
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bismuth gallate coordination networks inspired by an active pharmaceutical ingredient†
Erik
Svensson Grape
 a,
Victoria
Rooth
a,
Simon
Smolders
b,
Ambre
Thiriez
c,
Sofia
Takki
a,
Dirk
De Vos
b,
Tom
Willhammar
a and
A. Ken
Inge
*a
a,
Victoria
Rooth
a,
Simon
Smolders
b,
Ambre
Thiriez
c,
Sofia
Takki
a,
Dirk
De Vos
b,
Tom
Willhammar
a and
A. Ken
Inge
*a
aDepartment of Materials and Environmental Chemistry, Stockholm University, SE-106 91, Sweden. E-mail: andrew.inge@mmk.su.se
bCentre for Membrane Separations, Adsorption, Catalysis and Spectroscopy for Sustainable Solutions (cMACS), Department of Microbial and Molecular Systems (M2S), KU Leuven, 3001 Leuven, Belgium
cDépartement Sciences et Génie Des Matériaux, INSA, Lyon 69621, France
First published on 29th August 2022
Abstract
The effect of solvent has been investigated for the synthesis of bismuth gallate compounds, of which the water-based bismuth subgallate has been used as an active pharmaceutical ingredient (API) for over a century. Using methanol as a solvent, two new bismuth gallates were acquired: first a flexible 3-periodic metal–organic framework (MOF) forms, which transforms upon extended synthesis times into a layered 2-periodic coordination polymer of the same bismuth-to-gallate ratio. The structures were determined by three-dimensional electron diffraction. Synthesis in ethanol resulted in the formation of the MOF phase, but not the layered phase. The layered material of the methanol-based synthesis was used as a Lewis acid catalyst due to its higher stability, showing a comparatively quick and regiospecific conversion of styrene oxide to 2-methoxy-2-phenylethanol, indicating the presence of open metal sites in the material. The acquisition of bismuth gallate structures of varying periodicity highlights the prospect of acquiring novel MOFs and coordination polymers from the same components of APIs.
Introduction
Bismuth is often considered to be non-toxic compared to neighbouring elements in the periodic table, and compounds of bismuth containing organic ligands have been used as active pharmaceutical ingredients (APIs) for over a century.1 Formulations such as bismuth subsalicylate and colloidal bismuth subcitrate (CBS) have mainly been used for their antimicrobial properties, such as in the treatment of Helicobacter pylori infections and other gastrointestinal disorders.2–6 Recently, bismuth compounds have been shown to efficiently combat antibiotic resistant bacterial strains,7 and are gaining interest as catalysts8 for their low cost, as well as their non-toxic and non-corrosive nature.9 Despite their large-scale production and long history of use, the structures of these materials have largely remained unknown or have only recently been determined. This is due their tendency to form as nanocrystalline powders rather than large single crystals, which can prevent structure determination through conventional means such as single-crystal X-ray diffraction (SCXRD).10,11 Knowledge of a material's structure is fundamental for understanding its physical and chemical properties, which can in turn explain their function and behaviour in biological systems or inspire investigations of previously unexplored properties and applications. For example, the structure of CBS crystallized under acidic conditions was revealed to be a metal–organic framework (MOF),12 a class of porous materials of interest for applications such as separation and heterogeneous catalysis.13,14With three-dimensional electron diffraction (3DED), structures of single microcrystals can now be more readily investigated—largely made possible through recent advances in hardware, software, and overall methodology.15–19 Using 3DED, bismuth subgallate (BSG), which has been commercially sold as an API since the late 19th century, was revealed by some of us to be a microporous one-dimensional coordination polymer (CP).10 By finally realizing the structure of BSG, the high stability of the material could be understood, and ideas to investigate previously unexplored sorption properties were conceived. As the structure of a material (i.e. the arrangement of molecules/ions and their interactions) can substantially influence its properties, this piqued our interest in altering the structure of this API to develop novel bismuth gallates with varied properties. In BSG, only the phenolate groups of gallate ligands coordinate to Bi3+, not the carboxylic acid group, resulting in a 1D structure. It was envisioned that by extending coordination of the gallate linker through both its phenolate and carboxylate groups, 2-periodic or 3-periodic framework structures may be developed. In addition, the use of gallic acid (also known as 3,4,5-trihydroxybenzoic acid) is of interest for the synthesis of CPs and MOFs as an organic linker that is eco-bio-compatible, sourced from biomass,20–25 rather inexpensive and known for its antioxidant properties.
For the synthesis of CPs and MOFs, solvothermal synthesis is the most common approach, providing an opportunity to alter the polarity, acid–base properties, and solubility of the reagents used. In the synthesis of CPs and MOFs, the solvent used can have multiple roles, potentially acting as a reaction medium, structure-directing agent or a ligand coordinating to the metal cation.26 Pertaining to bismuth, it has been shown that the choice of solvent can greatly affect the structure of the acquired product and properties thereof, as was revealed for a series of compounds made from Bi3+ and 1,2,4,5-tetrakis-(4-carboxyphenyl)benzene.27 Overall, the formulation, processing, and resulting structure and properties of a material can be greatly influenced by the choice of solvent. Inspecting the recently determined structure of BSG,10 the presence of a coordinated water molecule that bridges the Bi3+ cations within the structure hints at a possibility of acquiring novel phases through syntheses in other solvents.
Results and discussion
In this work, two novel crystalline bismuth gallate phases (1MeOH and 2) were obtained through solvothermal synthesis in methanol using bismuth nitrate and gallic acid (Fig. 1, see ESI† for details on the syntheses). After 2 hours of heating, a bismuth gallate MOF (1MeOH), Bi(C7H3O5)(MeOH), is acquired as plank-shaped crystals (Fig. S1†). Maintaining the same synthesis conditions as 1MeOH but extending the heating time from 2 to 5 hours resulted in a layered phase of the composition, Bi(C7H3O5), denoted 2.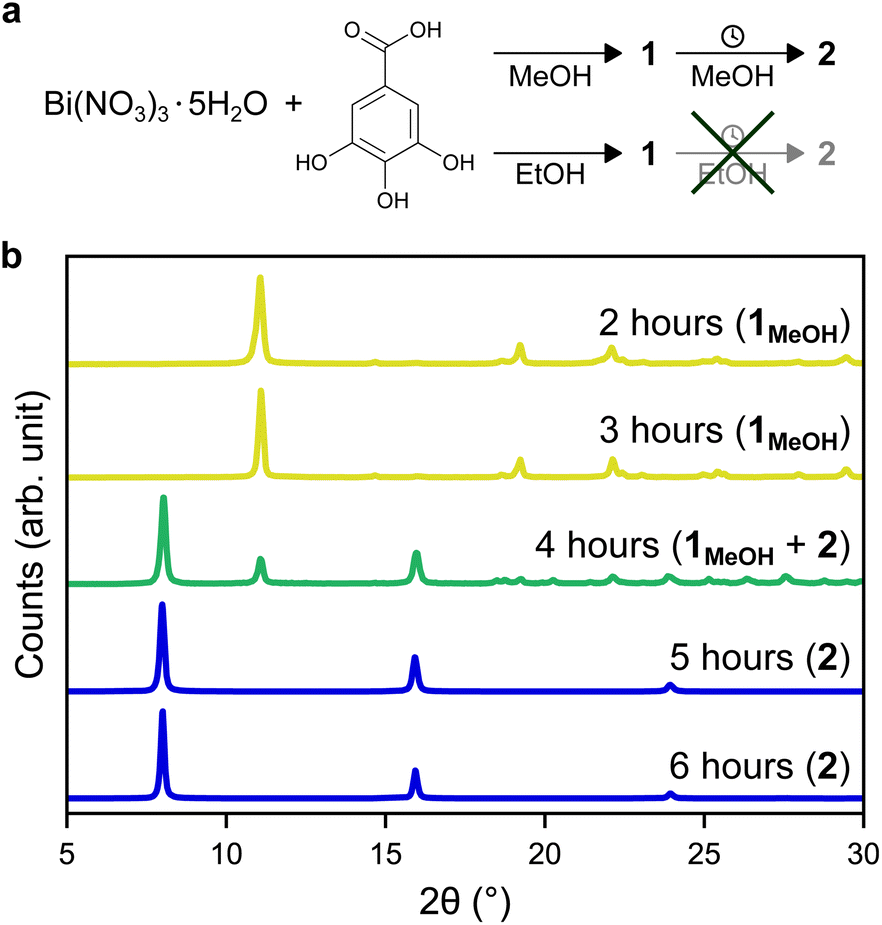 | ||
| Fig. 1 (a) Schematic representation of the synthesis of 1MeOH/1EtOH from Bi(NO3)3·5H2O and gallic acid in methanol/ethanol, where 1MeOH transforms into 2 upon continued heating in methanol. (b) X-ray powder diffraction patterns of bismuth gallates synthesized in methanol under identical conditions apart from different synthesis times. At short reaction times the MOF 1MeOH forms, which after a synthesis time of 4 hours begins to convert into the layered phase, 2. | ||
The structure of 1 was solved by 3DED (Fig. 2a, b and Table S1†).28,291MeOH (Fig. 3a) crystallizes in the polar orthorhombic space group P212121. The coordination number of bismuth in the structure is 6, with a skewed trapezoidal bipyramidal coordination geometry, and Bi–O bonds ranging from 2.1–2.8 Å, which is a typical range for carboxylate and phenolate interactions with Bi3+ cations.30,31 The hemidirected coordination environment is characteristic of the stereochemically active lone pair effect common to many Bi3+ compounds. Each bismuth cation is coordinated by four bridging phenolate groups and a carboxylate group from surrounding gallates, as well as a methanol molecule, resulting in a rod-shaped inorganic building unit (IBU, Fig. 3b). Each gallate linker coordinates to Bi3+ cations through two of the phenolate oxygen atoms in the 3- and 4-positions, as well as through one of the carboxylate oxygen atoms in a monodentate manner. This stands in contrast to the structure of BSG, where all three phenolate groups of the gallate anion coordinate to Bi3+ cations but not the carboxylic acid. As the gallates in 1MeOH coordinate on both ends of the molecule, this allows the linker to bridge IBUs into a 3-periodic framework structure, unlike the isolated rod IBUs found in BSG. Phase purity was assessed through a Pawley fit against X-ray powder diffraction (PXRD) data (Fig. 3f, S2 and Table S2†) and the suggested framework composition is further supported by thermogravimetric analysis (TGA), indicating a final sum formula of Bi(C7H3O5)(MeOH)·MeOH, i.e. for every Bi3+ cation there is one coordinated methanol and one disordered methanol molecule occupying the void space in the porous structure (Fig. S6†).
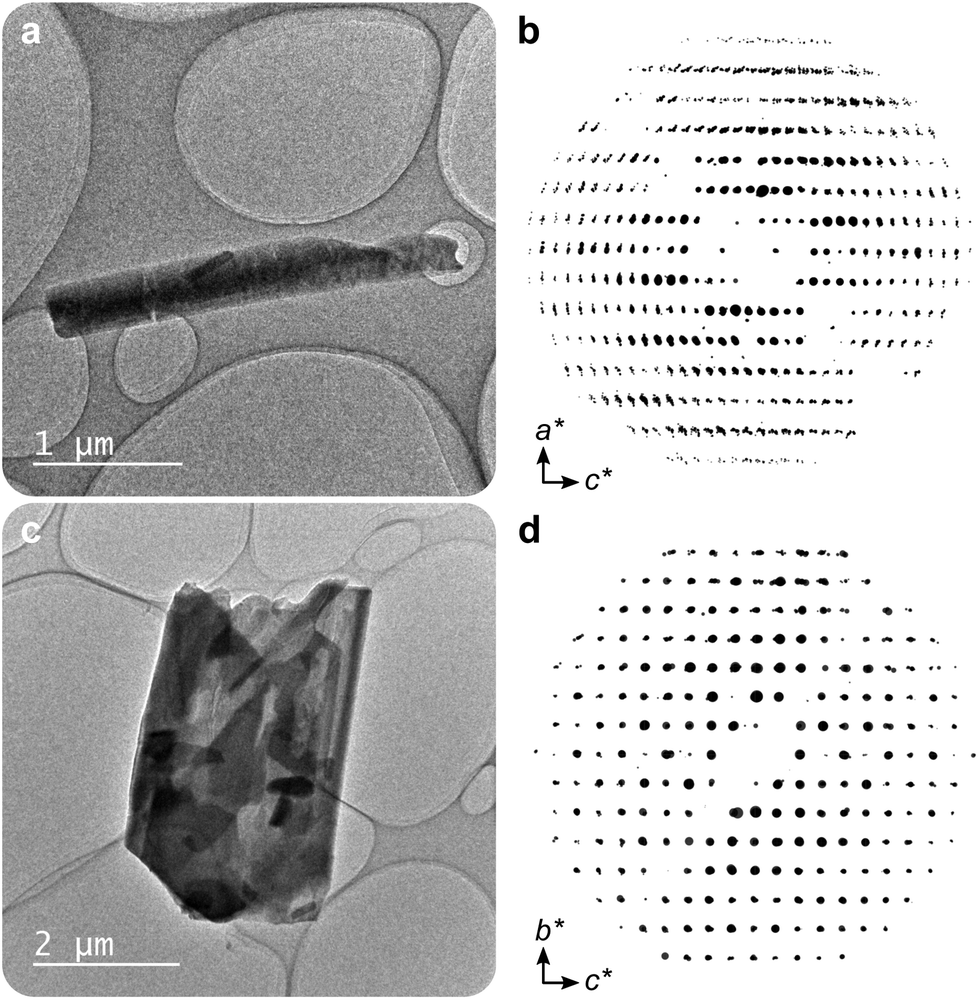 | ||
| Fig. 2 (a) A crystal of 1MeOH and (b) reconstructed reciprocal lattice based on the acquired 3DED data projected along b*. (c) A crystal of 2 and (d) reconstructed reciprocal lattice based on the 3DED data projected along a*. | ||
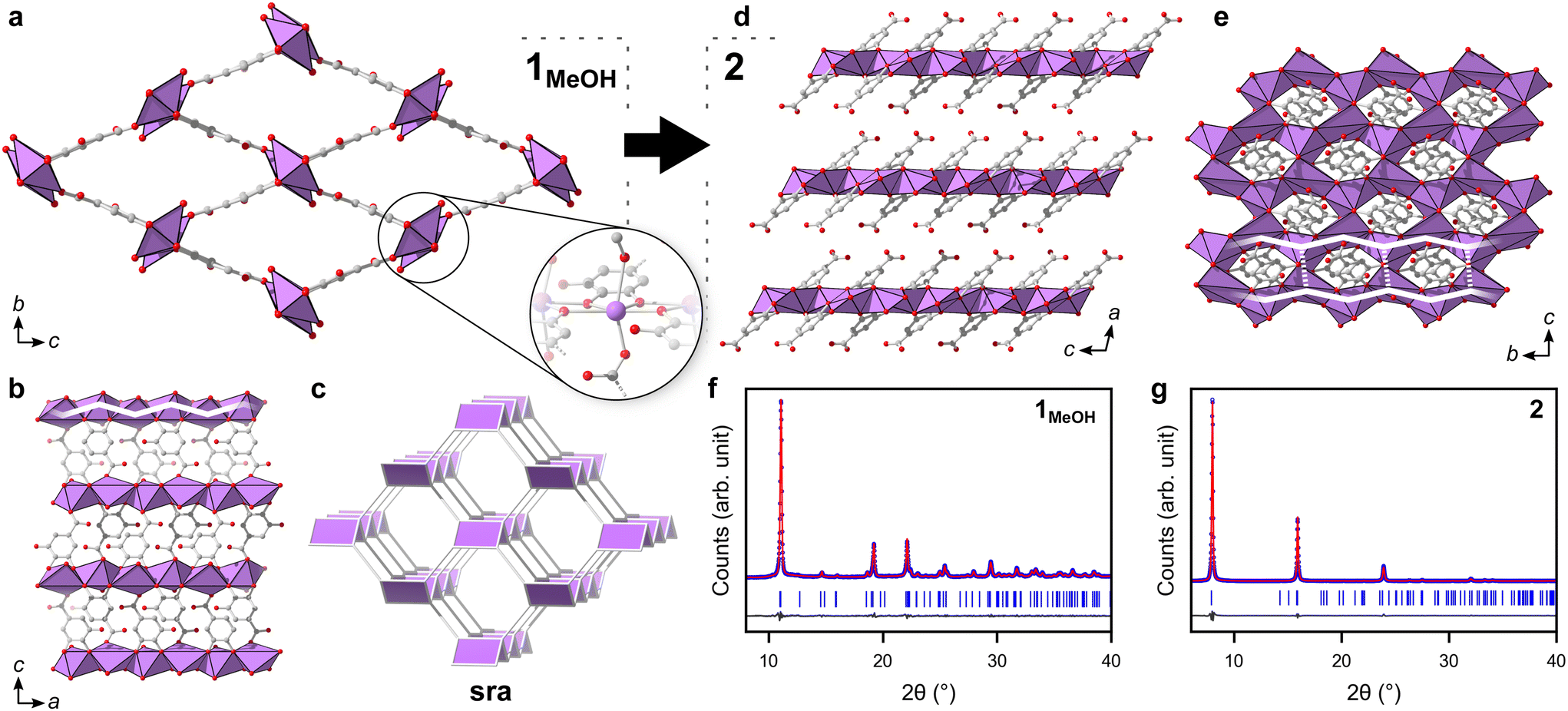 | ||
| Fig. 3 (a) The structure of 1MeOH as viewed along a. Bismuth, oxygen and carbon atoms are coloured purple, red and grey respectively. The methyl group of the coordinated methanol which points into the pores has been omitted for clarity. The insert shows the hemidirected coordination environment around the Bi3+ found in the structure. (b) The structure of 1MeOH as viewed along b. The partial overlay of a white line over the top row of Bi3+ cations shows the zig-zag shape of the inorganic building unit. (c) The underlying sra net of 1. (d) Structure of the layered phase 2 as viewed along b. (e) The structure of 2 as viewed along a, showing the honeycomb arrangement of bismuth cations. The partial overlay of white lines illustrates the relation to the IBU of 1 where the zig-zag chains are linked into a hcb net. (f and g) Difference plots for the Pawley fits of (f) 1MeOH and (g) 2, respectively, showing that the materials are acquired as pure phases. | ||
Upon inspecting the crystal structure of 1MeOH and assessing the collected FT-IR data (Fig. S9†), the coordinating carboxylate group within the structure is evidently deprotonated as indicated by the lack of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch band characteristic to carboxylic acids, typically found at 1690 cm−1 in the IR spectrum. This results in a charge-balanced material where the third phenolic oxygen on the 5-position of the gallate anion, which does not coordinate to the bismuth cations, remains protonated.
O stretch band characteristic to carboxylic acids, typically found at 1690 cm−1 in the IR spectrum. This results in a charge-balanced material where the third phenolic oxygen on the 5-position of the gallate anion, which does not coordinate to the bismuth cations, remains protonated.
The 3-periodic structure of 1MeOH can be deconstructed into an underlying sra net (Fig. 3c), the same as that of the archetypical flexible MOF MIL-53.32 Similarly to MIL-53, it was observed that 1MeOH is also highly flexible upon desorption/heating. The powder diffraction pattern of 1MeOH undergoes noticeable changes in peak positions upon exposing the sample to vacuum or slightly elevated temperatures as a result of the framework flexibility (Fig. S10†), a behaviour which will be discussed for the ethanol analogue, 1EtOH, at the end of this section. As 10.4% of the unit cell volume of 1MeOH is potential solvent-accessible voids, gas sorption experiments were performed. However, the material showed no porosity towards N2 or CO2 under the tested experimental conditions, possibly a consequence of a high energy barrier in re-opening the structure after emptying the framework, which remains crystalline after being exposed to reduced pressures, as was observed from variable temperature PXRD measurements (Fig. S10†). Immersing the solid in water yields the previously characterized BSG (Fig. S13†).10 Overall, this relatively small number of published metal-gallate structures are of particular interest, as gallic acid and related phenolic compounds are commonly utilized for their chelating abilities and biological roles pertaining to such properties.33
A second crystalline phase, 2, which is obtained under similar conditions as 1MeOH but with longer synthesis times (Fig. 1b), forms as plate-shaped crystals and its structure was also solved from 3DED data (Fig. 2c and d). The material crystallizes in the monoclinic space group P21/c and analysis of the 3DED data revealed a layered material (Fig. 3d). The coordination number of Bi3+ in the structure of 2 is 6 as well, with a hemidirected pentagonal pyramidal coordination geometry involving six bridging phenolates coordinating to each Bi3+ cation. The Bi–O distances found within 2 are in good agreement with previously characterized materials, spanning a range of 2.1–2.9 Å. The carboxylic acid group is not involved in coordination and points outwards from the layers. It is protonated, in contrast to what was found for 1MeOH, as validated by FT-IR data (Fig. S9†) by the clear presence of a band at 1690 cm−1 which is characteristic of carboxylic acid CO stretching. This preferential coordination of phenolate groups to the metal cation over carboxylate groups is in line with previous findings in similar materials,10,34 such as BSG, owing to the chelation of adjacent phenolate groups of the gallate anions.
The IBUs of 1 and 2 are related (Fig. 4). In 1 the Bi3+ coordination environment as aforementioned includes four phenolate, one terminal alcohol and one carboxylate oxygen, and the non-coordinating phenol has a long interatomic distance to the Bi3+ cation of 3.3 Å. In 2, the coordination by the alcohol is lost as well as the carboxylate, and the non-coordinating phenol oxygen in 1 is deprotonated in 2 and coordinates to Bi3+ with an interatomic distance of 2.8 Å. In 2 the layered IBU can be simplified into a 2-periodic honeycomb (hcb) net (Fig. 3e). This 2-periodic IBU of 2 is related to the zigzagging 1-periodic IBU of 1MeOH by directly linking the zigzagging IBUs into a layer as illustrated by the white lines in Fig. 3b and e. As such, 2 likely forms as a rearrangement of the 1MeOH structure: the monodentate carboxylate group coordinated to Bi3+ in 1MeOH detaches and the zig-zag rod-shaped IBUs in 1 are instead joined together via the third phenol oxygen of the gallate anions, resulting in the layered IBU found in 2.
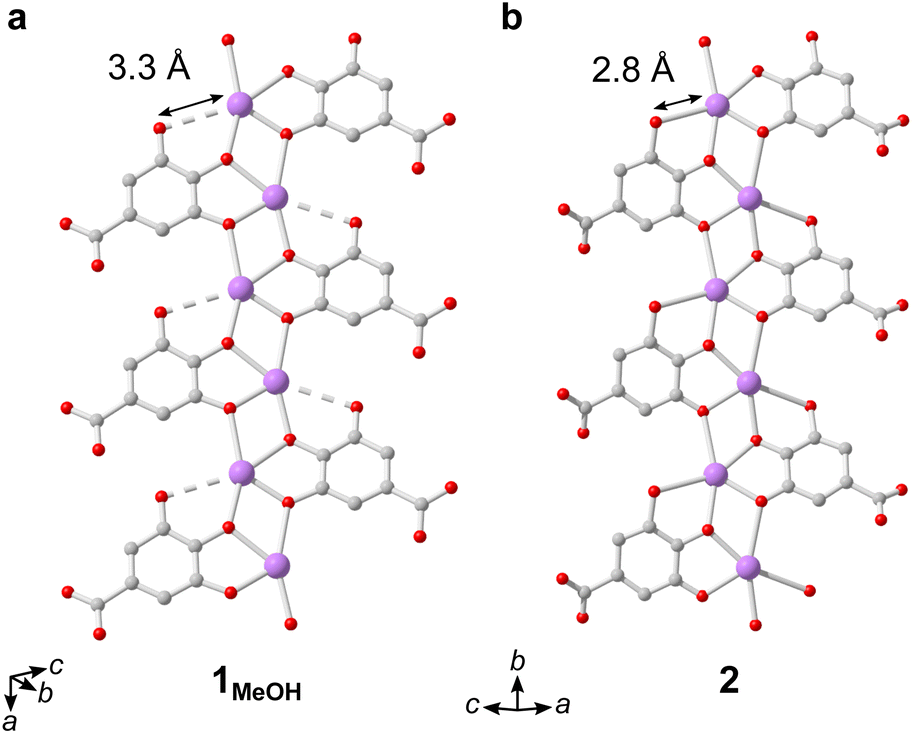 | ||
| Fig. 4 Select sections of the IBUs of (a) 1MeOH and (b) 2, showing similar zig-zag arrangement of Bi3+ cations bridged by coordinating phenolate groups. The structures of 1 and 2 thereby mainly differ in the linkage between these IBUs, being joined by carboxylate groups in 1 or phenol groups in 2. Hydrogen atoms are omitted for clarity. | ||
Phase purity of 2 was validated by PXRD (Fig. 1b, S3 and Table S3†) and the suggested formula for the material is Bi(C7H3O5)·MeOH, as was validated from TGA (Fig. S7†), i.e. a non-coordinated disordered methanol molecule occupies the void space between layers. Exposing 2 to higher temperatures gives a contraction of the interlayer distance (Fig. S11†), with the unit cell length a decreasing from 11.568(3) Å at room temperature in air to 11.390(7) Å at 160 °C, likely due to the removal of inter-layer methanol molecules.
The structure of 2 was shown to have higher thermal stability than 1, as indicated by variable temperature PXRD (Fig. S11†) and TGA (Fig. S7†), as well as increased stability to moisture, as would be expected based on Ostwald's step rule, which predicts the tendency of less thermodynamically stable phases to crystallize from solution prior to more stable phases.
Additionally, denser phases typically have higher thermodynamic stability due to their higher density of dispersion forces. The crystal structure of 2 is noticeably denser than 1MeOH (2.846 vs. 2.254 g cm−3, respectively) which is in line with a higher thermodynamic stability of 2 over 1MeOH. Attempts to synthesize the two materials using ethanol as an alternative solvent to methanol resulted in the acquirement of 1EtOH. 1EtOH is a pseudopolymorph of 1MeOH, as it only differs by the presence of ethanol rather than methanol. However, extending the synthesis time in ethanol did not yield 2 (Fig. 1a), but resulted in the continued presence of 1EtOH. It is therefore hypothesized that using ethanol as a solvent stabilizes the structure of 1 by better filling the pore space within the material. Exposing as-synthesized 1EtOH to vacuum (∼1 mbar) led to a shrinking of the unit cell by 13%, with unit cell parameters of a = 7.540(2) Å, b = 8.271(3) Å, c = 16.616(4) Å (Fig. S5, S12b and Table S5†). Further, storage of 1EtOH under ambient conditions for several days yielded a compressed but still crystalline material 1EtOH(stored), which, as determined from 3DED measurements (Table S1†) shows a decrease in unit cell volume by 24% (Fig. 5), with unit cell parameters of a = 7.73(4) Å, b = 6.84(3) Å, c = 16.87(8) Å (as synthesized 1EtOH: a = 7.89(4) Å, b = 8.61(4) Å, c = 17.38(9) Å). The contraction of the structure and narrowing of the pores of 1 occurs through the concerted rotation of the IBUs around the a-axis as well as a hinge-like rotation of the coordinated carboxylate groups, resulting in shrinkage of the b-axis by 21%. The phase purity of 1EtOH and unit cell parameters of the partially collapsed material were validated by a Pawley fit against PXRD data (Fig. S4, S5 and Tables S4, S5). The suggested overall composition of 1EtOH is Bi(C7H3O5)(EtOH)·0.5EtOH, as validated by TGA (Fig. S8†). For all three synthesized materials 1MeOH, 1EtOH, and 2 the sum formula, excluding solvent species, is Bi(C7H3O5).
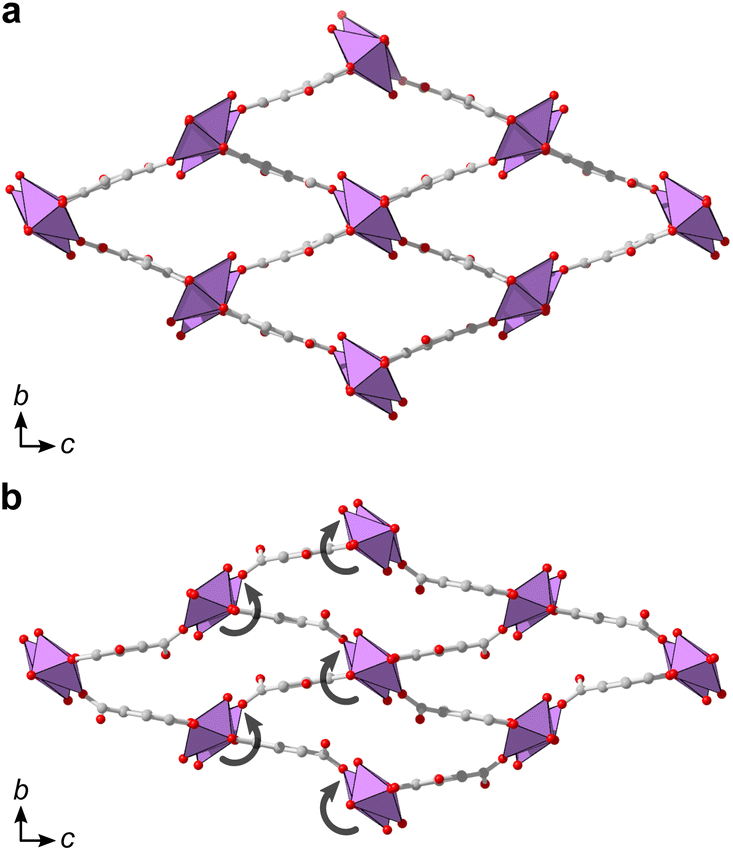 | ||
| Fig. 5 (a) The structure of as-synthesized 1EtOH as determined by 3DED viewed along a showing a similar pore shape as 1MeOH. (b) The structure of partially activated 1EtOH(stored) as viewed along a, showing an altered pore shape, resulting in a 24% decrease in unit cell volume (1181(10) Å3 to 891(8) Å3) upon loss of solvent. Semicircular arrows are drawn to illustrate the concerted rotation of the IBUs in the structure. The ethanol carbon atoms and hydrogen atoms are omitted for clarity. | ||
Synthesis in other primary alcohols but under otherwise similar conditions did not result in 1 or 2. In 1-propanol and 2-propanol a poorly crystalline unidentified phase formed, while for 1-butanol and 1-pentanol an amorphous phase or the basic bismuth nitrate [Bi6O4(OH)4](NO3)6 respectively resulted.35 Basic bismuth nitrates36 often form when using bismuth nitrate as a reagent, and their role in the synthesis or prevention of bismuth MOF formation is still under debate. It is most likely that 1 does not form with these larger alcohols as the coordinating alcohols would occupy too much space to fit within the pores.
Lewis acid catalysis
The potential presence of open metal sites in 2, the layered end-product of the reaction, was probed using an epoxide ring-opening reaction (Scheme 1, see ESI† for details). The same reaction has been studied using the previously described bismuth compound CAU-17,37,38 with similar epoxide ring-opening reactions being used to assess the presence of open metal-sites for other MOFs as well.39 As a comparison, two other bismuth phenolate materials, BSG10 and SU-101,34 were also studied.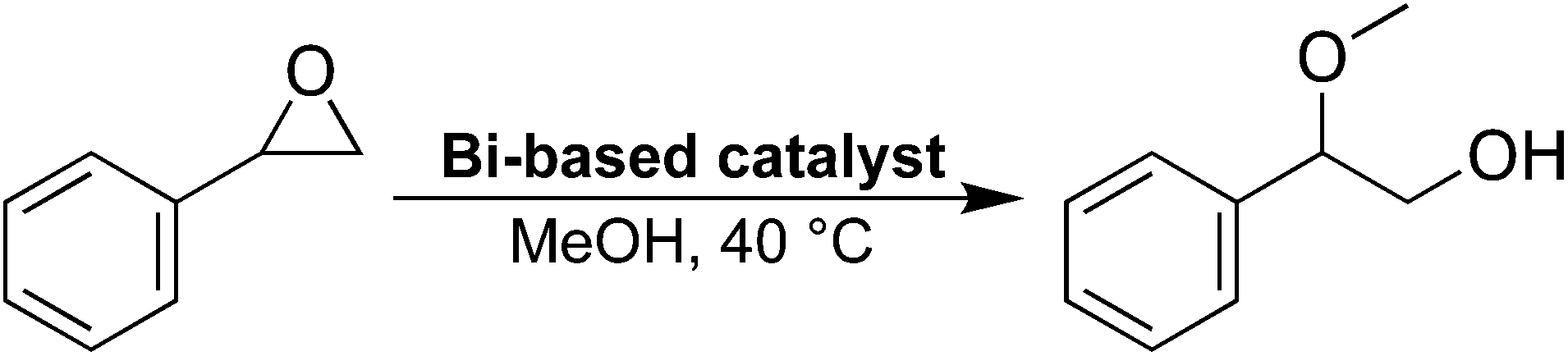 | ||
| Scheme 1 The epoxide ring-opening reaction studied using various bismuth-phenolate materials. | ||
In terms of composition, 2 is very similar to BSG, differing in the presence of a coordinated water molecule in the structure of BSG. SU-101, on the other hand, is a permanently porous MOF material made from bismuth cations and ellagic acid—a naturally occurring phenolic molecule resembling a dimer of gallic acid. Considering this, the metal-linker interactions are somewhat similar for the three materials, being composed of either coordinated galloyl or catechol groups.
The ring-opening reaction of styrene oxide with methanol was then studied using the materials as solid catalysts, acquiring 2-methoxy-2-phenylethanol as a product. The time-conversion plots for the three materials can be seen in Fig. 6a, showing a full conversion of styrene oxide in the presence of 2 after approximately 6 hours, while SU-101 and BSG only display conversions of about 82% and 20%, respectively, after the same time period. As such, both 2 and SU-101 show significantly faster reaction rates in comparison to CAU-17, which gave full conversion after approximately 100 hours under similar conditions.38
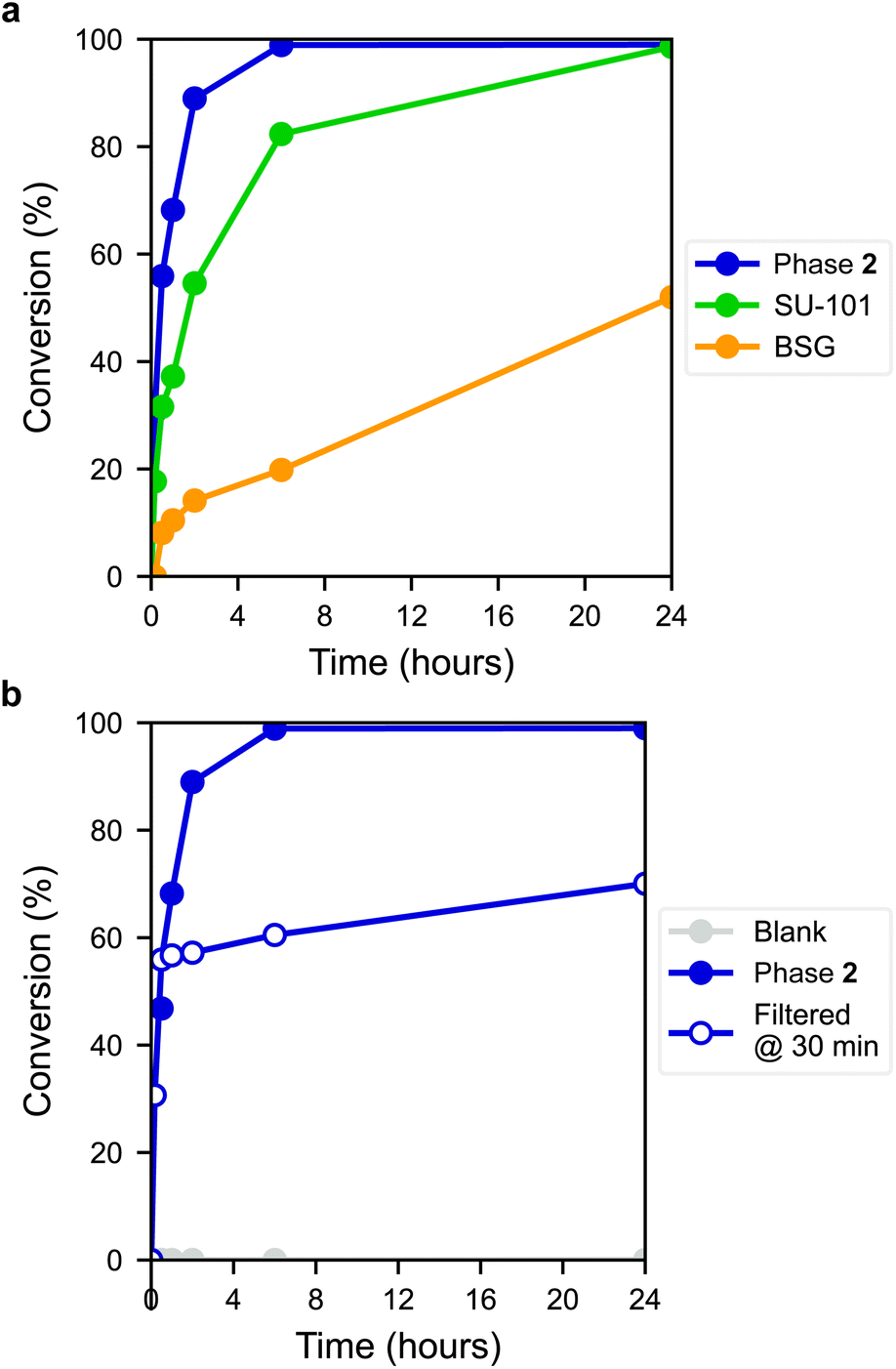 | ||
| Fig. 6 (a) Conversion of styrene oxide into 2-methoxy-2-phenylethanol using different bismuth-phenolate materials as heterogeneous catalysts, where 2 shows full conversion after approximately 6 hours. (b) Blank and filter tests of the conversion of styrene oxide into 2-methoxy-2-phenylethanol, showing no conversion if a catalyst is absent and little conversion upon removal of the solid catalyst, demonstrating that the catalyst is heterogeneous. | ||
The active sites in 2 are most likely the outer surface sites on the thin platelet-shaped crystals, which are thinnest in the a direction, likely providing a large number of active metal sites upon displacement of coordinated gallate anions found in the as-synthesized material. Coincidentally, thin layered structures are sought after in other Bi-based catalysts, such as bismuth oxyhalides.40 This may in part contribute to the faster conversion rate by the layered structure 2 with plate-like morphology, compared to the rod structure of BSG and the framework structure of SU-101, although other factors are also expected to influence performance. PXRD measurements of 2 post-catalysis showed that the material remains crystalline (Fig. S14†). Furthermore, a blank experiment showed that the reaction did not proceed without the presence of a catalyst (Fig. 6b) and leaching tests using 2 showed that the reaction proceeds at near-zero rate after filtering off the catalyst, indicating that the catalyst is indeed heterogeneous.
Conclusions
Solvothermal synthesis of bismuth gallate materials in methanol led to the successive acquisition of two novel bismuth gallate materials, 1 and 2, both of which are pseudopolymorphs of the long-used API bismuth subgallate, having the same bismuth-to-gallate ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, as determined from structural investigations using 3DED. The initially acquired metal–organic framework 1 exhibits a flexible behaviour, showing a decrease in unit cell volume of 24% upon storage and being subsequently exposed to the high-vacuum environment of the TEM. With continued heating of the reaction mixture in methanol, the layered structure 2 was acquired, consisting of honeycomb-shaped layers of bismuth cations coordinated to galloyl groups, likely forming as a change in coordinated species, as indicated from the related inorganic building units of the structures. In both 1 and 2, the framework composition is Bi(C7H3O5), excluding the solvent molecules. Attempts to synthesize the materials in other primary alcohols showed that 1 could be acquired in ethanol as well, resulting in a pseudopolymorph of 1MeOH, yet no formation of 2 was observed upon further heating, indicating a stabilizing role of ethanol for the structure of 1. Lastly, the layered end product 2 was used as a catalyst for the ring-opening reaction of styrene oxide in methanol, yielding 2-methoxy-2-phenylethanol, showing quicker conversion than previously studied bismuth-based materials.
:1, as determined from structural investigations using 3DED. The initially acquired metal–organic framework 1 exhibits a flexible behaviour, showing a decrease in unit cell volume of 24% upon storage and being subsequently exposed to the high-vacuum environment of the TEM. With continued heating of the reaction mixture in methanol, the layered structure 2 was acquired, consisting of honeycomb-shaped layers of bismuth cations coordinated to galloyl groups, likely forming as a change in coordinated species, as indicated from the related inorganic building units of the structures. In both 1 and 2, the framework composition is Bi(C7H3O5), excluding the solvent molecules. Attempts to synthesize the materials in other primary alcohols showed that 1 could be acquired in ethanol as well, resulting in a pseudopolymorph of 1MeOH, yet no formation of 2 was observed upon further heating, indicating a stabilizing role of ethanol for the structure of 1. Lastly, the layered end product 2 was used as a catalyst for the ring-opening reaction of styrene oxide in methanol, yielding 2-methoxy-2-phenylethanol, showing quicker conversion than previously studied bismuth-based materials.
The solvent-dependent formation of bismuth gallates 1 and 2 highlights the possibility of acquiring novel metal–organic frameworks and coordination polymers of variable periodicity by changing the solvent used. This extends the synthesis landscape of coordination polymers, as previously investigated combinations of metal cations and organic molecules could yield novel materials with significantly different structures, in this case taking inspiration from long-used metallodrugs.
Author contributions
Conceptualization, A. K. I. and E. S. G.; Investigation, E. S. G., V. R., S. S., A. T., S. T., and T. W.; Formal analysis, E. S. G. and T. W.; Writing – original draft, E. S. G.; Writing – review & editing, E. S. G., A. K. I., S. S. and D. D. V.; Visualization, E. S. G.; Funding acquisition, A. K. I., D. D. V., and T. W.; Resources, S. S., and D. D. V.; Supervision, A. K. I., D. D. V., T. W., and S. S.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
E. S. G. and A. K. I. acknowledge support from the Swedish Foundation for Strategic Research (SSF). A. K. I. also acknowledges support from the Knut and Alice Wallenberg Foundation (KAW 2016.0072). E. S. G. also acknowledges support from the jubilee donation of Knut and Alice Wallenberg foundation (SU FV-2.1.9-1894-18). T. W. acknowledges support from the Swedish Research Council (VR, 2019-05465). D. D. V. thanks FWO Vlaanderen for continued support. The authors thank Dr Ocean Cheung of Uppsala University for the collection and interpretation of gas sorption data, and Dr N. V. R. Aditya Dharanipragada for investigations on synthesis.References
- D. M. Keogan and D. M. Griffith, Molecules, 2014, 19, 15258–15297 CrossRef PubMed.
- H. Li and H. Sun, Curr. Opin. Chem. Biol., 2012, 16, 74–83 CrossRef CAS PubMed.
- P. C. Andrews, G. B. Deacon, C. M. Forsyth, P. C. Junk, I. Kumar and M. Maguire, Angew. Chem., Int. Ed., 2006, 45, 5638–5642 CrossRef CAS PubMed.
- P. J. Sadler, H. Li and H. Sun, Coord. Chem. Rev., 1999, 185–186, 689–709 CrossRef CAS.
- H. Sun and P. J. Sadler, in Metallopharmaceuticals II, Topics in Biological Inorganic Chemistry, ed. M. J. Clarke and P. J. Sadler, Springer, Berlin, Heidelberg, Bismuth Antiulcer Complexes, 1999, vol 2, pp. 159–185, DOI:10.1007/978-3-642-60061-6_5.
- K. D. Mjos and C. Orvig, Chem. Rev., 2014, 114, 4540–4563 CrossRef CAS PubMed.
- R. Wang, T. P. Lai, P. Gao, H. Zhang, P. L. Ho, P. C. Y. Woo, G. Ma, R. Y. T. Kao, H. Li and H. Sun, Nat. Commun., 2018, 9, 439 CrossRef PubMed.
- E. P. Gómez-Oliveira, D. Reinares-Fisac, L. M. Aguirre-Díaz, F. Esteban-Betegón, M. Pintado-Sierra, E. Gutiérrez-Puebla, M. Iglesias, Á. Monge and F. Gandara, Angew. Chem., Int. Ed., 2022 DOI:10.1002/anie.202209335.
- J. M. Bothwell, S. W. Krabbe and R. S. Mohan, Chem. Soc. Rev., 2011, 40, 4649–4707 RSC.
- Y. Wang, S. Takki, O. Cheung, H. Xu, W. Wan, L. Öhrström and A. K. Inge, Chem. Commun., 2017, 53, 7018–7021 RSC.
- E. Svensson Grape, V. Rooth, M. Nero, T. Willhammar and A. K. Inge, Nat. Commun., 2022, 13, 1–7 Search PubMed.
- W. Li, L. Jin, N. Zhu, X. Hou, F. Deng and H. Sun, J. Am. Chem. Soc., 2003, 125, 12408–12409 CrossRef CAS PubMed.
- D. Li, H. Q. Xu, L. Jiao and H. L. Jiang, EnergyChem, 2019, 1, 100005 CrossRef.
- C. He, J. Liang, Y.-H. Zou, J.-D. Yi, Y.-B. Huang and R. Cao, Natl. Sci. Rev., 2021, 9(6), nwab157 CrossRef PubMed.
- W. Wan, J. Sun, J. Su, S. Hovmöller and X. Zou, J. Appl. Crystallogr., 2013, 46, 1863–1873 CrossRef CAS PubMed.
- I. Nederlof, E. van Genderen, Y.-W. Li and J. P. Abrahams, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1223–1230 CrossRef CAS PubMed.
- B. L. Nannenga, D. Shi, A. G. W. Leslie and T. Gonen, Nat. Methods, 2014, 11, 927 CrossRef CAS PubMed.
- M. O. Cichocka, J. Ångström, B. Wang, X. Zou and S. Smeets, J. Appl. Crystallogr., 2018, 51, 1652–1661 CrossRef CAS PubMed.
- U. Kolb, Y. Krysiak and S. Plana-Ruiz, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2019, 75, 463–474 CrossRef CAS PubMed.
- R. Weber and G. Bergerhoff, Z. Kristallogr. – New Cryst. Struct., 1991, 195, 87–88 CAS.
- R. K. Feller and A. K. Cheetham, Solid State Sci., 2006, 8, 1121–1125 CrossRef CAS.
- P. J. Saines, H. H. M. Yeung, J. R. Hester, A. R. Lennie and A. K. Cheetham, Dalton Trans., 2011, 40, 6401–6410 RSC.
- L. Cooper, T. Hidalgo, M. Gorman, T. Lozano-Fernández, R. Simón-Vázquez, C. Olivier, N. Guillou, C. Serre, C. Martineau, F. Taulelle, D. Damasceno-Borges, G. Maurin, A. González-Fernández, P. Horcajada and T. Devic, Chem. Commun., 2015, 51, 5848–5851 RSC.
- T. Hidalgo, L. Cooper, M. Gorman, T. Lozano-Fernández, R. Simón-Vázquez, G. Mouchaham, J. Marrot, N. Guillou, C. Serre, P. Fertey, Á. González-Fernández, T. Devic and P. Horcajada, J. Mater. Chem. B, 2017, 5, 2813–2822 RSC.
- Z. Bao, J. Wang, Z. Zhang, H. Xing, Q. Yang, Y. Yang, H. Wu, R. Krishna, W. Zhou, B. Chen and Q. Ren, Angew. Chem., Int. Ed., 2018, 130, 16252–16257 CrossRef.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- M. Köppen, V. Meyer, J. Ångström, A. K. Inge and N. Stock, Cryst. Growth Des., 2018, 18, 4060–4067 CrossRef.
- M. Gemmi, E. Mugnaioli, T. E. Gorelik, U. Kolb, L. Palatinus, P. Boullay, S. Hovmöller and J. P. Abrahams, ACS Cent. Sci., 2019, 5, 1315–1329 CrossRef CAS PubMed.
- Z. Huang, E. Svensson Grape, J. Li, A. K. Inge and X. Zou, Coord. Chem. Rev., 2021, 427, 213583 CrossRef CAS.
- Q. X. Wang and G. Li, Inorg. Chem. Front., 2021, 8, 572–589 RSC.
- Z. Wang, Z. Zeng, H. Wang, G. Zeng, P. Xu, R. Xiao, D. Huang, S. Chen, Y. He, C. Zhou, M. Cheng and H. Qin, Coord. Chem. Rev., 2021, 439, 213902 CrossRef CAS.
- C. Serre, F. Millange, C. Thouvenot, M. Noguès, G. Marsolier, D. Louër and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519–13526 CrossRef CAS PubMed.
- B. Badhani, N. Sharma and R. Kakkar, RSC Adv., 2015, 5, 27540–27557 RSC.
- E. Svensson Grape, J. G. Flores, T. Hidalgo, E. Martínez-Ahumada, A. Gutierrez-Alejandre, A. Hautier, D. R. Williams, M. O’Keeffe, L. Öhrström, T. Willhammar, P. Horcajada, I. A. Ibarra and A. Ken Inge, J. Am. Chem. Soc., 2020, 142, 16795–16804 CrossRef PubMed.
- N. Henry, O. Mentré, F. Abraham, E. J. MacLean and P. Roussel, J. Solid State Chem., 2006, 179, 3087–3094 CrossRef CAS.
- L. Miersch, T. Rüffer, M. Schlesinger, H. Lang and M. Mehring, Inorg. Chem., 2012, 51, 9376–9384 CrossRef CAS PubMed.
- A. K. Inge, M. Köppen, J. Su, M. Feyand, H. Xu, X. Zou, M. O'Keeffe and N. Stock, J. Am. Chem. Soc., 2016, 138, 1970–1976 CrossRef CAS PubMed.
- M. Köppen, A. Dhakshinamoorthy, A. K. Inge, O. Cheung, J. Ångström, P. Mayer and N. Stock, Eur. J. Inorg. Chem., 2018, 2018, 3496–3503 CrossRef.
- Y. Liu, R. C. Klet, J. T. Hupp and O. Farha, Chem. Commun., 2016, 52, 7806–7809 RSC.
- L. Wang, L. Wang, Y. Du, X. Xu and S. X. Dou, Mater. Today Phys., 2021, 16, 100294 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2183351–2183354. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt02260e |
| This journal is © The Royal Society of Chemistry 2022 |