
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalyzed enantioselective α-heteroarylation of ketones via C–F bond activation to construct all-carbon quaternary stereocenters†
Xiaodong
Gu
a,
Kexin
Liu
a,
Limin
Yang
 c,
Chengyi
Xie
a,
Mingliang
Li
*b and
Jun (Joelle)
Wang
*ab
c,
Chengyi
Xie
a,
Mingliang
Li
*b and
Jun (Joelle)
Wang
*ab
aDepartment of Chemistry, Hong Kong Baptist University, Kowloon, Hong Kong, China. E-mail: junwang@hkbu.edu.hk
bDepartment of Chemistry, Southern University of Science and Technology (SUSTech), Shenzhen, 518055, China. E-mail: liml@mail.sustech.edu.cn
cCollege of Materials, Chemistry and Chemical Engineering, Hangzhou Normal University, Hangzhou 311121, China
First published on 12th October 2022
Abstract
Nickel-catalyzed asymmetric α-heteroarylation of ketones with fluorinated heteroarenes is reported via C–F bond activation. A series of ketones and 2-fluoropyridine derivatives with different functional groups proceed well to provide the corresponding products containing all-carbon quaternary stereocenters in good yields (up to 99% yield) and high ee values (up to 99% ee). In addition, drug molecule donepezil could also be compatible under the reaction conditions to afford late-stage diversification of pharmaceuticals.
Over the past decade, transition metal-catalyzed organic fluorine chemistry including C–F bond formation and cleavage has become a popular topic to be explored by organic chemists.1,2 Compared with other carbon–halogen (C–I, C–Br, and C–Cl) bonds, catalytic C–F bond cleavage is a highly regioselective strategy to achieve late-stage functionalization of pharmaceuticals and construct diversified complex molecules. Although the C–F bond has high bond dissociation energy, transition metal-catalyzed C–F bond activation has still made some progress through the unremitting effort of organic chemists.2,3 The transition metal-catalyzed C–F bond cleavage proceeds primarily through direct oxidative addition of low-valence metal complex to the C–F bond,2c–e,4 fluorine elimination of organic metal intermediates formed from fluorinated alkenes,2f,5 or photocatalytic SET-type radical defluorinative functionalization.2a,b,6
CAr-F bonds are arguably the strongest bonds that carbon can form. BDE (bond dissociation energy) of the CAr-F bond was calculated in the ESI† and is shown in Scheme 1a. Due to the high bond dissociation energy of the CAr-F bond (526 kJ mol−1 for fluorobenzene), early examples of defluorinative functionalization of fluoroarenes mainly focused on extremely electron deficient fluorides.7 Recently, transition metal-catalyzed C–F bond cleavage of (hetero)aryl fluoride has been widely developed to construct C–C and C–X (X = N, B, Si, etc) bonds with organometallic reagents, alkynes, amines, borylation reagents or silylboranes as the coupling partner (Scheme 1b).8–12 So far, there are rare examples of transition metal-catalyzed C(sp3)–H/CAr-F cross-coupling reactions,13 let alone asymmetric transformations with aryl or heteroaryl fluoride derivatives as coupling partners via C–F bond activation. Prompted by the nickel-catalyzed arylation of indanones with aryl triflates, chlorides, pivalates or pyrimidyl ether as arylation reagents,14 we develop herein the Ni-catalyzed asymmetric α-heteroarylation of ketones via C–F bond activation to deliver all-carbon chiral quaternary carbon centers (Scheme 1c).
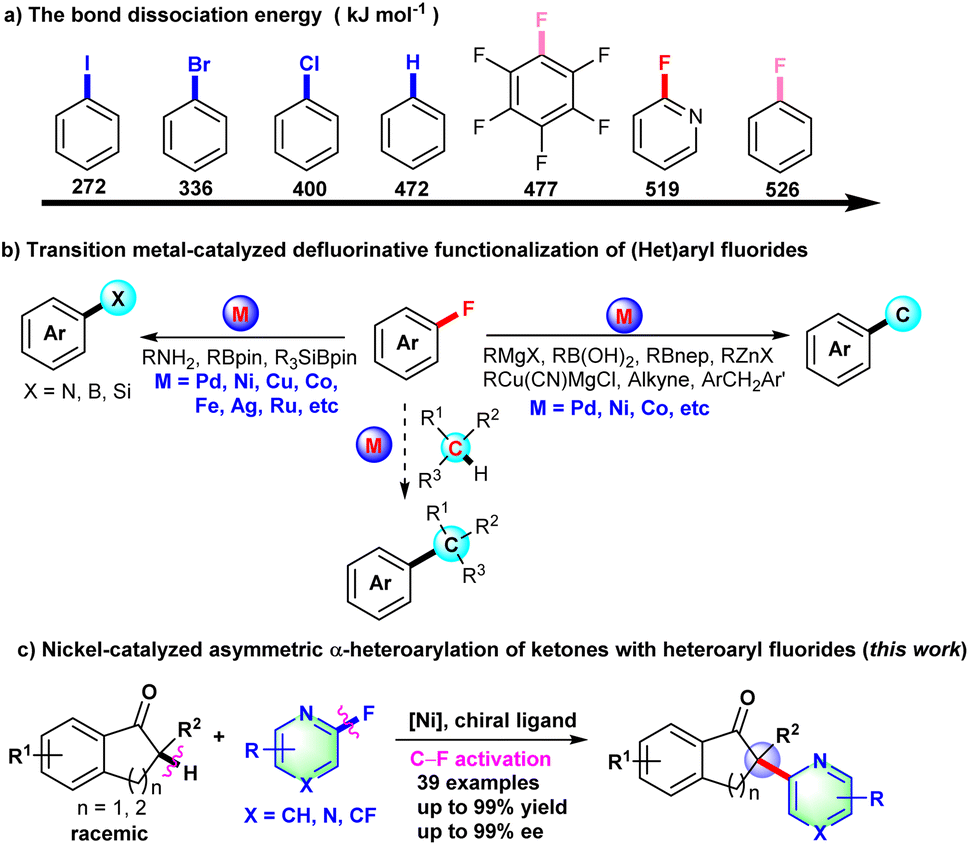 | ||
| Scheme 1 Transition metal-catalyzed C–F bond activation. | ||
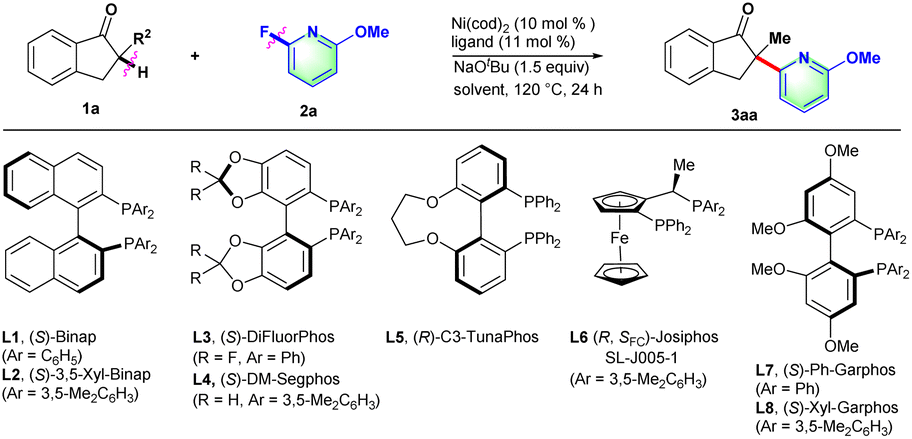
Initially, 2-methyl-2,3-dihydro-1H-inden-1-one (1a) was chosen to explore the nickel-catalyzed enantioselective α-heteroarylation with 2-fluoro-6-methoxypyridine (2a) including C–F and C–OMe bonds. To our delight, the single defluorinative product 3aa could be obtained in 39% yield and 68% ee in the presence of Ni(cod)2, (S)-Binap (L1) and NaOtBu (Table 1, entry 1). After the screening of a series of chiral phosphine ligands (Table 1, entries 2–8), (S)-Ph-GarPhos (L7) was found to afford the desired product in 42% yield and 92% ee. The reaction ee value could be improved to 97% with anisole as solvent, but the yield was just 44% (Table 1, entry 11). By adjusting the concentration ratio of substrates 1a and 2a, the reaction yield was improved to 77% yield (Table 1, entry 12). Finally, the defluorinative arylation product 3aa could be obtained in 77% yield and 97% ee in the presence of Ni(cod)2 (10 mol%), L7 (11 mol%) and NaOtBu (1.1 equiv.) in dry anisole.
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Solvent | Yieldb (%) | eec (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.15 mmol), Ni(cod)2 (10 mol%), ligand (11 mol%), NaOtBu (1.5 equiv.), dry solvent (1.0 mL), 120 °C, 24 h. b Yield of 3 was determined by 1H NMR using dibromomethane as the internal standard. c Determined by chiral HPLC. d 1a (0.2 mmol), 2a (0.1 mmol), and NaOtBu (1.1 equiv.). e Isolated yield. | ||||
| 1 | L1 | Toluene | 39 | 68 |
| 2 | L2 | Toluene | 21 | 65 |
| 3 | L3 | Toluene | NR | — |
| 4 | L4 | Toluene | 12 | 75 |
| 5 | L5 | Toluene | 17 | −74 |
| 6 | L6 | Toluene | 67 | −84 |
| 7 | L7 | Toluene | 42 | 92 |
| 8 | L8 | Toluene | 11 | 62 |
| 9 | L7 | 1,4-Dioxane | 84 | 90 |
| 10 | L7 | m-xylene | 27 | 93 |
| 11 | L7 | Anisole | 44 | 97 |
| 12d | L7 | Anisole | 78(77)e | 97 |
| 13d | L7 | 1,4-Dioxane | 85 | 91 |
Under the optimized reaction conditions, the scope of fluorinated heteroarene derivatives 2 with 2-methyl-2,3-dihydro-1H-inden-1-one 1a was examined (Scheme 2). A series of 2-fluoropyridines with an electron-donating group (e.g., OMe, OBn, OCH2R, OAr, PPh2 and Me) on the 6-position of the pyridine ring proceeded well to provide arylated products in moderate to good yields and excellent ee values (3aa–3ai). The arylated product 3ac involving a chiral morpholine moiety could be obtained in 48% yield, >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and 92% ee, which might be used as a potential dopamine receptor 4 (D4R) antagonist.15 Interestingly, the 2-fluoro-6-aryloxypyridines just underwent defluorinative arylation to deliver the corresponding products in good enantioselectivities (3ad–3ag), in which CAr-O bond cleavage did not occur as reported by our group recently.14d The diphenyl phosphine group was also compatible with reaction conditions to give the desired product in 47% yield and a 90% ee value with the addition of MgBr2 which probably assisted the coordination with pyridine or an indanone motif (3ah).16 2-Fluoro-6-aryl-pyridine derivatives involving a series of functional groups such as Me, OMe, OCF3, CF3 and CN all worked well to afford the corresponding products in good yields and excellent ee values (3aj–3ap). The absolute configuration of the major heteroarylation products was confirmed to be S-configuration by X-ray analysis of 3ap (CCDC 2156742).17 The 2-thienyl substituted 2-fluoropyridine derivative was also an effective coupling partner to deliver desired product 3aq in a good ee value. 2-Fluoropyridine derivatives without substituents on the 6-position could also couple well with 1a to provide the corresponding products (3ar). To our delight, the C–F bond activation is highly regioselective such that the cleavage only occurred at the 2-position of 2,4-difluorinated or 2,5-difluorinated pyridine, which is probably caused by the coordination-assistance of pyridine (3as–3at). 2-Fluoropyridine derivatives with functional groups such as CO2Me, OMe, Me or Ph on the 5-position of the pyridine ring were compatible with the reaction conditions to deliver the corresponding products in medium yields and good ee values, but 6-fluoronicotinonitrile afforded the desired product in poor yield and enantioselectivity probably caused by the coordination of the cyano group with nickel (3au–3ay). Besides, 2-methyl-2,3-dihydro-1H-inden-1-one 1a could also effectively react with 2-fluoropyrazine to provide the desired product in good enantioselectivity (3az).
:1 dr and 92% ee, which might be used as a potential dopamine receptor 4 (D4R) antagonist.15 Interestingly, the 2-fluoro-6-aryloxypyridines just underwent defluorinative arylation to deliver the corresponding products in good enantioselectivities (3ad–3ag), in which CAr-O bond cleavage did not occur as reported by our group recently.14d The diphenyl phosphine group was also compatible with reaction conditions to give the desired product in 47% yield and a 90% ee value with the addition of MgBr2 which probably assisted the coordination with pyridine or an indanone motif (3ah).16 2-Fluoro-6-aryl-pyridine derivatives involving a series of functional groups such as Me, OMe, OCF3, CF3 and CN all worked well to afford the corresponding products in good yields and excellent ee values (3aj–3ap). The absolute configuration of the major heteroarylation products was confirmed to be S-configuration by X-ray analysis of 3ap (CCDC 2156742).17 The 2-thienyl substituted 2-fluoropyridine derivative was also an effective coupling partner to deliver desired product 3aq in a good ee value. 2-Fluoropyridine derivatives without substituents on the 6-position could also couple well with 1a to provide the corresponding products (3ar). To our delight, the C–F bond activation is highly regioselective such that the cleavage only occurred at the 2-position of 2,4-difluorinated or 2,5-difluorinated pyridine, which is probably caused by the coordination-assistance of pyridine (3as–3at). 2-Fluoropyridine derivatives with functional groups such as CO2Me, OMe, Me or Ph on the 5-position of the pyridine ring were compatible with the reaction conditions to deliver the corresponding products in medium yields and good ee values, but 6-fluoronicotinonitrile afforded the desired product in poor yield and enantioselectivity probably caused by the coordination of the cyano group with nickel (3au–3ay). Besides, 2-methyl-2,3-dihydro-1H-inden-1-one 1a could also effectively react with 2-fluoropyrazine to provide the desired product in good enantioselectivity (3az).
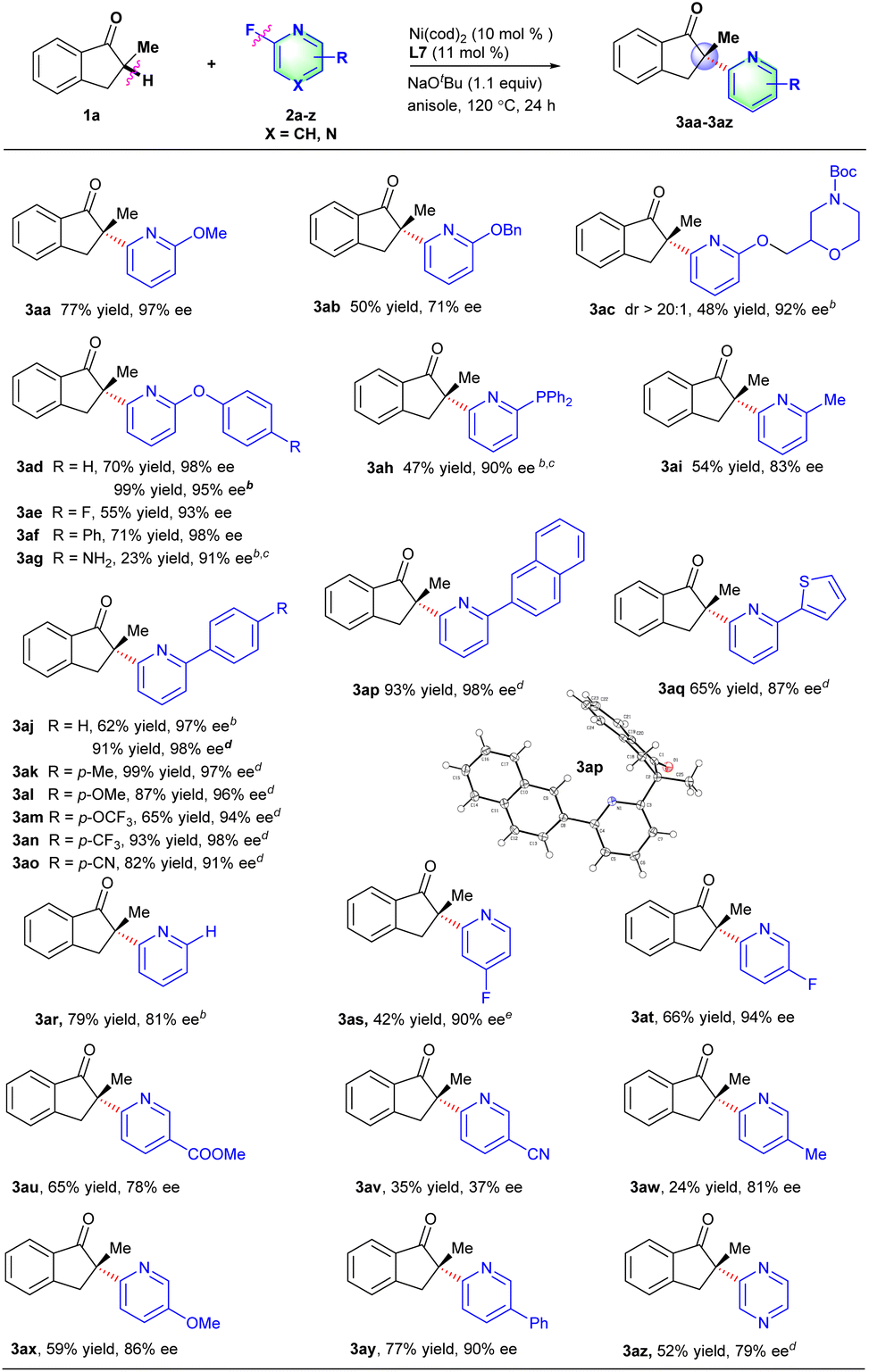 | ||
| Scheme 2 Substrate scope of fluorinated heteroarenes.a Reaction conditions: 1a (0.2 mmol), 2 (0.1 mmol), Ni(cod)2 (10 mol%), L7 (11 mol%), NaOtBu (0.11 mmol), anisole (1.0 mL), 120 °C, 24 h. Isolated yield. ee was determined by chiral HPLC.b 1,4-dioxane was used instead of anisole.c MgBr2 (0.55 equiv.) was added as an additive.d Reaction conditions: 1a (0.1 mmol), 2 (0.15 mmol), Ni(cod)2 (10 mol%), L7 (11 mol%), NaOtBu (0.15 mmol), 1,4-dioxane (1 mL), 120 °C, 24 h.e 96 h. | ||
Next, we turn our attention to investigate the scope of ketones with 2-fluoro-6-phenylpyridine 2j. Like described in Scheme 3, a series of 2-methyl-2,3-dihydro-1H-inden-1-one derivatives with electron-withdrawing or electron-donating groups (e.g., F, OMe and Me) on the phenyl ring all proceeded smoothly to deliver the corresponding products in good yields and excellent ee values (3bj–3gj). Indanone derivatives with various substituents (e.g., ethyl, n-pentyl, benzyl and allyl) at the α-position could well couple with 2-fluoro-6-phenylpyridine to afford the corresponding products in moderate to good yields and excellent ee values (3hj–3lj). In particular, acetylcholinesterase inhibitor donepezil 1m,18 for treating Alzheimer's and vascular dementia, was also compatible with the reaction conditions to provide the chiral α-heteroaryl substituted derivative 3mj in 65% yield and 93% ee. This transformation provided an effective methodology for the late-stage functionalization of donepezils. Besides, six-member cyclic ketones such as 2-methyl-3,4-dihydronaphthalen-1(2H)-one 1n could also proceed well with 2-fluoro-6-phenylpyridine to afford desired product 3nj in 88% yield and 97% ee.
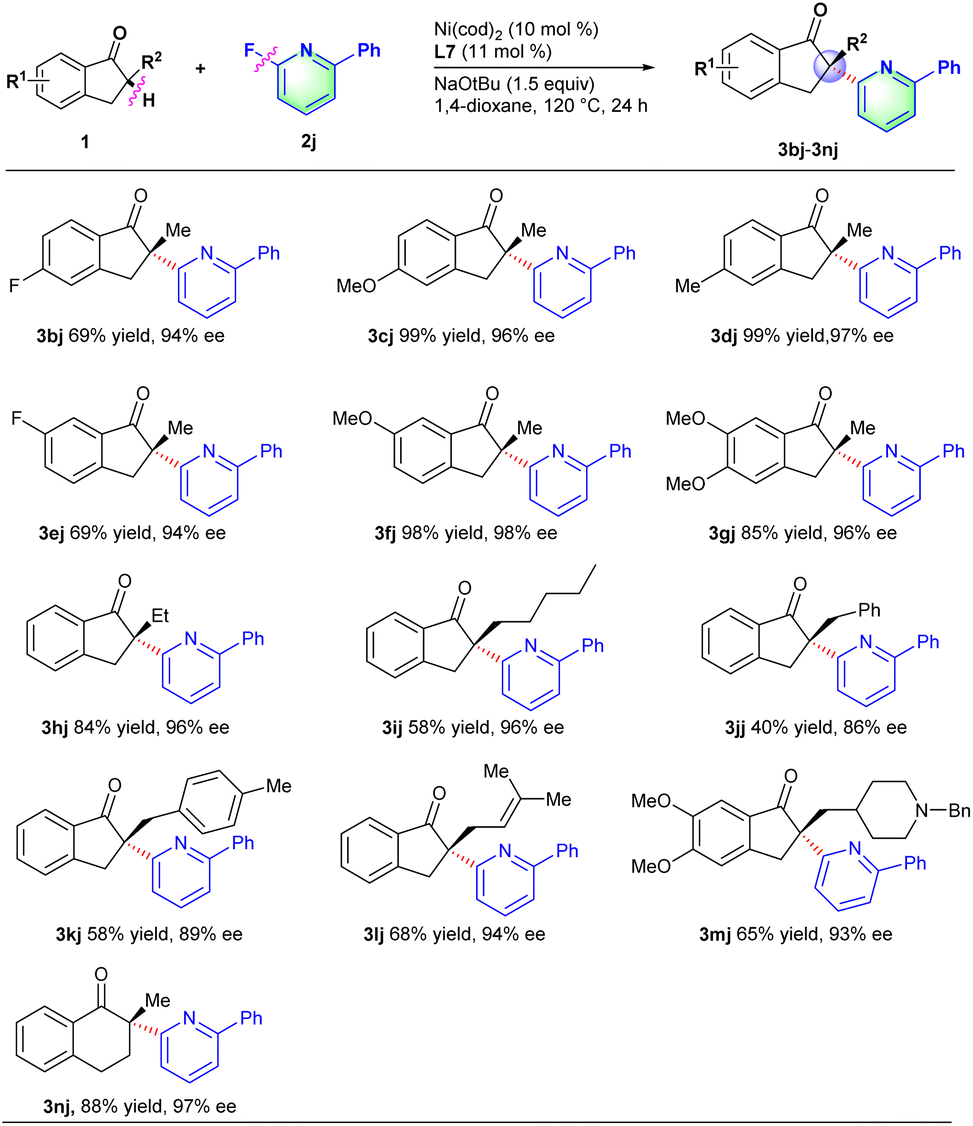 | ||
| Scheme 3 Substrate scope of indanone derivatives. Reaction conditions: 1 (0.1 mmol), 2j (0.15 mmol), Ni(cod)2 (10 mol%), L7 (11 mol%), NaOtBu (0.15 mmol), 1,4-dioxane (1 mL), 120 °C, 24 h. Isolated yield. ee was determined by chiral HPLC. | ||
To evaluate the utility of this approach, a gram-scale reaction was carried out with 3 mmol of indanone 1a, and the coupling product 3ap could be obtained in 83% yield and 99% ee under the standard conditions. When the loading of the nickel catalyst decreased to 2.5 mol%, product 3ap could also be provided in medium yield and good enantioselectivity after the optimization of the reaction conditions (Scheme 4a). The chiral α-heteroarylation product could be further transformed via functionalization of the carbonyl group (Scheme 4b). Coupling product 3ap could be reduced by NaBH4 to deliver secondary alcohol 4 in excellent yield and enantioselectivity. Tertiary alcohol 5 was obtained in good diastereoselectivity via nucleophilic addition with phenylmagnesium bromide. Besides, the carbonyl group could be transformed into the C = X bond (X = C, N) by condensation with the Wittig reagent or hydroxylamine hydrochloride.
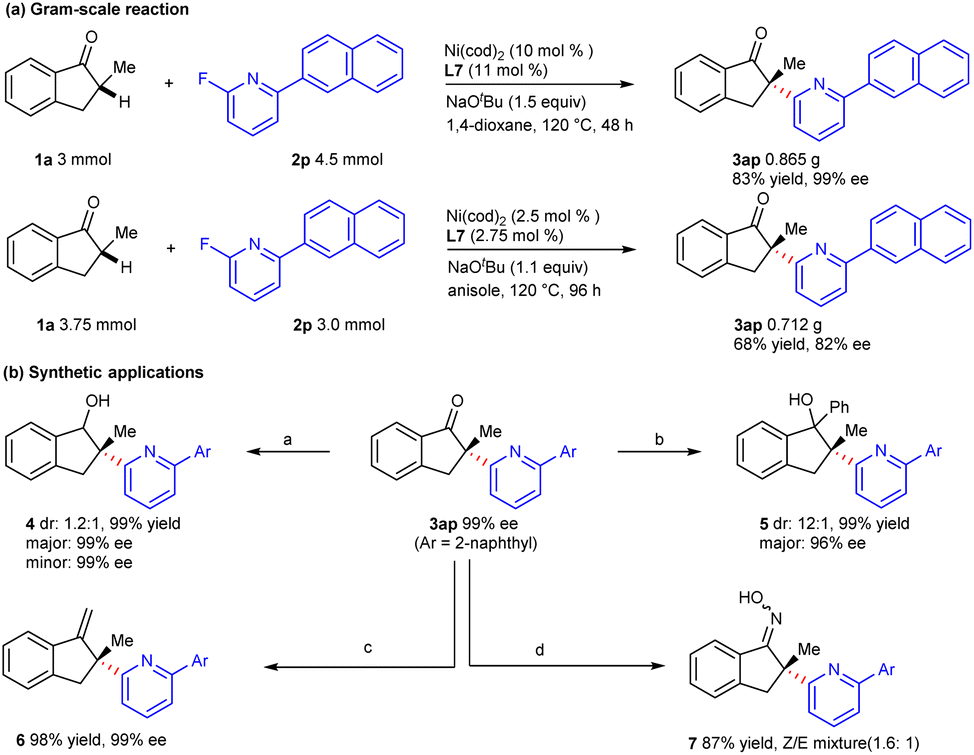 | ||
| Scheme 4 Gram-scale reaction and synthetic applications. (a) NaBH4 (2.0 equiv.), MeOH/THF = 1:1, 2 h, rt. (b) PhMgBr, −78 °C, 1 h, then rt, 2 h. (c) t-BuOK, PPh3CH3Br, rt, 5 min, THF, then rt, overnight. (d) NH2OH·HCl, pyridine, EtOH, reflux. | ||
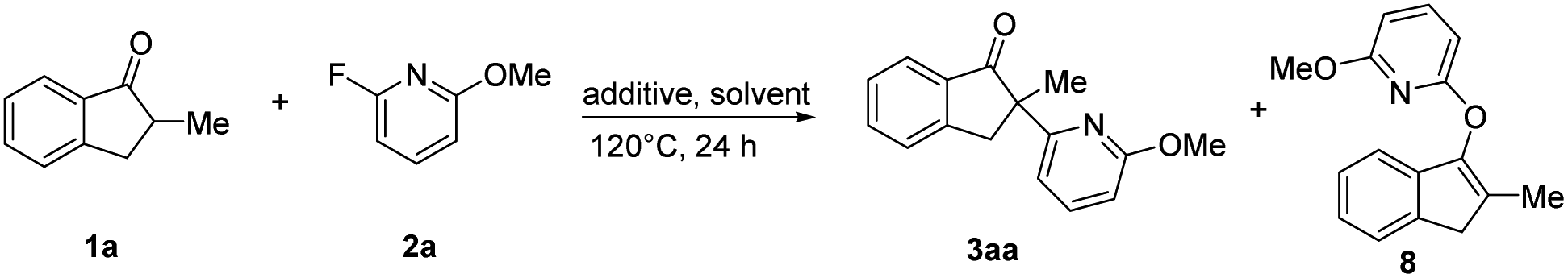
In order to explore the pathway of C–F bond cleavage, control experiments were explored (Table 2). When the reaction was carried out with 1a, 2a and NaOtBu in dry anisole, almost no racemic 3aa and O–heteroarylation by-product 8 were obtained, but 1,4-dioxane could deliver 3aa and 8 in 13% yield and 14% yield, respectively (Table 2, entry 4–5). In addition, by-product 8 could not be further transformed into product 3aa in the presence of Ni(cod)2 and L7 (Table S6†). Control experiments indicated that the excellent enantioselectivity of this methodology was probably derived from the faster reaction rate of oxidative addition of the chiral nickel complex to the C–F bond than that of nucleophilic substitution. Besides, we also explored the existence of possible intermediates via31P NMR and HRMS (ESI†). When 2-fluoropyridine was added to the mixture of Ni(cod)2 and (S)-Ph-Garphos (L7), chiral complex Ni(L7)(cod) (31P NMR, δ = 33.32) was absolutely transformed into intermediate Ni(L7)(2-fluoropyridine) (31P NMR, δ = 29.84). HRMS indicated that oxidative addition of nickel(0) complex Ni(L7)(2-fluoropyridine) to the C–F bond was observed in the presence of Ni(cod)2, L7 and 2-fluoropyridine at 120 °C for 4 h.
|
|
||||
|---|---|---|---|---|
| Entry | Additive (equiv.) | Solvent | 3aa Yieldb (%) | 8 Yieldb (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.1 mmol), additive, dry solvent (1.0 mL), 120 °C, 24 h. b Yield was determined by 1H NMR using dibromomethane as an internal standard. c 1a (0.2 mmol). d 2a (0.15 mmol). | ||||
| 1 | None | 1,4-Dioxane | ND | ND |
| 2 | Ni(cod)2 (0.1) | 1,4-Dioxane | ND | ND |
| 3 | NaOtBu (1.0) | 1,4-Dioxane | 14 | 17 |
| 4c | NaOtBu (1.1) | Anisole | Trace | Trace |
| 5c | NaOtBu (1.1) | 1,4-Dioxane | 13 | 14 |
| 6d | NaOtBu (1.5) | 1,4-Dioxane | 15 | 19 |
Based on the control experiments and related literature reported,2h,8a,19 a plausible mechanism is described in Scheme 5. Initially, intermediate I formed by nickel(0) catalyst and chiral bidentate phosphine ligand undergoes ligand exchange with 2-fluoropyridine derivatives to provide intermediate II confirmed by 31P NMR, which provided nickel(II) intermediate IIIvia oxidative addition of Ni(L7)(2-fluoropyridine) to the C–F bond detected by HRMS. Next, intermediate III is transformed into nickel(II) complex IVvia ligand exchange. Finally, reductive elimination of intermediate IV affords desired products, and the coordination of the resulting nickel complex with cycloocta-1,5-diene regenerates intermediate I to accomplish the catalytic cycle.
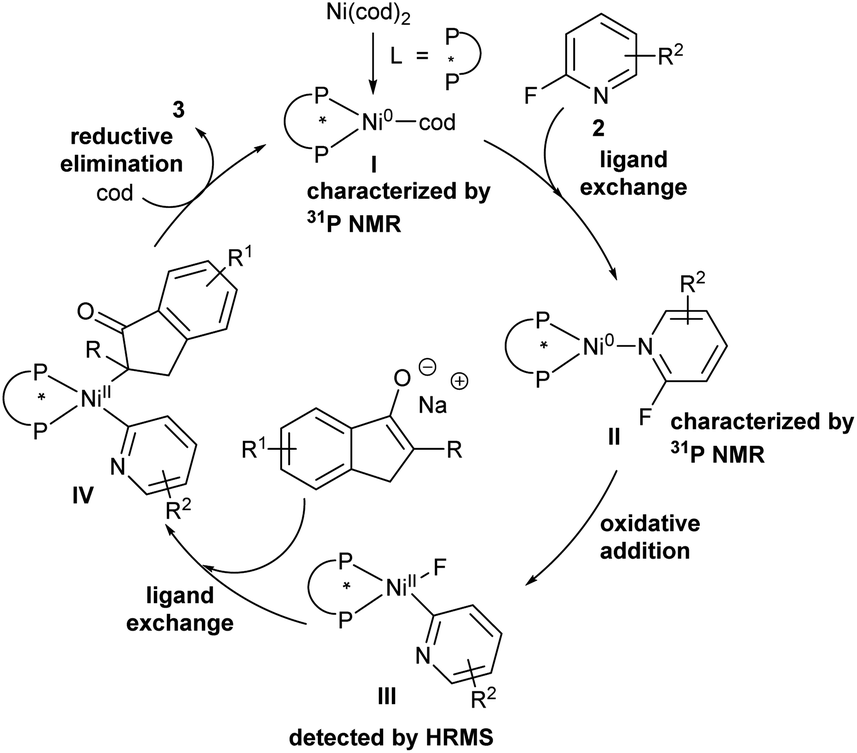 | ||
| Scheme 5 Proposed mechanism. | ||
In summary, we have developed the nickel-catalyzed asymmetric α-heteroarylation of indanone derivatives with 2-fluoropyridines via C–F bond activation. A series of ketones and 2-fluoropyridine derivatives proceed smoothly to deliver the corresponding products containing all-carbon quaternary stereocenters in good yields and high ee values. Drug molecule donepezil could also be compatible with the reaction conditions to afford the desired product in excellent enantioselectivity. Further research on asymmetric C–F activation of diverse compounds is still underway.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
X. Gu performed the experiments and prepared the ESI.† K. Liu repeated some experiments. L. Yang performed the DFT calculations. C. Xie ran some HRMS of intermediates. M. Li & J. W. conceived and directed the project, and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (NSFC 21902072) and HKBU RC-Tier 2 Start-up Grant for financial support.Notes and references
- (a) A. M. Remete, M. Nonn, J. Escorihuela, S. Fustero and L. Kiss, Eur. J. Org. Chem., 2021, 5946–5974 CrossRef CAS; (b) C. Chen, L. Fu, P. Chen and G. Liu, Chin. J. Chem., 2017, 35, 1781–1788 CrossRef CAS; (c) A. C. Sather and S. L. Buchwald, Acc. Chem. Res., 2016, 49, 2146–2157 CrossRef CAS; (d) Y. Li, Y. Wu, G.-S. Li and X.-S. Wang, Adv. Synth. Catal., 2014, 356, 1412–1418 CrossRef CAS; (e) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS; (f) V. V. Grushin, Acc. Chem. Res., 2010, 43, 160–171 CrossRef CAS.
- (a) F. Zhao, W.-L. Zhou and Z. Zuo, Adv. Synth. Catal., 2022, 364, 234–267 CrossRef CAS; (b) L. Zhou, Molecules, 2021, 26, 7051 CrossRef CAS PubMed; (c) J. Zhang, S. Geng and Z. Feng, Chem. Commun., 2021, 57, 11922–11934 RSC; (d) B. Zhao, T. Rogge, L. Ackermann and Z. Shi, Chem. Soc. Rev., 2021, 50, 8903–8953 RSC; (e) Y. Ogiwara and N. Sakai, Angew. Chem., Int. Ed., 2020, 59, 574–594 CrossRef CAS; (f) T. Fujita, K. Fuchibe and J. Ichikawa, Angew. Chem., Int. Ed., 2019, 58, 390–402 CrossRef CAS; (g) Q. Shen, Y.-G. Huang, C. Liu, J.-C. Xiao, Q.-Y. Chen and Y. Guo, J. Fluorine Chem., 2015, 179, 14–22 CrossRef CAS; (h) E. Clot, O. Eisenstein, N. Jasim, S. A. Macgregor, J. E. McGrady and R. N. Perutz, Acc. Chem. Res., 2011, 44, 333–348 CrossRef CAS PubMed.
- (a) T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931–972 CrossRef CAS PubMed; (b) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS; (c) G. Meier and T. Braun, Angew. Chem., Int. Ed., 2009, 48, 1546–1548 CrossRef CAS.
- (a) H. Iwamoto, H. Imiya, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2020, 142, 19360–19367 CrossRef CAS PubMed; (b) H. Dang, A. M. Whittaker and G. Lalic, Chem. Sci., 2016, 7, 505–509 RSC.
- (a) T. W. Butcher, J. L. Yang, W. M. Amberg, N. B. Watkins, N. D. Wilkinson and J. F. Hartwig, Nature, 2020, 583, 548–553 CrossRef CAS; (b) P. H. S. Paioti, J. del Pozo, M. S. Mikus, J. Lee, M. J. Koh, F. Romiti, S. Torker and A. H. Hoveyda, J. Am. Chem. Soc., 2019, 141, 19917–19934 CrossRef CAS PubMed; (c) S. Akiyama, K. Kubota, M. S. Mikus, P. H. S. Paioti, F. Romiti, Q. Liu, Y. Zhou, A. H. Hoveyda and H. Ito, Angew. Chem., Int. Ed., 2019, 58, 11998–12003 CrossRef CAS.
- (a) Y.-C. Luo, F.-F. Tong, Y. Zhang, C.-Y. He and X. Zhang, J. Am. Chem. Soc., 2021, 143, 13971–13979 CrossRef CAS PubMed; (b) X. Yuan, K.-Q. Zhuang, Y.-S. Cui, L.-Z. Qin, Q. Sun, X. Duan, L. Chen, N. Zhu, G. Li, J.-K. Qiu and K. Guo, Commun. Chem., 2020, 3, 98 CrossRef CAS; (c) D. B. Vogt, C. P. Seath, H. Wang and N. T. Jui, J. Am. Chem. Soc., 2019, 141, 13203–13211 CrossRef CAS.
- (a) H. Torrens, Coord. Chem. Rev., 2005, 249, 1957–1985 CrossRef CAS; (b) J. Burdeniuc, B. Jedicka and R. H. Crabtree, Chem. Ber., 1997, 130, 145–154 CrossRef CAS; (c) J. L. Kiplinger, T. G. Richmond and C. E. Osterberg, Chem. Rev., 1994, 94, 373–431 CrossRef CAS.
- (a) C. Wu, S. P. McCollom, Z. Zheng, J. Zhang, S.-C. Sha, M. Li, P. J. Walsh and N. C. Tomson, ACS Catal., 2020, 10, 7934–7944 CrossRef CAS; (b) M. Tobisu, T. Xu, T. Shimasaki and N. Chatani, J. Am. Chem. Soc., 2011, 133, 19505–19511 CrossRef CAS; (c) N. Yoshikai, H. Matsuda and E. Nakamura, J. Am. Chem. Soc., 2009, 131, 9590–9599 CrossRef CAS; (d) L. Ackermann, R. Born, J. H. Spatz and D. Meyer, Angew. Chem., Int. Ed., 2005, 44, 7216–7219 CrossRef CAS; (e) Y. M. Kim and S. Yu, J. Am. Chem. Soc., 2003, 125, 1696–1697 CrossRef CAS; (f) V. P. W. Bhm, C. W. K. Gstttmayr, T. Weskamp and W. A. Herrmann, Angew. Chem., Int. Ed., 2001, 40, 3387–3389 CrossRef.
- J. He, K. Yang, J. Zhao and S. Cao, Org. Lett., 2019, 21, 9714–9718 CrossRef CAS PubMed.
- (a) Q. K. Kang, Y. Lin, Y. Li and H. Shi, Synlett, 2020, 31, 1135–1139 CrossRef CAS; (b) Y. Wang, C. Wei, R. Tang, H. Zhan, J. Lin, Z. Liu, W. Tao and Z. Fang, Org. Biomol. Chem., 2018, 16, 6191–6194 RSC; (c) T. Harada, Y. Ueda, T. Iwai and M. Sawamura, Chem. Commun., 2018, 54, 1718–1721 RSC; (d) F. Zhu and Z.-X. Wang, Adv. Synth. Catal., 2013, 355, 3694–3702 CrossRef CAS.
- (a) S. Lim, D. Song, S. Jeon, Y. Kim, H. Kim, S. Lee, H. Cho, B. C. Lee, S. E. Kim, K. Kim and E. Lee, Org. Lett., 2018, 20, 7249–7252 CrossRef CAS PubMed; (b) X. Zhao, M. Wu, Y. Liu and S. Cao, Org. Lett., 2018, 20, 5564–5568 CrossRef CAS PubMed; (c) T. Niwa, H. Ochiai and T. Hosoya, ACS Catal., 2017, 7, 4535–4541 CrossRef CAS; (d) T. Niwa, H. Ochiai, Y. Watanabe and T. Hosoya, J. Am. Chem. Soc., 2015, 137, 14313–14318 CrossRef CAS; (e) X.-W. Liu, J. Echavarren, C. Zarate and R. Martin, J. Am. Chem. Soc., 2015, 137, 12470–12473 CrossRef CAS.
- (a) M. Sun, M. Tao, L. Zhao, W. Li, Z. Liu, C.-Y. He and Z. Feng, Org. Chem. Front., 2021, 8, 5322–5327 RSC; (b) S. Lim, H. Cho, J. Jeong, M. Jang, H. Kim, S. H. Cho and E. Lee, Org. Lett., 2020, 22, 7387–7392 CrossRef CAS; (c) B. Cui, S. Jia, E. Tokunaga and N. Shibata, Nat. Commun., 2018, 9, 4393 CrossRef.
- J. Li, C. Wu, B. Zhou and P. J. Walsh, J. Org. Chem., 2018, 83, 2993–2999 CrossRef CAS PubMed.
- (a) J. Cornella, E. P. Jackson and R. Martin, Angew. Chem., Int. Ed., 2015, 54, 4075–4078 CrossRef CAS; (b) S. Ge and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 16330–16333 CrossRef CAS; (c) X. Liao, Z. Weng and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 195–200 CrossRef CAS PubMed; (d) M. Li and J. Wang, CCS Chem., 2022 DOI:10.31635/ccschem.022.202101611.
- J. O. Witt, A. L. Mccollum, M. A. Hurtado, E. D. Huseman, D. E. Jeffries, K. J. Temple, H. C. Plumley, A. L. Blobaum, C. W. Lindsley and C. R. Hopkins, Bioorg. Med. Chem. Lett., 2016, 2481–2488 CrossRef CAS PubMed.
- (a) D. Yang, L. Wang, D. Li and R. Wang, Chem, 2019, 5, 1108–1166 CrossRef CAS; (b) X. Jusseau, H. Yin, A. T. Lindhardt and T. Skrydstrup, Chem.–Eur. J., 2014, 20, 15785–15789 CrossRef CAS; (c) G. E. Keck, S. M. Dougherty and K. A. Savin, J. Am. Chem. Soc., 1995, 117, 6210–6223 CrossRef CAS.
- CCDC 2156742 (3ap) contains the supplementary crystallographic data for this paper. These data can be obtained via the Cambridge Crystallographic Data Centre.
- A. H. Barfejania, M. Jafarvandc, S. M. Seyedsaadatd and R. T. Rasekhie, Life Sci., 2020, 263, 118575 CrossRef PubMed.
- A. Nova, R. Mas-Ballesté and A. Lledós, Organometallics, 2012, 31, 1245–1256 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2156742. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc03409c |
| This journal is © The Royal Society of Chemistry 2022 |