
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of phosphiranes via organoiron-catalyzed phosphinidene transfer to electron-deficient olefins†
Tiansi
Xin
 a,
Michael B.
Geeson
a,
Hui
Zhu
b,
Zheng-Wang
Qu
*b,
Stefan
Grimme
b and
Christopher C.
Cummins
*a
a,
Michael B.
Geeson
a,
Hui
Zhu
b,
Zheng-Wang
Qu
*b,
Stefan
Grimme
b and
Christopher C.
Cummins
*a
aDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: ccummins@mit.edu
bMulliken Center for Theoretical Chemistry, University of Bonn, Beringstr. 4, 53115 Bonn, Germany. E-mail: qu@thch.uni-bonn.de
First published on 14th October 2022
Abstract
Herein is reported the structural characterization and scalable preparation of the elusive iron–phosphido complex FpP(tBu)(F) (2-F, Fp = (Fe(η5-C5H5)(CO)2)) and its precursor FpP(tBu)(Cl) (2-Cl) in 51% and 71% yields, respectively. These phosphide complexes are proposed to be relevant to an organoiron catalytic cycle for phosphinidene transfer to electron-deficient alkenes. Examination of their properties led to the discovery of a more efficient catalytic system involving the simple, commercially available organoiron catalyst Fp2. This improved catalysis also enabled the preparation of new phosphiranes with high yields (tBuPCH2CHR; R = CO2Me, 41%; R = CN, 83%; R = 4-biphenyl, 73%; R = SO2Ph, 71%; R = POPh2, 70%; R = 4-pyridyl, 82%; R = 2-pyridyl, 67%; R = PPh3+, 64%) and good diastereoselectivity, demonstrating the feasibility of the phosphinidene group-transfer strategy in synthetic chemistry. Experimental and theoretical studies suggest that the original catalysis involves 2-X as the nucleophile, while for the new Fp2-catalyzed reaction they implicate a diiron–phosphido complex Fp2(PtBu), 4, as the nucleophile which attacks the electron-deficient olefin in the key first P–C bond-forming step. In both systems, the initial nucleophilic attack may be accompanied by favorable five-membered ring formation involving a carbonyl ligand, a (reversible) pathway competitive with formation of the three-membered ring found in the phosphirane product. A novel radical mechanism is suggested for the new Fp2-catalyzed system.
Phosphiranes, the phosphorus analogues of aziridines, have been used as ligands,1–8 as polymer precursors,9,10 and more recently as precursors to P,N-bidentate ligands via ring-opening reactions using amide nucleophiles.11 Given the possibility of chirality at both phosphorus and carbon in the three-membered ring,7,12 such ring-opening reactions have the potential to enable preparation of enantiomerically pure ligands from phosphiranes.
In contrast to the well-established alkene aziridination reactions that are facilitated by transition-metal catalysts,13,14 only a handful of transition-metal promoted phosphirane syntheses, namely “phosphiranation” reactions, have been reported,15–19 among which there are only two examples of catalytic alkene phosphiranation processes yielding free (unprotected) phosphiranes.20,21 We recently reported a catalytic method for preparing phosphiranes using a phosphinidene group-transfer strategy, mirroring the analogous aziridination reactions.20 The system consisted of dibenzo-7-phosphanorbornadienes (RPA, A = C14H10, anthracene), a class of compounds capable of transferring phosphinidene groups to unsaturated molecules upon loss of an aromatic moiety,22–30 as the phosphinidene source and styrene as the receptor (Fig. 1A). In addition, both a source of the cyclopentadienyliron dicarbonyl cation ([CpFe(CO)2]+, Fp+) and the fluoride anion were required as co-catalysts for phosphinidene group transfer. The reaction was postulated to proceed via an iron–phosphido intermediate (FpP(tBu)(F), 2-F, Fig. 1A) based on evidence provided by stoichiometric reactions, deuterium labeling studies, and a Hammett analysis. However, the intermediate 2-F eluded isolation and full characterization due to its instability and high solubility in organic solvents.
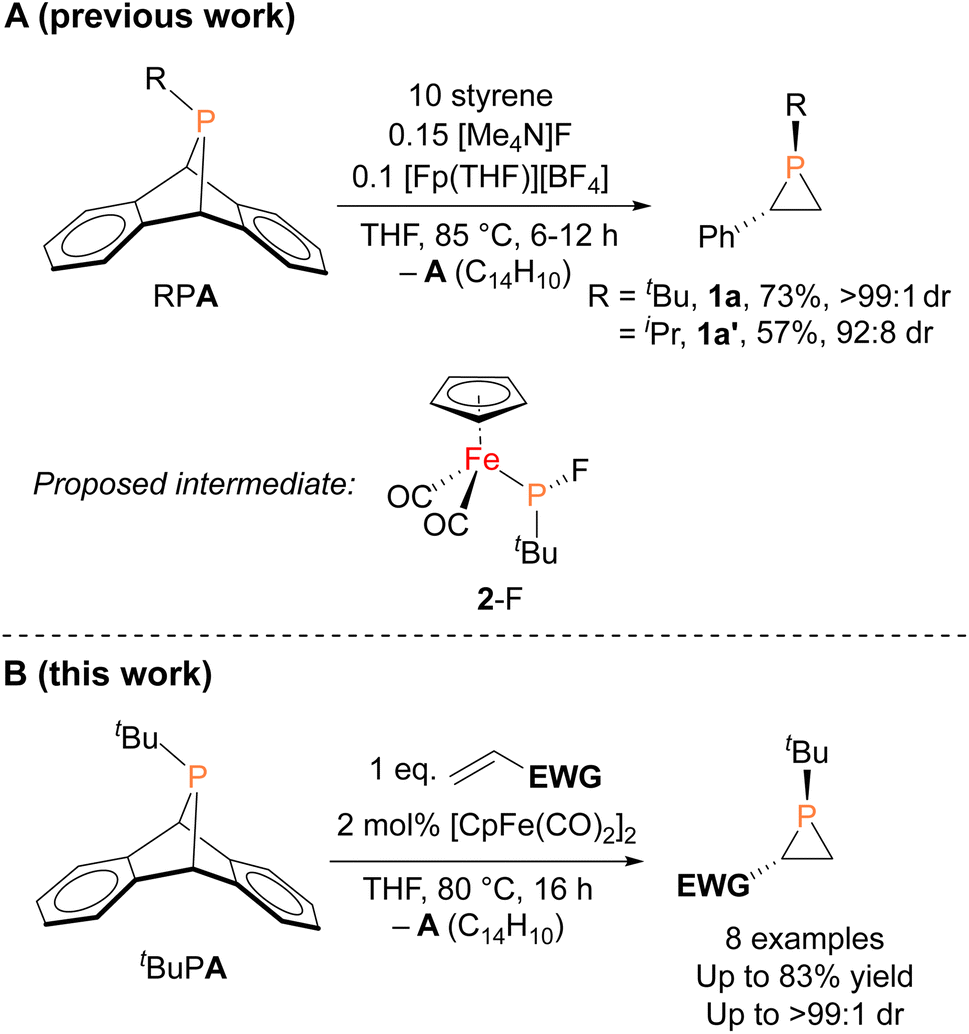 | ||
| Fig. 1 (A) Organoiron- and fluoride-catalyzed synthesis of phosphiranes by phosphinidene transfer and the proposed intermediate. (B) This work: organoiron-catalyzed phosphinidene transfer to electron-deficient olefins (EWG = electron-withdrawing group). | ||
Herein, we report the isolation and structural characterization of 2-F, thus providing a more complete picture of the catalytic cycle. In addition, we report the discovery of a more efficient catalytic system involving a simple, commercially available organoiron catalyst, the iconic cyclopentadienyliron dicarbonyl dimer ([CpFe(CO)2]2 or Fp2) for this phosphiranation reaction (Fig. 1B). This improved system was also used to prepare new phosphiranes bearing electron-withdrawing substituents, expanding the list of chemically accessible phosphiranes that may serve as useful ligands and synthetic building blocks.
The iron–phosphido complex 2-F was identified in our previous work as a plausible intermediate in the catalytic cycle,20 and therefore became the target of an independent synthesis. This species was previously generated in situ by treating [Fp(tBuPA)][BF4] with a source of fluoride to elicit anthracene elimination, though at the time it eluded purification and complete characterization. A new, scalable preparation of 2-F was achieved by utilizing the readily available tBuPCl2 and K[Fp]31 which, when combined in an equimolar ratio in THF, afforded 2-Cl in 71% yield (Scheme 1). Such halide displacement reactions at phosphorus were previously used to prepare analogous metal–phosphido complexes.32–34 In a subsequent step, halogen exchange with tetramethylammonium fluoride ([Me4N]F) in CH2Cl2 led to the isolation of 2-F in 51% yield (Scheme 2). In addition to characterization by NMR spectroscopy, both 2-Cl and 2-F were structurally characterized by X-ray crystallography (Fig. 2). Both compounds exhibit a pyramidal geometry at the phosphorus atom, with the sum of bond angles being 316° and 314° for 2-Cl and 2-F, respectively. The Fe–P distances in 2-Cl and 2-F are in the range of a Fe–P single bond, similar to other reported Fp–phosphido complexes.33,35,36
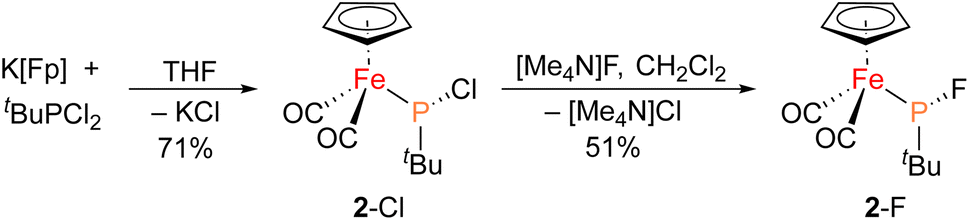 | ||
| Scheme 1 Synthesis of 2-Cl and 2-F starting from K[Fp] and tBuPCl2. | ||
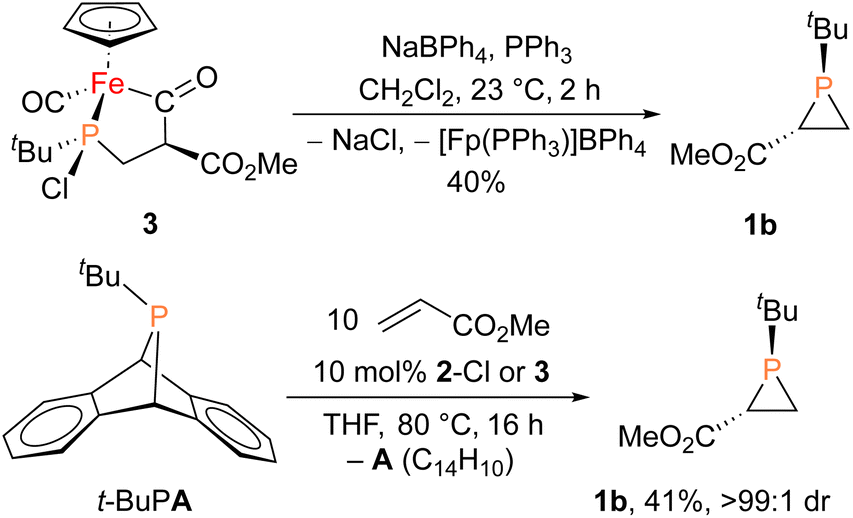 | ||
| Scheme 2 Stoichiometric (top) and catalytic (bottom) synthesis of 1b. | ||
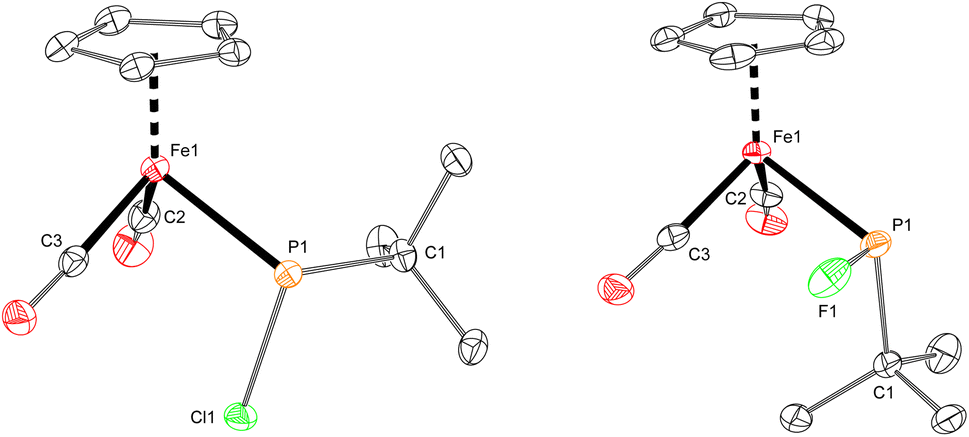 | ||
| Fig. 2 Solid-state structures of 2-Cl (left) and 2-F (right), with thermal ellipsoids shown at the 50% probability level and hydrogen atoms omitted for clarity. Selected bond lengths (Å) for 2-Cl: Fe1–P1: 2.2935(3); P1–Cl1: 2.1315(3). 2-F: Fe1–P1: 2.2898(4); P1–F1: 1.6455(11). | ||
With proposed catalytic intermediate 2-F in hand, it was tested as a catalyst in the phosphiranation reaction of tBuPA with styrene resulting in isolation of 1a with a yield similar to that originally reported, supporting the relevance of 2-F to the catalytic cycle. The chloride analogue 2-Cl was also found to catalyze the phosphiranation reaction, albeit with a lower yield (Table 1, entry 4). In fact, similar Fp–phosphido complexes have been reported as being nucleophilic at phosphorus; Fp*P(tBu)(Cl) (Fp* = Cp*Fe(CO)2) was reported to react with methyl iodide to give the phosphonium iodide [Fp*P(tBu)(Me)(Cl)][I],33 while FpP(Ph)2 could catalyze the isomerization of dimethyl maleate to dimethyl fumarate via reversible nucleophilic addition to the double bond.37 Similar reactions have also been reported for an iridium phosphido complex.38 When 2-Cl was treated with the electron-deficient olefin methyl acrylate, clean conversion to a single new compound 3 was observed (Fig. 3 top). Compound 3 features a five-membered organometallic ring, resulting from addition of methyl acrylate across the nucleophilic phosphorus center and into one carbonyl ligand of the Fp group. Two isomers were identified by NMR spectroscopy, and the major isomer was characterized by X-ray crystallography (Fig. 3 bottom). The formation of 3 from methyl acrylate raises the possibility that an analogous species may play a role in the catalytic cycle of the reaction employing styrene. Heating 2-Cl or 2-F in the presence of excess styrene, however, did not lead to any similar species, although it cannot be ruled out as a short-lived intermediate under the conditions of catalysis.
| Entry | Catalyst | 1a yield (%) |
|---|---|---|
| a Conditions: tBuPA (0.1 mmol), styrene (1 mmol), catalyst (10 mol%), THF (1 ml), 80 °C, 16 h. Yields were determined by 31P NMR analysis. b Isolated yield after vacuum distillation. c 2 mol%. d 0.5 mol%, 6 h reaction time. | ||
| 1 | [Fp(THF)][BF4]/1.5[Me4N]F | 91(73b) |
| 2 | [Fp(THF)][BF4]/1.5[nBu4N]Cl | 82 |
| 3 | 2-F | 90(75b) |
| 4 | 2-Cl | 72 |
| 5 | FpCl | 80 |
| 6 | FpI | 13 |
| 7 | FpOTf | 4 |
| 8 | FpPPh2 | 90 |
| 9 | FpPCy2 | 92(76b) |
| 10 | Fp2c | 99(78b) |
| 11 | Fp2d | 98 |
| 12 | Fe2(CO)9 | 63 |
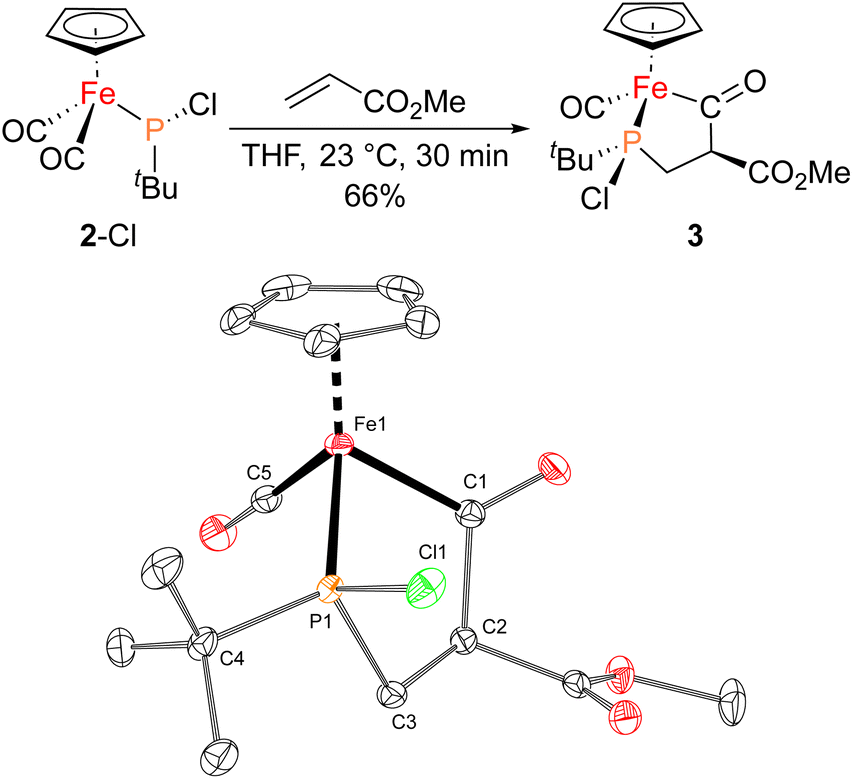 | ||
| Fig. 3 (Top) Synthesis of 3 from 2-Cl and methyl acrylate. (Bottom) Solid-state structure of 3 with thermal ellipsoids at the 50% probability level and hydrogen atoms omitted for clarity. Selected bond lengths (Å): Fe1–C1: 1.9625(8); C1–C2: 1.5960(10); C2–C3: 1.5316(12); P1–C3: 1.8277(8); Fe1–C5: 1.7393(8); Fe1–P1: 2.1335(2). | ||
Given the potential relevance of 3 to the catalytic cycle, stoichiometric reactions to effect phosphirane release were attempted. Treating isolated 3 with NaBPh4 and PPh3 in CH2Cl2 resulted in the formation of the corresponding phosphirane 1b in ca. 40% conversion within 2 h (Scheme 2), further supporting the proposed catalytic cycle. Employing a catalytic amount of 2-Cl, 1b can be prepared from tBuPA and methyl acrylate in 41% isolated yield as a single diastereomer, while a control experiment without any catalyst led to only minor amounts of phosphirane 1b.
These findings clearly revealed that fluoride is not essential for the catalytic phosphiranation reaction. We therefore set out to screen more catalysts (or precatalysts) and conditions with a view to optimizing and simplifying the reaction system (Table 1 and S1†). We first replaced the fluoride source ([Me4N]F) with a chloride source ([nBu4N]Cl), resulting in a slightly lower yield. Similarly, isolated FpCl gave a yield of ca. 80%. In contrast, FpI and FpOTf exhibited little catalytic reactivity, an observation explicable in terms of the poor nucleophilicity of the anions. Interestingly, Fp-phosphido complexes FpPPh2 and FpPCy2 were also found to be efficient (pre)catalysts. A closer inspection of these catalyst systems suggested that Fp2, likely generated in situ from Fp–phosphido compounds at 80 °C, was the actual species in play. Phosphiranation proceeded smoothly with Fp2 even at catalyst loading as low as 0.5 mol%. Moreover, Fp2 is readily available from commercial sources, making it user-friendly and attractive from a practical perspective.
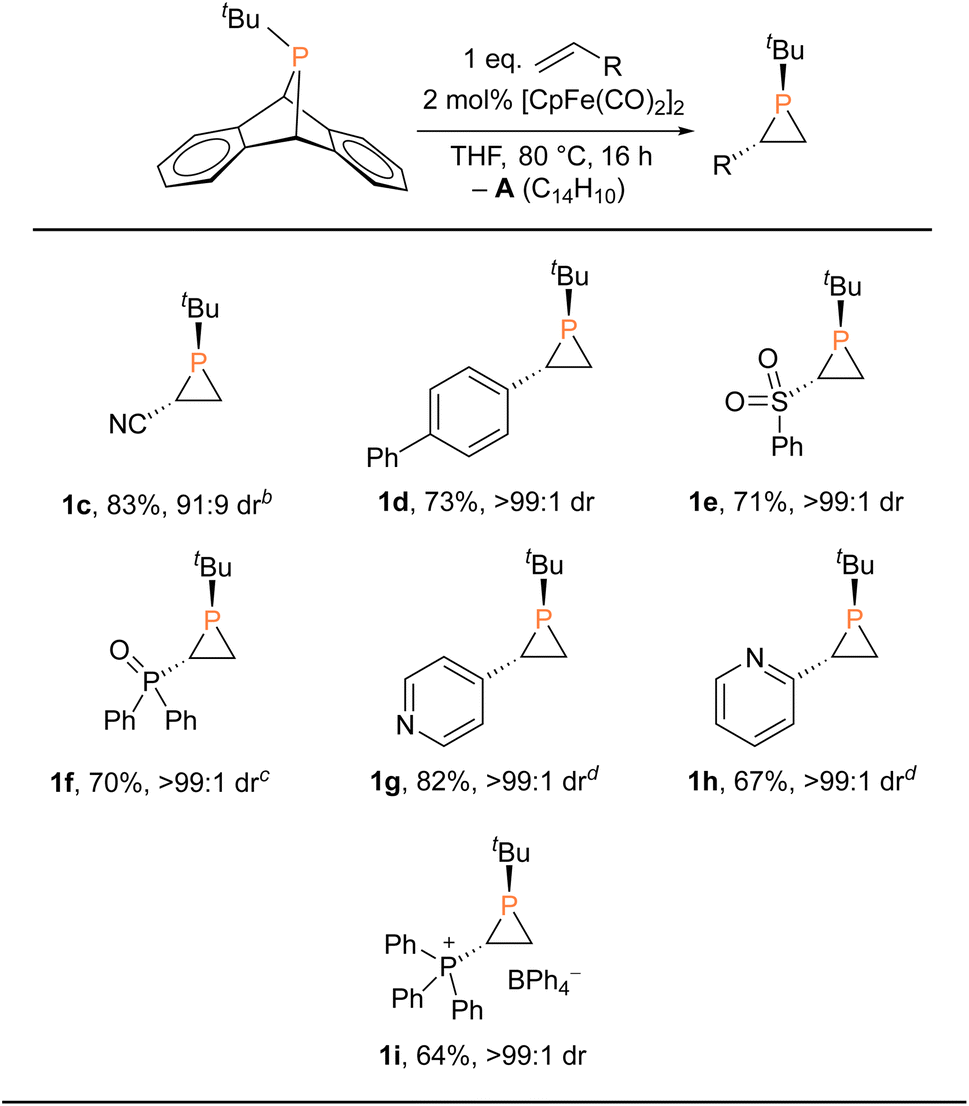
With the Fp2 catalyst system having been identified as optimal, we set out to expand the substrate scope (Table 2). Aryl-substituted olefins such as 4-vinylbiphenyl and vinylpyridines worked well in the phosphiranation reaction, giving the corresponding phosphiranes as a single diastereomer. 2-Pyridyl phosphiranes exemplified by 1h have the potential to function as chiral P,N-bidentate ligands in coordination chemistry.39–41 Olefins with electron-withdrawing groups such as sulfonyl, phosphine oxide, and phosphonium also gave acceptable yields with excellent diastereoselectivity. The Fp2 catalyst was essential for phosphirane formation, with the exception of 1c which was formed from tBuPA and acrylonitrile in the absence of any catalyst, albeit with diminished diastereoselectivity (91![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) compared to other substrates.
:9) compared to other substrates.
In order to shed light on a plausible mechanism for the new Fp2 catalyst system, some possible intermediates and other relevant species were isolated and characterized (Fig. 4). Treatment of tBuPA with Fp2 at 80 °C in the absence of an olefin trap led to the formation of anthracene and three new Fe–P containing species. Compound 4 (31P δ 166 ppm) corresponds to phosphinidene insertion into Fp2 and compound 5 (31P δ 623 ppm) results from decarbonylation of 4, while 6 (31P δ 55 ppm) corresponds to two phosphinidne units inserted into Fp2. Complexes 4–6 were initially assigned by comparing their chemical shifts to similar known compounds,42,43 and in the case of 4 and 6 verified by independent synthesis. Compound 4 was prepared using salt metathesis upon treatment of tBuPCl2 with K[Fp], or by treating tBuPH2 with benzylpotassium (KCH2Ph) followed by [Fp]I. Compound 6 was synthesized from [Fp(tBuPH2)][BF4] and 2-Cl in the presence of DBU. Interestingly, when treated with styrene at 80 °C, only 4 was found to afford phosphirane 1a, while 5 remained unreacted and 6 yielded the tetraphosphetane (P4(tBu)4) as the major product. These findings suggest that 4 could be a key intermediate in the catalytic alkene phosphiranation reaction. In addition, 4 was found to readily convert to 5via decarbonylation under reduced pressure or light exposure at room temperature, unlike the known phenyl derivative.42 Unfortunately, we were unable to obtain analytically pure 4 free of the Fp2 and 5 impurities due to their very similar solubilities, and we were also unable to obtain a crystal structure of 4 due to its high solubility and instability.
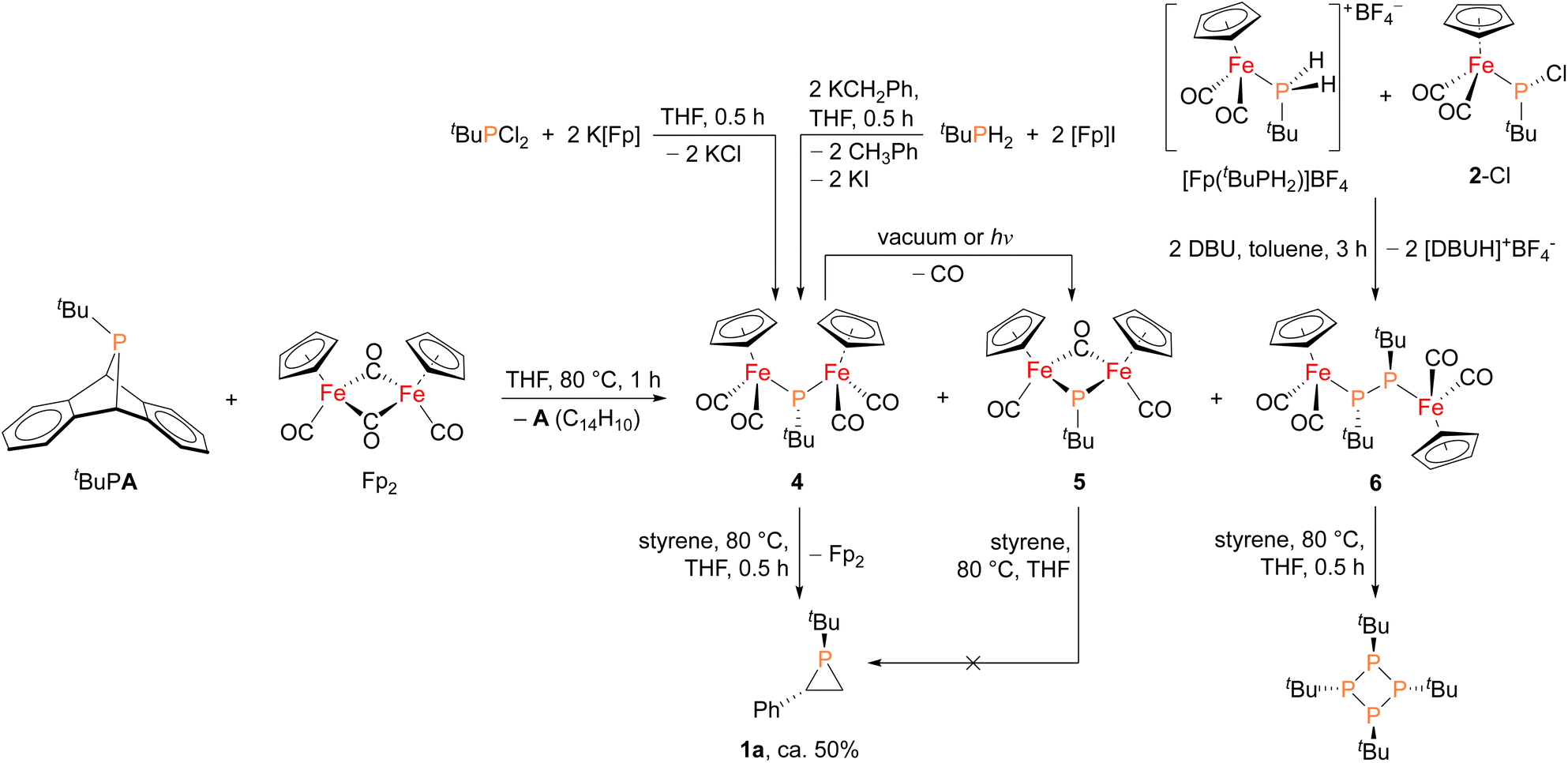 | ||
| Fig. 4 Synthesis of possible reaction intermediates 4, 5 and 6, and their reactivity studies towards styrene. | ||
Possible pathways for the catalytic formation of phosphirane were investigated using DFT calculations at the PW6B95-D3/def2-QZVP + COSMO-RS level of theory using TPSS-D3/def2-TZVP + COSMO optimized geometries in THF solution44–52 that has been well tested in recent mechanistic studies.53,54 We were able to locate many of the intermediates and transition states along the potential energy surface. The free energy change for the net phosphiranation reaction (tBuPA + styrene → 1a + anthracene) was calculated to be −10.9 kcal mol−1.
For the 2-Cl (or FpCl) catalyzed reaction, as shown in Fig. 5A, the Cl−/tBuPA ligand exchange at the iron center of mononuclear complex FpCl is 3.5 kcal mol−1 endergonic to form the cationic complex [Fp(tBuPA)]+, from which nucleophilic Cl− attack at the phosphorus center may lead to compound 2-Cl with loss of anthracene. Such formal phosphinidene transfer from tBuPA to FpCl is −10.7 kcal mol−1 exergonic over a free energy barrier of 19.1 kcal mol−1. Using styrene as the olefin substrate, the frustrated Lewis pair (FLP)-like alkene addition to 2-Cl across the Lewis basic phosphorus atom and a Lewis acidic CO ligand is −1.6 kcal mol−1 exergonic over a sizable barrier of 28.2 kcal mol−1 (viaTS2) to form the five-membered-ring adduct 3a, a result in agreement with the requirement of moderate heating at 80 °C under experimental conditions. Further ring-contraction of 3a is 1.4 kcal mol−1 endergonic over a 2.8 kcal mol−1 lower barrier of 25.4 kcal mol−1 (viaTS3 through transient ionic [Fp(1a)]+ and Cl− species) to release the phosphirane product 1a along with regenerated FpCl, consistent with the absence of 3a as an observable product despite its considerable kinetic stability. In contrast, a more electron-deficient olefin, methyl acrylate, turns out to be much more reactive due to more facile electrophilic alkene addition to 2-Cl, an addition now −5.6 kcal mol−1 exergonic over a low barrier of 17.2 kcal mol−1 (viabTS2; see ESI Fig. S52†), in good agreement with the formation of 3 observed experimentally at room temperature. Due to a higher barrier of 20.9 kcal mol−1 (via less stable 1b and bTS3), the regeneration of 2-Cl from FpCl and tBuPA becomes the rate-limiting step.
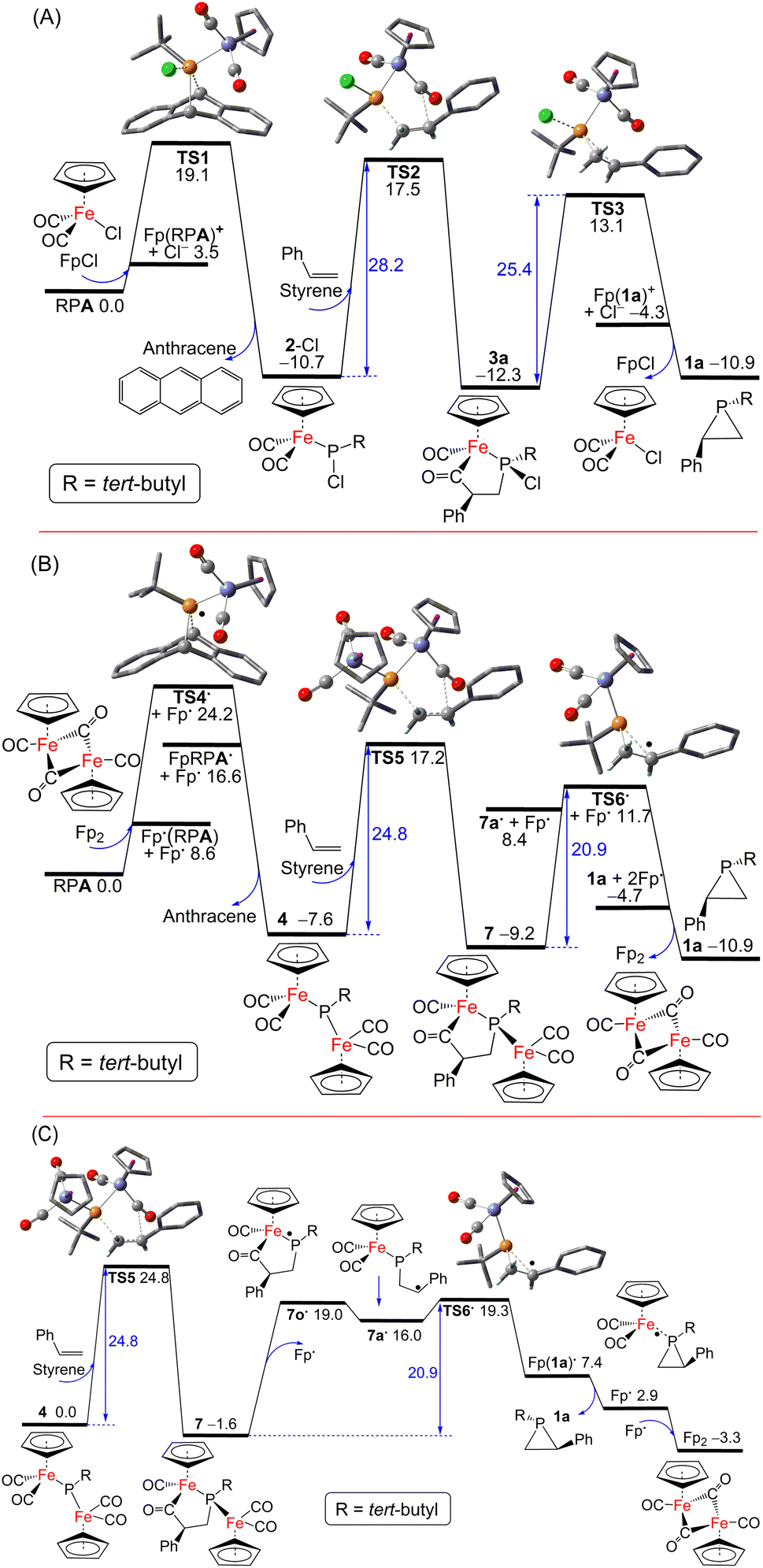 | ||
| Fig. 5 DFT computed free energy paths (in kcal mol−1, at 298 K and 1 M in THF solution) for (A) 2-Cl (or FpCl) and (B) 4 (or Fp2) catalyzed phosphinidene transfer from tBuPA to styrene, and (C) the key P–C bond formation steps from 4. Crucial C, O, P, Cl and Fe atoms in the transition state ball-and-stick models are highlighted as grey, red, orange, green and blue balls, respectively, with most H-atoms omitted for clarity. | ||
Interestingly, as shown in Fig. 5B, a [Fp]˙ radical is accessible from the Fp2 dimer complex to induce facile anthracene release from tBuPA, a microscopic step that is −7.6 kcal mol−1 exergonic over a barrier of 24.2 kcal mol−1 (viaTS4˙ and Fp(tBuPA)˙ radical) to form the diiron–phosphido complex 4. The phosphine adducts of the [Fp]˙ radical, formally 19-electron complexes, have been proposed in established experimental studies.55 Such a radical mechanism is also supported by the experimental observation of complex 6 resulting from radical–radical coupling of transient radical [FpP(tBu)]˙ intermediate. Subsequent addition of styrene to 4 in an FLP-like process is still −1.6 kcal mol−1 exergonic over a slightly higher barrier of 24.8 kcal mol−1 (viaTS5) to form the five-membered-ring adduct 7. The second rate-limiting barrier (TS5) is 3.4 kcal mol−1 lower than the first rate-limiting barrier (TS2), consistent with the evidently higher catalytic activity of 4 (or Fp2) than 2-Cl (or FpCl). Dissociation of the [Fp]˙ radical from 7 may induce the cyclic C–C bond cleavage of the transient P-centered radical 7o˙ to form the acyclic benzyl radical 7a˙, which is 17.6 kcal mol−1 endergonic without transition state (Fig. 5C). The final P–C bond formation proceeds viaTS6˙ and [Fp(1a)]˙, the phosphirane adduct of [Fp]˙, to produce phosphirane 1a and a second [Fp]˙ radical which recombines with the first to regenerate the closed-shell Fp2 catalyst, making the overall ring-contraction step 1.7 kcal mol−1 exergonic over a moderate barrier of 20.9 kcal mol−1. Alternatively, the reactive [FpP(tBu)]˙ radical could also be formed from 4via [Fp]˙ elimination and then react with styrene to afford 1a, which however encounters a high barrier of 36.2 kcal mol−1 (viaTS7 and TS7c; see ESI†). The decarbonylation of 4 is endergonic by 6.3 kcal mol−1 to form the complex 5 with a barrier of approximately 15 kcal mol−1, making 4 prone to decomposition under reduced pressure or light exposure. Much higher barriers are found for other conceivable ionic pathways for the first and the third catalytic steps, which are thus kinetically unfavorable.
In conclusion, we have achieved a scalable preparation and characterization of an elusive iron–phosphido complex 2-F, which was previously proposed as a key intermediate in the organoiron- and fluoride-catalyzed styrene phosphiranation reaction, from its precursor 2-Cl. Examination of the properties of 2-X (X = F, Cl) led to the isolation of another potential intermediate in the catalytic cycle, as well as the discovery of a more efficient catalytic system consisting of a simple, commercially available organoiron catalyst Fp2, RPA, and an electron-deficient olefin. In the new system, the catalyst loading could be lowered to 2 mol%, and only stoichiometric amounts of alkene substrate were required. The new and improved catalyst system also enabled the preparation of several new phosphiranes bearing electron-withdrawing groups with satisfying yields and excellent diastereoselectivity. Unlike the original catalytic system which is understood to proceed through 2-X and 3avia fully ionic pathways, this new reaction is postulated to proceed through a more reactive diiron–phosphido intermediate 4 and a five-membered iron–phosphorus–carbon ring intermediate 7via a novel radical mechanism involving [Fp]˙. The present findings enhance the expanding library of known phosphiranes, while further highlighting the feasibility of the transition-metal catalyzed phosphinidene group-transfer strategy in synthetic chemistry.
Data availability
The experimental details, characterization data, NMR spectra, crystallography and computational details associated with this article are provided in the ESI.† Crystallographic data for compound 2-Cl, 2-F and 3 has also been deposited at the CCDC under 2190120—2190122.Author contributions
T. X. and M. B. G. conducted experiments and wrote the manuscript. C. C. C. supervised the work and finalized the manuscript. H. Z., Z.-W. Q. and S. G. conducted DFT mechanistic study and partially wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. X., M. B. G. and C. C. C. would like to thank the National Science Foundation (NSF-CHE-1955612) for financial support of this research. H. Z., Z. W. Q. and S. G. are grateful to the German Science Foundation (DFG) for financial support (project 490737079).Notes and references
- J. C. Slootweg and K. Lammertsma, in Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, John Wiley & Sons, Ltd, 2012, ch. 9, pp. 309–320 Search PubMed.
- D. S. Glueck, in Comprehensive Heterocyclic Chemistry IV, ed. D. S. Black, J. Cossy and C. V. Stevens, Elsevier, Oxford, 2022, pp. 464–505 Search PubMed.
- N. Mézailles, P. E. Fanwick and C. P. Kubiak, Organometallics, 1997, 16, 1526–1530 CrossRef.
- F. Yang, P. E. Fanwick and C. P. Kubiak, Organometallics, 1999, 18, 4222–4225 CrossRef CAS.
- J. Liedtke, S. Loss, C. Widauer and H. Grützmacher, Tetrahedron, 2000, 56, 143–156 CrossRef CAS.
- J. Liedtke, H. Rüegger, S. Loss and H. Grützmacher, Angew. Chem., Int. Ed., 2000, 39, 2478–2481 CrossRef CAS PubMed.
- A. Marinetti, F. Mathey and L. Ricard, Organometallics, 1993, 12, 1207–1212 CrossRef CAS.
- A. Ficks, W. Clegg, R. W. Harrington and L. J. Higham, Organometallics, 2014, 33, 6319–6329 CrossRef CAS.
- S. Kobayashi and J.-i. Kadokawa, Macromol. Rapid Commun., 1994, 15, 567–571 CrossRef CAS.
- J.-I. Kadokawa and S. Kobayashi, Phosphorus, Sulfur Silicon Relat. Elem., 2002, 177, 1387–1390 CrossRef CAS.
- J. Gasnot, C. Botella, S. Comesse, S. Lakhdar, C. Alayrac, A.-c. Gaumont, V. Dalla and C. Taillier, Angew. Chem., Int. Ed., 2020, 59, 11769–11773 CrossRef CAS PubMed.
- X. Sava, A. Marinetti, L. Ricard and F. Mathey, Eur. J. Inorg. Chem., 2002, 2002, 1657–1665 CrossRef.
- P. Müller and C. Fruit, Chem. Rev., 2003, 103, 2905–2920 CrossRef PubMed.
- L. Degennaro, P. Trinchera and R. Luisi, Chem. Rev., 2014, 114, 7881–7929 CrossRef CAS PubMed.
- A. Marinetti and F. Mathey, Organometallics, 1984, 3, 456–461 CrossRef CAS.
- T. L. Breen and D. W. Stephan, J. Am. Chem. Soc., 1995, 117, 11914–11921 CrossRef CAS.
- J. B. Wit, G. T. van Eijkel, F. J. de Kanter, M. Schakel, A. W. Ehlers, M. Lutz, A. L. Spek and K. Lammertsma, Angew. Chem., Int. Ed., 1999, 38, 2596–2599 CrossRef CAS PubMed.
- R. Waterman and G. L. Hillhouse, J. Am. Chem. Soc., 2003, 125, 13350–13351 CrossRef CAS PubMed.
- P. Junker, Z.-W. Qu, T. Kalisch, G. Schnakenburg, A. E. Ferao and R. Streubel, Dalton Trans., 2021, 50, 739–745 RSC.
- M. B. Geeson, W. J. Transue and C. C. Cummins, J. Am. Chem. Soc., 2019, 141, 13336–13340 CrossRef CAS PubMed.
- M.-L. Y. Riu, A. K. Eckhardt and C. C. Cummins, J. Am. Chem. Soc., 2022, 144, 7578–7582 CrossRef CAS PubMed.
- A. Velian and C. C. Cummins, J. Am. Chem. Soc., 2012, 134, 13978–13981 CrossRef CAS PubMed.
- M.-A. Courtemanche, W. J. Transue and C. C. Cummins, J. Am. Chem. Soc., 2016, 138, 16220–16223 CrossRef CAS PubMed.
- W. J. Transue, A. Velian, M. Nava, C. García-Iriepa, M. Temprado and C. C. Cummins, J. Am. Chem. Soc., 2017, 139, 10822–10831 CrossRef CAS PubMed.
- K. M. Szkop, M. B. Geeson, D. W. Stephan and C. C. Cummins, Chem. Sci., 2019, 10, 3627–3631 RSC.
- M.-L. Y. Riu and C. C. Cummins, J. Org. Chem., 2020, 85, 14810–14816 CrossRef CAS PubMed.
- M.-L. Y. Riu, R. L. Jones, W. J. Transue, P. Müller and C. C. Cummins, Sci. Adv., 2020, 6, eaaz3168 CrossRef CAS PubMed.
- M.-L. Y. Riu, W. J. Transue, J. M. Rall and C. C. Cummins, J. Am. Chem. Soc., 2021, 143, 7635–7640 CrossRef CAS PubMed.
- A. K. Eckhardt, M.-L. Y. Riu, P. Müller and C. C. Cummins, J. Am. Chem. Soc., 2021, 143, 21252–21257 CrossRef CAS PubMed.
- A. K. Eckhardt, M.-L. Y. Riu, M. Ye, P. Müller, G. Bistoni and C. C. Cummins, Nat. Chem., 2022, 14, 928–934 CrossRef CAS PubMed.
- J. S. Plotkin and S. G. Shore, Inorg. Chem., 1981, 20, 284–285 CrossRef CAS.
- H. Nakazawa, W. E. Buhro, G. Bertrand and J. Gladysz, Inorg. Chem., 1984, 23, 3431–3433 CrossRef CAS.
- W. Malish, W. Angerer, A. H. Cowley and N. C. Norman, J. Chem. Soc., Chem. Commun., 1985, 1811–1812 RSC.
- K. Vaheesar, T. M. Bolton, A. L. East and B. T. Sterenberg, Organometallics, 2010, 29, 484–490 CrossRef CAS.
- L. Weber, K. Reizig and R. Boese, Chem. Ber., 1985, 118, 1193–1203 CrossRef CAS.
- W. F. McNamara, H. U. Reisacher, E. N. Duesler and R. T. Paine, Organometallics, 1988, 7, 1313–1317 CrossRef CAS.
- M. T. Ashby and J. H. Enemark, Organometallics, 1987, 6, 1323–1327 CrossRef CAS.
- A. L. Serrano, M. A. Casado, M. A. Ciriano, B. de Bruin, J. A. Lopez and C. Tejel, Inorg. Chem., 2016, 55, 828–839 CrossRef CAS PubMed.
- P. J. Guiry and C. P. Saunders, Adv. Synth. Catal., 2004, 346, 497–537 CrossRef CAS.
- S. Lühr, J. Holz and A. Boerner, ChemCatChem, 2011, 3, 1708–1730 CrossRef.
- J. Margalef, M. Biosca, P. de la Cruz Sanchez, J. Faiges, O. Pamies and M. Dieguez, Coord. Chem. Rev., 2021, 446, 214120 CrossRef CAS.
- I.-P. Lorenz, P. Mürschel, W. Pohl and K. Polborn, Chem. Ber., 1995, 128, 413–416 CrossRef CAS.
- M. A. Alvarez, M. E. García, R. Gonzalez, A. Ramos and M. A. Ruiz, Organometallics, 2011, 30, 1102–1115 CrossRef CAS.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- F. Eckert and A. Klamt, AIChE J., 2002, 48, 369–385 CrossRef CAS.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2005, 109, 5656–5667 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, Chem.–Eur. J., 2012, 18, 9955–9964 CrossRef CAS PubMed.
- L. Goerigk, A. Hansen, C. Bauer, S. Ehrlich, A. Najibi and S. Grimme, Phys. Chem. Chem. Phys., 2017, 19, 32184–32215 RSC.
- Z.-W. Qu, H. Zhu and S. Grimme, ChemistryOpen, 2019, 8, 807–810 CrossRef CAS PubMed.
- D. Astruc, Chem. Rev., 1988, 88, 1189–1216 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2190120–2190122. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05011k |
| This journal is © The Royal Society of Chemistry 2022 |