
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn integrated platform approach enables discovery of potent, selective and ligand-competitive cyclic peptides targeting the GIP receptor†
Bhaskar
Bhushan
a,
Daniele
Granata
*b,
Christian S.
Kaas‡
b,
Marina A.
Kasimova
b,
Qiansheng
Ren
c,
Christian N.
Cramer
 b,
Mark D.
White
a,
Ann Maria K.
Hansen
d,
Christian
Fledelius
d,
Gaetano
Invernizzi
b,
Kristine
Deibler
e,
Oliver D.
Coleman
f,
Xin
Zhao
c,
Xinping
Qu
c,
Haimo
Liu
c,
Silvana S.
Zurmühl
b,
Janos T.
Kodra
b,
Akane
Kawamura
*af and
Martin
Münzel
*b
b,
Mark D.
White
a,
Ann Maria K.
Hansen
d,
Christian
Fledelius
d,
Gaetano
Invernizzi
b,
Kristine
Deibler
e,
Oliver D.
Coleman
f,
Xin
Zhao
c,
Xinping
Qu
c,
Haimo
Liu
c,
Silvana S.
Zurmühl
b,
Janos T.
Kodra
b,
Akane
Kawamura
*af and
Martin
Münzel
*b
aDepartment of Chemistry, Oxford University, Chemistry Research Laboratory, 12 Mansfield Road, Oxford, OX1 3TA, UK. E-mail: Akane.Kawamura@chem.ox.ac.uk
bGlobal Research Technologies, Novo Nordisk A/S, Novo Nordisk Park, 2760 Måløv, Denmark. E-mail: myzm@novonordisk.com; dngt@novonordisk.com
cNovo Nordisk Research Center China, Novo Nordisk A/S, Shengmingyuan West Ring Rd, Changping District, Beijing, China
dGlobal Drug Discovery, Novo Nordisk A/S, Novo Nordisk Park, 2760 Måløv, Denmark
eNovo Nordisk Research Center Seattle, Novo Nordisk A/S, 530 Fairview Ave N # 5000, Seattle, WA 98109, USA
fSchool of Natural and Environmental Sciences, Chemistry, Newcastle University, Bedson Building, Kings Road Newcastle University Newcastle Upon Tyne, NE1 7RU, UK. E-mail: Akane.Kawamura@ncl.ac.uk
First published on 24th February 2022
Abstract
In any drug discovery effort, the identification of hits for further optimisation is of crucial importance. For peptide therapeutics, display technologies such as mRNA display have emerged as powerful methodologies to identify these desired de novo hit ligands against targets of interest. The diverse peptide libraries are genetically encoded in these technologies, allowing for next-generation sequencing to be used to efficiently identify the binding ligands. Despite the vast datasets that can be generated, current downstream methodologies, however, are limited by low throughput validation processes, including hit prioritisation, peptide synthesis, biochemical and biophysical assays. In this work we report a highly efficient strategy that combines bioinformatic analysis with state-of-the-art high throughput peptide synthesis to identify nanomolar cyclic peptide (CP) ligands of the human glucose-dependent insulinotropic peptide receptor (hGIP-R). Furthermore, our workflow is able to discriminate between functional and remote binding non-functional ligands. Efficient structure–activity relationship analysis (SAR) combined with advanced in silico structural studies allow deduction of a thorough and holistic binding model which informs further chemical optimisation, including efficient half-life extension. We report the identification and design of the first de novo, GIP-competitive, incretin receptor family-selective CPs, which exhibit an in vivo half-life up to 10.7 h in rats. The workflow should be generally applicable to any selection target, improving and accelerating hit identification, validation, characterisation, and prioritisation for therapeutic development.
In recent years, peptides have emerged as powerful therapeutic agents1 and the most prominent examples can match antibodies in selectivity and potency. This development has been greatly aided by breakthroughs in half life extension technologies by formulation or chemical modification. A key advantage of engineered peptides is their ease of production, and while antibodies are generally considered to be too large for efficient oral uptake in therapeutically relevant amounts, the small size of peptides allows the potential for oral dosing with significant progress made in recent years.2,3 Despite these advantages, most drug development projects heavily rely on antibodies, most likely due to the ease of de novo antibody discovery by evolutionarily optimised methods. Modern in vivo or in vitro display technologies, such as phage display and mRNA display, allow for the identification of potent and specific peptide ligands through iterative screening of peptide pools containing trillions of randomised sequences.4,5 These technologies have the potential to yield a plethora of de novo peptides which could be engineered into powerful therapeutics. However, these techniques are surprisingly scarce in clinical pipelines (compared to antibodies or small molecules) despite being known for decades and most peptides in late-stage discovery are derivatives of naturally occurring molecules.1 Challenges in hit prioritisation and time-consuming validation could at least partly be responsible for this observation. While classical approaches would rely on Sanger sequencing of the most abundant clones (yielding very few hits), the advent of next generation sequencing (NGS) has revolutionized the information content available from de novo peptide screens.4 In the majority of studies, however, this information is unused and only a handful of the most abundant sequences are followed up by chemical synthesis for testing of biological activity.5 We rationalized that powerful bioinformatic data mining methods would best utilise the vast information contained in peptide display datasets. To prove this, we combined chemical synthesis and high throughput binding analysis on data from mRNA display. We show that these methods identify more diverse series of hits with different modes of binding as starting points for peptide drug discovery efforts, especially when a functional binder to a specific binding pocket is desired (Scheme 1).
We chose the type 2 GPCR glucose-dependent insulinotropic peptide receptor (GIP-R) as a challenging and clinically relevant test case.6 Its natural ligand GIP and the close relative glucagon-like peptide 1 (GLP-1) are incretins, i.e., hormones which are secreted after oral nutrient uptake and which augment glucose-dependent insulin release from pancreatic beta cells.7 GLP-1 receptor agonists have been reported to not only have effects in metabolic disease through increasing insulin release, but also promote satiety by delaying gastric emptying, and are being investigated for their protective effects in obesity,8,9 heart disease,10 and diabetic kidney disease.11 Conversely, the biology of GIP is less clearly understood, and both GIP-R agonists12 as well as antagonists13,14 are under investigation as potential therapeutics in the fields of type 2 diabetes and obesity. Previous approaches to target the GIP-R thus far have made use of analogues of GIP itself,15,16 or GIP R specific antibodies14,17–20 and peptide display work was based on dual GIP-GLP-1 analogue libraries.14,19,20 As such, the GIP-R represented an ideal target for our mRNA based workflow as we strived to identify potent, ligand competitive, and subfamily selective de novo binders, which could provide valuable tools to understand GIP-R biology and serve as starting points for drug discovery efforts.
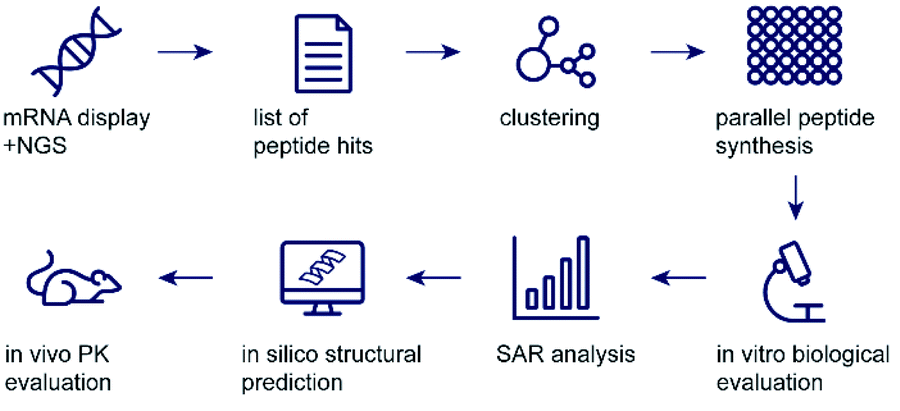 | ||
| Scheme 1 Workflow of peptide hit identification, prioritisation, and validation. | ||
We employed mRNA display to identify cyclic peptide ligands towards the biotinylated extracellular domain (ECD) of the human GIP receptor (residues 22–138 of hGIP-R), which has been shown to retain binding to GIP.21 Initially, we established conditions for efficient cyclisation by disulphide bond formation for representative library peptides (Fig. S3†), before conducting iterative rounds of mRNA display5,22 using a nucleotide library encoding two conserved cysteines for macrocyclization flanking a region of 4 to 12 random amino acid sequence. Negative selections were performed against biotin-loaded streptavidin-functionalised magnetic beads, followed by positive selection against bead-immobilised hGIP-R ECD. Sufficient library enrichment was obtained for hGIP-R after five rounds of selections (percentage recovery relative to input > 0.1%) (Fig. S4a†). NGS of enriched output cDNA was carried out and peptides were clustered by similarity for each selection round (R1–R5). All sequences exceeding six reads in R5 (the top 3160 sequences) were compared against each other using pairwise local alignments in order to derive a complete distance matrix. Single-linkage hierarchical clustering was then performed on the distance matrix, choosing a threshold for cutting the relative dendrogram into clusters based on a statistical optimality criterion.23 For this dataset, the sequence similarity threshold for assigning a sequence to a cluster was set at 0.38, and the fewest number of members that a cluster was allowed to contain was 20. This generated 13 clusters (assigned letters A through M), where the largest cluster, A, contained 1880 sequences, while the two least populated clusters L and M contained 21 sequences each, and the unclustered sequences were assigned to a “noise” cluster (termed cluster 0), encompassing 263 sequences (Fig. 1).
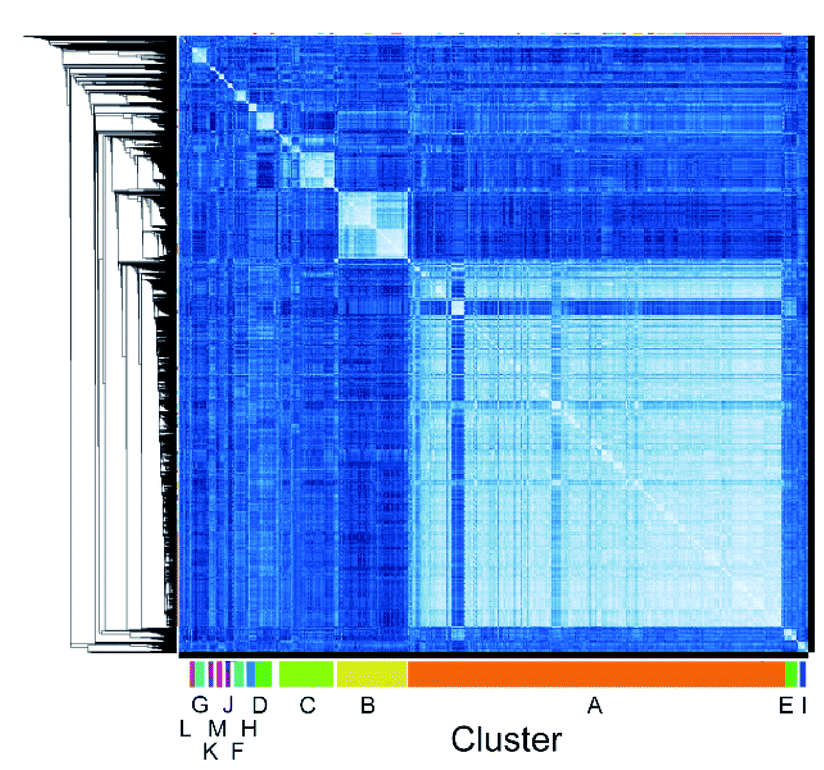 | ||
| Fig. 1 High throughput pairwise clustering analysis of 3160 Round 5 output sequences selected against hGIP-R ECD. Lighter colour indicates closer similarity between individual sequences (ESI Data 1†). | ||
In the next step, we selected peptides from each cluster (A-M, 0) based on sequence diversity and abundance in R5 for parallel solid-phase peptide synthesis (SPPS) in a 96 well format. The N-terminus was capped with an acetyl group (Ac) to mimic the fMet in mRNA-display and a C-terminal FLAG tag was added to each peptide to ensure peptide solubility in buffer by serving as a source of negative charge, analogous to C-terminal mRNA that is present during the selection screens.24 Peptides were assigned identifiers with cluster ID and their rank order based on their abundance in R5 (e.g. B_5). Following synthesis and cleavage, the peptides were macrocyclized via disulphide bond formation in 20% DMSO in buffer.25 Peptide identity, purity, and complete macrocyclization was confirmed by UPLC-MS and peptide concentrations were determined by UPLC-CAD (ESI Data 3†). For the initial high throughput characterisation, peptides were used without further purification (average purity 50–70%). To establish the binding properties, we performed high-throughput single-concentration biolayer interferometry (BLI) measurements, using biotinylated hGIP-R ECD immobilised on streptavidin-functionalised biosensors. Select peptides with favourable binding potencies were resynthesized, purified and their Kd determined by multiple concentration BLI measurements (Fig. 2). The results were generally in good agreement with the single concentration data.
We identified several peptides (>50% of our panel) with nanomolar binding affinities toward the hGIP-R from multiple sequence clusters, particularly from the two most abundant clusters A and B (Fig. 2a and b). However, potent binders were also identified in less abundant sequence families. In particular, peptide M_46 was the second most potent binder amongst the resynthesized peptide panel, while only accumulating 0.2% of the reads compared to the most abundant peptide A_0. This highlights the benefit of following up on peptide hits independent of their ranking based on sequencing reads. Encouragingly, weak-to-no binding was observed from peptide members of cluster 0 (Fig. S8†), corresponding to bioinformatic ‘noise’ in the NGS data, which likely consists of singleton sequences and possible sequencing errors. The FLAG tag itself did not bind to hGIP-R ECD. For members of clusters containing an internal cysteine, e.g. peptide C_24, we generated single cysteine-to-methionine mutants for each site (Met2, Met8, and Met13) and deduced the disulphide pattern from the binding data (i.e. a bond between Cys2 or Cys8 for C_24, see Fig. S8†). In the spirit of the high throughput nature of the workflow, we did not follow up on peptides which failed during parallel synthesis, as the goal was the identification of one hit sequence. As a consequence, we suggest that the pursuit of peptides with complex folds or unpaired cysteines is only indicated if no functional hits can be identified otherwise.
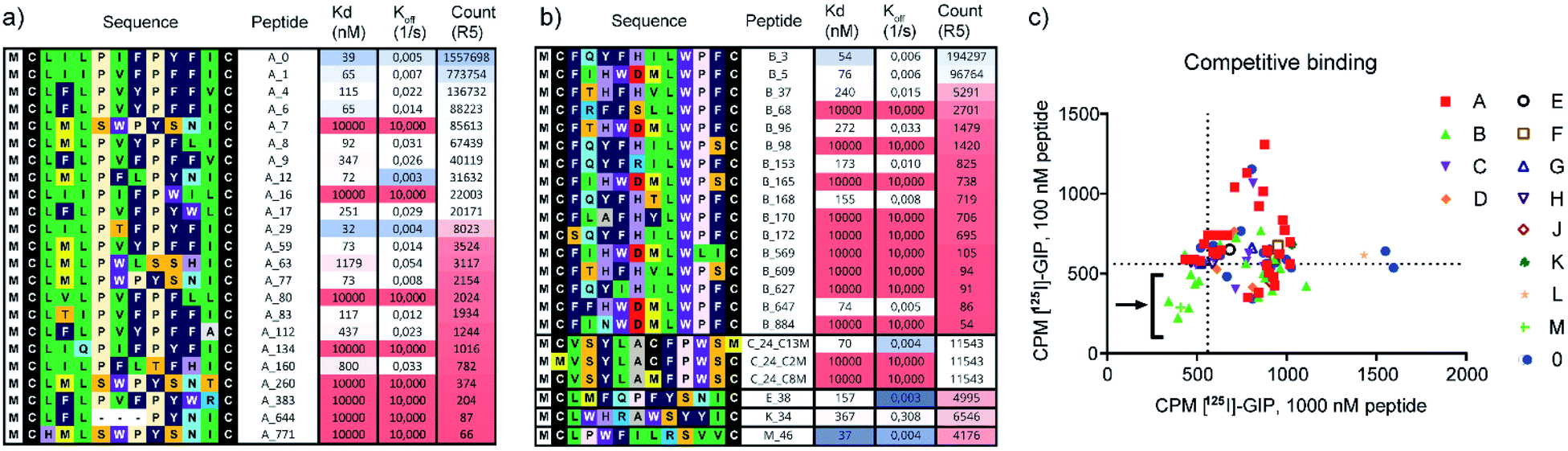 | ||
| Fig. 2 (a and b) Single concentration hGIP-R ECD binding Kd values and dissociation rates for the most abundant peptide clusters, and corresponding abundances/reads of peptide sequences in the final round of selection against GIP-R ECD. All peptides contain an N-terminal Ac group and C-terminal FLAG tag and are cyclised as disulphides. (c) Radiolabelled [125I]-GIP displacement from hGIPR#5/BHK Creluc 2P cells by cyclic peptides at a single concentration of 1000 nM CPs (x axis), and 100 nM CPs (y axis) (CPM = counts per minute). All peptides were tested crude without prior purification (ESI Data 2†). | ||
The non-biased selection process means that high affinity CPs can be generated against any sites on hGIP-R ECD. We hypothesized that each cluster represents peptides which bind at a specific target binding site, with the possibility of different clusters having overlapping or distinct binding sites on the receptor. In this study, we were interested in identifying inhibitory peptide families which are able to either compete or disrupt the natural ligand, whether through direct competition or allosteric binding. To investigate this, we selected representative CPs from each cluster and performed competitive displacement assays with 125I-GIP in BHK CreLuc 2P cells stably expressing hGIP-R. Notably, this assay shows (competitive) binding to the full length GIP-R, whereas our selection and initial screen had been performed against the ECD. As shown in Fig. 2c, only peptides from clusters B and M were found to displace 125I-GIP from hGIP-R (as measured at peptide concentrations of 100 nM and 1000 nM). Interestingly, cluster B contains a LWPF motif at the C-terminus, while cluster M has a related LPWF motif at the N-terminus, indicating a potentially conserved binding site. None of the members of other clusters were found to displace 125I-GIP, even those which were determined to have nanomolar binding affinities to the hGIP R ECD by BLI, including the most abundant peptide family of the selection (cluster A), which covered over 50% of the whole dataset. This highlights the value of the clustering approach, as enrichment of the sequences during mRNA display is driven only by binding affinities of encoded CPs to hGIP-R ECD, but our subsequent clustering analyses served to reveal different functionalities of these CPs. Thus, the high-throughput prioritisation approach is likely to have a higher chance of identifying functional ligands, rather than those prioritised based on observed amounts of NGS reads.
Encouraged by these results, we focussed our subsequent work on cluster B, namely the structurally most diverse members B_3, B_5, and B_68. B_68 had < 20% purity in the 96 well synthesis and did not reveal any binding in the initial high throughput binding assay but was included in the follow up. Further analysis of the sequence list showed another structurally diverse member of cluster B, B_1275, which we hypothesized would also exhibit potent binding and GIP competition, despite the low levels of sequencing reads. These four sequences were scaled up by traditional SPPS and further investigated in multi-concentration BLI and competitive binding assays. All purified peptides showed nanomolar binding and competition of the 125I-GIP from BHK cells stably expressing hGIP-R (Fig. 4). Furthermore, none of these ligands were found to displace radiolabelled GLP-1 from GLP-1R, nor glucagon from GCG-R (Fig. S11†). The parent CP sequences also do not bear any significant sequence homology with native GIP, nor any known interactors of hGIP-R, suggesting that these are the first reported examples of de novo incretin receptor selective and competitive peptides for hGIP-R.
To optimise the biophysical and physicochemical properties of the CPs, and to identify a suitable attachment point for half-life extending moieties, such as albumin binders, we sought to establish a detailed structure–activity relationship (SAR) on cluster B. We chose the most and least abundant of our cluster B lead peptides (B_3 and B_1275) as starting points for full amino acid mutation scans.24,26 We focussed on natural amino acids only to keep the option for (semi-)recombinant expression of the compounds, should large amounts be needed for further development stages. Single mutation variant peptides were synthesised in a 96 well format for each amino acid in the variable region contained between the two conserved cysteine residues C2 and C13. Methionine and cysteine were not included in the mutational scan due to potential oxidation, and asparagine was not included due to the risk of deamidation and isomerisation to iso-aspartate. Additionally, scrambled peptides were included in the panel. The binding affinities of these mutant peptides to hGIP-R ECD were then determined by single concentration BLI experiments.
As shown in Fig. 3a and b, the consensus sequence of cluster B (positions 9–12, LWPF) was found to be the most intolerant to mutation, confirming this region to be critical for binding. Proline at position 11 was found to be completely intolerant to replacement by any other amino acid, suggesting that this residue is critical for maintaining conformationally restricted binding interface of CPs. While residues 9–12 are hydrophobic in both parent peptides, it is unlikely that the interaction of the peptides to hGIP-R ECD is unspecific, as none of the scrambled peptides (e.g. LWPF) were found to bind, including the transposed mutants of B_3 and B_1275, with interchanged L9 and W10. The data shows some general trends for both B_3 and B_1275. In general, positions 3 and 6 (both F in the parent peptides) favour aromatic residues, position 8 favours aliphatic residues, while residues 4, 5, and 7 are fairly tolerant to mutation. The single amino acid scan did not reveal any substitutions that led to remarkable improvements in binding, suggesting that these peptides may either already be optimised for binding through several rounds of mRNA-display selection, or that further binding improvements would only be achieved by synergistic action of multiple substituted residues. The latter could ideally be investigated by mRNA display based affinity maturation experiments in follow up studies.24,26 To gain insight into the binding mode of CPs to hGIP-R on the atomistic level we employed a two-step modelling protocol that first generates multiple conformations of the complex and then selects the final conformation based on stability (for details, see ESI†). As our peptides were demonstrated to be 125I-GIP-competitive, we directly folded them inside the GIP binding site of hGIP-R using Rosetta (Fig. 3c and d; results for B_1275 shown). Briefly, each of the four crucial residues according to the SAR data (Leu9, Trp10, Pro11 or Phe12) was placed in the GIP binding site and the rest of the peptide was grown around it. The obtained conformations (24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 in total) were further clustered and the representative poses of the four most populated clusters were submitted to molecular dynamics (MD) simulations (600 ns for each pose) to assess their stability. The model with the lowest L_RMSD (Root Mean Square Deviation of the ligand backbone during MD trajectory) was selected for further characterization (Fig. 3c). In this model, the C-terminal half of the peptide faces the GIP binding site forming most of the interactions with hGIP-R. This is in line with the obtained SAR data that the C-terminal region is in general less tolerant to mutagenesis compared to the N-terminus. Furthermore, the Trp10 residue overlaps with the position of Phe22 in GIP which is known to be crucial for its binding to the receptor (Fig. 3d). Finally, overall conformation of the peptide resembles that of a hairpin with the turn located on Phe6 and His7, which are the only two positions (together with the peptide termini, see below) tolerating mutagenesis to a proline.
000 in total) were further clustered and the representative poses of the four most populated clusters were submitted to molecular dynamics (MD) simulations (600 ns for each pose) to assess their stability. The model with the lowest L_RMSD (Root Mean Square Deviation of the ligand backbone during MD trajectory) was selected for further characterization (Fig. 3c). In this model, the C-terminal half of the peptide faces the GIP binding site forming most of the interactions with hGIP-R. This is in line with the obtained SAR data that the C-terminal region is in general less tolerant to mutagenesis compared to the N-terminus. Furthermore, the Trp10 residue overlaps with the position of Phe22 in GIP which is known to be crucial for its binding to the receptor (Fig. 3d). Finally, overall conformation of the peptide resembles that of a hairpin with the turn located on Phe6 and His7, which are the only two positions (together with the peptide termini, see below) tolerating mutagenesis to a proline.
 | ||
| Fig. 3 Heat maps showing Kd (nM) values for binding of single-residue mutant peptides derived from B3 (a) and B1275 (b) to biotinylated hGIP-R ECD as determined by single-concentration BLI. The columns indicate the amino acid changes compared to the parent sequence (displayed at the top of the table). All peptides featured a C-terminal Cys and were tested in crude form. Peptides that were not tested or those where synthesis failed are marked as grey boxes. None of the scrambled mutant peptides showed any binding. (c) Atomistic model of the cyclic peptide B_1275 and GIP receptor complex suggested by molecular modelling. The LWPF sequence of the peptide is shown in red, while the rest of the molecule is coloured by the atom name (carbon in yellow, nitrogen in blue, oxygen in red, and sulphur in orange). The receptor is show in grey; the binding site residues (L35, W39, M67 and Y87) are highlighted in green. (d) Overlay of the crystal structure of GIP complexed with hGIP receptor (PDB 2QKH) and atomistic model of cyclic peptide B_1275 in the binding site of the GIP receptor. Note that the positions of W10 of the cyclic peptide and F22 of GIP overlap. | ||
Next, we wanted to use the accrued binding and SAR information to design peptides for in vivo studies. For these, peptides need to fulfil two crucial requirements. Firstly, they need to be stable in plasma and secondly, they need to be protected from renal clearance. The latter is crucial to the development of any peptide therapeutic as even protease resistant peptides usually exhibit plasma half lives in the range of a few minutes. Attachment of a fatty acid-based albumin binder (termed protractor) is one of the most employed strategies to extend peptide half-life and we opted for a 2xOEG-gGlu-C18 diacid albumin binder for our peptide hits.3,27–29 The binding model suggested that the addition of an albumin binding moiety is tolerated at the N- or C-terminus, as these are solvent exposed. Further evidence that replacement of the Met in position 1 with Ala is possible for both B_3 and B_1275 without loss of affinity and the ease of synthesis prompted us to design four peptides with a protractor attached to the N-terminus (B_3.1, B_5.1, B_68.1 and B_1275.1). Furthermore, we synthesized linear versions of the parent peptides in order to investigate the importance of macrocyclization on binding and stability (B_3.2, B_5.2, B_68.2 and B_1275.2). Additionally, we synthesized peptides without FLAG tag to prove that binding indeed is mediated by the macrocycle and not the tag (B_1275.3). During the purification of the latter, we realized that solubility was low (as expected from the rather hydrophobic sequence), complicating purification and analysis. Thus, we utilized our SAR understanding to design variants with improved biophysical properties by reducing the hydrophobicity and increasing the charge at positions where mutation was tolerated (especially positions 1, 4 and 7). Furthermore, we replaced the Met residues to avoid oxidation of the sulphur atom. We focussed on B_1275 as we anticipated from the sequence that this peptide is most hydrophilic, as also indicated by the shortest retention time in HPLC chromatography (Table S1†). All resulting peptides containing a FLAG tag (B_1275.7 through B_1275.11) were soluble at the relevant conditions, however, only B_1275.5 showed high solubility without the tag, as measured by nephelometry at different PEG concentrations (Fig. S14†). Pleasingly, addition of an albumin binder (B_1275.6) did not negatively affect solubility. Analysis of the peptides by multi-concentration BLI and radio-GIP displacement showed that indeed N-terminal modification was allowed for most peptides (Fig. 4). None of the linear variants (B_3.2, B_5.2, B_68.2, B_1275.2) showed binding, suggesting that macrocyclization is crucial for the interaction. Most mutant peptides, however, retained binding, including the triple mutant B_1275.9 (M1A, T4R, H7E; Kd at 31 nM) and quadruple mutants B_1275.7 (M1A, F3W, H5E, H7E, Kd at 101 nM) and B_1275.8 (M1S, T4R, H5A, H7Q, Kd at 184 nM), showing the power of having detailed SAR information available for multi-factorial optimisation of hits. Interestingly, in contrast to the binding data obtained from F11P mutants from high throughput mutagenesis studies (Fig. 3), B_1275.10 with a F11P substitution did not show any binding, which exemplifies the limits of high throughput SAR analysis using crude peptides, as the terminal P possibly interferes with cyclisation and might lead to multi- or polymers which interfere with the BLI assay.
Finally, we tested a panel of the optimised peptides for stability in human plasma and determined in vivo pharmacokinetic parameters in rats. Most cyclic peptides (B_3.1, B_1275.3, B_1275.4, B_1275.5 and B_1275.6) showed no decrease in plasma stability over the course of 5 h, including in the presence of a FLAG tag or of a protractor (Fig. 4b). While parent B_1275 had a t1/2 of 3.5 h, the two linear peptides tested (B_3.2 and B_1275.2) exhibited low levels of degradation (t1/2ca. 2 h, see Table S2 in ESI† for exact values), and the biologically active linear peptides of both GIP and GLP-1 were more rapidly degraded (t1/2ca. 35–45 min), which highlights the benefits of cyclic peptides in terms of druggability. Having established the plasma stability, we turned our attention to the in vivo half-life. We chose two protracted parent peptides (B_3.1 and B_1275.1) and the optimised B_1275 variants with FLAG tag (B_1275.4), without FLAG tag (B_1275.5) and with protractor but without FLAG tag (B_1275.6). The two non-protracted peptides exhibit very short half-lives (B_1275.4 = 3.5 min, B_1275.5 = 1.8 min), whereas the peptides carrying an albumin binder show significant plasma exposure over a long period of time (t1/2, B_3.1 = 9.8 h,B_1275.1 = 10.7 h B_1275.6 = 4.2 h), with half-lives being increased by more than 100 fold (Fig. 4d) (as a comparison, the GLP-1 analogue semaglutide, which is dosed once-weekly in humans has a t1/2 in rats of 7 h.)28
 | ||
| Fig. 4 (a) BLI binding Kd values and radiolabelled [125I]-GIP displacement IC50 data for purified peptides. Multiple concentrations of CPs were used to determine these values. (*) uncertain values due to limited solubility of CP. (b) Stability of selected peptides upon incubation with human plasma at 37 °C. (c) In vivo plasma exposure levels of selected peptides upon i.v. dosing to rats. All data are presented as mean ± SEM of three independent experiments. | ||
Conclusions
In conclusion, we have developed a powerful and efficient workflow to identify and prioritize hit peptides from display screens and to rapidly progress them to functional lead molecules. These can be used to decipher biological questions in vitro and in vivo or be used as starting points to initiate drug development programmes. Our results show that peptide display technologies coupled to NGS-guided clustering and high-throughput hit validation offers a fast, powerful, and robust strategy for identification of de novo ligands against targets of interest, and additionally allows the rapid construction of meaningful and data based structural models in silico. As the crucial aspect of our workflow, it allows holistic and unbiased analysis of binding sequence mapping of CPs against the target of interest. This enables thorough investigation of the different possible binding motifs – and possibly different binding sites – in the selection for further validation, rather than limiting the follow up to the most enriched sequences (which could be dominated by a handful of binding motifs). We believe that this prioritisation and optimisation approach holds great promise for future lead identification campaigns both in an academic and industrial setting.Data availability
Next generation sequencing data including cluster information, measured kd values and experimental data on synthesized peptides are available as ESI.†Author contributions
B. B. investigation and data curation, D. G. software and data curation, C. S. K. investigation and data curation, M. A. K. software, data curation and visualisation, Q. R. investigation, C. N. C. investigation, M. D. W. investigation, A. M. K. H. investigation, C. F. investigation, G. I. investigation, K. D. software and data curation, O. D. C. investigation, X. Z. investigation, X. Q. investigation, H. L. investigation, S. S. Z. investigation, J. T. K supervision, A. K. conceptualisation, methodology, supervision, funding acquisition, M. M. conceptualisation, funding acquisition, data curation and visualisation, methodology, supervision, writing original draft. All authors have reviewed the final draft.Conflicts of interest
All authors apart from B. B., O. D. C., and A. K. are or were employees and shareholders of Novo Nordisk A/S. A. K has received consultancy fees from Novo Nordisk A/S.Acknowledgements
This work was supported by a NovoNordisk STAR postdoc fellowship and the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (grant agreement No. 679479). The authors would like to thank Johnny Madsen, Jesper Damsgaard, Kira Meyhoff-Madsen, and Vibeke Nielsen (Novo Nordisk) for excellent technical assistance.Notes and references
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707 CrossRef CAS PubMed.
- D. J. Drucker, Nat. Rev. Drug Discovery, 2020, 19, 277–289 CrossRef CAS PubMed.
- F. Hubalek, H. H. F. Refsgaard, S. Gram-Nielsen, P. Madsen, E. Nishimura, M. Munzel, C. L. Brand, C. E. Stidsen, C. H. Claussen, E. M. Wulff, L. Pridal, U. Ribel, J. Kildegaard, T. Porsgaard, E. Johansson, D. B. Steensgaard, L. Hovgaard, T. Glendorf, B. F. Hansen, M. K. Jensen, P. K. Nielsen, S. Ludvigsen, S. Rugh, P. W. Garibay, M. C. Moore, A. D. Cherrington and T. Kjeldsen, Nat. Commun., 2020, 11, 3746 CrossRef CAS PubMed.
- S. Goodwin, J. D. McPherson and W. R. McCombie, Nat. Rev. Genet., 2016, 17, 333–351 CrossRef CAS PubMed.
- A. Kawamura, M. Munzel, T. Kojima, C. Yapp, B. Bhushan, Y. Goto, A. Tumber, T. Katoh, O. N. King, T. Passioura, L. J. Walport, S. B. Hatch, S. Madden, S. Muller, P. E. Brennan, R. Chowdhury, R. J. Hopkinson, H. Suga and C. J. Schofield, Nat. Commun., 2017, 8, 14773 CrossRef CAS PubMed.
- J. J. Holst and M. M. Rosenkilde, J. Clin. Endocrinol. Metab., 2020, 105, E2710–E2716 CrossRef PubMed.
- J. J. Holst, Metabolism, 2019, 96, 46–55 CrossRef CAS PubMed.
- J. Blundell, G. Finlayson, M. Axelsen, A. Flint, C. Gibbons, T. Kvist and J. B. Hjerpsted, Diabetes, Obes. Metab., 2017, 19, 1242–1251 CrossRef CAS PubMed.
- J. P. H. Wilding, R. L. Batterham, S. Calanna, M. Davies, L. F. Van Gaal, I. Lingvay, B. M. McGowan, J. Rosenstock, M. T. D. Tran, T. A. Wadden, S. Wharton, K. Yokote, N. Zeuthen, R. F. Kushner and S. S. Group, N. Engl. J. Med., 2021, 384, 989 CrossRef CAS PubMed.
- M. Husain, S. C. Bain, O. K. Jeppesen, I. Lingvay, R. Sorrig, M. B. Treppendahl and T. Vilsboll, Diabetes, Obes. Metab., 2020, 22, 442–451 CrossRef PubMed.
- J. L. Gorriz, M. J. Soler, J. F. Navarro-Gonzalez, C. Garcia-Carro, M. J. Puchades, L. D’Marco, A. Martinez Castelao, B. Fernandez-Fernandez, A. Ortiz, C. Gorriz-Zambrano, J. Navarro-Perez and J. J. Gorgojo-Martinez, J. Clin. Med., 2020, 9(4), 947 CrossRef CAS PubMed.
- J. Rosenstock, C. Wysham, J. P. Frías, S. Kaneko, C. J. Lee, L. Fernández Landó, H. Mao, X. Cui, C. A. Karanikas and V. T. Thieu, The Lancet, 2021, 398, 143–155 CrossRef CAS.
- E. A. Killion, M. Chen, J. R. Falsey, G. Sivits, T. Hager, L. Atangan, J. Helmering, J. Lee, H. Li, B. Wu, Y. Cheng, M. M. Veniant and D. J. Lloyd, Nat. Commun., 2020, 11, 4981 CrossRef CAS PubMed.
- E. A. Killion, J. Wang, J. Yie, S. D. Shi, D. Bates, X. Min, R. Komorowski, T. Hager, L. Deng, L. Atangan, S. C. Lu, R. J. M. Kurzeja, G. Sivits, J. Lin, Q. Chen, Z. Wang, S. A. Thibault, C. M. Abbott, T. Meng, B. Clavette, C. M. Murawsky, I. N. Foltz, J. B. Rottman, C. Hale, M. M. Veniant and D. J. Lloyd, Sci. Transl. Med., 2018, 10, eaat3392 CrossRef CAS PubMed.
- P. A. Mroz, B. Finan, V. Gelfanov, B. Yang, M. H. Tschop, R. D. DiMarchi and D. Perez-Tilve, Mol. Metab., 2019, 20, 51–62 CrossRef CAS PubMed.
- P. K. Norregaard, M. A. Deryabina, P. Tofteng Shelton, J. U. Fog, J. R. Daugaard, P. O. Eriksson, L. F. Larsen and L. Jessen, Diabetes, Obes. Metab., 2018, 20, 60–68 CrossRef CAS PubMed.
- X. Min, J. Yie, J. Wang, B. C. Chung, C. S. Huang, H. Xu, J. Yang, L. Deng, J. Lin, Q. Chen, C. M. Abbott, C. Gundel, S. A. Thibault, T. Meng, D. L. Bates, D. J. Lloyd, M. M. Veniant and Z. Wang, MAbs, 2020, 12, 1710047 CrossRef PubMed.
- P. Ravn, C. Madhurantakam, S. Kunze, E. Matthews, C. Priest, S. O'Brien, A. Collinson, M. Papworth, M. Fritsch-Fredin, L. Jermutus, L. Benthem, M. Gruetter and R. H. Jackson, J. Biol. Chem., 2013, 288, 19760–19772 CrossRef CAS PubMed.
- A. Demartis, A. Lahm, L. Tomei, E. Beghetto, V. Di Biasio, F. Orvieto, F. Frattolillo, P. E. Carrington, S. Mumick, B. Hawes, E. Bianchi, A. Palani and A. Pessi, Sci. Rep., 2018, 8, 585 CrossRef PubMed.
- Y. Wu, T. Ji, J. Lv and Z. Wang, Life Sci., 2020, 257, 118025 CrossRef CAS PubMed.
- C. Parthier, M. Kleinschmidt, P. Neumann, R. Rudolph, S. Manhart, D. Schlenzig, J. Fanghanel, J. U. Rahfeld, H. U. Demuth and M. T. Stubbs, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13942–13947 CrossRef CAS PubMed.
- R. W. Roberts and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12297–12302 CrossRef CAS PubMed.
- M. Marsili, I. Mastromatteo and Y. Roudi, J. Stat. Mech.: Theory Exp., 2013, 2013 Search PubMed.
- J. M. Rogers, T. Passioura and H. Suga, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10959–10964 CrossRef CAS PubMed.
- J. P. Tam, C. R. Wu, W. Liu and J. W. Zhang, J. Am. Chem. Soc., 2002, 113, 6657–6662 CrossRef.
- T. Passioura, B. Bhushan, A. Tumber, A. Kawamura and H. Suga, Bioorg. Med. Chem., 2018, 26, 1225–1231 CrossRef CAS PubMed.
- T. Coskun, K. W. Sloop, C. Loghin, J. Alsina-Fernandez, S. Urva, K. B. Bokvist, X. Cui, D. A. Briere, O. Cabrera, W. C. Roell, U. Kuchibhotla, J. S. Moyers, C. T. Benson, R. E. Gimeno, D. A. D'Alessio and A. Haupt, Mol. Metab., 2018, 18, 3–14 CrossRef CAS PubMed.
- J. Lau, P. Bloch, L. Schaffer, I. Pettersson, J. Spetzler, J. Kofoed, K. Madsen, L. B. Knudsen, J. McGuire, D. B. Steensgaard, H. M. Strauss, D. X. Gram, S. M. Knudsen, F. S. Nielsen, P. Thygesen, S. Reedtz-Runge and T. Kruse, J. Med. Chem., 2015, 58, 7370–7380 CrossRef CAS PubMed.
- T. B. Kjeldsen, F. Hubalek, C. U. Hjorringgaard, T. M. Tagmose, E. Nishimura, C. E. Stidsen, T. Porsgaard, C. Fledelius, H. H. F. Refsgaard, S. Gram-Nielsen, H. Naver, L. Pridal, T. Hoeg-Jensen, C. B. Jeppesen, V. Manfe, S. Ludvigsen, I. Lautrup-Larsen and P. Madsen, J. Med. Chem., 2021, 64, 8942–8950 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc06844j |
| ‡ Novo Nordisk Bio Innovation HubNovo Nordisk A/S255 Main St 10th Floor, Cambridge, MA 02142, United States |
| This journal is © The Royal Society of Chemistry 2022 |