
Effects of cyclic and acyclic amidine side-chains on the properties of polysiloxane ionomers constructed in situ from three uncharged components†
Louis
Poon
,
Jacob R.
Hum
and
Richard G.
Weiss
 *
*
Department of Chemistry and Institute for Soft Matter Synthesis and Metrology, Georgetown University, Washington, DC 20057-1227, USA. E-mail: weissr@georgetown.edu
First published on 18th July 2022
Abstract
Ionomers, polysiloxanes with imidazolinium dithiocarbamate side chains, have been synthesized in situ from three uncharged components—a polysiloxane with imidazole side chains, CS2, and hexylamine or octadecylamine. Their structural and dynamic properties are compared over a temperature range of 0–50 °C with those of the analogous ionomers in which the polysiloxanes have amidinium side chains. The results, primarily from differential scanning calorimetry, powder X-ray diffraction measurements, and rheology show that the small structural (and smaller electronic) differences between the cyclic 5-membered ring imidazolinium and acyclic amidinium groups have marked effects on the bulk properties of the ionomers. These include their shear strengths and the manner in which the microcrystalline portions of the ionomers with dithiooctadecylcarbamate anions are packed. Thus, it is possible to finely tune the natures of the ionomers from one polysiloxane by changing temperature, the chain length of the alkylamine, and the nature of the base attached to the polysiloxane chain. Why these changes occur to the various properties is discussed.
1. Introduction
Ionomers are copolymers with small amounts of charged or ionizable groups.1 In low polarity environments, they undergo microphase separation into clusters of charged centers within the larger amorphous phase contributed by the polymer backbone. The dual nature of ionomers, one part charged and another part lipophilic, has been exploited in applications that require materials to be relatively water resistant but also contain groups that can interact with hydrophilic species.2–7 Key to these and other applications is understanding how the polymer backbone and charged groups affect an ionomer and its solution properties and how the solvent properties affect the ionomer.Previously, we investigated the in situ construction of neat ionomeric materials constituted from two uncharged components–polysiloxanes with amino side chains and carboxylic acids or simple triatomic molecules—and from three uncharged components–polysiloxanes with amidine side chains, an alkylamine, and one of several small gaseous molecules, principally CO2 or CS2.5,8,9 Whereas the ionomers employing CO2 are stable under one atmosphere of that gas, they revert slowly to their non-ionomeric polysiloxane and alkylamine mixtures in air at room temperature. However, the ionomers employing CS2 are stable at room and at elevated temperatures in the air for reasons dealing with the different thermodynamic stabilities of CO2 and CS2; vide infra.
Here, we report the syntheses and properties of analogous ionomers, constructed again in situ from three uncharged components–hexylamine or octadecylamine, CS2, and a polysiloxane with imidazole side chains. Because a major focus of the research is to probe differences between how cyclic 5-membered ring imidazolinium and acyclic amidinium groups (Fig. 1A) attached to the side chains influence the structural and dynamic properties of the ionomers at different temperatures, the only triatomic gas employed is CS2. An additional impetus for using imidazolinium is its important role in influencing the properties of materials, such as pharmaceuticals,10–14 corrosion inhibitors,15–18 reaction catalysts,19 and switchable surfactants.20–22 Also, substituted polysiloxanes have been used widely as tunable hydrophilic–lipophilic matrixes for many other applications.7,23,24 We find here that although the amidinium and imidazolinium functional groups are very similar electronically, their influences on the properties of the polysiloxane-based ionomers are quite different in several important respects. For reasons that will be alluded to below, we prefer to attribute those difference to entropic differences rather than π-stacking. In fact, these ionomers are different from many others because of their ease of structural modification that allows the various structural factors to be tuned and related to the bulk properties.
 | ||
| Fig. 1 (A) Amidinium (left) and imidazolinium (right) groups; (B) choosing the triatomic CX2 and alkyl chain length; (C) ionic (coulombic) attractions among the ionic centers along the chains and van der Waals (dispersive) interactions along the alkyl chains of the anions; and (D) one of the resulting materials applied as an adhesive to borosilicate glass surfaces. | ||
Like amidine, imidazoline is known to be a stronger organo-base than mono-substituted alkylamines.25 The pKa of imidazoline is similar to that of amidine. For example, the pKa of the acyclic amidine, N,N-dimethyl-N′-octanamide, is 12.2 while that of the cyclic imidazoline analogue. 1-Methyl-2-octyl-4,5-dihydro-1H-imidazole, has a pKa of 11.0.22 Amidine and imidazoline functional groups are polar and uncharged; in the presence of an acid, they can be converted to protonated ionic forms. Thus, a polysiloxane with amidine or imidazoline pendant groups can be changed into an ionomer26 with amidinium or imidazolinium pendant groups. Also, depending on the specific nature of an amidine or imidazoline, the material in which it resides may be hygroscopic or hydrophobic.27,28 Regardless, a hydrophobic backbone like that of a polysiloxane can retard water from entering the system, making water sensitive applications involving electrolyte media possible.23,29
Amidinium salts can be generated easily through the reaction of an amidine with an alkylamine and either CO2 (forming an amidinium carbamate) or CS2 (forming an amidinium dithiocarbamate);30,31 the same procedure generates the analogous imidazolinium alkylcarbamates and alkyldithiocarbamates. As noted in our previous work,9 amidinium alkyldithiocarbamate ionomers are thermally stable at room temperature and allow the bulk properties of these ionomers to be studied. The same is not true for amidinium alkylcarbamate-based systems which revert to a mixture of amidine, amine, and CO2 when mild heat is applied.30,31 This reversibility is advantageous for applications in switchable materials but not in measuring bulk properties that require thermal stability.
The reversibility of the alkylcarbamates is, in large part, a consequence of the strongly negative heat of formation of CO2 (−393 kJ mol−1).31 The irreversibility of the alkyldithiocarbamates when heated can be attributed mainly to the very positive heat of formation of CS2 (+115 kJ mol−1)31 and the conversion of the alkyldithiocarbamates into thioureas at elevated temperatures.31 Thus, according to the Hammond postulate,32 adducts between CO2 and an amine are more prone to dissociate than those between CS2 and an amine. Regardless, amidinium alkyldithiocarbamate ionomers are a good model for the less thermally stable amidinium alkylcarbamate ionomers in many respects. Previously, we demonstrated that both the bulk and solution properties of amidinium–alkylcarbamate, room-temperature ionic liquids are similar in magnitude to their amidinium alkyldithiocarbamate counterparts.31
In that regard, studies of neat imidazolinium alkyldithiocarbamate ionomer systems can indicate which aspects of their amidinium ionomer counterparts are most important in imparting desired properties for different future applications. Although the structural and electronic differences between an imidazoliniun group and an amidinium group are small, the lower conformational lability of the former was expected, and has been found, to influence the polymer chain packing and, thus, affect several microscopic and bulk properties.
Similar to their amidinium analogues, polysiloxane-based ionomers with imidazolinium hexyldithiocarbamate pendant groups are viscous liquids at room temperature, while the 20% imidazolinium ionomers with longer octadecyldithiocarbamate chains show structurally rich morphologies with regions of crystallinity that are different from those of their amidinium counterparts. By combining the body of DSC and PXRD results, it has been possible to probe the phase transitions and assign the packing geometries of the partially-crystalline phases of the polysiloxane with 20% imidazolinium octadecyldithiocarbamate side chain groups.
2. Experimental
Syntheses of the imidazoline polymer and the imidazolinium alkyldithiocarbamate ionomers are detailed in the ESI† of this manuscript. Syntheses of the analogous amidine and amidinium alkyldithiocarbamate ionomers have been reported in a recent publication.9 Many details of the synthetic steps and characterization protocols are presented in ESI.† Structural analyses of the neat materials include differential scanning calorimetry (DSC) measurements, variable temperature powder X-ray diffraction (PXRD), and polarizing optimal microscopy (POM). A general reaction pathway for the synthesis of the imidazolium materials is shown in Fig. 2 and acronyms for them and some of their basic structural properties are summarized in Table 1. | ||
| Fig. 2 General pathways for syntheses of PDMS-based ionomers (R = –C6H13 or –C18H37) from PDMS-based copolymers with randomly distributed N-propylethylenediamine groups (AEAP): (i) 2.0 M dimethylamine and N,N-dimethylacetamide dimethyl acetal (DMADA) in THF under N2 at room temperature; (ii) hexylamine or octadecylamine and CS2 in CHCl3 at 0 °C. The filled black sphere represents an imidazolinium group on a PDMS backbone; n = 18–24% of the monomer units within a chain. | ||
| Amidine or imidazoline-substituted PDMS copolymers and their acronyms | Anion chain lengths and ionomer acronyms | ||
|---|---|---|---|
| Amidine or imidazoline substituted PDMS | Calcd. amidine or imidazoline Mw rangea (KDa) and amidine or imidazoline/imidate ratios (in parentheses)b | R = −C6H13 | R = −C18H37 |
| a Average molecular weight ranges of amidine or imidazoline-substituted PDMS calculated from viscosity data. b Ratio of amidine or imidazoline and imidate ester groups along a polymer chain after reaction of amino groups. c See ref. 34 for details about 1 and its ionomers. | |||
| 1c | 29.5–32.0 (85/15) | 1–6 | 1–18 |
| 2 | 15.4–20.5 (88/12) | 2–6 | 2–18 |
3. Results and discussion
3.1. DSC and PXRD studies of imidazolinium alkyldithiocarbamate-based salts
Although there are similarities between the rheological properties of 2–18 and 1–18, their thermal and structural properties are markedly different. There are at least 4 unique first-order transitions in the heating portions of the two DSC thermograms belonging to 2–18 (Fig. 3). During the first cooling, a first-order broad exotherm and a peak temperature of 34.0 °C was observed. In the second heating, two corresponding endotherms in the same temperature region were detected at 21 and 30 °C. Other enthalpy values are in collected Table S1 of the ESI.†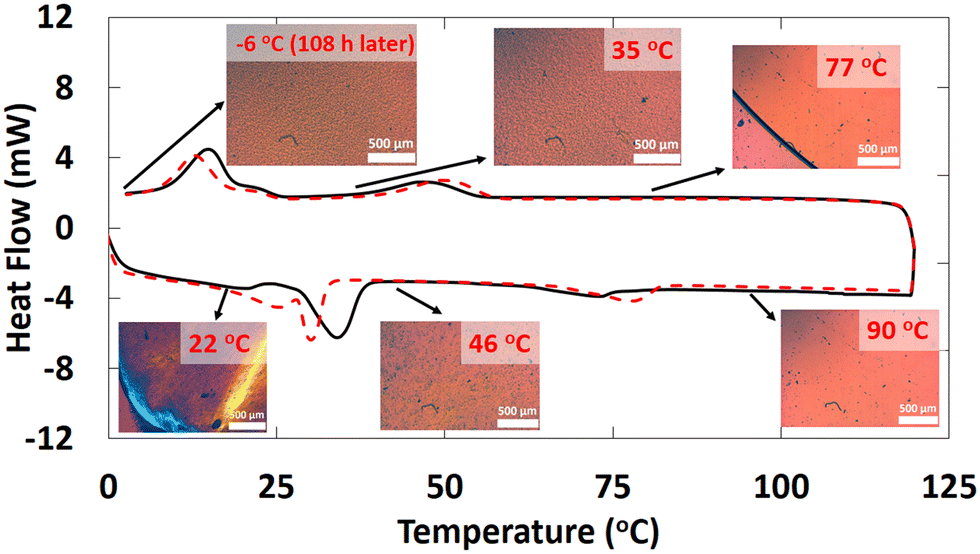 | ||
| Fig. 3 DSC thermogram of 2–18 for the first (black) and second heating–cooling cycle (red dashed). Polarized optical micrographs taken during the first heating and cooling are shown at the indicated temperatures. | ||
The species responsible for only two of the multiple transitions could be assigned using DSC and PXRD. One of the unknown states (Y-state, Fig. 4) may exhibit crystalline domains like a triclinic lattice (δ-state) that undergo a transition at 33.7 °C and that does not have a detectable corresponding endotherm during the second heating.33 Instead, two endotherms with onset temperatures at ∼22 °C (broad; peak at 25.4 °C) and ∼30 °C (sharper; peak at 30.1 °C) were found.
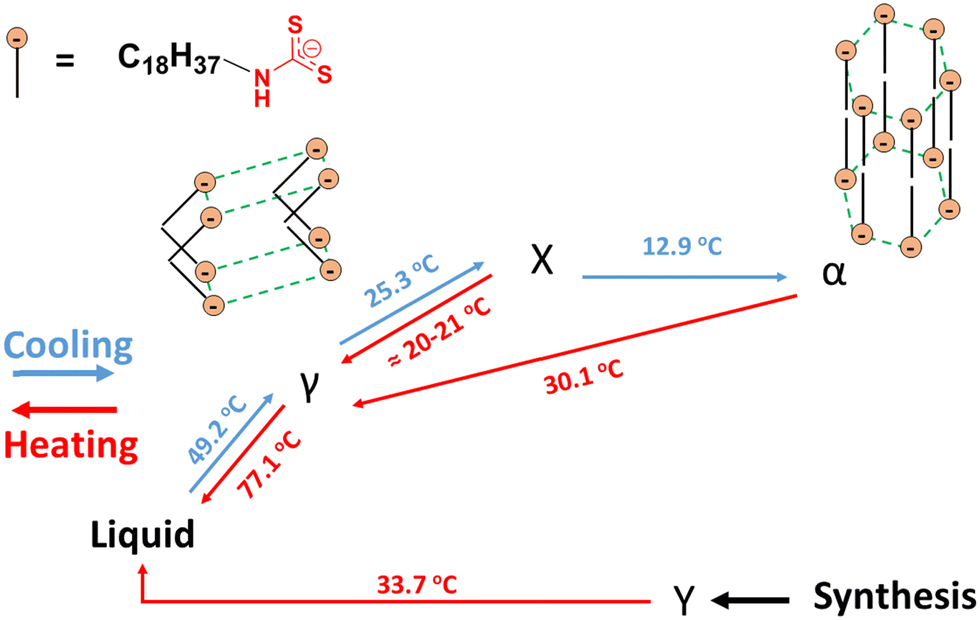 | ||
| Fig. 4 Cartoon representations of the proposed packing arrangements of the anions and designations of transition types and their peak temperatures for morphs of the 2–18 ionomer, based on DSC and PXRD data. ‘Synthesis’ refers to the initially isolated material from a reaction mixture in which the solvent was chloroform. | ||
The proposed assignments of the structures within the phases of the 2–18 ionomer, based in part on the data shown in Fig. 5 and in Table S2, (ESI†) are summarized in Fig. 4. Packing of the octadecyl chains is a result of the net contributions of van der Waals (i.e., dispersive) and Coulombic (i.e., electrostatic) interactions. In the absence of single-crystal type information about the packing within the ionic aggregates, additional detail about the side-chain orientations is conjectural. In fact, electrostatic repulsions among vicinal anionic groups and separately among proximal cations attached to the polysiloxane chains may force the crystalline centers within a layer to adopt crenellated arrangements. Regardless, when 2–18 was cooled after initial heating to its isotropic phase, three transitions, two of which are overlapping, were observed. The broader exotherm (centered at 12.9 °C) is consistent with a solid–solid transition whose heat is similar to an endotherm (centered at 30 °C) and observed upon heating during the second cycle. The broadness of these DSC peaks is typical of those from neat long-chain ionic surfactants such as n-alkyltrimethylammonium bromides.34 Thus, the second unknown transition (X-state, Fig. 4) is perhaps from an orthorhombic rotator phase to a hexagonal phase (α-state).33 A second, smaller cooling transition of the ionomer, a shoulder on the exotherm peak centered at 12.9 °C, is estimated to be centered at ∼20–21 °C. It appears to be the reverse of the heating transition from the α-state to the γ-state. On this basis, the large exotherm at 46.6 °C is due to crystallization to the γ-state. Similar to 1–18, ion association within the polymeric network slows the α–γ transition of 2–18.
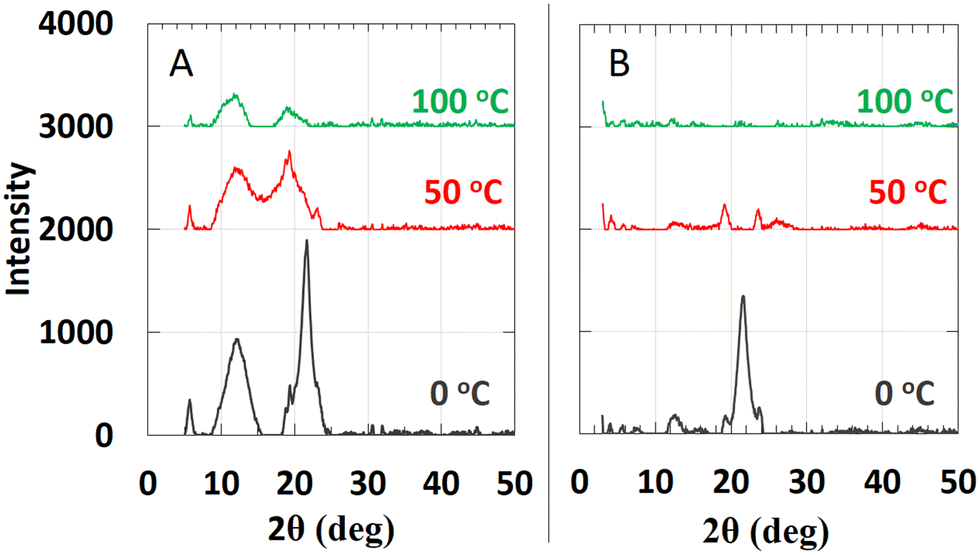 | ||
| Fig. 5 Variable temperature PXRD diffractograms of 2–18: (A) starting at 0 °C, 2–18 was heated from 0 to 100 °C; (B) after 5 days at room temperature, 2–18 was again heated from 0 °C to 100 °C. | ||
The attribution to the α-state and γ-state from the DSC data is supported by PXRD data. Unlike, 1–18, there are intense diffraction peaks at 21.56° and 21.64° in 2θ (d = 4.12 Å and 4.11 Å, respectively), characteristic of hexagonally-packed ionic centers within 2–18 at 0 °C. The two peaks are assigned to the (110) and (111) planes, respectively. The d-spacing for the (110) plane is similar to the ones found for octadecyl acrylate (4.13 Å) and polymethacrylates with octadecyl side chains;33,35 it is responsible for the (110) and (200) crystal planes. Knowing the reciprocal plane for (110) facilitated calculations for the axes, a = b = 8.24 Å. For the γ-state, the a- and b-axes are calculated to be 4.16 Å and 9.08 Å, respectively. Because the α- and γ-states share similar crystal planes for the c-axis, the calculated distance for it, 65.0 Å, using the (003) crystal plane, suggests that it is from two extended octadecyldithiocarbamate anions and a part of the associated imidazolinium cations.36,37 Crystalline domains can be observed at 46 °C in optical micrographs (Fig. 3). They resemble those found throughout the sample when the sample was cooled from the completely amorphous state to 35 °C. At 50 °C, the increase in thermal energy transforms the phases of both 1–18 and 2–18 to herringbone orthorhombic (Table 2). Both 1–18 and 2–18 share similar a and b dimensions but there is greater interdigitation or tilt angle38 for 1–18 than for 2–18.
| Ionomer | Temperature (°C) | Crystal system | a (Å) | b (Å) | c (Å) |
|---|---|---|---|---|---|
| 1–18 | 0 | Orthorhombic | 7.76 | 9.69 | 55.6 |
| 50 | Herringbone orthorhombic | 4.25 | 9.17 | 46.8 | |
| 2–18 | 0 | Hexagonal | 8.24 | 8.24 | 65.0 |
| 50 | Herringbone Orthorhombic | 4.16 | 9.08 | 64.3 | |
Thus, 2–18 at room temperature consists of hexagonally-packed and (a smaller amount) orthorhombic–herringbone crystalline types, with varying degrees of interdigitation of the octadecyl chains (Table 2).
3.2. Bulk rheological and adhesive properties of imidazolinium alkyldithiocarbamate salts
The quality of the fits to the power law is similar for 2–6 and 1–6 in the frequency range explored (Fig. 6), 10–100 rad s−1. Thus, 2–6 behaves like 1a–6, where we postulate that there is no delay to formation or loss of chain entanglements caused by ionic interactions between the polymer backbone and the counter ions. It is possible that the similar behavior is due to the size of the cations. In studies involving polystyrene sulfonate with varying sizes of cations (Li+ to Cs+), the onset of polymer entanglement occurred at a higher frequency39 for larger cations, such as Rb+ or Cs+ than for smaller ones, such as Li+ or Na+.
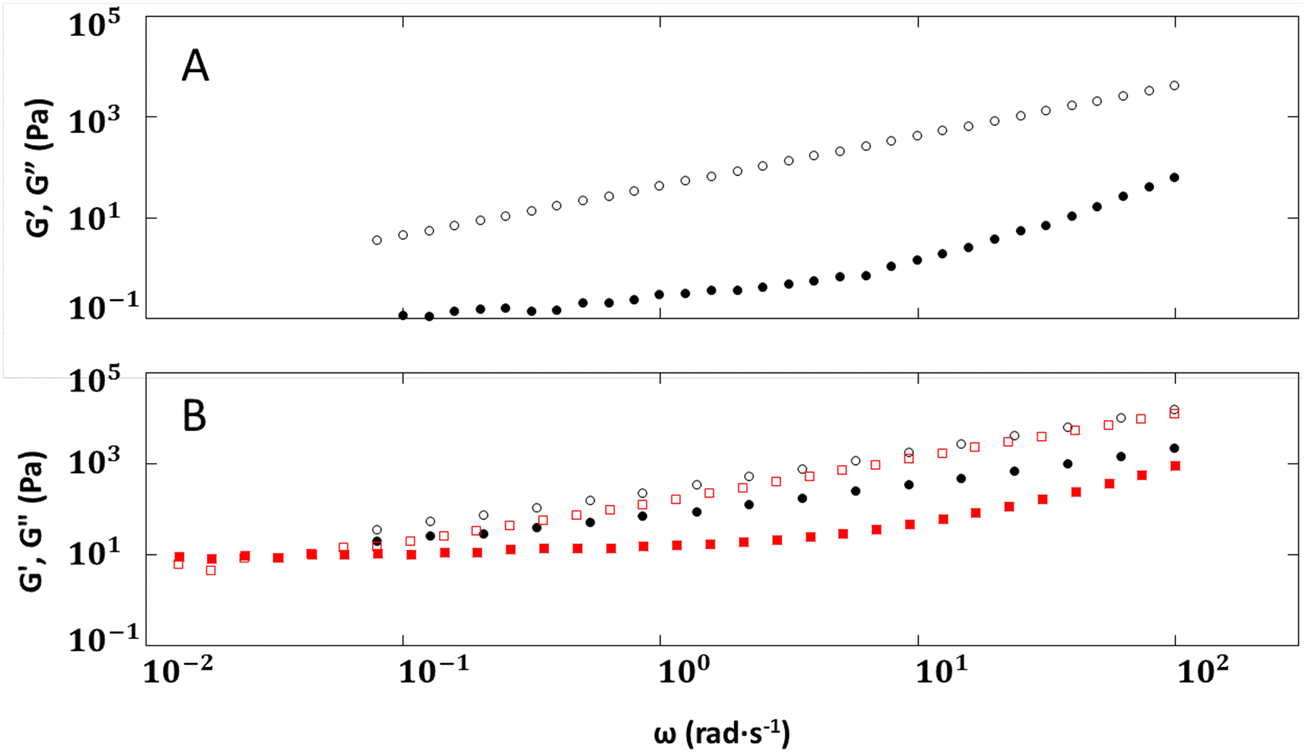 | ||
Fig. 6 Moduli versus angular frequency sweeps (ω) of imidazolinium ionomers with hexyldithiocarbamate and octadecyldithiocarbamate anions: (A) 2–6 (• = G′, ○ = G′′), at 25 °C and 1% strain; (B) 2–18 (• = G′, ○ = G′′) at 40 °C and 0.5% strain and 2–18 ( = G′, = G′,  = G′′) at 50 °C and 0.5% strain. = G′′) at 50 °C and 0.5% strain. | ||
Although the overall shapes of the frequency sweep curves for 1–18 and 2–18 are similar, the delay in segmental relaxation for the imidazoline is again different. Both 1–18 and 2–18 at 40 °C exhibit frequency sweeps showing a transition region, suggesting a highly dynamic microstructure even after a 30 min equilibration. A crossover point can be observed at 40 °C at ω = 0.54 rad s−1 (G′ and G′′ = 248 Pa) for 1–18, while no observable transitions can be observed for 2–18 at the same temperature. Only at a higher temperature, 50 °C, can a crossover point at ω = 0.04 rad s−1 (G′ and G′′ = 10 Pa) be observed for 2–18. Molecular weight differences of the 2 ionomers play a role in chain and segmental relaxation, but 2–18, with the lower molecular weight, was expected to have the shorter relaxation time.
Thus, summarizing the comparison of results between 2–6 and 1–6 and between 2–18 and 1–18, we observe that there are clear consequences to switching from an amidinium to an imidazolinium group in the ionomers. While the overall shapes of the curves are similar for both amidinium and imidazolinium ionomers, the latter exhibit a delay in chain and segmental relaxation, likely due to the greater steric hindrance and the decreased conformational lability of the imidazolinium head group.
3.3. Adhesion studies of 2–18
At room temperature, where ionic centers of 2–18 are fully-extended in their microcrystalline domains, the torque on our rheometer exceeded the acceptable maximum. Thus, to probe the bulk properties within the crystalline domains, single-lap shear experiments were performed. As observed in Fig. 7, the hexagonal phase generates a ductile material, as indicated by the lack of a sharp drop in shear stress (as found for brittle materials) when the ultimate shear strength was achieved. The presence of crystalline domains also provided some resistance to shearing, which reduced the elasticity of the material. The strains at ultimate shear strength for 2–18 were reached at small values, 5% and 3% for trials 1 and 2, respectively.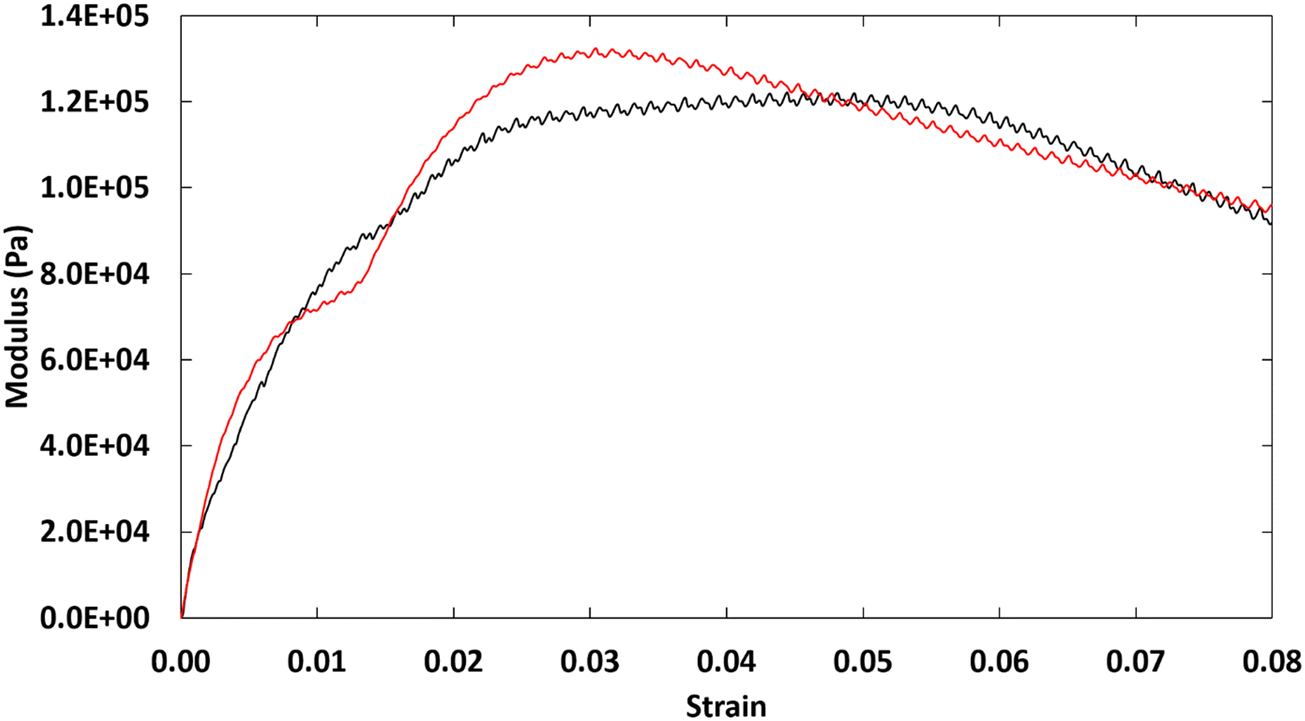 | ||
| Fig. 7 Stress–strain curves for 2–18 at room temperature: trial 1 (black) and trial 2 (red). | ||
When compared to 1–18, we note some intriguing results which show the effect of changing the amidinium side chain to an imidazolinium group. The ultimate shear strength of 2–18 (126 KPa) is nearly 4 times that of 1–18 (39.9 ± 13.3 KPa), but both are within the same order of magnitude. If the shear strength were dependent solely on the molecular weights of 2–18 and 1–18, we would have anticipated 1–18 to have the greater ultimate shear strength. Thus, changing the head group from an amidinium to an imidazolinium does result in some important changes in the mechanical properties of the materials.
A similar difference can be observed in the shear strain. The ionomer 1–18 has an ultimate shear strain of 0.008–0.011; for 2–18, the value is 0.03–0.05. Again, 2–18 is 4 times more elastic than 1–18. From the oscillatory rheological studies at 40 °C, 2–18 has the longer segmental relaxation time, which signifies that its viscoelastic properties remain to a higher temperature. Thus, the larger ductility of 2–18 than 1–18 can be explained if the trend in polymer dynamics at 40 °C is extrapolatable to room temperature. Extension of this explanation to the microstructural crystalline domains is only speculative at this time given the data which are available; it is not known whether the differences in the shear modulus are also be due to the known differences between the packing of 1–18 (extended orthorhombic) and 2–18 (hexagonal). One explanation may relate to the prolonged segmental relaxation in 2–18. As a result of the larger size of the imidazolinium ring, the polymer chains of 2–18 may relax to the rubbery plateau at a slower rate and increase adhesion. In that regard, Turdyn and coworkers40 found that increasing the cation size from lithium to sodium in a polyethylene oxide-based ionomer led to a higher glass transition temperature and better adhesion to some surfaces. Another possible explanation involves the ion aggregates that are formed by 1–18 and 2–18 and their interactions with the amorphous region or glass substrate. As such, while additional experiments are needed, the relationship between the polymer dynamics results from rheology and structural details from PXRD provide a potential path to understanding connections between the bulk mechanical properties of ionomers and their viscoelastic and structural properties; some of these ideas are summarized in cartoon format in Fig. S10 (ESI†).
2–18 and 1–18 do exhibit similar properties in the linear viscoelastic region of their lap-shear curves (Fig. S9, ESI†). This region is highly relevant to the frequency sweeps in the oscillatory rheology studies because both materials have been studied in their linear viscoelastic regions. Thus, the similarities between the shear moduli of 1–18 (0.020 GPa) and 2–18 (0.020 GPa) based on their frequency sweeps (Table S3, ESI†) are not surprising.
4. Conclusions
The subtle structural differences between the amidine and imidazole functional groups attached to the polysiloxane backbones clearly influence the bulk properties of the ionomers; they allow those properties to be ‘fine-tuned’ without introducing significant electronic differences.Thus, careful analyses of the structural and bulk properties of poly(dimethyl)siloxane-based materials with imidazolinium (i.e., cyclic amidinium) alkyldithiocarbamate side chains have uncovered subtle, yet important, differences between their ionomers and analogous materials with acyclic amidinium groups. The differences have been explored principally using DSC and PXRD over temperature ranges where prominent microstructural changes are observed. Based on the evidence in hand and the paucity of data from the literature (N.B., the much lower epoxide ring opening efficiency by an imidazolinium catalyst than by a homologous 7-membered 1,3-diazepinium cation that offers much more conformational lability),41 we attribute those differences primarily to the greater entropy content (i.e., lower conformational constraints) of the amidinium groups than of the cyclic imidazolinium groups. Although π-stacking might be a dominant factor in explaining differences between polysiloxanes with uncharged amidine and imidazoline side groups, it is less likely in polymers with amidinium or imidazolinium, where electrostatic repulsion among the positive charges should make π-stacking much less favorable.
Whereas the imidazolinium ionomer, 2–6, with short hexyldithiocarbamate side chains, remains a viscous liquid at 0 °C, and exhibits no detectable peaks in both DSC curves and PXRD diffractograms over the temperature range explored, the corresponding ionomer with octadecyldithiocarbamate groups, 2–18, exhibits a much more complex thermal and morphological profile due to the much greater van der Waals interactions among its longer chains. At 0 °C, 2–18 packs in a hexagonal phase where the alkyl chains in the octadecyldithiocarbamate are fully extended. This is in contrast to the analogous acyclic amidinium octadecyldithiocarbamate ionomer, 1–18, which is in an orthorhombic phase at the same temperature. However, both 2–18 and 1–18, when heated to 40 °C, form a herringbone orthorhombic phase. Thus, it is not surprising that both 1–18 and 2–18 exhibited similar frequency sweep rheology profiles at 40 °C.
The rheological properties of 2–18 indicate a very dynamic system at 40 °C, even after 30 min of temperature equilibration. Although the values of G′ and G′′ are similar in magnitude for 2–18 and 1–18, 2–18 exhibits the longer segmental relaxation time. This longer relaxation time at 40 °C may be important also in explaining the results from the lap-shear studies performed at room temperature where the imidazolinium ionomer, 2–18, contains crystalline microdomains in which the side chains are hexagonally packed. Surprisingly, the ultimate shear strength (from extrapolation of the relaxation times to room temperature) predicts a shear strain that is ca. four times greater for 2–18 than for 1–18. Although we conjecture that the differences between conformational labilities and the packing of the octadecyldithiocarbamate chains in 2–18 and 1–18 (orthorhombic phase) play a central role here, as well, further investigation is needed to ascertain whether other factors are also important.
The structure–property relationships of the imidazolinium alkyldithiocarbamate ionomers described here provide the foundation for future studies that will exploit the tunability of the PDMS backbones with amidinium- or imidazolinium-based side chains for the development of new ionomeric materials and ionic polymers in a variety of applications, including as pharmaceuticals.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the US National Science Foundation for financial support through Grant CHE-1502856. We are grateful to Dr Xinran Zhang for aid in obtaining some of the rheology and adhesion data and the microscope images, and to Dr Jeffrey A. Bertke for assistance in obtaining and analyzing the diffractograms. We are indebted to Prof. Daniel Blair for advice concerning the interpretation of some of the rheological data.References
- A. D. Jenkins, P. Kratochvíl, R. F. T. Stepto and U. W. Suter, Pure Appl. Chem., 2007, 68, 2287–2311 Search PubMed.
- X. Meng, S. L. Perry and J. D. Schiffman, ACS Macro Lett., 2017, 6, 505–511 CrossRef CAS PubMed.
- S. Magalhães, L. Alves, B. Medronho, A. C. Fonseca, A. Romano, J. F. J. Coelho and M. Norgren, Polymers, 2019, 11, 1–20 CrossRef PubMed.
- C. Creton and M. Ciccotti, Rep. Prog. Phys., 2016, 79, 046601 CrossRef PubMed.
- Y. He, H. Zhao, M. Yao and R. G. Weiss, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3851–3861 CrossRef CAS.
- F. P. García de Arquer, C. T. Dinh, A. Ozden, J. Wicks, C. McCallum, A. R. Kirmani, D. H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef PubMed.
- Y. Lei, S. Zhou, C. Dong, A. Zhang and Y. Lin, React. Funct. Polym., 2018, 124, 20–28 CrossRef CAS.
- T. Yu, K. Wakuda, D. L. Blair and R. G. Weiss, J. Phys. Chem. C, 2009, 113, 11546–11553 CrossRef CAS.
- L. Poon and R. G. Weiss, J. Polym. Sci., 2021, 59, 2345–2354 CrossRef CAS.
- B. Szabo, Pharmacol. Ther., 2002, 93, 1–35 CrossRef CAS PubMed.
- B. K. Yeh, A. Nantel and L. I. Goldberg, Arch. Intern. Med., 1971, 127, 233–237 CrossRef CAS PubMed.
- P. Bousquet, A. Hudson, J. A. García-Sevilla and J. X. Li, Pharmacol. Rev., 2020, 72, 50–79 CrossRef CAS PubMed.
- M. Pigini, P. Bousquet, A. Carotti, M. Dontenwill, M. Giannella, R. Moriconi, A. Piergentili, W. Quaglia, S. K. Tayebati and L. Brasili, Bioorg. Med. Chem., 1997, 5, 833–841 CrossRef CAS PubMed.
- P. Hurwitz and J. M. Thompson, Arch. Ophthalmol., 1950, 43, 712–717 CrossRef CAS PubMed.
- D. Turcio-Ortega, T. Pandiyan and E. M. García-Ochoa, Mater. Sci., 2007, 13, 163–166 Search PubMed.
- L. Xiao, W. Qiao, H. Guo and J. Qu, Tenside, Surfactants, Deterg., 2008, 45, 244–248 CrossRef CAS.
- W. E. G. Hansal, S. Hansal, M. Pölzler, A. Kornherr, G. Zifferer and G. E. Nauer, Surf. Coat. Technol., 2006, 200, 3056–3063 CrossRef CAS.
- G. Chen, J. Feng, W. Qiu and Y. Zhao, RSC Adv., 2017, 7, 55967–55976 RSC.
- H. Liu and D. M. Du, Adv. Synth. Catal., 2009, 351, 489–519 CrossRef CAS.
- W. Qiao, Z. Zheng and Q. Shi, J. Surfactants Deterg., 2012, 15, 533–539 CrossRef CAS.
- M. F. Cunningham and P. G. Jessop, Macromolecules, 2019, 52, 6801–6816 CrossRef CAS.
- A. Darabi, P. G. Jessop and M. F. Cunningham, Chem. Soc. Rev., 2016, 45, 4391–4436 RSC.
- S. Liang, M. V. Oreilly, U. H. Choi, H. S. Shiau, J. Bartels, Q. Chen, J. Runt, K. I. Winey and R. H. Colby, Macromolecules, 2014, 47, 4428–4437 CrossRef CAS.
- V. S. Cobb, W. W. Rauscher, M. A. Stanga, R. E. Stevens, R. H. Whitmarsh and K. D. Wiese, US Pat., 5830970, 1998 Search PubMed.
- J. Y. Quek, T. P. Davis and A. B. Lowe, Chem. Soc. Rev., 2013, 42, 7326 RSC.
- E. S. Wilks, P. Kratochvíl, P. Kubisa, W. Mormann, M. Hess, J. Vohlídal, D. Tabak, R. G. Jones, J. Kahovec, T. Kitayama and R. F. T. Stepto, Pure Appl. Chem., 2006, 78, 2067–2074 Search PubMed.
- U. S. Mahajan, R. R. Godinde and P. N. Mandhare, Synth. Commun., 2011, 41, 2195–2199 CrossRef CAS.
- G. A. Shvekhgeimer and V. I. Kelarev, Chem. Heterocycl. Compd., 1973, 9, 1515–1516 CrossRef.
- A. Jourdain, A. Serghei and E. Drockenmuller, ACS Macro Lett., 2016, 5, 1283–1286 CrossRef CAS PubMed.
- T. Yamada, P. J. Lukac, T. Yu and R. G. Weiss, Chem. Mater., 2007, 19, 4761–4768 CrossRef CAS.
- T. Yu, T. Yamada and R. G. Weiss, Chem. Mater., 2010, 22, 5492–5499 CrossRef CAS.
- G. S. Hammond, J. Am. Chem. Soc., 1955, 77, 334–338 CrossRef CAS.
- M. Yao, J. Nie and Y. He, Macromolecules, 2018, 51, 3731–3737 CrossRef CAS.
- J. K. Cockcroft, A. Shamsabadi, H. Wu and A. R. Rennie, Phys. Chem. Chem. Phys., 2019, 21, 25945–25951 RSC.
- H. Shi, Y. Zhao, X. Zhang, S. Jiang, D. Wang, C. C. Han and D. Xu, Macromolecules, 2004, 37, 9933–9940 CrossRef CAS.
- Lengths were estimated using Avogadro (Universal force field) geometry optimization program. The lengths of two extended octadecyldithiocarbamate anions (2 × 25.05 Å) and 2-methyl-imidazolinium (2 × 3.22 Å) portions of 2-methyl-1-propylimidazolinium summed together.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeerschd, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 476–483 Search PubMed.
- M. George and R. G. Weiss, Langmuir, 2003, 19, 1017–1025 CrossRef CAS.
- Q. Chen, G. J. Tudryn and R. H. Colby, J. Rheol., 2013, 57, 1441–1462 CrossRef CAS.
- G. J. Tudryn, M. V. O’Reilly, S. Dou, D. R. King, K. I. Winey, J. Runt and R. H. Colby, Macromolecules, 2012, 45, 3962–3973 CrossRef CAS.
- O. Sereda, N. Clemens, T. Heckel and R. Wilhelm, Beilstein J. Org. Chem., 2012, 8, 1798–1803 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sm00382a |
| This journal is © The Royal Society of Chemistry 2022 |