
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thiocarbonyl chemistry in polymer science
Nathaniel M.
Bingham
 a,
Zahra
Abousalman-Rezvani
b,
Kyle
Collins
a and
Peter J.
Roth
*a
a,
Zahra
Abousalman-Rezvani
b,
Kyle
Collins
a and
Peter J.
Roth
*a
aDepartment of Chemistry, School of Chemistry and Chemical Engineering, University of Surrey, Guildford, Surrey, GU2 7XH, UK. E-mail: p.roth@surrey.ac.uk
bInstitute of Pharmaceutical Sciences, Monash University, 339 Royal Parade, VIC 3052, Australia
First published on 4th May 2022
Abstract
Organised by reaction type, this review highlights the unique reactivity of thiocarbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S) groups with radicals, anions, nucleophiles, electrophiles, in pericyclic reactions, and in the presence of light. In the polymer chemistry arena, thiocarbonyl compounds have been used as monomers, polymerization catalysts, reversible and irreversible chain transfer agents, and in post-polymerization modification reactions. Past and ongoing applications are reviewed including iniferters, radical and cationic RAFT, switchable RAFT agents, cyclic RAFT agents, chain transfer, thiocarbonyl addition-ring-opening, CS radical and anionic polymerization, acyl substitution, cationic, anionic/organo-catalytic ring-opening, Diels–Alder additions, thermolysis, and photo reactions. The review discusses the mechanisms of these reactions and highlights how the reactivity differs from oxocarbonyl analogues. Emphasis is put on the development of novel thiocarbonyl monomers which, uniquely, undergo polymerization through different mechanisms.
S) groups with radicals, anions, nucleophiles, electrophiles, in pericyclic reactions, and in the presence of light. In the polymer chemistry arena, thiocarbonyl compounds have been used as monomers, polymerization catalysts, reversible and irreversible chain transfer agents, and in post-polymerization modification reactions. Past and ongoing applications are reviewed including iniferters, radical and cationic RAFT, switchable RAFT agents, cyclic RAFT agents, chain transfer, thiocarbonyl addition-ring-opening, CS radical and anionic polymerization, acyl substitution, cationic, anionic/organo-catalytic ring-opening, Diels–Alder additions, thermolysis, and photo reactions. The review discusses the mechanisms of these reactions and highlights how the reactivity differs from oxocarbonyl analogues. Emphasis is put on the development of novel thiocarbonyl monomers which, uniquely, undergo polymerization through different mechanisms.
 Nathaniel M. Bingham | Nathaniel M. Bingham gained an MChem degree from the University of Southampton in 2016 before moving to the University of Surrey for his PhD under the supervision of Dr Peter Roth. His thesis surrounded the synthesis of novel monomers for the production of degradable polymers, specifically working on the first thionolactone that would undergo radical ring-opening polymerization (RROP, TARO). In 2019, he was appointed as a Teaching Fellow in the Department of Chemistry at the University of Surrey. His research interests include novel degradable polymers, smart polymers, and game-based learning. |
 Zahra Abousalman-Rezvani | Zahra Abousalman-Rezvani received her bachelor's degree in polymer engineering in 2016. Her master's studies on drug delivery systems were carried out at Sahand University of Technology in the research group of Dr Roghani-Mamaqani in Tabriz, Iran. Afterward, she started as a PhD candidate under the supervision of Prof. Voelcker and Dr Esser with the focus on the synthesis of stimuli-responsive polymers via reversible addition–fragmentation chain transfer polymerization at Monash University and the Commonwealth Scientific and Industrial Research Organization (CSIRO). Her research interests involve the self-assembly of block copolymers, stimulus-responsive copolymers as well as their potential applications in gene delivery systems. |
 Kyle Collins | Kyle Collins began his studies at the Florida Institute of Technology, located in Melbourne, Florida, where he graduated in 2020 with his BSc in Chemistry. Following this, he furthered his studies at the University of Surrey in Guildford, UK, studying the synthesis, polymerization, and degradation of novel biodegradable polymers under the guidance of Dr Peter Roth and Dr Nathaniel Bingham. |
 Peter J. Roth | Peter J. Roth received his PhD in Chemistry in 2009 from the University of Mainz, Germany, and subsequently moved to the University of New South Wales, Sydney, to work in the Centre for Advanced Macromolecular Design (CAMD) first as a postdoc, then as a senior research associate and ARC DECRA researcher. Following a brief stint at Curtin University (Perth, Western Australia) as a lecturer, Peter took up an academic post at the University of Surrey in 2016. His research interests include post-polymerization modification, stimulus-responsive polymers, and the development of novel monomers for radical-ring-opening polymerization. |
1. Introduction
Stable π double bonds are formed between carbon, nitrogen, oxygen, phosphorous, and sulfur atoms. Of these 2/3pπ–2/3pπ systems, the CS double bond has a uniquely multifaceted reactivity. Thiocarbonyl groups are reactive toward nucleophiles (similar to CO groups) and electrophiles (more so than CO groups), but also radicals and dienophiles (similar to CC bonds), making many of the known thiocarbonyl compounds versatile reagents, see Scheme 1.
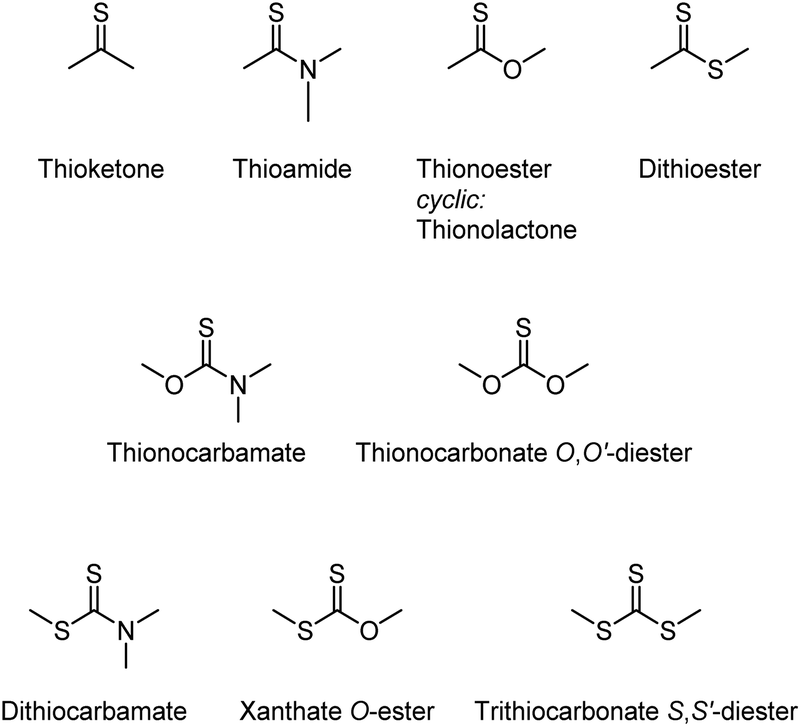 | ||
| Scheme 1 Structures and names of common thiocarbonyl compounds. | ||
While several radical CS-based reactions (such as the Barton-McCombie reduction)1 are now a staple in organic chemistry,2–4 it was the polymer science arena that has witnessed the perhaps most ground-breaking applications of thiocarbonyl compounds. Even before Herrmann Staudinger conceptualised macromolecules in the 1920s,5 the reaction of cellulose with carbon disulfide had already developed into a major industry to meet the growing demand for regenerated cellulose.6 The resulting cellulose xanthates can be solution-processed before being treated with acid, which regenerates the virtually insoluble cellulose. With various (at times ambiguous) names including Rayon, Viscose, and Cellophane, regenerated cellulose remains an important polymer material today and is used in textiles, food packaging and adhesive tape.
The more recent history of thiocarbonyl chemistry on polymers is undoubtedly dominated by reversible addition–fragmentation chain transfer (RAFT) polymerization, the arguably most versatile reversible deactivation radical polymerization method.7–9 Applications of RAFT and its success in smart materials,10 nanohybrid materials,11 functional surfaces,12 and biomedical applications,13,14 specifically drug delivery15 and gene delivery16 can be followed in recent review articles.
However, there are further established and emerging applications of CS-functional compounds in the polymer field. While thiol (SH)-based chemistries in the polymer arena,17 thioester-functional polymers18 and sulfur-containing polymers19 have been reviewed, the current review focusses specifically on thiocarbonyl chemistry in polymer science. Organised by reaction type, this review attempts to explain the success of CS-functional compounds from a mechanistic point of view. RAFT polymerization is covered within this mechanistic context, for example, to demonstrate how substituents influence the reactivity of thiocarbonyl groups. Finally, the scope for future development among the manifold applications of CS compounds in the polymer arena are discussed.
2. Radical reactions of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) S compounds in polymer science
S compounds in polymer science
2.1 Radical reactivity
With a low overlap coefficient between the C2pz and S3pz orbitals, the CS double bond is weaker than the oxocarbonyl CO analogue, which is also reflected in their lengths (∼1.6 Å for CS versus ∼1.25 Å for CO) and bond dissociation energies (573 kJ mol−1 for CS versus 602 kJ mol−1 for CC and 745 kJ mol−1 for CO).20,21 Carbon and sulfur have very similar electronegativities making the thiocarbonyl group less polar than its oxygen analogue and prone to homolytic cleavage. The large size of the S3sp2 lone pairs facilitate an orbital-controlled attack onto the sulfur atom, while radical addition onto the carbon centre is prevented by a large energy barrier.22
The prevalent mechanism for radical chemistry on thiocarbonyl groups involves reversible23 addition onto the sulfur atom, formation of a radical intermediate of, ideally,24 low reactivity due to steric crowding, followed (for the forward reaction) by β-scission and formation of a new double bond and a different active radical,25 see Scheme 2.
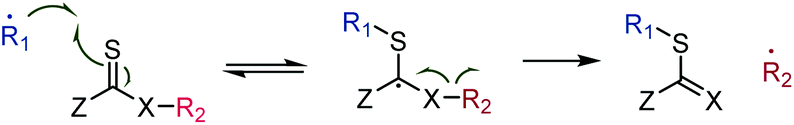 | ||
| Scheme 2 Common radical reaction pathway of thiocarbonyls involving reversible addition, an intermediate radical stabilised by the Z group, and β-scission. | ||
The β-scission can involve several (or all) of the following driving forces: (i) increase of entropy due to fragmentation; (ii) formation of a more stable double bond than CS (for X ≠ S); (iii) formation of a stabilised, lower energy, radical R2˙; and (iv) release of ring strain (Z–R2 cyclic compounds only). Higher reaction temperatures increase the influence of entropy, can drive the expulsion of more reactive radicals, R2˙, and prevent radical reactions of the intermediate.26
The radical thiocarbonyl reactions can be classified according to the chemical nature of the groups R1, R2, and especially X (Scheme 2). The following sections discuss the details of the cases X = SSC(S)Ph, X = S (RAFT), and X = O (chain transfer and thiocarbonyl addition-ring-opening (TARO)).
2.2 Photo iniferters
Over a decade before the development of RAFT, the key concept of “living” radical polymerization—an equilibrium reaction that reversibly deactivates growing polymer-based radicals—was proposed in 1982, see Scheme 3.27–29 Several thiocarbonyl species (together with diphenyldisulfide) were explored as iniferters (a portmanteau of initiator–transfer agent–terminator), some of which had previously been shown to act as photoinitiators.30 Photopolymerization of styrene in the presence of tetraethyl thiuram disulfide (structure 1), for example, proceeded with a nearly linear increase of molar mass with conversion, which provided evidence of a successive addition of monomer units onto active radicals before a reversible deactivation through recombination. The concept was expanded to the formation of diblock copolymers.31,32 Mechanistically, the method relies on the weak S–S bond enabling photocleavage, as well as the stability of the thiocarbonylthiyl radical.33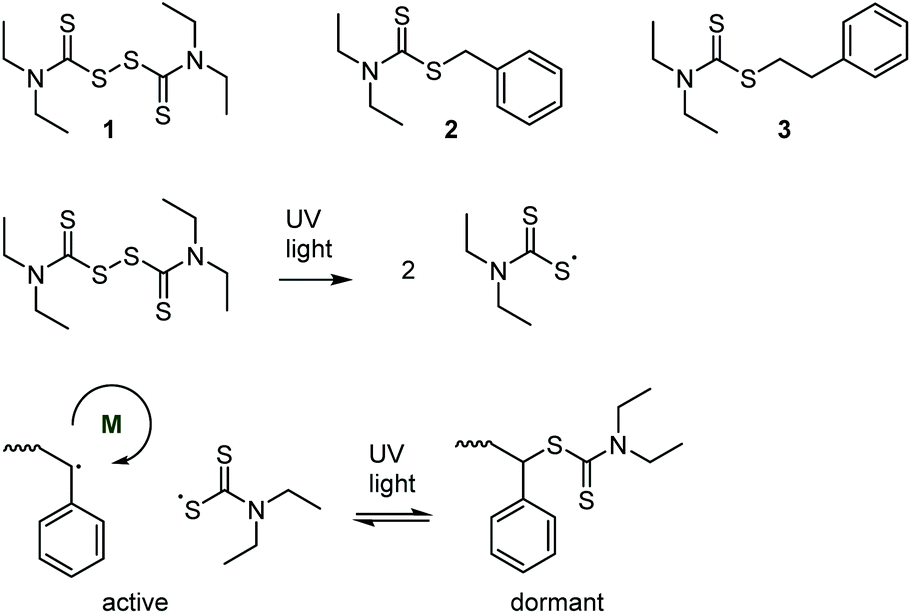 | ||
| Scheme 3 Structures of the thiocarbonyl compounds tetraethyl thiuram disulfide (1), S-benzyl-N,N-diethyldithiocarbamate (2), and 2-S-phenylethyl N,N-diethyldithiocarbamate (3) explored as initiator–transfer agent–terminators (iniferters) (top); photodissociation of 1 (middle); and reversible photodeactivation equilibrium (bottom). | ||
2.3 RAFT
The literature on thiocarbonyl chemistry in the polymer arena is clearly dominated by the unprecedented success and paradigm shift associated with the RAFT process. Briefly, it involves the addition of a thiocarbonylthio compound (the RAFT agent, X in Scheme 2 = S) to a radical polymerization. β-Scission recreates a thiocarbonyl group and therefore allows for reversible addition and β-scission. This mechanism (Scheme 4) is exploited to share radicals between chains. When terminated with the thiocarbonylthio moiety (the “RAFT end group”), a chain is dormant. It awakens when a radical (another growing chain as in the main equilibrium or an initiator-derived radical as in the pre-equilibrium) adds onto the CS bond and the chain is released through β-scission. In an ideal RAFT polymerization, chains become active for a short period of time during which only few monomer units are added before the growing chain becomes dormant again through addition onto another RAFT agent or RAFT end group. The commonly depicted RAFT mechanism suggests deceptively that one radical is shared between two chains. In reality, the concentration of radicals (which, in classic RAFT, are continuously provided through the decomposition of a thermal initiator) is low and a single radical is shared between tens or hundreds of chains. As a consequence, all chains grow at a similar rate and the average molar mass increases linearly with conversion (in contrast to uncontrolled radical polymerization where long chains are formed from the start of the polymerization). All active radicals are eventually terminated through the same recombination or disproportionation mechanisms as in radical polymerization. The predominant product of a RAFT polymerization is the dormant species, i.e., a polymer chain carrying an initiator- or RAFT agent-derived α end group and a thiocarbonylthio ω end group.7,34
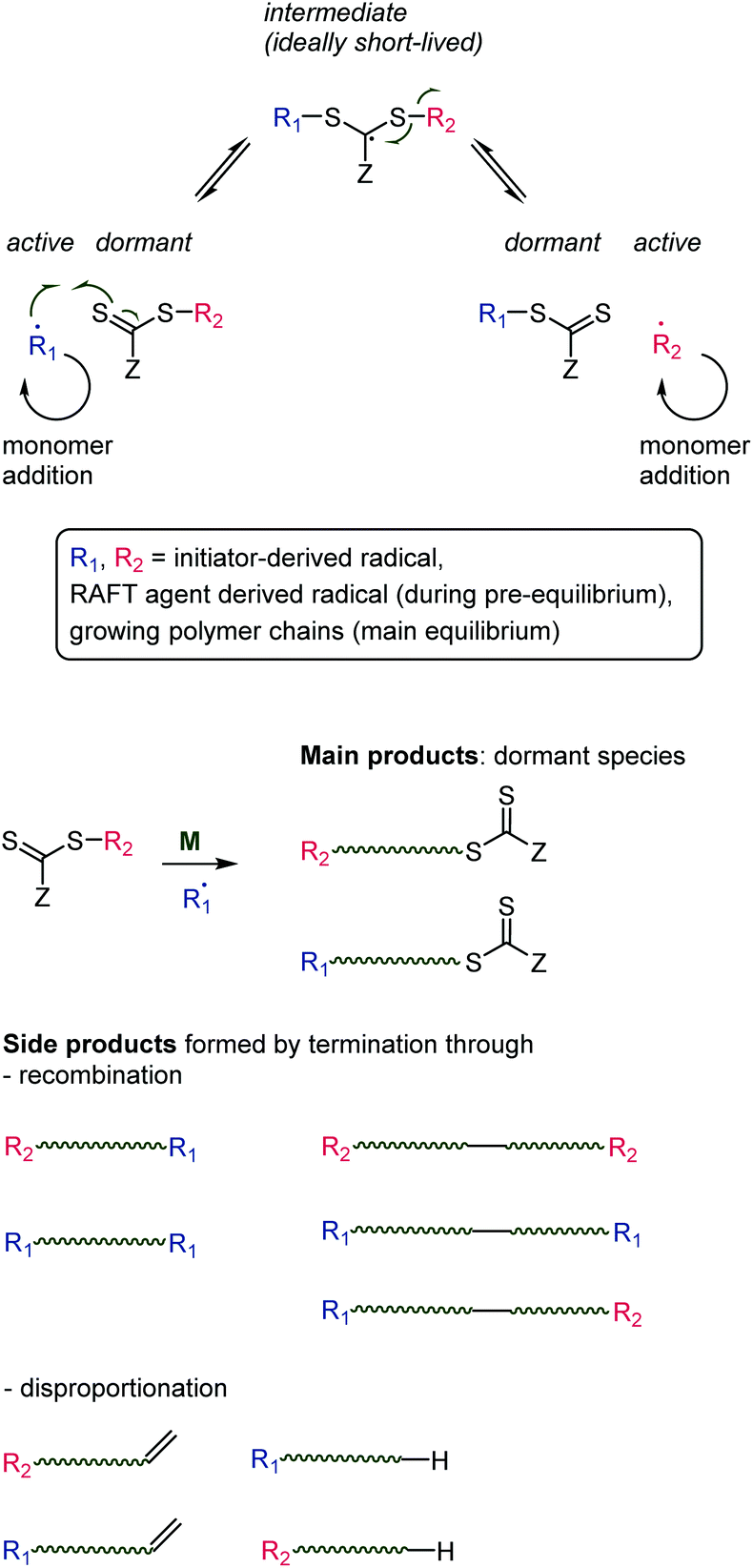 | ||
| Scheme 4 Reversible addition–fragmentation chain transfer (RAFT) mechanism (top) and products (bottom). | ||
There are two main requirements for RAFT to work well. Firstly, the R-group on the RAFT agent needs to resemble (in terms of radical stabilisation) the structure of the growing chain. This is necessary to ensure a balanced pre-equilibrium. RAFT agents with varying R groups suitable for different monomer types have been presented35 and are commercially available. Secondly, and more relevant to the current review, the energies of the intermediate radical (top of Scheme 4) and the growing polymer chain radical need to be similar. If the intermediate species has too high energy, it is less likely to form and the growing polymer chain undergoes uncontrolled radical polymerization. If the intermediate species is too low in energy, radicals become trapped and eventually terminate; in this case the would-be RAFT agent acts as a retarder or inhibitor.
Vinyl monomers are typically grouped into two categories. Less activated monomers (LAMs, such as vinyl acetate) form higher energy radicals. This means the monomers are relatively unreactive, while their radicals, if formed, are highly reactive. More activated monomers (MAMs, such as methyl methacrylate) form radicals more easily because of better radical stabilisation, but the radicals are less reactive, see Scheme 5. Additionally, sterics play an important role in polymerization kinetics.
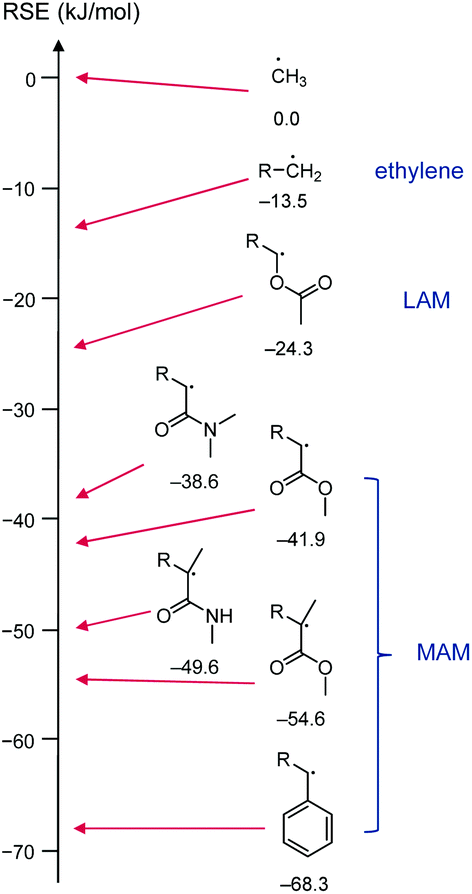 | ||
| Scheme 5 Selection of structures resembling growing vinyl chains with literature radical stabilisation energies (RSE, calculated at G3 (MP2)RAD level, values in kJ mol−1)36 for R = methyl. The radical stabilisation energy of a species R1˙ is defined as the energy difference of the reaction CH3˙ + R1–H → CH4 + R1˙, i.e., a radical is more stable the larger negative its RSE is, with the methyl radical set to RSE = 0 kJ mol−1. | ||
For the benefit of understanding RAFT polymerization and for later sections of this review, it is worthwhile to summarise the three structural effects that contribute to radical stabilisation. The strongest stabilising effect is π conjugation which leads to spin delocalisation. This effect is strong in allylic and benzylic (including styryl) radicals but also applies in α-carbonyl-based radicals including (meth)acrylic species. The second strongest contribution to the radical stabilisation energy (RSE) is π donation through an adjacent heteroatom with lone pairs, e.g., in the R1–CH2CH˙OR2 species derived from attack of a vinyl ester (R2 = C(O)R′) or vinyl ether (R2 = R′) with a radical, R1˙. The interaction with the heteroatom-based lone pair increases the energy of the radical (makes it more nucleophilic) but lowers the lone pair electron energy which results in an overall energy gain. The third and weakest effect to stabilise radicals is hyperconjugation with adjacent C(sp3)–H(s) bonds and is the reason for the general order tertiary > secondary > primary > methyl for the stability of radicals. The combination of these effects leads to the (calculated) radical stabilisation energies shown in Scheme 5.
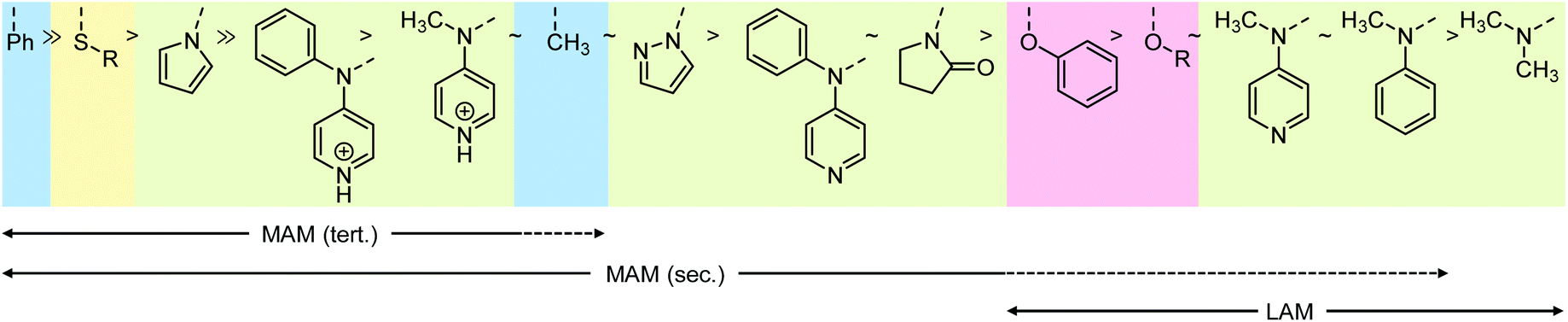 | ||
| Scheme 6 Z groups of RAFT agents (RS–CS–Z) in order of decreasing stabilisation of the intermediate radical with shading indicating the nature of the atom adjacent to the thiocarbonyl group (C blue; S yellow, N green, O red). Solid (dashed) arrows beneath indicate good (partial) compatibility with the respective monomer classes, where MAM (tert.) refers to more activated monomers with tertiary radicals (methacrylates, methacrylamides), MAM (sec.) to more activated monomers with secondary radicals (acrylates, acrylamides, styrenes, acrylonitrile), and LAM to the less activated monomers vinyl esters and vinyl amides. | ||
It may seem contradictory to cite the oxygen lone pairs in vinyl acetate as stabilising a radical, while the oxygen and nitrogen atoms in xanthates and dithiocarbamates are shown to increase the energy of intermediate radical compared to an alkyl Z group. The discrepancy comes from the fact that Z groups with a lone pair contribute strongly to resonance stabilisation of the intact thiocarbonyl group, lowering its energy and making it less reactive to radicals, see Scheme 7A.37,38 This resonance stabilisation is lost upon radical attack, making the formation of the radical intermediate less energetically favourable (in other words, the radical is less stable) and only feasible with the most reactive, LAM-derived, radicals. The intermediate radical is indeed stabilised through lone pair donation from the two adjacent S atoms (Scheme 7B).38 With smaller lone pairs and higher electronegativity, the contribution to stabilising the intermediate radical of oxygen and nitrogen-based Z groups are, however, minimal36 and the stabilisation of the intact RAFT agents dominates.
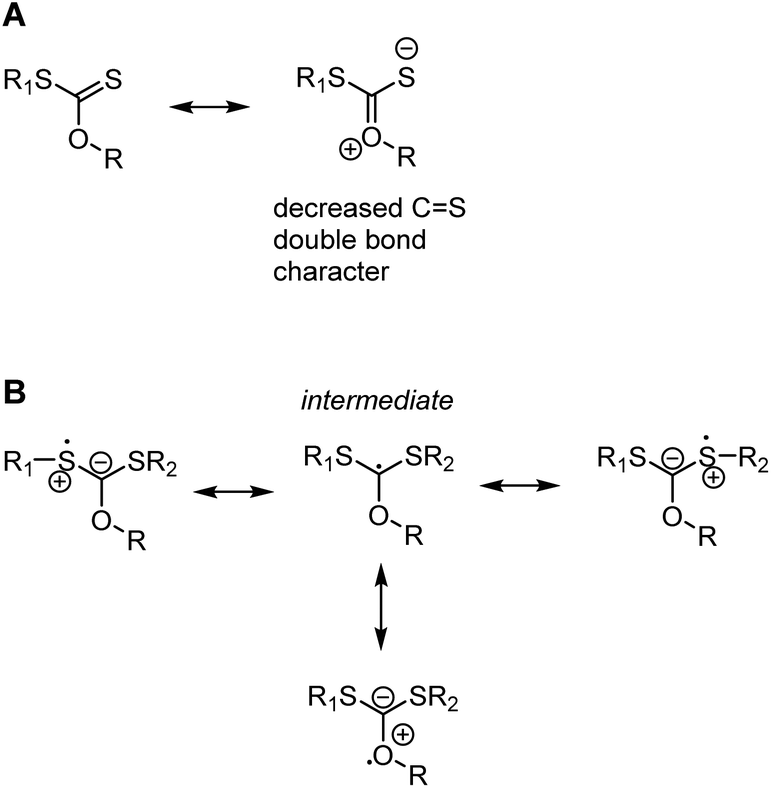 | ||
| Scheme 7 Resonance form of a xanthate RAFT agent showing the decrease of CS double bond character (and double bond reactivity) as a result of Z group lone pair donation (A) and resonance forms of the xanthate-based intermediate radical where the stabilisation through the two sulfur atoms (top row) outweighs the influence of the smaller and less polarisable O-based lone pairs (bottom structure) (B). While only shown for xanthates (Z = OR), the same considerations apply to dithiocarbamates (Z = NR2). | ||
The resonance stabilisation of the intact thiocarbonylthio compound is strongest with N and O-based lone pairs, with a less pronounced effect of S-based Z groups, explaining the trend in Scheme 6. Likewise, the order of the N-based Z groups is also based on the extent that the lone pair is able to conjugate into the CS bond ranging from no conjugation in the case of pyrrole (the N lone pair is part of the aromatic system), partial conjugation into CS in the case of 2-pyrrolidone (where the N lone pair can also conjugate into the CO bond), to full conjugation in the case of the dialkylamino Z group.
The development of thiocarbonyl-based radical chemistries25 and reversible deactivation radical polymerization methods provide excellent examples to demonstrate that radicals are not indiscriminately reactive but can undergo very selective reactions. One such example is the successful RAFT polymerization mediated by a dithiobenzoate of methacrylic monomers carrying xanthate or dithiocarbamate side group functionality. During RAFT polymerization, only the Ph-stabilised intermediate radicals on the RAFT agent form while the xanthate or dithiocarbamate side groups are not attacked since they are not able to stabilise intermediate radicals as well. Upon completed RAFT polymerization, the xanthate and dithiocarbamate side groups were removed through aminolysis, providing access to thiol side groups.39
In the context of choosing the correct Z group for a RAFT agent, it is worth mentioning the special case of monomers involving primary radicals such as ethylene40 or 2-methylene-1,3-dioxepane41 (a cyclic ketene acetal that undergoes radical ring-opening polymerization). Despite the high energy of primary radicals (see Scheme 5), RAFT polymerization of these monomers is possible with a moderate degree of control using xanthate RAFT agents.40,41 In case of the most commonly used O-alkyl xanthates, however, Z group fragmentation can occur which likewise produces a primary radical (in the Barton-McCombie deoxygenation this is the desired fragmentation).23 In RAFT, this undesirable side reaction produces mid-chain S,S′-dipolymer dithiocarbonate functionality. An O-aryl xanthate RAFT agent was shown to prevent this side reaction41 because the associated aryl sp2-based radical is even more energetic than a primary alkyl-based radical (the calculated radical stabilisation energy of the C6H5˙ radical is +37.0 kJ mol−1),36 see Scheme 8.
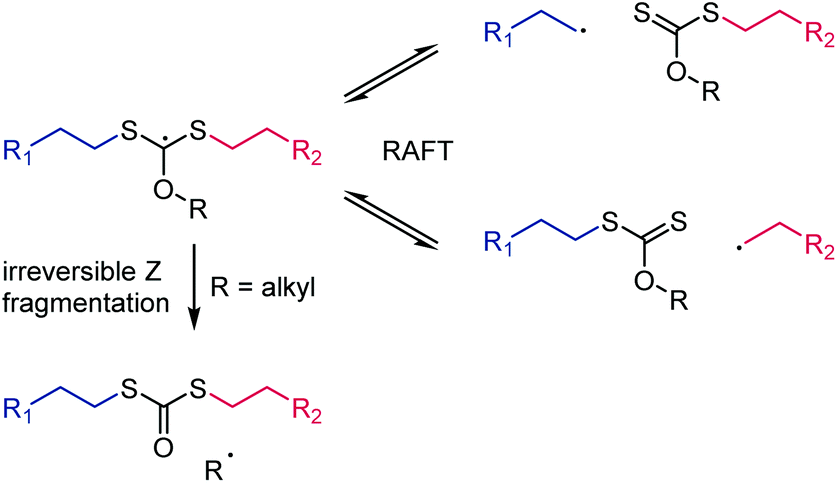 | ||
| Scheme 8 Irreversible Z fragmentation during the use of primary radical-forming monomers for O-alkyl functional xanthates. This side reaction is prevented by using O-aryl xanthates.41 | ||
S group lowers the reactivity toward radical attack (lowers the stabilisation of the intermediate radical), N-4-pyridineamino-functional Z groups were explored. Under neutral conditions, the N-based lone pair conjugates into the CS bond and lowers its reactivity, requiring reactive LAM-derived radicals to add to the CS bond (Scheme 9, top). Upon protonation of the pyridine ring under acidic conditions, the N-based lone pair can still conjugate into the CS bond (Scheme 9 bottom right), but this resonance form is less favourable due two positive charges on/in an aromatic. Instead, the dominating resonance form involves conjugation of the N-lone pair into the pyridine ring to neutralise the pyridinium charge. With nearly unimpeded CS bond activity, the protonated RAFT agent is compatible with MAMs. Following simple pH control, block copolymers involving LAMs and MAMs can thus be produced from switchable RAFT agents.44–46
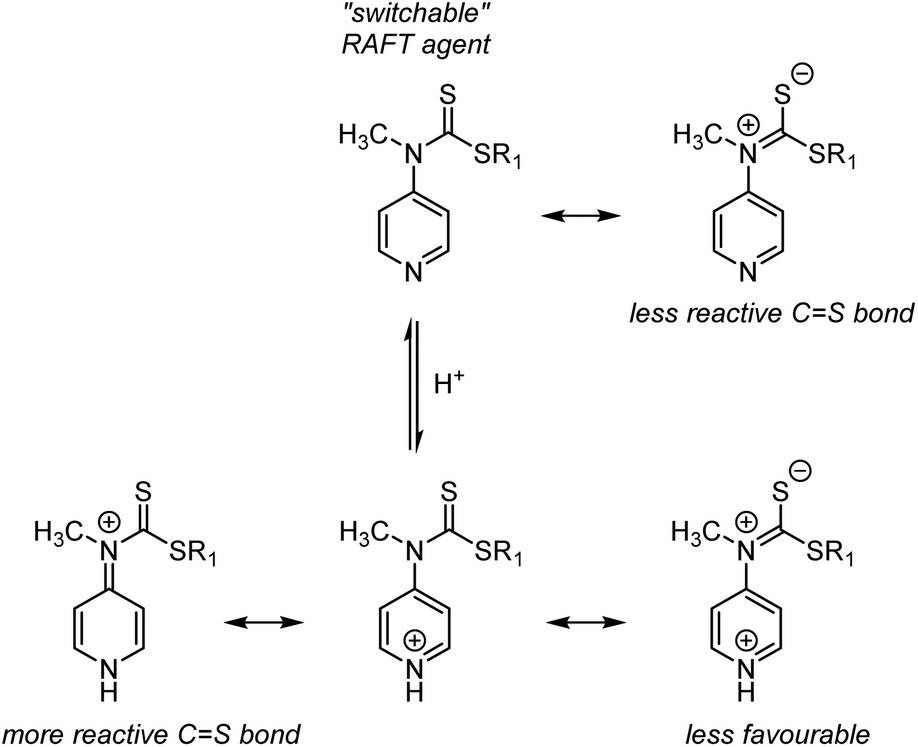 | ||
| Scheme 9 Resonance forms of a “switchable” N-4-pyridineamine-based RAFT agent under neutral (top) and acidic (bottom) conditions. | ||
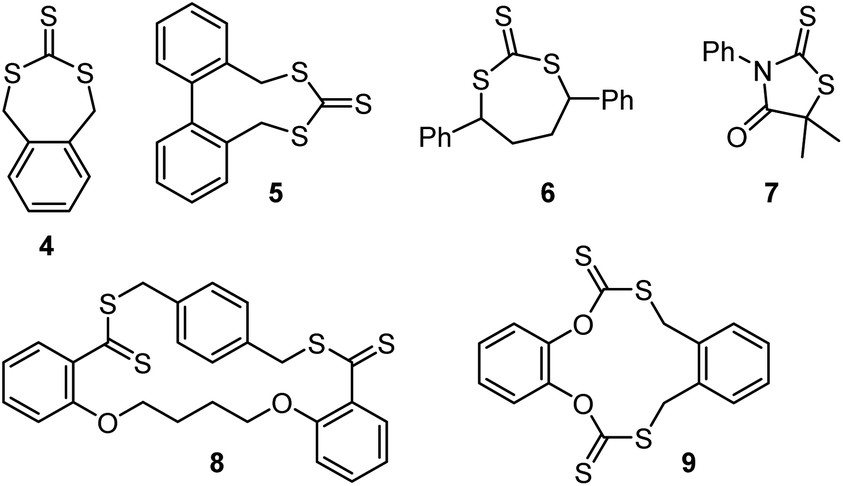 | ||
| Scheme 10 Structures of cyclic RAFT agents. | ||
Cyclic RAFT agents 4,47,485,49 and 648,50–52 have been used in radical polymerizations of methyl acrylate,52n-butyl acrylate,48,50 styrene,47,49,50 and 4-vinylpyridine,51 while compound 7 was used with vinyl acetate.53 In these polymerizations, the cyclic RAFT agent undergoes ring-opening copolymerization leading to the incorporation of trithiocarbonate (for 4, 5, 6), or dithiocarbamate (7) functionality into the backbone, similar to other cases of radical ring-opening polymerization (RROP). These backbone groups can be cleaved through aminolysis (see section 3.2.1) leading to the degradation into shorter chains.47–53 However, unlike other RROP systems, the products of these cyclic RAFT agents are not usually described as copolymers but as multiblock systems because the in-chain thiocarbonylthio groups remain active toward radical attack. While the dispersity of the individual blocks (which can be isolated through aminolysis) was typically low, the multiblock systems were usually found to have higher dispersities (of or above 2) because radical addition and β-scission of an in-chain thiocarbonylthio group can lead the formation of terminated chains.50,52 Interestingly, the diseleno analogue of 7 was found to lead to slightly lower multiblock dispersities (Đ ∼ 1.9).53
Cyclic RAFT agents 4,548,55 and 956 (Scheme 10) have been used for the production of cyclic polymers of methyl acrylate,54,56 methyl methacrylate55 and methyl methacrylate–N-isopropyl acrylamide copolymers.55 These polymerizations required low temperatures (−30 to −76 °C) to reduce diffusion and chain transfer, i.e., for the monomers to be inserted into the same combination of R and Z group, which, in these cases, were connected by a cycle. To reduce the number of linear polymers formed, initiation was done through γ-ray irradiation. However, termination events still led to linear chain side products.
2.4 Thiocarbonyl chain transfer agents
With the prevalence of RAFT, regular, i.e., irreversible, chain transfer has been receiving less attention, although there has been renewed interest in the field of emulsion polymerization.57 Several types of thiocarbonyl compounds have been investigated as (irreversible) chains transfer agents. In this case, X (in Scheme 2) can be a nitrogen, as in the N-acyloxy derivatives of 2-thiopyridone, 10, and 4-methylthiazole-2(3H)thione, 11, see Scheme 11.58,59 Radical CS addition followed by β-scission produces an acyloxy radical that decomposes to carbon dioxide and a carbon-based radical60 that can, ideally, re-initiate the polymerization. Unlike RAFT, the β-scission is irreversible; the SX species (see Scheme 2) is aromatic and not prone to radical attack. In another type of thiocarbonyl chain transfer agent, X is oxygen, as in the thionoester species, 12 (Scheme 11).61 The transfer of the radical from one chain onto a new initiating species is likewise irreversible due to the formation of a stable thioester (SCO) group that is not susceptible to radical attack.
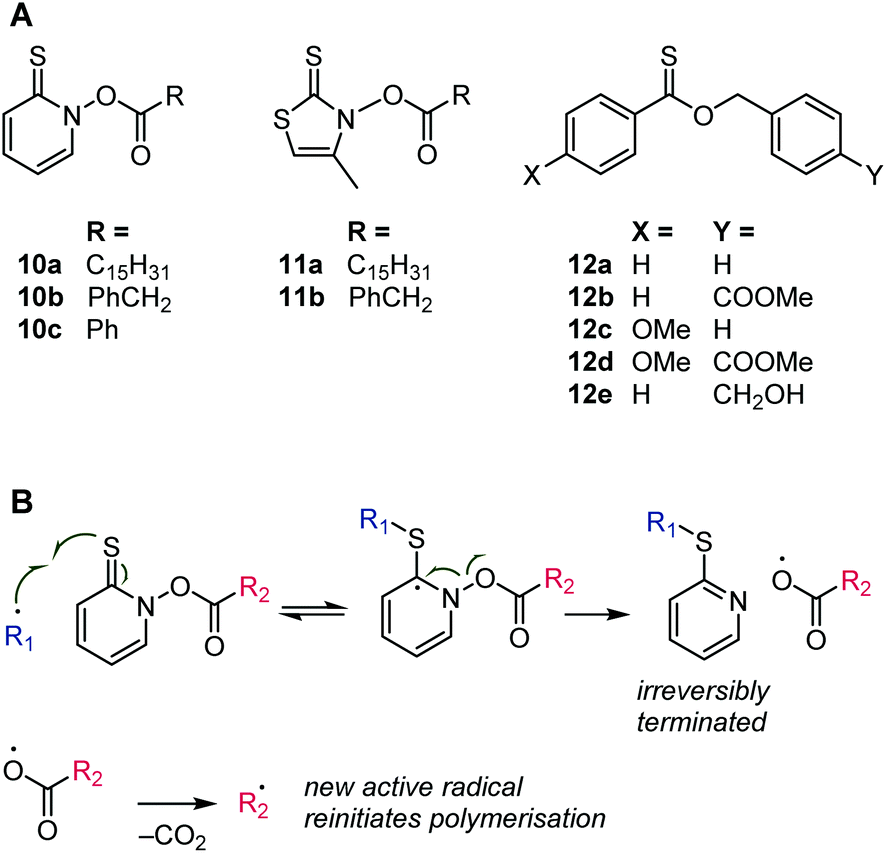 | ||
| Scheme 11 Structures of thiocarbonyl chain transfer agents (A) and addition–β-scission mechanism for compound 10 (B). | ||
The thiopyridone-based thiohydroxamic esters, 10, were highly efficient chain transfer agents (lowered the molecular weight of polymers without affecting the rate of monomer consumption) for methyl methacrylate and styrene but were too reactive to be used with methyl acrylate and vinyl acetate. For the latter comonomers, retardation and broad molar mass distributions were observed. Species 11a was found to be a less active chain transfer agent compared to species 10, while 11b showed ideal behaviour (chain transfer constant = 1, i.e., a growing chain has equal preference between the addition of another monomer and the attack of a chain transfer agent) with methyl methacrylate and styrene.58,59 These observations agree with the above discussion that dithiocarbamates are highly reactive toward radicals (i.e., form stabilised intermediate radicals) if their nitrogen lone pairs are not able to conjugate into the CS bond. In the case of species 10 and 11, the nitrogen lone pair is part of an aromatic system.
The thionoesters, 12, showed effective chain transfer (with transfer constants around 1) in polymerizations of methyl acrylate and styrene, but were inactive for methacrylates,61 which can be rationalised through the destabilising effect of the oxygen atom compared to dithiobenzoates (which are highly effective RAFT agents in the presence of methacrylates). On the other hand, the thionoesters, 12, were too reactive for vinyl acetate and caused substantial retardation, presumably because the benzylic radical formed by β-scission is too stable to effectively re-initiate vinyl esters.62 Functionalisation (12b–12e) allowed for the introduction of chain end functionality. Interestingly, substitution on either aromatic ring reduced the chain transfer efficiency for styrene, while, except for species 12e, slightly higher chain transfer constants were found with methyl acrylate.61
2.5 Thiocarbonyl–addition ring-opening (TARO) polymerization
Recently, thionolactones (featuring in-cycle –C(S)O– functionality) were shown to undergo radical ring-opening polymerization.63,64 In this case, the atom X (Scheme 2) is an oxygen and the groups Z and R2 (Scheme 2) are connected by a cycle. The addition–β-scission mechanism therefore leads to backbone thioester functionality, see Scheme 12. At the time of writing, four cyclic thiocarbonyl species had been shown to undergo thiocarbonyl addition-ring-opening polymerization, see Table 1.
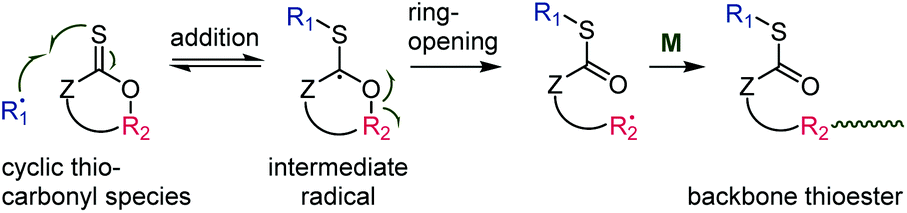 | ||
| Scheme 12 Mechanism of thiocarbonyl addition-ring-opening (TARO) radical polymerization. | ||
| Thiocarbonyl species | Vinyl comonomer | Comments |
|---|---|---|
|
Homopolymerization 65 | Low rate; yields oligomers only |
| Methyl acrylate63 | Thiocarbonyl species is incorporated at a faster rate than the vinyl comonomer | |
| mPEG acrylate63,66,67 | ||
| n-Butyl acrylate68,69 | ||
| t-Butyl acrylate64 | ||
| Acrylamide66 | ||
| N-Isopropylacrylamide66 | ||
| N-2-Methoxyethoxyacrylamide67 | ||
| N,N-Dimethylacrylamide63,66,70 | ||
| N,N-Diethylacrylamide66 | ||
| N-3-(N-4-Sulfobutyl-N,N-dimethylammonium)propylacrylamide66 | ||
| Acrylonitrile63,66 | ||
| Pentaerythritol triacrylate71 | ||
| Styrene69,72 | ||
| N-Methylmaleimide65 | Alternating copolymerization between the thiocarbonyl species and the vinyl comomomer | |
| N-Phenylmaleimide65 | ||
| N-Pentafluorophenylmaleimide65 | ||
|
Homopolymerization 73 | Low yielding |
| Styrene73 | The thiocarbonyl species polymerizes faster than styrene, but slower than t-butylmethacrylate | |
| t-Butylmethacrylate73 | ||
|
Vinyl acetate70 | Thiocarbonyl species is incorporated at a faster rate than the vinyl comonomer |
|
Homopolymerization 70 | In the presence of 2-methylene-1,3-dioxepane |
| Vinyl acetate70,74 | Thiocarbonyl species is incorporated at a faster rate than the vinyl comonomer | |
| Vinyl pivalate74 | ||
| 2-Methylene-1,3-dioxepane70 | ||
| N-Vinylpyrrolidone74 | Retardation; low molar mass products |
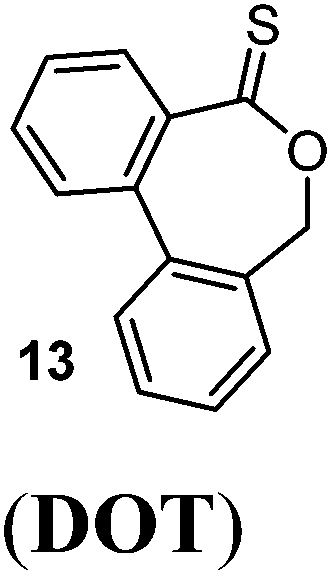
As described above, dithiobenzoate (Ar–C(S)S–)-based RAFT agents are well-suited to mediate the polymerization of MAMs including methacrylates. In comparison, the analogous thionolactone DOT (13) (featuring an Ar–C(S)O– group) has lower CS double bond reactivity due to conjugation of the oxygen lone pair (see section 2.3.1 above). DOT was found to be incorporated rapidly during polymerizations of acrylate, acrylamide and styrene comonomers, leading to gradient copolymers at 70 and 80 °C.63,64,66,72 With styrene, however, conditions could be tweaked (150 °C, Trigonox 301 initiator, 5 mol% DOT) to lead to a randomly distributed copolymer, and a closer to ideal copolymerization.72
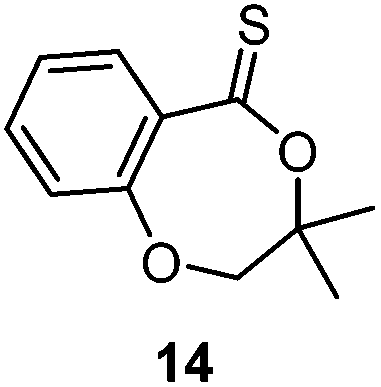
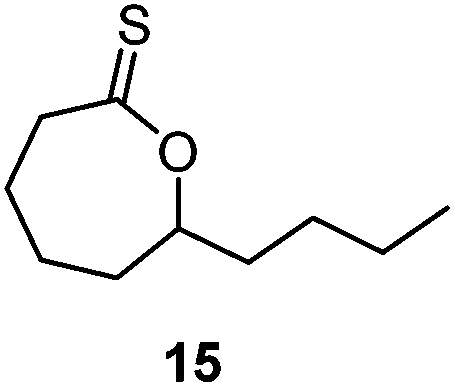
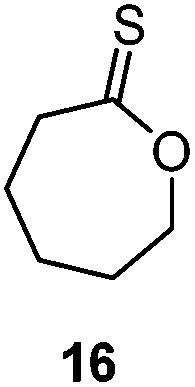
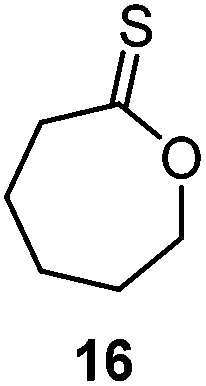
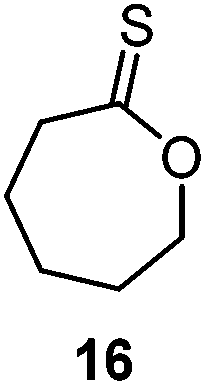
A second generation thionobenzoate, 3,3-dimethyl-2,3-dihydro-5H-benzo[e][1,4]dioxepine-5-thione (14) with an ortho alkoxy substituent was shown to provide slightly better stabilisation of the intermediate radical and copolymerized, albeit sluggishly, with t-butylmethacrylate.73 The thiocaprolactones 15 and 16, lacking the stabilisation of the intermediate radical by the aryl group, were shown to polymerize rapidly with the LAMs vinyl acetate, vinyl pivalate, and, though with retardation, N-vinylpyrrolidone.70,74
Emerging applications of TARO-made copolymers exploit the fact that the backbone thioesters are weaker than (oxo)ester linkages available through conventional radical ring-opening methods. TARO-made copolymers were shown to degrade rapidly under mild conditions through aminolysis,63,66 cysteinolysis,64,66 thiolysis,66,67 and oxidative cleavage.66 The degradation was shown to cause insoluble–soluble transitions (including of thermally responsive polymers with LCST or UCST behaviour)66 in water, which was exploited to release an encapsulated dye (as a drug model) from core-degradable PEG-based diblock copolymer micelles.67 As the thioester linkages were shown not to contribute to cytotoxicity, the TARO method is promising for intracellular drug delivery applications with glutathione-triggered degradation and release.67
TARO monomers have also found a use in producing degradable cross-linked materials through uncontrolled radical polymerization.68,71 Incorporation of only 4 mol% of DOT into acrylate-based networks led to the ability to completely degrade the insoluble materials. The low incorporation allowed for the retention of the physical properties of the network, compared to non-degradable controls, and 3D-print some of the structures. Additionally, TARO-made co-/ter-polymers have been synthesised through emulsion polymerization. Here, degradable latexes have been produced by polymerization of DOT with n-butyl acrylate and styrene, producing polymers with variable glass transition temperatures.69
2.6 Radical polymerization of the CS group
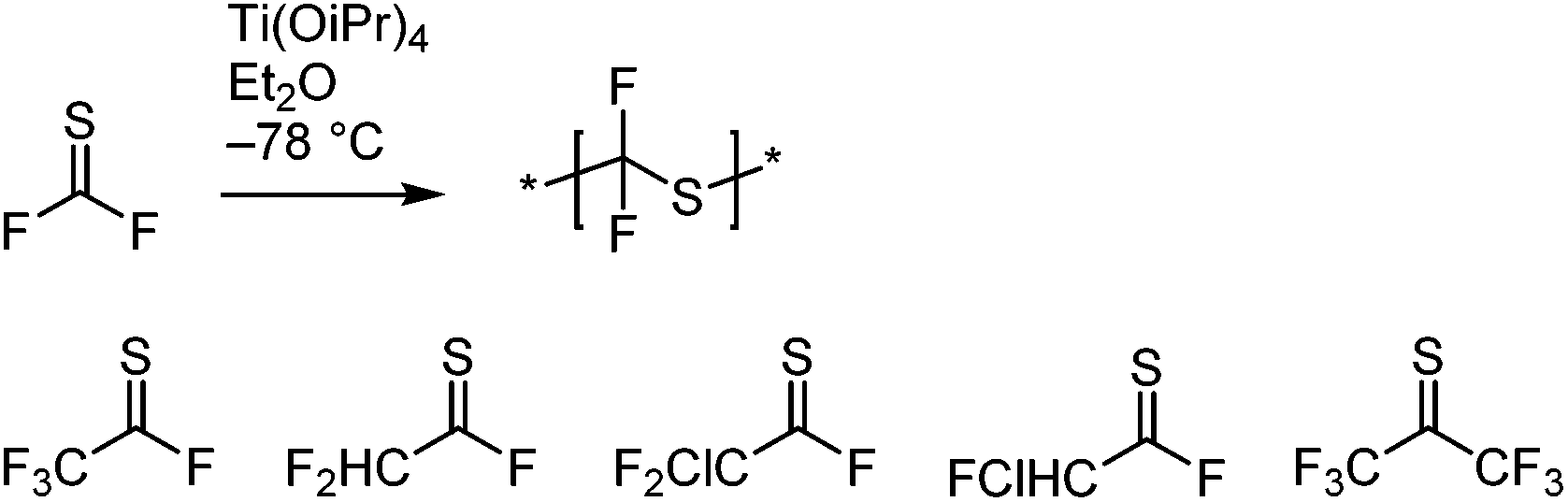
The above systems involve β-scission. Conversely, thiocarbonyl fluoride (F2CS) and thiocarbonyl chlorofluoride (ClFCS), which do not have β-bonds, undergo radical polymerization of the CS bond, see Scheme 13A.75 Although the thiocarbonyl fluoride homopolymer has excellent chemical stability (see section 3.4.3),76 this synthesis has not found much interest since the original publications in the 1960s. Challenges involve the laborious low-yielding synthesis of the monomer through fluorination of the toxic thiophosgene and the necessity to polymerize it at temperatures between −60 °C and −120 °C. Radical initiation at these temperatures was achieved by using the trialkylboron–oxygen redox couple. In addition to poly(thiocarbonyl fluoride) homopolymer, copolymers with a variety of olefins and vinyl monomers were prepared, see Scheme 13B. While the authors believed that the mechanism involved the addition of the initiating radical onto the carbon centre, the above considerations make it more likely that radicals added onto the sulfur atom. The observation that copolymers with vinyl acetate showed rapid backbone degradation upon cleavage of the acetate side groups supports the formation of thioacetal groups through radical attack onto the sulfur atom, see Scheme 13C.
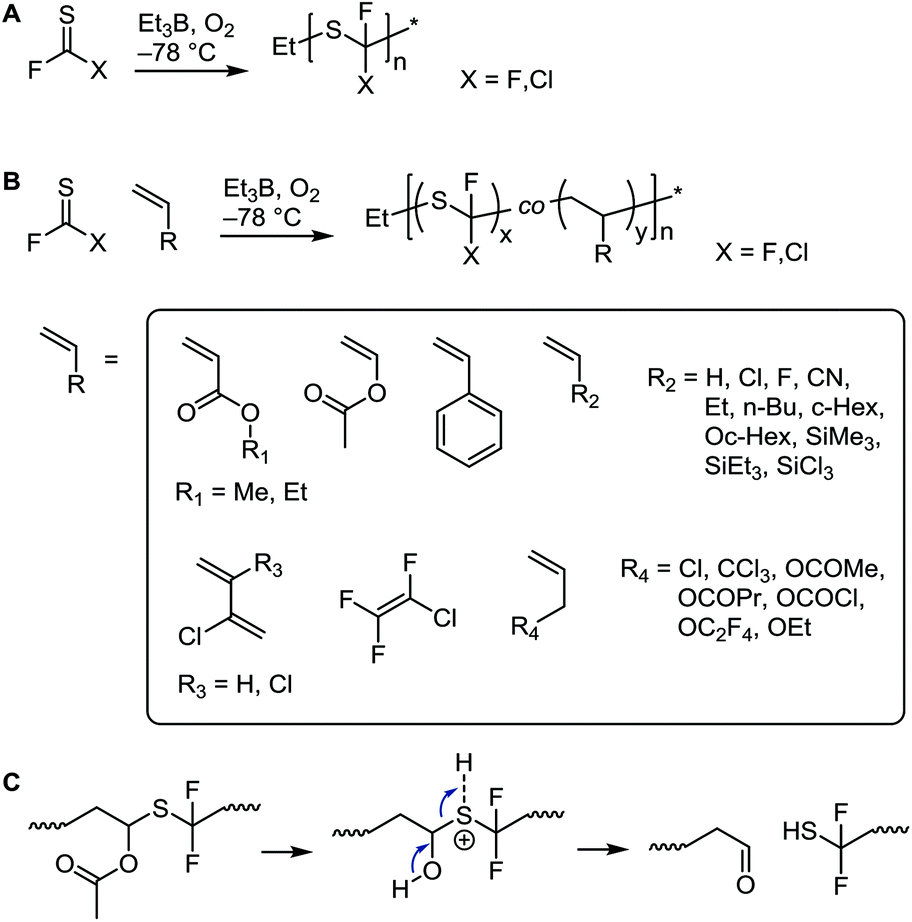 | ||
| Scheme 13 Radical polymerization of thiocarbonyl fluoride and thiocarbonyl chlorofluoride (A), copolymerization with a selection of vinyl monomers and olefins (B), and proposed backbone degradation mechanism of thiocarbonyl fluoride–vinyl acetate copolymers (C).75,76 | ||
Another polymerization that is thought to involve radical species is the copolymerization of carbon disulfide with ethylene to produce poly(dithioesters) with ethyl spacers, see Scheme 14.77 However, the polymers were also found to contain other functional groups such as alkene and disulfide links. The polymerization was undertaken in the gas phase and initiated with UV light to produce a biradical dimer of CS2, which was then free to propagate with either itself or ethylene units. The polymer product formed a deposit and was sparingly soluble in chloroform, which was explained through extensive cross-linking, perhaps through the creation of similar units to CS2 homopolymerizations, see section 3.4.1.78 Unfortunately, the same reaction could not be carried out with other alkene monomers, such as acrylates.79
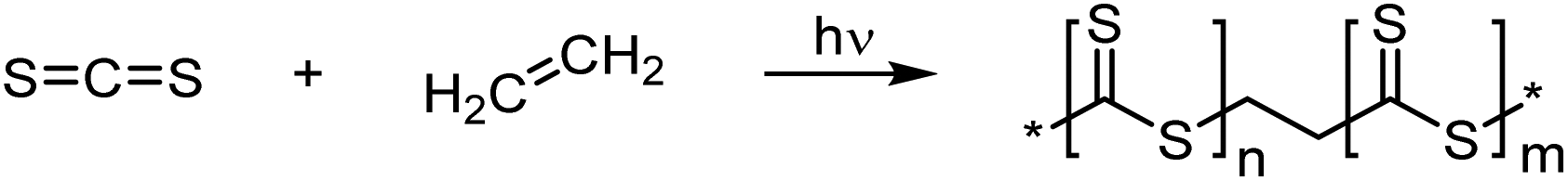 | ||
| Scheme 14 Polymerization of CS2 with ethylene through a radical mechanism, initiated by UV-light.77 | ||
3. Reactions with nucleophiles
3.1 Reactivity
Thioesters, thionoesters, and dithioesters undergo the same nucleophilic acyl substitutions as their all-oxygen analogues.80,81 Due to the larger polarizability of the sulfur atom(s), substitutions typically follow a step-wise mechanism with a/several tetrahedral transition state(s).80,82 For the same reason, the relative reactivity in nucleophilic substitutions depends strongly on the hardness/softness of the nucleophile, with sulfur-containing esters reacting drastically faster than oxoesters with soft nucleophiles, see Scheme 15. This reactivity difference is sufficient for the selective cleavage of thioesters in the presence of oxoesters, as recently shown on TARO-made copolymers (see section 2.5).63 In the context of RAFT agent stability, dithiobenzoates are more hydrolytically labile than trithiocarbonates,8 while the aminolysis rate of thiocarbonyl species increases in the order dithiocarbamate < thioester < xanthate.39 In agreement with the reactivity order of carbonyl compounds against hard nucleophiles, organic isothiocyanates (R–NCS) are stable in neutral water at RT, while the corresponding isocyanates (R–NCO) hydrolyse quickly to the amine and CO2. As mentioned above, the CO bond is more stable than the CS bond. This can lead to isomerisation reactions, for example of cyclic compounds where a nucleophilic attack expels a thionocarboxylate, see Scheme 16.83
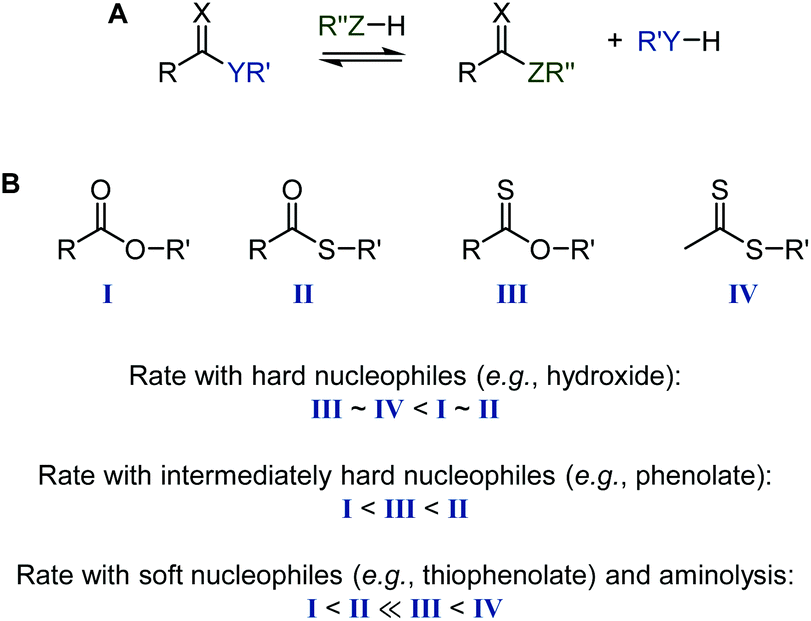 | ||
| Scheme 15 Acyl substitution (A) and relative rates of ester (I), thioester (II), thionoester (III), and dithioester (IV) cleavage (hydrolysis, trans(thio)esterification, aminolysis) (B), depending on the type of nucleophile. Note that the specific rates may differ from this guide based on the basicity of the nucleophile and nucleofuge, the nature of group R, and the solvent polarity.67,80,84–87 | ||
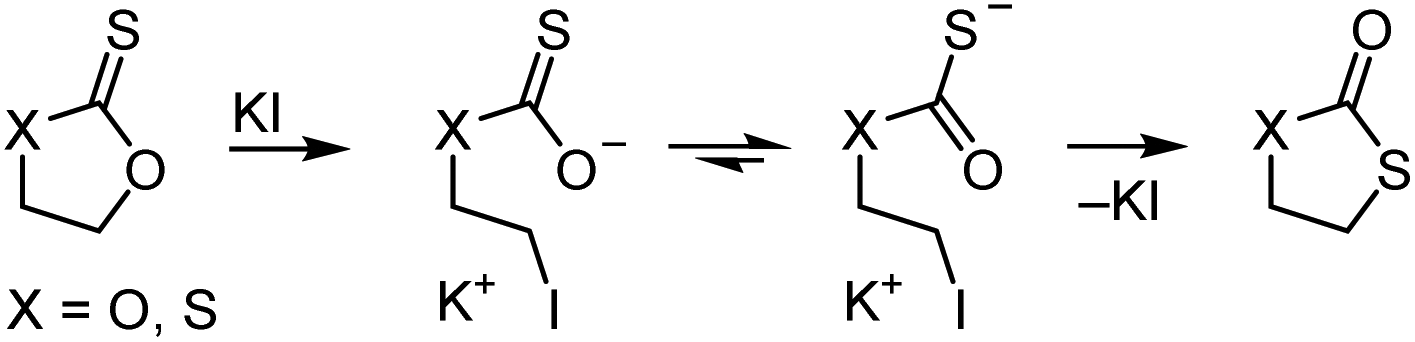 | ||
| Scheme 16 Isomerisation of ethylene thionocarbonate (X = O) and 1,3-oxathiolane-2-thione (X = S) in the presence of halides at 60 °C to form the more stable oxocarbonyl derivative.83 | ||
3.2 Postpolymerization modification
More recently, small molecule94 and RAFT end group95 dithiobenzoates were shown to release hydrogen sulfide in the presence of cysteine or homocysteine95 (in addition to a small molecule/polymer thiol). Hydrogen sulfide finds pharmaceutical application, for example to lower blood pressure,96 making dithiobenzoate-terminated polymers promising for controlled release applications.
Xanthate side group functionality in (meth)acrylic monomers was shown to be stable during RAFT polymerization mediated by a dithiobenzoate (due to better stabilisation of the intermediate; see section 2.3.1), to allow for a simple release of pendant thiol groups through selective aminolysis without harming the (meth)acrylic oxoesters and to enable postpolymerization side group functionalisation.39
3.3 Anionic ring-opening polymerization (AROP)
The nucleophilic ring-opening of lactones and carbonates is a major route toward backbone-degradable polymers such as poly(lactic acid) and poly(ε-caprolactone).99 Similarly, a range of cyclic thionocarbonates, xanthates, and thionolactones has been polymerized through anionic or organocatalytic ring-opening polymerization methods, see Table 2.| Thiocarbonyl species | Mechanism/polymer product | Conditions | Comments |
|---|---|---|---|
|
Anionic Retention of thiocarbonyl and/or S–O isomerism | MeLi, n/sec/t-BuLi, PhLi, t-BuMgCl, t-BuOLi, t-BuOK, DBU, 100 °C (ref. 100) | Product depends on initiator used: poly(thionoesters) produced when a strong nucleophile is used (organolithium, Grignard); mainly poly(thioester) produced with poor nucleophile (DBU), but with low conversions |
| Organocatalytic Retention of thiocarbonyl | Benzyl alcohol, DBU or BEMP, a thiourea, r.t.–100 °C (ref. 101) | Solely produces the thionoester polymer, even when using DBU, due to the inclusion of a thiourea; larger thionolactones have been investigated (8, 9 and 16 membered, a 17-membered dithionolactone), showing the same retention of thionoester moiety, with high conversion (62–92%)102 | |
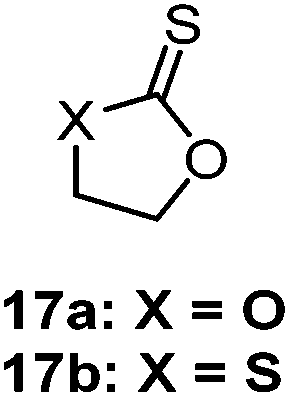
|
Anionic S–O isomerism | Anhydrous acid83 | Similar polymerization of ethylene carbonate leads to ether linkages due to loss of carbon dioxide103 |
|
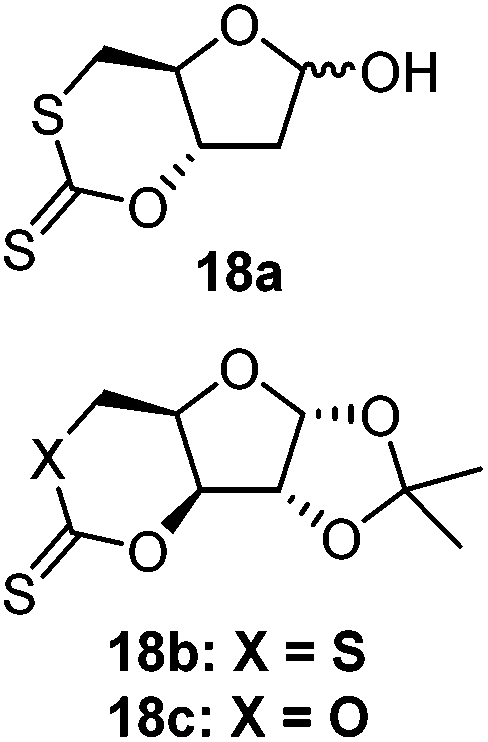
Organocatalytic Retention of thiocarbonyl | 1,5,7-Triazabicyclo[4.4.0]dec-5-ene (TBD, organocatalyst) and 4-methyl benzyl alcohol (initiator)104 | Proceeds via retention of the thiocarbonyl bond, without an S–O isomerism, different to 17, reportedly due to the use of an organocatalyst; 18a was shown to polymerize the fastest due the additional ring strain of the trans configuration of the xanthate ring; 18b was thought to be faster than 18c due to C–S bonds being weaker and easier to break than C–O bonds |
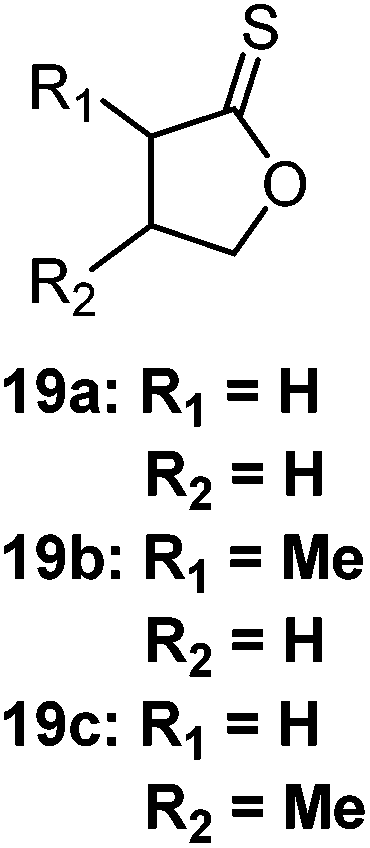
|
Organocatalytic S–O isomerism | BEMP, r.t.105 | Produces only the thioester polymer, through an irreversible mechanism; similar to the DBU ring-opening of ε-thionocaprolactone 16, however no trace of the thionoester is found |
|
Organocatalytic Retention of thiocarbonyl | DMAP, DBU, thiourea106 | Poor yields and conversions achieved with this specific monomer, although retention of the thionoester was found; further promising results have been reported using an aluminium catalyst, leading to isolatable polymers (89% conversion after 3 h) |
The AROP of ε-thionocaprolactone, 16, has been demonstrated to produce two different classes of polymers with either retention of the thionoester units in the backbone or the production of thioester units through an S–O isomerisation.100,107 Here, the initiator/catalyst dictate which mechanism is prevalent. Direct attack of the initiator/anionic chain end onto the thiocarbonyl carbon produced thionoester containing polymers (see Scheme 17, Pathway A), whilst attack via addition onto the ε-carbon produced thioester containing polymers (see Scheme 17, Pathway B).100 Pathway A was referred to as the formation of the kinetic product,100 due to the greater electrophilic nature of the thiocarbonyl carbon compared to the ε-carbon, leading to a faster reaction. This route could be selected by using strongly nucleophilic initiators, such as organolithium and Grignard reagents, which consequently produced only thionoester repeat units with up to 100% conversion. The mechanism resembles that of the AROP of the oxoester analogue, ε-caprolactone.108 Conversely, Pathway B involves a substitution reaction, followed by S–O isomerisation to form the more thermodynamically stable CO bond (as opposed to the CS bond of Pathway A) and thiocarboxylate anion. This route is also followed by monomer 17 (see Table 2) and was achieved with ε-thionocaprolactone when using DBU as the ‘initiator’. However, this pathway was shown to be much slower than pathway A, leading to only 25% conversion, and a maximum of 89% thioester units, whilst still incorporating 11% of the thionoester units.100
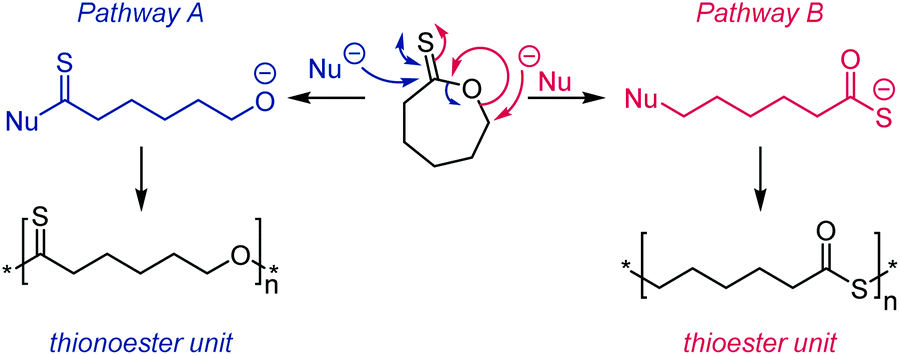 | ||
| Scheme 17 The two potential mechanisms for AROP of ε-thionocaprolactone, 16. Pathway A: use of a strong nucleophile, such as an organolithium reagent, formed thionoester polymers; Pathway B: use of a poor nucleophile, such as DBU, formed thioester polymers through an S–O isomerisation. Adapted from ref. 100 with permission from the American Chemical Society, copyright 1999. | ||
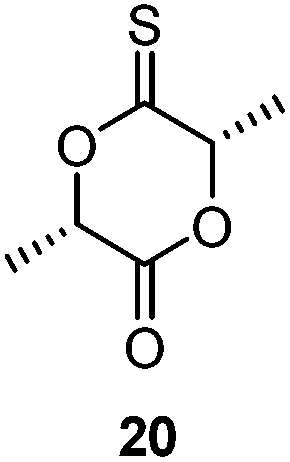
The thionoester containing polymer has also been synthesised through organocatalytic ROP of ε-thionocaprolactone, using a non-nucleophilic base (such as DBU and 2-t-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine (BEMP)), benzyl alcohol and a thiourea.101 Here, the thiourea is thought to hydrogen-bond to the sulfur atom of the thionolactone, further activating the thiocarbonyl carbon toward nucleophilic attack, see Scheme 18.99 This allowed selectivity of the polymer makeup when using DBU, as both thionoester and thioester polymers can be produced, depending on the inclusion, or absence, of a thiourea. Likewise, monomer 20 could be organocatalytically polymerized with retention of the thiocarbonyl function.106
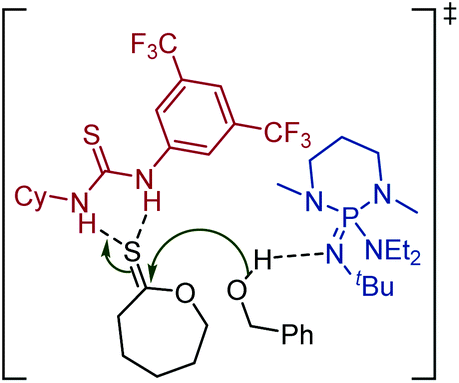 | ||
| Scheme 18 Organocatalytic ROP of ε-thionocaprolactone using a thiourea to help selectively produce the thionoester polymer, poly(ε-thionocaprolactone). Shown is the transition state.99,101 Reproduced from ref. 101 with permission from the American Chemical Society, copyright 2016. | ||
This use of a thiourea as a polymerization catalyst demonstrates another application of CS functional molecules in polymer science.101 Several similar catalysts (along with their oxo-urea analogues) have been investigated for the ring-opening of δ-valerolactone, ε-caprolactone, and lactide.109,110 For thiourea catalysts, the rate of polymerization has been demonstrated to be highly dependent on the functional groups attached to the nitrogen atoms and solvent choice, with faster rates found for non-polar solvents. A second, (thio)imidate-mediated, mechanism has been shown to be in competition with the mechanism shown in Scheme 18.111 This second mechanism is preferred with higher monomer concentrations, polar solvents, higher temperatures, and stronger base cocatalysts.
A recent contribution demonstrated that even 5-membered thionolactones (19) can undergo ring-opening polymerization, using a basic phosphorous catalyst (such as BEMP) and ambient conditions.105 Here, the thioester product is the only polymer produced, utilising the S–O isomerisation as the main driving force and overcoming the lack of ring-strain.112–114 However, two competing side reactions occur. These are the S–O isomerisation reaction from the thionolactone to the thiolactone, and the production of dimers from the thionolactones. Maximum monomer conversions were limited with this method, achieving 46–51%, with a 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :36:4 ratio of polymer:isomerisation product:dimer. Notably, the thionolactones monomer was synthesised from succinic acid derived from biomass, demonstrating the ability to produce degradable polymers from sustainable sources.105
:36:4 ratio of polymer:isomerisation product:dimer. Notably, the thionolactones monomer was synthesised from succinic acid derived from biomass, demonstrating the ability to produce degradable polymers from sustainable sources.105
3.4 Anionic polymerization of thiocarbonyls
The homopolymerization of CS2 involved the formation of a black solid,120 which did not only contain the expected linear dithioester polymer repeat unit (Scheme 19, structure I), but instead produced a complex concoction of many different sulfur moieties, such as trithiocarbonates,78 and disulfides (structure II), along with several cyclic polymers (structure III)121 and small molecules, for example the cyclic dimer of CS2 (structure IV). Reported conditions for the homopolymerization are typically harsh and have included sodium initiators (anionic polymerization),122 high pressures,78 ultraviolet123 and gamma irradiation,124 and, most recently, electroreduction,125 all of which led to the production of a very similar product mixtures.
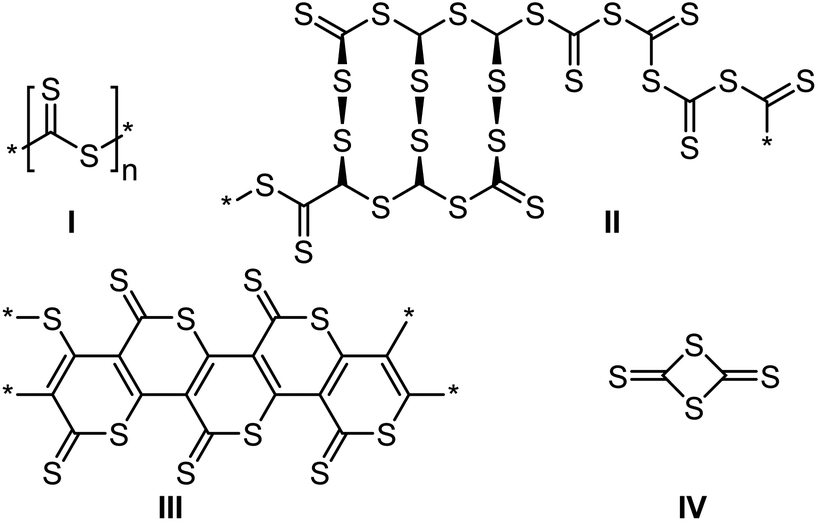 | ||
| Scheme 19 Multiple products of the homopolymerization of carbon disulfide.121,123 | ||
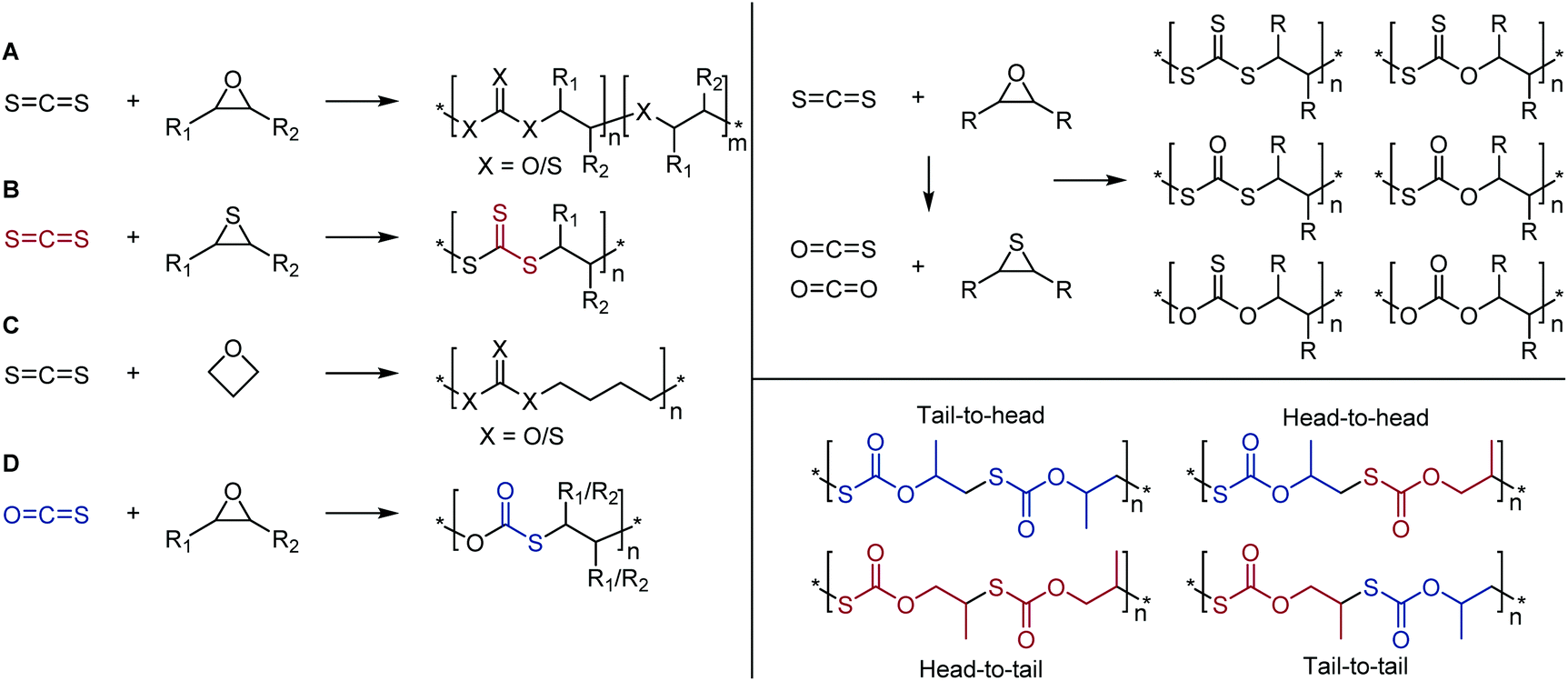 | ||
| Scheme 20 Left: Copolymerization reactions of carbon disulfide with (A) epoxides, (B) thiiranes, (C) oxetane, and (D) copolymerization with carbonyl sulfide and epoxides; Right top: O/S scrambling of CS2 and epoxides leading to a large range of backbone polymer moieties; Right bottom: copolymerization of COS and propylene oxide leading to different polymer microstructures. Adapted from ref. 133 with permission from the American Chemical Society, copyright 2016. | ||
In addition to epoxide comonomers, the copolymerization of CS2 can be achieved with other highly strained ring systems such as thiiranes and the 4-membered oxetane. With methyl thiirane (see Scheme 20B), the poly(trithiocarbonate) was exclusively produced when using (salen)CrCl as the catalyst. This was due to the absence of oxygen-containing species in the reaction mixture, hindering the formation of thiocarbonates and xanthates.115 Here, the polymer is produced in an entirely alternating fashion, with the major by-product being the cyclic trithiocarbonate small molecule. It was found that using a greater ratio of CS2 to methyl thiirane (2:1 as opposed to 1:1) led to the greatest ratio of polymer compared to the cyclic by-product (92% polymer). More recently other sulfur-rich polymers were produced by using cyclohexene sulfide, forming the same trithiocarbonate containing polymer, along with some of the cyclic trithiocarbonate by-product (>80% polymer), see Scheme 21.134
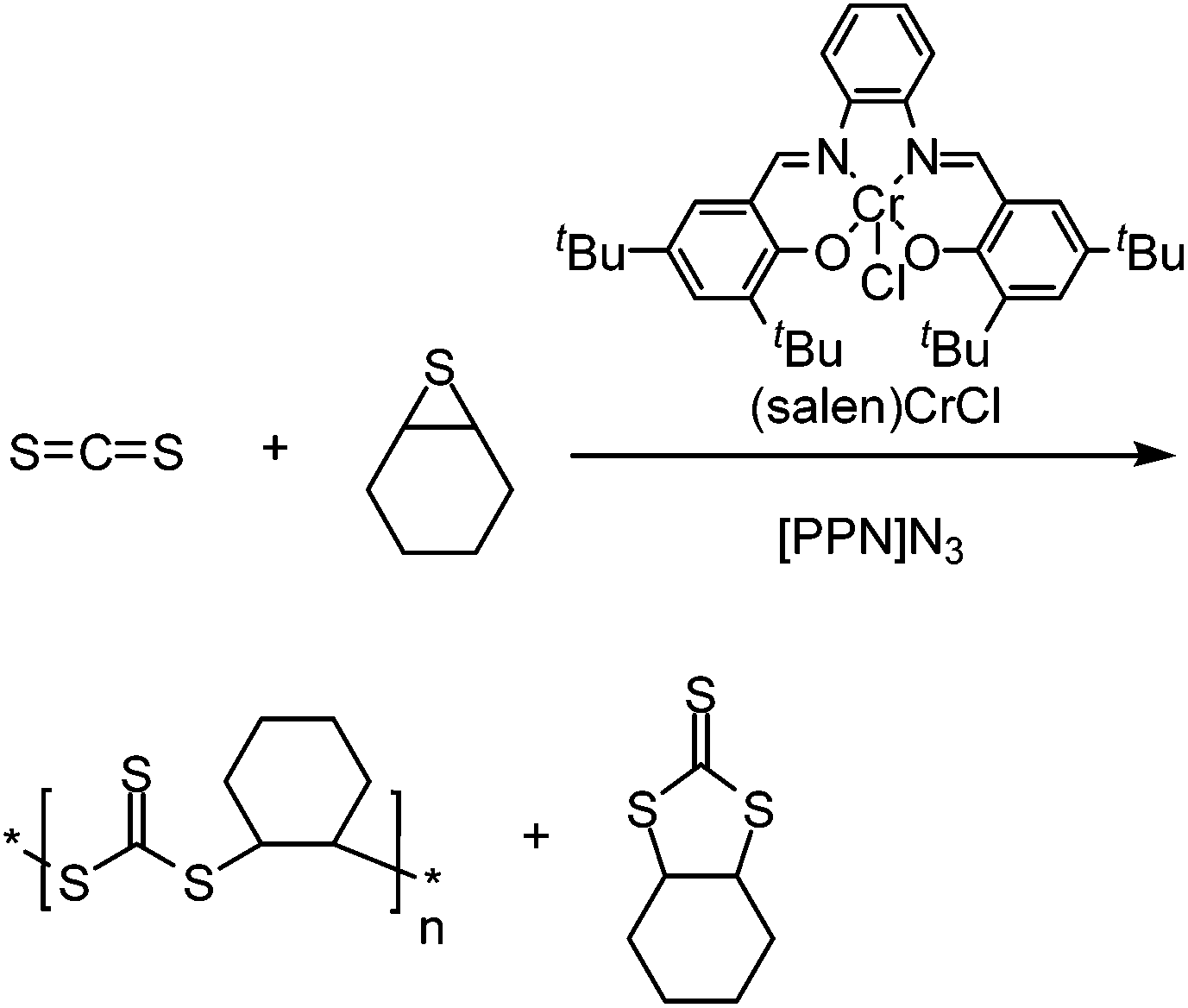 | ||
| Scheme 21 Production of poly(trithiocarbonates) using thiiranes and CS2.134 | ||
With oxetane as comonomer, multiple repeat units can be produced, similar to epoxide copolymerizations, see Scheme 20C.116 In this case, however, the main units incorporated were the thiocarbonate and trithiocarbonate moieties, with the ratios being dictated by the ratio of CS2, the temperature, and the cocatalyst. It was found that a greater CS2–oxetane ratio led to the formation of repeat units with a higher sulfur content (e.g., as xanthates), and higher likelihood of CS bond incorporation. Increasing the temperature made CS to CO switching more likely, leading to the formation of more carbonate and thiocarbonate repeat units. The mechanism for the O/S-scrambling was also discussed in this article.116
An analogous reaction occurs when using carbonyl sulfide (COS) and epoxides. However, this combination exclusively leads to the poly(thiocarbonate) product, preferentially retaining the more thermodynamically stable CO bond over the CS bond, see Scheme 20D.20,21,135 Two potential units still can form however, depending on whether the thiocarbonate anion prefers to attack the carbon atom of R1 or R2. If one of the groups is a hydrogen and the other an electron-donating group, then attack will occur on the least hindered end of the epoxide, forming the tail-to-head polymer, see Scheme 20.135 If, however, a sterically hindered but also electron withdrawing group is used, such as in the case of styrene oxide, attack will occur on either carbon atom, due to both a steric driving force and an electronic driving force. In this case, the metal catalyst can dictate the predominance of either unit forming.130 Carbonyl sulfide can also be reacted with oxetane to produce the poly(thiocarbonate) exclusively, in a similar fashion to Scheme 20C.133
Another copolymer stemming from carbon disulfide involves ketene cyclic N,O-acetals. This copolymerization leads to the formation of polymer repeat units containing both dithioesters and amides, see Scheme 22.117 The resulting polymer is zwitterionic, with ionic groups found at the two polymer terminals; containing a cyclic N,O-acetal cation at the α-end and a dithioester anion at the ω-end. Polymerization involved the enamine attacking the CS2 carbon, followed by nucleophilic attack of the formed sulfur anion on another ketene cyclic N,O-acetal, leading to ring-opening and propagation. Polymerization without the inclusion of the two methyl groups in the ketene cyclic N,O-acetal led to a tautomerism with the acidic protons between the thiocarbonyl and carbonyl group. The sulfur atom from the in-chain dithioester can then react with further ketene cyclic N,O-acetals, leading to the production of two different polymer repeat units.
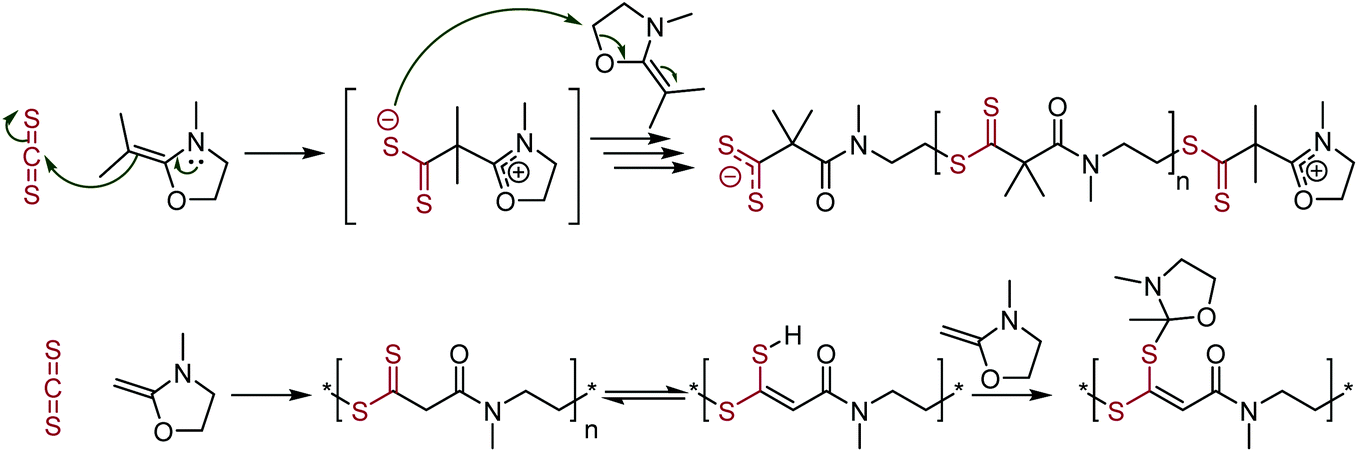 | ||
| Scheme 22 Polymerization of ketene cyclic N,O-acetals, forming dithioester- and amide-containing polymers. The top scheme shows the mechanism of the polymerization. The bottom scheme shows side reactions when using the hydrogen species, rather than the dimethyl analogue.117 | ||
Another route that does not involve an epoxide is the use of a trithiocarbonate ion reacting with a dihalogen species using a tetraalkyl ammonium catalyst for phase-transfer.136,137 Here, the trithiocarbonate ion (CS32−) can be synthesised from CS2 and NaOH, and the dihalogen species can be either an alkyl chain or aromatic, as long as the halogen can be substituted. For an alkyl chain, if n ≥ 2 (Scheme 23) a polymer will form, however for n = 0, 1, it is speculated that the intramolecular reaction is preferred, forming 5/6-membered trithiocarbonates.137 Including a stoichiometric amount of a diol compared to the dihalogen species, and aqueous sodium hydroxide resulted in the formation of the xanthate polymer, instead of the trithiocarbonate.138
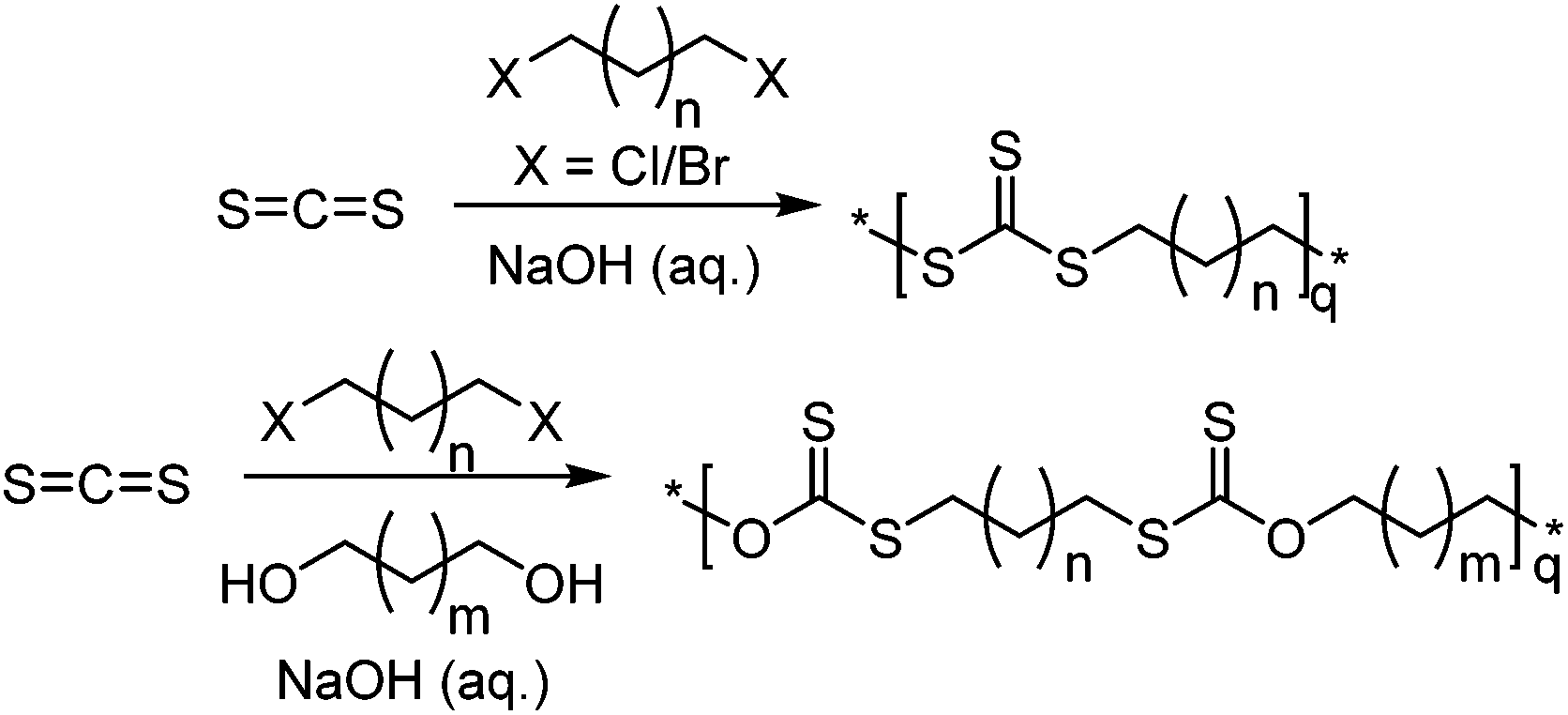 | ||
| Scheme 23 Top: Production of trithiocarbonate anions from CS2 and sodium hydroxide and their use to produce alternating copolymers with a dihalogen species. Bottom: Production of poly(xanthate) through the use of a dihalogen and diol with carbon disulfide and aqueous sodium hydroxide. | ||
S) undergoes anionic polymerization in anhydrous solvents at low temperatures when initiated by metal alkoxides, amines, or even DMF, see Scheme 24. The resulting homopolymer was a chemically resistant elastomer that survived immersion into boiling fuming nitric acid for 1 h and showed little change when immersed in boiling 40% aq NaOH for 24 hours. Conversely, exposure to organic amines at RT or temperatures above 175 °C caused rapid depolymerization of the native polymers. This degradability was prevented by capping the ends with CF3 groups through antimony pentafluoride. Although the very low glass transition temperature of −118 °C is promising for application as a low temperature elastomer, the slow crystallisation at temperatures below its melting point of 35 °C and associated loss of its elastic properties, however, may hamper this use.76,139 Under similar conditions, other fluorinated thiocarbonyl compounds were found to undergo anionic polymerization, although the resulting materials were not as chemically resistant. Poly(hexafluorothioacetone) produced at −110 °C underwent depolymerization at RT.76,139
 | ||
| Scheme 24 Anionic polymerization of thiocarbonyl fluoride (top) and structures of other thiocarbonyl compounds to undergo this polymerization (bottom). | ||
4. Reactions with electrophiles
Due to the large S-lone pairs, thiocarbonyls are more nucleophilic than the corresponding O-analogues. For example, thiourea reacts under mild conditions with alkyl halides to form isothiouronium salts which can conveniently be hydrolysed into alkyl thiols.140 In the polymer field, the most important reactions of thiocarbonyls with electrophiles are cationic ring-opening polymerization (CROP) and cationic RAFT.4.1 Cationic ring-opening polymerization (CROP)
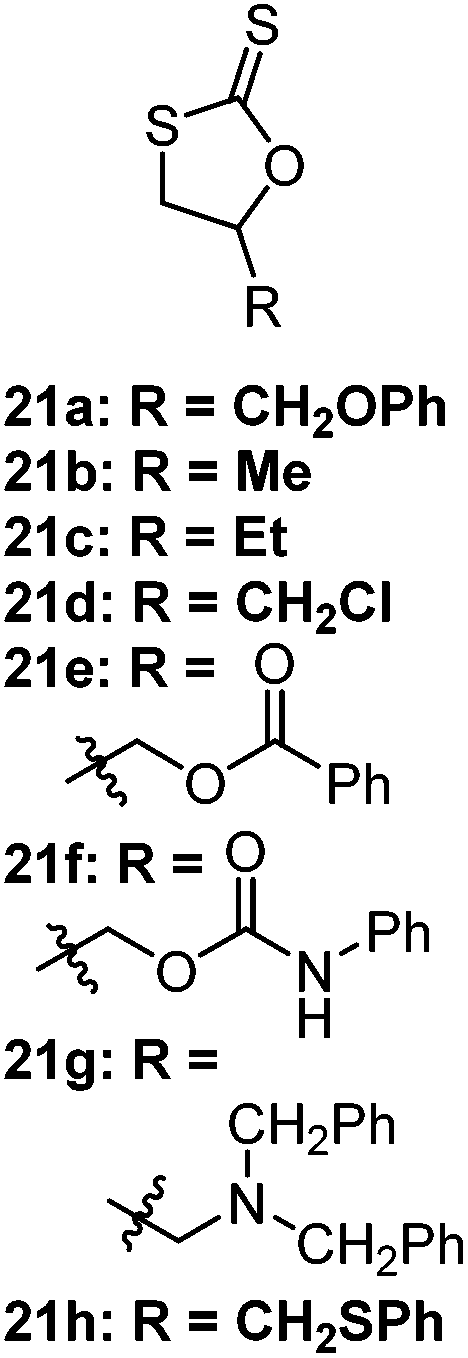
The CROP mechanism can be demonstrated on the cyclic xanthates 21a–d, see Scheme 25 and Table 3.141 There are several potential pathways that can occur depending on the conditions used. Using the Lewis acids TfOEt or BF3OEt2 to activate the xanthate group has two possible outcomes: (i) an S–O isomerisation of the cyclic xanthate 21, converting the monomer into a cyclic dithiocarbonate (path A, A′) in the absence of polymerization; (ii) polymerization can occur through an S–O isomerisation, forming polythiocarbonates (path B, B′). It was shown that increasing the amount of Lewis acid catalyst led to greater amounts of polymer being formed (path B, B′). Interestingly, depolymerization was also shown to occur through a backbiting reaction when using TfOEt (path E), demonstrated by a decrease of polymer molecular weight throughout the polymerization. Attempts to polymerize the dithiocarbonate isomer were not successful under any conditions used (path F). Conversely, when triflic acid was used as the Lewis acid no polymer was formed (path D, D′), but exclusively the cyclic dithiocarbonate isomer (path C, C′). Monomers 22 (see Table 3) followed the same pathways, producing polymers through path B, B′, the cyclic isomer by path A, A′, and depolymerization through path E.142 Cationic polymerization of ε-thionocaprolactone, 16, led exclusively to the thioester containing polymer, through path B, B′.107,141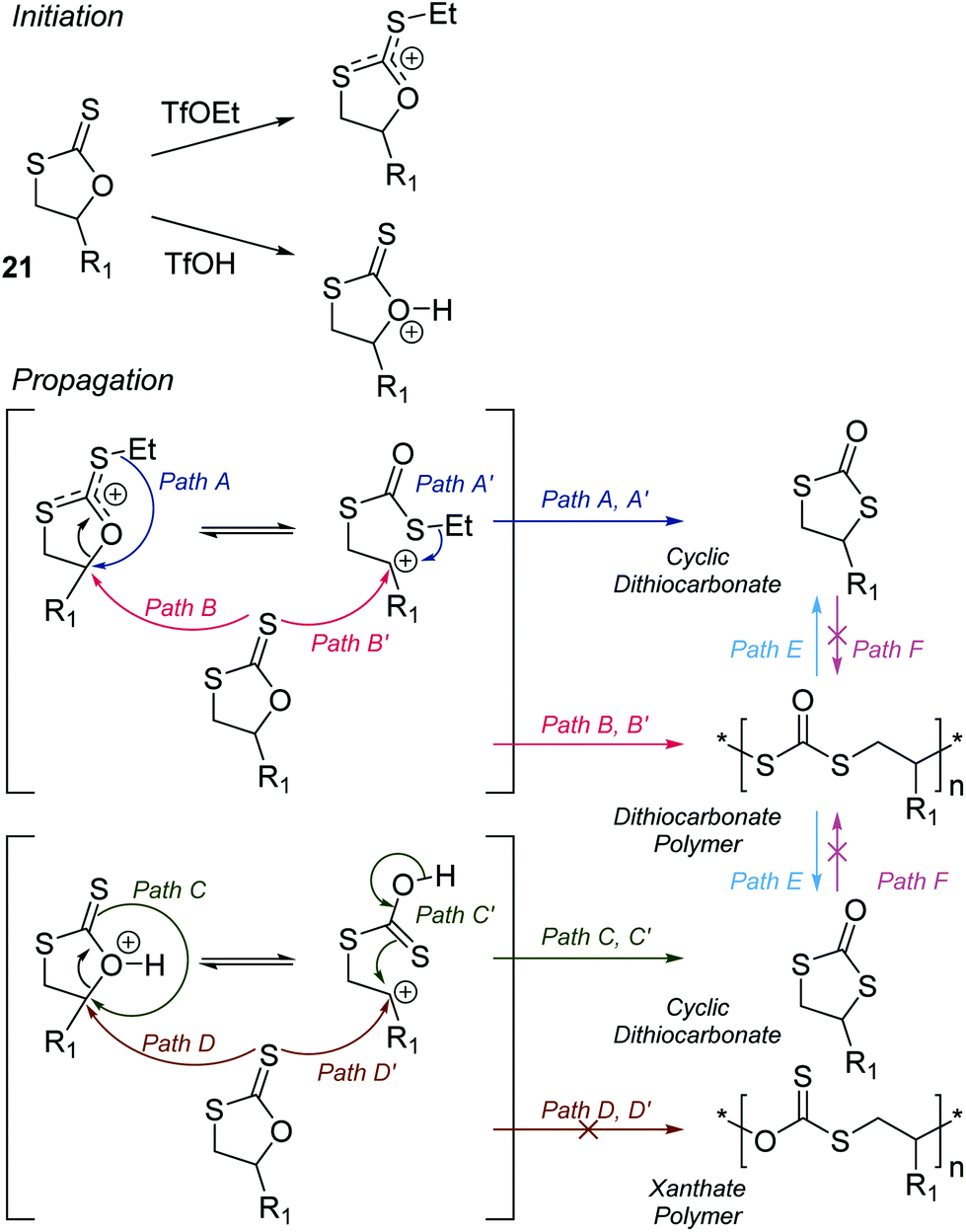 | ||
| Scheme 25 CROP mechanism of cyclic xanthates (21) as a representative cyclic thiocarbonyl compound. The product of the polymerization is Lewis acid and concentration dependent. Reproduced from ref. 141 with permission from the American Chemical Society, copyright 1998. | ||
| Thiocarbonyl species | Mechanism/polymer product | Conditions | Comments |
|---|---|---|---|
|
Cationic S–O isomerism | TfOMe or TfOEt, r.t.–60 °C; BF3OEt2 at 60–100 °C (ref. 143 and 144) |
21a–d = high polymerization conversions with dispersities as low as Đ = 1.23; isomerisation to the cyclic thiocarbonate in the presence of other Lewis acids (ZnCl2, SnCl4, TfOH); greater polymer:cyclic thiocarbonate ratios were achieved with higher concentrations of Lewis acid (2% = 38:62; 20% = 82:18); reactivity followed 21b/c > 21a > 21d, reportedly due to electron donating ability of the R-group |
| 21e–h undergo ring-opening through neighbouring group participation,141,145–149 leading to living polymerizations and different polymer backbones to monomers 21a–d | |||
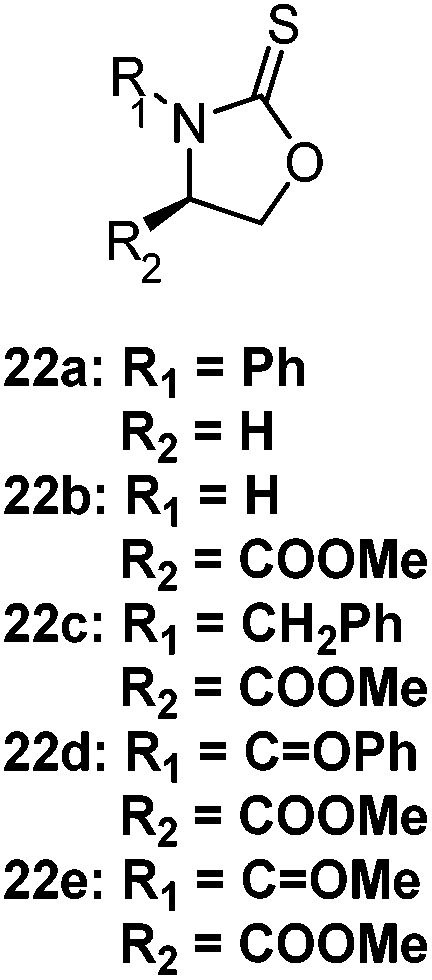
|
Cationic S–O isomerism | 22a = P(OEt)3, 140–160 °C (ref. 142) | 22a: depolymerization occurs at higher temperatures (179 °C) and longer reaction times (20 h versus 3 h); analogous O-analogue did not undergo polymerization under the same conditions142 |
| 22b–e = TfOMe, 30 °C (ref. 150) | Reactivity followed 22b > 22c > 22d > 22e, in agreement with the nucleophilicity of the sulfur atoms in each monomer150 | ||
|
Cationic S–O isomerism | BF3OEt2, TfOH, TfOMe, TfOEt, 0–28 °C (ref. 107) | Solely produces the thioester polymer, opposite to the preferred product of the anionic mechanism |
The CROP of cyclic xanthates has been explored in producing degradable crosslinked polymers, see Scheme 26.151 Here, the use of a bis(cyclic xanthate) allowed ring-opening and polymerization through both units, whereby each produced dithiocarbonate units whilst forming the polymer network. This could then be degraded or ‘depolymerized’ through the addition of t-BuOK, leading solely to the cyclic dithiocarbonate isomer that can also form as a side-product through polymerization.
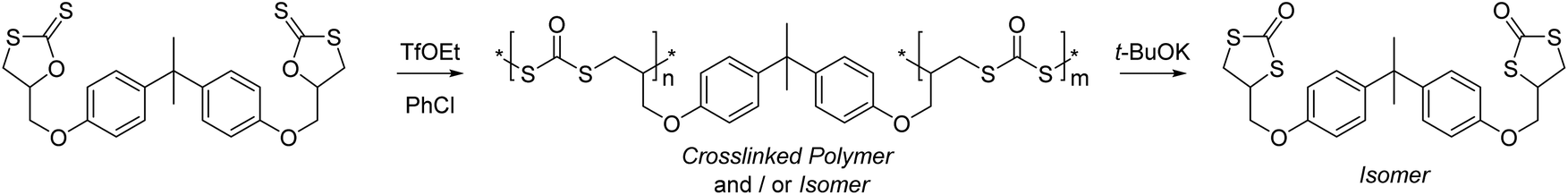 | ||
| Scheme 26 CROP of a bis(cyclic xanthate) leading to crosslinked polymers. Reproduced from ref. 151 with permission from the American Chemical Society, copyright 2005. | ||
Similarly to the cyclic xanthates 21a–d, monomers 21e–h undergo an S–O isomerisation and form polydithiocarbonates. However, their backbones varied slightly due to neighbouring group participation.149 Demonstrated in Scheme 27 with 21e as an example, the oxygen of the ester side group can encourage ring-opening to form intermediate III. This intermediate was observable by NMR spectroscopy due to its stability and led to a living polymerization (Đ < 1.2), due to its long lived nature. Upon ring-opening, path G was then exclusively followed, instead of path A or B which was favoured by monomers 21a–d in Scheme 25. This led to migration of the ester group to the adjacent carbon atom, and instillation of an additional CH2 group in the polymer backbone. The absence of any of the cyclic isomer (IV) and the expected polymer (V) for 21e, was suggested to be due to steric factors making path A/B less favourable than path G. The cyclic isomer was also not produced when TfOH was used as the initiator, suggesting suppression of the intramolecular isomerisation of intermediate I seen with 21a–d, due to the ability to form the stable intermediate III.
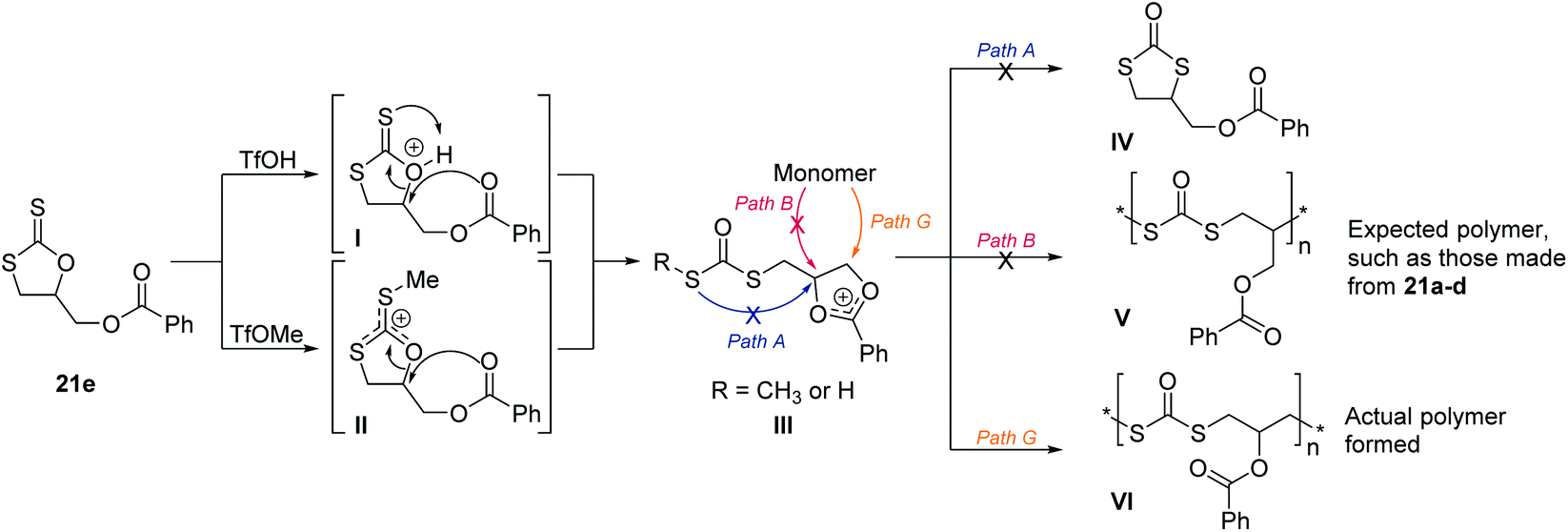 | ||
| Scheme 27 CROP mechanism of cyclic xanthates featuring neighbour group participation, demonstrated on monomer 21e, and followed analogously by monomers 21f–h. The ester oxygen encourages ring-opening, forming a more stable carbenium cation through the two oxygen atoms and a phenyl group. Path G was shown to be the selective route for 21e. Reproduced from ref. 149 with permission from John Wiley and Sons, copyright 2001. | ||
This has been replicated with other neighbouring groups such as other functionalised esters,149 carbamates (21f),148 thioethers (21h),145,146 and most recently, a tertiary amine (21g).147 Path G was exclusively followed by the monomers 21e and 21f. Livingness was observed for all monomers 21e–h. For monomers 21g and 21h, some of the polymer product (<35%) from path B (Scheme 27) was also observed along with the predominant ‘path G’ product. 21h led to the production of some of the cyclic isomer through path A.146 Despite this, the polymerization of the thioether containing monomers (21h) were reported to be living as well. Other cyclic xanthates, containing a spiro thia-heterocycle, have led to similar results along with ring contraction, see Scheme 28.145
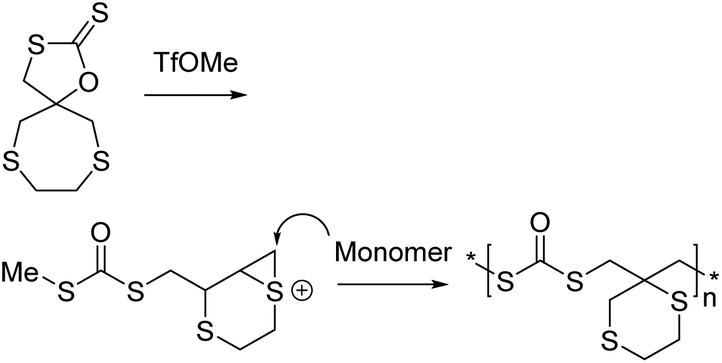 | ||
| Scheme 28 CROP of a cyclic xanthate containing a spiro thia-heterocycle. Reproduced from ref. 145 with permission from Elsevier, copyright 2020. | ||
4.2 Cationic RAFT
Living cationic polymerization was developed in the 1980s. The concept (which is still one of the most-used ones today) relies on the reversible deactivation of the carbocationic chain end through the reversible transfer of an anion from a suitable Lewis acid catalyst.152 In 2015, metal-free living cationic polymerization through a reversible addition–fragmentation chain transfer (RAFT) process was described153 following earlier work that involved a Lewis acid catalyst and additives.154 The new concept has recently been reviewed155 and we limit this section to a brief account of thiocarbonylthio compounds for cationic chain transfer. An example of cationic RAFT using a dithiocarbamate is shown in Scheme 29. Efficient transfer of the cation onto the chain transfer agent requires +M substituents that stabilise the intermediate cation and enhance the nucleophilicity of the thiocarbonyl sulfur (Scheme 7A). Thus, dithiocarbamates (Z = NR2), trithiocarbonates (Z = SR), and xanthates (Z = OR) were found to work well, with the lowest dispersities (Đ = 1.08) observed for the polymerization of isobutyl vinyl ether mediated by a dithiocarbamate chain transfer agent.153 To transfer the cation onto the R-group of the chain transfer agent, it likewise needs to stabilise cations and was conveniently derived from the same vinyl ether monomer.153 Interestingly, the authors described diblock copolymers prepared through a switch from cationic to radical RAFT polymerization.153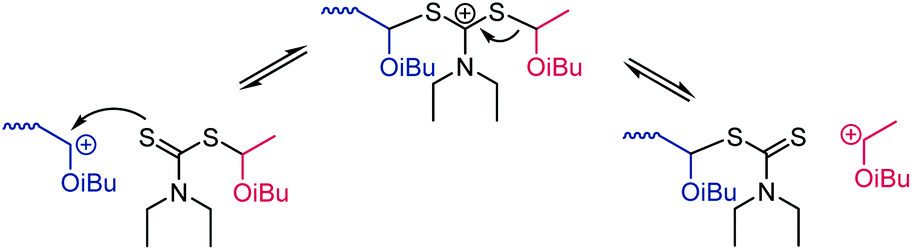 | ||
| Scheme 29 Mechanism of cationic RAFT polymerization shown for the example of poly(isobutyl vinyl ether) (blue) mediated by a dithiocarbamate chain transfer agent carrying an isobutyl vinyl ether-derived R-group (red). The initiator and counterion are triflic acid and triflate, respectively (not shown). | ||
5. Other
5.1 Thiocarbonyl groups in hetero-Diels–Alder reactions
Thiocarbonyl groups can function as dienophiles in hetero-Diels–Alder (HDA) reactions, allowing for the construction of substituted thianes. Work in the late nineties156,157 established a computationally-based understanding of these reactions and concluded that (excluding solvent effects) electron-withdrawing substituents on the thiocarbonyl dienophile promote higher conversions to the HDA product by lowering the LUMO. Electron poor CS compounds employed in HDA cyclo-additions have thus included cyanodithioformates,158–160 thioaldehydes,160 polyfluoroalkyl thionoesters and dithioesters,161 phosphonodithioformates under Lewis acid catalysis,162,163 2, 3, and 4-pyridinedithioesters (activated through protonation, Lewis acid complexation, or formation of the N-oxides) in the synthesis of the drug Aprikalim,164 and monothiono oxalic derivatives,163,165 including with high enantioselectivity, see Scheme 30.163,165,166 Thiabutadiene derivatives, conversely, have been used as dienes in HDA reactions.167
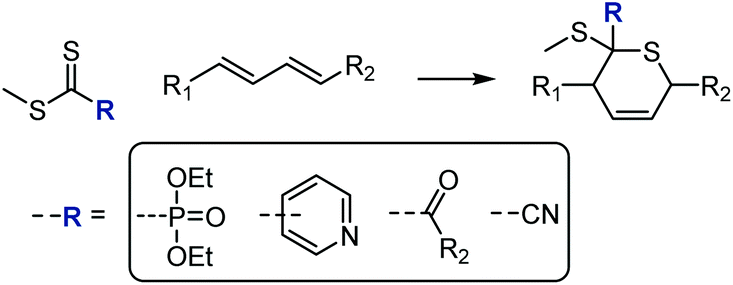 | ||
| Scheme 30 Hetero-Diels–Alder [4 + 2] cycloadditions on thiocarbonyls carrying electron-withdrawing groups. | ||
The concept has been triumphantly exploited in the polymer arena on RAFT end groups.168–170 Inspired by the high reactivity of 2-pyridine and phosphono-functional dithioformates in HDA reactions, RAFT agents 23 and 24 (Scheme 31) were prepared and shown to successfully mediate RAFT polymerizations. HDA reactions with a variety of diene- and cyclopentadiene-end capped polymers resulted in highly efficient conjugation and the preparation of diblock copolymers even at RT171–173 and in water.174 Star-shaped polymers with two, three, or four arms were efficiently prepared by grafting arms onto a multi diene-functional core.175 The high efficiency of the reaction was also exploited to graft diene-end terminal chains onto divinylbenzene microspheres prepared with a phosphonodithioformate 23.176 Like most (hetero-)Diels–Alder cycloadditions, the thiocarbonyl–diene reaction is thermally reversible. This concept was exploited to anchor a cyclopentadiene-protected RAFT agent onto a surface through catechol binding. Heating with a cyclopentadiene end-functional polymer resulted in retro-Diels–Alder loss of the cyclopentadiene protecting group and concurrent Diels–Alder binding of the cyclopentadiene-terminal polymers, resulting in surface grafting.177
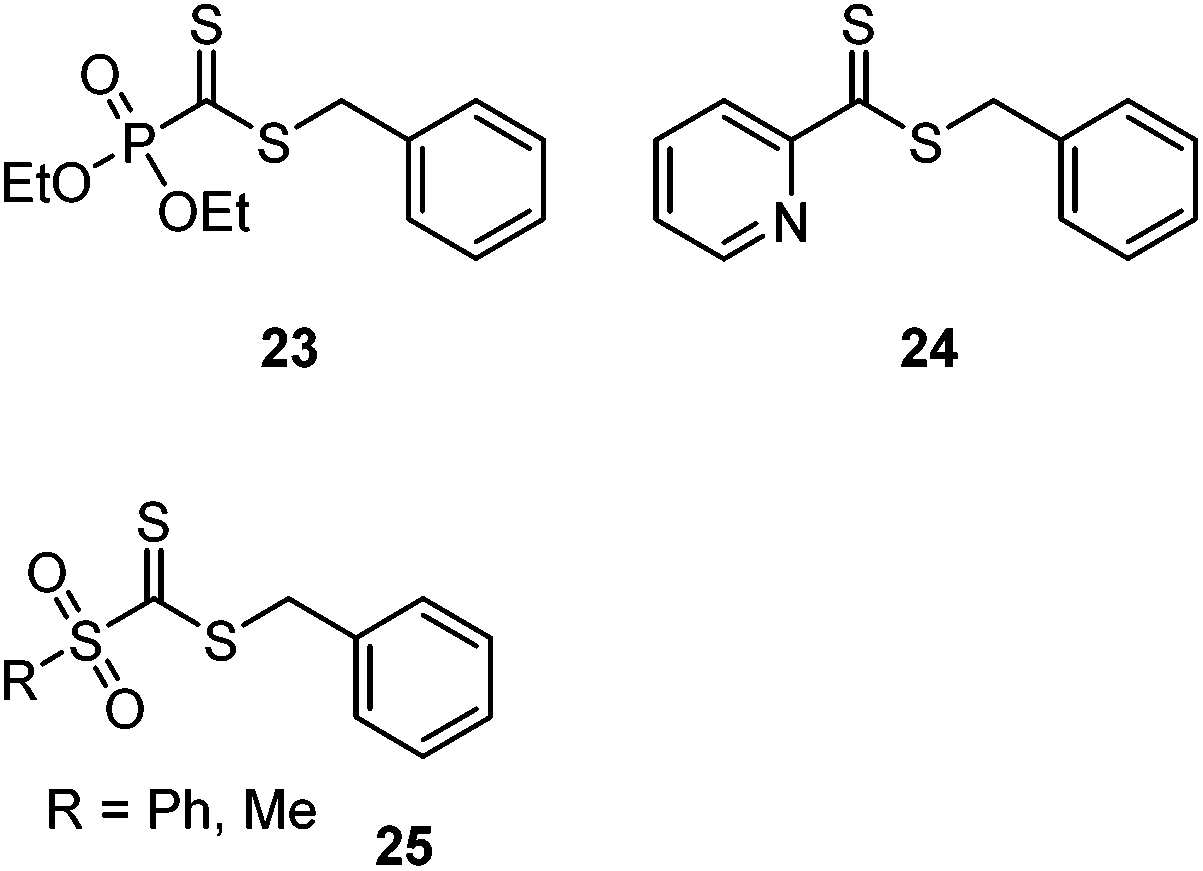 | ||
| Scheme 31 Structures of RAFT agents with electron-poor CS groups suitable for (post-polymerization) hetero-Diels–Alder cycloadditions with diene-functional polymers or surfaces. | ||
Following computational predictions, a novel RAFT agent, 25, was prepared which features a very electron poor sulfonyl-functional thiocarbonyl group.178 This chain transfer agent successfully mediated the RAFT polymerization of an acrylate and underwent HDA cycloadditions with dienes even in the absence of a catalyst. The dienophile was so reactive it even reacted with styrene. This reaction was subsequently used to modify silica surfaces pre-treated with a trialkoxysilyl-functional styrene derivative.179
5.2 Thermolysis of RAFT end groups
As this review documents, thiocarbonyl groups undergo an impressive range of reactions. Their high reactivity, however, can result in thermal lability. Several studies have investigated the thermal stability of RAFT agents.180–182 The onset of thermal degradation depended heavily on the structure, with an aliphatic xanthate decomposing above 75 °C, while dithiobenzoates were generally stable until around 180 °C, with degradation usually involving a combination of pathways.180 The decomposition of a RAFT agent during polymerization may result in loss of control and retardation through the formation of side products.182 Likewise, the thermal stability of a RAFT-made polymer and the degradation pathway depend on both the RAFT agent Z group and the type of repeat unit. Two mechanisms have been observed. Temperature-triggered radical C–S bond scission, followed by backbiting, radical decomposition of the polymer backbone and formation of multiple products was observed on trithiocarbonate-terminated poly(butyl acrylate)183 and poly(methyl methacrylate).184 Conversely, “clean” Chugaev eliminations were found for dithiobenzonate-terminated poly(methyl methacrylate) (Scheme 32A)184 and trithiocarbonate-terminated polystyrene (Scheme 32B).183 As such, thermal eliminations have been employed as a means to remove RAFT agent end groups.89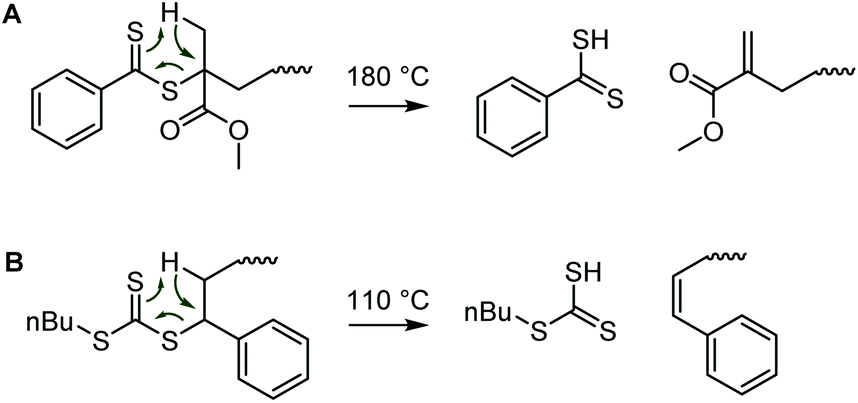 | ||
| Scheme 32 (A) Thermal Chugaev eliminations on dithiobenzoate-terminated poly(methyl methacrylate)184 and (B) trithiocarbonate-terminated polystyrene.183 Both reactions provide intact polymers with terminal unsaturation. The respective RCSSH products are unstable and degrade further. | ||
5.3 UV-Vis absorbance of thiocarbonyl compounds
Most thiocarbonyl compounds have two UV-visible absorbance maxima. A weak n–π* transition in the visible range is responsible for the characteristic colour of many compounds; trithiocarbonates, thionolactones, xanthates, and dithiocarbamates are typically yellow (absorbance around λ = 430 nm)63,185 while most dithiobenzoates and thiophosgene are dark red (absorbance around λ = 520 nm).185 This colour is very useful, for example, during chromatographic separation of thiocarbonyl compounds or to judge crudely and quickly whether RAFT end groups are still present following a polymerization or post-polymerization modification reaction, or qualitatively monitor the disappearance of thionolactones during TARO polymerization.64 The much stronger π–π* transition lies in the UV range with peaks around λ = 300–310 nm for dithioester, trithiocarbonate, dithiocarbamate RAFT agents and is exploited for the determination of number-average molecular weights (assuming that all chains are terminated by RAFT agent end groups),185 or to quantitatively monitor reactions of RAFT end groups.169Sufficiently strong UV irradiation can cause photolytic cleavage of a C–S bond, which can be exploited for photoinitiation of RAFT reactions, in the absence of traditional radical initiators.186
In the realm on non-vinyl polymers, a conjugated thioquinacridone-based polymer was presented as an efficient photosensor for mercury ions. In its thiocarbonyl state, the polymer was only weakly fluorescent and appeared dark green. Micro-molar quantities of Hg2+ cations catalysed the dethionation toward the oxocarbonyl analogue which was bright red with strong fluorescence originating from the quinacridone fluorophore, see Scheme 33.187
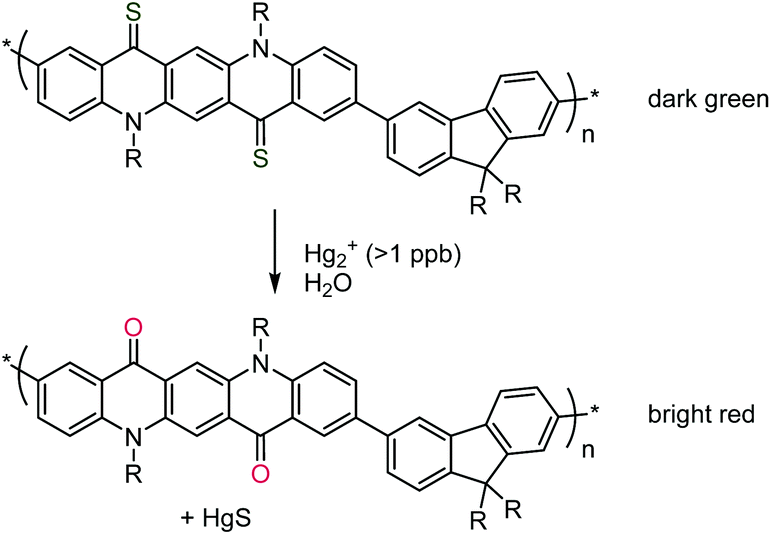 | ||
| Scheme 33 Mercury-sensing conjugated polymer that changes colour based on the catalysed dethionation of two thiocarbonyl groups. Reproduced from ref. 187 with permission from the Royal Society of Chemistry, copyright 2014. | ||
6. Outlook
Thiocarbonyl compounds have been exploited in polymer science for over a century. But the recent increase of work in several fields promises that more fascinating times lie ahead. In particular, the reaction of dithiobenzoate RAFT end groups with (homo)cysteine to produce hydrogen sulfide is a promising avenue to release and deliver this pharmaceutically important small molecule. Increasing the control over macromolecular synthesis hinges mainly on achieving monomer sequence control and the insertion of single monomer units.188 For RAFT, these goals require an excellent understanding of the stability and behaviour of thiocarbonyl-derived radicals. The reversibility of the thiocarbonyl Diels–Alder adducts is promising for the development of reversible RAFT agent protecting groups that could survive a postpolymerization modification of side groups. The radical ring-opening of thionolactones has the advantage over conventional systems that the resulting thioester backbone units can be cleaved selectively under mild conditions. It is foreseeable that the TARO concept will be expanded from thionolactones to other cyclic thiocarbonyl monomers (cyclic xanthates, cyclic thionocarbonates) to develop novel monomers compatible with all classes of vinyl comonomers. The organo-catalytic ring-opening polymerization of thionolactones can likewise be driven by the CS → CO isomerization; its application to nature-derived 5-membered rings is promising for the development of sustainable materials. Notably, the fact that the same thionolactone monomers can undergo radical, cationic, anionic, and organo-catalytic ring-opening polymerizations increases their potential for future industrial significance.
Conflicts of interest
There are no conflicts to declare.References
- D. H. R. Barton and S. W. McCombie, J. Chem. Soc., Perkin Trans. 1, 1975, 1574–1585, 10.1039/P19750001574.
- D. Crich and L. Quintero, Chem. Rev., 1989, 89, 1413–1432 CrossRef CAS.
- S. Z. Zard, Helv. Chim. Acta, 2019, 102, e1900134 CrossRef.
- S. Z. Zard, in Handbook of RAFT Polymerization, 2008, DOI:10.1002/9783527622757.ch5.
- R. Mülhaupt, Angew. Chem., Int. Ed., 2004, 43, 1054–1063 CrossRef PubMed.
- G. W. Blanco, Ind. Eng. Chem., 1926, 18, 1257–1259 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- G. Moad, in Controlled Radical Polymerization: Mechanisms, American Chemical Society, 2015, vol. 1187, ch. 12, pp. 211–246 Search PubMed.
- M. D. Nothling, Q. Fu, A. Reyhani, S. Allison-Logan, K. Jung, J. Zhu, M. Kamigaito, C. Boyer and G. G. Qiao, Adv. Sci., 2020, 7, 2001656 CrossRef CAS PubMed.
- G. Moad, Polym. Chem., 2017, 8, 177–219 RSC.
- M. Beija, J.-D. Marty and M. Destarac, Prog. Polym. Sci., 2011, 36, 845–886 CrossRef CAS.
- Y. Zhao and S. Perrier, in Controlled Radical Polymerization at and from Solid Surfaces, ed. P. Vana, Springer International Publishing, Cham, 2016, pp. 77–106. DOI:10.1007/12_2015_316.
- B. D. Fairbanks, P. A. Gunatillake and L. Meagher, Adv. Drug Delivery Rev., 2015, 91, 141–152 CrossRef CAS PubMed.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef CAS PubMed.
- A. Gregory and M. H. Stenzel, Expert Opin. Drug Delivery, 2011, 8, 237–269 CrossRef CAS PubMed.
- D. Smith, A. C. Holley and C. L. McCormick, Polym. Chem., 2011, 2, 1428–1441 RSC.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- S. Aksakal, R. Aksakal and C. R. Becer, Polym. Chem., 2018, 9, 4507–4516 RSC.
- H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef PubMed.
- G. Rindorf and L. Carlsen, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 1179–1182 CrossRef.
- K. B. Wiberg and Y. Wang, ARKIVOC, 2010, 2011, 45–56 Search PubMed.
- D. J. Henry, M. L. Coote, R. Gómez-Balderas and L. Radom, J. Am. Chem. Soc., 2004, 126, 1732–1740 CrossRef CAS PubMed.
- D. H. R. Barton, D. Crich, A. Löbberding and S. Z. Zard, Tetrahedron, 1986, 42, 2329–2338 CrossRef CAS.
- G. Moad, Macromol. Chem. Phys., 2014, 215, 9–26 CrossRef CAS.
- S. W. McCombie, B. Quiclet-Sire and S. Z. Zard, Tetrahedron, 2018, 74, 4969–4979 CrossRef CAS.
- D. H. R. Barton, W. B. Motherwell and A. Stange, Synthesis, 1981, 743–745 CrossRef CAS.
- T. Otsu and M. Yoshida, Makromol. Chem., Rapid Commun., 1982, 3, 127–132 CrossRef CAS.
- T. Otsu, M. Yoshida and A. Kuriyama, Polym. Bull., 1982, 7, 45–50 CAS.
- T. Otsu, M. Yoshida and T. Tazaki, Makromol. Chem., Rapid Commun., 1982, 3, 133–140 CrossRef CAS.
- T. Otsu and K. Nayatani, Makromol. Chem., 1958, 27, 149–156 CrossRef CAS.
- T. Otsu, J. Polym. Sci., 1957, 26, 236–239 CrossRef CAS.
- T. Otsu and A. Kuriyama, J. Macromol. Sci., Part A: Pure Appl.Chem., 1984, 21, 961–977 CrossRef.
- T. Otsu, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2121–2136 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS.
- J. Hioe and H. Zipse, in Encyclopedia of Radicals in Chemistry, Biology and Materials, 2012. DOI:10.1002/9781119953678.rad012.
- J. Chiefari, R. T. A. Mayadunne, C. L. Moad, G. Moad, E. Rizzardo, A. Postma and S. H. Thang, Macromolecules, 2003, 36, 2273–2283 CrossRef CAS.
- M. L. Coote and D. J. Henry, Macromolecules, 2005, 38, 1415–1433 CrossRef CAS.
- M. Le Neindre, B. Magny and R. Nicolaÿ, Polym. Chem., 2013, 4, 5577–5584 RSC.
- C. Dommanget, F. D'Agosto and V. Monteil, Angew. Chem., Int. Ed., 2014, 53, 6683–6686 CrossRef CAS PubMed.
- C. A. Bell, G. G. Hedir, R. K. O'Reilly and A. P. Dove, Polym. Chem., 2015, 6, 7447–7454 RSC.
- C. Bergerbit, B. Farías-Mancilla, L. Seiler, V. Monteil, S. Harrisson, F. D'Agosto and M. Destarac, Polym. Chem., 2019, 10, 6630–6640 RSC.
- J. Zeng, J. Zhu, X. Pan, Z. Zhang, N. Zhou, Z. Cheng, W. Zhang and X. Zhu, Polym. Chem., 2013, 4, 3453–3457 RSC.
- M. Benaglia, J. Chiefari, Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, J. Am. Chem. Soc., 2009, 131, 6914–6915 CrossRef CAS PubMed.
- G. Moad, D. Keddie, C. Guerrero-Sanchez, E. Rizzardo and S. H. Thang, Macromol. Symp., 2015, 350, 34–42 CrossRef CAS.
- G. Moad, J. Polym. Sci., Part A: Polym. Chem., 2018, 57, 216–227 CrossRef.
- J. Hong, Q. Wang, Y. Lin and Z. Fan, Macromolecules, 2005, 38, 2691–2695 CrossRef CAS.
- P. Lei, Q. Wang, J. Hong and Y. Li, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6600–6606 CrossRef CAS.
- Q. Wang, Y.-X. Li, J. Hong and Z.-Q. Fan, Chin. J. Polym. Sci., 2006, 24, 593–597 CrossRef CAS.
- J. Hong, Q. Wang and Z. Fan, Macromol. Rapid Commun., 2006, 27, 57–62 CrossRef CAS.
- L. Zhang, Q. Wang, P. Lei, X. Wang, C. Wang and L. Cai, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2617–2623 CrossRef CAS.
- Y. Wu and Q. Wang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2425–2429 CrossRef CAS.
- Y. Zhan, Z. Zhang, X. Pan, J. Zhu, N. Zhou and X. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1656–1663 CrossRef CAS.
- D. Hua, X. Ge, J. Tang, X. Zhu and R. Bai, Eur. Polym. J., 2007, 43, 847–854 CrossRef CAS.
- T. He, G.-H. Zheng and C.-Y. Pan, Macromolecules, 2003, 36, 5960–5966 CrossRef CAS.
- D. Hua, X. Ge, J. Tang, X. Zhu, R. Bai and C. Pan, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2847–2854 CrossRef CAS.
- L. An, C. Gao, X. Yan, Z. Fu, W. Yang and Y. Shi, Colloid Polym. Sci., 2012, 290, 719–729 CrossRef CAS.
- G. F. Meijs and E. Rizzardo, Polym. Bull., 1991, 26, 291–295 CrossRef CAS.
- G. F. Meijs, E. Rizzardo and T. P. T. Le, Polym. Int., 1991, 26, 239–244 CrossRef CAS.
- D. H. R. Barton, D. Crich and W. B. Motherwell, J. Chem. Soc., Chem. Commun., 1983, 939–941, 10.1039/C39830000939.
- G. F. Meijs, E. Rizzardo, T. P. T. Le and Y.-C. Chen, Makromol. Chem., 1992, 193, 369–378 CrossRef CAS.
- M. Benaglia, M. Chen, Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2009, 42, 9384–9386 CrossRef CAS.
- N. M. Bingham and P. J. Roth, Chem. Commun., 2019, 55, 55–58 RSC.
- R. A. Smith, G. Fu, O. McAteer, M. Xu and W. R. Gutekunst, J. Am. Chem. Soc., 2019, 141, 1446–1451 CrossRef CAS PubMed.
- M. P. Spick, N. M. Bingham, Y. Li, J. de Jesus, C. Costa, M. J. Bailey and P. J. Roth, Macromolecules, 2020, 53, 539–547 CrossRef CAS.
- N. M. Bingham, Q. u. Nisa, S. H. L. Chua, L. Fontugne, M. P. Spick and P. J. Roth, ACS Appl. Polym. Mater., 2020, 2, 3440–3449 CrossRef CAS.
- N. M. Bingham, Q. u. Nisa, P. Gupta, N. Young, E. Velliou and P. J. Roth, Biomacromolecules, 2022 DOI:10.1021/acs.biomac.1022c00039 , accepted.
- H. Elliss, F. Dawson, Q. u. Nisa, N. M. Bingham, P. J. Roth and M. Kopeć, ChemRxiv, 2022, DOI:10.26434/chemrxiv-2022-f1hv5, This content is a preprint and has not been peer-reviewed.
- P. Galanopoulo, N. Gil, D. Gigmes, C. Lefay, Y. Guillaneuf, M. Lages, J. Nicolas, M. Lansalot and F. D'Agosto, Angew. Chem., Int. Ed., 2022, e202117498, DOI:10.1002/anie.202117498.
- C. M. Plummer, N. Gil, P.-E. Dufils, D. J. Wilson, C. Lefay, D. Gigmes and Y. Guillaneuf, ACS Appl. Polym. Mater., 2021, 3, 3264–3271 CrossRef CAS.
- N. Gil, C. Thomas, R. Mhanna, J. Mauriello, R. Maury, B. Leuschel, J.-P. Malval, J.-L. Clément, D. Gigmes, C. Lefay, O. Soppera and Y. Guillaneuf, Angew. Chem., Int. Ed., 2022, e202117700, DOI:10.1002/anie.202117700.
- N. Gil, B. Caron, D. Siri, J. Roche, S. Hadiouch, D. Khedaioui, S. Ranque, C. Cassagne, D. Montarnal, D. Gigmes, C. Lefay and Y. Guillaneuf, ChemRxiv, 2022, DOI:10.26434/chemrxiv-2022-78t5s, This content is a preprint and has not been peer-reviewed.
- M. F. I. Rix, S. Higgs, E. Dodd, S. Coles, N. M. Bingham and P. J. Roth, ChemRxiv, 2022, DOI:10.26434/chemrxiv-2022-cdt52, This content is a preprint and has not been peer-reviewed.
- O. Ivanchenko, U. Authesserre, G. Coste, S. Mazières, M. Destarac and S. Harrisson, Polym. Chem., 2021, 12, 1931–1938 RSC.
- A. L. Barney, J. M. Bruce Jr., J. N. Coker, H. W. Jacobson and W. H. Sharkey, J. Polym. Sci., Part A-1: Polym. Chem., 1966, 4, 2617–2636 CrossRef CAS.
- W. H. Sharkey, J. Macromol. Sci., Part A: Pure Appl.Chem., 1967, 1, 291–299 CrossRef.
- R. Tomovska, M. Urbanová, R. Fajgar, Z. Bastl, J. Šubrt and J. Pola, Macromol. Rapid Commun., 2004, 25, 587–591 CrossRef CAS.
- J. J. Colman and W. C. Trogler, J. Am. Chem. Soc., 1995, 117, 11270–11277 CrossRef CAS.
- H. Morita and H. Haga, J. Photopolym. Sci. Technol., 1995, 8, 475–480 CrossRef CAS.
- E. A. Castro, J. Sulfur Chem., 2007, 28, 401–429 CrossRef CAS.
- E. A. Castro, Chem. Rev., 1999, 99, 3505–3524 CrossRef CAS PubMed.
- R. Ormazábal-Toledo, E. A. Castro, J. G. Santos, D. Millán, A. Cañete, R. Contreras and P. R. Campodónico, J. Phys. Org. Chem., 2012, 25, 1359–1364 CrossRef.
- F. N. Jones and S. Andreades, J. Org. Chem., 1969, 34, 3011–3014 CrossRef CAS.
- P. J. Roth, C. Boyer, A. B. Lowe and T. P. Davis, Macromol. Rapid Commun., 2011, 32, 1123–1143 CrossRef CAS PubMed.
- S.-E. Chun, I.-H. Um and D.-S. Kwon, Bull. Korean Chem. Soc., 1991, 12, 510–514 Search PubMed.
- D.-S. Kwon, H.-S. Park and I.-H. Um, Bull. Korean Chem. Soc., 1991, 12, 93–97 CAS.
- D.-S. Kwon, H.-S. Park and I.-H. Um, Bull. Korean Chem. Soc., 1994, 15, 860–864 CAS.
- Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2007, 40, 4446–4455 CrossRef CAS.
- M. A. Harvison, P. J. Roth, T. P. Davis and A. B. Lowe, Aust. J. Chem., 2011, 64, 992–1006 CrossRef CAS.
- A. B. Lowe, in Thiol-X Chemistries in Polymer and Materials Science, The Royal Society of Chemistry, 2013, pp. 28–58. 10.1039/9781849736961-00028.
- D. J. Lunn, E. H. Discekici, J. Read de Alaniz, W. R. Gutekunst and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2903–2914 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polym. Int., 2011, 60, 9–25 CrossRef CAS.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- M. M. Cerda, T. D. Newton, Y. Zhao, B. K. Collins, C. H. Hendon and M. D. Pluth, Chem. Sci., 2019, 10, 1773–1779 RSC.
- M. C. Urquhart, N. V. Dao, F. Ercole, B. J. Boyd, T. P. Davis, M. R. Whittaker and J. F. Quinn, ACS Macro Lett., 2020, 9, 553–557 CrossRef CAS.
- E. Zaorska, T. Hutsch, M. Gawryś-Kopczyńska, R. Ostaszewski, M. Ufnal and D. Koszelewski, Bioorg. Chem., 2019, 88, 102941 CrossRef CAS PubMed.
- R. Seto, K. Matsumoto and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4522–4529 CrossRef CAS.
- R. Seto, K. Matsumoto and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1832–1842 CrossRef CAS.
- A. P. Dove, ACS Macro Lett., 2012, 1, 1409–1412 CrossRef CAS.
- F. Sanda, D. Jirakanjana, M. Hitomi and T. Endo, Macromolecules, 1999, 32, 8010–8014 CrossRef CAS.
- P. P. Datta and M. K. Kiesewetter, Macromolecules, 2016, 49, 774–780 CrossRef CAS PubMed.
- U. L. D. Inush Kalana, P. P. Datta, R. S. Hewawasam, E. T. Kiesewetter and M. K. Kiesewetter, Polym. Chem., 2021, 12, 1458–1464 RSC.
- R. Abdul-Karim, A. Hameed and M. I. Malik, RSC Adv., 2017, 7, 11786–11795 RSC.
- E. M. López-Vidal, G. L. Gregory, G. Kociok-Köhn and A. Buchard, Polym. Chem., 2018, 9, 1577–1582 RSC.
- P. Yuan, Y. Sun, X. Xu, Y. Luo and M. Hong, Nat. Chem., 2022, 14, 294–303, DOI:10.1038/s41557-021-00817-9.
- A. Mangalum, F. Boadi, S. A. Masand, R. A. Lalancette and A. Pietrangelo, RSC Adv., 2016, 6, 74250–74253 RSC.
- F. Sanda, D. Jirakanjana, M. Hitomi and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4057 CrossRef CAS.
- H. Keul and H. Höcker, Macromol. Rapid Commun., 2000, 21, 869–883 CrossRef CAS.
- N. U. Dharmaratne, J. U. Pothupitiya and M. K. Kiesewetter, Org. Biomol. Chem., 2019, 17, 3305–3313 RSC.
- R. S. Hewawasam, U. L. D. Inush Kalana, N. U. Dharmaratne, T. J. Wright, T. J. Bannin, E. T. Kiesewetter and M. K. Kiesewetter, Macromolecules, 2019, 52, 9232–9237 CrossRef CAS.
- J. U. Pothupitiya, R. S. Hewawasam and M. K. Kiesewetter, Macromolecules, 2018, 51, 3203–3211 CrossRef CAS.
- A. Duda and A. Kowalski, in Handbook of Ring–Opening Polymerization, 2009, pp. 1–51. DOI:10.1002/9783527628407.ch1.
- K. N. Houk, A. Jabbari, H. K. Hall and C. Alemán, J. Org. Chem., 2008, 73, 2674–2678 CrossRef CAS PubMed.
- C. G. Overberger and J. K. Weise, J. Am. Chem. Soc., 1968, 90, 3533–3537 CrossRef CAS.
- K. Nakano, G. Tatsumi and K. Nozaki, J. Am. Chem. Soc., 2007, 129, 15116–15117 CrossRef CAS PubMed.
- M. Luo, X.-H. Zhang and D. J. Darensbourg, Macromolecules, 2015, 48, 5526–5532 CrossRef CAS.
- H. Fukuda, M. Oda and T. Endo, Macromolecules, 1996, 29, 3043–3045 CrossRef CAS.
- B. Ochiai and T. Endo, Prog. Polym. Sci., 2005, 30, 183 CrossRef CAS.
- M. Luo, Y. Li, Y.-Y. Zhang and X.-H. Zhang, Polymer, 2016, 82, 406–431 CrossRef CAS.
- P. W. Bridgman, J. Appl. Phys., 1941, 12, 461–469 CrossRef CAS.
- S. F. Agnew, R. E. Mischke and B. I. Swanson, J. Phys. Chem., 1988, 92, 4201–4204 CrossRef CAS.
- J. Tsukamoto and A. Takahashi, Jpn. J. Appl. Phys., 1986, 25, L338–L340 CrossRef CAS.
- F. Cataldo, Inorg. Chim. Acta, 1995, 232, 27–33 CrossRef CAS.
- F. Cataldo, Radiat. Phys. Chem., 2000, 58, 217–222 CrossRef CAS.
- Y. Matsumura and B. Ochiai, Chem. Lett., 2021, 50, 1856–1858 CrossRef CAS.
- N. Adachi, Y. Kida and K. Shikata, J. Polym. Sci., Polym. Chem. Ed., 1977, 15, 937–944 CrossRef CAS.
- D. J. Darensbourg, J. R. Andreatta, M. J. Jungman and J. H. Reibenspies, Dalton Trans., 2009, 41, 8891 RSC.
- X. H. Zhang, F. Liu, X. K. Sun, S. Chen, B. Y. Du, G. R. Qi and K. M. Wan, Macromolecules, 2008, 41, 1587 CrossRef CAS.
- D. J. Darensbourg, S. J. Wilson and A. D. Yeung, Macromolecules, 2013, 46, 8102 CrossRef CAS.
- M. Luo, X.-H. Zhang and D. J. Darensbourg, Macromolecules, 2015, 48, 6057–6062 CrossRef CAS.
- J. Diebler, H. Komber, L. Häußler, A. Lederer and T. Werner, Macromolecules, 2016, 49, 4723–4731 CrossRef CAS.
- J.-L. Yang, Y. Wang, X.-H. Cao, C.-J. Zhang, Z. Chen and X.-H. Zhang, Macromol. Rapid Commun., 2021, 42, 2000472 CrossRef CAS PubMed.
- M. Luo, X.-H. Zhang and D. J. Darensbourg, Acc. Chem. Res., 2016, 49, 2209–2219 CrossRef CAS PubMed.
- S. Silvano, C. F. Carrozza, A. R. de Angelis, I. Tritto, L. Boggioni and S. Losio, Macromolecules, 2020, 53, 8837–8846 CrossRef CAS.
- M. Luo, X. H. Zhang, B. Y. Du, Q. Wang and Z. Q. Fan, Macromolecules, 2013, 46, 5899 CrossRef CAS.
- A. W. M. Lee, W. H. Chan and H. C. Wong, Synth. Commun., 1988, 18, 1531–1536 CrossRef CAS.
- Y.-Z. You, C.-Y. Hong and C.-Y. Pan, Macromol. Rapid Commun., 2002, 23, 776–780 CrossRef CAS.
- L. M. Leung, W. H. Chan, S. K. Leung and S. M. Fung, J. Macromol. Sci., Part A: Pure Appl.Chem., 1994, 31, 495–505 CrossRef.
- W. J. Middleton, H. W. Jacobson, R. E. Putnam, H. C. Walter, D. G. Pye and W. H. Sharkey, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 4115–4129 CrossRef CAS.
- L. E. Iglesias, A. Baldessari and E. G. Gros, Org. Prep. Proced. Int., 1996, 28, 319–324 CrossRef CAS.
- W. Choi, F. Sanda and T. Endo, Macromolecules, 1998, 31, 9093–9095 CrossRef CAS.
- T. Mukaiyama, I. Kuwajima and K. Mizui, J. Org. Chem., 1966, 31, 32–34 CrossRef CAS.
- W. Choi, F. Sanda and T. Endo, Macromolecules, 1998, 31, 2454–2460 CrossRef CAS.
- W. Choi, F. Sanda, N. Kihara and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 3853–3856 CrossRef CAS.
- J. Y. Do, S. B. Shin, S. M. Jeong and M.-Y. Jung, Eur. Polym. J., 2020, 131, 109689 CrossRef CAS.
- C. G. Kim, M. J. Son and J. Y. Do, Eur. Polym. J., 2021, 156, 110611 CrossRef CAS.
- S. Krishnamurthy, Y. Yoshida and T. Endo, Polym. Chem., 2022, 13, 267–274 RSC.
- S. Motokucho, A. Sudo and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4459–4464 CrossRef CAS.
- A. Steblyanko, W. Choi, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3967–3980 CrossRef CAS.
- A. Nagai, B. Ochiai and T. Endo, Macromolecules, 2004, 37, 7538–7542 CrossRef CAS.
- T. Nakamura, B. Ochiai and T. Endo, Macromolecules, 2005, 38, 4065–4066 CrossRef CAS.
- M. Kamigaito and M. Sawamoto, Macromolecules, 2020, 53, 6749–6753 CrossRef CAS.
- M. Uchiyama, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2015, 54, 1924–1928 CrossRef CAS PubMed.
- S. Kumagai, K. Nagai, K. Satoh and M. Kamigaito, Macromolecules, 2010, 43, 7523–7531 CrossRef CAS.
- M. Uchiyama, K. Satoh and M. Kamigaito, Prog. Polym. Sci., 2022, 124, 101485 CrossRef CAS.
- V. Barone and R. Arnaud, Chem. Phys. Lett., 1996, 251, 393–399 CrossRef CAS.
- V. Barone, R. Arnaud, P. Y. Chavant and Y. Vallée, J. Org. Chem., 1996, 61, 5121–5129 CrossRef CAS.
- J. D. Friedrich, J. Org. Chem., 1987, 52, 2442–2446 CrossRef CAS.
- Y. Qin, A. J. P. White, D. J. Williams and P.-H. Leung, Organometallics, 2002, 21, 171–174 CrossRef CAS.
- E. Vedejs, J. S. Stults and R. G. Wilde, J. Am. Chem. Soc., 1988, 110, 5452–5460 CrossRef CAS.
- V. M. Timoshenko, S. A. Siry, A. B. Rozhenko and Y. G. Shermolovich, J. Fluorine Chem., 2010, 131, 172–183 CrossRef CAS.
- B. Heuzé, R. Gasparova, M. Heras and S. Masson, Tetrahedron Lett., 2000, 41, 7327–7331 CrossRef.
- H. Dentel, I. Chataigner, F. Le Cavelier and M. Gulea, Tetrahedron Lett., 2010, 51, 6014–6017 CrossRef CAS.
- R. Bastin, H. Albadri, A.-C. Gaumont and M. Gulea, Org. Lett., 2006, 8, 1033–1036 CrossRef CAS PubMed.
- H. Jiang, D. C. Cruz, Y. Li, V. H. Lauridsen and K. A. Jørgensen, J. Am. Chem. Soc., 2013, 135, 5200–5207 CrossRef CAS PubMed.
- H. Dentel, I. Chataigner, J.-F. Lohier and M. Gulea, Tetrahedron, 2012, 68, 2326–2335 CrossRef CAS.
- T. Saito, K. Takekawa, J.-I. Nishimura and M. Kawamura, J. Chem. Soc., Perkin Trans. 1, 1997, 2957–2960, 10.1039/A703590J.
- G. Delaittre, N. K. Guimard and C. Barner-Kowollik, Acc. Chem. Res., 2015, 48, 1296–1307 CrossRef CAS PubMed.
- A. S. Goldmann, M. Glassner, A. J. Inglis and C. Barner-Kowollik, Macromol. Rapid Commun., 2013, 34, 810–849 CrossRef CAS PubMed.
- A. J. Inglis and C. Barner-Kowollik, Macromol. Rapid Commun., 2010, 31, 1247–1266 CrossRef CAS PubMed.
- A. J. Inglis, S. Sinnwell, M. H. Stenzel and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2009, 48, 2411–2414 CrossRef CAS PubMed.
- A. J. Inglis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2009, 30, 1792–1798 CrossRef CAS PubMed.
- S. Sinnwell, A. J. Inglis, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Chem. Commun., 2008, 2052–2054, 10.1039/B718180A.
- M. Glassner, G. Delaittre, M. Kaupp, J. P. Blinco and C. Barner-Kowollik, J. Am. Chem. Soc., 2012, 134, 7274–7277 CrossRef CAS PubMed.
- A. J. Inglis, S. Sinnwell, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 4120–4126 CrossRef CAS.
- L. Nebhani, S. Sinnwell, A. J. Inglis, M. H. Stenzel, C. Barner-Kowollik and L. Barner, Macromol. Rapid Commun., 2008, 29, 1431–1437 CrossRef CAS.
- C. M. Preuss, M. M. Zieger, C. Rodriguez-Emmenegger, N. Zydziak, V. Trouillet, A. S. Goldmann and C. Barner-Kowollik, ACS Macro Lett., 2014, 3, 1169–1173 CrossRef CAS.
- L. Nebhani, S. Sinnwell, C. Y. Lin, M. L. Coote, M. H. Stenzel and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6053–6071 CrossRef CAS.
- L. Nebhani, P. Gerstel, P. Atanasova, M. Bruns and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 7090–7095 CrossRef CAS.
- T. M. Legge, A. T. Slark and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6980–6987 CrossRef CAS.
- Y. Liu, J. He, J. Xu, D. Fan, W. Tang and Y. Yang, Macromolecules, 2005, 38, 10332–10335 CrossRef CAS.
- J. Xu, J. He, D. Fan, W. Tang and Y. Yang, Macromolecules, 2006, 39, 3753–3759 CrossRef CAS.
- A. Postma, T. P. Davis, G. Moad and M. S. O'Shea, Macromolecules, 2005, 38, 5371–5374 CrossRef CAS.
- B. Chong, G. Moad, E. Rizzardo, M. Skidmore and S. H. Thang, Aust. J. Chem., 2006, 59, 755–762 CrossRef CAS.
- K. Skrabania, A. Miasnikova, A. M. Bivigou-Koumba, D. Zehm and A. Laschewsky, Polym. Chem., 2011, 2, 2074–2083 RSC.
- T. G. McKenzie, Q. Fu, M. Uchiyama, K. Satoh, J. Xu, C. Boyer, M. Kamigaito and G. G. Qiao, Adv. Sci., 2016, 3, 1500394 CrossRef PubMed.
- Y. Qu, X. Zhang, Y. Wu, F. Li and J. Hua, Polym. Chem., 2014, 5, 3396–3403 RSC.
- J. Xu, Macromolecules, 2019, 52, 9068–9093 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |