DOI:
10.1039/D2SC04507A
(Review Article)
Chem. Sci., 2022,
13, 13690-13707
Organothianthrenium salts: synthesis and utilization
Received
12th August 2022
, Accepted 7th October 2022
First published on 7th October 2022
Abstract
Organothianthrenium salts are a class of compounds containing a positively charged sulfur atom and a neutral sulfur atom. Over the past years, organothianthrenium salts have been emerging as attractive precursors for a myriad of transformations to forge new C–C and C–X bonds due to their unique structural characteristics and chemical behaviors. The use of the thianthrenation strategy selectively transforms C–H, C–O, and other chemical bonds into organothianthrenium salts in a predictable manner, providing a straightforward alternative for regioselective functionalizations for arenes, alkenes, alkanes, alcohols, amines and so on through diverse reaction mechanisms under mild conditions. In this review, the preparation of different organothianthrenium salts is summarized, including aryl, alkenyl and alkyl thianthrenium salts. Moreover, the utilization of organothianthrenium salts in different catalytic processes and their synthetic potentials are also discussed.
1. Introduction
Direct functionalization of inert chemical bonds, such as C–H, C–C, C–O, and C–N bonds in organic molecules, into other chemical bonds in a selective manner is appealing and represents a long-term challenge for chemists. Over the past few decades, considerable progress has been achieved in the selective functionalization of the C–H bond for the synthesis of C–C and C–X bonds.1–15 In particular, successful applications of direct C–H bond functionalizations in the synthesis of complex target molecules, including natural products, lead drug molecules, and functional materials have been achieved, demonstrating the significance and great potential of selective C–H bond functionalizations in organic synthesis and related areas.16–30 However, selective functionalization of specific C–H bonds in a chemo- and regioselective fashion remains a formidable challenge due to the massive existence of different C–H bonds and their similar reactivity. Typically, chemo- and regioselectivity can be achieved in substrates with specific substitution patterns or facilitated by an appropriate directing group. The use of a directing group significantly improves the selectivity of C–H activation, thus increasing the synthetic potential of C–H bond functionalizations. However, the installation and removal of directing groups from the substrates need tedious steps and impose extra challenges for this strategy. Accordingly, strategies enabling the C–H functionalization reaction proceed with high positional selectivity and install a functional group, which could directly serve as a synthetic linchpin for further elaboration to access a variety of well-defined molecular complexities, are ideal alternatives for selective C–H functionalizations. To date, a few highly selective C–H functionalization reactions which do not require directing groups, including the para-selective C–H TEDAylation (TEDA, N-(chloromethyl) triethylenediamine) reaction, have been achieved.31 The TEDA could serve as a chemical handle for further elaboration. Unfortunately, these methods suffer from limited types of substrates.
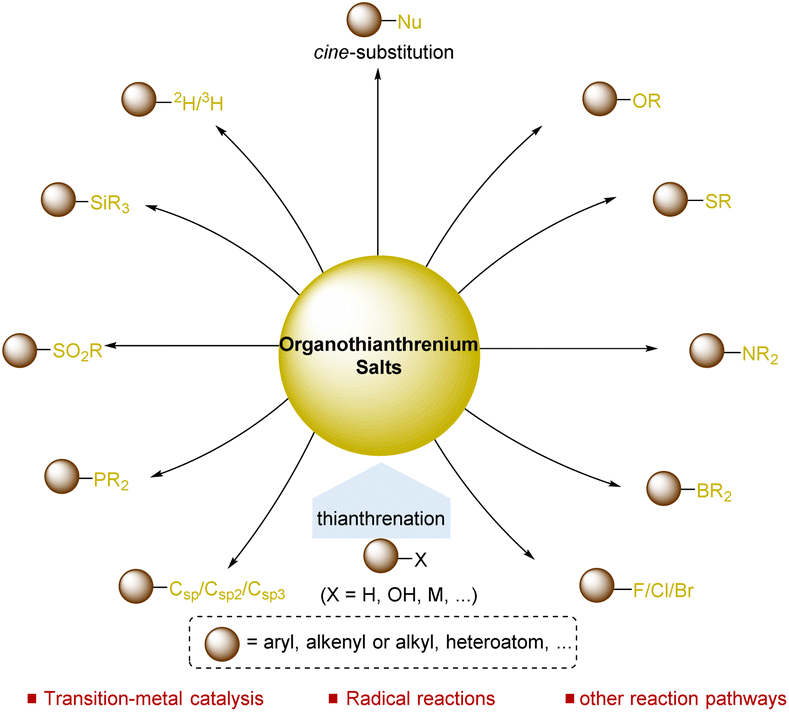
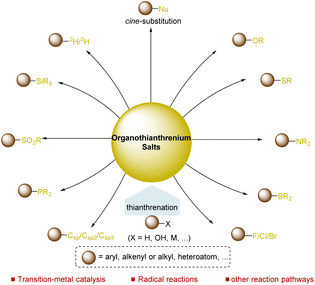
Over the past few decades, organosulfur compounds played a vital role in the development of synthetic chemistry,32 including sulfur ylides,33 sulfoxides,34 sulfonates,35 and sulfonium salts.36,37 As a typical type of organosulfur compound, organothianthrenium salts have immense potential in organic synthesis. As a typical type of organosulfur compound, organothianthrenium salts offer additional potential in organic synthesis due to their unique structural properties and reactivities. Thianthrenation provides an opportunity for direct access to thianthrenium salts in the absence of any directing groups via C–H, C–O, or C–M bond functionalizations. Furthermore, thianthrenation achieves positional selectivity by combining the electronic effect and steric effect together, providing additional chemical space for the selective formation of C–C, C–X bonds through different mechanisms under mild conditions.38 To date, organothianthrenium salts have been developed as versatile precursors for diverse chemical bond-forming processes.39–54 In 2022, Wang summarized recent advances in the thianthrenation-enabled C(sp2)–C/X bond formation reactions of aryl thianthrenium salts.50 However, a comprehensive review covering the synthesis and application of organothianthrenium salts, including aryl, alkenyl, and alkyl thianthrenium salts, is necessary. This review summarizes the synthesis of different types of organothianthrenium salts from various precursors by converting C–H and C–O bonds to C–S bonds. Moreover, the applications of organothianthrenium salts in organic synthesis to forge new C–C, C–H and C–heteroatom bonds by C–S bond cleavage are discussed with mechanistic considerations (Fig. 1).
 |
| | Fig. 1 Synthesis and utilization of organothianthrenium salts. | |
2. Synthesis of organothianthrenium salts
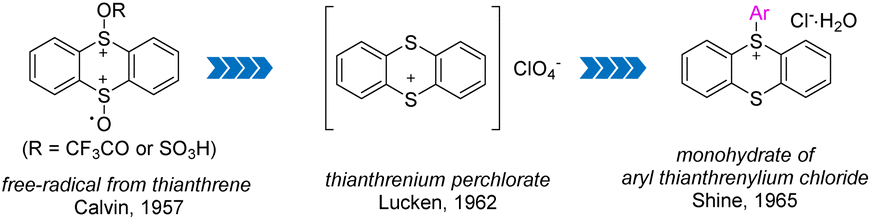
In 1957, Calvin and co-workers detected free cationic radical species by dissolving thianthrene or its monosulphoxide or disulphoxide in trifluoroacetic acid (Fig. 2).55 In 1962, Lucken isolated and characterized the formation of perchlorate of thianthrenium from perchloric acid and thianthrene for the first time.56 In 1965, Shine and co-workers synthesized and identified aryl thianthrenium salts for the first time by treating the thianthrene cationic radical intermediate with simple electron-rich aromatics.57 This seminal work has stimulated great effort into the development of efficient synthesis of different organothianthrenium salts.
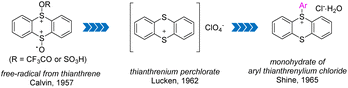 |
| | Fig. 2 The landmarks for the discovery of organothianthrenium salts. | |
2.1 Synthesis of aryl thianthrenium salts
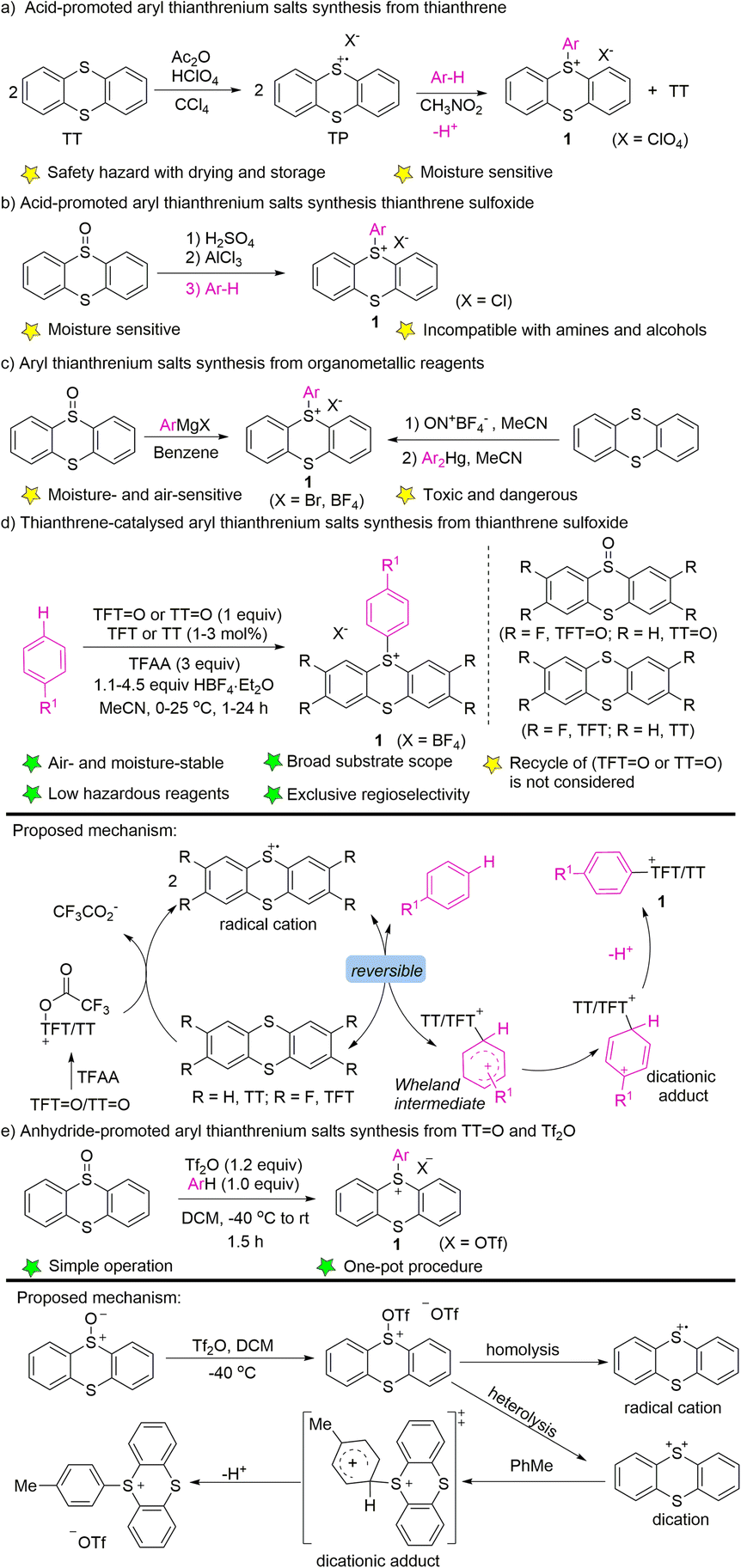
In 1971, Shine reported the electrophilic aromatic substitution of thianthrenium perchlorate with electron-rich aromatics to form the corresponding perchlorate aryl thianthrenium perchlorates (1) (Scheme 1a).40–42 Thianthrenium perchlorate (TP) was prepared from thianthrene, by treating it with perchloric acid and acetic anhydride. An excess amount of arenes was added to a solution of TP in nitromethane to generate the corresponding aryl thianthrenium perchlorates (1). The conditions worked well for anisole and toluene, but were not applicable to benzene, chlorobenzene and nitrobenzene. Later, the aryl thianthrenium chlorides (1) were obtained by the reaction of AlCl3 with thianthrene S-oxide, followed by treatment with arenes (Scheme 1b).42,44 However, the above-mentioned aryl thianthrenium salts have the propensity to undergoing detonation during drying, rendering the thianthrenium salts intrinsically unstable and dangerous. Later, methods using aryl Grignard reagents or Ar2Hg to synthesize aryl thianthrenium salts from thianthrene sulfoxide or thianthrene were developed (Scheme 1c). However, the organometallic reagents are toxic as well as sensitive to air and moisture.58,59 To this end, the development of organothianthrenium salts was suspended for decades. In 2019, Ritter and co-workers developed an elegant example of aryl thianthrenium tetrafluoroborate synthesis from the tetrafluorothianthrene sulfoxide (TFT![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) or thianthrene sulfoxide (TTO) reacting with arenes in the presence of a catalytic amount of tetrafluorothianthrene (TFT) or thianthrene (TT) (Scheme 1d).38A plausible reaction pathway involving thianthrene radical cations, which resulted in the formation of dicationic adducts and deprotonation to form the desired products was proposed. Further elucidation of the reaction mechanism by additional experimental and computational studies suggested a direct attack of arenes to O-trifluoracetylthianthrene S-oxide (TT+–TFA) or to the thianthrene dication (TT2+) via electron transfer under acidic conditions. A reversible interconversion of the different Wheland-type intermediates before an irreversible deprotonation is proposed to be responsible for the high para-selectivity of the reaction.52 Wang developed the synthesis of aryl thianthrenium salts from thianthrene sulfoxide (TTO) and arenes in the presence of triflic anhydride.60 The electrophilicity of thianthrene dication intermediates was proposed to account for the high regioselectivity of C–H thianthrenation (Scheme 1e).
O) or thianthrene sulfoxide (TTO) reacting with arenes in the presence of a catalytic amount of tetrafluorothianthrene (TFT) or thianthrene (TT) (Scheme 1d).38A plausible reaction pathway involving thianthrene radical cations, which resulted in the formation of dicationic adducts and deprotonation to form the desired products was proposed. Further elucidation of the reaction mechanism by additional experimental and computational studies suggested a direct attack of arenes to O-trifluoracetylthianthrene S-oxide (TT+–TFA) or to the thianthrene dication (TT2+) via electron transfer under acidic conditions. A reversible interconversion of the different Wheland-type intermediates before an irreversible deprotonation is proposed to be responsible for the high para-selectivity of the reaction.52 Wang developed the synthesis of aryl thianthrenium salts from thianthrene sulfoxide (TTO) and arenes in the presence of triflic anhydride.60 The electrophilicity of thianthrene dication intermediates was proposed to account for the high regioselectivity of C–H thianthrenation (Scheme 1e).
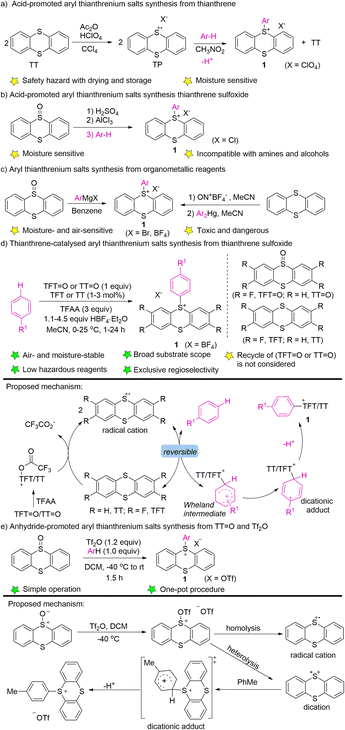 |
| | Scheme 1 Synthesis of aryl thianthrenium salts. | |
2.2 Synthesis of alkenyl thianthrenium salts
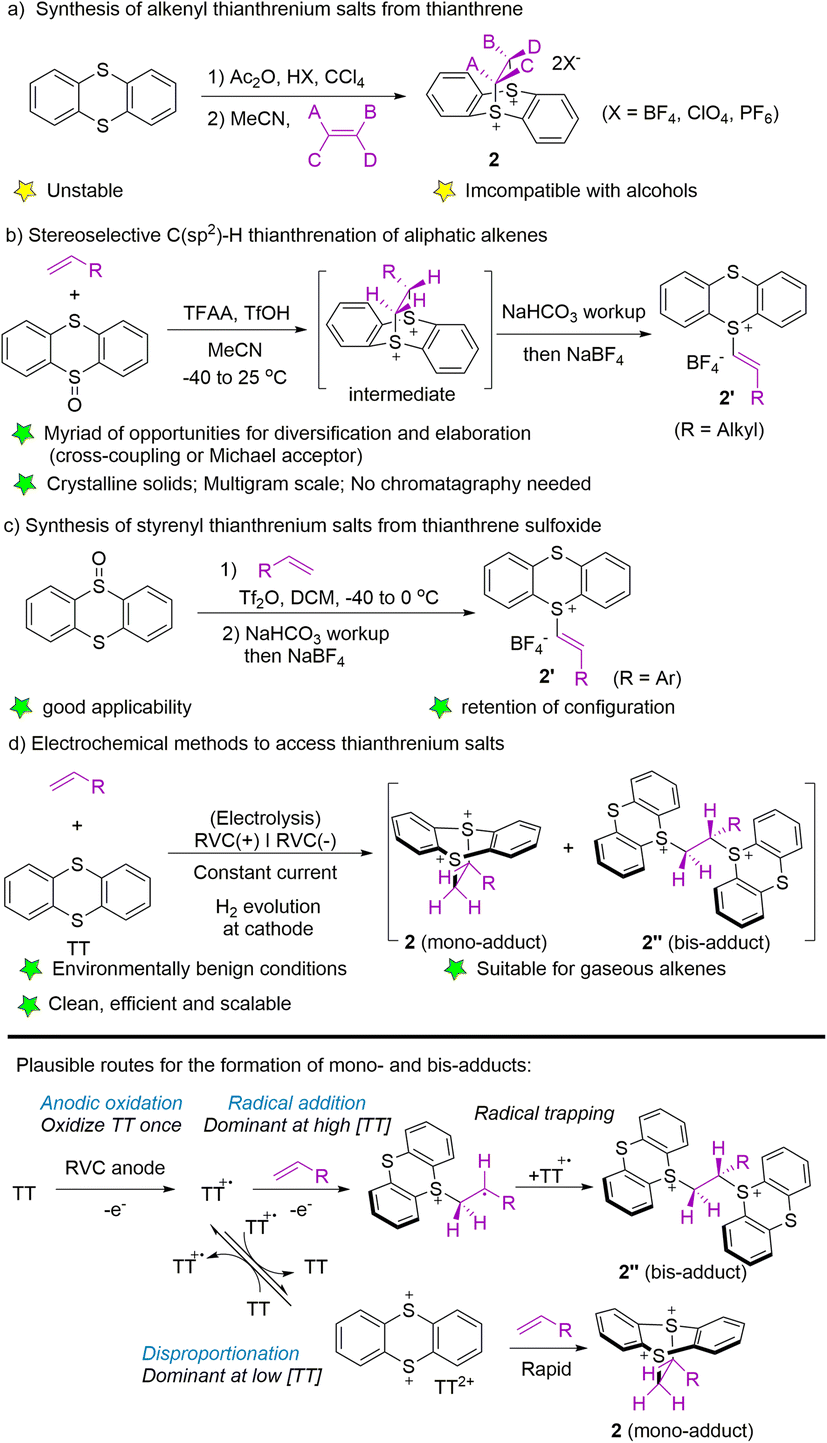
To date, three typical synthetic methods have been established for the synthesis of alkenyl thianthrenium salts. In 2004, Shine and co-workers utilized thianthrene to form thianthrene cation radical perchlorate in the presence of acetic acid and perchloric acid, followed by treatment with alkenes in acetonitrile to form alkenyl thianthrenium salts (2) (Scheme 2a).53 In 2019, Ritter employed trifluoroacetic anhydride to activate thianthrene oxide, followed by the reaction with alkenes to generate cyclic adducts, which furnished the alkenyl thianthrenium tetrafluoroborates (2′, R = alkyl) by base workup and anion exchange.54 The isolated alkenyl thianthrenium salts are thermal- and bench-stable, and insensitive to moisture and air (Scheme 2b). In 2021, the synthesis of styrenyl thianthrenium salts (2′) from styrenes and thianthrene oxide was developed (Scheme 2c).61 In 2021, Wickens and co-workers employed an electrochemical strategy to produce a mixture of dicationic adducts (2 and 2′′) from alkenes with thianthrene, which could serve as an equivalent to alkenyl thianthrenium salts (Scheme 2d).62 Although the reactivity of the two dicationic adducts may be high, the electrochemical formation of these double cation adducts are stable, insensitive to exposure to air, and even tolerate addition of exogenous water.
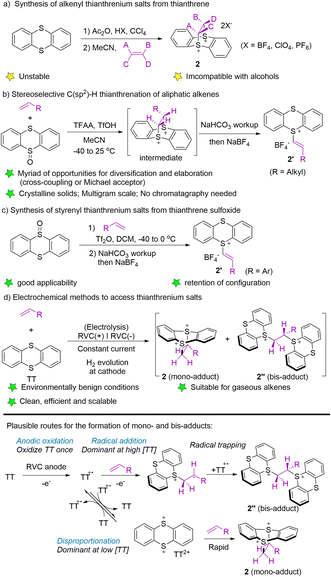 |
| | Scheme 2 Synthesis of alkenyl thianthrenium salts. | |
2.3 Synthesis of alkyl thianthrenium salts
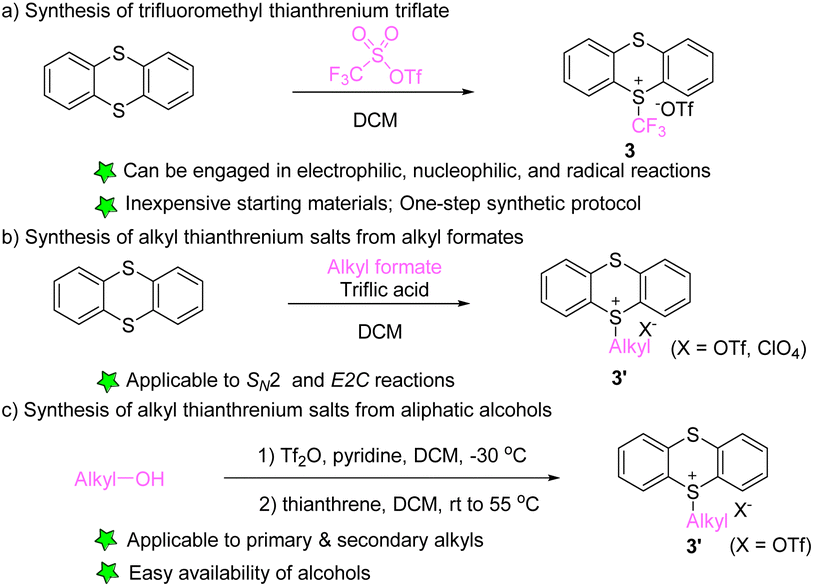
In 2021, Ritter’s group developed the synthesis of trifluoromethyl thianthrenium salt (3) by mixing thianthrene with trifluoromethyl sulfonic anhydride (Scheme 3a).63 Trifluoromethyl thianthrenium salt has the advantages of simple preparation, easy handling, high reactivity, and broad tolerance of functional groups during the reaction. Trifluoromethyl thianthrenium salt can be employed as the precursor of the trifluoromethyl cation, radical and anion to participate in diverse trifluoromethylation reactions. In 2001, Shine reported the synthesis of alkyl thianthrenium salts (3′) by treating thianthrene with alkyl formates and triflic acid (Scheme 3b).64 Recently, Shi and co-workers reported the synthesis of regular alkyl thianthrenium salts (3′) from the corresponding aliphatic alcohols (Scheme 3c). This protocol utilizes triflic anhydride to activate alcohol followed by treatment with thianthrene (TT), and is applicable to the synthesis of primary and secondary alkyl thianthrenium salts.65 Due to the easy availability of alcohols, this method provides access to a wide range of alkyl thianthrenium salts.
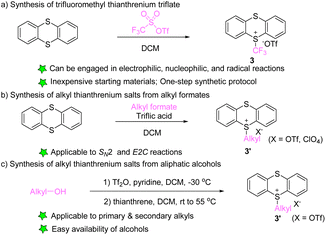 |
| | Scheme 3 Synthesis of alkyl thianthrenium salts. | |
3. Utilization of thianthrenium salts in organic synthesis
Since substantial progress in the synthesis of organothianthrenium salts (R–TT+X−, R = aryl, alkenyl, alkyl) has been achieved, more and more attention has been paid to utilize thianthrenium salts as precursors for diverse bond-forming processes in order to ultimately transform C–H or C–O bonds to carbon–carbon bonds or carbon–heteroatom bonds.
3.1. Application of aryl thianthrenium salts
(Hetero)aryl thianthrenium salts (Ar–TT+X−) could be accessed from the selective transformation of the C–H bonds of (hetero)arenes to C–S bonds in the absence of any directing group. The use of aryl thianthrenium salts for new bond-forming reactions provides a promising alternative to functionalize Csp2–H bonds without the assistance of directing groups.66–69 To date, aryl thianthrenium salts have been demonstrated to construct C–C, C–N, C–O, C–X, and C–S bonds through different mechanistic pathways.
3.1.1 Transition-metal-catalysed cross-coupling of aryl thianthrenium salts.
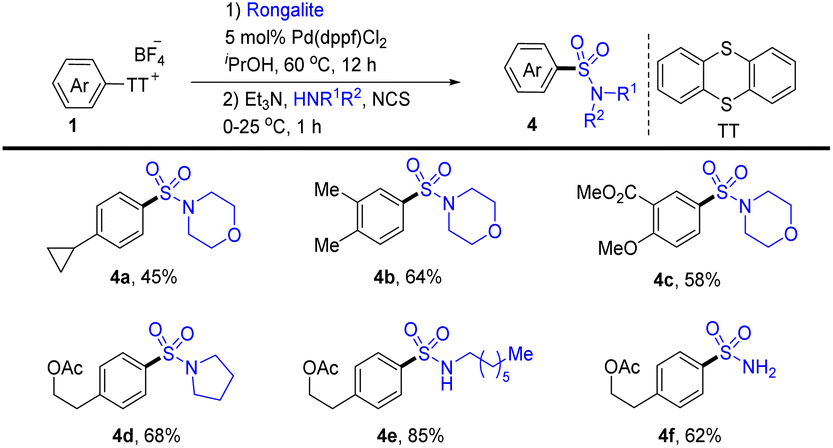
In 2020, Ritter’s group reported a two-step regioselective C–H sulfination of arenes at room temperature enabled by the formation of aryl thianthrenium salts (Scheme 4).70 Using sodium hydroxymethylsulfinate (rongalite) as the source of SO22−, palladium-catalysed C–S coupling of aryl thianthrenium salts (1) introduces the hydroxymethyl sulfonyl group onto arenes. The hydroxymethyl sulfone intermediates generated from the catalytic process can be employed as a chemical handle to furnish different types of sulfonamides (4) by dealing with amines. Notably, this method directly employs rongalite as a readily available and cost-effective surrogate of SO22−, circumventing the use of a toxic gas (SO2).
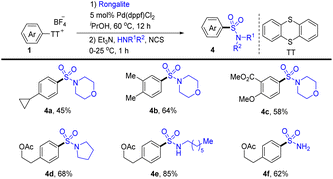 |
| | Scheme 4 Two-step sulfination of arenes by thianthrenation. | |
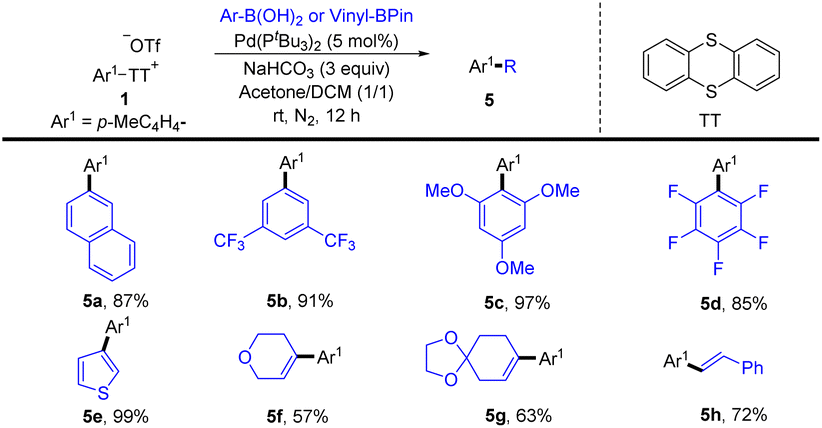
In 2020, Wang and co-workers reported a palladium-catalysed highly site-selective Suzuki–Miyaura coupling reaction to realize para-arylation and alkenylation of the C–H bond of monosubstituted aromatic compounds by coupling with aryl and vinyl boronic acids were achieved in good yields with excellent regioselectivity (Scheme 5).71 Notably, this strategy was compatible with both electron-deficient and electron-rich aromatics, as well as (hetero)aromatics. Moreover, this protocol was successfully applied to the direct synthesis of bioactive molecules.
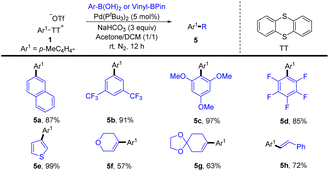 |
| | Scheme 5 Palladium-catalysed Suzuki–Miyaura coupling reaction of aryl thianthrenium salts. | |
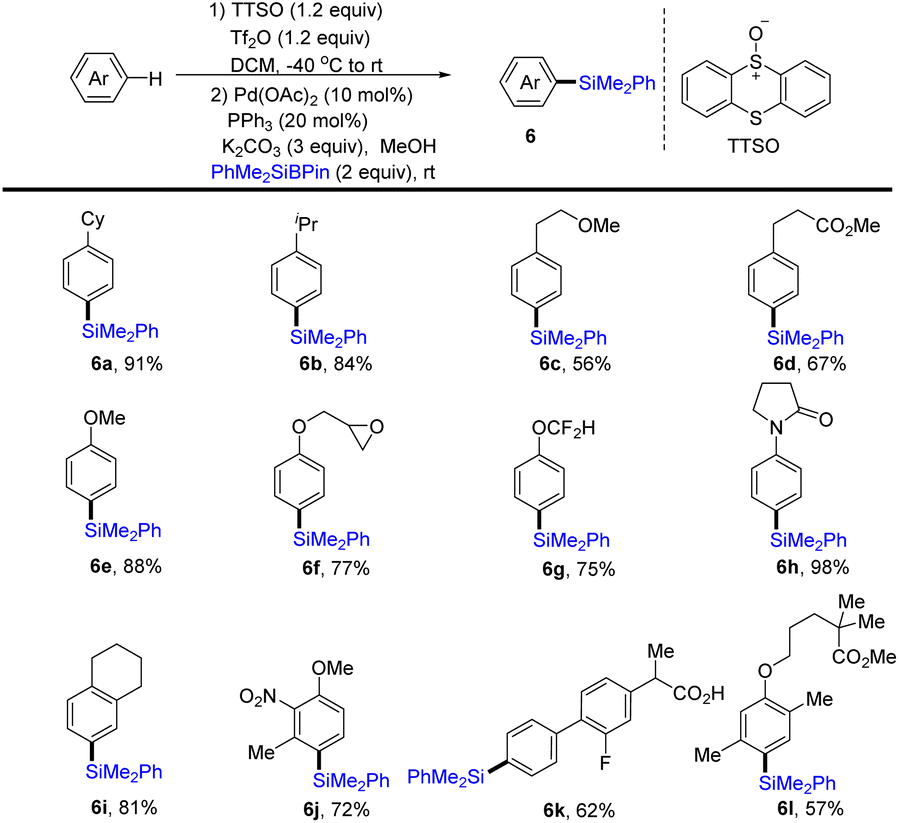
In 2020, Wang and co-workers realized the one-pot procedure to transform the C–H bond of arenes into a C–Si bond through the formation of aryl thianthrenium salts from arenes followed by palladium-catalysed C–Si formation to furnish arylsilane (6) (Scheme 6).72 This strategy is suitable for both monosubstituted and multi-substituted aromatics to deliver highly regioselective silylation products. Unfortunately, application of the silylation of a C–H bond to (hetero)arenes is not demonstrated.
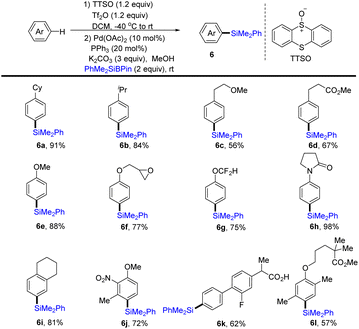 |
| | Scheme 6 Regioselective silylation of arenes by thianthrenation. | |
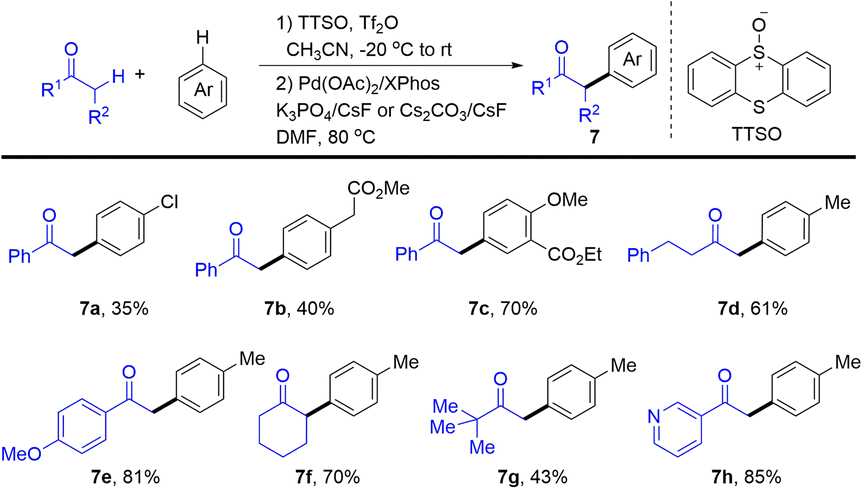
In 2020, Wang and co-workers developed Pd-catalysed α-arylation of carbonyl compounds with aromatics enabled by thianthrenation of arenes to provide the desired α-arylated ketones (7) (Scheme 7).73 Aryl thianthrenium salts were generated in situ in a regioselective manner under mild conditions followed by Pd-catalysed arylation of ketones. Notably, thianthrene or phenoxathiine could be recovered after the completion of the C–C cross-coupling reaction, increasing the atom-economy of the reaction of organothianthrenium salts. The reaction condition is compatible with heterocycle-containing carbonyl compounds and complex bioactive molecules, which is appealing in medicinal chemistry.
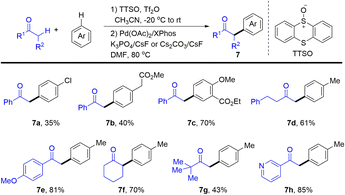 |
| | Scheme 7
α-Arylation of carbonyl compounds using aryl thianthrenium salts. | |
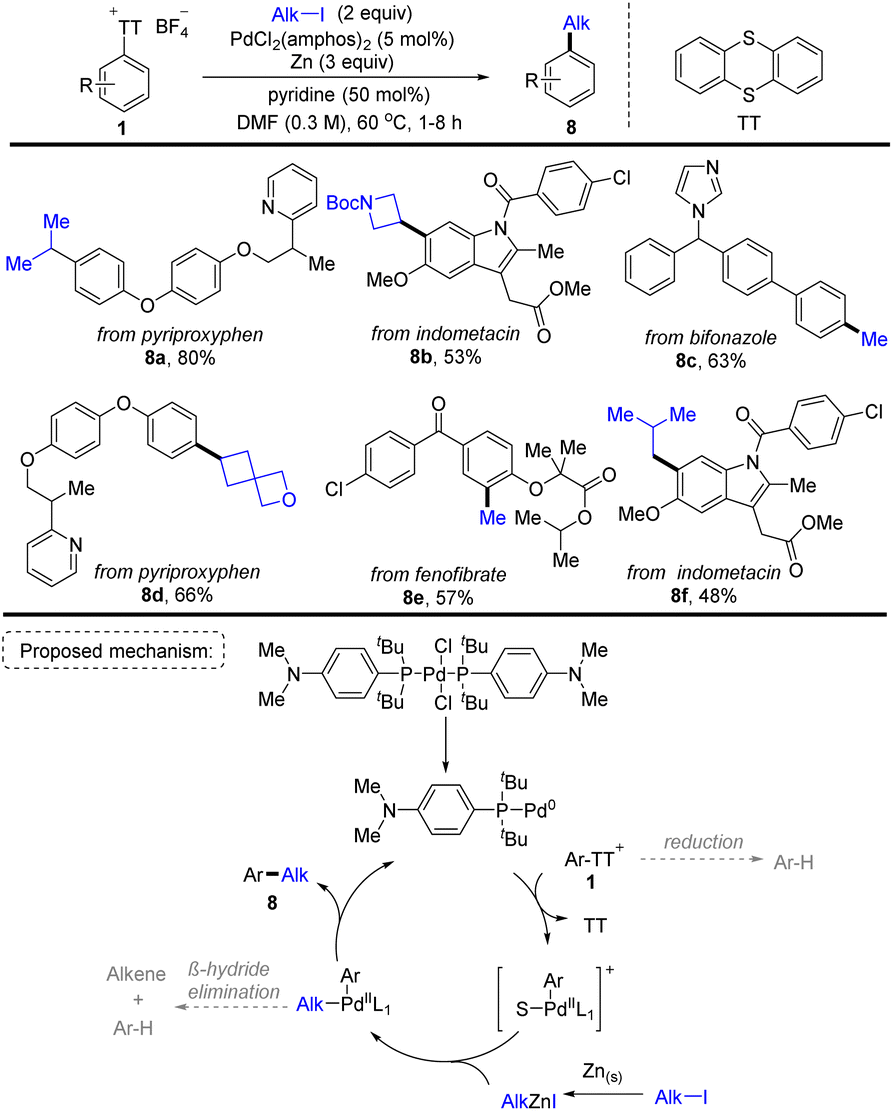
In 2021, Ritter and co-workers discovered a Pd-catalysed two-step aryl thianthrenation/reductive alkylation sequence of arenes using alkyl halides as the alkylating reagents, providing a highly site-selective one-pot strategy for C–H alkylation of non-directed aromatic compounds (Scheme 8).74 A plausible reaction pathway was proposed. First, a monoligated Pd0 catalyst was formed from the resting state PdCl2(amphos)2. The Pd0 catalyst underwent oxidative addition with Ar–TT+ to afford an aryl PdII cation. Meanwhile, zinc reacted with alkyl iodide to form an alkyl zinc reagent. The aryl PdII cation underwent transmetalation with the alkyl zinc reagent to obtain an alkyl aryl PdII intermediate, followed by reductive elimination to deliver the final product (8) and regenerate the Pd0 catalyst. This strategy furnished a wide range of synthetically useful alkylated arenes which are inaccessible otherwise. The robustness and practicality of this method is further demonstrated by the reductive coupling of two complex fragments.
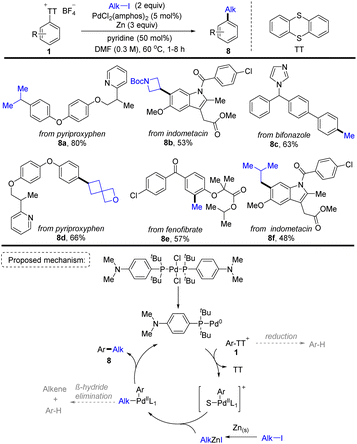 |
| | Scheme 8 Pd-catalysed reductive alkylation of aryl thianthrenium salts. | |
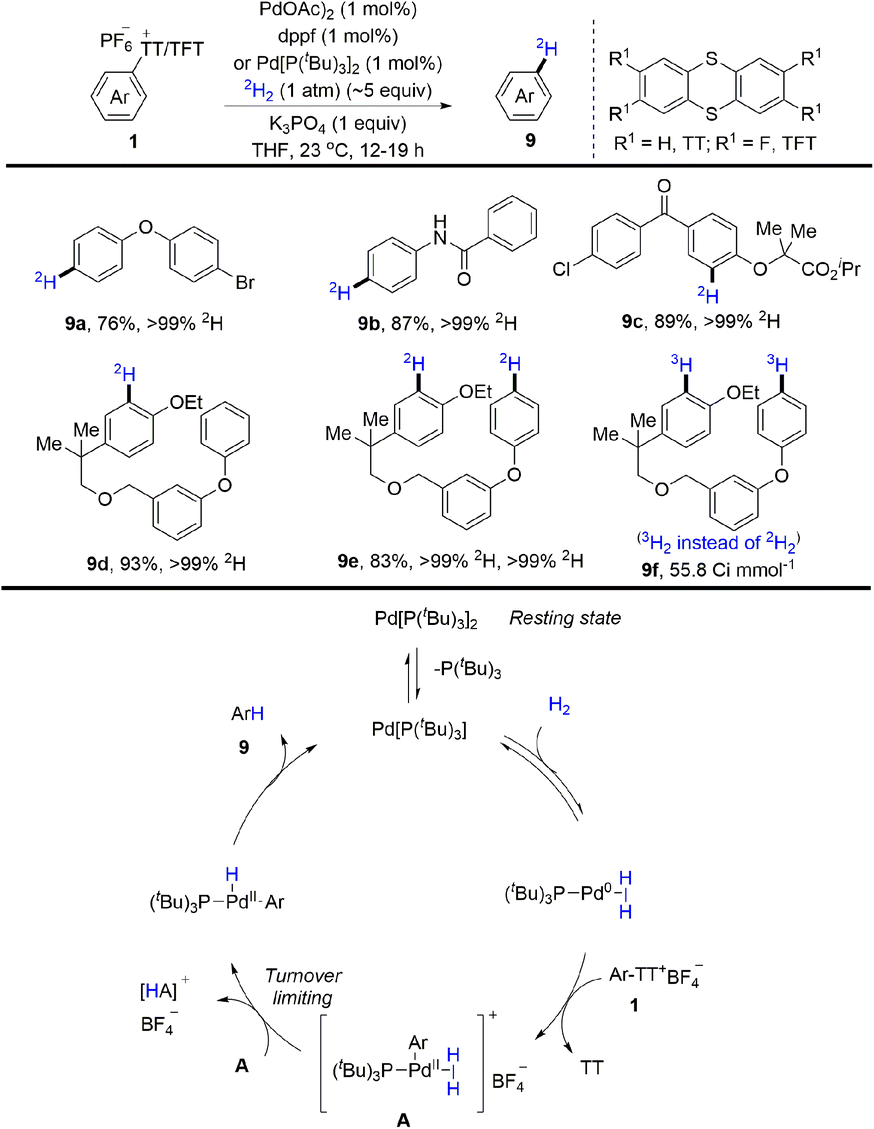
Tritium and deuterium labeling are critical tools to introduce radioactive tags into drugs without altering the chemical and physical properties as well as biological activity of the molecules. In 2021, radioactive tags were introduced into drugs without altering the chemical and physical properties as well as biological activity of the molecules. In 2021, Ritter and co-workers reported palladium-catalysed reductive deuteration and tritiation of aryl thianthrenium salts (1) (Scheme 9).75 Ritter used aryl thianthrenium salts on the micromole scale in 2H2/3H2 gas at atmospheric pressure. A proposed mechanism showed that a monoligated Pd0 catalyst generated from Pd[(PtBu3)]2 would undergo reversible association with H2 and oxidative addition with aryl thianthrenium salts (1) to afford A. Then A went through dihydrogen splitting and reductive elimination to furnish the final product (9) and regenerate the Pd0 catalyst. This reaction tolerates a wide range of substitution patterns, providing more opportunities for drug design in the future.
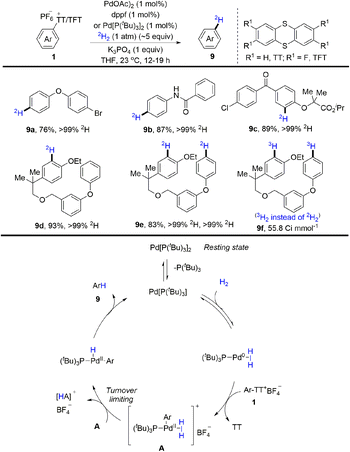 |
| | Scheme 9 Reductive deuteration/tritiation of aryl thianthrenium salts. | |
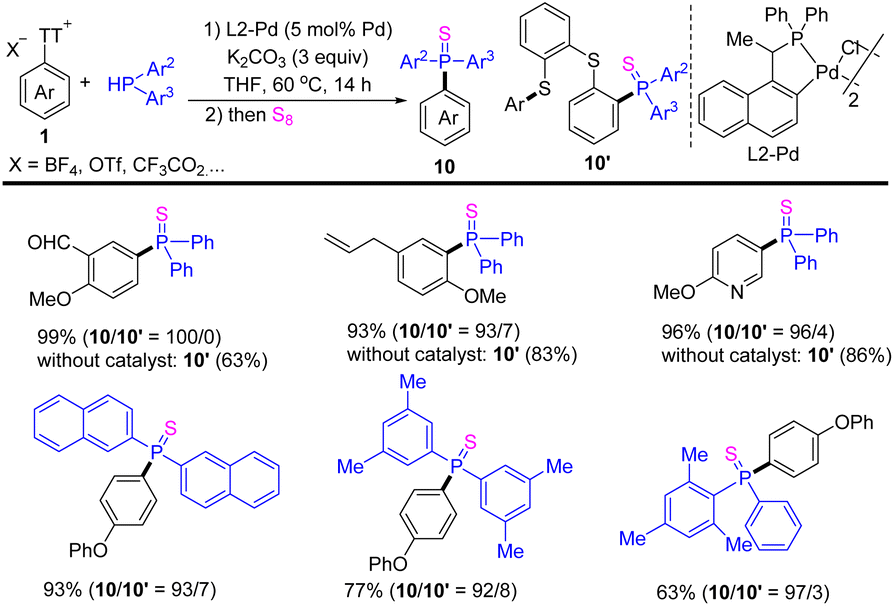
In 2021, Huang reported the cross-coupling reaction of aryl thianthrenium salts 1 with diarylphosphine to form a C–P bond (Scheme 10).76 The use of palladacycle catalysts significantly accelerated the cleavage rate of the exocyclic C–S bond, introducing phosphine groups on to arenes to access diverse substituted triarylphosphine sulfide (10). In contrast, the reaction of aryl thianthrenium salts underwent a phosphinative ring-opening process via the cleavage of the endocyclic C–S bond to afford (10′) in the absence of a catalyst. This strategy is suitable for all kinds of substituted aromatic and heteroaromatic compounds.
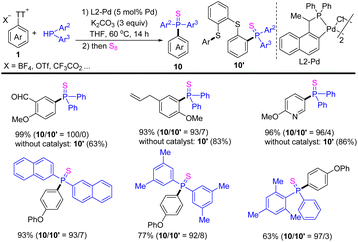 |
| | Scheme 10 Palladium-catalysed phosphination of aryl thianthrenium salts with diarylphosphine. | |
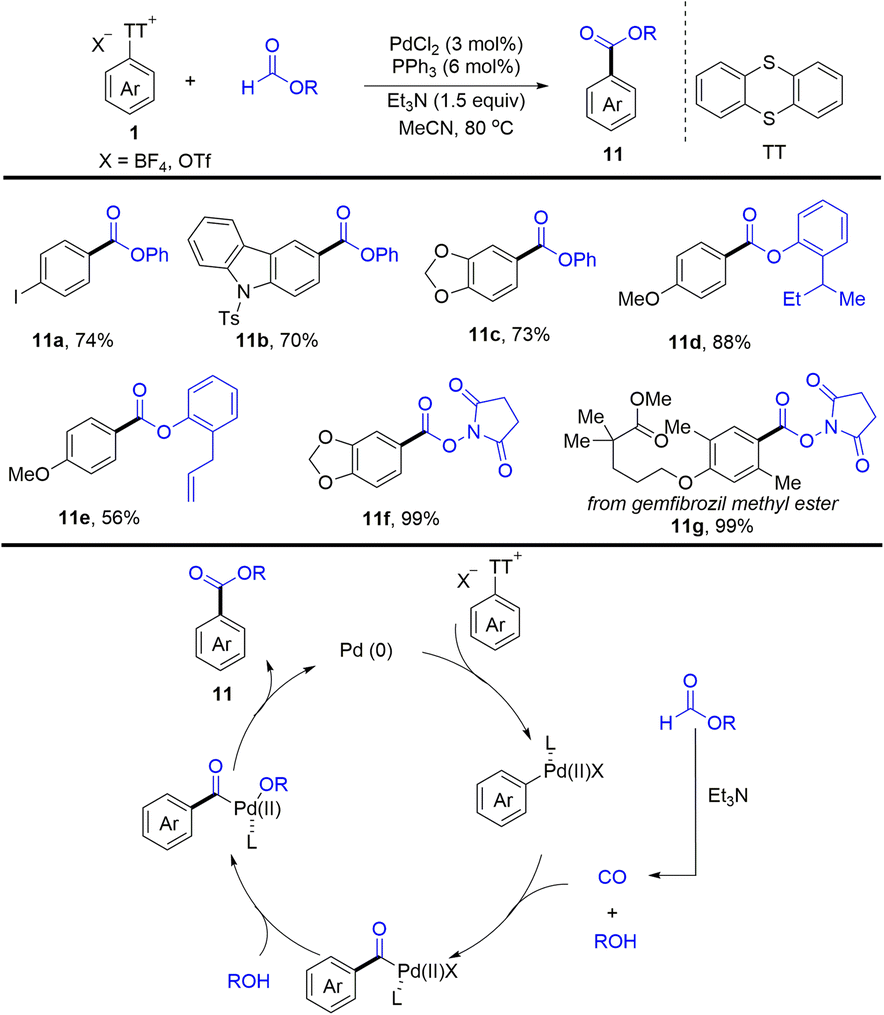
In 2022, Zhao and co-workers reported an efficient palladium-catalysed esterification of aryl thianthrenium salts (Scheme 11).77 The reaction provided a straightforward method for the esterification of C–H of arenes by thianthrenation with good functional group tolerance. The proposed mechanism showed that Pd0 was converted into Ar–PdII complexes via oxidative addition with aryl thianthrenium salts (Ar–TT+). Carbon monoxide and phenol were formed in situ from phenol formate in the presence of Et3N. The insertion of CO to Ar–PdII complexes afforded acyl palladium complexes, which underwent anion exchange with phenol to afford Ar–PdII–OR complexes. Reductive elimination of Ar–PdII–OR afforded the desired esters (11) and regenerated Pd0.
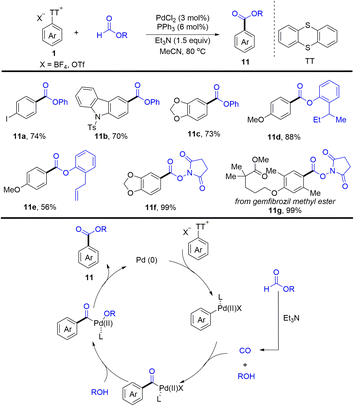 |
| | Scheme 11 Palladium-catalysed esterification of aryl thianthrenium salts. | |
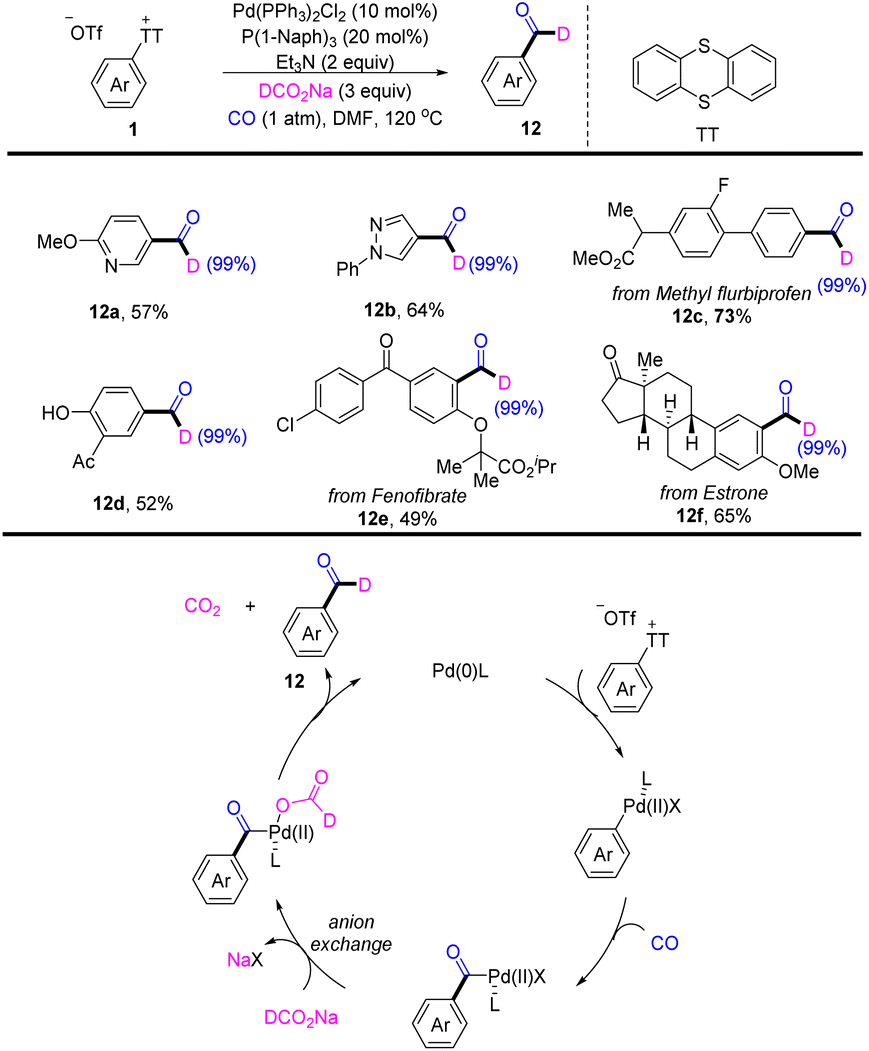
In 2022, Zhao reported a palladium-catalysed deuterated formylation of aryl sulfonium salts (Scheme 12).78 A proposed mechanism showed that the Pd0 catalyst underwent oxidative addition with Ar–TT+ to afford an aryl PdII cation. Coordination and insertion of CO into an aryl PdII cation generating a benzoyl PdII intermediate. A new benzoyl PdII intermediate was generated, which underwent anion exchange. After decarboxylation and reductive elimination, the desired product (12) was obtained. This reaction tolerates a wide range of substitution patterns, such as (hetero)aryl, ester, and hydroxyl. This reaction is made available for bioactive molecules.
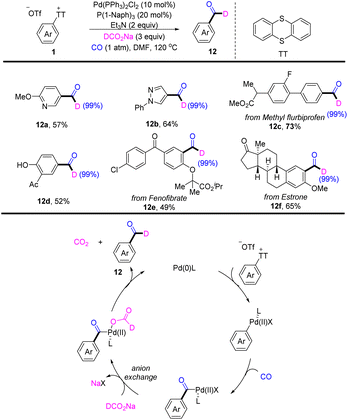 |
| | Scheme 12 Palladium-catalysed deuterated formylation of aryl thianthrenium salts. | |
3.1.2 Free radical pathway.
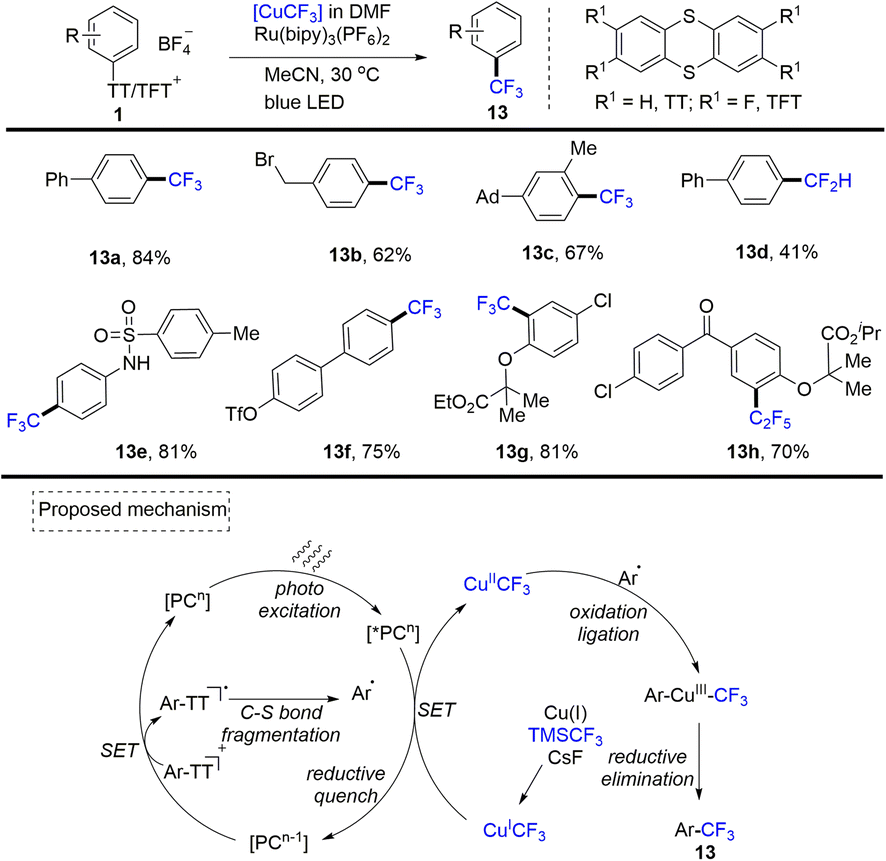
In 2019, Ritter reported the first photoredox-catalysed cross-coupling of aryl thianthrenium salts (1) with copper-based trifluoromethyl reagents to achieve regioselective trifluoromethylation of aromatics (Scheme 13).79 A plausible mechanism indicated that the photocatalyst PC was converted to excited *PCn through photo-excitation. *PCn was quenched by the [CuICF3] reagent forming [CuIICF3] and PCn−1. Meanwhile, single electron transfer between aryl sulfonium salts and PCn−1 resulted in C–S bond fragmentation to generate aryl radicals and PC. Aryl radicals could recombine with [CuIICF3] to form an aryl–CuIII–CF3 intermediate. Then, reductive elimination of the CuIII intermediate could deliver the desired product (13) and CuI. This strategy can also be further applied to the pentafluoroethylation and difluoromethylation of aryl thianthrenium salts.
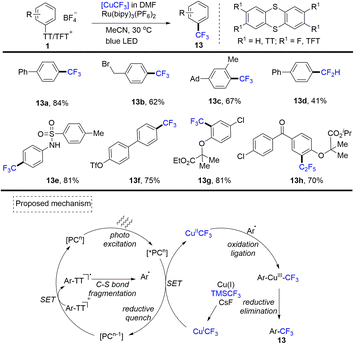 |
| | Scheme 13 Cross-coupling of aryl thianthrenium salts with copper-based trifluoromethyl reagents. | |
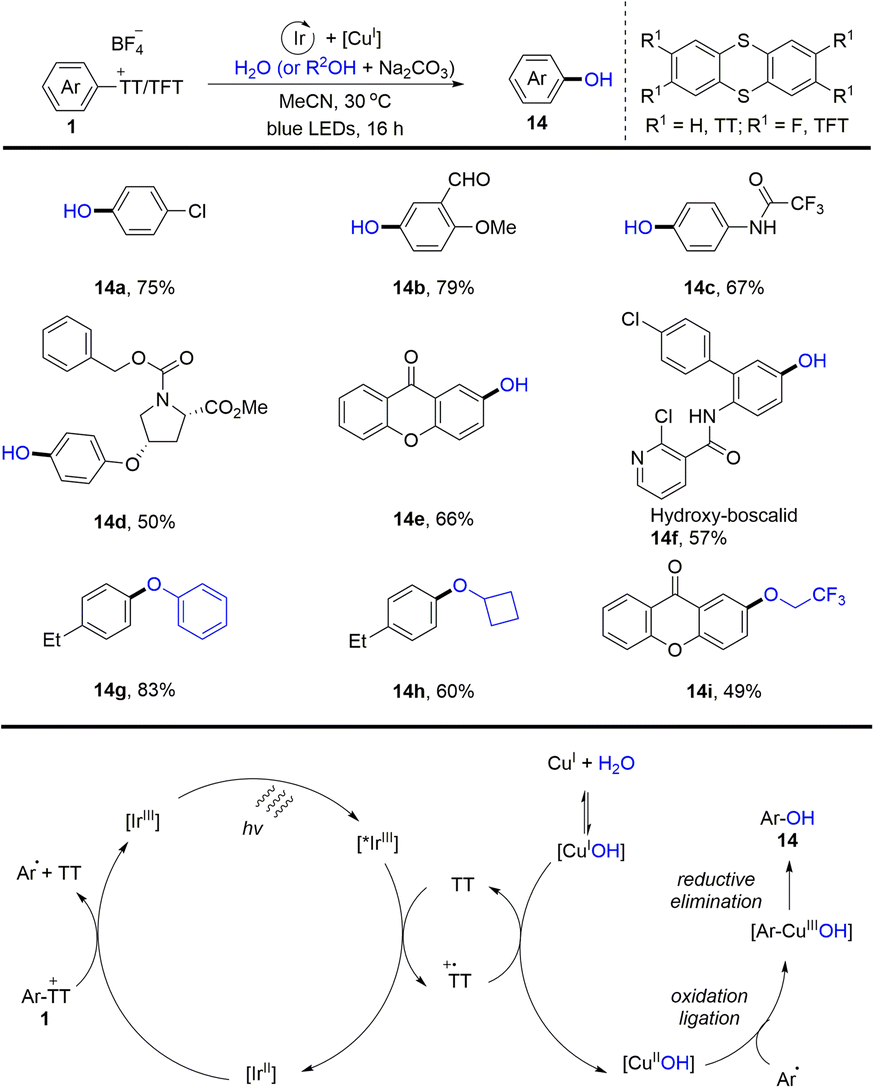
In the same year, Ritter and co-workers reported a two-step procedure for regioselective C–O bond cross-coupling from the C–H bond of arenes by the formation of aryl thianthrenium salts (1) (Scheme 14).80 A series of phenols and aryl ethers (14) were synthesized by photoredox and transition metal-catalysed coupling of aryl thianthrenium salts with water or alcohols. The proposed mechanism shows that the excited photocatalyst *IrIII was quenched by thianthrene (TT) to form a thianthrenium radical cation (TT˙+) and reduced photocatalyst IrII. IrII interacted with aryl thianthrenium salt (Ar–TT+) to generate an aryl radical, thianthrene (TT) along with IrIII. On the other hand, CuIOH could be generated from CuITC or Cu2O in the presence of water, which underwent SET with a thianthrenium radical cation (TT˙+) to form CuIIOH. Aryl radicals recombined with CuIIOH species to afford Ar–CuIIIOH species, which underwent reductive elimination to furnish the final product (14) by C–O bond formation. Using this strategy, both electron-rich and electron-deficient aromatics could be successfully converted into the corresponding phenols and aryl ethers.
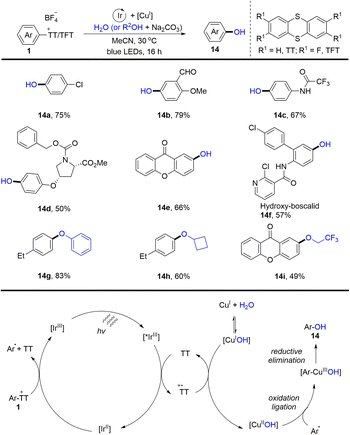 |
| | Scheme 14 Regioselective hydroxylation of aryl thianthrenium salts. | |
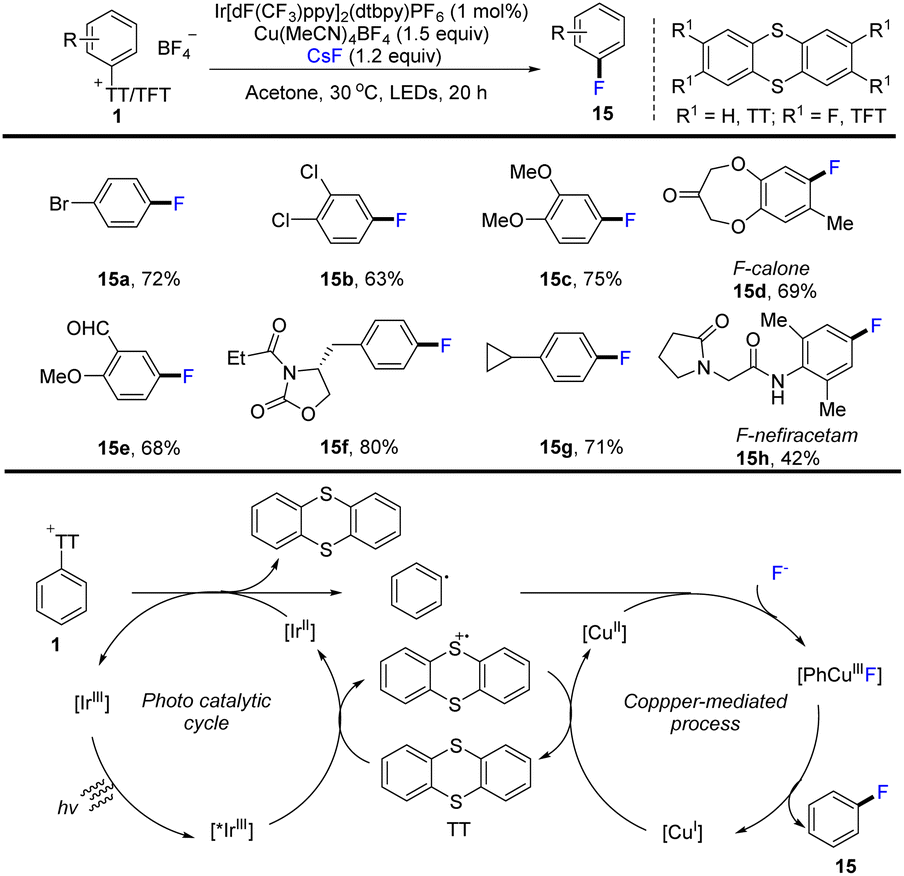
In 2020, Ritter and co-workers developed the use of aryl thianthrenium salts (1) as a mediator to convert aromatics into aryl fluorides by thianthrenation. This strategy successfully constructed an aryl C–F bond through CuIII metallophotoredox catalysis (Scheme 15).81 The proposed mechanism shows that an excited photoredox catalyst *IrIII was formed through photo-excitation. The excited photocatalyst *IrIII was quenched by thianthrene (TT) to form a thianthrenium radical cation (TT˙+) and reduced photocatalyst IrII. Reduction of the aryl thianthrenium salts with IrII resulted in aryl radicals, TT, and IrIII. CuI species reacted with the aryl thianthrenium cationic radical via SET to form CuII species. Then CuII species underwent oxidative ligation with aryl radicals and anion-exchange with fluoride to afford Ar–CuIII–F complexes, followed by reductive elimination to generate product (15) by C–F bond formation. Compared with the corresponding halides, aryl thianthrenium salts (1) have higher reduction potentials. The dissociation rate of aromatic radical anions is obviously sped up, and thus the formation process of C–F bond via metallophotoredox catalysis can be realized. This strategy is compatible with electron-deficient aromatics, electron-rich aromatics, aldehydes, amides, phenols, benzyl ethers and halides.
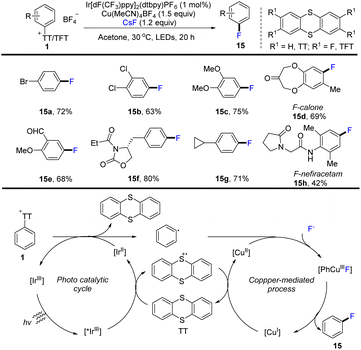 |
| | Scheme 15 Photoredox and Cu-catalysed fluorination of aryl thianthrenium salts. | |
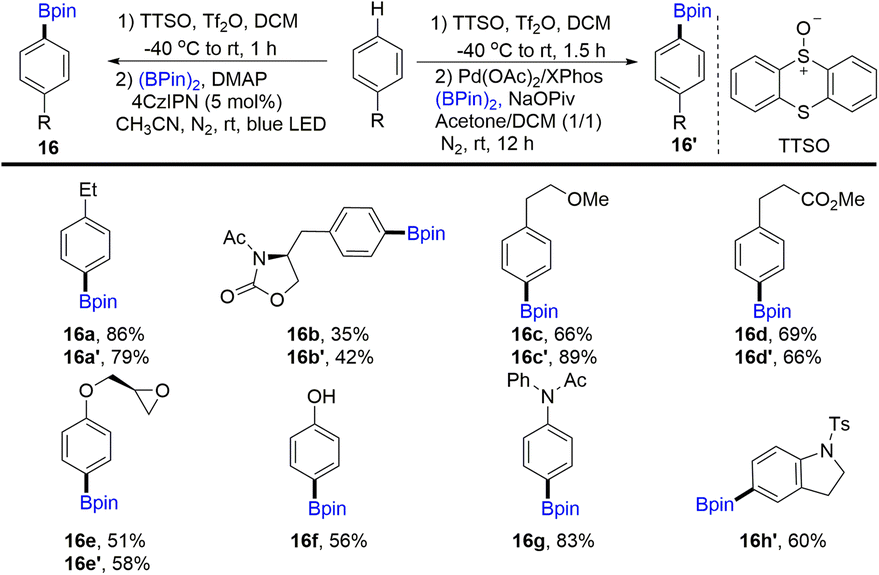
In 2020, Wang and co-workers discovered the para-selective borylation of monosubstituted benzenes via the formation of aryl thianthrenium salts (Scheme 16).60,82 Borylation products (16) can be obtained by photocatalysis or metal catalysis. Various electron-rich mono-substituted arenes could be site-selectively borylated to deliver functionalized aryl borylation. Mechanistic investigation indicated that para-selectivity might be correlated to the high electrophilicity of thianthrene dication intermediates.
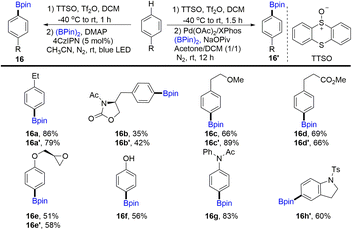 |
| | Scheme 16
para-Selective borylation of monosubstituted benzenes with aryl thianthrenium salts. | |
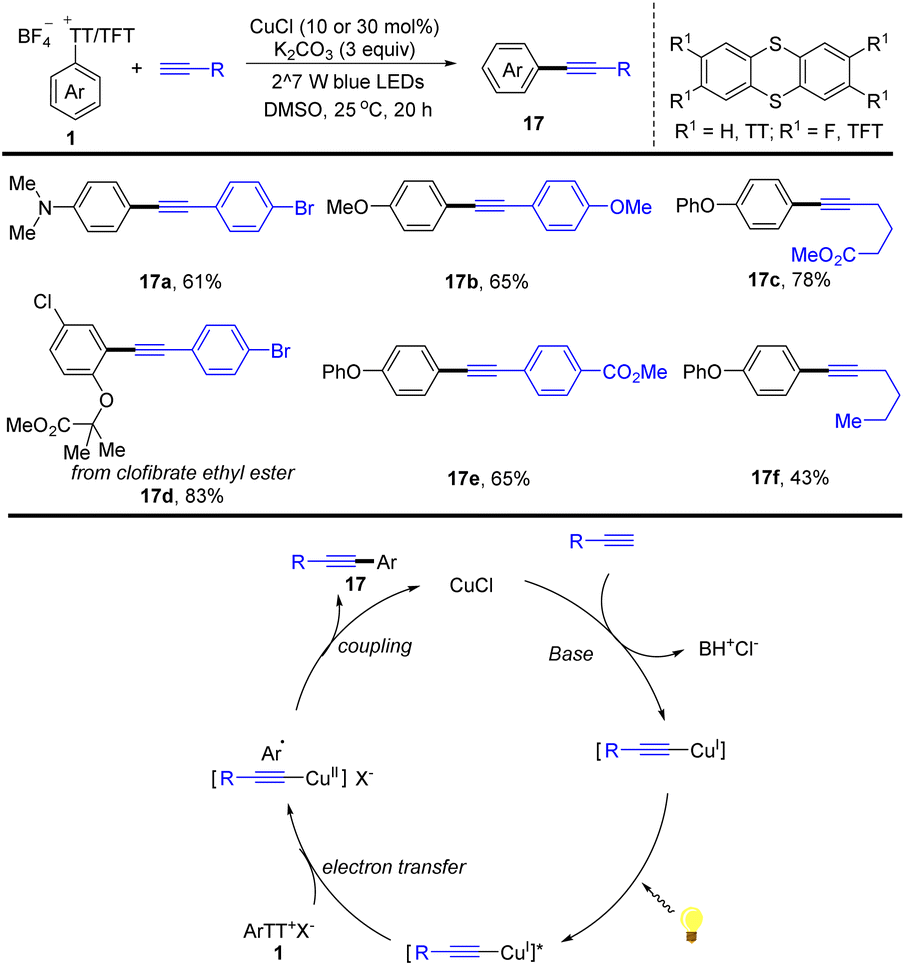
In the same year, Guo reported an alternative for the synthesis of arylalkynes (17) enabled by visible-light promoted copper-catalysed Sonogashira reaction of aryl thianthrenium salts with alkynes (Scheme 17).83 This strategy selectively transformed arenes into arylalkynes by thianthrenation with different aryl sulfonates, providing a convenient synthetic strategy for the construction of functionalized arylalkynes. The proposed mechanism shows that a Cu acetylide intermediate is formed from CuICl with a terminal alkyne. The Cu acetylide intermediate could be excited by visible light irradiation to form a photoexcited complex, which could undergo an SET process with aryl thianthrenium salts (1) and reductive elimination to afford the arylalkynes (17) and regenerate CuICl. This method can be applied to late-stage alkynylation of drugs, which is difficult to achieve otherwise, indicating the potential of this work in rapid drug modification.
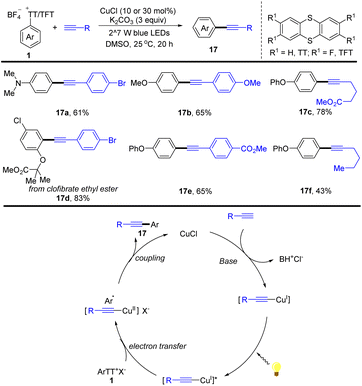 |
| | Scheme 17 Cu-catalysed Sonogashira-type reaction of aryl thianthrenium salts. | |
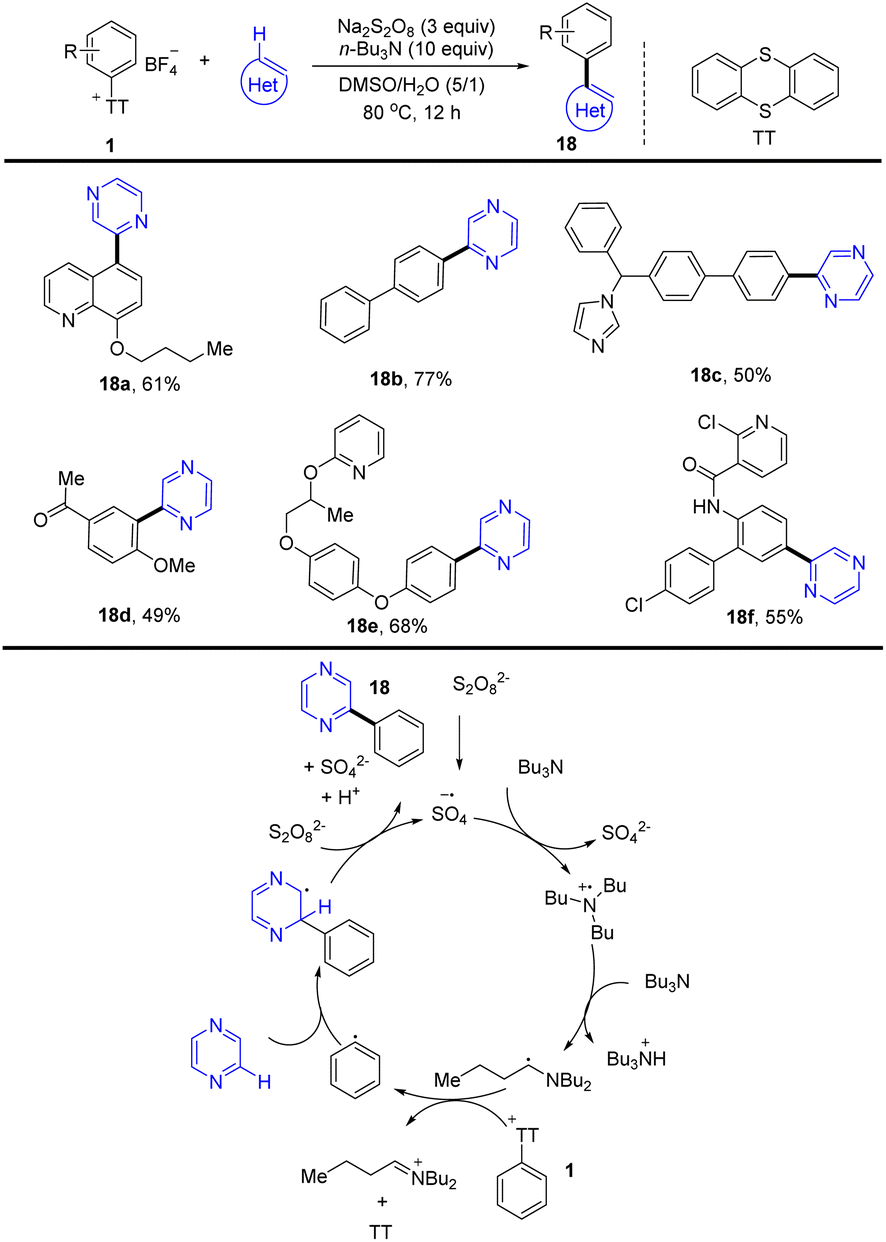
(Hetero)biaryls are ubiquitous substructures in medical substances, functional materials, and agrochemicals. In 2021, Ritter and co-workers reported a late-stage heteroarylation of aryl thianthrenium salts (1) activated by a α-amino alkyl radical (Scheme 18).84 The activated mode is different from halogen-atom transfer (XAT), delivering a wide range of biaryls with heteroarenes. The proposed mechanism shows that an amine radical cation was formed from an amine via single-electron oxidation with a sulfate radical anion, which further generated an α-aminoalkyl radical by deprotonation. The α-amino alkyl radical interacted with aryl thianthrenium salt (1) to form thianthrene and an aryl radical. Then the aryl radical was added to a heteroarene followed by single-electron oxidation and deprotonation to generate the biaryl product (18) (Scheme 18). The reaction proceeded without any additives, light, or transition metals and was compatible with air and water.
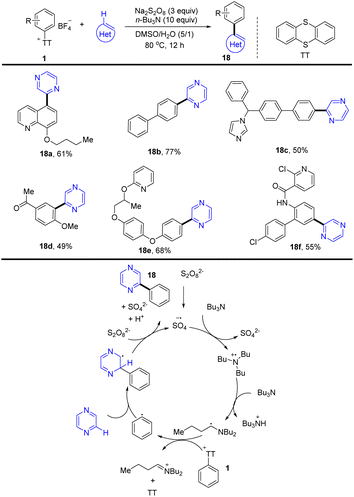 |
| | Scheme 18 Late-stage heteroarylation of aryl thianthrenium salts. | |
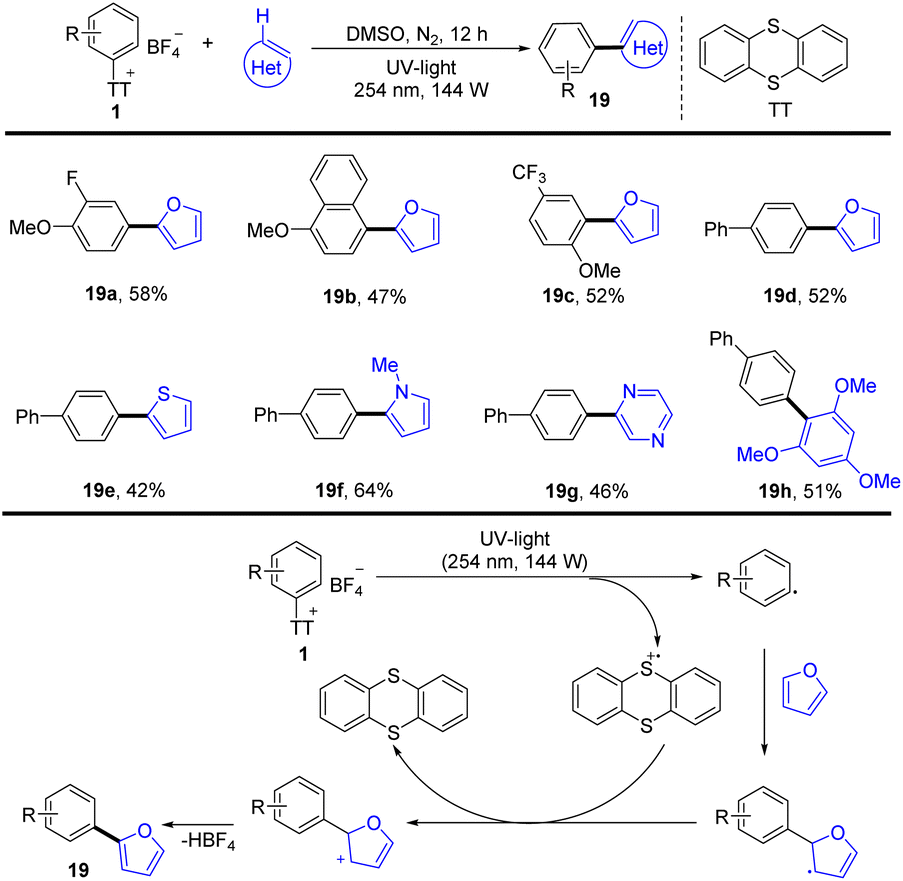
In 2021, Patureau and co-workers reported a photoinduced and catalyst-free (hetero)arylation of aryl thianthrenium salts (Scheme 19).85 The proposed mechanism shows that the key to success is the use of UV-light, which could disrupt the C–S bond of aryl thianthrenium salts (1) to form thianthrene radical cations and aryl radicals. Aryl radicals were added to heteroarene to form a new radical intermediate, which underwent single electron oxidation by thianthrene radical cations to deliver the final biaryls (19) by deprotonation.
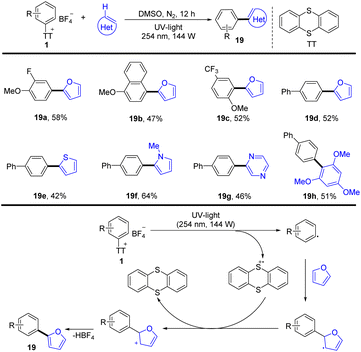 |
| | Scheme 19 Photochemical (hetero)arylation of aryl thianthrenium salts. | |
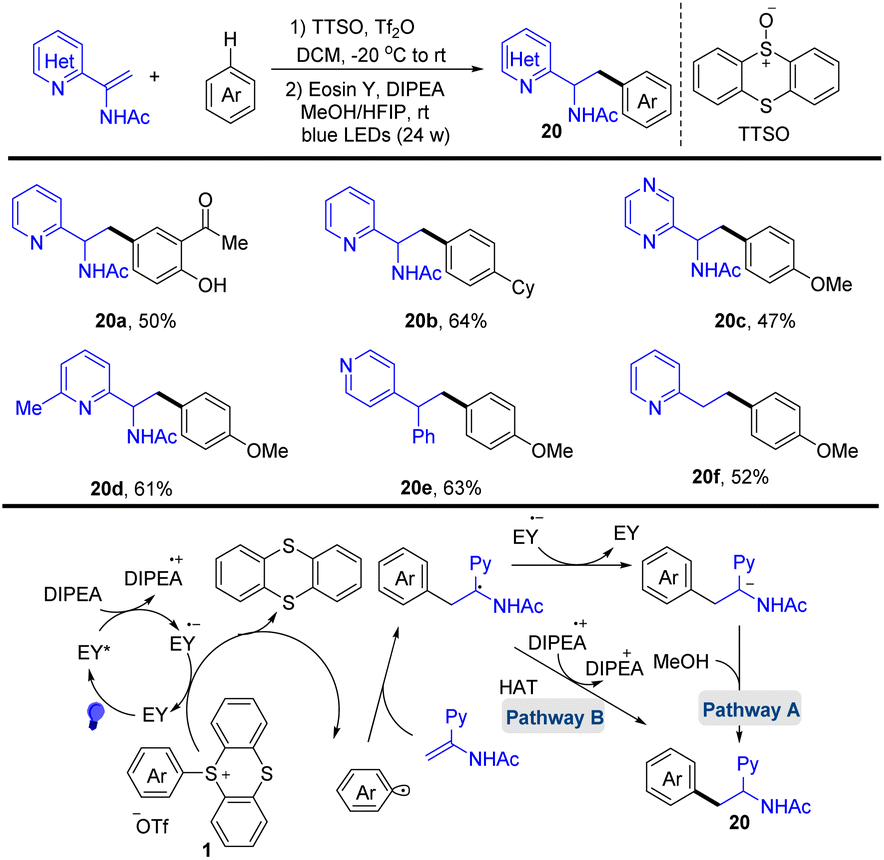
In 2020, Wang and co-workers reported the photocatalysed synthesis of α-amino azines from azine substituted enamides with simple aromatics via the formation of aryl thianthrenium salts (Scheme 20).86 The proposed mechanism shows that Eosin Y (EY) was converted to EY* through photo-excitation. EY* was quenched by DIPEA to form DIPEA˙+ and the reduced photocatalyst EY˙−. Reduction of the aryl thianthrenium salts (1) by EY˙− resulted in aryl radicals, TT, and EY. The addition of aryl radicals to azine-substituted enamides afforded new carbon-centered alkyl radical intermediates, which underwent SET or HAT to afford the desired product (20). This protocol tolerates a wide range of sensitive functional groups.
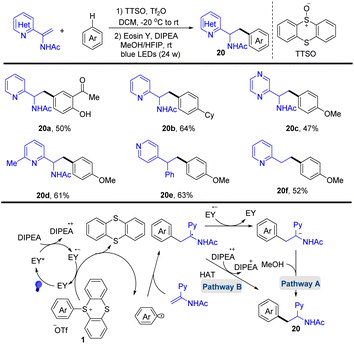 |
| | Scheme 20 Photocatalysed radical hydroarylation of azine-substituted enamides with aryl thianthrenium salts. | |
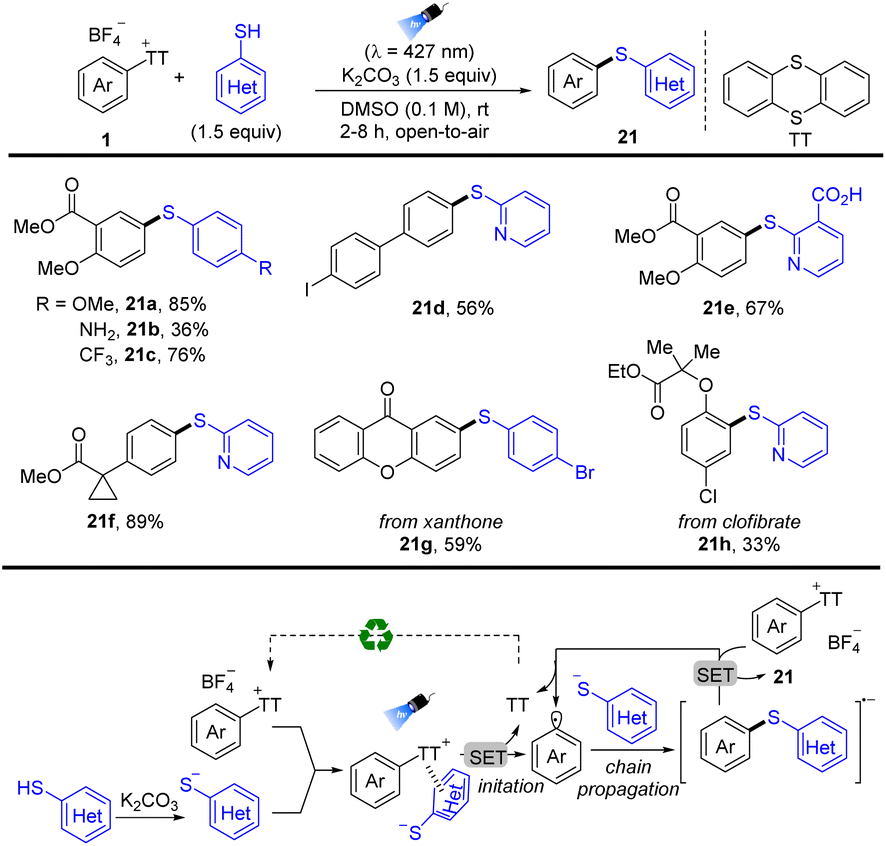
In 2022, Molander reported photoinduced regioselective C–H thioetherification of arenes by thianthrenation (Scheme 21).87 The reaction tolerates a wide range of functional groups, such as free boronic acids, carboxylic acids, and halogens. Moreover, the sulfide motif was incorporated in highly functionalized biomolecules and pharmaceuticals. The proposed mechanism shows that an EDA complex was formed between aryl thianthrenium salts (1) and in situ formed aryl thiolate anions, which induced unimolecular nucleophilic substitution (SRN1) free radical chain reaction. Irradiation and intramolecular electron transfer of the EDA complex generated disulfides, aryl radicals, and TT. Aryl radicals coupled with another aryl thiolate anion to afford diaryl sulfide radical anions, which underwent subsequent single electron reduction of 1 to form the final product (21) and regenerate aryl radicals.
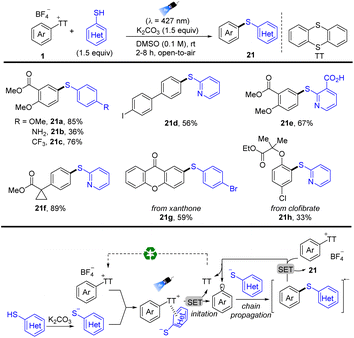 |
| | Scheme 21 Thioetherification of aryl thianthrenium salts via electron donor–acceptor photoactivation. | |
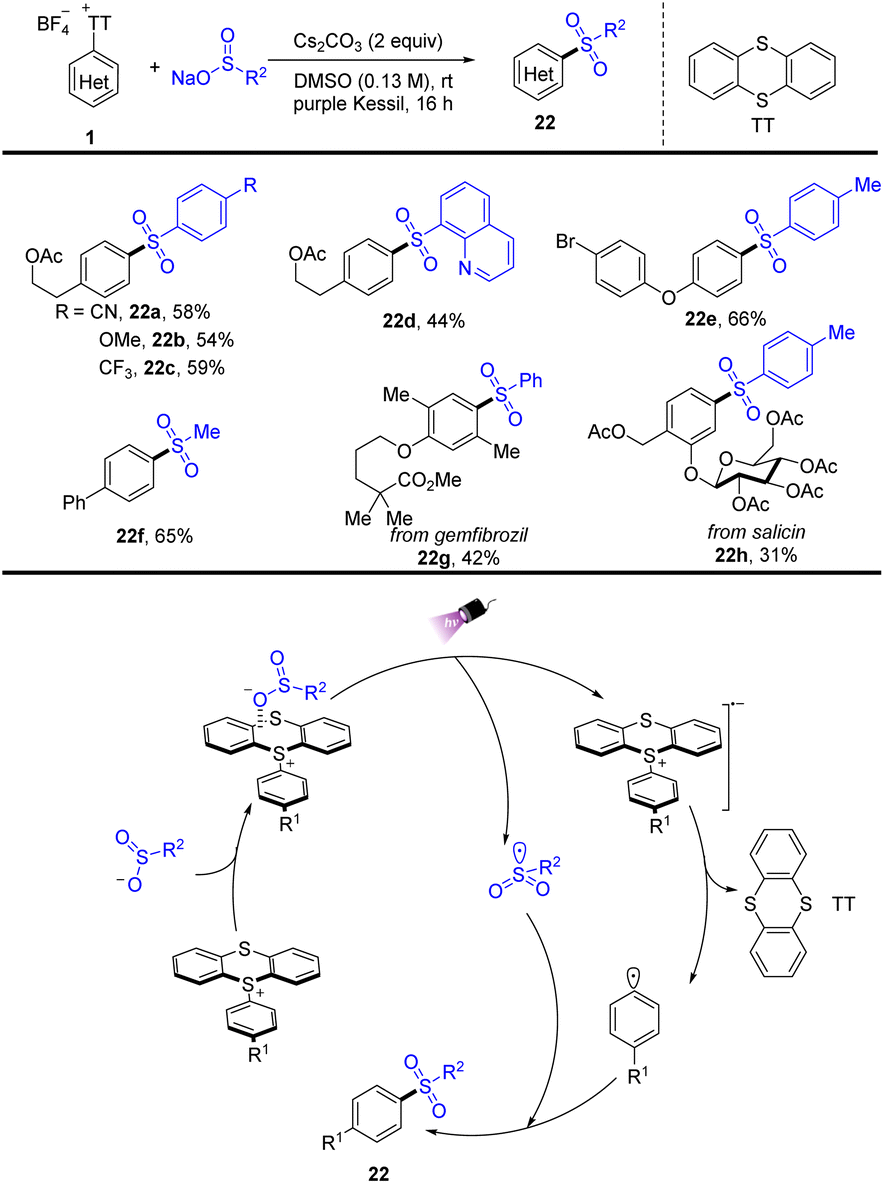
In 2022, Molander reported a visible-light promoted thianthrenium-enabled sulfonylation of arenes to furnish arylsulfone-containing compounds (22) (Scheme 22).88 The EDA complex enabled intra-complex charge transfer to generate transient aryl and sulfonyl radicals that underwent selective coupling to generate the corresponding sulfones under light irradiation conditions. The proposed mechanism shows that a sulfinate anion combined with aryl thianthrenium salts (1) to form an EDA intermediate, generating a radical anion and a persistent sulfonyl radical under irradiation of purple kessil. The radical anion produced an aryl radical and thianthrene by irreversible fragmentation. The aryl radical and sulfonyl radical underwent radical–radical coupling to furnish the target sulfones.
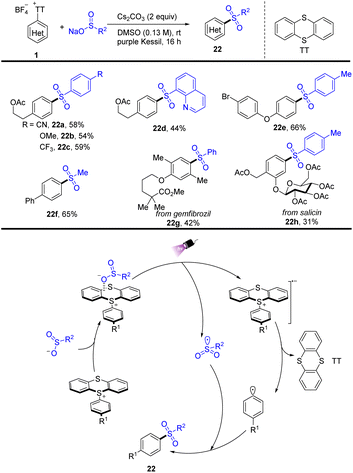 |
| | Scheme 22 Photoinduced sulfonylation of aryl thianthrenium salts. | |
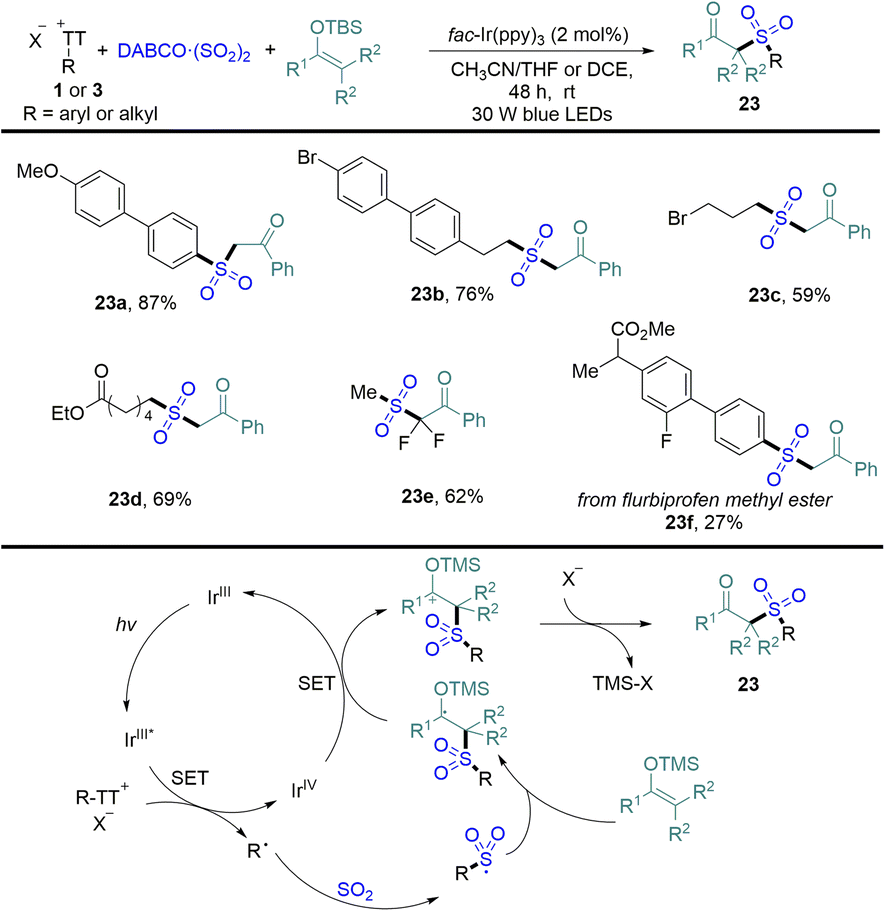
In 2022, Wu reported an efficient photocatalytic sulfonation of silyl enol ethers with DABCO·(SO2)2 and organothianthrenium salts, providing a variety of β-keto sulfones in medium to good yields (Scheme 23).89 Both aryl and alkyl thianthrenium salts were tolerated in the reaction. The proposed mechanism showed that photoexcited IrIII* reduced organothianthrenium salts to form radical species and the oxidized photocatalyst IrIVvia SET. Radical species was trapped by sulfur dioxide and converted into sulfonyl radicals. Sulfonyl radicals were added to silyl enol ethers to form carbon-centered radical intermediates, which underwent single-electron oxidization and desilylation to form β-keto sulfones (23).
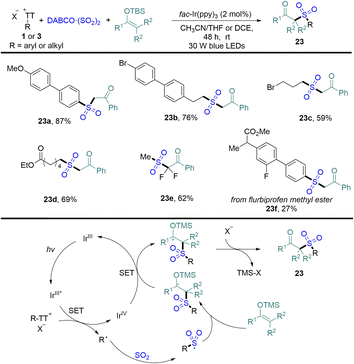 |
| | Scheme 23 Photoredox-catalysed α-sulfonylation of ketones with aryl/alkyl thianthrenium salts. | |
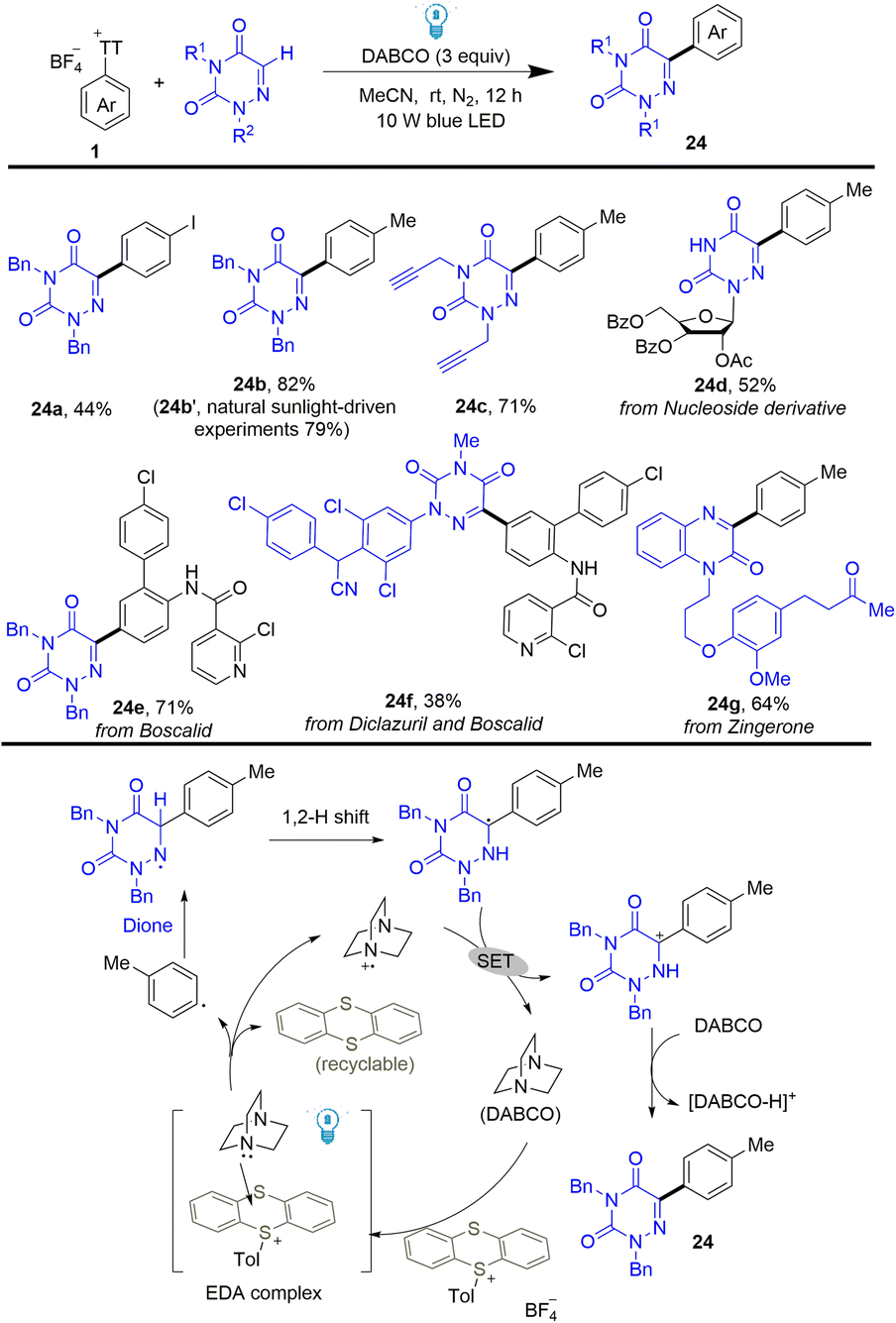
Using the same strategy, Yu developed a photoinduced C–H arylation of N-containing heterocycles using arenes by thianthrenation (Scheme 24).90 The reaction efficiently and selectively functionalized bioactive molecules with structurally complex drugs or agricultural pharmaceuticals. The proposed mechanism shows that DABCO interacted with thianthrenium salts to form an EDA complex, which generated aryl radicals, thianthrene, and DABCO˙+via single electron transfer. Aryl radicals were added to N-heterocycles to give N-centered radicals, which underwent a 1,2 H shift to form C-centered radicals. Carbon cations were formed after single electron oxidation of C-centered radicals by DABCO˙+, which delivered the final coupling product (24) after deprotonation.
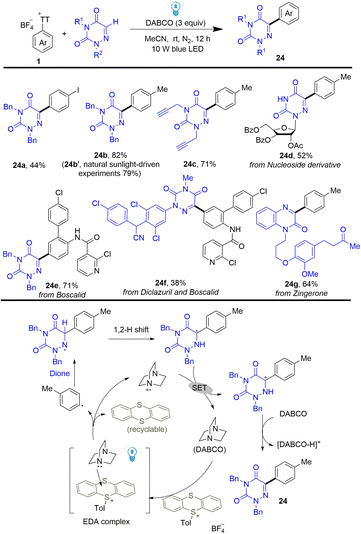 |
| | Scheme 24 Photoinduced C–H arylation of heterocycles with aryl thianthrenium salts. | |
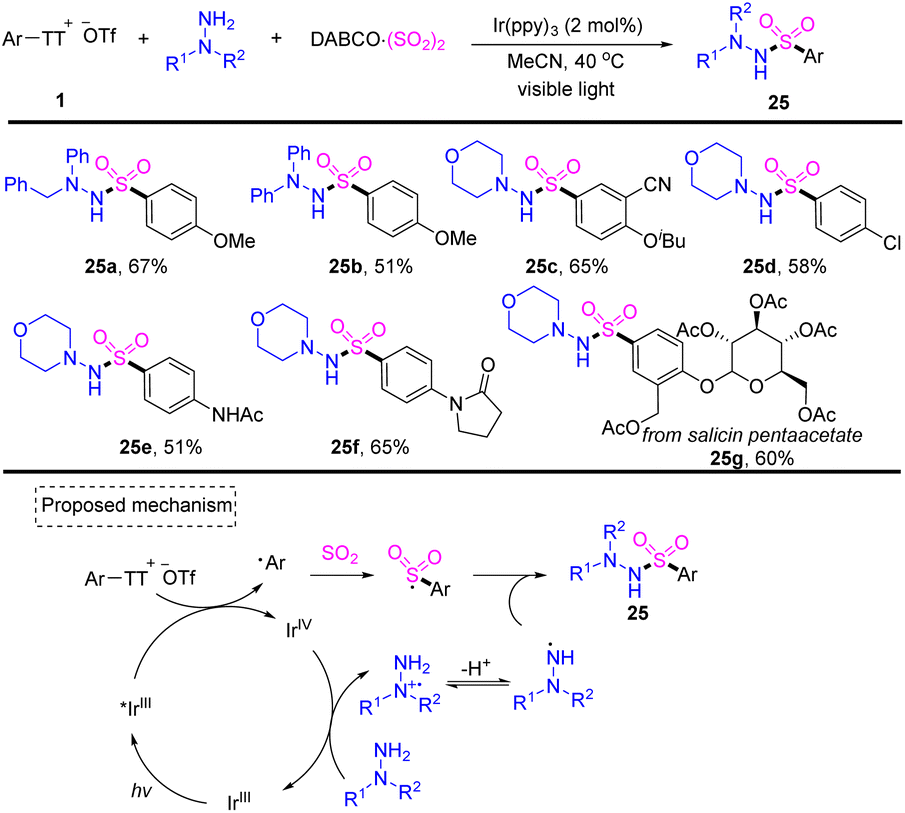
In 2022, Wu reported a photoredox-catalysed three-component reaction of aryl thianthrenium salts with hydrazines and DABCO·(SO2)2, providing facile access to diverse aryl sulfonohydrazides (Scheme 25).91 Both aryl and alkyl hydrazines worked well for this reaction and tolerated a wide range of functional groups. Photoexcited IrIII* would reduce aryl thianthrenium salts 1 to generate aryl radicals and IrIVvia SET. Aryl radicals would be trapped by sulfur dioxide to generate aryl sulfonyl radicals. A SET process would occur between IrIV and hydrazine to deliver cationic radical intermediates, which afforded N-centered radical intermediates after deprotonation. Aryl sulfonyl radicals rebounded with an N-centered radical to afford the desired product (25) by S–N bond formation.
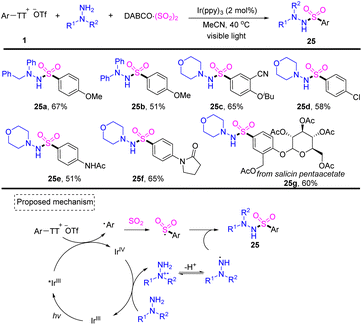 |
| | Scheme 25 Photoredox-catalysed three-component synthesis of aryl sulfonohydrazides using aryl thianthrenium salts. | |
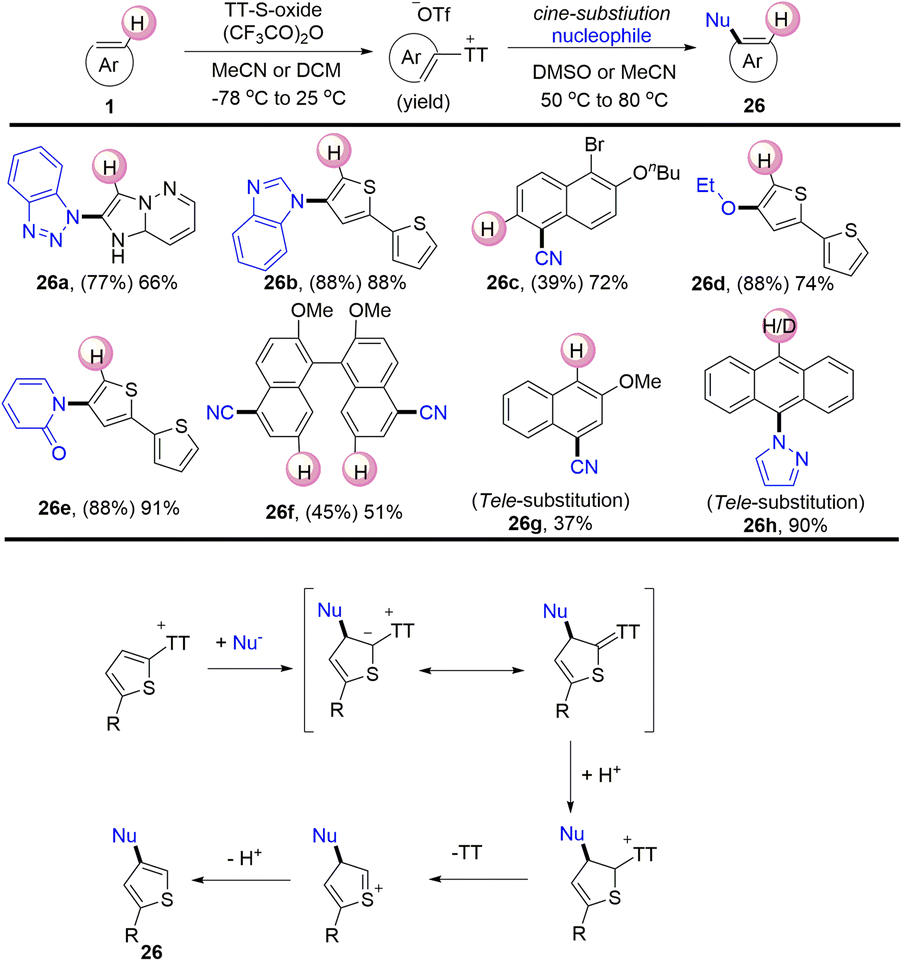
In 2020, Ritter reported a nucleophilic cine-substitution reaction of (hetero)aryl sulfonium salts (Scheme 26).92 The nucleophiles were added to the (hetero)aryl thianthrenium salts to generate the corresponding sulfur ylides. Following this, the protonation of the ylides generated dearomatized sulfonium salts. Subsequently, the compounds (26) were generated by elimination of thianthrene and deprotonation. The reaction utilized (hetero)aryl sulfonium salts as pseudo-Michael acceptors to deliver cine-substitutions difficult to access otherwise.
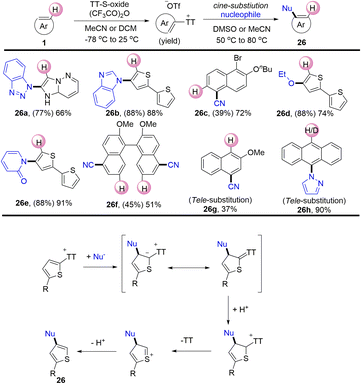 |
| | Scheme 26
cine-Substitution of (hetero)aryl thianthrenium salts. | |
3.2 Application of alkenyl thianthrenium salts
Alkenes are one of the most powerful synthetic blocks for the synthesis of value-added molecules. The use of alkenyl electrophiles represents one of the most accessible and straightforward means to introduce alkenes into target molecules.48 To this end, alkenyl thianthrenium salts have the advantages of easy preparation and bench-stable, and serve as a promising alternative for alkenylation reagents. Moreover, alkenyl thianthrenium salts have found applications in alkylation, allylation and other reactions.
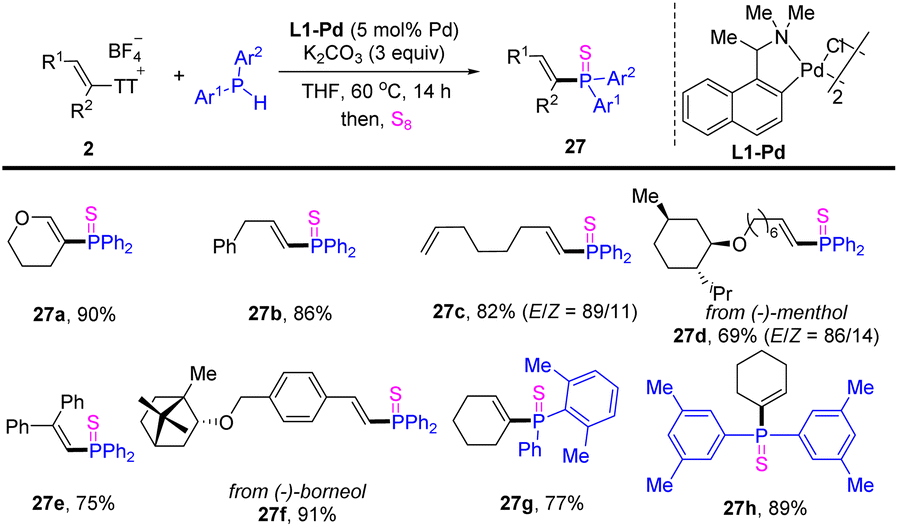
In 2022, Huang reported olefinic C–P cross-coupling of diarylphosphines with alkenyl thianthrenium salts (Scheme 27).93 A variety of alkenes with diverse substitution patterns could be converted to the corresponding alkenyl phosphines. Moreover, unsymmetrical diaryl phosphines could also be coupled smoothly with alkenyl thianthrenium salts.
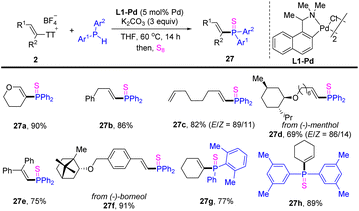 |
| | Scheme 27 Pd-catalysed olefinic C–P cross-coupling of alkenyl thianthrenium salts with diarylphosphine. | |
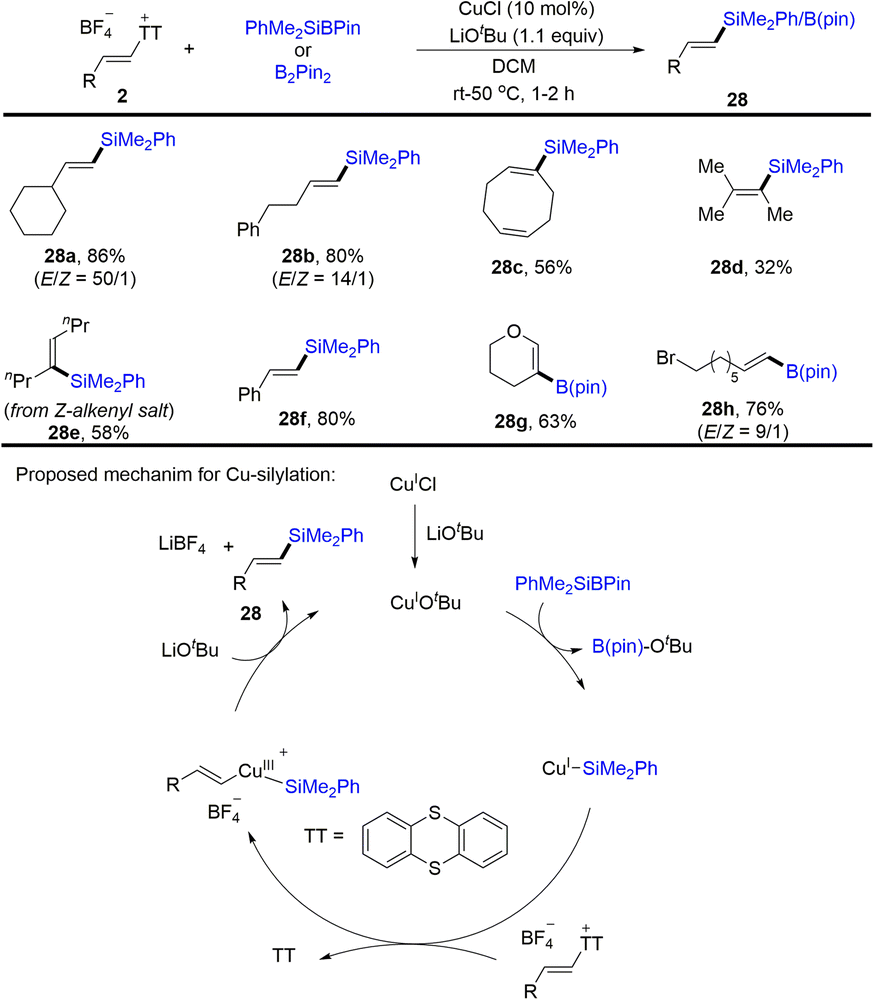
Later, Cu-catalysed silylation and borylation of alkenyl sulfonium salts was developed (Scheme 28).61 A variety of alkenyl silanes and borates with diverse substitution patterns were successfully obtained from alkenyl sulfonium salts. CuICl was converted to active species CuIOtBu in the presence of lithium tert-butoxide, undergoing transmetalation with PhMe2SiB(pin) to form a CuI–SiMe2Ph intermediate. Oxidative addition occurred between CuI–SiMe2Ph and alkenyl sulfonium salts to afford CuIII species, which underwent reductive elimination to afford the desired product (28) with regeneration of CuIOtBu in the presence of a base. In these cases, thianthrene could be recovered in quantitative yield.
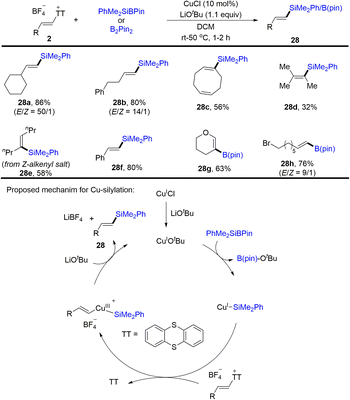 |
| | Scheme 28 Cu-catalysed silylation and borylation of alkenyl thianthrenium salts. | |
In 2021, Ritter reported developed a synthesis of alkenyl thianthrenium salts (Scheme 29).54 The alkenyl thianthrenium salts are used as excellent electrophiles in subsequent conversion; thus, they differ from other alkenyl sulfonium salts that have narrower reactivity. Palladium catalysed cross-coupling reactions of alkenyl thianthrenium salts are usually carried out under the condition of retaining the geometry of the double bond. The alkenyl thianthrenium salts are also suitable for ruthenium-based catalysis reactions, which enables the synthesis of alkenyl halides.
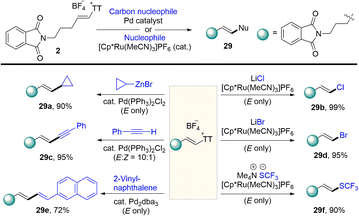 |
| | Scheme 29 Derivatizations of alkenyl thianthrenium salts. | |
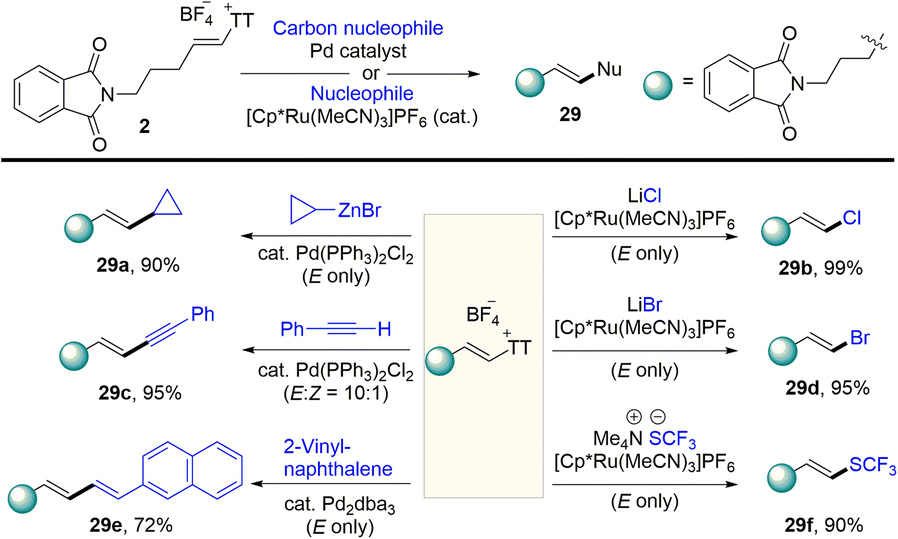
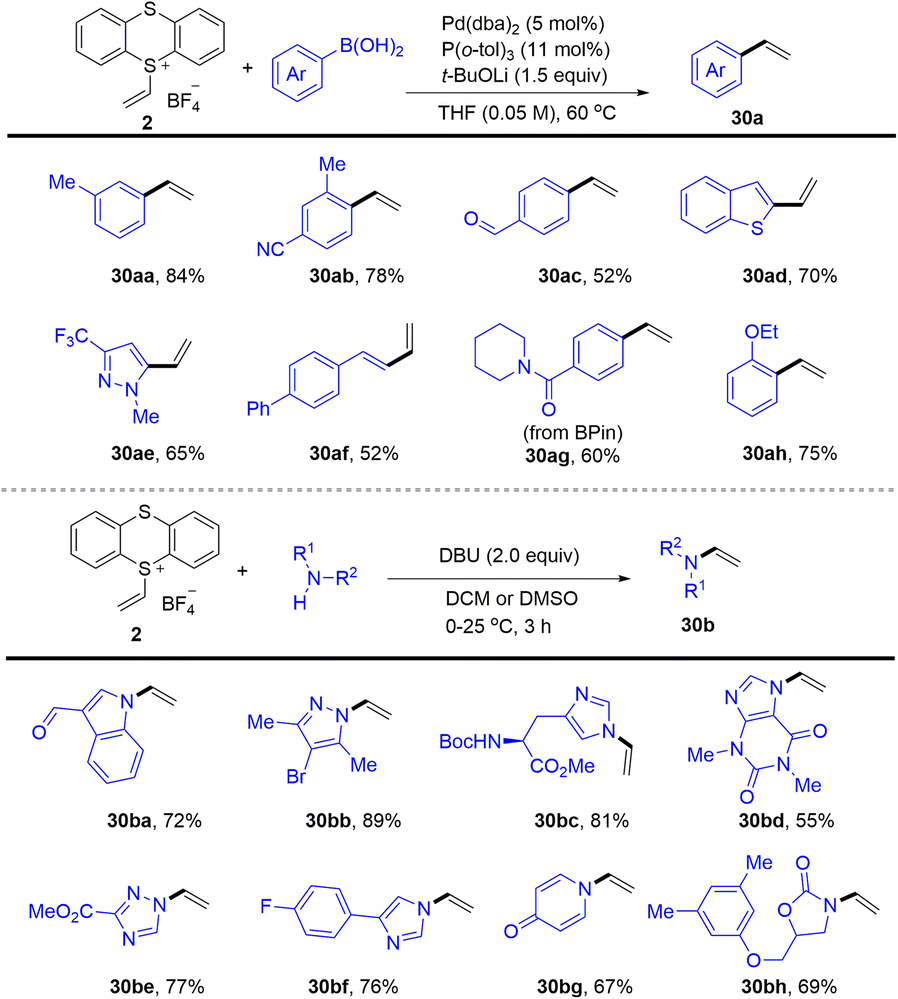
In 2021, Ritter reported the synthesis of vinyl thianthrenium salt (2) from ethylene at atmospheric pressure in one step, which is a bench-stable and crystalline reagent (Scheme 30).94 The vinyl thianthrenium salt was successfully applied to palladium-catalysed Suzuki-type vinylation of aryl boronic acids (30), N-vinylation of heterocyclic compounds, and annulation chemistry of (hetero)cycles. Aziridines are ubiquitous substructures in bioactive molecules and serve as versatile precursors for amine synthesis.
 |
| | Scheme 30 Vinylation reaction using alkenyl thianthrenium salts. | |
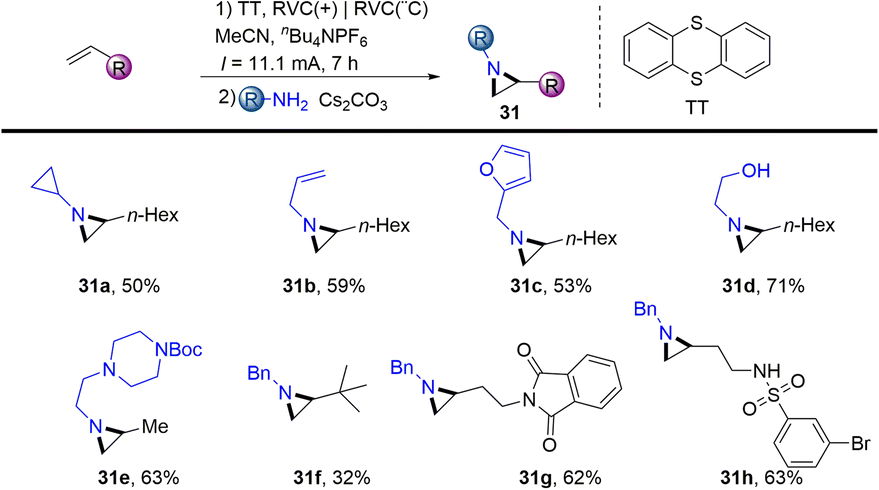
In 2021, Wickens reported an electrochemical synthesis of aziridines from alkenes and primary amines in the presence of thianthrene (Scheme 31).62 The reaction formed metastable dicationic intermediates from unactivated aliphatic alkenes and thianthrene, which underwent two-fold substitution by primary amines to afford the desired aziridines (31). This scalable procedure bears sensitive functional groups such as ethanolamine, esters, sulfonamides, aryl halides and phthalimides. However, this protocol is limited to aliphatic terminal alkenes and aliphatic amines.
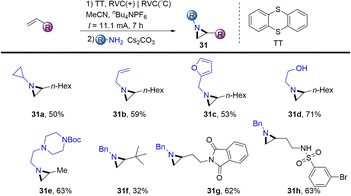 |
| | Scheme 31 Electrochemical synthesis of aziridines from alkenyl thianthrenium salts. | |
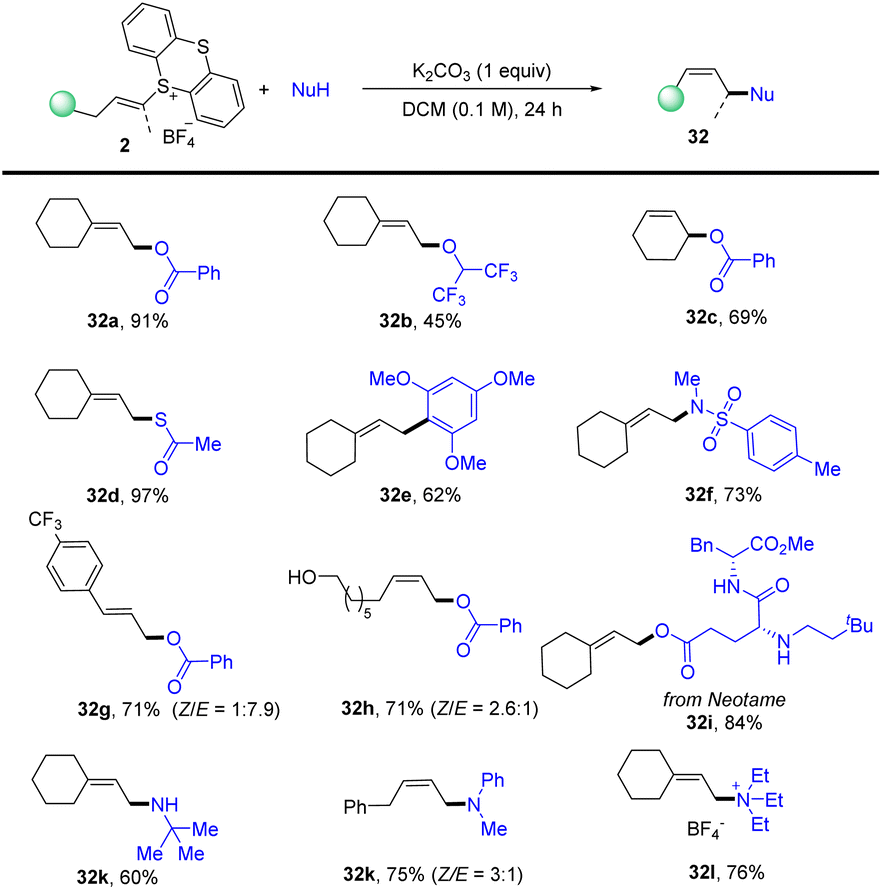
In 2021, Shu’s group isolated the alkenyl thianthrenium salts and subjected them to nucleophiles. Notably, formal allylic C–H amination of alkenes was observed instead of aziridination in the presence of primary amines (Scheme 32).95 A variety of nucleophiles, including primary, secondary, and tertiary amines, sulfonamides, acids, ethers, and electron-rich arenes could be involved to afford the corresponding allylation products (32). This strategy can be applied to the late allylation of natural products, drug molecules and peptides with excellent chemical selectivity.
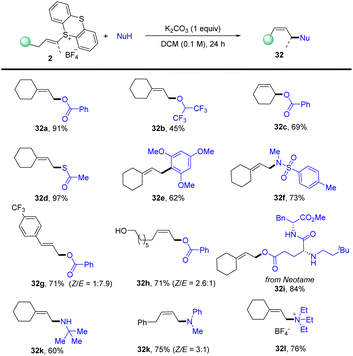 |
| | Scheme 32 Metal-free allylic C–H functionalizations of alkenyl thianthrenium salts. | |
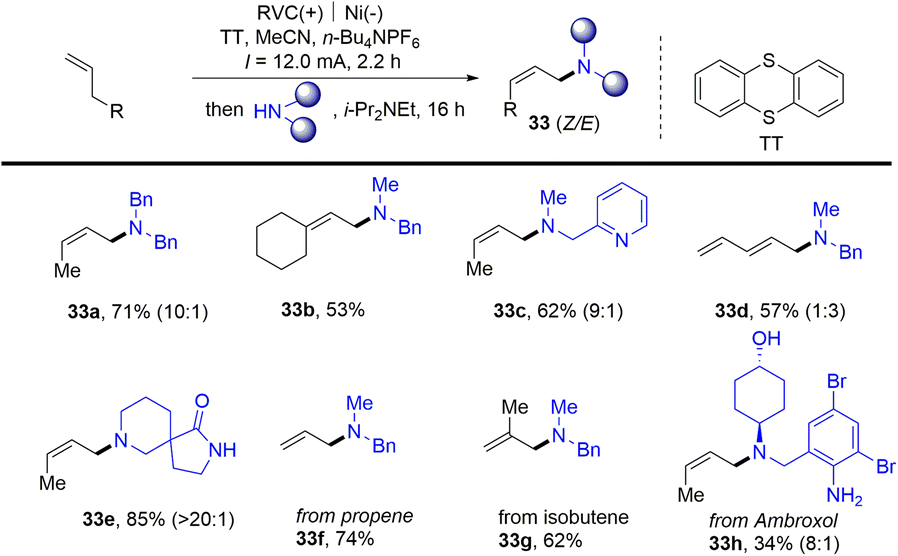
Meanwhile, Wickens reported an electrochemical strategy for the preparation of allyl amines (33) from alkenes and primary amines in the presence of thianthrene (Scheme 33).96 Alkenyl thianthrenium salts (2) serve as a key intermediate for the reaction, affording allyl amines with preferred Z-selectivity. This linear selective process shows good functional group tolerance for gas raw materials and complex molecular derivatization. However, only terminal aliphatic alkenes and secondary aliphatic amines are tolerated in this reaction.
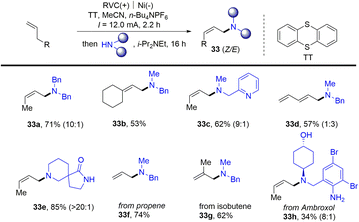 |
| | Scheme 33 Electrochemical synthesis of aliphatic allylamines from alkenes by thianthrenation. | |
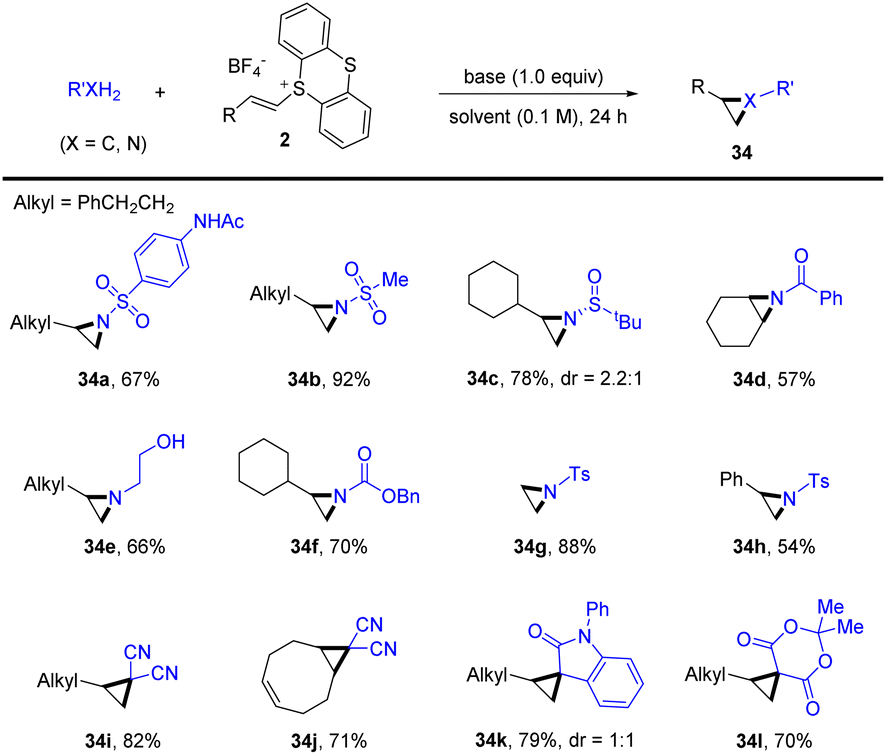
Recently, Shu’s group reported a general protocol for intermolecular aziridination and cyclopropanation of alkenes with XH2 (X = C, N) by thianthrenation under transition-metal-free conditions (Scheme 34).97 The reaction tolerates a wide range of C- and N-precursors, such as free sulfonamides, amides, carbamates, amines, and methylene with acidic protons, for three-membered ring cyclization. This strategy provided an attractive alternative for straightforward synthesis of aziridines and cyclopropanes from easily available starting materials.
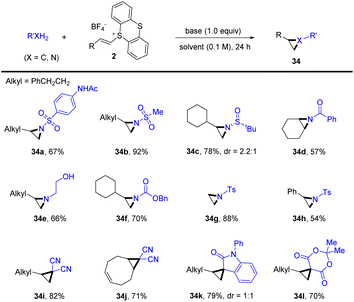 |
| | Scheme 34 Metal-free intermolecular aziridination and cyclopropanation of alkenyl thianthrenium salts. | |
3.3 Application of alkyl thianthrenium salts
Due the highly polarized C(sp3)–S bond in alkyl thianthrenium salts, alkyl thianthrenium salts have the potential to serve as alkyl electrophiles, alkyl radicals as well as sulfur ylides through α-deprotonation.
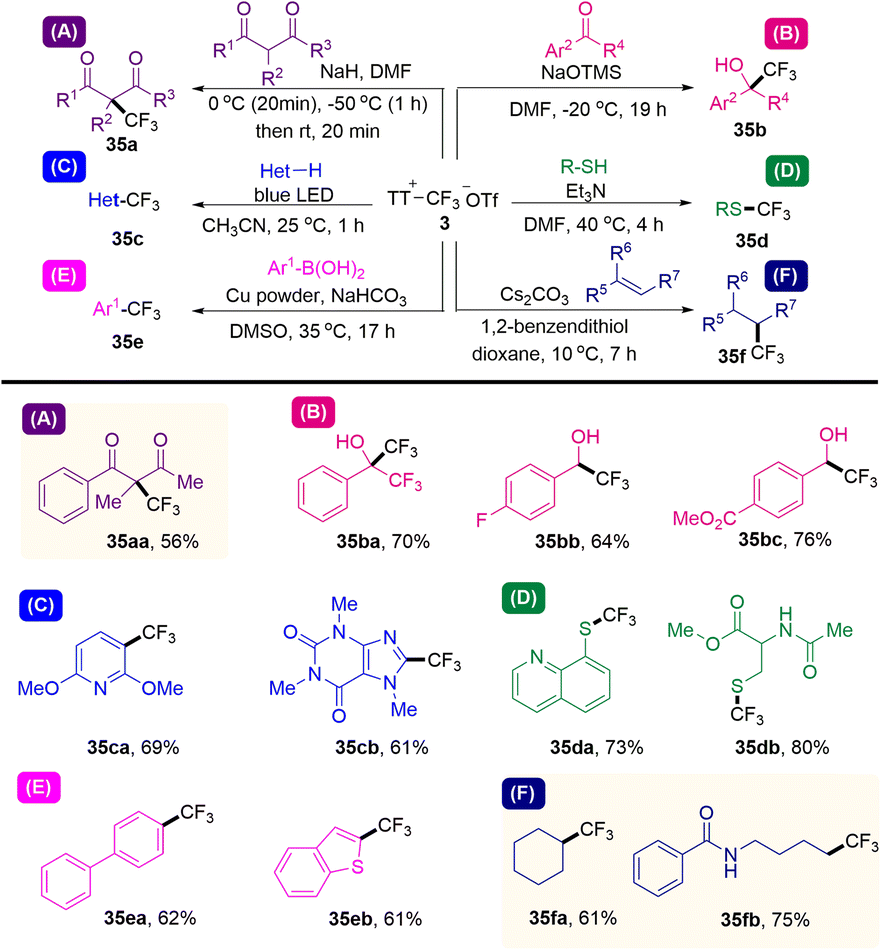
In 2021, Ritter and co-workers reported that a kind of trifluoromethyl thianthrenium triflate (3) was developed (Scheme 35).63 Trifluoromethyl thianthrenium triflate can be easily prepared from commercially available reagents with excellent stability. Thianthrenium (3) could be used as an equivalent reagent for three active trifluoromethyl species (CF3·, CF3−, and CF3+). Trifluoromethyl thianthrenium triflate can carry out types of reactions providing trifluoromethylation products (35), such as Cu-mediated aryl boric acid coupling reaction, light-mediated CF3 substitution reaction of a heterocycle, trifluoromethylation of mercaptan/thiophenol, hydrotrifluoromethylation of olefins, nucleophilic trifluoromethylation of ketones and electrophilic trifluoromethylation of 1,3-diketone. Trifluoromethyl thianthrenium triflate (3) is an all-powerful trifluoromethylating reagent. It shows great potential in the synthesis of trifluoromethyl compounds.
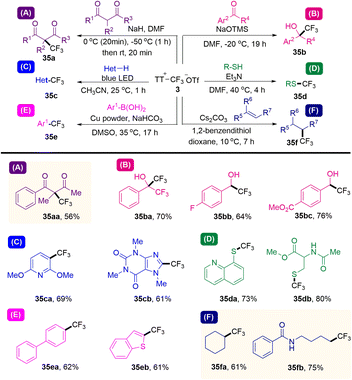 |
| | Scheme 35 Trifluoromethylations using trifluoromethyl thianthrenium salts. | |
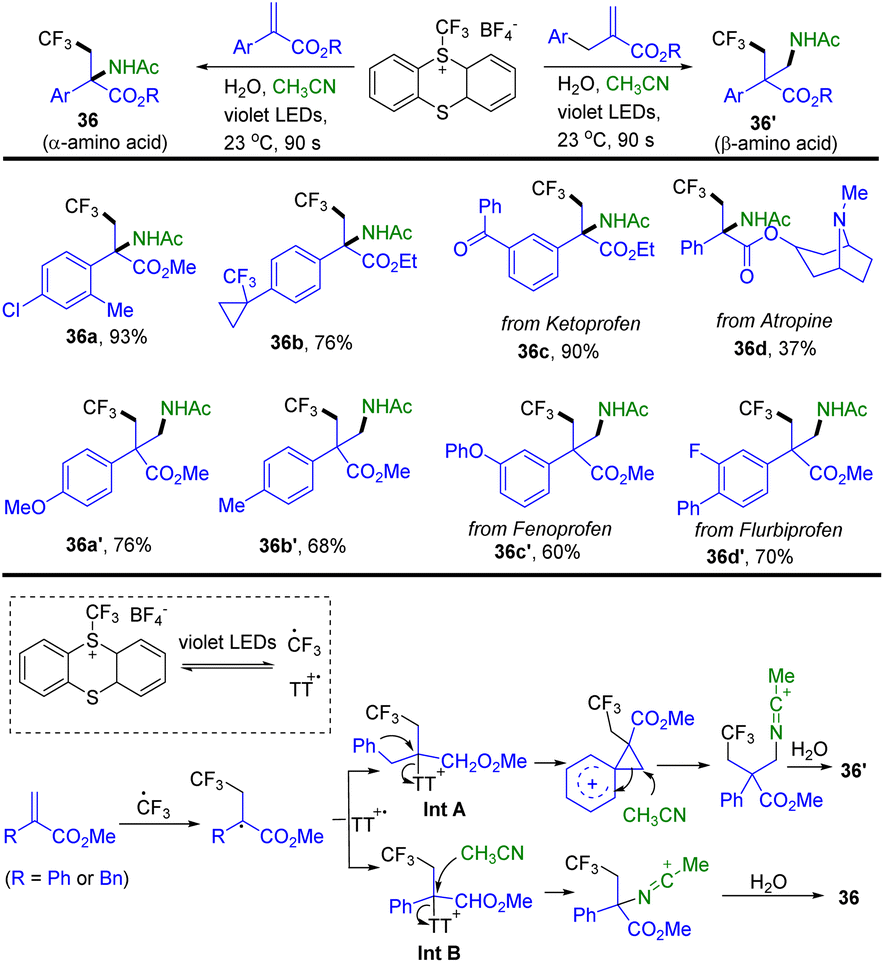
In 2022, Ritter and co-workers reported a synthesis of Cα-tetrasubstituted α- and β-amino acid analogues through the reaction of trifluoromethyl thianthrenium salts with Michael acceptors and acetonitrile (Scheme 36).98a TT–CF3+BF4− generated CF3 radical and thianthrene radical cations (TT˙+) via light induced homolysis. Radical addition of the CF3 radical to acrylate forms a carboxyl α-carbon radical. The carboxyl α-carbon radical recombined with TT˙+ to afford α-thianthrenium carbonyl species Int A or Int B. Intramolecular rearrangement of Int A generates a carbocation intermediate. An acetonitrile attacked carbocation intermediate or Int B results in the formation of a nitrilium ion by in situ hydrolysis. The nitrilium ion combined with hydroxide to afford 36 or 36′. The reaction is tolerant towards ketones, cyclopropyl substituents, ethers, esters, and chlorines. Alkyl substitution instead of aryl or benzyl substitution and methyl acrylate were not tolerated. Recently, Ritter reported a photocatalysed Meerwein-type bromoarylation of electron-deficient alkenes using aryl thianthrenium salts and tetrabutylammonium bromide.98b
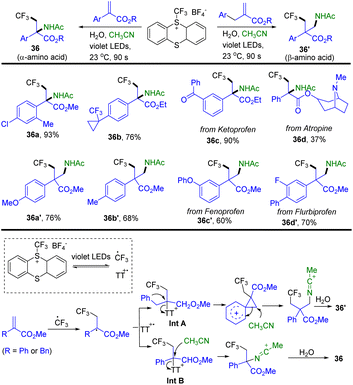 |
| | Scheme 36 Synthesis of trifluoromethyl compounds from trifluoromethyl thianthrenium salts. | |
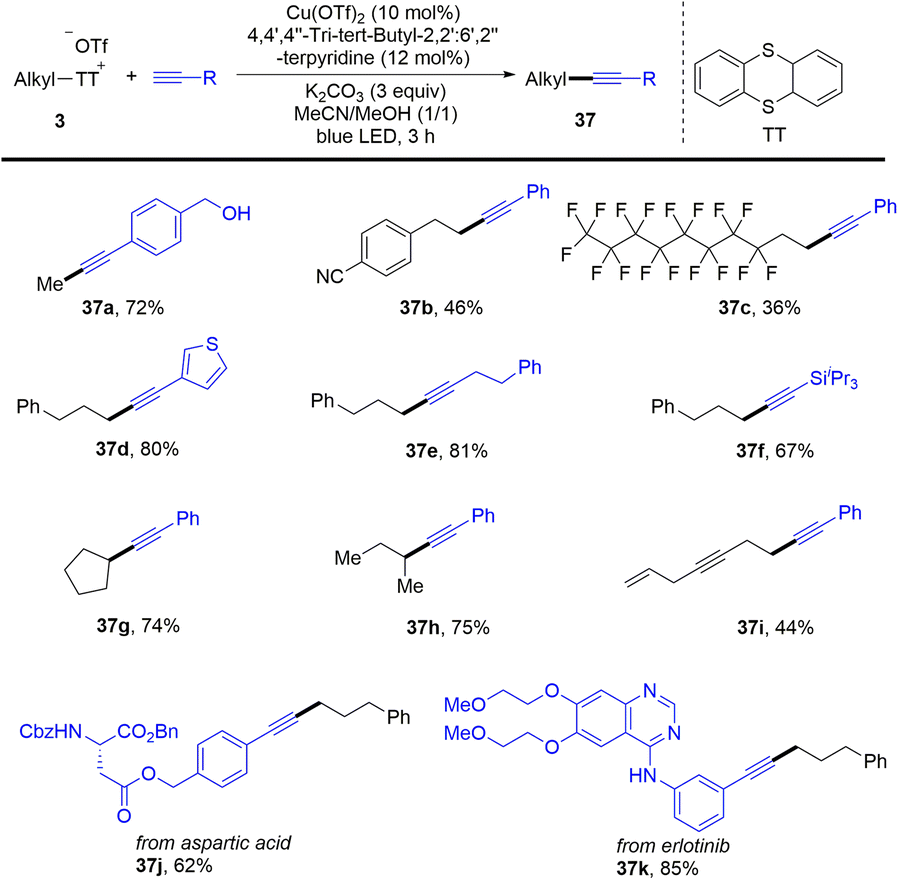
In 2021, Shi and co-workers reported a photoinduced copper-catalysed Sonogashira coupling reaction of alkyl thianthrenium salts (3) with alkynes (Scheme 37).65 Alkyl thianthrenium salts were easily prepared from the corresponding aliphatic alcohols. The protocol successfully converted primary and secondary alcohols into alkylating reagents for alkynes. Both aryl and alkyl alkynes are tolerated to afford the C(sp)–C(sp3) cross-coupling products (37). Traditional synthesis of alkyl borates includes the use of stoichiometric organometallic reagents to react with borates or hydroboration of alkenes.
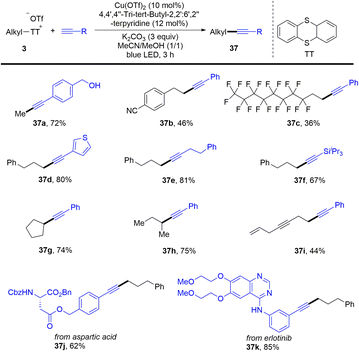 |
| | Scheme 37 Photoinduced Cu-catalysed Sonogashira coupling reaction of alkyl thianthrenium salts. | |
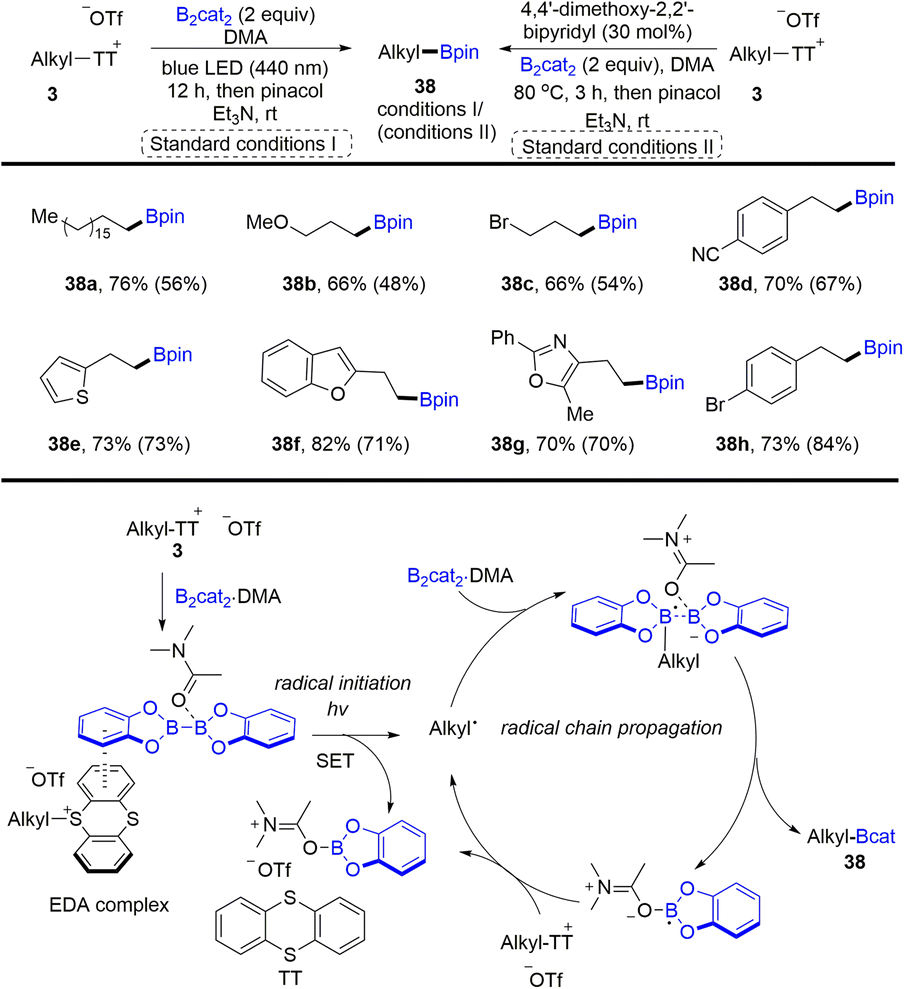
In 2021, Shi and co-workers developed the synthesis of alkyl borates (38) through desulfurative borylation of alkyl thianthrenium salts (3) under either photo- or thermo-induced conditions (Scheme 38).99 The photochemical strategy has better reactivity than the Lewis base strategy. The proposed mechanism shows that an EDA complex was formed between alkyl thianthrenium salts and the DMA·(Bcat)2 adduct. Photoexcitation or heating induced intramolecular single electron transfer to form alkyl radicals, the DMA·Bcat adduct and TT. The alkyl radical reacted with the B2cat2·DMA adduct to produce a boron–centered radical complex. Cleavage of the B–B bond generated alkyl borates (38) and a DMA·Bcat radical, which could undergo radical chain propagation. Primary and secondary alcohols are good substrates to form the corresponding alkyl borates by thianthrenation.
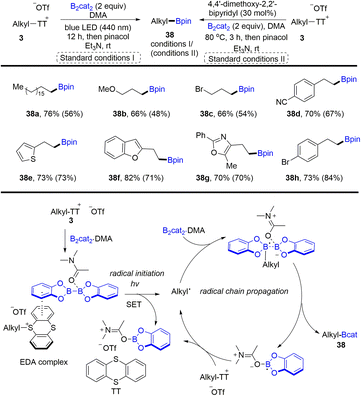 |
| | Scheme 38 Borylation of alkyl thianthrenium salts under photo-and thermo-induced conditions. | |
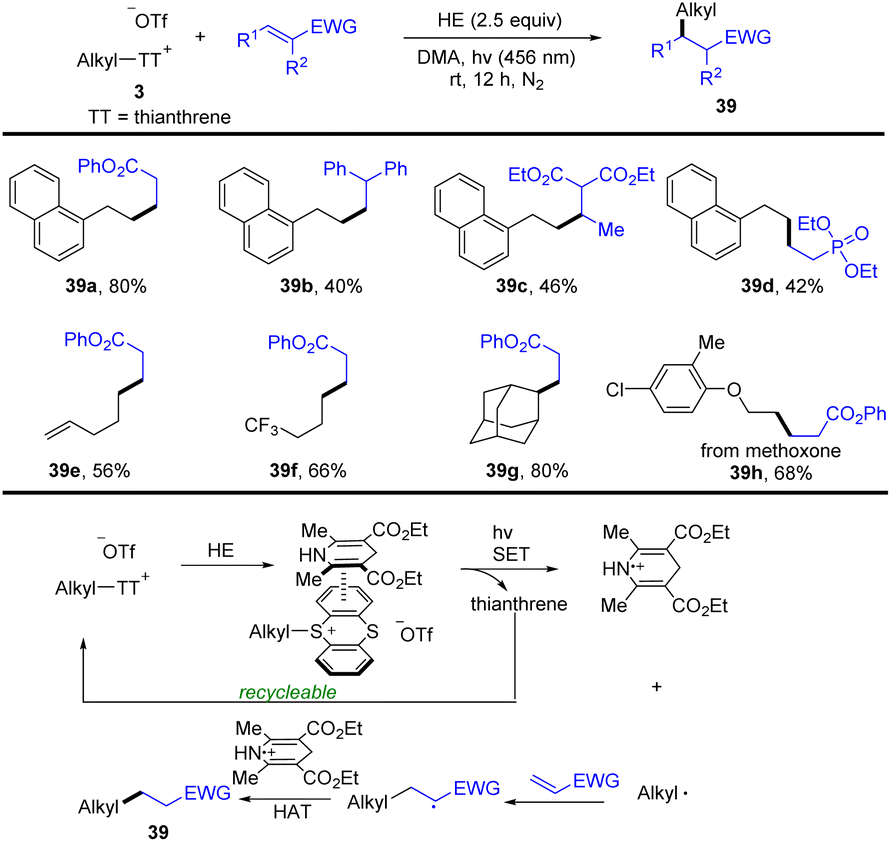
In 2022, Zhang and co-workers developed a photocatalyst- and metal-free approach for the conversion of a C–OH bond to a C–C bond via an alkyl thianthrenium salt/Hantzsch ester electron donor–acceptor complex (Scheme 39).100 Alkyl thianthrenium salt 3 interacted with a Hantzsch ester to form an alkyl thianthrenium salt/Hantzsch ester electron donor–acceptor complex (EDA complex). The EDA complex was converted to an alkyl radical, thianthrene and an oxidized Hantzsch ester via SET by using blue LEDs. The alkyl radical reacted with activated olefin to form a new carbon radical. The new carbon radical abstracts a hydrogen atom from the oxidized Hantzsch ester via the HAT process to obtain product (39). This reaction tolerates a wide range of functional groups and substitution patterns with respect to alkyl thianthrenium salts and electron-deficient alkenes.
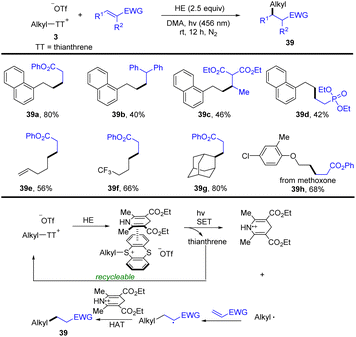 |
| | Scheme 39 Visible-light-promoted reductive hydroalkylation of alkenes using alkyl thianthrenium salts. | |
4. Miscellaneous
In 1974, Shine reported the reaction of thianthrene radical cationic perchlorate with dimethylamine, carbazole and cyanoacetamide to give 5-(dimethylamino)-and 5-carbazol-9-yl thianthrenium perchlorate and 5-[(cyanoacetyl) imino]-5,5-dihydrothianthrene.101 In 2005, Shine reported that the addition of thianthrene- and phenoxathiine cation radical tetrafluoroborates to alkyne gave 1-(5-thianthreniumyl)alkyne tetrafluoroborates.47
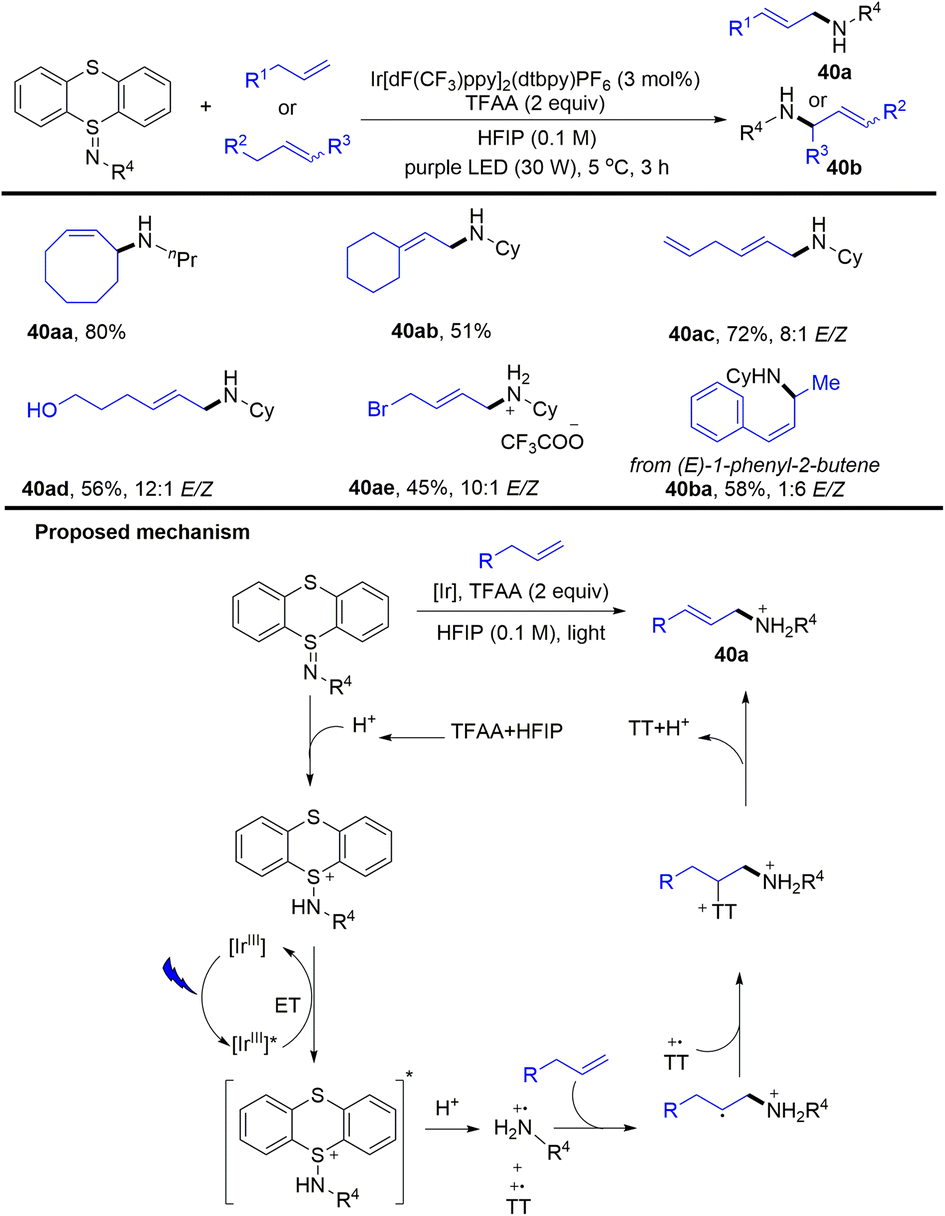
In 2020, Ritter reported a photocatalytic allylic C–H amination reaction of alkenes and iminothianthrene to prepare aliphatic allylamines (Scheme 40).102 The proposed mechanism shows that the excited iridium photocatalyst was quenched by protonated iminothianthrene to generate an excited complex via energy transfer, which underwent further protonation to afford a cationic nitrogen radical intermediate and thianthrenium radical cation. The cationic nitrogen radical was added to alkenes regioselectively to afford an alkyl radical, which recombined with a thianthrenium radical cation to afford the allyl amines (40) by elimination. The reaction tolerates imines, esters, oxysilanes, alcohols, carboxylic acids and ketones. For substrates with multiple alkenes, the reaction occurs selectively at the more electron-rich alkenes.
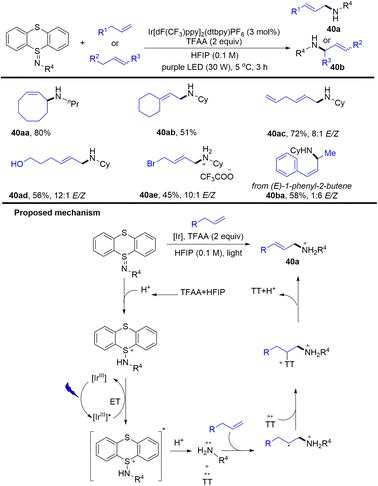 |
| | Scheme 40 Photocatalysed allylic C–H aminations with iminothianthrenes. | |
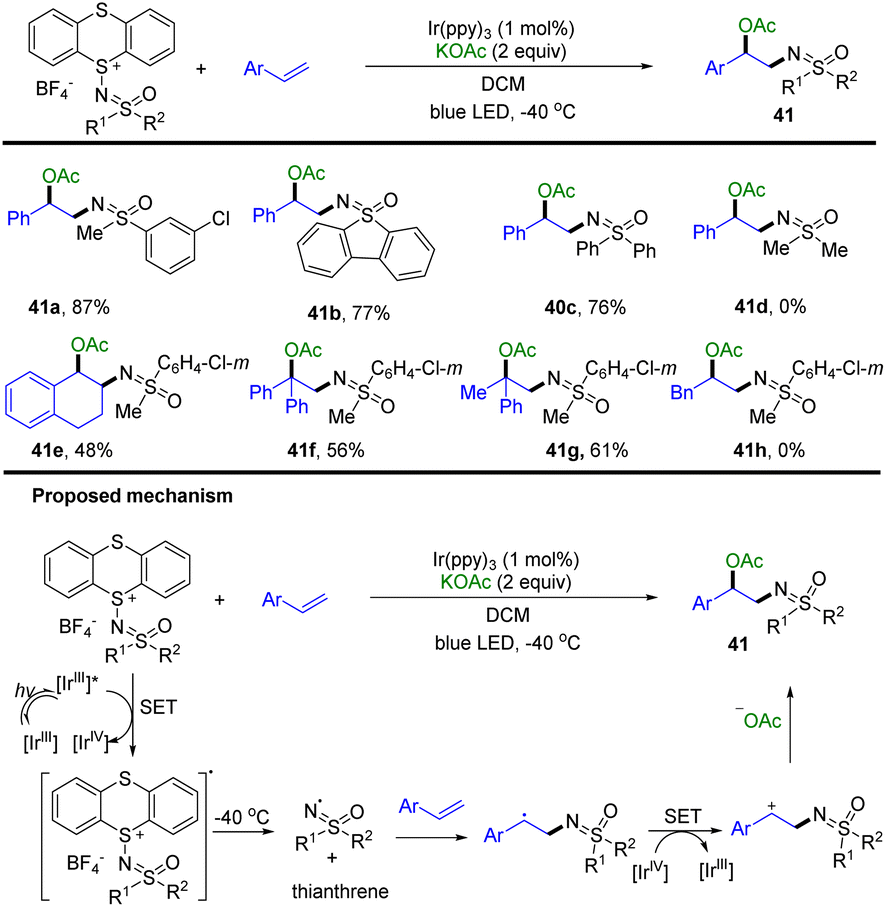
In 2022, Tang and co-workers developed the synthesis of sulfoximidoyl thianthrenium salts as a redox-active sulfoximination reagent (Scheme 41).103 The use of sulfoximidoyl thianthrenium enabled the photocatalysed acetoxysulfoximination of styrenes (41). [IrIII] was excited to [IrIII]* by blue LED irradiation. Sulfoximidoyl thianthrenium tetrafluoroborate was converted to the corresponding anion radical and [IrIV] via SET with [IrIII]*. The sulfoximidoyl radical and TT were generated from the mesolytic cleavage of the sulfide-sulfoximine bond. The sulfoximidoyl radical reacted with styrenes to generate benzylic radicals, which were converted to benzylic cations via single-electron oxidation by [IrIV]. The benzylic cations are trapped with acetate to generate the desired products.
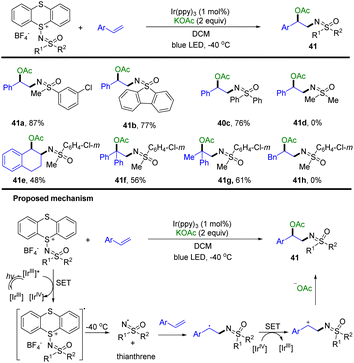 |
| | Scheme 41 Photocatalysed acetoxysulfoximination of styrenes using sulfoximidoyl thianthrenium tetrafluoroborate. | |
5. Conclusion
In summary, a couple of reliable methods have been developed for the preparation of organothianthrenium salts, including aryl, alkenyl, and alkyl thianthrenium salts and other analogues, from easily available starting materials via functionalization of C–H, C–O or other chemical bonds over the past few decades. The easy access to organothianthrenium salts in a selective manner has stimulated great efforts to develop transformations to forge new C–C and C–X bonds under mild conditions using organothianthrenium salts as precursors and synthetic intermediates. In principle, organothianthrenium salts can serve different roles, such as electrophiles and radical precursors, in different bond-forming processes. As a result, selective functionalizations of arenes, alkenes, alkanes, and alcohols have been achieved via thianthrenation through different pathways. Although organothianthrenium salts have been successfully involved in transition-metal-catalysed cross-coupling reactions, radical reactions and substitution reactions, the transformations of organothianthrenium salts remain limited. Future efforts need be devoted to both preparation and synthetic utilization of organothianthrenium salts. First, diverse straightforward and scalable methods to access different types of organothianthrenium salts under mild conditions with great functional group tolerance need to be developed. Moreover, the use of organothianthrenium salts to develop new reactions as well as explore new reactivity and new reaction modes of organothianthrenium salts is crucial in the future. Furthermore, the incorporation of organothianthrenium salts in asymmetric synthesis and catalysis is a potential area for organothianthrenium salts. In addition, the recycling of thianthrene after reaction is another issue for the application of thianthrenium salts due to the heavy mass of thianthrene. Ideally, the use of a catalytic amount of thianthrene forms organothianthrenium salts in situ to mediate the reaction. We anticipate that organothianthrenium salts will evolve into one of the reliable, universal, and practical alternatives for different bond-formation processes.
Author contributions
The manuscript was written through contributions of all authors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from the NSFC (21971101 and 22171127), the Guangdong Basic and Applied Basic Research Foundation (2022A1515011806), the Department of Education of Guangdong Province (2021KTSCX106 and 2022JGXM054), the Pearl River Talent Recruitment Program (2019QN01Y261), the Stable Support Plan Program of Shenzhen Natural Science Fund (No. 20200925152608001), the Thousand Talents Program for Young Scholars, and the Guangdong Provincial Key Laboratory of Catalysis (No. 2020B121201002) is sincerely acknowledged.
Notes and references
- J. Cornella, C. Zarate and R. Martin, Chem. Soc. Rev., 2014, 43, 8081–8097 RSC.
- Q. Wang, Y. Su, L. Li and H. Huang, Chem. Soc. Rev., 2016, 45, 1257–1272 RSC.
- D.-Y. Wang, M. Kawahata, Z.-K. Yang, K. Miyamoto, S. Komagawa, K. Yamaguchi, C. Wang and M. Uchiyama, Nat. Commun., 2016, 7, 12937 CrossRef PubMed.
- S. Shi, S. P. Nolan and M. Szostak, Acc. Chem. Res., 2018, 51, 2589–2599 CrossRef CAS PubMed.
- Z. Qiu and C.-J. Li, Chem. Rev., 2020, 120, 10454–10515 CrossRef CAS PubMed.
- X. Pang, R.-D. He and X.-Z. Shu, Synlett, 2020, 31, 635–640 CrossRef CAS.
- T. R. McDonald, L. R. Mills, M. S. West and S. A. L. Rousseaux, Chem. Rev., 2021, 121, 3–79 CrossRef CAS PubMed.
- X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed.
- J. Wang, S. A. Blaszczyk, X. Li and W. Tang, Chem. Rev., 2021, 121, 110–139 CrossRef CAS PubMed.
- R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086–9139 CrossRef CAS PubMed.
- Y. Wang, P. Hu, J. Yang, Y.-A. Zhu and D. Chen, Chem. Soc. Rev., 2021, 50, 4299–4358 RSC.
- X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed.
- N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236–10254 CrossRef CAS PubMed.
- P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed.
- M. Wang and Z. Shi, Chem. Rev., 2020, 120, 7348–7398 CrossRef CAS PubMed.
- N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326–1330 CrossRef CAS PubMed.
- M. P. Paudyal, A. M. Adebesin, S. R. Burt, D. H. Ess, Z. Ma, L. Kürti and J. R. Falck, Science, 2016, 353, 1144–1147 CrossRef CAS PubMed.
- D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174–238 CrossRef CAS PubMed.
- G. P. McGlacken and L. M. Bateman, Chem. Soc. Rev., 2009, 38, 2447–2464 RSC.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- C. Cheng and J. F. Hartwig, Science, 2014, 343, 853–857 CrossRef CAS PubMed.
- Y. Saito, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2015, 137, 5193–5198 CrossRef CAS PubMed.
- W.-J. Zhou, G.-M. Cao, G. Shen, X.-Y. Zhu, Y.-Y. Gui, J.-H. Ye, L. Sun, L.-L. Liao, J. Li and D.-G. Yu, Angew. Chem., Int. Ed., 2017, 56, 15683–15687 CrossRef CAS PubMed.
- K. Godula and D. Sames, Science, 2006, 312, 67–72 CrossRef CAS PubMed.
- C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed.
- C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780–1824 CrossRef CAS PubMed.
- C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Commun., 2010, 46, 677–685 RSC.
- Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC.
- R.-J. Tang, T. Milcent and B. Crousse, J. Org. Chem., 2018, 83, 930–938 CrossRef CAS PubMed.
- G. B. Boursalian, W. S. Ham, A. R. Mazzotti and T. Ritter, Nat. Chem., 2016, 8, 810–815 CrossRef CAS PubMed.
- L. Zhang and T. Ritter, J. Am. Chem. Soc., 2022, 144, 2399–2414 CrossRef CAS PubMed.
- D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Chem. Rev., 2019, 119, 8701–8780 CrossRef CAS PubMed.
-
(a) Q.-Q. Cheng, L. A. Massey, B. S. Willett, Y. Deng, H. Arman and M. P. Doyle, Angew. Chem., Int. Ed., 2018, 57, 10343–10346 CrossRef CAS PubMed;
(b) Q. Wang, T.-R. Li, M.-M. Li, K. Zhang and W.-J. Xiao, J. Am. Chem. Soc., 2016, 138, 8360–8363 CrossRef CAS PubMed.
-
(a) Q. Chen, S. Wu, S. Yan, C. Li, H. Abduhulam, Y. Shi, Y. Dang and C. Cao, ACS Catal., 2020, 10, 8168–8172 CrossRef CAS;
(b) K.-S. Du and J.-M. Huang, Green Chem., 2018, 20, 1405–1411 RSC.
-
(a) T. Kui, C. Chardin, J. Rouden, S. Livi and J. Baudoux, ChemSusChem, 2022, 15, e202200198 CrossRef CAS PubMed;
(b) Q. Yan, H. Huang and X. Zhang, ACS Catal., 2022, 12, 4825–4832 CrossRef CAS.
-
(a) V. G. Nenaidenko and E. S. Balenkova, Russ. J. Org. Chem., 2003, 39, 291–330 CrossRef CAS;
(b) S. I. Kozhushkov and M. Alcarazo, Eur. J. Inorg. Chem., 2020, 2486–2500 CrossRef CAS PubMed;
(c) Á. Péter, G. J. P. Perry and D. J. Procter, Adv. Synth. Catal., 2020, 362, 2135–2142 CrossRef;
(d) Z.-Y. Tian, Y.-T. Hu, H.-B. Teng and C.-P. Zhang, Tetrahedron Lett., 2018, 59, 299–309 CrossRef CAS.
- Z.-Y. Tian and C.-P. Zhang, Org. Chem. Front., 2022, 9, 2220–2227 RSC.
- F. Berger, M. B. Plutschack, J. Riegger, W. Yu, S. Speicher, M. Ho, N. Frank and T. Ritter, Nature, 2019, 567, 223 CrossRef CAS PubMed.
- H. J. Shine and D. R. Thompson, Tetrahedron Lett., 1966, 7, 1591–1597 CrossRef.
- Y. Murata and H. J. Shine, J. Org. Chem., 1969, 34, 3368–3372 CrossRef CAS.
- H. J. Shine and Y. Murata, J. Am. Chem. Soc., 1969, 91, 1872–1874 CrossRef CAS.
- J. J. Silber and H. J. Shine, J. Org. Chem., 1971, 36, 2923–2926 CrossRef.
- H. J. Shine and L. Piette, J. Am. Chem. Soc., 1962, 84, 4798–4806 CrossRef CAS.
- K. Kim, V. J. Hull and H. J. Shine, J. Org. Chem., 1974, 39, 2534–2537 CrossRef CAS.
- D.-Q. Qian, H. J. Shine, I. Y. Guzman-Jimenez, J. H. Thurston and K. H. Whitmire, J. Org. Chem., 2002, 67, 4030–4039 CrossRef CAS PubMed.
- H. J. Shine, P. Rangappa, J. N. Maxr, D. C. Shelly, T. Ould-Ely and K. H. Whitmire, J. Org. Chem., 2005, 70, 3877–3883 CrossRef CAS PubMed.
- P. Rangappa, H. J. Shine, J. N. Maxr, T. Ould-Ely, A. T. Kelly and K. H. Whitmire, J. Org. Chem., 2005, 70, 9764–9770 CrossRef CAS PubMed.
- B.-J. Zhao, D. H. Evans, N. A. Macías-Ruvalcaba and H. J. Shine, J. Org. Chem., 2006, 71, 3737–3742 CrossRef CAS PubMed.
- B.-J. Zhao, H. J. Shine, J. N. Marx, C. Hofmann and K. H. Whitmire, J. Org. Chem., 2007, 72, 6154–6161 CrossRef CAS PubMed.
- X.-Y. Chen, Y. Wu and P. Wang, Synthesis, 2022, 54, 3928–3940 CrossRef CAS.
- P. S. Engl, A. P. Häring, F. Berger, G. Berger, A. Pérez-Bitrián and T. Ritter, J. Am. Chem. Soc., 2019, 141, 13346–13351 CrossRef CAS PubMed.
- F. Juliá, Q. Shao, M. Duan, M. B. Plutschack, F. Berger, J. Mateos, C. Lu, X.-S. Xue, K. N. Houk and T. Ritter, J. Am. Chem. Soc., 2021, 143, 16041–16054 CrossRef PubMed.
- H. J. Shine, B.-J. Zhao, J. N. Marx, T. Ould-Ely and K. H. Whitmire, J. Org. Chem., 2004, 69, 9255–9261 CrossRef CAS PubMed.
- J. Chen, J. Li, M. B. Plutschack, F. Berger and T. Ritter, Angew. Chem., Int. Ed., 2020, 59, 5616–5620 CrossRef CAS PubMed.
- A. Fava, P. B. Sogo and M. Calvin, J. Am. Chem. Soc., 1957, 79, 1078–1083 CrossRef CAS.
- E. A. C. Lucken, J. Chem. Soc., 1962, 4963–4965 RSC.
- H. J. Shine and C. F. Dais, J. Org. Chem., 1965, 30, 2145–2148 CrossRef CAS.
- D.-Q. Qian, B. Liu, H. J. Shine, I. Y. Guzman-Jimenez and K. H. Whitmire, J. Phys. Org. Chem., 2002, 15, 139–147 CrossRef CAS.
- B. Boduszek and H. J. Shine, J. Org. Chem., 1988, 53, 5142–5143 CrossRef CAS.
- J. Wu, Z. Wang, X.-Y. Chen, Y. Wu, D. Wang, Q. Peng and P. Wang, Sci. China Chem., 2020, 63, 336–340 CrossRef CAS.
- R. Xie, J. Zhu and Y. Huang, Org. Chem. Front., 2021, 8, 5699–5704 RSC.
- D. E. Holst, D. J. Wang, M. J. Kim, I. A. Guzei and Z. K. Wickens, Nature, 2021, 596, 74–79 CrossRef CAS PubMed.
- H. Jia, A. P. Häring, F. Berger, L. Zhang and T. Ritter, J. Am. Chem. Soc., 2021, 143, 7623–7628 CrossRef CAS PubMed.
- B. Liu and H. J. Shine, J. Phys. Org. Chem., 2001, 14, 81–89 CrossRef CAS.
- C. Chen, M. Wang, H. Lu, B. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2021, 60, 21756–21760 CrossRef CAS PubMed.
- F. Berger and T. Ritter, Synlett, 2022, 32, 339–345 Search PubMed.
- X.-Q. Chu, Z.-L. Shen and T.-P. Loh, Chem, 2019, 5, 1025–1027 CAS.
- C. B. Kelly and R. Padilla-Salinas, Chem. Sci., 2020, 11, 10047–10060 RSC.
- J. Tian, W.-C. Gao and X.-F. Jiang, Chem. Reagents, 2021, 43, 447–453 Search PubMed.
- E. M. Alvarez, M. B. Plutschack, F. Berger and T. Ritter, Org. Lett., 2021, 22, 4593–4596 CrossRef PubMed.
- X.-Y. Chen, X.-X. Nie, Y. Wu and P. Wang, Chem. Commun., 2020, 56, 5058–5061 RSC.
- Y. Wu, Y.-H. Huang, X.-Y. Chen and P. Wang, Org. Lett., 2020, 22, 6657–6661 CrossRef CAS PubMed.
- X.-X. Nie, Y.-H. Huang and P. Wang, Org. Lett., 2020, 22, 7716–7720 CrossRef CAS PubMed.
-
(a) B. Lansbergen, P. Granatino and T. Ritter, J. Am. Chem. Soc., 2021, 143, 7909–7914 CrossRef CAS PubMed;
(b) B. Lansbergen, P. Granatino and T. Ritter, J. Am. Chem. Soc., 2021, 143, 10477–10478 CrossRef CAS PubMed.
- D. Zhao, R. Petzold, J. Yan, D. Muri and T. Ritter, Nature, 2021, 600, 444–449 CrossRef CAS PubMed.
- Y. Ye, J. Zhu and Y. Huang, Org. Lett., 2021, 23, 2386–2391 CrossRef CAS PubMed.
- M. Wang, X. Zhang, M. Ma and B. Zhao, Org. Lett., 2022, 24, 6031–6036 CrossRef CAS PubMed.
- B. Zhao, Q. Wang, T. Zhu, B. Feng and M. Ma, Org. Lett., 2022, 24, 5608–5613 CrossRef CAS PubMed.
- F. Ye, F. Berger, H. Jia, J. Ford, A. Wortman, J. Börgel, C. Genicot and T. Ritter, Angew. Chem., Int. Ed., 2019, 58, 14615–14619 CrossRef CAS PubMed.
- R. Sang, S. E. Korkis, W. Su, F. Ye, P. S. Engl, F. Berger and T. Ritter, Angew. Chem., Int. Ed., 2019, 58, 16161–16166 CrossRef CAS PubMed.
- J. Li, J. Chen, R. Sang, W.-S. Ham, M. B. Plutschack, F. Berger, S. Chabbra, A. Schnegg, C. Genicot and T. Ritter, Nat. Chem., 2020, 12, 56–62 CrossRef CAS PubMed.
- X.-Y. Chen, Y.-H. Huang, J. Zhou and P. Wang, Chin. J. Chem., 2020, 38, 1269–1272 CrossRef CAS.
- L. Liang, H.-Y. Niu, R.-L. Li, Y.-F. Wang, J.-K. Yan, C.-G. Li and H.-M. Guo, Org. Lett., 2020, 22, 6842–6846 CrossRef CAS PubMed.
- E. M. Alvarez, T. Karl, F. Berger, L. Torkowski and T. Ritter, Angew. Chem., Int. Ed., 2021, 60, 13609–13613 CrossRef CAS PubMed.
- Y. Zhao, C. Yu, W. Liang and F. W. Patureau, Org. Lett., 2021, 23, 6232–6236 CrossRef CAS PubMed.
- Y.-L. Zhang, G.-H. Wang, Y. Wu, C.-Y. Zhu and P. Wang, Org. Lett., 2021, 23, 8522–8526 CrossRef CAS PubMed.
- M. J. Cabrera-Afonso, A. Granados and G. A. Molander, Angew. Chem., Int. Ed., 2022, 61, e202202706 CrossRef CAS PubMed.
- A. Granados, M. J. Cabrera-Afonso, M. Escolano, S. O. Badir and G. A. Molander, Chem. Catal., 2022, 2, 898–907 CrossRef PubMed.
- F.-S. He, P. Bao, Z. Tang, F. Yu, W.-P. Deng and J. Wu, Org. Lett., 2022, 24, 2955–2960 CrossRef CAS PubMed.
- K. Sun, A. Shi, Y. Liu, X. Chen, P. Xiang, X. Wang, L. Qu and B. Yu, Chem. Sci., 2022, 13, 5659–5666 RSC.
- Q. Li, J. Huang, Z. Cao, J. Zhang and J. Wu, Org. Chem. Front., 2022, 9, 3781–3785 RSC.
- F. Berger, E. M. Alvarez, N. Frank, K. Bohdan, M. Kondratiuk, L. Torkowski, P. S. Engl, J. Barletta and T. Ritter, Org. Lett., 2020, 22, 5671–5674 CrossRef CAS PubMed.
- J. Zhu, Y. Ye and Y. Huang, Organometallics, 2022, 41, 2342–2348 CrossRef CAS.
- F. Juliá, J. Yan, F. Paulus and T. Ritter, J. Am. Chem. Soc., 2021, 143, 12992–12998 CrossRef PubMed.
- M.-S. Liu, H.-W. Du and W. Shu, Chem. Sci., 2022, 13, 1003–1008 RSC.
- D. J. Wang, K. Targos and Z. K. Wickens, J. Am. Chem. Soc., 2021, 143, 21503–21510 CrossRef CAS PubMed.
- M. S. Liu, H.-W. Du and W. Shu, Angew. Chem., Int. Ed., 2022, 61, e202209929 CAS.
-
(a) H. Jia and T. Ritter, Angew. Chem., Int. Ed., 2022, 61, e202208978 CAS;
(b) Y. Cai and T. Ritter, Angew. Chem., Int. Ed., 2022, 61, e202209882 Search PubMed.
- C. Chen, Z.-J. Wang, H. Lu, Y. Zhao and Z. Shi, Nat. Commun., 2021, 12, 4526 CrossRef CAS PubMed.
- X. Li, W. Si, Z. Liu, H. Qian, T. Wang, S. Leng, J. Sun, Y. Jiao and X. Zhang, Org. Lett., 2022, 24, 4070–4074 CrossRef CAS PubMed.
- K. Kim and H. J. Shine, J. Org. Chem., 1974, 39, 2537–2539 CrossRef CAS.
- Q. Cheng, J. Chen, S. Lin and T. Ritter, J. Am. Chem. Soc., 2020, 142, 17287–17293 CrossRef CAS PubMed.
- Y. M. Yang, C. Zhang, H. Yang and Z.-Y. Tang, Chem. Commun., 2022, 58, 8580–8583 RSC.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*