
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight-driven reduction of aromatic olefins in aqueous media catalysed by aminopyridine cobalt complexes†
Carla
Casadevall
 a,
David
Pascual
a,
Jordi
Aragón
a,
Arnau
Call
a,
Alicia
Casitas
a,
Irene
Casademont-Reig
b and
Julio
Lloret-Fillol
*ac
a,
David
Pascual
a,
Jordi
Aragón
a,
Arnau
Call
a,
Alicia
Casitas
a,
Irene
Casademont-Reig
b and
Julio
Lloret-Fillol
*ac
aInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology, Avinguda Països Catalans 16, 43007 Tarragona, Spain. E-mail: jlloret@iciq.es
bDonostia International Physics Center (DIPC), Polimero eta Material Aurreratuak: Fisika, Kimika eta Teknologia, Kimika Fakultatea, Euskal Herriko Unibertsitatea UPV/EHU, P.K. 1072, 20080 Donostia, Euskadi, Spain
cCatalan Institution for Research and Advanced Studies (ICREA), Passeig Lluïs Companys, 23, 08010, Barcelona, Spain
First published on 14th March 2022
Abstract
A catalytic system based on earth-abundant elements that efficiently hydrogenates aryl olefins using visible light as the driving-force and H2O as the sole hydrogen atom source is reported. The catalytic system involves a robust and well-defined aminopyridine cobalt complex and a heteroleptic Cu photoredox catalyst. The system shows the reduction of styrene in aqueous media with a remarkable selectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) versus water reduction (WR). Reactivity and mechanistic studies support the formation of a [Co–H] intermediate, which reacts with the olefin via a hydrogen atom transfer (HAT). Synthetically useful deuterium-labelled compounds can be straightforwardly obtained by replacing H2O with D2O. Moreover, the dual photocatalytic system and the photocatalytic conditions can be rationally designed to tune the selectivity for aryl olefin vs. aryl ketone reduction; not only by changing the structural and electronic properties of the cobalt catalysts, but also by modifying the reduction properties of the photoredox catalyst.
000) versus water reduction (WR). Reactivity and mechanistic studies support the formation of a [Co–H] intermediate, which reacts with the olefin via a hydrogen atom transfer (HAT). Synthetically useful deuterium-labelled compounds can be straightforwardly obtained by replacing H2O with D2O. Moreover, the dual photocatalytic system and the photocatalytic conditions can be rationally designed to tune the selectivity for aryl olefin vs. aryl ketone reduction; not only by changing the structural and electronic properties of the cobalt catalysts, but also by modifying the reduction properties of the photoredox catalyst.
Introduction
The use of sunlight as driving force is a promising but challenging strategy towards sustainability in chemical production.1–4 Artificial photosynthesis (AP), as a technology to provide a clean source of reductive equivalents,4–8 has been mainly studied in the context of H2O9–13 and CO2 (ref. 14–18) reduction, with still limited progress towards organic transformations.1,2,4,8,19–32 A challenging transformation in the context of AP is the hydrogenation of double bonds.8 Among difficulties, most developed protocols employing the activation of H2, silanes or alcohols are not suitable for operation under aqueous conditions.33–37 And only a few studies used light as a driving force and most of them have been carried out in the absence of water as a solvent. Moreover, selectivity of olefin reduction versus water reduction is needed for achieving a practical use in the context of AP. In the case of aromatic olefins, they are prompted to form radicals, tending to polymerize under such conditions.38In the context of AP, semiconductor materials such as TiO2 and CdS were initially used as catalysts for the light-driven hydrogenation of electron-deficient olefins. These systems required UV light (λ = 365 nm) and noble metals to obtain moderate selectivity for alkane formation (Scheme 1a).39–41 Another remarkable example employed a B12–TiO2 hybrid for the UV light-driven reduction of alkenes to alkanes.42 Such highly energetic conditions led to the formation of dimeric products compromising the selectivity.43
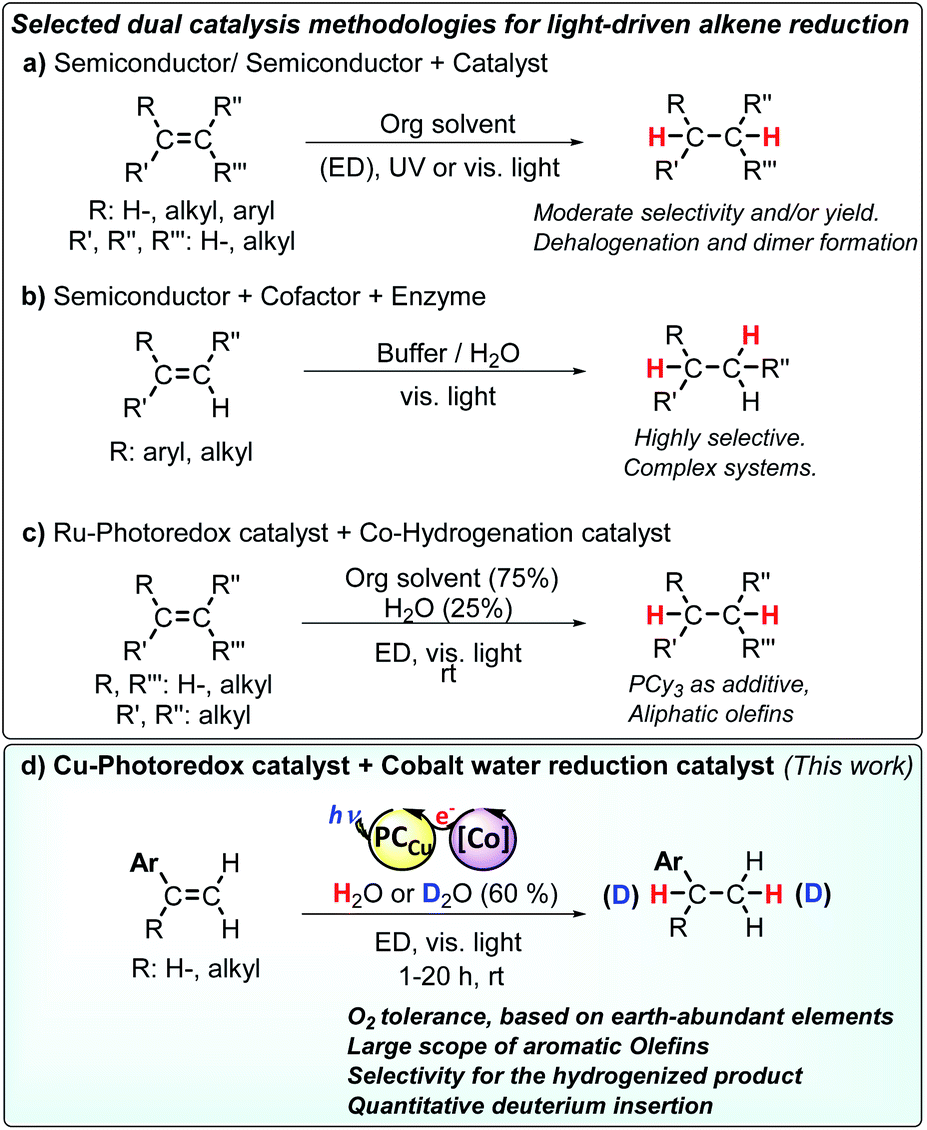 | ||
| Scheme 1 Selected dual catalysis methodologies for the light driven reduction of olefins. Abbreviations: ED: electron donor. Selected references: (a) ref. 8 and 10, (b) ref. 11, (c) ref. 12 and (d) the developed methodology in this study. | ||
Holland, Corma and co-workers introduced the stereoselective hydrogenation of the conjugated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of ketoisophorone by combining the photocatalytic activity of Au nanoparticles supported on TiO2 with the enzymatic activity of oxidoreductases by means of FAD+ as a mediator and cofactor (Scheme 1b).22 Likewise, Hartwig, Zhao and co-workers reported a cooperative chemoenzymatic reaction that combines the isomerization of CC by an iridium photoredox catalyst with the hydrogenation activity of ene-reductases to selectively reduce electron-deficient aromatic olefins to their corresponding alkanes in high enantiomeric excess (88–99% ee).44 All of these previous studies show that light-driven reduction of alkenes is feasible, albeit limited in substrate scope, selectivity and yield.
C bond of ketoisophorone by combining the photocatalytic activity of Au nanoparticles supported on TiO2 with the enzymatic activity of oxidoreductases by means of FAD+ as a mediator and cofactor (Scheme 1b).22 Likewise, Hartwig, Zhao and co-workers reported a cooperative chemoenzymatic reaction that combines the isomerization of CC by an iridium photoredox catalyst with the hydrogenation activity of ene-reductases to selectively reduce electron-deficient aromatic olefins to their corresponding alkanes in high enantiomeric excess (88–99% ee).44 All of these previous studies show that light-driven reduction of alkenes is feasible, albeit limited in substrate scope, selectivity and yield.
Very recently, X. C. Cambeiro and co-workers reported the use of an iridium photosensitizer in combination with a Hantzsch ester for the reduction of aromatic 1,2-disubstituted alkenes, albeit good yields were obtained only for activated olefins.45 At the same time, M. Kojima, S. Matsunaga and co-workers found that glyoxime type complexes with a PCy3 ligand and a ruthenium photoredox catalyst reduce aliphatic alkenes via a hydrogen atom transfer (HAT) mechanism.46 However, aromatic alkenes have additional difficulties such as their tendency to engage in radical polymerization38 or isomerization reactions47–49 under reductive conditions, which makes their reduction under photocatalytic conditions more challenging (Scheme 1). By a conceptually different approach, X. Guo, O. S. Wenger and co-workers recently reported the reduction of olefins by the photo-HAT reactivity of the organometallic iridium hydride complex [Cp*IrII(phen)–H] in acetonitrile.50 Polyzos and co-workers showed the reduction of aromatic olefins via direct photoinduced electron transfer under highly reductive conditions and light intensity.51
Ideally, AP systems should employ water as a source of hydrogen atoms and efficiently use the photoredox catalyst in combination with molecular catalysts to facilitate the selective reduction of alkenes. Such an approach should identify catalysts' hits to develop more complex AP schemes. Towards this goal, we previously reported a dual catalytic system formed by [Co(OTf)(Py2Tstacn)](OTf) (1) (Py2Tstacn = 1,4-di(picolyl)-7-(p-toluenesulfonyl)-1,4,7-triazacyclononane, OTf = trifluoromethanesulfonate anion) as a reduction catalyst and [Cu(bathocuproine)(xantphos)](PF6)52 (PCCu) as a photoredox catalyst, which efficiently reduces water to hydrogen53 and carbonyl groups to alcohols, using water as a hydrogen source.29 Our mechanistic studies suggested that a cobalt(II) hydride ([Co(H)(Py2Tstacn)]+, [CoII–H]) was a common intermediate in both reduction reactions. Based on these precedents, we hypothesized that [Co(H)(Py2Tstacn)]+ should also be reactive enough to engage in the reduction of other organic functionalities without the need for strong reducing agents.31
Herein, we present a dual catalytic system consisting of cobalt complexes based on nitrogenated ligands (1–15), different Ir and Cu photoredox catalysts and an electron donor for the reduction of styrene derivatives using light as an energy source. The reduction operates without the use of typical reducing agents such as silanes, H2, HCO2H or alcohols. The best performing catalytic system is obtained by combining [Co(OTf)(Py2Tstacn)](OTf) (1) and PCCu under light irradiation employing H2O/Et3N as a hydride source (Scheme 2).29–32 This synthetic methodology also allows for replacing H2O by D2O to produce the analogous deuterated alkanes. Reactivity and mechanistic studies based on kinetics, isotopic labelling and radical clock experiments suggest that the reduction of aromatic olefins occurs via a HAT mechanism, most likely through a [Co–H] intermediate. According to our mechanistic findings, the interplay between the catalyst/substrate redox potential, together with the light intensity, provides a way to control the selectivity for the reduction of aromatic olefins versus aromatic ketones and vice versa. This unique behaviour is rationalized by the different reduction mechanisms that each substrate undergoes.
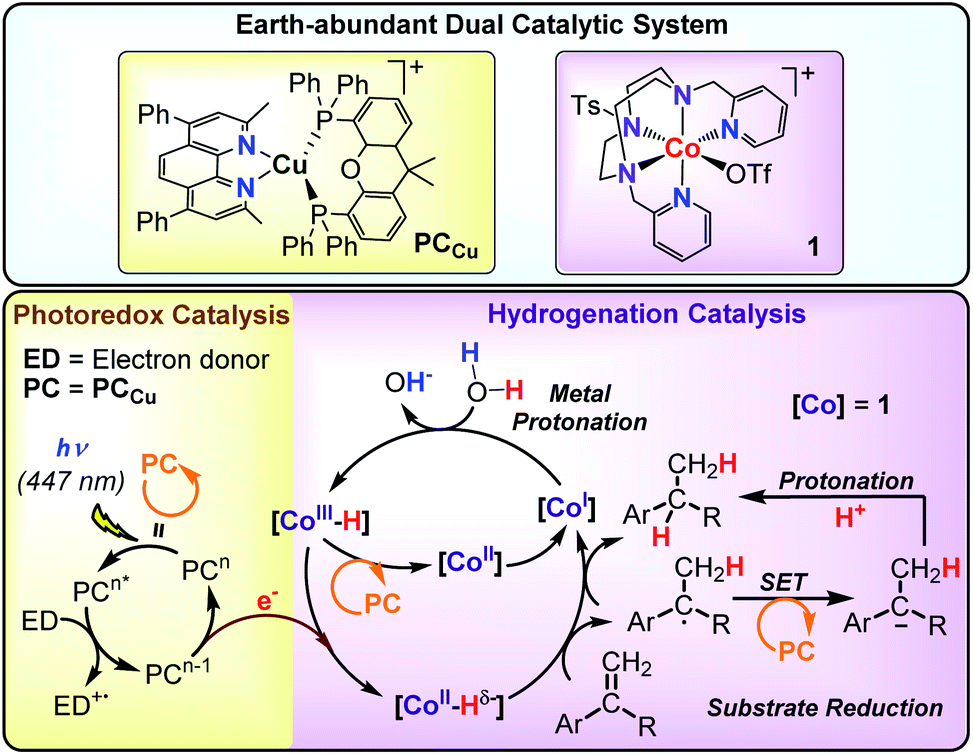 | ||
| Scheme 2 Earth-abundant dual catalytic system for the photoreduction of aromatic olefins. | ||
Results and discussion
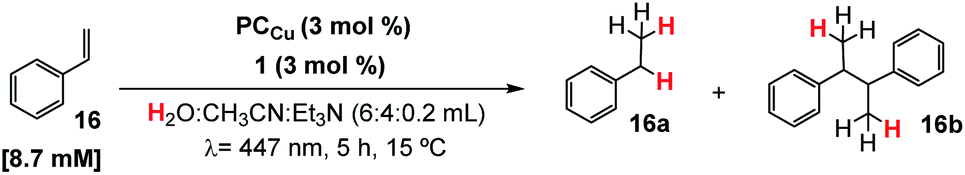
Based on previous studies,29,30 we hypothesized that the putative [Co(H)(Py2Tstacn)]+ intermediate formed under photochemical conditions may engage in the reduction of more challenging substrates, such as styrene (16). The irradiation (λ = 447 ± 20 nm ; T = 35 °C) of a solution containing styrene (16, 16.5 mM), complex 1 (1 mol%) as a reduction catalyst, PCCu (1.5 mol%) as a photoredox catalyst, and Et3N (0.2 mL, 8.5 equiv.) as a sacrificial electron donor in a H2O:CH3CN (6:4 mL) solvent mixture, produced ethylbenzene (16a) in good yield (67%) (Table S.8, entry 1†). The reduction of styrene is accompanied by small amounts of styrene dimerization (butane-2,3-diyldibenzene, 16b, 11% yield). The detection of this homocoupling product suggests the formation of benzylic radicals during the reaction, whose mechanistic implications will be discussed later (see the Mechanistic studies section).
(I) Screening of conditions and the catalytic system
The formation of homocoupling product 16b was significantly minimized (4%) by increasing the catalyst and photoredox catalyst loadings (from 1.5 to 3 mol%) and reducing the styrene concentration (from 16.5 to 8.7 mM), yielding ethylbenzene (16a) in 86% yield. Further refinement of the reaction conditions was achieved by adjusting the reaction temperature. It is worth noticing that the reaction yield and selectivity are sensitive to the temperature. At 35 °C or higher, the yield of the reduced product drops in favour of the reduced homocoupling product. Therefore, the reaction temperature should be effectively controlled to prevent warming by light irradiation. Despite the common LED intensity (∼2.1 W at 447 nm) used, the produced heat is enough to warm up the reactions to 45 °C if the temperature is not controlled.We have used an in-house developed parallel photoreactor to precisely control the temperature and light intensity (see the ESI for further details†).29,54 At 25 °C, the formation of the homocoupling product was only 3% (Table 1, entry 4) and negligible at 15 °C (Table 1, entry 5 and Table S8†). At lower temperatures, the homocoupling product was detected only in traces. We rationalized that the reduction of the selectivity at higher temperature is most likely due to accumulation of benzylic radicals, which then favour dimerization. Under optimized conditions, the hydrogenation of styrene yields 16a in 91% yield without detection of dimeric products (Table 1, entry 5).
|
|
|||
|---|---|---|---|
| Entry | Deviation from conditions | % 16a | % 16b |
|
a Conditions: 1 (mol%), PC (mol%), and 16 (mM) as indicated in the table in H2O:CH3CN:Et3N (6:4:0.2 mL), irradiation at λ = 447 nm for 5 h at 35, 25 or 15 °C under N2. Yields were determined by GC analysis after workup of the reaction and relative to a calibrated internal standard. Values are average of triplicates. [Ni(OTf)(Py2Tstacn)](OTf) (1Ni). See the ESI for the synthesis and characterization of metal complexes.
|
|||
| 1 | 16.5 mM 16, 35 °C, 1 (1 mol%), and PCCu (1.5 mol%) | 67 | 11 |
| 2 | 16.5 mM 16 and 35 °C | 81 | 6 |
| 3 | 35 °C | 86 | 4 |
| 4 | 25 °C | 90 | 3 |
| 5 | None | 91 | n.d. |
| 6 | No light | n.d. | n.d. |
| 7 | No Et3N | n.d. | n.d. |
| 8 | No PCCu | n.d. | n.d. |
| 9 | No 1 | n.d. | n.d. |
| 10 | Co(OTf)2(MeCN)2 instead of 1 | n.d. | n.d. |
| 11 | [Co(OTf)2(MeCN)2 + Py2Tstacn] instead of 1 | 91 | n.d. |
| 12 | IPCCu instead of PCCu | 21 | n.d. |
| 13 | SO3PCCu instead of PCCu | 78 | n.d. |
| 14 | HPCIr instead of PCCu | 11 | 8 |
| 15 | NMe2PCIr instead of PCCu | 67 | n.d. |
| 16 | CO2HPCIr instead of PCCu | 47 | 8 |
| 17 | 1Ni instead of 1 | n.d. | n.d. |
Control experiments determined that all components of the photocatalytic system (light, the cobalt catalyst, photoredox catalyst and electron donor) are necessary for the formation of 16a or 16b. In the absence of light, 1, PCCu, or Et3N (Table 1, entries 6–9), neither 16a nor 16b was observed. To discard the formation of benzylic radicals from a direct single electron transfer (SET) reduction from the reduced photoredox catalyst ([Cu(bathocuproine)(xantphos)], [PCCu]0) to styrene, we performed photocatalytic studies in the absence of complex 1. Even when the reaction was performed at 55 °C using 32 mM 16 and 10 mol% PCCu, no dimeric products were observed, discarding direct SET from [PCCu]0 to styrene (Table S.9†). Indeed, [PCCu]0 is not reductive enough to transfer an electron to 16 (E(PCCuI/0) = −1.60 and Ered = −2.31 V vs. SCE for 16). Additionally, no reduced products were detected when 1 was replaced by cobalt salts such as the starting cobalt(II) triflate (Co(OTf)2(MeCN)2, Table 1 entry 10). We found practically that the reduction yield of 16 was equivalent when complex 1 was synthesized in situ by adding the Py2Tstacn ligand with an equimolar amount of Co(OTf)2(MeCN)2 (Table 1 entry 11) under catalytic conditions.
|
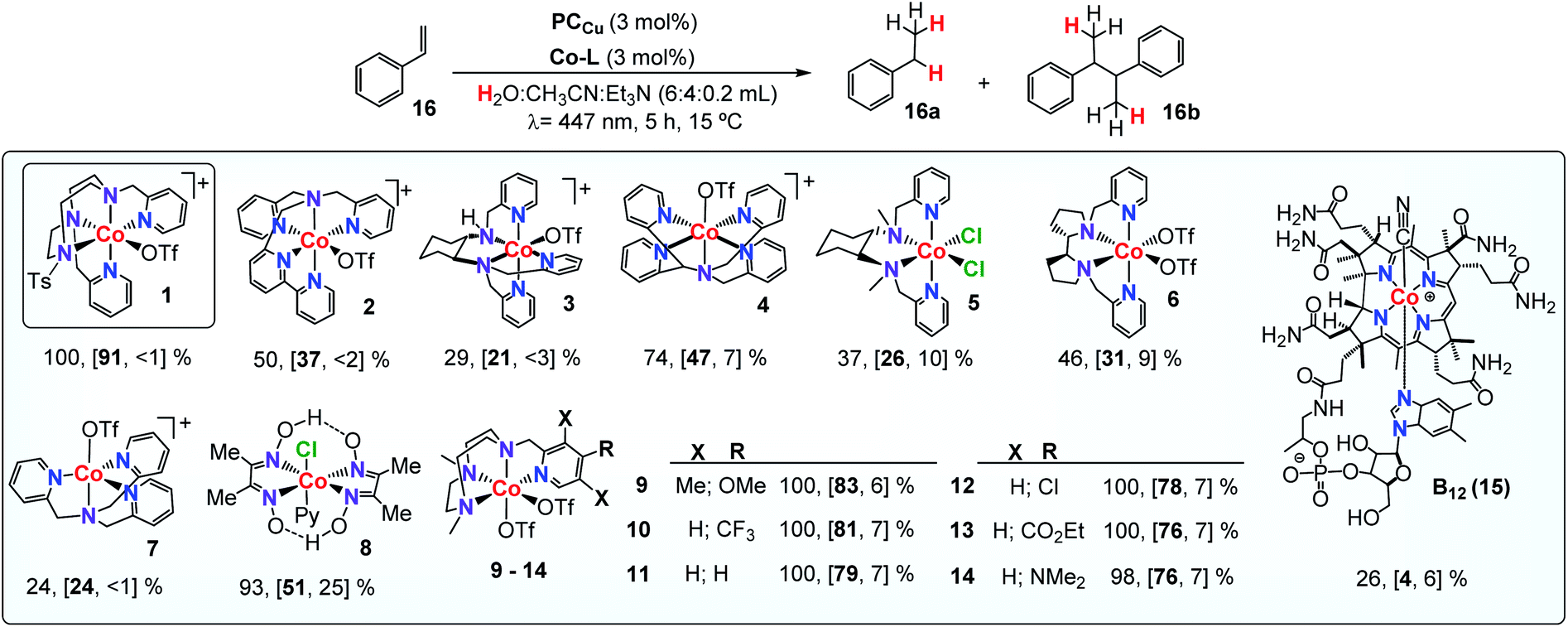
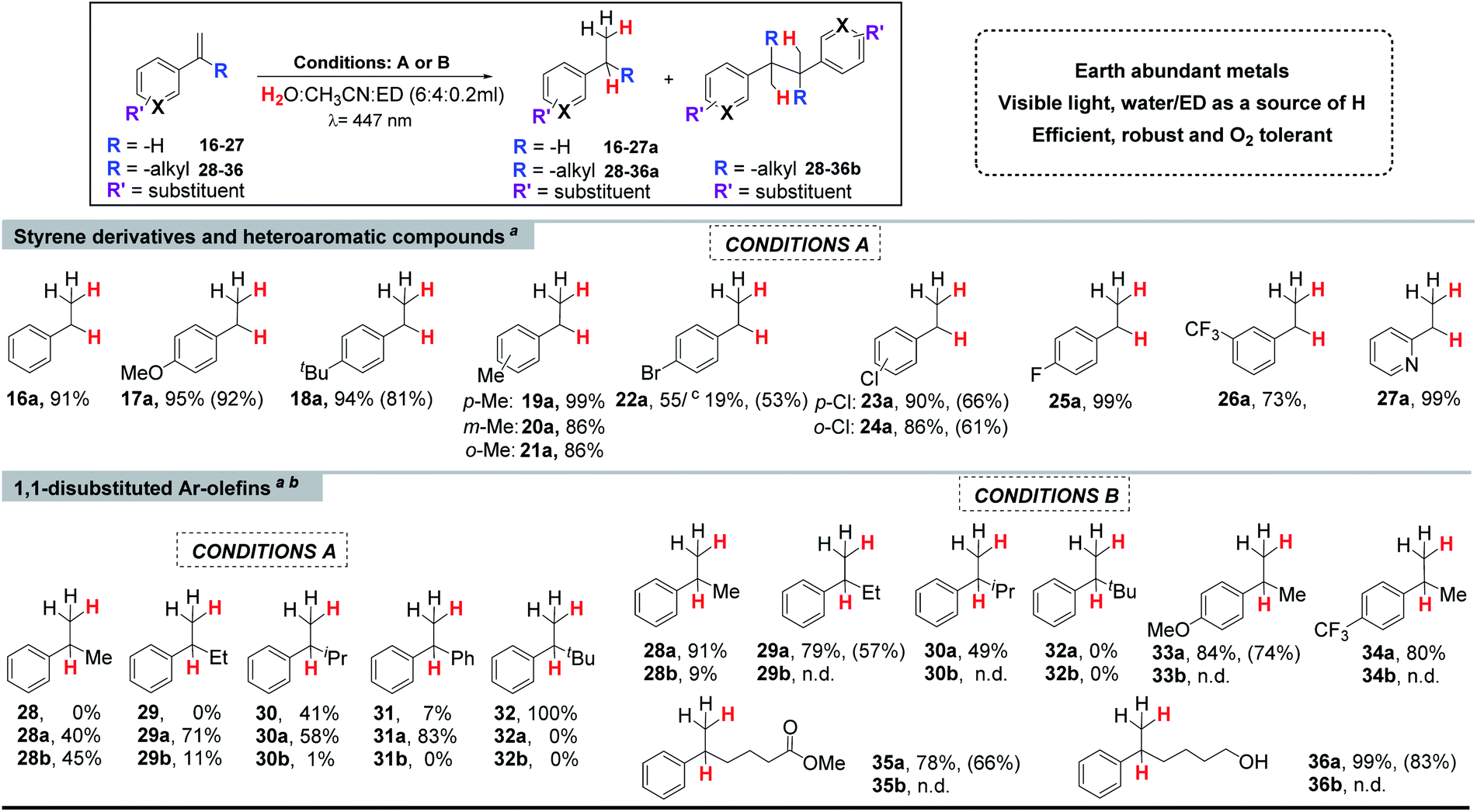
a Selected WR complexes: 1: [CoII(OTf)(Py2Tstacn)](OTf), 2:[CoII(OTf)(DPA-Bpy)](OTf), 3: [CoII(OTf)(N4Py)](OTf), 4: [CoII(OTf)(H-CDPy3)](OTf), 5:[CoII(OTf)2(PDP)], 6: [CoII(OTf)2(TPA)], 7: [CoII(Cl)2(BpcMe)], 8: [CoIII(Cl)(Py)(Glioxim)], 9: [CoII(OTf)2(Me,OMePyMe2tacn)], 10: [CoII(OTf)2(H,CF3PyMe2tacn)], 11: [CoII(OTf)2(H,HPyMe2tacn)], 12: [CoII(OTf)2(H,ClPyMe2tacn)], 13: [CoII(OTf)2(H,CO2EtPyMe2tacn)], 14: [CoII(OTf)2(H,NMe2PyMe2tacn)] and 15: vitamin B12. Conditions A: Co-Cat (261 μM, 3 mol%), PCCu (261 μM, 3 mol%), and 16 (17.4 μmol, 8.7 mM) in H2O:CH3CN:Et3N (6:4:0.2 mL), irradiation (447 nm) for 5 h at 15 °C under N2. Yields were determined by GC analysis after workup relative to a calibrated internal standard. Values were average of triplicates and correspond to conversion and [16a and 16b yield].
|
|---|
|
Although a correlation between molecular properties of the cobalt complex and reaction yield is not straightforward, we noticed that complexes with the tacn ligand scaffold are among the best catalysts. For instance, the lowest yield was obtained by complex 7, which has also the lowest reduction potential in Table 2, whereas the highest yield was obtained by complex 1 (EII/I = −0.97 and −1.15 V vs. SCE, respectively, Fig. S.26†). Finally, replacing the cobalt centre by nickel in complexes 1, 2 and 6 did not yield any reduced product.
Co complexes with aminopyridyl-tacn derived ligands [CoII(OTf)2(Y,XPyMetacn)] (series 9–14Y,XPyMetacn = 1-[(4-X-3,5-Y-2-pyridyl)methyl]-4,7-dimethyl-1,4,7-triazacyclononane) have been reported among the fastest catalysts for H2 production with TOF, TON, and quantum efficiencies as high as 52000 h−1, 9000, and 9.7% ± 1.0 for complex 13.55 In this regard, the photocatalytic reduction of 16 was accompanied by the formation of H2 as a side-product, in analogy to ketone and aldehyde reduction reactivity.29 The formation of H2 during the styrene reduction was monitored using pressure sensors29,56 and quantified by analysis of the reaction headspace using Gas Chromatography-thermal conductivity detector (GC-TCD, see the ESI for further details†). As expected, the selectivity of the studied Co complexes for the substrate vs. water reduction (Sel16a/H2 calculated as n(16a)/n(H2)) strongly depends on the ligand scaffold. Under optimized conditions, complex 1 is also the most active catalyst for H2 evolution with a Sel16a/H2 of 1.04 (Table 3 and Fig. S.30†). Nonetheless, considering the concentrations of water and 16 in catalysis (ratio H2O/16 ∼ 19100), the normalized selectivity for the olefin reduction is about 20000. Reducing the water in the reaction decreased the formation of H2 at the expense of diminishing the TON of olefin reduction.
|
a Conditions A: 1 (3 mol%), PCCu (3 mol%), and the substrate (8.7 mM) in H2O:CH3CN:Et3N (6:4:0.2 mL), irradiated for 5 h (447 nm) at 15 °C under N2.
b Conditions B: 1 (6 mol%), PCCu (6 mol%), and the substrate (4.4 mM) in H2O:CH3CN:iPr2EtN (6:4:0.2 mL), irradiated for 24 h (447 nm) at −3 °C under N2. Yields after workup (average of triplicates) were determined by GC analysis relative to a calibrated internal standard. Isolated yields between parentheses (average of 16 parallel reactions).
c Yield of the reduced dehalogenated product.
|
|---|
|
The produced H2 was lower for the non-tacn ligand-based catalysts (2–8), providing an excellent selectivity for 16versus water reduction and also the yields (Sel16a/H2 = 5.4–9.0, Fig. S.30†). For tetradentate tacn-based complexes 9–14, increasing the electron donating character of the ligand disfavours the H2 evolution, complex 10 being the most efficient for H2 evolution in the presence of the alkene (Sel16a/H2 = 0.8 for 10, Fig. S.31†).30 The 15-fold selectivity difference between 9 and 10 (p-MeO- versus p-CF3-) illustrates the ligand control in the selective olefin/water reduction.
(II) Scope of the reaction
With the optimal conditions in hand, we examined the scope of aromatic olefins.Such benzylic radicals could be formed by a HAT reaction from the [Co–H] intermediate to the less hindered carbon of the olefin, in agreement with the previously observed selectivity for M–H species.59–61 These results are also in agreement with recent reactivity reported on the reduction and homo- and heterocoupling of alkenes using silanes as reducing agents;62,63 and on the reactivity of putative [Co–H] species with olefins by J. Norton and co-workers,64,65 as well as the recent studies of M Kojima, S. Matsunaga and co-workers.46 Nevertheless, it is well-known that cobalt hydride complexes can undergo HAT to alkenes, and it was recently shown that it can be photo-induced.35,66
We noted that the yield of the reductive homocoupling product decreased with the alkyl substituent size at the olefin (from –Me to iPr), suggesting that the steric effect of the substituent influences the product selectivity, as expected for radical couplings. Moreover, the conversion of the –Me and –Et substituted alkenes (28and 29) was quantitative, but not for the one with the iPr group (30). With the aim to give an explanation for the lack of reactivity, we calculated and compared first the redox potentials of the olefins. As expected, the one electron reduction potential of the studied olefins is very negative (see Fig. S.26 and S.48†). However, the reaction yields are excellent for para-substituted arenes with a methoxide 17 and a tert-butyl group 18, despite having E0/−I1/2 redox potentials of −2.50 and −2.45 V vs. SCE, respectively. These redox potentials are more negative than that calculated for the 1,1′-disubstituted Ph, iPr alkenes (30) (−2.40 V, V vs. SCE), suggesting that the lower reactivity for 30 could be related to steric effects. Indeed, for the bulkier tBu-substituted aryl-alkyl olefin (32), there was no conversion. This agrees with the HAT mechanism since it is very sensitive to steric effects.67
On the other hand, the catalytic system is inefficient towards the reduction of 1,2-disubstituted styrene derivatives (37–41), yielding the trans–cis isomerization reaction as previously reported,47–49 and only a trace amount of the reduced product was observed (Table S.16†). This is an indication of a triplet energy transfer from PCCu to the disubstituted alkene. Moreover, the isomerization also occurs without the presence of the Co(II) complex 1. However, we can reasonably propose that the energy transfer (ET) path is minor to olefins because the reductive quenching of PCCu considering the concentrations is 10-fold larger by Et3N than by our model olefin 16. Moreover, when increasing the amount of Et3N added to the reaction, the reduction of 16 was also accelerated. If the ET mechanism was the productive path for the reduction of 16, an increase in the Et3N concentration should have decreased the production rate of the reduced product; however, the opposite effect was observed (Fig. S.36†).
It is worth noting that in the absence of the cobalt catalyst, neither olefin reduction nor homocoupling products were detected for the 1,1-disubstituted alkenes studied (28–31, Table S.15†). These results agree with the low redox potentials of 28–31 (Ered −2.36, −2.37, −2.40 and −2.29 V vs. SCE, respectively, Fig. S.26 and S.48†), that are ≥690 mV more negative than the [PCCu]0 reduction redox potential (E(PCCuI/0) = −1.60 V vs. SCE). This supports that for the studied olefins, the generation of benzylic radicals by single electron transfer (SET) from [PCCu]0 is not feasible.
At this point, we hypothesized that the selectivity towards olefin reduction could be improved by decreasing the temperature of the reaction. Such a decrease should have an impact on the rate of formation of [Co–H] species and then the following formation of benzylic radical species. Preventing the radical accumulation, thus, avoids the formation of homocoupling products since the reaction is second order with respect to the concentration of radicals (eqn (1) and (2), where vrad and krad are the rate and the formation rate constant of the benzylic radical, respectively; [R] and [PC−] are the concentration of the substrate and reduced photoredox catalyst, and vdim and kdim are the rate and the formation rate constant of the dimeric product, respectively). Consequently, at higher temperatures (>35 °C), the yield of the reduced product drops in favour of the homocoupling product. In addition, small quantities of trimers or higher order coupling products can be also detected by GC-MS, indicating a higher formation of radicals. In contrast, at low temperatures (15 to −3 °C, as specified per case), the dimeric products are negligible (Fig. S.32†).
| vrad = krad[R] × [PCn−1] | (1) |
| vdim = kdim[R]2 | (2) |
Further optimization showed that a bulkier electron donor such as diisopropylethylamine (DIPEA) favours the selectivity towards the reduced product (Tables S.8†), since it avoids formation of by-products between the electron donor and the substrate due to steric hindrance.68 In addition, a finer tuning of the concentration of the substrate, catalyst 1 and photoredox catalyst PCCu (Tables S.11 to S.13†) allowed establishing optimal reaction conditions. Thus, by increasing the loading of 1 and PCCu (6 mol%), reducing the substrate concentration (4.4 mM), and using DIPEA at −3 °C, we achieved excellent selectivity for the reduction of 1,1-disubstituted aromatic alkenes (Table S.13† entry 5 and Table 1 conditions B).
:CH3CN mixture. We observed nearly quantitative olefin deuteration (see Fig. S.50–S.65†). Analytics showed the incorporation of one deuterium atom at each carbon atom of the double bond (α and β) (Scheme 3). Moreover, D-labelling studies of α-methylstyrene (28) under catalytic conditions that favour the reduced homocoupling product (high temperature and concentration of the substrate) showed the incorporation of only two deuterium atoms – each one at a different methyl group of the coupling product (Fig. S.56 and S.57†).
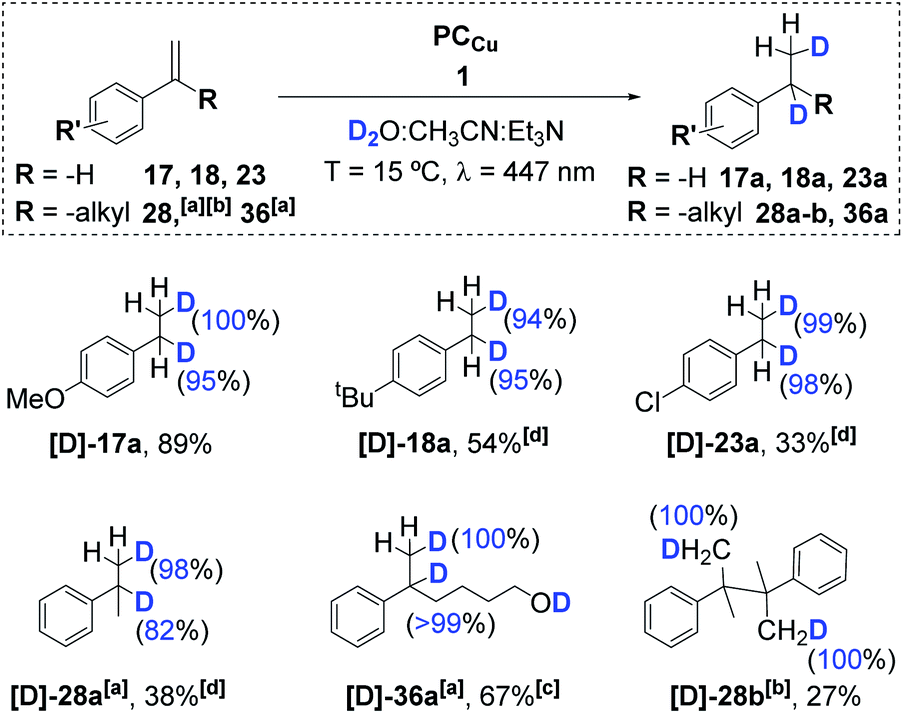 | ||
| Scheme 3 Deuterium labelling studies of aromatic olefins (17, 18, 23, 28 and 36). Conditions: 1 (3 mol%), PCCu (3 mol%), the substrate (8.7 mM) in H2O:CH3CN:Et3N (6:4:0.2 mL) irradiated for 5 h (447 nm) at 15 °C under N2. [a] Conditions: 1 (6 mol%), PCCu (6 mol%), and the substrate (4.4 mM) in D2O:CH3CN:iPr2EtN (6:4:0.2 mL), irradiated (447 nm) for 24 h at −3 °C under N2. [b] Conditions [a] modifying [Subs.] to 16 mM and 5 h at 30 °C. [c] NMR yield. [d] Low isolated yields were obtained due to the high volatility of the products. Isolated yields (average of 16 reactions). D-insertion analysed by NMR. | ||
This agrees with the formation of M-deuteride species ([Co–D]) that are involved in a deuterium atom transfer (DAT) mechanism with the olefin, forming the corresponding benzylic radical, which dimerizes or ends up reduced and deuterated, affording the deuterated product. Moreover, we did not observe double deuteration at the same carbon nor H/D scrambling. Therefore, if organometallic cobalt species are formed after the insertion of Co–D/H in the olefin, the reversibility of the reaction via beta-hydride elimination should be negligible. Thus, it is more likely that benzylic radicals are directly formed via DAT/HAT from Co–D/H, supported by the observation of benzylic radical species. Nevertheless, an irreversible metal hydride insertion cannot be fully discarded.
(III) Mechanistic studies
To gain insight into the nature of the active species and the mechanism, we performed kinetic and reactivity studies, including D-labelling and radical clock experiments (Fig. 1).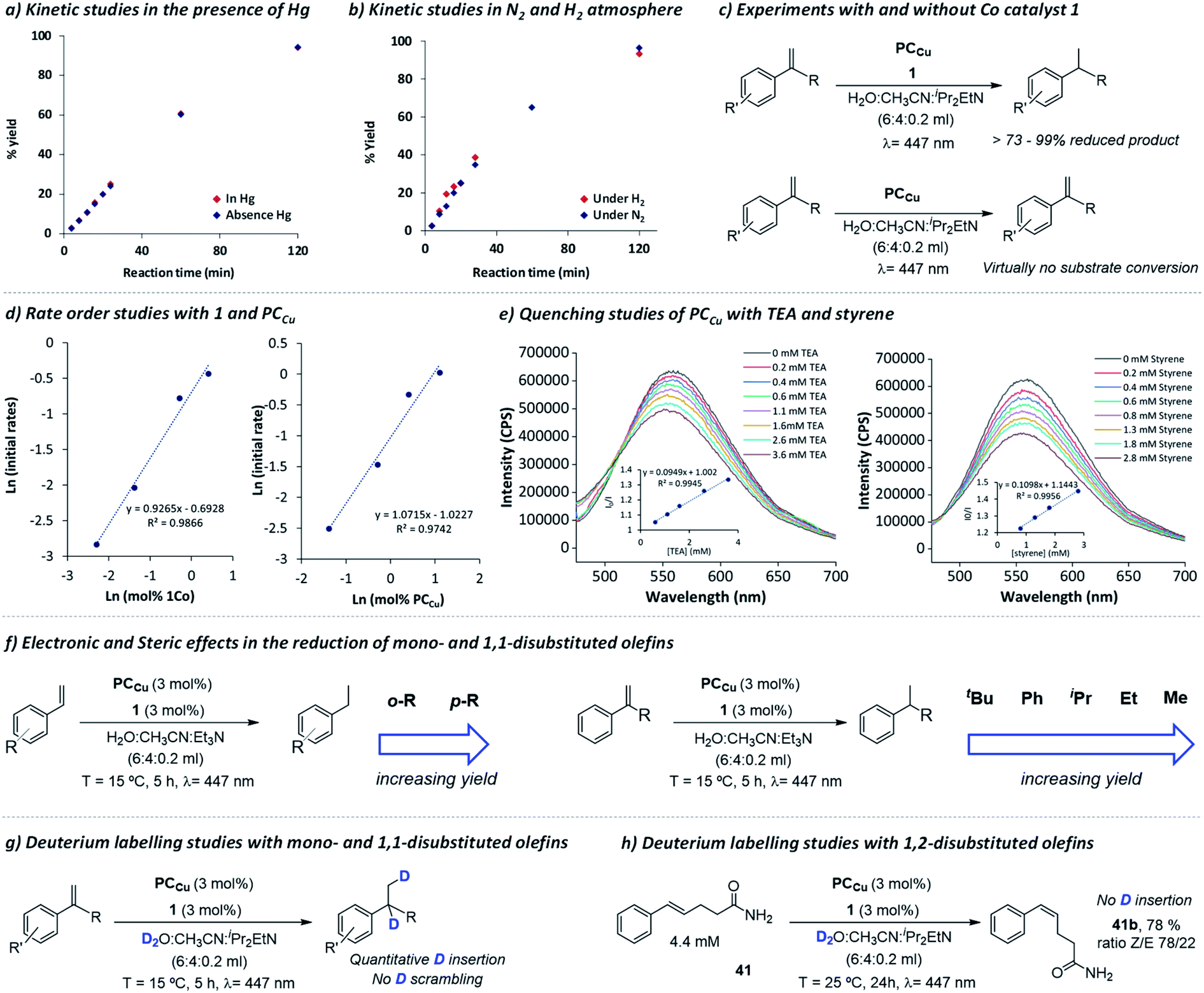 | ||
| Fig. 1 Mechanistic studies. Formation of 16a in (a) the absence (red diamonds) and presence of Hg(0) (>2000 eq.) (blue diamonds); and in (b) H2 (red diamonds) or N2 (blue diamonds) atmosphere. (c) Experiments with and without complex 1. (d) Reaction rate kinetics regarding complex 1 and PCCu. (e) Reductive quenching studies of PCCu with TEA and styrene. (f) Steric effects in alkene reduction. Deuterium labelling studies of mono- and 1,1-disubstituted olefins (g) and 1,2-disubstituted olefin 41 (h, Z/E ratio analysed by NMR). See Experimental section 3 for the procedure details.† | ||
First, kinetic studies in the presence of mercury showed that the photocatalytic activity is not inhibited, even in the presence of a large amount of mercury (>2000 eq. vs.1, Fig. 1a). This suggests that the catalysis is not related to metal (0) nanoparticles, and thus, agrees with the homogeneous nature of the active species. We also studied the influence on the reduction rate for 16 in both H2 and N2 atmospheres (Fig. 1b). The kinetic traces were identical regardless of the atmosphere used, indicating that the source of hydrogen in the reduced products does not come from the H2 generated under photocatalytic conditions. Indeed, both H2 evolution and olefin reduction are competing pathways that share a common intermediate, since with catalyst 1, the H2 produced decreased at higher concentrations of styrene (Fig. S.33†).
By initial reaction rate analyses, we found that both 1 and PCCu have a first-order reaction (Fig. 1d, S.34 and S.35†), in agreement with the bimolecular reduction mechanism of the cobalt catalyst by PCCu. On the other hand, the reaction order found in olefin is close to zero (Fig. S.33†). Quenching studies with the copper photoredox catalyst (PCCu) in the presence of TEA and styrene as a model substrate show similar quenching constants (KSV = 94.9 M−1 and 109.8 M−1, respectively). However, since the concentration of TEA under catalytic conditions is 10 times higher than that of styrene, we can consider the energy transfer pathway to the organic substrate to be negligible (Fig. 1e).
As introduced in the previous section, the exchange of H2O by D2O led to the formation of deuterated olefins with exclusive deuteration on the carbons of the olefin, and only two deuterium atoms were introduced one on each of the olefinic carbon atoms (Scheme 3 and ESI section 13† for NMR analyses). The absence of scrambling and double deuterium insertions indicates that regardless of the mechanistic path, the reaction is not reversible. On the other hand, when we employed 1,2 disubstituted olefin 41, we did not observe deuterium insertion (Fig. S.62 and S.63†), indicating that although isomerization takes place, it is most likely to proceed through a photochemically generated exited state without the intervention of [Co–H] insertion–elimination steps.50 As introduced above, we discard the energy transfer as a significant path responsible for the reduction of olefins since the increase of the Et3N concentration increased the reaction rate, despite both showing a similar quenching rate of the photoredox catalyst.
Experiments with radical clocks were performed to evaluate the formation of benzyl radicals via a HAT mechanism as already suggested by the formation of dimeric products. Olefins containing a 2-aryl-cyclopropyl moiety at the α-position are commonly used as probes for the formation of radicals in organic transformations.50,73 These radical clocks are very convenient because if a radical is generated in the benzylic position, it will trigger the cyclopropane ring opening with a high rate constant. For the (1-(2-phenylcyclopropyl)vinyl)benzene (42), cyclopropane ring opening is about 108 s−1.74,75 Moreover, the presence of an aromatic group at the olefin is necessary to stabilize the benzylic radical after the cyclopropane ring-opening. Without the stabilizing group, the resulting product from the cyclopropane ring-opening is endergonic and could yield wrong conclusions (Fig. S.49†).74,75 Under catalytic conditions, the reduction of 42 only affords the ring-opening product (42b) without detection of 42a (Scheme 4).
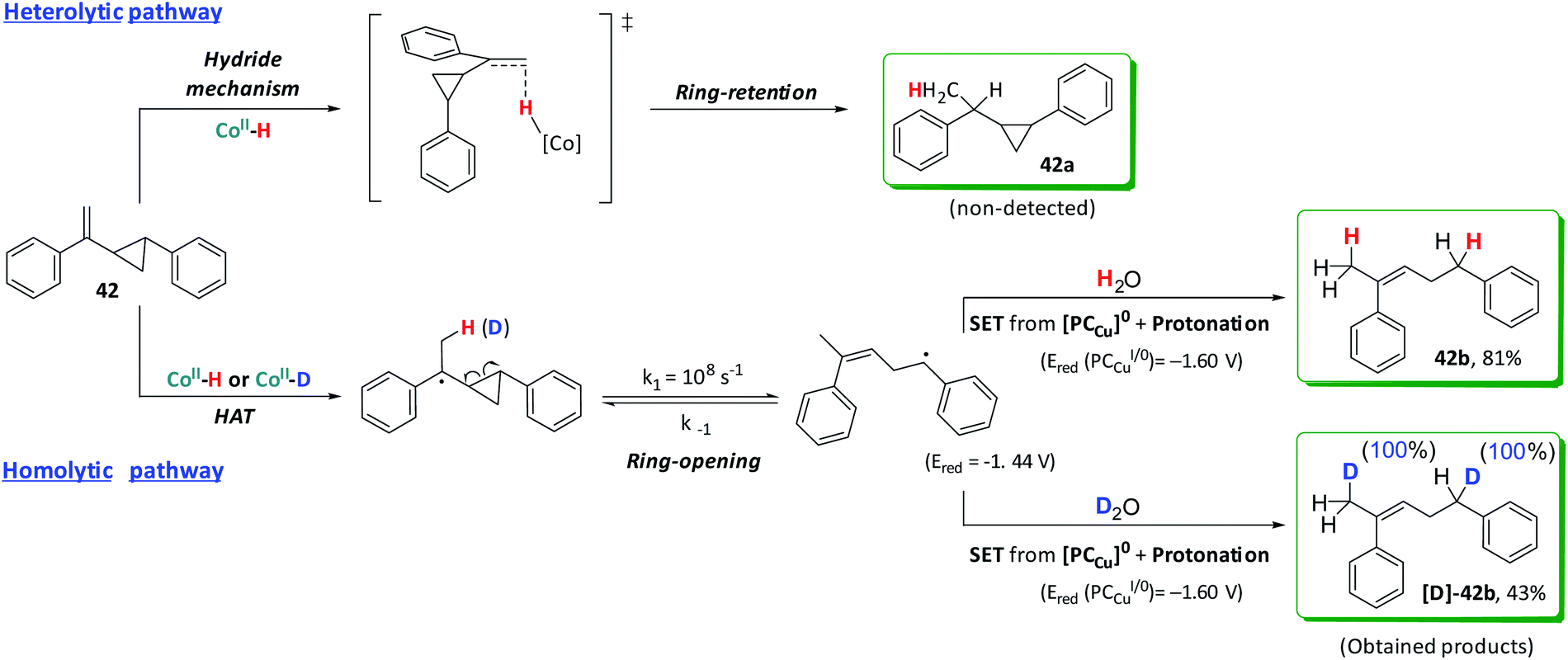 | ||
| Scheme 4 Two possible reduction pathways for (1-(2-phenylcyclopropyl)vinyl)benzene (42). | ||
When the reaction was performed in D2O, quantitative D-insertion at the β and benzylic positions was observed ([D]-42b, see Fig. S.64–S.70†). With the lack of the cobalt catalyst 1, no ring-opening products were detected, fully recovering the substrate after irradiation. Both 42b and [D]-42b can be formed via initial HAT (or DAT) from [Co–H] (or [Co–D]) followed by ring-opening and rearrangement to the most stable radical (Scheme 4).
(IV) Mechanistic discussion
Mechanisms in Scheme 5 were proposed considering the collection of experimental results. It is well-established that light irradiation (447 nm) of [Cu(bathocuproine)(xantphos)]+ ([PCCu]+) leads to the formation of the *[PCCu]+ excited state, which is reductively quenched in the presence of an excess of aliphatic amine (Et3N or iPr2EtN was used as quenchers in our study).52 The reaction then affords the one-electron reduced [PCCu]0 species (E(PCCuI/0) = −1.60 V vs. SCE).76,77 The reduction potentials of the studied olefins are in the range of −2.1 to −2.6 V vs. SCE50 (Fig. S.48† for the theoretical values). Therefore, based on the thermodynamics of the process, the direct reduction of the studied olefins by the singly reduced species [PCCu]0 could be discarded (ΔG of the reaction is endergonic by 12 to 23 kcal mol−1 depending on the olefin).29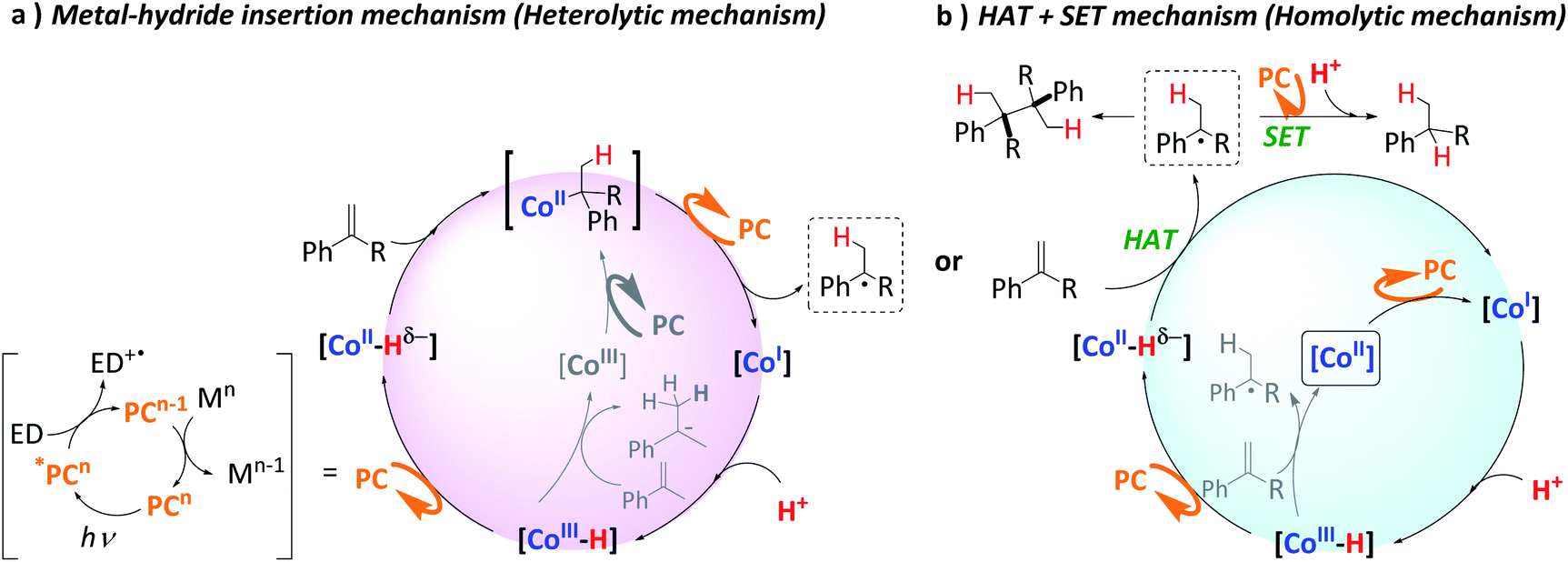 | ||
| Scheme 5 Possible mechanistic scenarios for the reduction of aromatic olefins by the light-driven dual-copper–cobalt catalytic system in aqueous media. The highlighted [CoII] intermediate indicates the beginning of the catalytic cycle. Acronyms stand for PC = photoredox catalyst; ED = electron donor; SET = single electron transfer and HAT = hydrogen atom transfer. | ||
This is supported by the lack of reduced, dimerized or ring-opening products formed when radical clock 42 (E(420/−) = −2.36 V vs. SCE) was tested in the absence of complex 1. However, [PCCu]0 is reductive enough to reduce complex 1 (E(CoII/I) = −1.15 V vs. SCE) affording CoI species.53 We previously studied the formation of similar CoI species under photo- and electrochemical conditions.55,78 These low-valent intermediates can be protonated by water to form a putative [CoIII–H]. We previously reported that the reduction of [CoIII–H] to [CoII–H] by [PCCu]0 is largely exergonic (−17.6 kcal mol−1),29,53 and therefore, we can reasonably consider a fast ET to yield [CoII–H]. Therefore, under such reducing photochemical conditions, [CoIII–H] should be easily converted into [CoII–H] species.29,55
On the other hand, it is interesting that the reaction order found in the olefin is close to zero (Fig. S.33†). The fact that the reaction rate does not depend on the alkene concentration is consistent with our previous studies on hydrogen evolution and the reduction of ketones and aldehydes.29,55 Thus, we interpreted this result as the formation of the [CoIII–H] complex as a rate-determining step, since the following reduction to [CoII–H] species and their subsequent HAT to the olefin are fast. Moreover, as discussed above, this [CoIII–H] is a common intermediate for both H2 evolution and olefin reduction.
The formation of homocoupling products and the isotopic D-labelling pattern observed, together with the radical clock experiments support that the reaction proceeds via the formation of benzylic radical species. As explained above, the most likely pathway to form the benzylic radicals is a HAT from [CoII–H] to the β carbon of the olefin, whereas a potential metal hydride insertion is less likely, but cannot be discarded.
Although it cannot be completely ruled out, since no doubly-deuterated terminal-carbons in the reduced products were detected, this indicates that neither β-hydride elimination nor reversible HAT occurs (Scheme 5a). Then, the fastest and most likely scenario is that benzylic radicals undergo a reduction (Etheorred = −1.44 to −1.69 V vs. SCE) by [PCCu]0 (E = −1.60 V vs. SCE), which after subsequent protonation gives the alkane (Scheme 5b). We discard the HAT from the oxidized amine because the deuteration of the benzylic position is virtually quantitative.
(V) Mechanistically designed ketone/olefin reduction selectivity
The postulated HAT mechanism for olefin reduction (Scheme 5b) implies that the selectivity could be modulated against functional groups that are reduced via a different mechanism such as SET. This is the case for our previously reported reduction of aromatic ketones using the dual catalytic system.29To test our hypothesis, we used styrene (16) and acetophenone (43) as competing substrates. We previously determined, that under equivalent photocatalytic conditions, 43 is reduced to 1-phenylethanol via a SET mechanism from [PCCu]0 yielding the ketyl radical anion (KRA) as an intermediate (Ered = −1.65 V vs. SCE).29,30 Then the generated KRA is trapped by [Co–H] to form the reduced product via HAT (Scheme 6). Therefore, it should be possible to differentiate the KRA formation via SET from the HAT mechanism of the olefin, designing rationally the photocatalytic conditions.
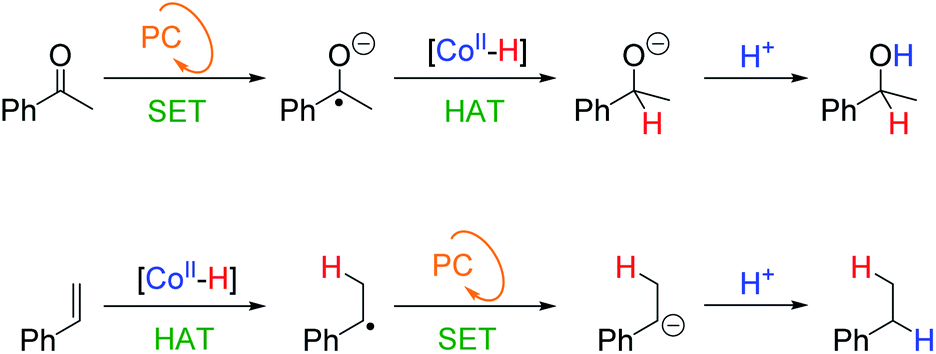 | ||
| Scheme 6 Different substrate-dependent mechanisms for the observed selectivity. KRA stands for the ketyl radical anion and PC for the photoredox cycle. | ||
This rationalization suggests that the counterintuitive idea of increasing the redox potential of the photoredox catalyst should make the catalytic system more selective. For instance, under more reducing conditions, a SET mechanism should be favoured, and therefore, the reduction of the ketone would be preferent, while the opposite would favour otherwise.
First, when similar catalytic conditions to those reported for the selective reduction of aromatic ketones vs. aliphatic aldehydes29 were used (1 (1 mol%), PCCu (1.5 mol%), total substrates (16 + 43, 16.5 mM, 1:1) in H2O:CH3CN:Et3N (6:4:0.2 mL) irradiated for 4 h at 25 °C under N2), acetophenone (43) was preferentially reduced in the presence of 16 (Sel43a/(16a+43a) = 65%, Fig. 2a, Table S.18 and Fig. S.37†). Then, we modified the reaction conditions to test the hypothesis of favouring either the SET to 43 or the HAT to 16. Under stronger reducing conditions, which favour the SET initial step, we sequentially increased the reduction potential, concentration of PC and the light irradiation intensity. Moreover, we also decreased the concentration of complex 1 to disfavour the HAT process. To our delight, an improvement in the selectivity was observed by using NMe2PCIr instead of PCCu (from 65 to 72% selectivity).
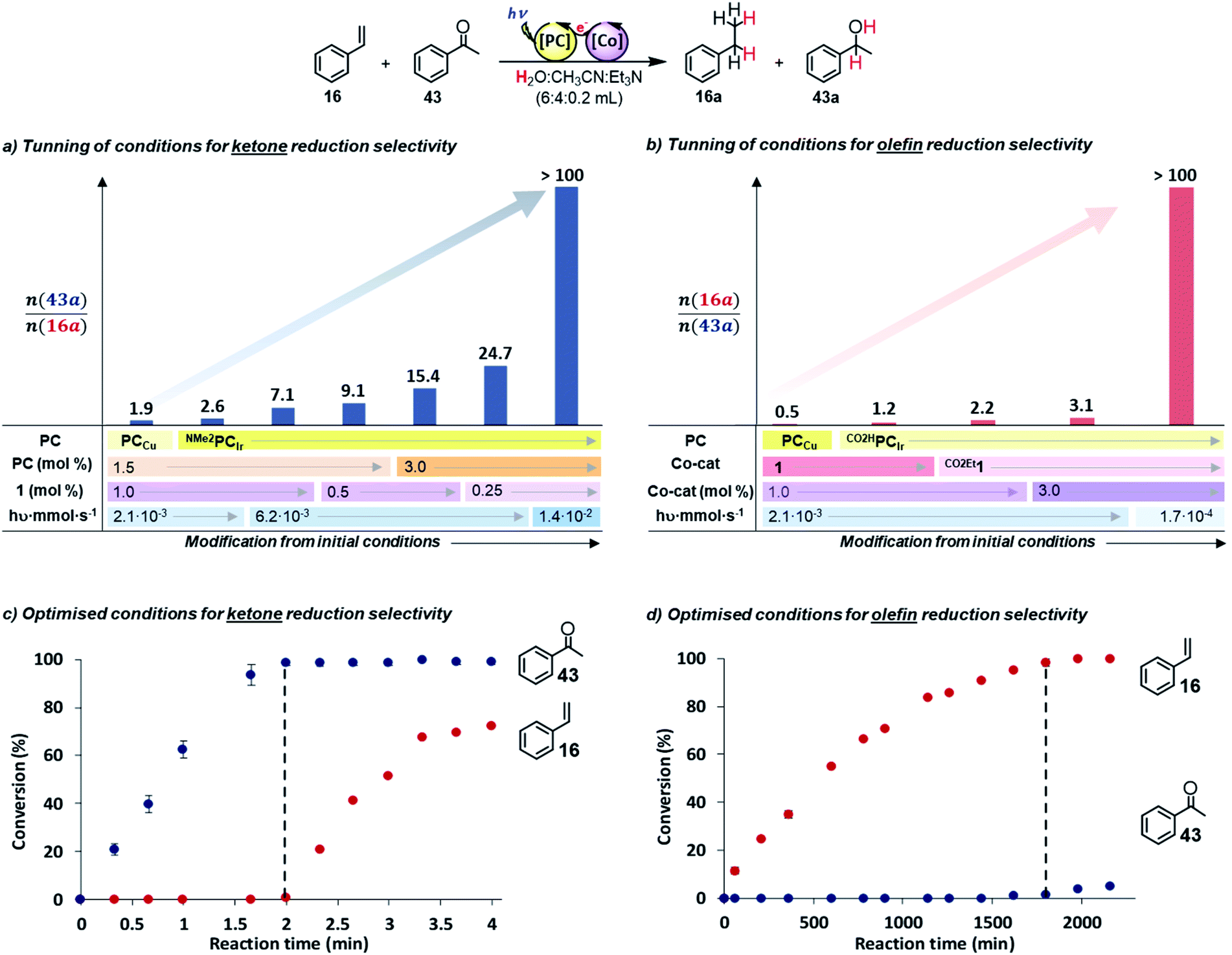 | ||
| Fig. 2 Competition studies between styrene (16) and acetophenone (43). (a and b) Optimization of the photocatalytic conditions for selective reduction of 43 and 16, respectively. Conditions: (c) 1 (0.25 mol%), NMe2PCIr (3 mol%), total substrate concentration (16 + 43, 16.5 mM, 1:1) in a H2O:CH3CN:Et3N (3:2:0.1 mL) mixture, irradiated at 447 nm (7 LED at 700 mA, 1.44 × 10−2 mmol hν s−1 of photons55) for 4 min at 25 °C under N2. (d) CO2Et1 (3 mol%), CO2HPCIr (1.5 mol%), total substrate concentration (16.5 mM) in a H2O:CH3CN:Et3N (3:2:0.1 mL) mixture, irradiated at 447 nm (1 LED at 50 mA 1.67 × 10−4 mmol hν s−1 of photons55) for 36 h (2160 min) at 25 °C under N2. The black dotted line indicates where substrates 16 (c) and 43 (d) start reacting. | ||
Further improvement was obtained by increasing the light intensity up to 6.2 × 10−3 mmol hν s−1vs. the normal 2.1 × 10−3 mmol hν s−1,55 which presumably increased the concentration of the reduced photoredox catalyst ([NMe2PCIr]0), and thus, accelerating the SET reaction (from 72 to 88% Table S.18 and Fig. S.38 and S.39†). In contrast, the effect of complex 1 concentration was minor (from 88 to 90%). As a result, 100% selectivity for ketone 43 reduction (in 2 min) was obtained in the presence of styrene 16 using NMe2PCIr (3 mol%, E1/2(IrIII/II) = −1.80 V vs. SCE), complex 1 (0.25 mol%, E(CoII/I) = −1.15 V vs. SCE) and irradiation at higher light intensity (1.4 × 10−2 mmol hν s−1) (Fig. 2a and c).
To favour the HAT, we modified the reaction conditions to make them less reducing. First, we used the less reductive photoredox catalyst CO2HPCIr (E1/2(IrIII/II) = −1.01 V vs. SCE), and the selectivity for styrene reduction was increased by 20% (Fig. 2b, Table S.19 and Fig. S.44†). Likewise, using a less reductive Co catalyst [(CoII(OTf)(H,CO2EtPy2Tstacn)] (CO2Et1, E(CoII/I) = −0.96 V vs. SCE), the selectivity also improved by 14%. Then, we increased the CO2Et1 concentration with the idea to increase [Co–H] and thus the HAT product (Sel16a/(43a+16a) = 76%, Fig. S.45 and S.46†).
Finally, lowering the light intensity (1.7 × 10−4 mmol hν·s−1), 16 was selectively reduced over 43 with 100% selectivity (Fig. 2d and Table S.19†). GC monitoring of the reactions showed that 16 was consumed and converted to 16a, whereas 43 remained virtually intact and vice versa (Fig. 2c and d).
As a summary of this section, the observed selectivity agrees with the existence of two competitive mechanistic scenarios involving: (i) SET + HAT for substrates than can be directly reduced such as acetophenone, and (ii) HAT for substrates with low reduction potentials that cannot be directly reduced by one electron from the PC such as aromatic olefins.
Conclusions
We developed a dual cobalt–copper catalytic system capable of reducing aromatic olefins under simple operational conditions, solely using H2O and an amine (Et3N or iPr2EtN) as the source of hydrides and visible light as the driving force. Our mechanistic studies based on reactivity and selectivity studies, catalyst design, deuterium-labelling and radical-clock experiments support a well-defined cobalt hydride as the intermediate responsible for the reductions, most likely through a HAT mechanism, discarding free radical diffusion as the main pathway. These results show that the selectivity of the metal hydrides in basic media can be controlled and directed to the reduction of organic functionalities. For this reason, we envision that other readily available H2O reduction catalysts could also be active in the reduction of other functional groups and more complex organic structures. These results pave the way for the development of selective organic reductions and solar-chemical generation using artificial catalytic systems that operate entirely with earth-abundant elements, using visible light as the driving force and H2O as a source of hydrides.Data availability
All the data is provided in the ESI.†Author contributions
AC identified the potential of olefin reduction at an early satge; CC performed most of the experiments as well as the DFT calculations, worked with AC and ICR on the synthesis of the photoredox catalysts, with AC on the optimisation studies, with DP and JA on the selectivity studies, wrote the manuscript together with JL and brought the project to completion. JL oversaw the project, discussed the results, and edited the manuscript. Moreover, all authors contributed with fruitful discussions to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the European Commission for the ERC-CG-2014-648304 (J. Ll.-F.) project. The Spanish Ministry of Science is acknowledged for a FPU fellowship to C. C. (FPU14/02550) and A. C. and a Juan de la Cierva contract to A. C. The Spanish Ministry of Science is acknowledged for projects (PID2019-110050RB-I00 and CTQ2016-80038-R) and the MCIN/AEI/10.13039/501100011033 (CEX2019-000925-S). We also thank Catexel for a generous gift of tritosyl-1,4,7-triazacyclononane. The financial support from ICIQ Foundation and CELLEX Foundation through the CELLEX-ICIQ high throughput experimentation platform is acknowledged. We also thank CERCA Programme (Generalitat de Catalunya) for financial support. We thank Dr Ferran Acuña-Parés for fruitful discussions.Notes and references
- J. Barber and P. D. Tran, J. R. Soc., Interface, 2013, 10, 20120984 CrossRef PubMed.
- R. E. Galian and J. Pérez-Prieto, Energy Environ. Sci., 2010, 3, 1488 RSC.
- C. R. Stephenson and T. P. Yoon, Acc. Chem. Res., 2016, 49, 2059–2060 CrossRef CAS PubMed.
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave and V. Artero, Photochem. Photobiol., 2011, 87, 946–964 CrossRef CAS PubMed.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- Z. Han and R. Eisenberg, Acc. Chem. Res., 2014, 47, 2537–2544 CrossRef CAS PubMed.
- V. S. Thoi, Y. Sun, J. R. Long and C. J. Chang, Chem. Soc. Rev., 2013, 42, 2388–2400 RSC.
- D. Z. Zee, T. Chantarojsiri, J. R. Long and C. J. Chang, Acc. Chem. Res., 2015, 48, 2027–2036 CrossRef CAS PubMed.
- N. Kaeffer, M. Chavarot-Kerlidou and V. Artero, Acc. Chem. Res., 2015, 48, 1286–1295 CrossRef CAS PubMed.
- N. Queyriaux, R. T. Jane, J. Massin, V. Artero and M. Chavarot-Kerlidou, Coord. Chem. Rev., 2015, 304–305, 3–19 CrossRef CAS PubMed.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. Kenis, C. A. Kerfeld, R. H. Morris, C. H. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- C. Costentin, M. Robert and J. M. Saveant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- S. Boddu, S. T. Nishanthi and K. Kailasam, in Visible Light-Active Photocatalysis, 2018, pp. 421–446, DOI:10.1002/9783527808175.ch15.
- F. Franco, S. Fernández and J. Lloret-Fillol, Curr. Opin. Electrochem., 2019, 15, 109–117 CrossRef CAS.
- G. Palmisano, V. Augugliaro, M. Pagliaro and L. Palmisano, Chem. Commun., 2007, 3425–3437, 10.1039/b700395c.
- S. Choudhury, J. O. Baeg, N. J. Park and R. K. Yadav, Angew. Chem., Int. Ed., 2012, 51, 11624–11628 CrossRef CAS PubMed.
- S. H. Lee, J. H. Kim and C. B. Park, Chem.–Eur. J., 2013, 19, 4392–4406 CrossRef CAS PubMed.
- M. Mifsud, S. Gargiulo, S. Iborra, I. W. Arends, F. Hollmann and A. Corma, Nat. Commun., 2014, 5, 3145 CrossRef PubMed.
- J. H. Kim, D. H. Nam and C. B. Park, Curr. Opin. Biotechnol., 2014, 28, 1–9 CrossRef CAS PubMed.
- J. Liu, J. Huang, H. Zhou and M. Antonietti, ACS Appl. Mater. Interfaces, 2014, 6, 8434–8440 CrossRef CAS PubMed.
- A. Bachmeier, B. J. Murphy and F. A. Armstrong, J. Am. Chem. Soc., 2014, 136, 12876–12879 CrossRef CAS PubMed.
- E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave and V. Artero, Photochem. Photobiol., 2011, 87, 946–964 CrossRef CAS PubMed.
- J. A. Macia-Agullo, A. Corma and H. Garcia, Chem.–Eur. J., 2015, 21, 10940–10959 CrossRef CAS PubMed.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- A. Call, C. Casadevall, F. Acuna-Pares, A. Casitas and J. Lloret-Fillol, Chem. Sci., 2017, 8, 4739–4749 RSC.
- A. Call and J. Lloret-Fillol, Chem. Commun., 2018, 54, 9643–9646 RSC.
- C. Casadevall, PhD thesis, Universitat Rovira i Virgili, 2019.
- J. Lloret-Fillol, A. Casitas, A. Call and C. Casadevall, Photocatalytic Reduction Process and catalytic composition used in the process, Spain Patent, Submitted as: PCT. Fundació privada Institut Català d’Investigació Química, Application No PCT/ES2017/070314, application date 10.05.2017.
- L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed.
- M. Trincado, J. Bösken and H. Grützmacher, Coord. Chem. Rev., 2021, 443, 213967 CrossRef CAS.
- W. Ai, R. Zhong, X. Liu and Q. Liu, Chem. Rev., 2019, 119, 2876–2953 CrossRef CAS PubMed.
- J. Wen, F. Wang and X. Zhang, Chem. Soc. Rev., 2021, 50, 3211–3237 RSC.
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 683–733 CrossRef CAS PubMed.
- A. J. Perkowski, W. You and D. A. Nicewicz, J. Am. Chem. Soc., 2015, 137, 7580–7583 CrossRef CAS PubMed.
- K. Imamura, Y. Okubo, T. Ito, A. Tanaka, K. Hashimoto and H. Kominami, RSC Adv., 2014, 4, 19883–19886 RSC.
- J. Li, J. Yang, F. Wen and C. Li, Chem. Commun., 2011, 47, 7080–7082 RSC.
- H. Yamataka, N. Seto, J. Ichihara, T. Hanafusa and S. Teratani, J. Chem. Soc., Chem. Commun., 1985, 788–789, 10.1039/c39850000788.
- H. Shimakoshi and Y. Hisaeda, ChemPlusChem, 2014, 79, 1250–1253 CrossRef CAS.
- T. Shiragami, C. Pac and S. Yanagida, J. Phys. Chem., 1990, 94, 504–506 CrossRef CAS.
- Z. C. Litman, Y. Wang, H. Zhao and J. F. Hartwig, Nature, 2018, 560, 355–359 CrossRef CAS PubMed.
- N. A. Larionova, J. M. Ondozabal and X. C. Cambeiro, Adv. Synth. Catal., 2021, 363, 558–564 CrossRef CAS.
- Y. Kamei, Y. Seino, Y. Yamaguchi, T. Yoshino, S. Maeda, M. Kojima and S. Matsunaga, Nat. Commun., 2021, 12, 966 CrossRef CAS PubMed.
- J. B. Metternich and R. Gilmour, J. Am. Chem. Soc., 2015, 137, 11254–11257 CrossRef CAS PubMed.
- J. Saltiel and G. S. Hammond, J. Am. Chem. Soc., 1963, 85, 2515–2516 CrossRef CAS.
- G. S. Hammond and J. Saltiel, J. Am. Chem. Soc., 1962, 84, 4983–4984 CrossRef CAS.
- M. R. Schreier, B. Pfund, X. Guo and O. S. Wenger, Chem. Sci., 2020, 11, 8582–8594 RSC.
- M. L. Czyz, M. S. Taylor, T. H. Horngren and A. Polyzos, ACS Catal., 2021, 5472–5480, DOI:10.1021/acscatal.1c01000.
- S. P. Luo, E. Mejia, A. Friedrich, A. Pazidis, H. Junge, A. E. Surkus, R. Jackstell, S. Denurra, S. Gladiali, S. Lochbrunner and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 419–423 CrossRef CAS PubMed.
- A. Call, Z. Codolà, F. Acuña-Parés and J. Lloret-Fillol, Chem.–Eur. J., 2014, 20, 6171–6183 CrossRef CAS PubMed.
- J. Lloret-Fillol, C. Casadevall, J. León, A. Call, A. Casitas, J. J. Pla, P. J. Hernández and X. F. Caldentey, Photoreactor, Submitted as, European Patent, Fundació privada Institut Català d’Investigació Química, Application No 17382313.9-1370, application date 31.05.2017 Search PubMed.
- A. Call, F. Franco, N. Kandoth, S. Fernández, M. González-Béjar, J. Pérez-Prieto, J. M. Luis and J. Lloret-Fillol, Chem. Sci., 2018, 9, 2609–2619 RSC.
- J. Lloret-Fillol, Z. Codolà, I. Garcia-Bosch, L. Gómez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807–813 CrossRef PubMed.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- The bond dissociation energy (BDE) is lower for aryl bromides than for other aryl halides (DFT calculated bond dissociation energies are as follows: BDEC–Br = 81.9 kcal mol−1 for substrate 22, BDEC–Cl = 93.8 kcal mol−1 for 23, and BDEC–F = 126.0 kcal mol−1 for 25).
- E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock, A. J. Miller and A. M. Appel, Chem. Rev., 2016, 116, 8655–8692 CrossRef CAS PubMed.
- Y. Hu, A. P. Shaw, D. P. Estes and J. R. Norton, Chem. Rev., 2016, 116, 8427–8462 CrossRef CAS PubMed.
- S. W. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Chem. Rev., 2016, 116, 8912–9000 CrossRef CAS PubMed.
- J. C. Lo, J. Gui, Y. Yabe, C. M. Pan and P. S. Baran, Nature, 2014, 516, 343–348 CrossRef CAS PubMed.
- J. C. Lo, Y. Yabe and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 1304–1307 CrossRef CAS PubMed.
- G. Li, A. Han, M. E. Pulling, D. P. Estes and J. R. Norton, J. Am. Chem. Soc., 2012, 134, 14662–14665 CrossRef CAS PubMed.
- D. C. Eisenberg and J. R. Norton, Isr. J. Chem., 1991, 31, 55 CrossRef CAS.
- S. Sang, T. Unruh, S. Demeshko, L. I. Domenianni, N. P. van Leest, P. Marquetand, F. Schneck, C. Würtele, F. J. de Zwart, B. de Bruin, L. González, P. Vöhringer and S. Schneider, Chem.–Eur. J., 2021, 27, 16978–16989 CrossRef CAS PubMed.
- G. Litwinienko and K. U. Ingold, Acc. Chem. Res., 2007, 40, 222–230 CrossRef CAS PubMed.
- H. Seo, M. H. Katcher and T. F. Jamison, Nat. Chem., 2017, 9, 453–456 CrossRef CAS PubMed.
- J.-W. Wang, K. Yamauchi, H.-H. Huang, J.-K. Sun, Z.-M. Luo, D.-C. Zhong, T.-B. Lu and K. Sakai, Angew. Chem., Int. Ed., 2019, 58, 10923–10927 CrossRef CAS PubMed.
- M. R. Friedfeld, H. Zhong, R. T. Ruck, M. Shevlin and P. J. Chirik, Science, 2018, 360, 888–893 CrossRef CAS PubMed.
- M. R. Friedfeld, M. Shevlin, J. M. Hoyt, S. W. Krska, M. T. Tudge and P. J. Chirik, Science, 2013, 342, 1076–1080 CrossRef CAS PubMed.
- W. Liu, B. Sahoo, K. Junge and M. Beller, Acc. Chem. Res., 2018, 51, 1858–1869 CrossRef CAS PubMed.
- J. P. Stevenson, W. F. Jackson and J. M. Tanko, J. Am. Chem. Soc., 2002, 124, 4271–4281 CrossRef CAS PubMed.
- R. Hollis, L. Hughes, V. W. Bowry and K. U. Ingold, J. Org. Chem., 1992, 57, 4284–4287 CrossRef CAS.
- J. Masnovi, E. G. Samsel and R. M. Bullock, J. Chem. Soc., Chem. Commun., 1989, 1044–1045, 10.1039/C39890001044.
- E. Mejía, S.-P. Luo, M. Karnahl, A. Friedrich, S. Tschierlei, A.-E. Surkus, H. Junge, S. Gladiali, S. Lochbrunner and M. Beller, Chem.–Eur. J., 2013, 19, 15972–15978 CrossRef PubMed.
- S. Fischer, D. Hollmann, S. Tschierlei, M. Karnahl, N. Rockstroh, E. Barsch, P. Schwarzbach, S.-P. Luo, H. Junge, M. Beller, S. Lochbrunner, R. Ludwig and A. Brückner, ACS Catal., 2014, 4, 1845–1849 CrossRef CAS.
- S. Fernández, F. Franco, C. Casadevall, V. Martin-Diaconescu, J. M. Luis and J. Lloret-Fillol, J. Am. Chem. Soc., 2020, 142, 120–133 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc06608k |
| This journal is © The Royal Society of Chemistry 2022 |