ni(OH)2 nanoholes and nanomeshes towards the UOR. They obtained a Tafel slope of 80 mV dec−1 for the Ni(OH)2 nanomesh and 231 mV dec−1 for Ni(OH)2 nanoholes. Nickel oxide supported on activated carbon black showed a Tafel slope of 79.57 mV dec−1 as compared to nickel oxide supported on multiwalled carbon nanotubes (85.35 mV dec−1), graphene (90.87 mV dec−1), and graphite (116.08 mV dec−1).9 The above studies are all based on NiO, while our research shows that Ni2O3 is a potential catalyst for the UOR.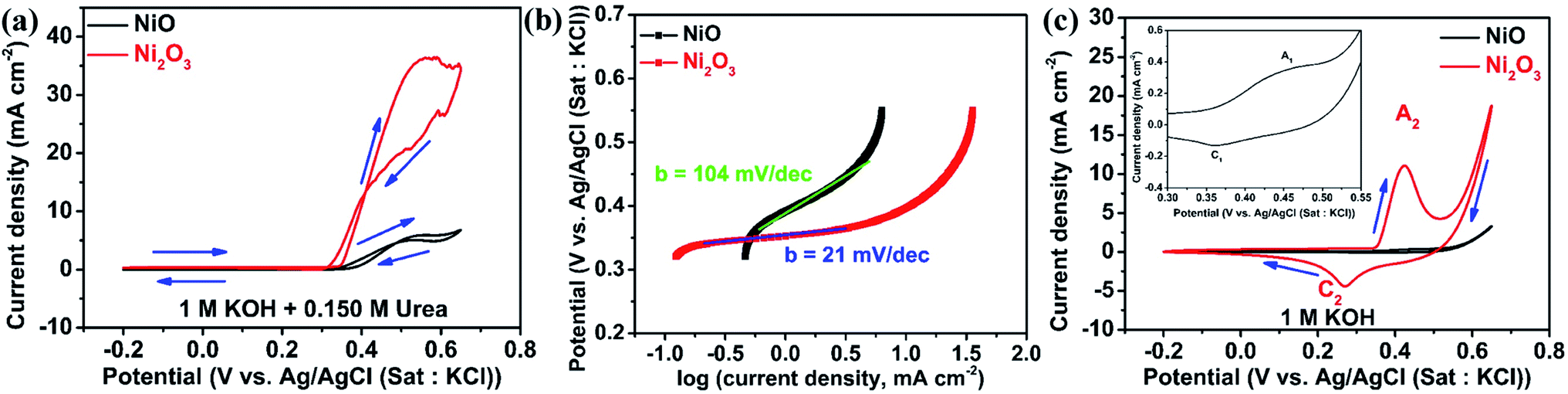 | ||
| Fig. 4 (a) CV of NiO and Ni2O3 in 1 M KOH containing 0.150 M urea at a scan rate of 10 mV s−1, (b) Tafel slope of NiO and Ni2O3 in 1 M KOH + 0.150 M urea, and (c) CV of NiO and Ni2O3 in 1 M KOH at a scan rate of 10 mV s−1 along with the magnified view of the redox region of NiO in the inset. | ||
The CVs of both samples are acquired in 1 M KOH in the absence of urea to study the active species (NiO(OH)) formation leading to UOR catalysis. Fig. 4(c) shows redox peaks (A1) at 0.45 V in the forward scan resulting from the oxidation of Ni2+ (Ni(OH)2) to Ni3+ (NiO(OH)) and the cathodic peak (C1) at 0.36 V belonging to the reduction of Ni3+ to Ni2+ in 1 M KOH solution.30 Similarly, Ni2O3 gives an anodic peak (A2) at 0.424 V with a higher current density and a cathodic peak (C2) in the reverse scan at 0.268 V. The anodic peak current density for Ni2O3 is 14 times higher as compared to NiO in 1 M KOH solution, which implies a greater number of oxidized Ni3+ ions that are significant active sites for the UOR. For a better comparison, the CVs of NiO and Ni2O3 in the presence (0.150 M urea) and absence of urea are shown in Fig. S5(a) and (b),† respectively. The electrochemical active surface area (ECSA) is calculated from double-layer capacitive current (Cdl), given in Fig. S6.† The Ni2O3 catalyst showed an ECSA of 0.74 cm2, 2.5 times higher than that of NiO (0.29 cm2). The higher ECSA value estimated for Ni2O3 also supports the improved UOR activity. The oxidized Ni3+ ions in Ni2O3 are more favorable for the adsorption of reactants, resulting in a high current for urea electro-oxidation. A detailed explanation is given below with theoretical modeling. The UOR performance is seen increasing with urea concentration in terms of current density achieved, and it shows a maximum in 0.150 M urea and 1 M KOH electrolyte solution. The effect of urea concentration on UOR performance is explained below.
Fig. 5(a) and (b) show the CV of NiO and Ni2O3 catalysts in 1 M KOH with varying molar concentrations of urea at a scan rate of 10 mV s−1. For the NiO sample, the anodic peak current density attributed to the UOR performance increases with urea concentration (0 M to 0.150 M urea) and gets saturated near 0.150 M of urea. This current density increment is due to the increased accessibility of urea molecules for oxidation near the working electrode. This behavior signifies that the UOR is a diffusion-limited process when the urea concentration is less than 0.150 M. At lower concentrations, the surface coverage of the urea molecule on the catalyst is lower and with gradual increment in urea concentration, the surface coverage is enhanced, contributing to increase in current density. Once the urea concentration reaches 0.150 M, the catalyst lacks OH− ions on the electrode surface for Ni2+ to Ni3+ oxidation (active species for urea oxidation) and as a result, a saturation of UOR current is observed. This speculation is confirmed by performing the UOR in 0.150 M urea with varying KOH concentrations from 1 to 8 M. It is observed that the urea electro-oxidation current increases gradually and saturates near an electrolyte concentration of 6 M KOH containing 0.150 M urea for NiO as can be noticed in Fig. 5(c) and accordingly, the redox peak current improvement of Ni2+ to Ni3+ is also observed (Fig. 5(c)). Even though the anodic peak current density saturated at 6 M KOH containing 0.150 urea, the onset potential continued to decrease towards a favorable lower potential from 0.34 V to 0.26 V during the increase in KOH concentration from 1 to 8 M.
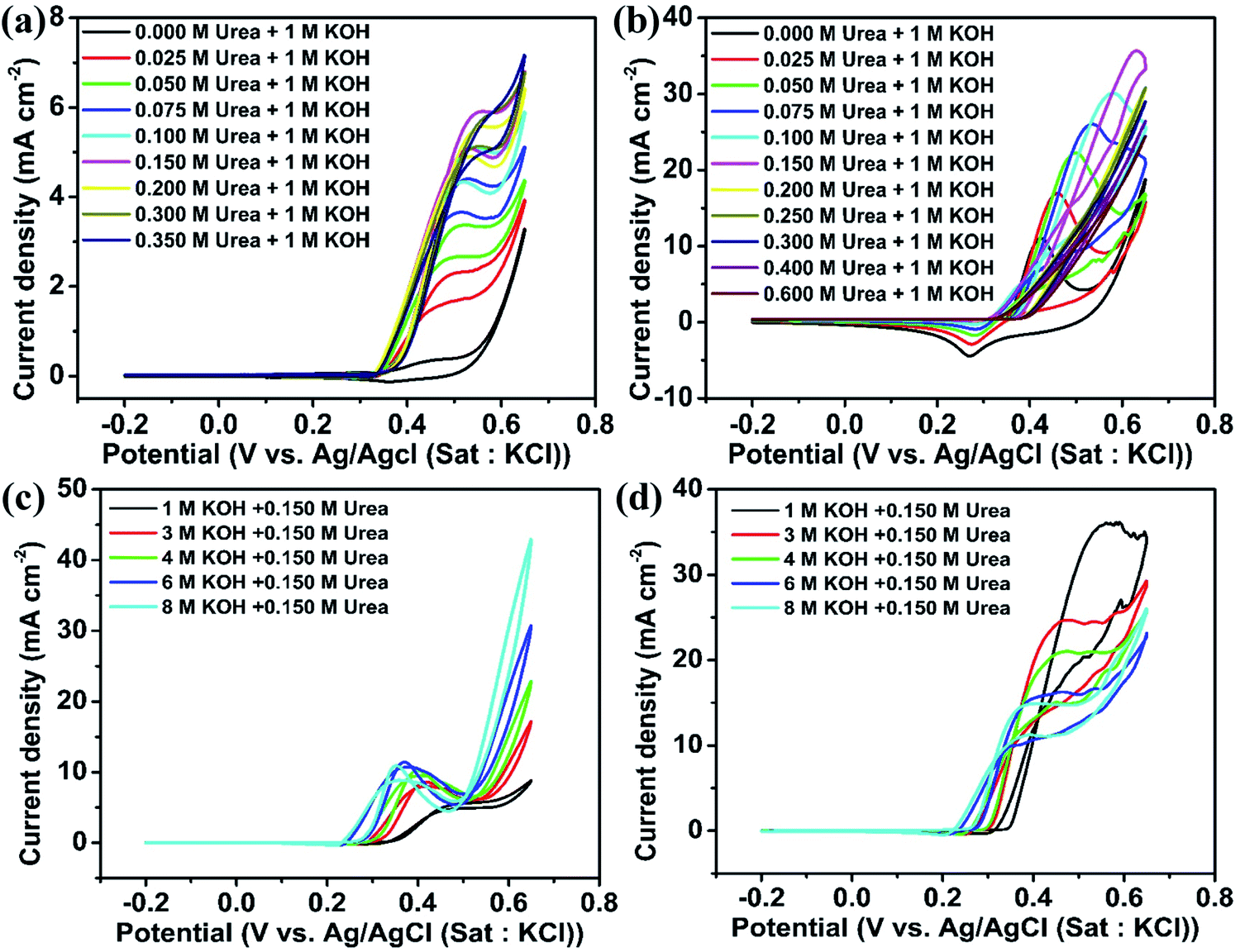 | ||
| Fig. 5 CV of (a) NiO and (b) Ni2O3 modified GCEs in 1 M KOH containing various concentrations of urea: (c) NiO and (d) Ni2O3 in different molar concentrations of KOH with a fixed urea concentration of 0.150 M at a scan rate of 10 mV s−1. | ||
Ni2O3 also shows a similar UOR current improvement with the urea concentration to the case of NiO (Fig. 5(b)). The UOR current achieves saturation at a urea concentration of 0.150 M. However, the maximum current obtained is much higher in this case, 35.85 mA cm−2 for 0.150 M urea in 1 M KOH, which is only 5.5 mA cm−2 for NiO and supports the effective adsorption of reactants on Ni2O3. We have also increased KOH concentration (1 to 8 M) concerning fixed urea concentration to enhance the UOR current, similar to the case of the NiO sample. However, current reduction is observed in this case, as seen in Fig. 5(d). A detailed CV of GCEs modified with NiO and Ni2O3 at different scan rates in various concentrations of urea and KOH is given additionally (Fig. S7–S10†). Fig. S7 and S9† show the effect of urea concentration (in 1 M KOH) and the effect of KOH concentration (containing 0.150 M urea) for NiO, respectively, whereas Fig. S8 and S10† show the effect of urea concentration (in 1 M KOH) and effect of KOH concentration (containing 0.150 M urea) for Ni2O3, respectively.
In order to understand the UOR current reduction observed for Ni2O3 with increasing KOH, cyclic voltammetry is performed in 1 to 8 M KOH solutions in the absence of urea (Fig. S11†). The decrease in the anodic peak current density may arise due to NiOO− ion formation on the surface resulting from strong OH− interaction at very high pH. Initially, 1 M KOH facilitated NiO(OH) formation via oxidation of surface species Ni(OH)2 on Ni2O3. As the KOH concentration increases, the chemical deprotonation of the active species NiO(OH) forms a negatively charged species superoxide type NiOO− ions (NiOOH + OH− → NiOO−+ H2O) that inhibit further OH− adsorption.31 As a result, a reduction in CV current with KOH concentration is observed. However, similar to what we observed for NiO, a decrease in onset potential towards a favorable direction is seen for Ni2O3 as well when the concentration of KOH is increased (0.34 V for 1 M KOH and 0.150 M urea to 0.25 V for 8 M KOH and 0.150 M urea).
Impedance analysis
Electrochemical impedance spectroscopy (EIS) is an efficient technique to study the number of electrochemical processes during the electrochemical reaction. The Nyquist plot is obtained by plotting real (Z′) impedance vs. imaginary (Z′′) impedance and gives information about solution resistance, and charge transfer resistances related to electrochemical reactions. In contrast, the Bode plot obtained by plotting log(frequency) vs. −phase angle gives details about the number of processes that occur during the electrochemical reaction.32 For complex electrochemical reactions such as urea and methanol oxidation, the Nyquist plot with a negative real impedance value directly indicates the catalyst active site, which undergoes competitive phenomena such as surface poisoning by COx and its removal through OH− ions that slow down the UOR.33,34Fig. 6(a) shows the Nyquist plot of the NiO catalyst at UOR potentials from 0.34 to 0.64 V in 1 M KOH containing 0.150 M urea. Initially, the impedance is monitored at 0.34 V vs. Ag/AgCl(sat.KCl), where the UOR starts. The spectra show a broad peak in the Nyquist plot that arises from the overlap of two small semicircles, which is identified through the appearance of two relaxation processes in the Bode plot (Fig. 6(c)). The higher frequency relaxation process (102.5 to 101.9 Hz), as seen in the Bode plot, belongs to the Ni2+/Ni3+ redox process and is related to an indirect UOR. Similarly, the occurrence of a low-frequency relaxation process near (101.7 to 10−0.90 Hz) represents direct urea electro-oxidation.35–37 When the applied potential is increased gradually from 0.34 to 0.44 V (Fig. 6(a)), a reduction in the diameter of the two semicircles is observed. It can be seen in the enlarged plot given in Fig. 6(b). In the potential range of 0.34 to 0.44 V, all data points with positive real impedance values indicate CO2 production as a rate-determining step for the UOR. At 0.48 V applied potential, the impedance data points in the low-frequency region undergo reversal with negative real impedance values (10−0.50 to 10−2.0 Hz), and this behavior pertains till 0.52 V (100.42 to 10−2.0 Hz). The appearance of negative real impedance values is probably due to electrochemical surface oscillatory phenomena. This arises from competition between COx adsorption (blocks the active sites of the catalyst) and its removal by surface covered OH− ions (release the blocked active sites) and makes CO2 desorption a rate-determining step for the UOR.33,34 The flip of impedance spectra (appearance of data points in II and III quadrants) with negative real impedance values is also reflected in the Bode plot as the −phase angle has values between ±90° to ±180° in the potential range 0.48 to 0.52 V (Fig. 6(c)) for NiO in 1 M KOH + 0.150 M urea. The flip of the impedance spectra explains that the catalyst is not poison tolerant. The produced CO2 during urea electro-oxidation gets strongly adsorbed on the NiO surface without effective removal. As the potential increases from 0.56 to 0.64 V, the CO2 production returns as the rate-determining step with positive real impedance values. Impedance analysis reveals CO2 production as the rate-determining step at lower UOR potentials that alters to CO2 desorption in the mid potential range (0.48 to 0.52 V). Finally, in the higher potential range, the CO2 production returns as the rate-determining step. This behavior of NiO indicates that CO2 surface poisons have a significant role in the electrochemical UOR and it is not entirely removed from the catalyst surface. The impedance spectra acquired in 3 M KOH + 0.150 M urea solution at various UOR potentials are shown in Fig. 6(d). It may be recalled from the CV (Fig. 5(c)) that the onset potential gets shifted to a lower potential value (0.30 V) with increase in KOH concentration. The impedance spectra of NiO in 3 M KOH + 0.150 M urea give positive real impedance values from 0.30 to 0.38 V, which change to negative real impedance values at 0.40 V (10−0.83 to 10−2.0 Hz) remaining till 0.48 V (100.75 to 10−2.0 Hz). On further increasing the potential to 0.52 V (Fig. 6(e) and (f)), positive real impedance returns and is retained till 0.64 V. Fig. 6(g) shows the impedance spectra of NiO in 6 M KOH + 0.150 M urea. The positive real impedance values with CO2 production as the rate-determining step are observed from 0.26 to 0.34 V and negative real impedance values with CO2 desorption as a rate-determining step are observed from 0.36 (10−0.25 to 10−0.20 Hz) to 0.44 V (100.67 to 10−0.20 Hz) (Fig. 6(i)). Further increase of potential from 0.48 to 0.64 V leads to CO2 production as the rate-determining step with positive real impedance values (Fig. 6(h)). The shifting of the UOR onset potential and rate-determining step (CO2 production and desorption) with KOH concentration is probably due to the concentration dependent potential shift38 of the Ni2+/Ni3+ redox (KOH concentration improves the pH of the electrolyte), which is confirmed by cyclic voltammetry of samples at various KOH concentrations (1 to 8 M) (Fig. 5(c) and S11†).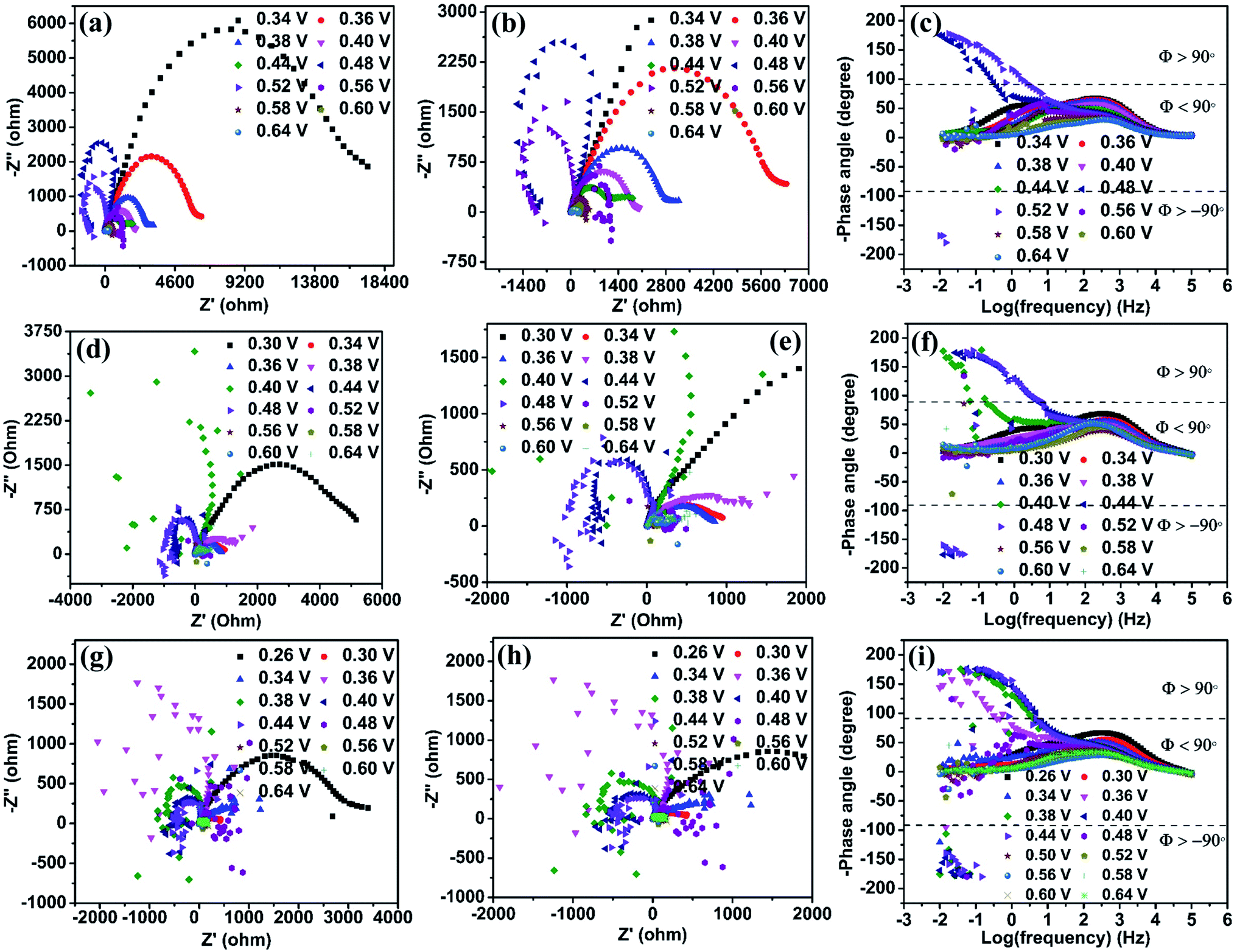 | ||
| Fig. 6 Nyquist plots of the NiO catalyst in various electrolyte solutions: (a) 1 M KOH with 0.150 M urea, (d) 3 M KOH with 0.150 M urea and (g) 6 M KOH with 0.150 M urea. (b), (e) and (h) Zoomed-in view of the semicircle in the high-frequency region in (a), (d) and (g), respectively. Corresponding Bode plots of NiO in (c) 1 M KOH with 0.150 M urea, (f) 3 M KOH with 0.150 M urea and (i) 6 M KOH with 0.150 M urea. | ||
Ni2O3 impedance studies are carried out in 1 M KOH + 0.150 M urea solution at an initially applied potential of 0.34 V (onset potential for the UOR). The spectra show positive real impedance values from 0.34 to 0.44 V (Fig. 7(a)), which indicates CO2 production as the rate-determining step and is confirmed by the Bode plot (−phase angle below 90°). When the applied potential is increased to 0.48 V, the impedance spectra start giving negative real impedance values (Fig. 7(b)), which appear in the frequency range of 10−1.5 to 10−2.0 Hz as observed from the Bode plot (−phase angle between ±90° and ±180°, Fig. 7(c)). In the case of Ni2O3, though the impedance data corresponding to the UOR potential range, 0.48 to 0.60 V, start giving negative real impedance, the data points are seen to vary between I and III quadrants. Accordingly, both negative and positive real impedance values are noted and might be taken as an indication of competition in the rate-determining step between CO2 production and its desorption during the relaxation time between 10−1.5 to 10−2.0 Hz. The above may occur when competition between OH− ions and COx for the active sites causes surface oscillatory behavior resulting in positive and negative real impedance in EIS. But once the surface is free from COx or in other words, if the catalyst is completely poison tolerant and active sites are covered/saturated by OH− ions rather than COx, then positive real impedance should be observed. This can be achieved if the catalyst has weak adsorption for CO2. To signify these processes (competition of CO2 production and its desorption), we have counted the number of data points with negative real impedance values among the total points in those frequency ranges. The calculated points showing negative real impedance at a potential of 0.48 V from frequency 10−1.5 to 10−2.0 Hz are 6 out of 7, and further increasing potential to 0.50 V gives 4 out of 14 points in the frequency range of 10−0.92 to 10−2.0 Hz, 5 out of 14 points in the frequency range of 10−0.92 to 10−2.0 at 0.52 V, etc. (Table S4†). This is indicative of the high tolerance of the Ni2O3 catalyst against COx poisons.
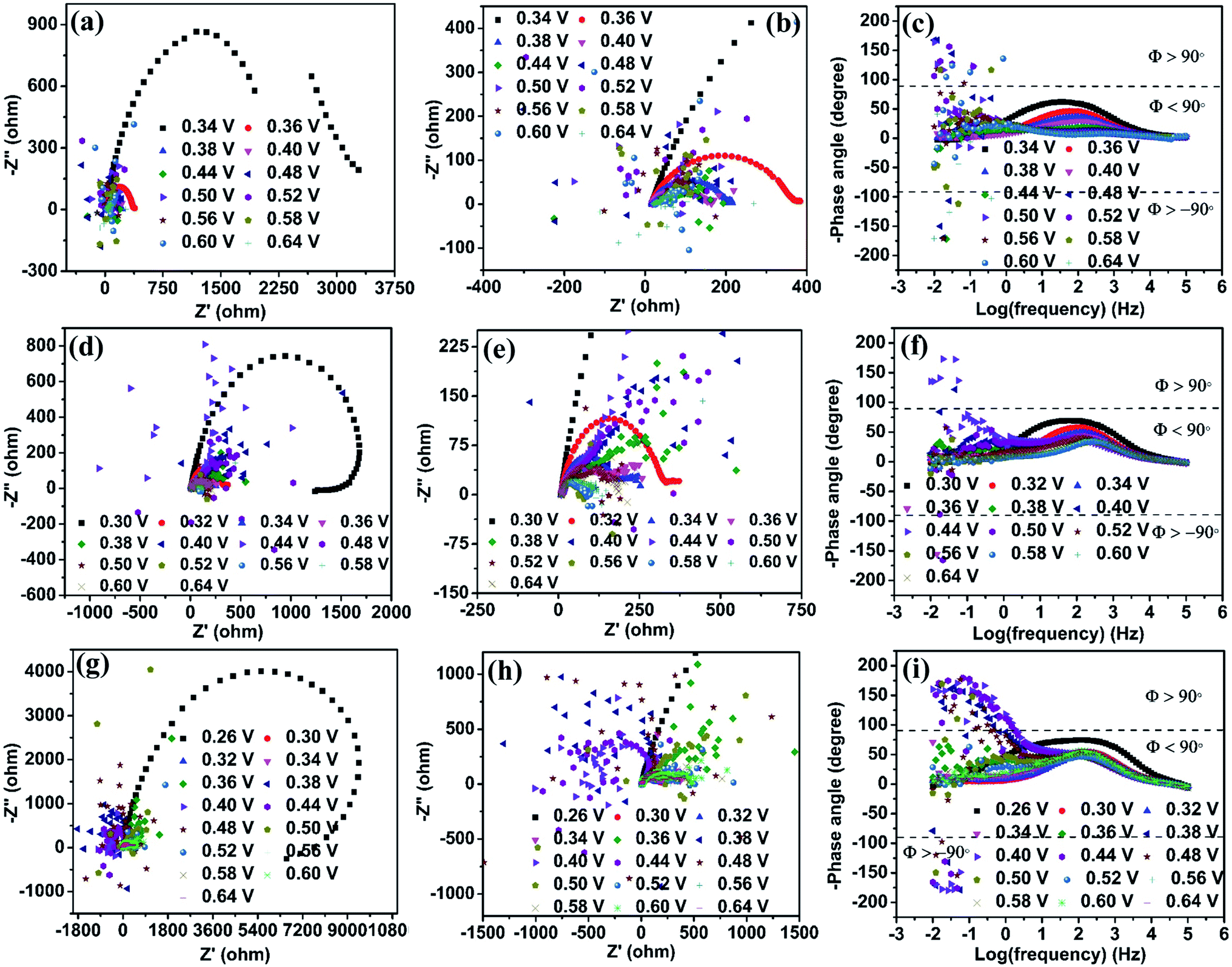 | ||
| Fig. 7 Nyquist plots of the Ni2O3 catalyst in various electrolyte solutions: (a) 1 M KOH + 0.150 M urea, (d) 3 M KOH + 0.150 M urea and (g) 6 M KOH + 0.150 M urea. (b), (e) and (h) Enlarged views of the semicircle in the high-frequency region of (a), (d) and (g). Corresponding Bode plots in (c) 1 M KOH + 0.150 M urea, (f) 3 M KOH + 0.150 M urea and (i) 6 M KOH + 0.150 M urea. | ||
In the case of NiO, the analysis of data points at 0.48 V in the frequency range of 10−0.50 to 10−2.0 Hz shows 18 continuous points out of 19 with negative real impedance under the same electrolyte conditions revealing that CO2 production and its desorption competition is predominant for NiO. At increasing potential (0.52 V) for NiO, there is no significant difference in its impedance data behavior (Table S4†). These observations are supported by the Bode plot with the −phase angle varying between ±90° to ±180° (corresponding to Nyquist plot data points in negative real impedance values). This observation confirms that electrochemical oscillatory behavior is predominated by CO2 poison in NiO and less in Ni2O3. The increase in KOH concentration till 3 M does not significantly change the impedance behavior for both the samples (Fig. 7(d)–(f)). The impedance data of Ni2O3 in 6 M KOH + 0.150 M urea at various UOR potentials show continuous negative real impedance data points similar to NiO as shown in Fig. 7(g) and (h) (20 out of 22) in the frequency range of 10−0.33 to 10−2.0 Hz at 0.38 V (Fig. 7(i)), and further increasing potential exhibits the same behavior as NiO. This might probably be due to the lack of OH− coverage (confirmed by CV) and its availability on the Ni2O3 surface resulting in the equal number of surface adsorbed CO2 and OH− leading to surface oscillatory behavior33 (Table S4†). The flipping of impedance data points with real impedance values from negative to positive is explained before and shown in Table S3.†
The data points with positive real impedance values are fitted considering the equivalent circuit in Fig. S12.† The Rs represents the solution resistance, and R1 and CPE1 are the charge transfer resistance and constant phase elements for an indirect electrochemical UOR, while R2 and CPE2 are the charge transfer resistance and constant phase elements for a direct electrochemical UOR.39 The values of these fitted data are given in Table S5.† In 1 M KOH + 0.150 M urea solution, Ni2O3 shows comparatively lower charge transfer resistance for indirect (R1 = 5.171 Ω) and direct (R2 = 3093 Ω) reactions than NiO (R1 = 1049 Ω and R2 = 15862 Ω) indicating fast kinetics for the UOR, which is supported by the estimated low Tafel slope value. The impedance data points obtained for NiO and Ni2O3 at higher pH in 0.150 M urea + 3 M KOH and 6 M KOH solutions are also fitted using the equivalent circuit. The R1 and R2 values are seen reducing with increasing potential indicating a fast electrochemical UOR with applied potentials. It can be noticed that Ni2O3 shows lower resistance at all applied potentials than NiO for both direct and indirect reactions.
The electrochemical stability of both the catalysts during the UOR is studied using chronoamperometry in their respective electrolytes in which they exhibit high activity, 0.150 M urea + 1 M KOH for Ni2O3 and 0.150 M urea + 6 M KOH for NiO samples and at applied potentials at which the catalyst gives maximum current for the UOR, 0.40 and 0.58 V for NiO and Ni2O3, respectively. Stability studies of both samples are performed for 25 hours (Fig. 8). NiO shows an initial current density of 5.1 mA cm−2, which reduced to 1.9 mA cm−2 after 25 hours (37% retention in current density). The up and down fluctuation of UOR current values indicates the blockage (reduction in current) and regeneration (current enhancement) of active sites (Fig. 8(a) and (b) inset). Fen Guo et al.37 have shown that the current hike in i–t measurement is due to catalyst active site regeneration by OH− ions and COx removal. They have also pointed out that the magnitude of current fluctuation increases with KOH concentration, implying that OH− ions are effective in removing the poison from the catalyst surface. The magnitude of current fluctuation in the Ni2O3 sample (2.30 mA cm−2) is higher than that of NiO (0.045 mA cm−2), indicating effective COx poison removal with active site regeneration, supported by impedance spectra and complemented by the low Tafel slope value. Furthermore, effective active site regeneration and COx removal are the main factors in Ni2O3 for COx tolerance and sustained activity. The stability of Ni2O3 is compared with that of nickel oxide and hydroxide-based compounds available in the literature (Table S8†) and it shows that Ni2O3 is highly active for a longer duration of catalysis at a high current density (25 mA cm−2). After the stability test (25 h), the Ni2O3 coating was scratched off from the GCE and washed several times with ethanol for TEM analysis to understand any structural and morphological change. The sheet-like morphology of Ni2O3 persists even after 25 h (Fig. S13†). Elemental mapping (Fig. S14†) and EDS analysis (Fig. S15†) also show the presence of nickel and oxygen. The above studies by LSV, CV, Tafel slope analysis, i–t curve analysis and EIS reveal that Ni2O3 is capable of generating efficient active species NiO(OH) and resultant high urea electro-oxidation current with significant CO2 tolerance compared to NiO. In order to obtain more insights into the reaction mechanism as well as the activity difference exhibited by NiO and Ni2O3 surfaces towards the electrochemical urea oxidation reaction (UOR), we have performed DFT modeling. The adsorption energies of key reactants such as urea, hydroxyl, and CO2 poison on the catalyst surface are calculated from the model.
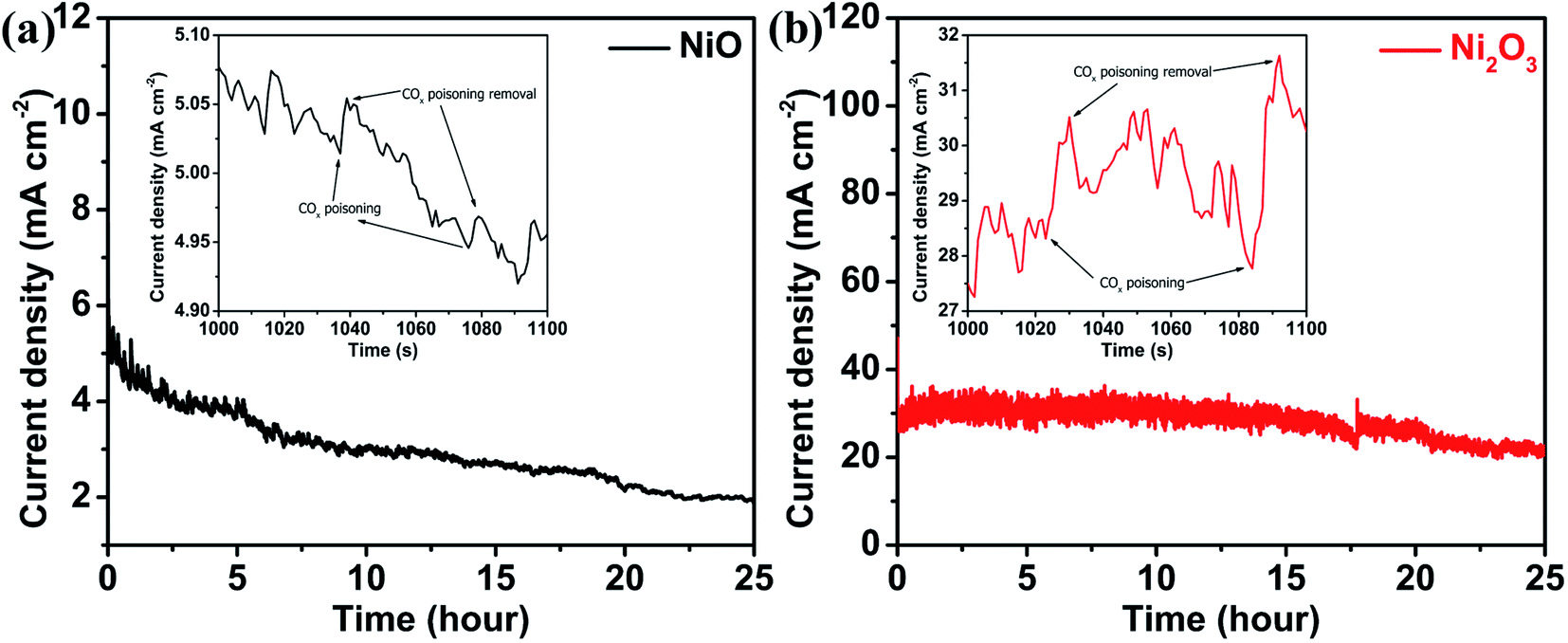 | ||
| Fig. 8 i–t curves monitored for 25 hours for (a) NiO in 6 M KOH + 0.150 M urea at 0.40 V vs. Ag/AgCl(sat.KCl) and (b) the Ni2O3 catalyst in 1 M KOH + 0.150 M urea at 0.58 V vs. Ag/AgCl(sat.KCl); insets show the enlarged view of current density fluctuations. | ||
Theoretical analysis on NiO and Ni2O3
DFT study has been performed based on first-principles calculations using the VASP simulation package.40 As revealed by HRTEM analysis, the surface-exposed planes namely (111) and (002) of NiO and Ni2O3, respectively, are the active surfaces towards the electrochemical UOR. In this DFT study, both the Ni and O-terminated NiO (111) surfaces are considered, while Ni2O3 (002) can have only one stable surface termination as suggested by earlier studies.41,42 The contribution from Ni(OH)2 in Ni2O3 (002) was taken into account by hydrogenating a few Ni–O bonds in Ni2O3. However, as evident from experimental studies, under the influence of applied electrode potential in alkaline medium the catalyst surface will undergo surface reconstruction resulting in a hydroxylated active NiOOH surface before the UOR and this phenomenon is well explored in the literature.11,43 Accordingly, we have constructed in situ generated dynamic hydroxylated surfaces and found out that the formation of a hydroxylated surface is more facile for Ni2O3 compared to NiO due to the presence of active Ni(III) centers on the Ni2O3 surface (Fig. S16†). The OH adsorption energies per OH species are −0.75 eV for the pristine NiO surface and −0.98 eV for the pristine Ni2O3 surface, indicating stronger OH adsorption on the Ni2O3 surface.In a typical UOR process, the primary step is the adsorption of CO(NH2)2 (urea) on the surface of the electrocatalyst. Then the subsequent deprotonation of CO(NH2)2 occurs in an alkaline medium in the presence of OH− ions resulting in CO2 as the oxidized product.11,43,44 The detailed mechanism of urea electro-oxidation is given in the ESI (Table S6†). However, as proposed earlier, the adsorption of CO(NH2)2 and desorption of CO2 are the key processes in the electrochemical UOR.11,43,44 The stronger the adsorption of CO(NH2)2, the higher will be the UOR activity. According to DFT calculations, urea prefers to adsorb on the active hydroxylated Ni2O3 surface with an adsorption energy of −0.85 eV, while the adsorption energy on both the hydroxylated NiO (111) surfaces is −0.12 eV (Ni-termination) and −0.14 eV (O-termination) (Fig. 9 and S17†). Due to the unique surface structure of the hydroxylated Ni2O3 (002) surface, urea adsorbs on the Ni2O3 surface through strong covalent bonding interaction (between the Ni atom of the surface and the O atom of urea), while on hydroxylated NiO surfaces the adsorption occurs due to weak hydrogen bonding interactions resulting in stronger urea adsorption on the Ni2O3 surface. Besides adsorption energy, the stronger adsorption of urea on the Ni2O3 surface can also be rationalized through Bader charge population analysis and charge density difference (CDD) plots (Fig. 9 and S17†). Bader charge analysis quantifies that 0.18|e| charge is transferred from the Ni2O3 surface to urea, while merely 0.04|e| charge transfer occurred from the NiO surface to adsorbed urea, justifying stronger urea adsorption on Ni2O3. Bader charge analysis further supports CDD plots indicating enhanced charge transfer interaction and charge accumulation around the interacting region for the urea adsorbed Ni2O3 surface compared to the NiO surface.
 | ||
| Fig. 9 Optimized structures and corresponding charge density difference (CDD) plots: (a and b) urea adsorbed, (c and d) CO2 adsorbed Ni-terminated hydroxylated NiO (111) surface, (e and f) urea adsorbed, (g and h) CO2 adsorbed hydroxylated Ni2O3 (002) surface. Isodensity value at surfaces is ±0.004 e a.u.−3 (positive: cyan and negative: olive). | ||
Another important parameter for the electrochemical UOR is the facile desorption of CO2 from the catalyst surface, which is considered the rate-limiting step for the UOR.11,43,44 A weaker CO2 adsorption is essential for an efficient UOR catalyst.11,43,44 As evident from Fig. 9(c) and S17(c),† NiO surfaces strongly adsorb CO2 through bonding interaction of the C atom of CO2 and the O atom of hydroxylated surfaces as well as hydrogen bonding with an adsorption energy of −0.98 and −0.92 eV. The stronger adsorption of CO2 can be vividly visualized from the bent structure of adsorbed CO2. The linear CO2 molecule (with ∠OCO angle 180° and C–O bond length 1.16 Å) bends to ∠OCO = ∼164° with the increase of the average C–O bond distance (1.22 Å) upon interaction with NiO surfaces, while relatively weaker adsorption of CO2 (adsorption energy −0.45 eV) occurs on the Ni2O3 surface with bonding interaction with the O atom of CO2 resulting in less bending of adsorbed CO2 (∠OCO = ∼178° and average C–O distance 1.17 Å). Moreover, Bader charge analysis and CDD plots further exhibit more charge transfer from the NiO surface (0.16|e|) to CO2, while for Ni2O3 0.07|e| charge is transferred (Fig. 9(d), (h) and S17(d)†). The adsorption energies of reactants and CO2 for both the catalysts are compared in Table S7.† From this study, it can be envisaged that the Ni2O3 surface retards the CO2 poisoning effect more efficiently compared to the NiO surface. Hence, based on two important factors namely the adsorption of urea and CO2, it can be concluded that the Ni2O3 surface is superior to NiO for the electrochemical UOR with relatively stronger urea adsorption and weaker CO2 adsorption ability.
Conclusions
This study projects Ni2O3 as a promising electrocatalyst for the urea oxidation reaction with its high activity and sustained urea oxidation reaction, which are correlated with the higher Ni3+/Ni2+ active species formation, effective reactant adsorption, and better tolerance towards CO2 poison. These are reflected in the observed higher UOR performance and lower Tafel slope for Ni2O3 compared to the conventional NiO catalyst at the same electrolyte concentration of urea and KOH. Detailed voltammetry and impedance studies are undertaken to get better insight into the catalyst activity and poison tolerance. A perfect reversal of impedance from the first quadrant to the second/third quadrants is observed for the NiO catalyst in a given applied potential window, implying that NiO is prone to COx poisoning to a large extent, while for the Ni2O3 catalyst, a complete reversal is not observed and this is an indication of COx tolerance in Ni2O3 as there is significant surface saturation by OH− ions even in 1 M KOH. The surface saturation is associated with effective COx removal during the UOR and confirmed by cyclic voltammetry and impedance. A 70% UOR activity retention at a current density of 25 mA cm−2 for a 25 h duration is shown for Ni2O3, while the NiO catalyst can only retain 37% of the initial low current density for the same duration. The sustained UOR performance for Ni2O3 is due to better COx tolerance. Moreover, as gleaned from theoretical calculations, Ni2O3 has better reactant (urea and hydroxyl) adsorption as compared to NiO. At the same time, NiO has strong adsorption towards CO2, which poisons the catalyst surface and affects its long-term activity. The Ni2O3 catalyst presents weaker adsorption to CO2 facilitating better tolerance and prolonged UOR activity. This study provides a correlation between prominent hydroxylation and COx tolerance of the catalyst and gives insights into designing a UOR catalyst with optimum adsorption of reactants and poisons on its surface.Conflicts of interest
There are no conflicts of interests to declare.Acknowledgements
MS and CA thank CeNS for the research fellowship. The authors also thank CRF, CeNS for experimental facilities. The authors acknowledge MNIT, Jaipur for XPS analysis. R. J. thanks IACS and DST for research fellowship and A. D. thanks TRC-DST and SERB grant no. DIA/2018/000013 and CRG/2020/000301 for partial funding.References
- H. Zhou, F. Yu, Q. Zhu, J. Sun, F. Qin, L. Yu, J. Bao, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2018, 11, 2858–2864 RSC.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- The United Nations World Water Development Report 2017. Wastewater: the Untapped Resource, UNESCO. Paris, 2017, vol. 53 Search PubMed.
- J. H. Meessen, Urea in Ullmann's Encyclopedia of Industrial Chemistry, 2010, DOI:10.1002/14356007.a27_333.pub2.
- V. Vedharathinam and G. G. Botte, Electrochim. Acta, 2013, 108, 660–665 CrossRef CAS.
- V. Vedharathinam and G. G. Botte, Electrochim. Acta, 2012, 81, 292–300 CrossRef CAS.
- G. Gnana, A. Farithkhan and A. Manthiram, Adv. Energy Sustainable Res., 2020, 2000015, 1–17 Search PubMed.
- F. Guo, K. Ye, K. Cheng, G. Wang and D. Cao, J. Power Sources, 2015, 278, 562–568 CrossRef CAS.
- R. M. Abdel Hameed and S. S. Medany, J. Colloid Interface Sci., 2018, 513, 536–548 CrossRef CAS PubMed.
- H. M. Abd El-Lateef, M. M. Khalaf and I. M. A. Mohamed, Ceram. Int., 2020, 46, 20376–20384 CrossRef CAS.
- S. Lu, M. Hummel, Z. Gu, Y. Wang, K. Wang, R. Pathak, Y. Zhou, H. Jia, X. Qi, X. Zhao, B. Bin Xu and X. Liu, ACS Sustainable Chem. Eng., 2021, 9, 1703–1713 CrossRef CAS.
- Z. Yue, W. Zhu, Y. Li, Z. Wei, N. Hu, Y. Suo and J. Wang, Inorg. Chem., 2018, 57, 4693–4698 CrossRef CAS PubMed.
- B. Zhang, S. Wang, Z. Ma and Y. Qiu, Appl. Surf. Sci., 2019, 496, 143710 CrossRef CAS.
- L. Wang, S. Zhu, N. Marinkovic, S. Kattel, M. Shao, B. Yang and J. G. Chen, Appl. Catal. B Environ., 2018, 232, 365–370 CrossRef CAS.
- C. Alex, G. Shukla and N. S. John, Electrochim. Acta, 2021, 385, 138425 CrossRef CAS.
- D. S. Hall, D. J. Lockwood, C. Bock and B. R. MacDougall, Proc. R. Soc. A, 2015, 471, 20140792, DOI:10.1098/rspa.2014.0792.
- C. Li and S. Liu, J. Nanomater., 2012, 1–6, 648012, DOI:10.1155/2012/648012.
- S. Dey, S. Bhattacharjee, M. G. Chaudhuri, R. S. Bose, S. Halder and C. K. Ghosh, RSC Adv., 2015, 5, 54717–54726 RSC.
- M. Fingerle, S. Tengeler, W. Calvet, T. Mayer and W. Jaegermann, J. Electrochem. Soc., 2018, 165, H3148–H3153 CrossRef CAS.
- R. E. Dietz, G. I. Parisot and A. E. Meixner, J. Appl. Phys., 1971, 42, 1484 CrossRef.
- B. Zhao, X. K. Ke, J. H. Bao, C. L. Wang, L. Dong, Y. W. Chen and H. L. Chen, J. Phys. Chem. C, 2009, 113, 14440–14447 CrossRef CAS.
- M. A. Peck and M. A. Langell, Chem. Mater., 2012, 24, 4483–4490 CrossRef CAS.
- A. F. Carley and S. Rassias, Surf. Sci., 1983, 135, 35–51 CrossRef CAS.
- H. A. E. Hagelin-Weaver, J. F. Weaver, G. B. Hoflund and G. N. Salaita, J. Electron Spectrosc. Relat. Phenom., 2004, 134, 139–171 CrossRef CAS.
- K. S. Kim and N. Winograd, Surf. Sci., 1974, 43, 625–643 CrossRef CAS.
- E. L. Ratcliff, J. Meyer, K. X. Steirer, A. Garcia, J. J. Berry, D. S. Ginley, D. C. Olson, A. Kahn and N. R. Armstrong, Chem. Mater., 2011, 23, 4988–5000 CrossRef CAS.
- S. Hu, S. Wang, C. Feng, H. Wu, J. Zhang and H. Mei, ACS Sustainable Chem. Eng., 2020, 8, 7414–7422 CrossRef CAS.
- D. Liu, T. Liu, L. Zhang, F. Qu, G. Du, A. M. Asiri and X. Sun, J. Mater. Chem. A, 2017, 5, 3208–3213 RSC.
- Y. Ding, Y. Li, Y. Xue, B. Miao, S. Li, Y. Jiang, X. Liu and Y. Chen, Nanoscale, 2019, 11, 1058–1064 RSC.
- B. K. Boggs, R. L. King and G. G. Botte, Chem. Commun., 2009, 4859–4861 RSC.
- O. Diaz-Morales, D. Ferrus-Suspedra and M. T. M. Koper, Chem. Sci., 2016, 7, 2639–2645 RSC.
- S. M. Park and J. S. Yoo, Anal. Chem., 2003, 75, 455–461 CrossRef.
- F. Seland, R. Tunold and D. A. Harrington, Electrochim. Acta, 2006, 51, 3827–3840 CrossRef CAS.
- G. X. Cai, J. W. Guo, J. Wang and S. Li, J. Power Sources, 2015, 276, 279–290 CrossRef CAS.
- I. M. A. Mohamed, P. Kanagaraj, A. S. Yasin, W. Iqbal and C. Liu, J. Alloys Compd., 2020, 816, 152513 CrossRef CAS.
- I. M. A. Mohamed and C. Liu, Appl. Surf. Sci., 2019, 475, 532–541 CrossRef CAS.
- F. Guo, K. Ye, M. Du, X. Huang, K. Cheng, G. Wang and D. Cao, Electrochim. Acta, 2016, 210, 474–482 CrossRef CAS.
- G. Jerkiewicz, ACS Catal., 2020, 10, 8409–8417 CrossRef CAS.
- S. Qian, Z. Rao, Y. Liu, J. Yan, B. Fan, Y. Gui and F. Guo, Electrochim. Acta, 2020, 330, 135211 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS PubMed.
- W. Zhao, M. Bajdich, S. Carey, A. Vojvodic, J. K. Nørskov and C. T. Campbell, ACS Catal., 2016, 6(11), 7377–7384 CrossRef CAS.
- W. B. Zhang and B. Y. Tang, J. Chem. Phys., 2008, 128, 124703 CrossRef PubMed.
- S. Lu, M. Hummel, S. Kang, R. Pathak, W. He, X. Qi and Z. Gu, ACS Omega, 2021, 6(22), 14648–14654 CrossRef CAS PubMed.
- Z. Ji, J. Liu, Y. Deng, S. Zhang, Z. Zhang, P. Du, Y. Zhao and X. Lu, J. Mater. Chem. A, 2020, 8, 14680–14689 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta05753g |
| This journal is © The Royal Society of Chemistry 2022 |