
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkylboronic acids as alkylating agents: photoredox-catalyzed alkylation reactions assisted by K3PO4†
Fuyang
Yue
,
Henan
Ma
,
Hongjian
Song
 *,
Yuxiu
Liu
,
Jianyang
Dong
* and
Qingmin
Wang
*
*,
Yuxiu
Liu
,
Jianyang
Dong
* and
Qingmin
Wang
*
State Key Laboratory of Elemento-Organic Chemistry, Research Institute of Elemento-Organic Chemistry, College of Chemistry, Frontiers Science Center for New Organic Matter, Nankai University, Tianjin 300071, People's Republic of China. E-mail: wangqm@nankai.edu.cn; songhongjian@nankai.edu.cn; jydong@mail.nankai.edu.cn
First published on 1st November 2022
Abstract
Despite the ubiquity of alkylboronic acids in organic synthesis, their utility as alkyl radical precursors in visible-light-induced photocatalytic reactions is limited by their high oxidation potentials. In this study, we demonstrated that an inorganophosphorus compound can modulate the oxidation potentials of alkylboronic acids so that they can act as alkyl radical precursors. We propose a mechanism based on the results of fluorescence quenching experiments, electrochemical experiments, 11B and 31P NMR spectroscopy, and other techniques. In addition, we describe a simple and reliable alkylation method that has good functional group tolerance and can be used for direct C–B chlorination, cyanation, vinylation, alkynylation, and allylation, as well as late-stage functionalization of derivatized drug molecules. Notably, alkylboronic acids can be selectively activated in the presence of a boronic pinacol ester.
Introduction
Alkyl radicals (R˙), which are among the most fundamental intermediates in organic chemistry, can be generated from feedstock chemicals such as alkanes, alkenes, amines, carboxylic acids, alcohols, ketones, aldehydes and their derivatives and can be used for various synthetic transformations.1–14 The reactivity of alkyl radicals is complementary to that of other alkyl intermediates, such as carbanions, carbocations, and carbenes; and taken together, these intermediates offer flexible synthetic routes to structurally complex, C(sp3)-rich scaffolds. Traditionally, alkyl radicals were rarely used in reaction design because they are usually generated by means of energy-intensive or complicated procedures. In addition, their reactivity is difficult to control, which can lead to the formation of byproducts.15 However, over the past 10 years, as a result of pioneering work by the groups of MacMillan, Stephenson, Yoon, and others, visible-light-induced free radical reactions have emerged as useful tools for accomplishing synthetic transformations under mild, environmentally benign conditions.16–25 Unlike traditional methods for generating alkyl radicals, visible-light-induced photocatalysis can produce low concentrations of free radicals. Although photochemical approaches have been employed to accomplish economical, scalable, and unique transformations, chemists have continued to search for novel alkyl radical precursors and methods for production of alkyl radicals.26–31We reasoned that organoboron species might be useful for this purpose. Organoborons have long been recognized as valuable synthetic intermediates and have recently become more accessible as highly functionalized building blocks because of the development of flexible synthetic approaches from various functional groups.32,33 Visible-light-induced photoredox reactions involving alkyl radicals generated from organoboron species have been shown to be an important tool for the preparation of high-value organic compounds.32–38 Since the pioneering work reported by the Molander37 and Akita38 groups, trifluoroborates, which are synthesized from boronic esters or boronic acids, have been widely used for various organic transformations, mainly because of their low oxidation potentials [∼1.2 V vs. the saturated calomel electrode (SCE)] and bench stability. In recent years, other boric acid derivatives, such as borate esters, borooctene, and their metal salts (e.g., lithium alkyl borates based on boron ferrocene), have also been explored to enrich their applications (Fig. 1A).39–54
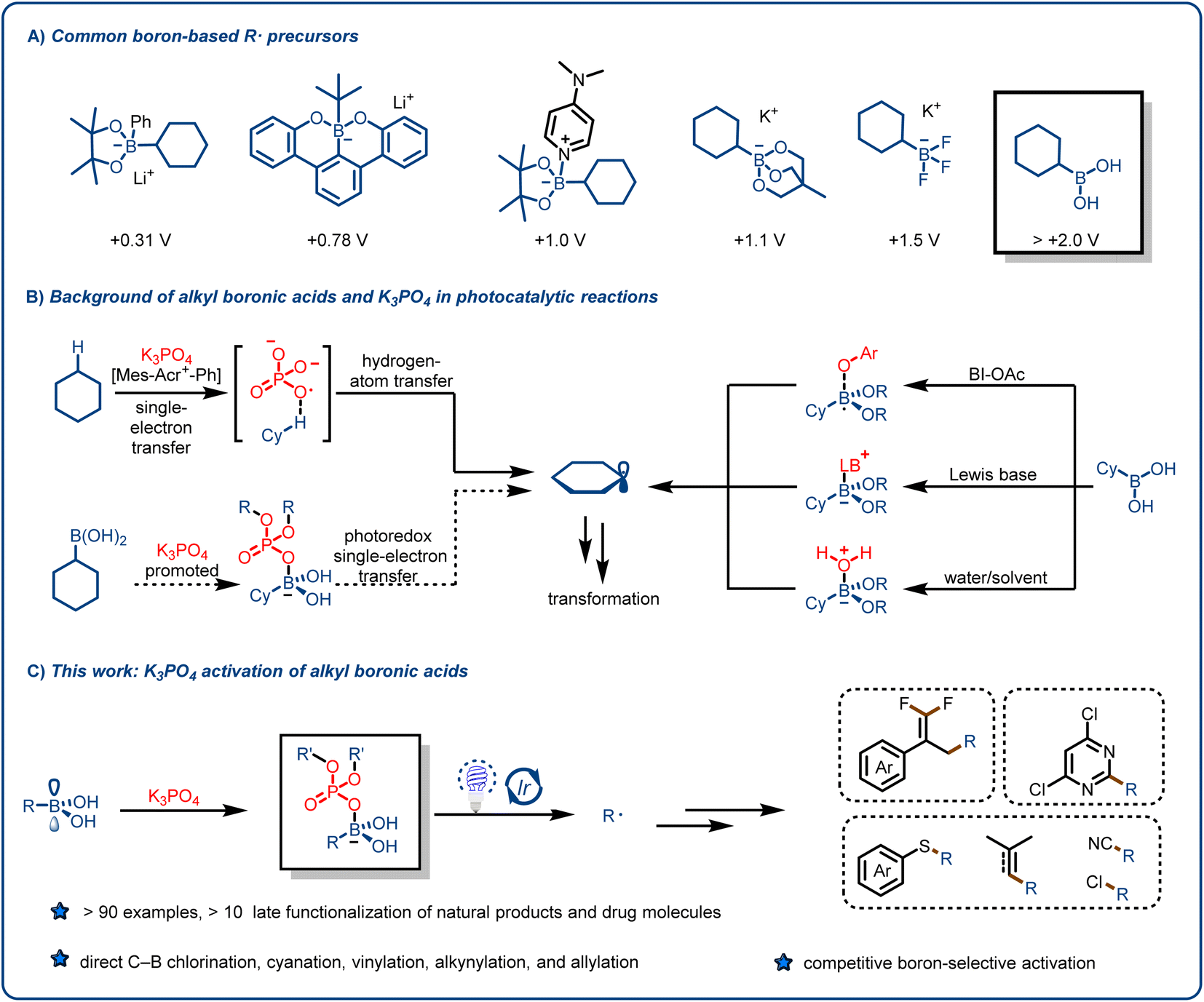 | ||
| Fig. 1 From inspiration to reaction design. (A) Typical boronic acid derivatives used as alkyl radical sources, along with their oxidation potentials (vs. SCE). (B) Background of alkyl boronic acids and K3PO4 in photocatalytic reactions. BI-OAc = acetoxybenziodoxole. (C) Activation of alkylboronic acids by K3PO4. | ||
Although organoboron species have been widely used as sources of alkyl radicals, boronic acids are more suitable precursors because they are widely available from commercial and synthetic sources. In addition, they have a vacant p orbital centered on the B atom, which facilitates the formation reversible-lattice bonds with O or N nucleophiles.55–57 Although the number of applications of boron-containing molecules in the fields of medicine (e.g., tavaborole and crisaborole), materials chemistry, and biomedical engineering has increased substantially in the past decade, maintaining their high commercial availability,39–54 only a few studies have focused on the use of boronic acids as alkyl radical precursors, primarily because of the high oxidation potentials of boronic acids (>2.0 V vs. SCE).39–48 To overcome this challenge, Chen's group activated boronic acids with a mild oxidant (acetoxybenziodoxole) to generate alkyl radical intermediates (Fig. 1B, right),55 but the use of the oxidant limits the applications of this method. Ley and coworkers40 reported the formation of Lewis base adducts of boronic acids, and this mode of activation increased the range of applications of boronic acids and their derivatives (Fig. 1B, right). However, rapid protonation of the carbanion intermediates formed by Giese-type addition limits the opportunity for intramolecular elimination reactions that proceed via polar crossing. The groups of Bloom42 and Sharma56 recently demonstrated the generation of alkyl radicals by means of proton-coupled electron transfer or direct oxidation of boronic acids using water or dimethylformamide as both a solvent and an activating agent (Fig. 1B, right). Chen's group57 reported a method whereby the reaction substrate activates the alkylboronic acids. It is clear from the above-described literature that boronic acids can be used to form various types of C–C and C–heteroatom bonds.
We speculated that we could unlock the full potential of alkylboronic acids as radical precursors and overcome their limitations by introducing a simple reagent such as an organophosphorus compound. These compounds not only have important biological functions but also are widely used in biomedicine, the agrochemical industry, agriculture, materials science, and other fields.58–70 Moreover, Alexanian et al. found that K3PO4 can act as hydrogen-atom-transfer reagents in a highly oxidizing photocatalytic system: specifically, an oxygen-centered radical generated from a phosphate salt can abstract hydrogen atoms from unactivated aliphatic C–H bonds71 (Fig. 1B, left top). Inspired by this work, we hypothesized that the interaction between a suitable activation reagent and the vacant p orbital of a boronic acid would reduce the oxidation potential of the acid and thus result in the release of an alkyl radical under mild photocatalytic conditions (Fig. 1B, left bottom). Herein, we report that our hypothesis was confirmed, and that we were able to develop a widely applicable method for the formation of alkyl radicals from various alkylboronic acids and participation of the generated radicals in functionalization reactions of electron-deficient olefins and other compounds (Fig. 1C).
Results and discussion
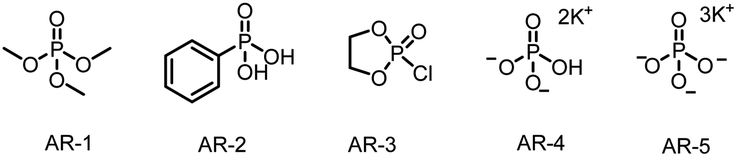
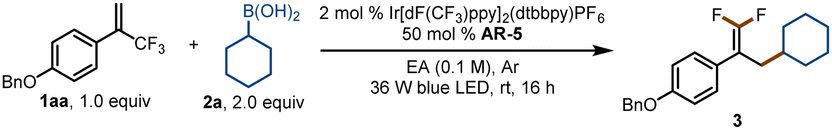
For our initial experiments, we chose 1-(benzyloxy)-4-(3,3,3-trifluoroprop-1-en-2-yl)benzene (1aa) and cyclohexylboronic acid (2a) as model substrates (Table 1). A mixture of the substrates in dry ethyl acetate ([1aa] = 0.1 M) containing Ir[dF(CF3)ppy]2(dtbbpy)PF6 (2 mol%) as a photocatalyst and K3PO4 (AR-5, 0.5 equiv.) as an activation reagent was irradiated with a 36 W blue LED at room temperature under argon for 16 h. To our delight, the desired defluorinative alkylation product 3 was obtained in 82% yield (entry 1). Various other activation reagents were then tested. AR-4 gave a slightly lower yield of 3 (70%, entry 2), but all the other tested activation reagents gave yields of <50% (entry 3). Decreasing the amount of AR-5 decreased the yield of 3 considerably (entry 4), but increasing the amount improved the yield (entries 5–7). The control experiments proved that the photocatalyst, the activation reagent, light, and an oxygen-free atmosphere were essential for the transformation (entries 8–11; see the ESI† for details). Thus, the optimal conditions identified for this defluorinative alkylation involved the use of [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 as a photocatalyst and K3PO4 as an activation reagent in dry ethyl acetate.|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield (%)b |
| a Standard conditions: 1aa (0.2 mmol), 2a (0.4 mmol), photocatalyst (0.004 mmol), activation reagent (0.1 mmol), ethyl acetate (EA, 2 mL), Ar, 36 W blue LED, rt, 16 h. b Yields were determined by 19F NMR spectroscopy with fluorobenzene as an internal standard. NR = no reaction. c Isolated yield. | ||
| 1 | None | 82 (80c) |
| 2 | AR-4 as the activation reagent | 70 |
| 3 | AR-1–AR-3 as the activation reagent | <50 |
| 4 | 0.2 equiv. of AR-5 | 34 |
| 5 | 1.0 equiv. of AR-5 | 95 |
| 6 | 1.5 equiv. of AR-5 | 94 |
| 7 | 2.0 equiv. of AR-5 | 97 |
| 8 | No photocatalyst | NR |
| 9 | No activation reagent | NR |
| 10 | No light | NR |
| 11 | In air | 32 |
|
||
To gain insight into the mechanism underlying the observed reactivity, we performed UV-vis spectroscopy and a fluorescence quenching experiment and prepared a Stern–Volmer plot (Fig. 2A). UV-vis spectroscopy confirmed our assumption that the proposed transient intermediate acted as a reductive quencher for excited-state [Ir(dF(CF3)ppy)2(dtbbpy)]PF6. Although the boronic acid and K3PO4 alone did not quench the photocatalyst, when we mixed the two reagents together, obvious quenching of the photocatalyst was observed (see the ESI† for more details). Next, we carried out a light/dark experiment (Fig. 2B), which showed that when there was no light, the reaction ceased; this result indicates that any chain-propagation process was short-lived and that light was essential for product formation. We carried out electrochemical analyses of each reaction component with Ag/AgNO3 as a reference electrode. We detected a new, low-potential oxidation peak; and the cyclic voltammetry results suggested that coordination between the activation reagent and boronic acid lowered the oxidation potential of the acid (Fig. 2C). As a result, the K3PO4-activated acid could undergo efficient single-electron oxidation by excited-state [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 (E1/2 = +1.21 V vs. SCE in MeCN). These preliminary observations were reinforced by NMR spectroscopy (Fig. 2D). When we added the activation reagent to cyclohexylboronic acid, the signal for the boron atom showed an obvious upfield shift (the spectrum was recorded in D2O). In addition, we found that mixing the activation reagent with boronic acid resulted in a slight downfield shift in the 31P NMR spectrum of the former. Finally, we found that reaction was prevented by radical scavengers (Fig. 2E), and radical-trapping product 1-(cyclohexyloxy)-2,2,6,6-tetramethylpiperidine or (2-cyclohexylethene-1,1-diyl)dibenzene was detected by high-resolution mass spectrometry. This experiment clearly indicates that the reaction proceeded through a free radical pathway.
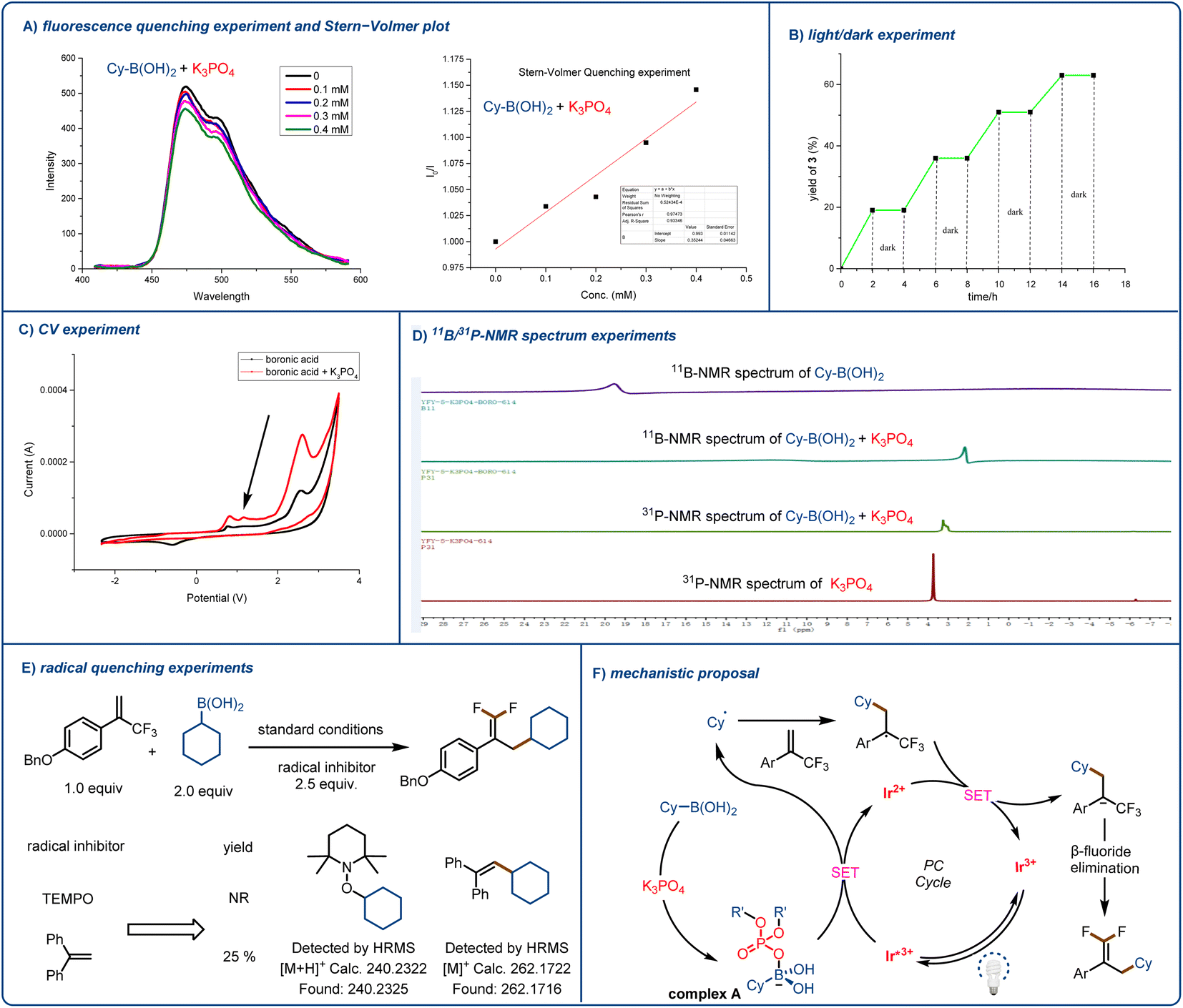 | ||
| Fig. 2 Mechanistic experiments. (A) Fluorescence quenching experiment and Stern–Volmer plot confirming that the excited state of the photocatalyst was quenched by the cyclohexylboronic acid–K3PO4 complex. (B) Light/dark experiment. (C) Cyclic voltammograms showing lowering of the cyclohexylboronic acid oxidation potential by K3PO4. (D) 11B NMR and 31P NMR spectra in D2O (0.5 mL). (E) Radical quenching experiments. (F) Proposed mechanism, R′ = cyclohexylboronic acid. | ||
In light of these observations, we propose the mechanism illustrated in Fig. 2F. First, the photocatalyst is excited by blue light, and the resulting excited state is quenched by complex A to form an alkyl radical. The radical adds to the α-trifluoromethyl arylalkene to afford a CF3-substituted styrene carbon radical. This radical undergoes single-electron reduction by the reduced photocatalyst to form a carbanion, which undergoes an E1cb elimination reaction to afford the desired product.
With the optimized conditions in hand, and convincing evidence for the proposed mechanism, we set out to study the scope of the reaction with respect to alkylboronic acid (Fig. 3). The conditions were broadly applicable for defluorinative alkylation reactions of 1aa and showed outstanding selectivity and functional group compatibility. Various primary boronic acids, including halogenated long-chain alkylboronic acids and an iodine-substituted aromatic alkylboronic acid, were suitable substrates; the desired products (4–9) were obtained in 72–82% yields. Two secondary boronic acids gave 10 and 11 in 87% and 85% yields, respectively, which were slightly higher than the yields obtained with primary alkylboronic acids. Secondary alkylboronic acids bearing five- and six-membered rings, including bulky, sterically demanding groups, gave 3 and 12–15 in excellent yields. In addition, we were pleased to find that a tertiary alkylboronic acid gave desired products 16 and 17 in good yield (75% and 82%).
 | ||
| Fig. 3 Substrate scope of the defluorinative alkylation reaction. Reaction conditions, unless otherwise noted: 1 (0.2 mmol), 2 (0.4 mmol), K3PO4 (0.1 mmol, 0.5 equiv.), Ir[dF(CF3)ppy]2(dtbbpy)PF6 (2 mol%), ethyl acetate (EA, 2.0 mL), Ar, 36 W blue LED, rt, 16 h. aK3PO4 (0.2 mmol, 1.0 equiv.). | ||
Next, we explored the scope with respect to the α-trifluoromethyl arylalkene. We found that arylalkenes with a para electron-donating group afforded the corresponding products 18 and 19 in 72% and 78% yields, respectively. However, when the para substituent was a substituted aryl ring, the yields were somewhat lower (20–24, 57–68%). The position of a phenyl group on the arylalkene had little effect on the yield; products 25–27 were obtained in 85–88% yields. α-Trifluoromethyl arylalkenes bearing an electron-withdrawing group gave somewhat lower yields (28–32). Substrates containing functionality that is useful for further synthetic manipulations, such as a pyridine ring (33, 54%), a quinoline ring (34, 52%), or a naphthalene ring (35, 72%; 36, 60%), were well tolerated. Disubstituted arylalkenes gave 37–42 in moderate yields, and a trisubstituted arylalkene was tolerated as well (43, 77%). Interestingly, when the substrate had a strongly electron-withdrawing substituent, pairs of products were obtained (44–46). We speculated that in these reactions, the carbanion intermediate was stabilized by the electron-withdrawing group, which made elimination of a fluoride ion more difficult.
Finally, we explored the use of sulfur-containing radical acceptors for this activation mode (Fig. 4). Sulfur-containing motifs are present in chiral ligands, catalysts, bioactive molecules, and natural products and are used in asymmetric catalysis; and the formation of C(sp3)–S bonds has long been of interest to chemists.72–75 We began by exploring the use of SOMO-philes 1b (SOMO = singly occupied molecular orbital) as radical acceptors. After optimizing the reaction conditions, we found that sulfide 47 could be obtained in 80% yield from S-phenyl benzenethiol-sulfonate and cyclohexylboronic acid (2a). S-Phenyl benzenethiol-sulfonates with an electron-donating (48 and 49) or electron-withdrawing (50–52) substituent provided the corresponding products in moderate yields. When the substituent was in the meta or ortho position, the yield decreased (53–58). A disubstituted benzenethiol-sulfonate and a pyridyl benzenethiol-sulfonate were also suitable, affording the desired products 59 (62%) and 60 (42%), respectively. We also found that various primary and secondary alkylboronic acids reacted with S-phenyl benzenethiol-sulfonate to provide the corresponding sulfides (61–66) in 66–80% yields. We tested SOMO-philes 1c and 1d, which allowed us to achieve the products of direct C–B chlorination (67), cyanation (68), vinylation (69–76), alkynylation (77), and allylation (78). Finally, when we used heteroaryl sulfone 1e as the radical acceptor, alkylation products 79–81 were obtained in moderate yields.
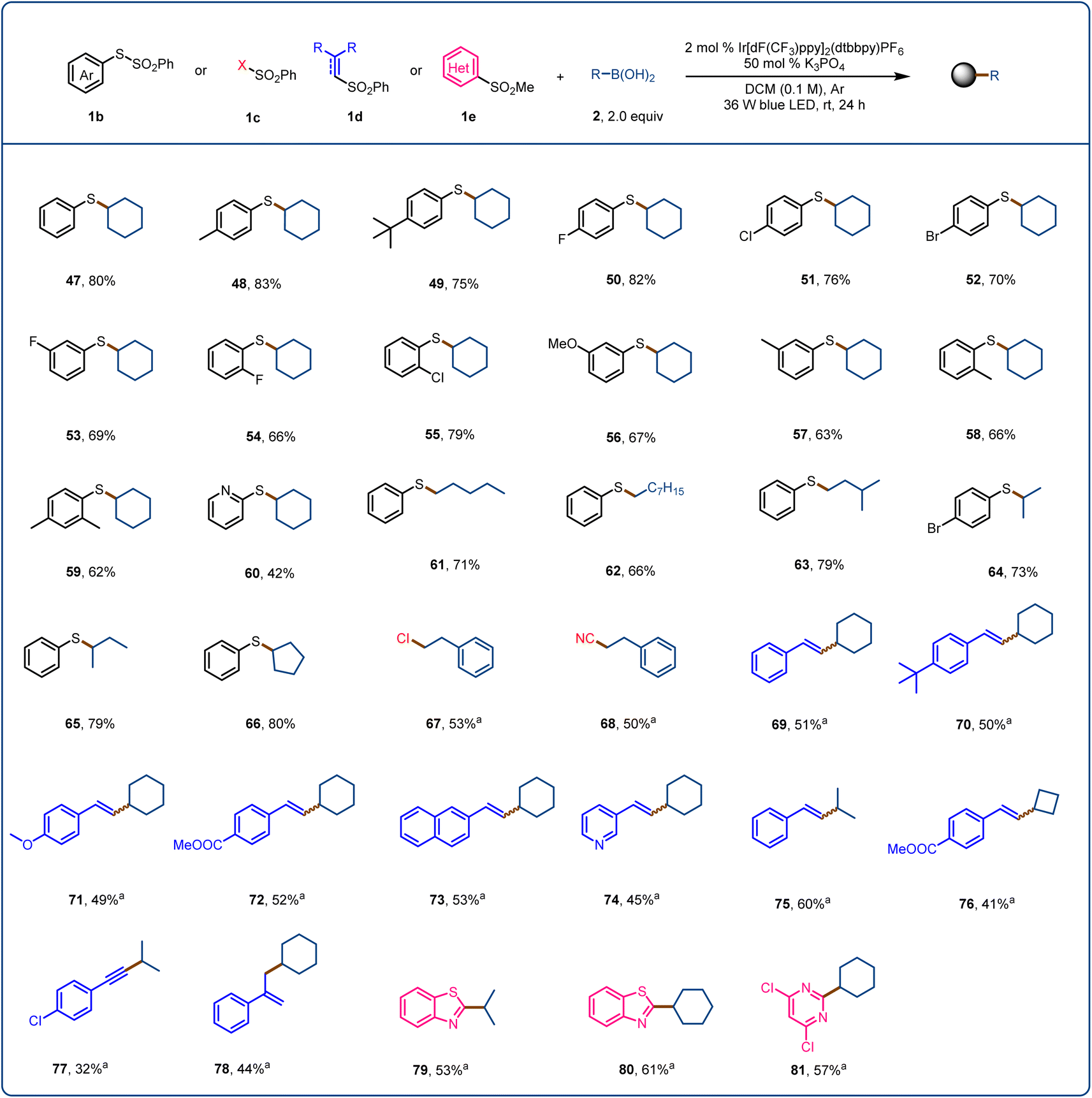 | ||
| Fig. 4 Scope with respect to the radical acceptor. Reaction conditions, unless otherwise noted: 1 (0.2 mmol), 2 (0.4 mmol), K3PO4 (0.1 mmol, 0.5 equiv.), Ir[dF(CF3)ppy]2(dtbbpy)PF6 (2 mol%), dichloromethane (DCM, 2.0 mL), Ar, 36 W blue LEDs, rt, 24 h. aK3PO4 (0.2 mmol, 1.0 equiv.). | ||
The broad substrate scope and promising functional group compatibility of these reactions encouraged us to evaluate their utility for late-stage modification of natural products, pharmaceuticals, and other bioactive molecules (Fig. 5A). Because of their lipophilicity, metabolic stability, binding selectivity, and bioabsorption characteristics, organofluorine motifs are widely used in polymers and pharmaceuticals.76 Such motifs are useful for overcoming a major challenge in drug discovery and development, that is, the need to replace metabolically reactive groups without affecting biological activity.70–82 With this in mind, we performed late-stage defluorinative alkylation of various bioactive molecules to obtain products 82–90. In addition, we were pleased to find that ketoprofen-, ibuprofen-, and L-menthol-derived alkenylation products (91–93) could be obtained in good yields. Moreover, we performed a gram–scale reaction of 94 to give 1.1 g (62% yield) of 21 (Fig. 5B). The bromine substituent allowed us to convert 21 to 96 in high yield via a Suzuki reaction, underscoring the synthetic utility of the defluorinative alkylation method. When the benzene ring of the α-trifluoromethyl arylalkene had a strongly electron-withdrawing carbomethoxy (97) or ketone (98) group, we could obtain products 99 and 100, respectively, by increasing the reaction temperature, prolonging the reaction time, and increasing the amount of the activation reagent (which also acted as a base). Because of the importance of selective activation in the presence of other boron-based species, we tested our method with a boronic pinacol ester (101) and found that it was not activated by K3PO4 (Fig. 5D). In addition, only the boronic acid was activated when a boronic pinacol ester (102) and a boronic acid (2a) were present in the same reaction mixture.
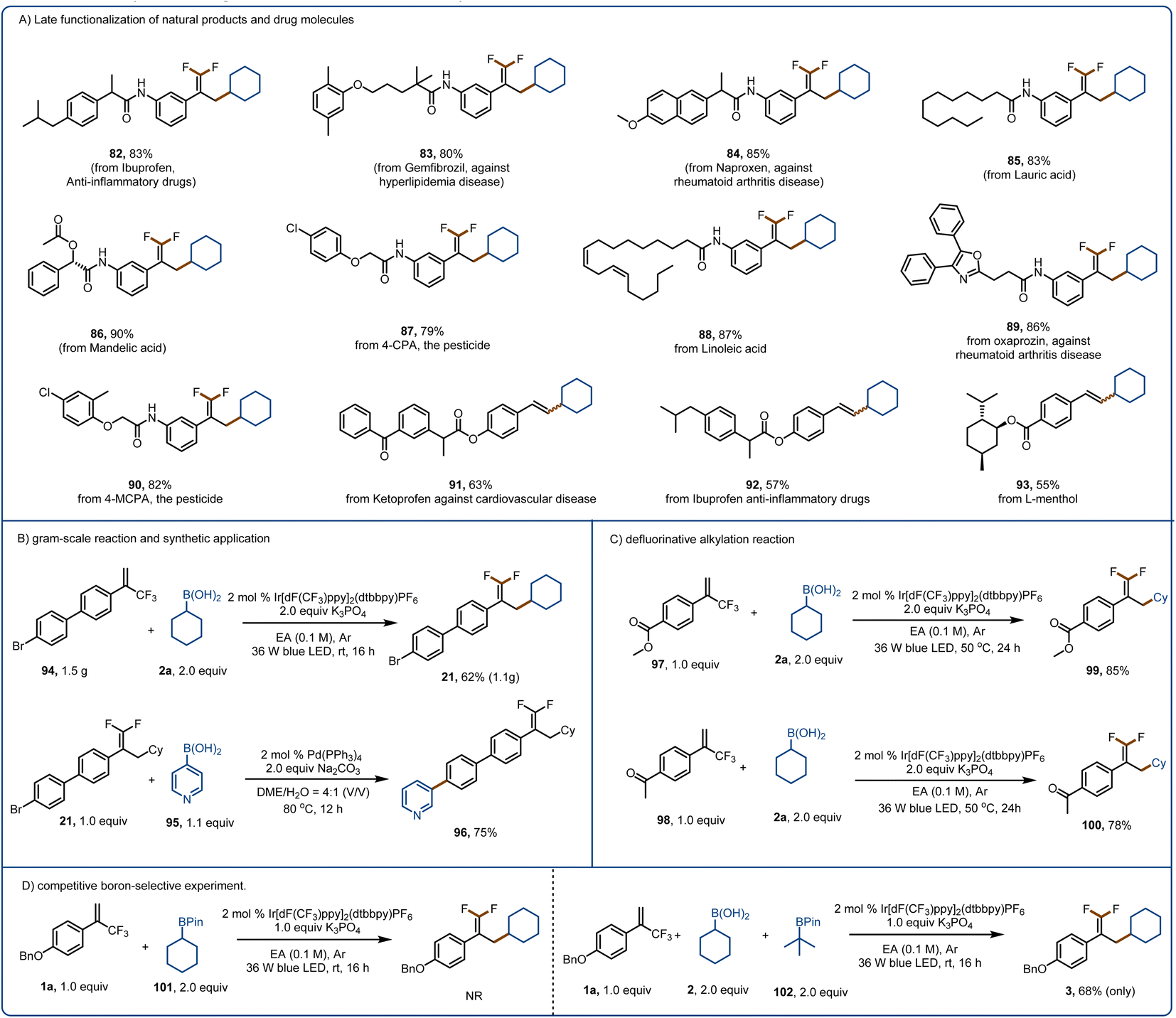 | ||
| Fig. 5 Late-stage functionalization of bioactive molecules and synthetic applications of the defluorinative alkylation method. (A) Late-stage functionalization of natural products and drugs. (B) Gram-scale reaction and synthetic applications. (C) Defluorinative alkylation reactions of substrates with electron-withdrawing substituents. (D) Competitive experiment to assess boron selectivity. | ||
Conclusions
In conclusion, we have demonstrated that activation by an inorganophosphorus compound can lower the oxidation potentials of alkylboronic acids. In addition, we found that boronic acids can be selectively activated in the presence of a boronic ester, indicating that our method may find utility for building complex molecular scaffolds. We carried out late-stage functionalization of some natural products and drugs. On the basis of the findings reported herein, we believe that our approach will facilitate the efficient use of boronic acids as alkyl radical sources in the evolving field of photoredox catalysis.Date availability
The ESI† includes all experimental details, including optimization of the synthetic method, synthesis and characterization of all starting materials and products reported in this study, and mechanistic studies. NMR spectra of all products reported are included as well.Author contributions
F. Y. conceived the chemistry and designed the experiments under the guidance of Professor Q. W., Dr H. S. and Dr J. D.; F. Y., H. M. and Y. L. conducted the experiments or analyzed the data. F. Y. wrote the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (21732002 and 22077071) and Frontiers Science Center for New Organic Matter, Nankai University (63181206) for generous financial support for our programs.Notes and references
- D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- W. Ding, L. Lu, Q. Zhou, Y. Wei, J. Chen and W.-J. Xiao, J. Am. Chem. Soc., 2017, 139, 63–66 CrossRef CAS PubMed.
- X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed.
- X. Wu and C. Zhu, Acc. Chem. Res., 2020, 53, 1620–1636 CrossRef CAS PubMed.
- K. J. Romero, M. S. Galliher, D. A. Pratt and C. R. J. Stephenson, Chem. Soc. Rev., 2018, 47, 7851–7866 RSC.
- L. Buzzetti, A. Prieto, S. R. Roy and P. Melchiorre, Angew. Chem., Int. Ed., 2017, 56, 15039–15043 CrossRef CAS PubMed.
- R. A. Garza-Sanchez, A. Tlahuext-Aca, G. Tavakoli and F. Glorius, ACS Catal., 2017, 7, 4057–4061 CrossRef CAS.
- P. Liu, W. Liu and C.-J. Li, J. Am. Chem. Soc., 2017, 139, 14315–14321 CrossRef CAS PubMed.
- W. Liu, P. Liu, L. Lv and C.-J. Li, Angew. Chem., Int. Ed., 2018, 57, 13499–13503 CrossRef CAS PubMed.
- R. S. J. Proctor, H. J. Davis and R. J. Phipps, Science, 2018, 360, 419–422 CrossRef CAS PubMed.
- D. Zheng and A. Studer, Angew. Chem., Int. Ed., 2019, 131, 15950–15954 CrossRef.
- Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 5257–5260 CrossRef CAS PubMed.
- C. Huang, J. Li and C.-J. Li, Chem. Sci., 2022, 13, 5465–5504 RSC.
- D. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed.
- D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS PubMed.
- M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef CAS PubMed.
- J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed.
- S. Li and Z. Xie, J. Am. Chem. Soc., 2022, 144, 7960–7965 CrossRef CAS PubMed.
- Y. Yin, Y. Dai, H. Jia, J. Li, L. Bu, B. Qiao, X. Zhao and Z. Jiang, J. Am. Chem. Soc., 2018, 140, 6083–6087 CrossRef CAS PubMed.
- A. Hu, J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef CAS PubMed.
- M. Silvi, C. Sandford and V. K. Aggarwal, J. Am. Chem. Soc., 2017, 139, 5736–5739 CrossRef CAS PubMed.
- A. Bunescu, Y. Abdelhamid and M. J. Gaunt, Nature, 2021, 598, 597–603 CrossRef PubMed.
- T. Constantin, M. Zanini, A. Regni, N. S Sheikh, F. Juliá and D. Leonori, Science, 2020, 367, 1021–1026 CrossRef CAS PubMed.
- J. D. Bell and J. A. Murphy, Chem. Soc. Rev., 2021, 90, 9540–9685 RSC.
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485–1542 CrossRef CAS PubMed.
- S. Crespi and M. Fagnoni, Chem. Rev., 2020, 120, 9790–9833 CrossRef CAS PubMed.
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed.
- (a) J. Chen, X. Hu, L. Lu and W. Xiao, Acc. Chem. Res., 2016, 49, 1911–1923 CrossRef CAS PubMed; (b) J. Xie, J. Yu, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 9416–9421 CrossRef CAS PubMed; (c) W. Xing, J. Wang, M. Fu and Y. Fu, Chin. J. Chem., 2022, 40, 323–328 CrossRef CAS; (d) L. Xi, Chin. J. Chem., 2020, 38, 897–898 CrossRef CAS.
- H. Cao, X. Tang, H. Tang, Y. Yuan and J. Wu, Chem. Catal., 2021, 1, 523–598 CrossRef.
- G. Duret, R. Quinlan, P. Bisseret and N. Blanchard, Chem. Sci., 2015, 6, 5366–5382 RSC.
- K. Duan, X. Yan, Y. Liu and Z. Li, Adv. Synth. Catal., 2018, 360, 2781–2795 CrossRef CAS.
- J. Chen, X. Hu, L. Lua and W. Xiao, Chem. Soc. Rev., 2016, 45, 2044–2056 RSC.
- M. Li, Y. Wei, J. Liu, H. Chen, L. Lu and W. Xiao, J. Am. Chem. Soc., 2017, 139, 14707–14713 CrossRef CAS PubMed.
- B. Lu, M. Xu, X. Qi, M. Jiang, W. Xiao and J. Chen, J. Am. Chem. Soc., 2022, 144, 14923–14935 CrossRef CAS PubMed.
- J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433–436 CrossRef CAS PubMed.
- Y. Yasu, T. Koike and M. Akita, Adv. Synth. Catal., 2012, 354, 3414–3420 CrossRef CAS.
- C. Shu, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2019, 58, 3870–3874 CrossRef CAS PubMed.
- F. Lima, U. K. Sharma, L. Grunenberg, D. Saha, S. Johannsen, J. Sedelmeier, E. V. Van der Eycken and S. V. Ley, Angew. Chem., Int. Ed., 2017, 56, 15136–15140 CrossRef CAS PubMed.
- Y. Iwata, Y. Tanaka, S. Kubosaki, T. Morita and Y. Yoshimi, Chem. Commun., 2018, 54, 1257–1260 RSC.
- M. Chilamari, J. R. Immel and S. Bloom, ACS Catal., 2020, 10, 12727–12737 CrossRef CAS.
- I. B. Seiple, S. Su, R. A. Rodriguez, R. Gianatassio, Y. Fujiwara, A. L. Sobel and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 13194–13196 CrossRef CAS PubMed.
- L. Zhang and Z.-Q. Liu, Org. Lett., 2017, 19, 6594–6597 CrossRef CAS PubMed.
- Y. Sato, K. Nakamura, Y. Sumida, D. Hashizume, T. Hosoya and H. Ohmiya, J. Am. Chem. Soc., 2020, 142, 9938–9943 CrossRef CAS PubMed.
- A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283–286 CrossRef CAS PubMed.
- Y. Cheng, C. Mück-Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2018, 140, 6221–6225 CrossRef CAS PubMed.
- H. Huang, K. Jia and Y. Chen, Angew. Chem., Int. Ed., 2015, 54, 1881–1884 CrossRef CAS PubMed.
- H. Huang, G. Zhang, L. Gong, S. Zhang and Y. Chen, J. Am. Chem. Soc., 2014, 136, 2280–2283 CrossRef CAS PubMed.
- H. Huo, K. Harms and E. Meggers, J. Am. Chem. Soc., 2016, 138, 6936–6939 CrossRef CAS PubMed.
- E. E. Stache, T. Rovis and A. G. Doyle, Angew. Chem., Int. Ed., 2017, 56, 3679–3683 CrossRef CAS PubMed.
- F. Lima, M. A. Kabeshov, D. N. Tran, C. Battilocchio, J. Sedelmeier, G. Sedelmeier, B. Schenkel and S. V. Ley, Angew. Chem., Int. Ed., 2016, 55, 14085–14089 CrossRef CAS PubMed.
- D. Kaiser, A. Noble, V. Fasano and V. K. Aggarwal, J. Am. Chem. Soc., 2019, 141, 14104–14109 CrossRef CAS PubMed.
- D. Shi, C. Xia and C. Liu, CCS Chem., 2021, 3, 1718–1728 CrossRef CAS.
- G.-X. Li, C. A. Morales-Rivera, Y. Wang, F. Gao, G. He, P. Liu and G. Chen, Chem. Sci., 2016, 7, 6407–6412 RSC.
- P. Ranjan, S. Pillitteri, G. Coppola, M. Oliva, E. V. Van der Eycken and U. K. Sharma, ACS Catal., 2021, 11, 10862–10870 CrossRef CAS.
- S. Xie, D. Li, H. Huang, F. Zhang and Y. Chen, J. Am. Chem. Soc., 2019, 141, 16237–16242 CrossRef CAS PubMed.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS PubMed.
- D. G. Hall, Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine, WileyVCH, Weinheim, 2007 Search PubMed.
- A. H. Dardir, P. R. Melvin, R. M. Davis, N. Hazari and M. Mohadjer Beromi, J. Org. Chem., 2018, 83, 469–477 CrossRef CAS PubMed.
- M. J. West, J. W. B. Fyfe, J. C. Vantourout and A. J. B. Watson, Chem. Rev., 2019, 119, 12491–12523 CrossRef CAS PubMed.
- B. C. Das, P. Thapa, R. Karki, C. Schinke, S. Das, S. Kambhampati, S. K. Banerjee, P. Van Veldhuizen, A. Verma, L. M. Weiss and T. Evans, Med. Chem., 2013, 5, 653–676 CAS.
- Y. Shoji, Y. Ikabata, Q. Wang, D. Nemoto, A. Sakamoto, N. Tanaka, J. Seino, H. Nakai and T. Fukushima, J. Am. Chem. Soc., 2017, 139, 2728–2733 CrossRef CAS PubMed.
- V. D. Nguyen, V. T. Nguyen, S. Jin, H. T. Dang and O. V. Larionov, Tetrahedron, 2019, 75, 584–602 CrossRef CAS PubMed.
- U. Pradere, E. C. Garnier-Amblard, S. J. Coats, F. Amblard and R. F. Schinazi, Chem. Rev., 2014, 114, 9154–9218 CrossRef CAS PubMed.
- M. D. Sørensenet, L. K. A. Blæhr, M. K. Christensen, T. Høyer, S. Latini, P. V. Hjarnaa and F. Björkling, Bioorg. Med. Chem., 2003, 11, 5461–5484 CrossRef PubMed.
- M. Sawaet, T. Kiyoi, K. Kurokawa, H. Kumihara, M. Yamamoto, T. Miyasaka, Y. Ito, R. Hirayama, T. Inoue, Y. Kirii, E. Nishiwaki, H. Ohmoto, Y. Maeda, E. Ishibushi, Y. Inoue, K. Yoshino and H. Kondo, J. Med. Chem., 2002, 45, 919–929 CrossRef PubMed.
- A. Babbset, et al. , Tetrahedron, 2020, 76, 130819–130827 CrossRef PubMed.
- A. Nocentiniet, V. Alterio, S. Bua, L. Micheli, D. Esposito, M. Buonanno, G. Bartolucci, S. M. Osman, Z. A. ALOthman, R. Cirilli, M. Pierini, S. M. Monti, L. D. C. Mannelli, P. Gratteri, C. Ghelardini, G. D. Simone and C. T. Supuran, J. Med. Chem., 2020, 63, 5185–5200 CrossRef PubMed.
- M. Dutartre, J. Bayardon and S. Jugé, Chem. Soc. Rev., 2016, 45, 5771–5794 RSC.
- K. A. Margrey, W. L. Czaplyski, D. A. Nicewicz and E. J. Alexanian, J. Am. Chem. Soc., 2018, 140, 4213–4217 CrossRef CAS PubMed.
- P. Johannesson, G. Lindeberg, A. Johansson, G. V. Nikiforovich, A. Gogoll, B. Synnergren, M. Le Grèves, F. Nyberg, A. Karlén and A. Hallberg, J. Med. Chem., 2002, 45, 1767–1777 CrossRef CAS PubMed.
- N. Wang, P. Saidhareddy and X. Jiang, Nat. Prod. Rep., 2020, 37, 246–275 RSC.
- S. Ni, L. Zhang, W. Zhang, H. Mei, J. Han and Y. Pan, J. Org. Chem., 2016, 81, 9470–9475 CrossRef CAS PubMed.
- Y. Wang, L. Deng, X. Wang, Z. Wu, Y. Wang and Y. Pan, ACS Catal., 2019, 9, 1630–1634 CrossRef CAS.
- J. Fried and E. Sabo, J. Am. Chem. Soc., 1954, 76, 1455–1456 CrossRef CAS.
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- C. Alonso, E. M. de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847–1935 CrossRef CAS PubMed.
- T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed.
- J. Hu, W. Zhang and F. Wang, Chem. Commun., 2009, 7465–7478 RSC.
- H. Xiao, Z. Zhang, Y. Fang, L. Zhu and C. Li, Chem. Soc. Rev., 2021, 50, 6308–6319 RSC.
- G. Tarantino and C. Hammond, Green Chem., 2020, 22, 5195–5209 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05521j |
| This journal is © The Royal Society of Chemistry 2022 |