DOI:
10.1039/D2DT00391K
(Paper)
Dalton Trans., 2022,
51, 7889-7898
Synthesis, structure and reactivity with phosphines of Hg(II) ortho-cyano-aminothiophenolate complexes formed via C–S bond cleavage and dehydrogenation of 2-aminobenzothiazoles†
Received
8th February 2022
, Accepted 21st April 2022
First published on 25th April 2022
Abstract
Addition of 2-aminobenzothiazole (abt) and substituted derivatives to Hg(OAc)2 leads to the high yield formation of ortho-cyano-aminothiophenolate (ocap) complexes [Hg{SC6H3XN(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N)}]n (X = H, Me, Cl, Br, NO2) resulting from dehydrogenation and C–S bond cleavage. The reaction appears to be unique to Hg(OAc)2 and with HgCl2 the product [HgCl2(abt)]n contains an intact abt ligand, but reacts with acetate to afford the ocap complex [Hg{SC6H4N(CN)}]n. Binding of abt to Hg(II) has previously been probed in molecular structures of [Hg(sac)2(abt)L] (L = MeOH, DMSO) and these have been reexamined to understand the perturbation of abt upon coordination. When the reaction of abt and Hg(OAc)2 was carried out at low temperatures the intermediate [Hg(κ2-OAc)(EtOH)(μ-HNCNSC6H4)]2 was isolated resulting from a single ligand deprotonation thus allowing a mechanism for ring-opening to be proposed. Reactions of [Hg{SC6H3XN(CN)}]n with mono- and bidentate phosphines have been studied, affording a series of complexes in which the ocap ligands adopt four different binding modes in the solid state, as shown by a number of crystallographic studies. In all, the ligand chelates to a single mercury centre but spans to the second via either: (i) a simple S,N-chelate, (ii) coordination through nitrogen of the CN group, (iii) the sulfur acting as a thiolate-bridge, (iv) both thiolate bridging and cyanide coordination. With PPh3 two different binding modes are seen in complexes [Hg{SC6H3XN(CN)}(PPh3)]2 being dependant upon the nature of the arene-substituent, while addition of excess PPh3 affords mononuclear [Hg{SC6H3XN(CN)}(PPh3)2]. With dppm, binuclear [Hg{SC6H3XN(CN)}(κ1-dppm)]2 result in which the diphosphine binds in a monodentate fashion. With the more flexible diphosphines, dppe and dppb, coordination polymers [Hg{SC6H3XN(CN)}(κ1,κ1-diphosphine)]n result in which ocap binds in a simple chelate fashion. Somewhat unexpectedly, with dppp, binuclear complexes [Hg2{SC6H3XN(CN)}2(μ,κ1,κ1-dppp)] result in which two diphosphines bridge the Hg2 centre, while with dppf mononuclear chelates are proposed to result. Thus, the simple and high-yielding ring-opening of 2-aminobenzothiazole and substituted derivatives by mercuric acetate provides ready access to a range of novel ortho-cyano-aminothiophenolate complexes, being shown to be a highly versatile ligand that can adopt a number of different coordination modes.
N)}]n (X = H, Me, Cl, Br, NO2) resulting from dehydrogenation and C–S bond cleavage. The reaction appears to be unique to Hg(OAc)2 and with HgCl2 the product [HgCl2(abt)]n contains an intact abt ligand, but reacts with acetate to afford the ocap complex [Hg{SC6H4N(CN)}]n. Binding of abt to Hg(II) has previously been probed in molecular structures of [Hg(sac)2(abt)L] (L = MeOH, DMSO) and these have been reexamined to understand the perturbation of abt upon coordination. When the reaction of abt and Hg(OAc)2 was carried out at low temperatures the intermediate [Hg(κ2-OAc)(EtOH)(μ-HNCNSC6H4)]2 was isolated resulting from a single ligand deprotonation thus allowing a mechanism for ring-opening to be proposed. Reactions of [Hg{SC6H3XN(CN)}]n with mono- and bidentate phosphines have been studied, affording a series of complexes in which the ocap ligands adopt four different binding modes in the solid state, as shown by a number of crystallographic studies. In all, the ligand chelates to a single mercury centre but spans to the second via either: (i) a simple S,N-chelate, (ii) coordination through nitrogen of the CN group, (iii) the sulfur acting as a thiolate-bridge, (iv) both thiolate bridging and cyanide coordination. With PPh3 two different binding modes are seen in complexes [Hg{SC6H3XN(CN)}(PPh3)]2 being dependant upon the nature of the arene-substituent, while addition of excess PPh3 affords mononuclear [Hg{SC6H3XN(CN)}(PPh3)2]. With dppm, binuclear [Hg{SC6H3XN(CN)}(κ1-dppm)]2 result in which the diphosphine binds in a monodentate fashion. With the more flexible diphosphines, dppe and dppb, coordination polymers [Hg{SC6H3XN(CN)}(κ1,κ1-diphosphine)]n result in which ocap binds in a simple chelate fashion. Somewhat unexpectedly, with dppp, binuclear complexes [Hg2{SC6H3XN(CN)}2(μ,κ1,κ1-dppp)] result in which two diphosphines bridge the Hg2 centre, while with dppf mononuclear chelates are proposed to result. Thus, the simple and high-yielding ring-opening of 2-aminobenzothiazole and substituted derivatives by mercuric acetate provides ready access to a range of novel ortho-cyano-aminothiophenolate complexes, being shown to be a highly versatile ligand that can adopt a number of different coordination modes.
1. Introduction
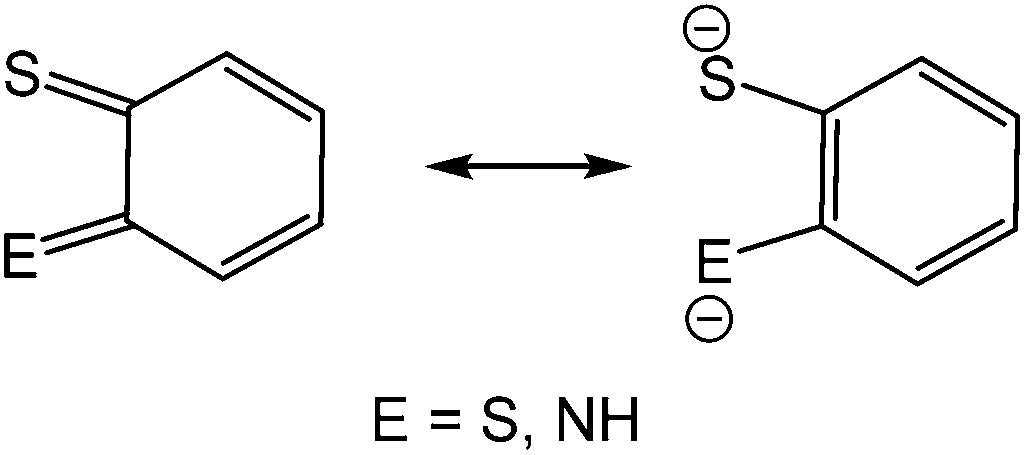
The coordination chemistry of so-called non-innocent ligands1 is an area of intense research interest of which dithiolenes2 and to a lesser extent, ortho-aminothiophenolates3–6 (Chart 1) are good examples. Both can adopt different resonance forms, rendering them redox active and capable of stabilising metals in a wide range of oxidation states. The vast majority of ortho-aminothiophenolate chemistry focuses on the parent ligand (E = NH) as it is easily accessible from ortho-aminothiophenol or bis(ortho-aminophenyl)disulfide.3–6 In contrast, N-functionalised derivatives have received less attention despite allowing for fine-tuning of steric and electronic properties. A rare amino-substituent is the cyano group,7 yet such species could potentially exhibit interesting electronic and redox properties resulting from the potential delocalisation of electron-density over the N–CN moiety. A few years ago, we reported our preliminary observations of the serendipitous synthesis of ortho-cyano-aminothiophenolate (ocap) complexes of mercury via the ring-opening dehydrogenation of 2-aminobenzothiazole (abt) and some derivatives.8 Herein we develop this chemistry, showing that a range of such ligands are readily accessible in a simple one-pot reaction, and these complexes react with phosphines to give complexes displaying varying coordination geometries of the ortho-cyano-aminothiophenolate ligand.
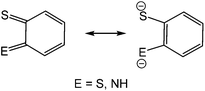 |
| | Chart 1 Resonance hybrids of dithiolene (E = S) and ortho-aminothiophenolate (E = NH) ligands. | |
2. Results and discussion
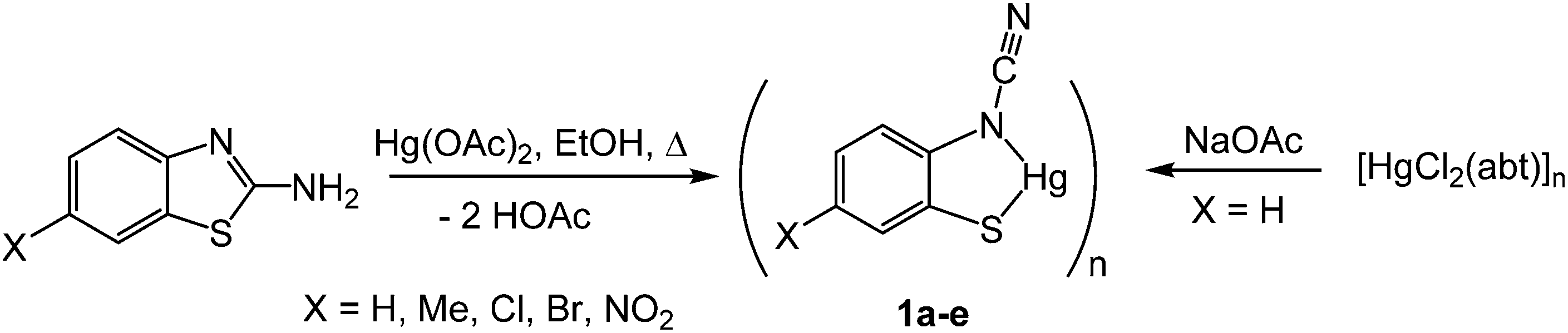
(i) Ring-opening of 2-aminobenzothiazoles (abt) upon reaction with Hg(OAc)2: synthesis of ortho-cyano-aminothiophenolate (ocap) complexes [Hg{SC6H3XN(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) N)}]n (1a–e)
N)}]n (1a–e)
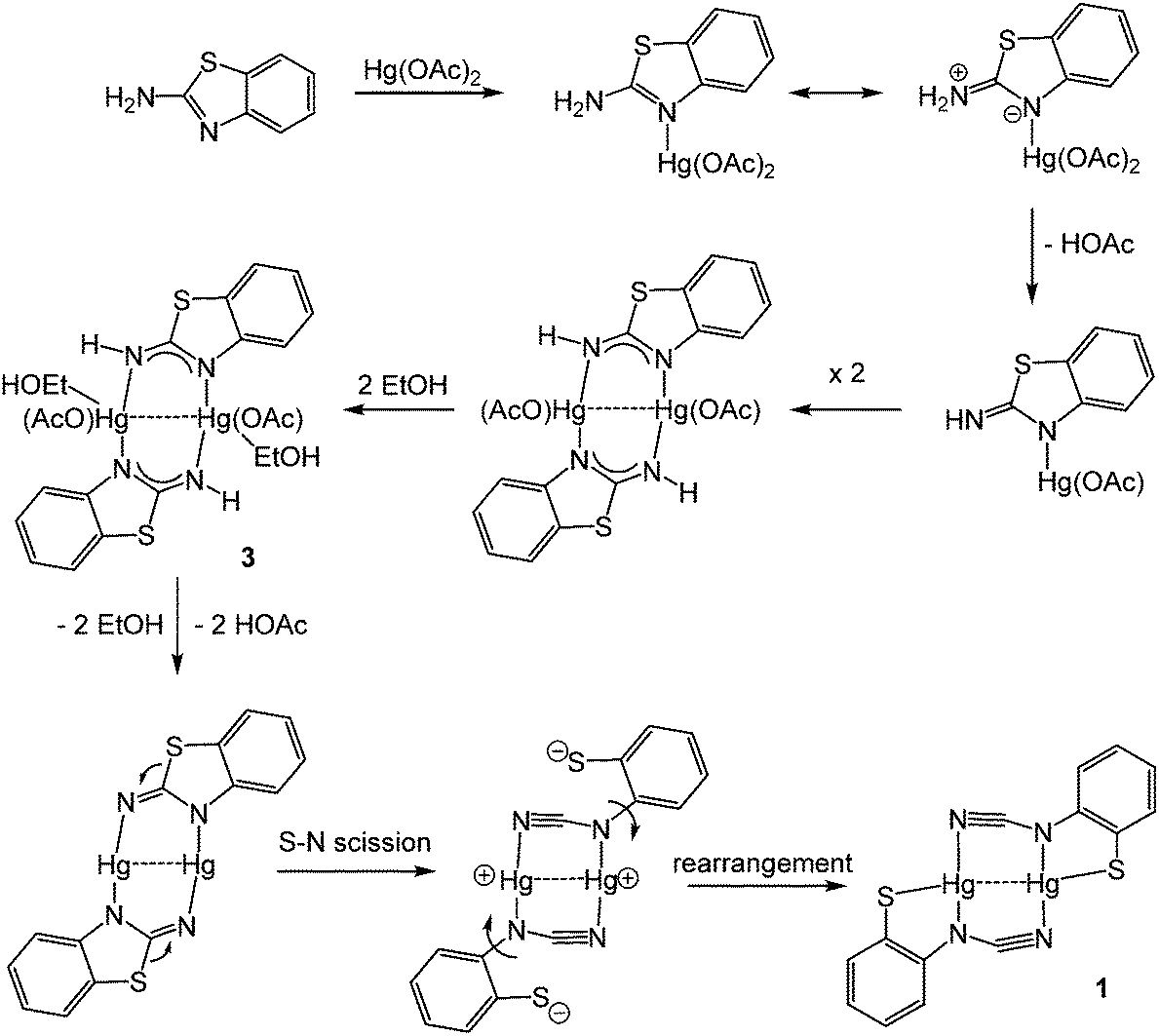
Addition of 2-aminobenzothiazole (abt) and substituted derivatives to Hg(OAc)2 in warm ethanol led to the high yield (70–90%) formation of ring-opened ortho-cyano-aminothiophenolate complexes formulated as [Hg{SC6H3XN(CN)}]n (1a–e) (Scheme 1). A major ligand rearrangement was immediately apparent from the disappearance of the amine protons in the 1H NMR spectrum, being confirmed by the appearance of a strong absorption at ca. 2120 cm−1 associated with a CN group in IR spectra. Complexes 1a–e are only sparingly soluble in organic solvents, suggesting that they are probably coordination polymers. We also prepared 1a in similar yields upon addition of NaOAc to an ethanol solution of [HgCl2(abt)]n.9
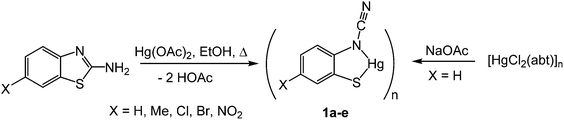 |
| | Scheme 1 Synthesis of ocap complexes 1a–evia ring-opening of abt upon reaction with Hg(OAc)2. | |
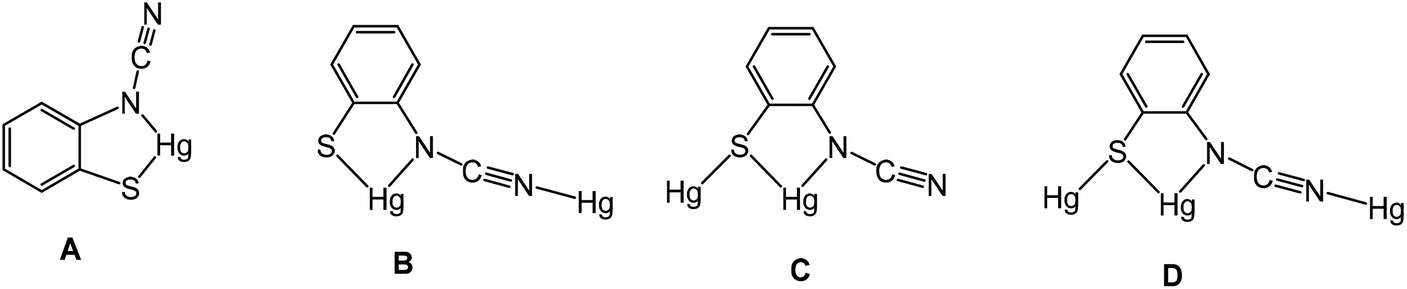
We have been unable to crystallographically characterise these species and thus unambiguously assign the ocap coordination mode, although based on the different binding modes seen in phosphine adducts (A–CChart 2 see later) we speculate that they most likely act as a tetra-coordinating ligand (D) binding through both nitrogen atoms and with a bridging sulfur, giving rise to a coordination polymer.
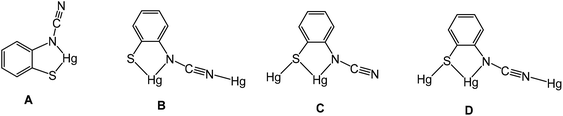 |
| | Chart 2 Binding modes (A–D) of the ortho-cyano-aminothiophenolate (ocap) ligand. | |
(ii) Probing the mechanism of the ring-opening reaction: characterisation of intermediate [Hg(κ2-OAc)(EtOH)(μ-HNCNSC6H4)]2 (3)
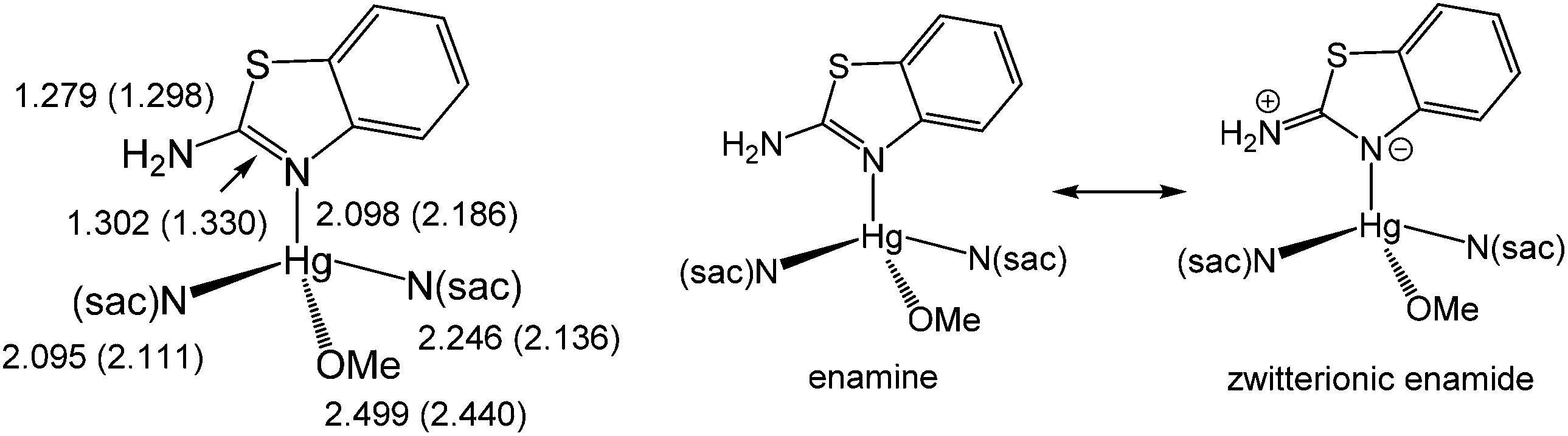
The reaction appears to be limited to Hg(OAc)2, since other mercury salts give simple adducts in which abt is bound through the endocyclic nitrogen.9 For example, we recently crystallographically characterised [Hg(sac)2(abt)L] (2a–b) (L = MeOH, DMSO) (Chart 3) formed upon addition of two equivalents of sodium saccharinate (Nasac) to [HgCl2(abt)]n.10Chart 3 gives a comparison of important bond lengths within these two closely related molecules. Consideration of the C–N bond distances within the abt ligands provides insight into the nature of its bonding to mercury. Thus, while errors are high and differences are not with the accepted 3σ range, there is nevertheless a clear trend. Thus, the putative C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond [2a N(3)–C(3) 1.302(10); 2b N(3)–C(15) 1.330(8) Å] is longer than the supposed single C–N bond to the amine [2a N(4)–C(3) 1.279(10); 2b N(4)–C(15) 1.298(8) Å] suggesting that the zwitterionic resonance form cannot be discounted (Chart 3). This suggests that binding of abt to the Hg(II) centre significantly perturbs its electronic structure leading to a favouring of the zwitterionic enamide over the enamine form.
N double bond [2a N(3)–C(3) 1.302(10); 2b N(3)–C(15) 1.330(8) Å] is longer than the supposed single C–N bond to the amine [2a N(4)–C(3) 1.279(10); 2b N(4)–C(15) 1.298(8) Å] suggesting that the zwitterionic resonance form cannot be discounted (Chart 3). This suggests that binding of abt to the Hg(II) centre significantly perturbs its electronic structure leading to a favouring of the zwitterionic enamide over the enamine form.
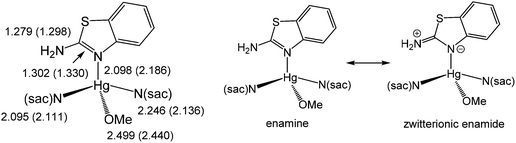 |
| | Chart 3 A summary of important bond lengths in [Hg(sac)2(abt)L] (2a–b) (L = MeOH, DMSO) and resonance hybrids for Hg(II)-abt complexes. | |
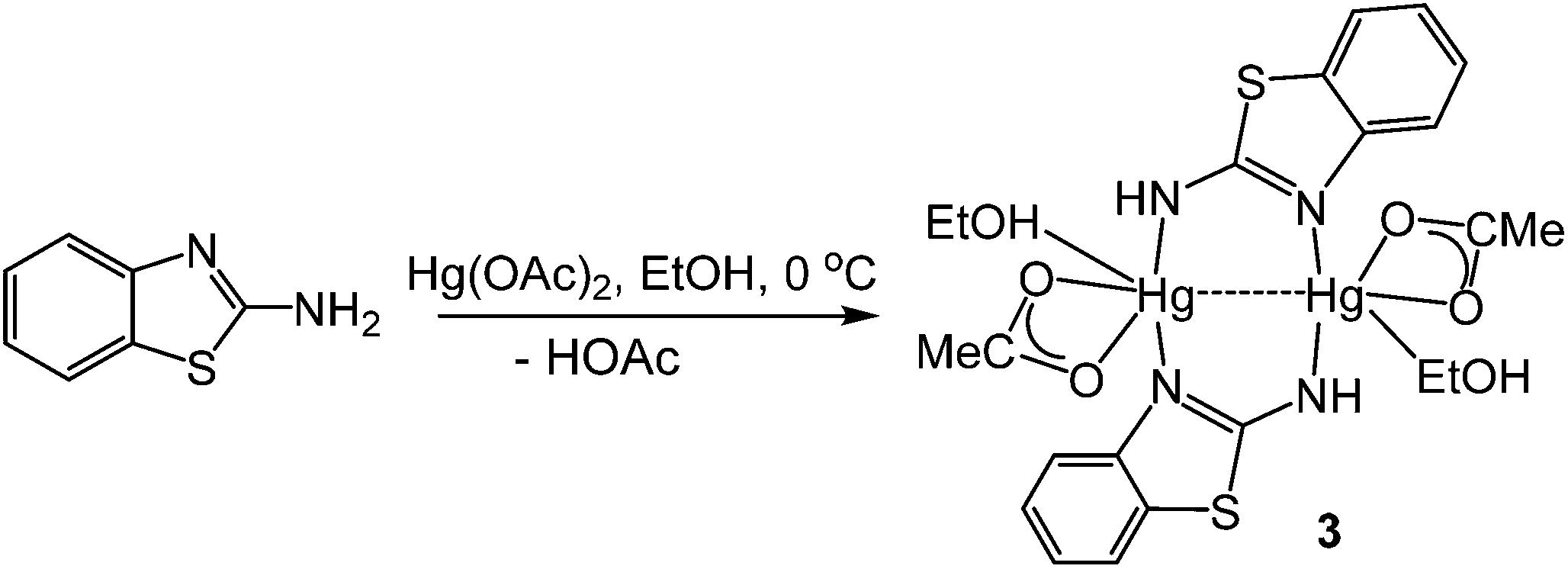
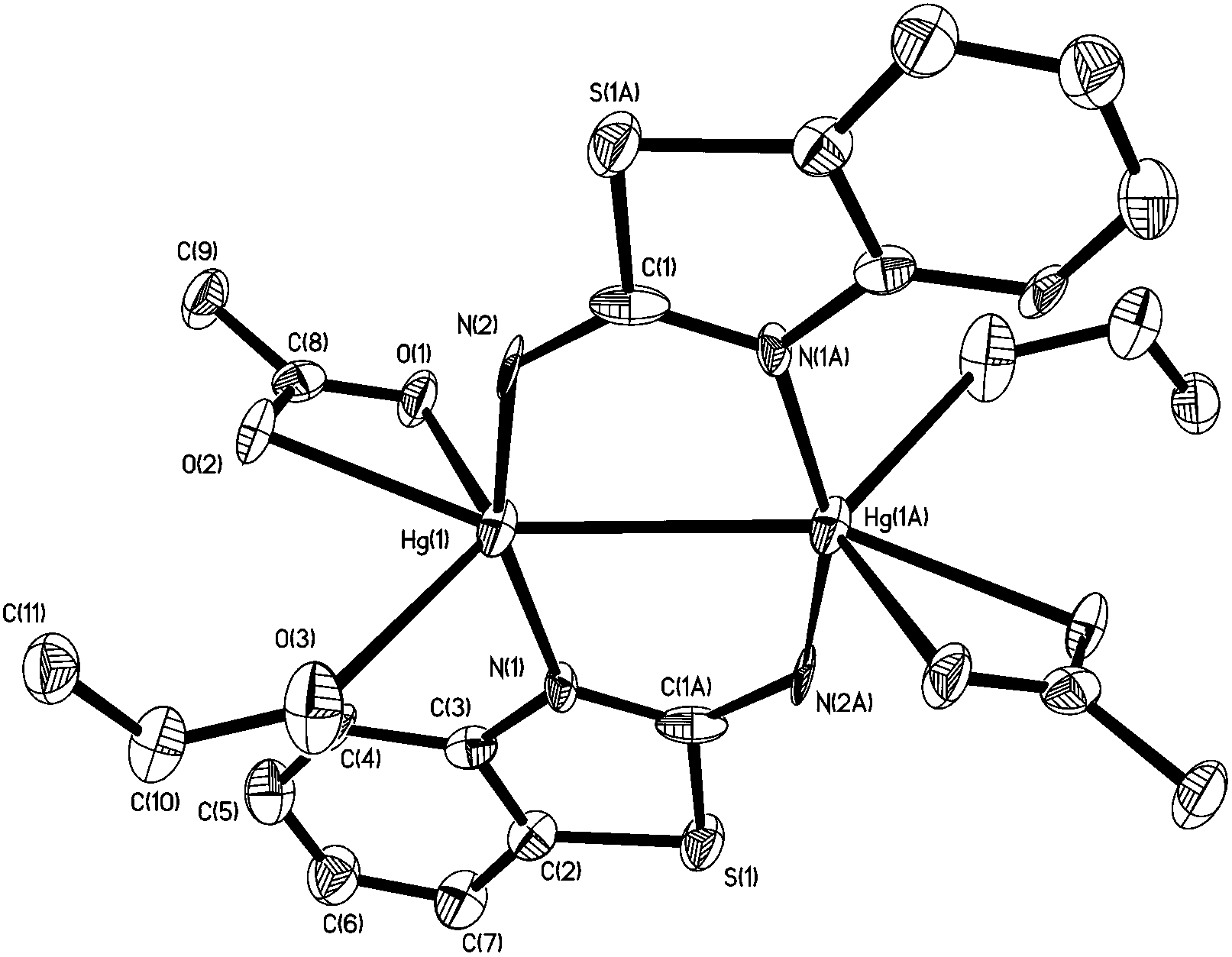
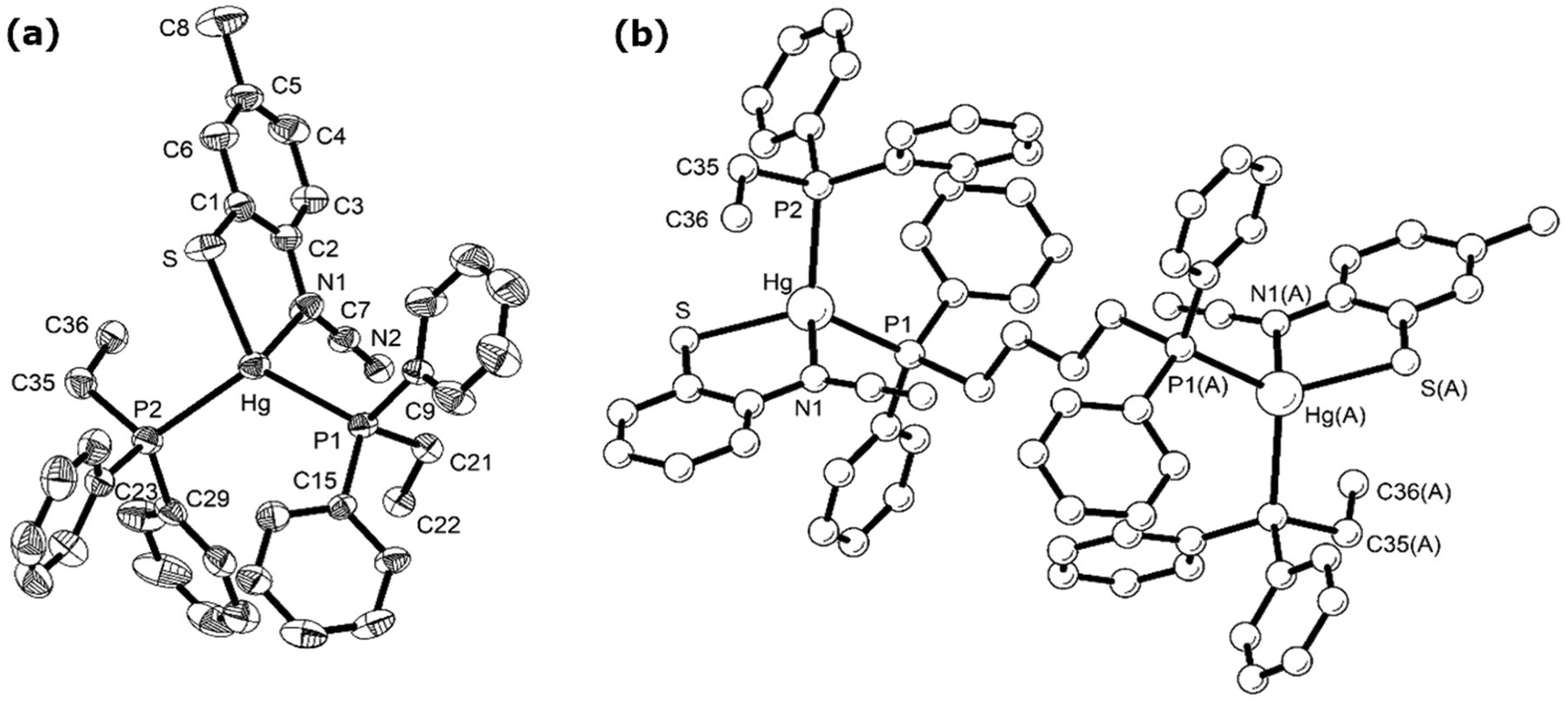
In order to further probe the ring-opening process, we carried out the reaction of abt and Hg(OAc)2 at 0 °C and were able to isolate intermediate [Hg(κ2-OAc)(EtOH)(μ-HNCNSC6H4)]2 (3) in good yield (Scheme 2). An X-ray crystallographic study was undertaken the results of which are displayed in Fig. 1.
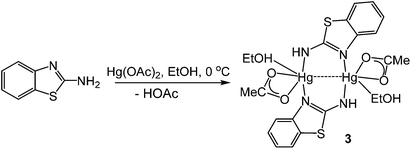 |
| | Scheme 2 Formation of 3 from reaction of abt and Hg(OAc)2 at 0 °C. | |
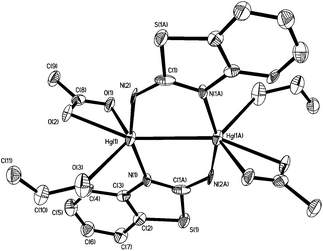 |
| | Fig. 1 Molecular structure of 3. Thermal ellipsoids are displayed at 50% probability and hydrogen atoms omitted for clarity. Selected bond lengths (Å) and bond angles (°): Hg1–Hg1A 3.094(1), Hg1–N1 2.067(6), Hg1–N2 2.040(8), Hg1–O1 2.560(7), Hg1–O2 2.481(6), Hg1–O3 2.592(8), N1–C1A 1.348(11), N2–C1 1.286(10), C1–S1A 1.738(8); N1A–C1–N2 126.5(7), N1–Hg–N2 158.5(2), O1–Hg–O2 114.5(2), N1–Hg1–O3 91.1(2). | |
The molecule consists of a Hg(II)–Hg(II) centre spanned by two 2-aminobenzothiazolate ligands, resulting from deprotonation of abt. Binding to mercury is through the two nitrogen atoms, the distance to the endocyclic nitrogen of 2.071(6) Å being slightly longer than that of 2.040(8) Å to the exocyclic amide. The two N–C bonds differ significantly, that to the amide [N2–C1 1.286(10) Å] being shorter than the endocyclic bond [N1–C1 1.348(11) Å]. The coordination sphere of each mercury atom is completed by a chelating acetate ligand [O1–Hg1–O2 51.38(2)°] and a molecule of ethanol. Thus, ignoring the metal–metal interaction, each mercury center is five-coordinate and the coordination geometry is best described as a highly distorted trigonal bipyramid with the two nitrogen atoms occupying axial sites [N1–Hg1–N2 158.8(2)°]. Two resonance structures can be drawn for the bridging 2-aminobenzothiazolate ligand (Chart 4) and on the basis of the bond lengths and angles found in 3 we conclude that actual structure lies closer to the zwitterionic form.
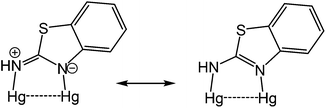 |
| | Chart 4 Resonance hybrids for the bridging 2-aminobenzothiazolate ligand. | |
Redissolving 3 in ethanol and heating afforded 1a. This allows a plausible mechanistic pathway for the formation of [Hg{SC6H3XN(CN)}]n (1a–e) (Scheme 3) to be suggested. Initial generation of a simple abt adduct is proposed, two resonance hybrids of which are possible. As a result of the change in electronegativity of the endocyclic nitrogen upon binding to Hg(II), the zwitterionic hybrid is populated which has an acidic proton and can eliminate acetic acid. This leads to formation of an uncharged mercuric amide (ketimide); dimerisation and addition of ethanol to which afford intermediate 3. Upon heating, elimination of a second equivalent of acetic acid affords a highly strained bridging diamide, which undergoes C–S bond scission, and rotation about the single N–Ar bond allows sulfur to find to bind to chalcogenophilic Hg(II) resulting in formation of 1.
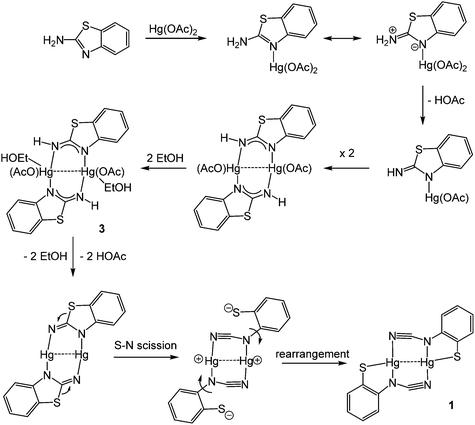 |
| | Scheme 3 Suggested pathway for the ring-open dehydrogenation of abt upon reaction with Hg(OAc)2. | |
(iii) Reactions with PPh3: synthesis of [Hg{SC6H3XN(CN)}(PPh3)]2 (4) and identification of two structural types
We note at the outset of our reactivity studies that Hg(II) is a d10 metal centre and thus, lacking any crystal field stabilisation energy, is labile. This, coupled with the large size and relatively soft nature of Hg(II) suggests that the structures of the phosphine adducts shown below are likely those which have the lowest ground state energies in the solid-state. In solution there may well be a number of interconverting species, and this is validated by the broad nature of the 31P{1H} NMR spectra. Thus, we do not suggest that the different coordination geometries shown in the solid-state are also those adopted in solution.
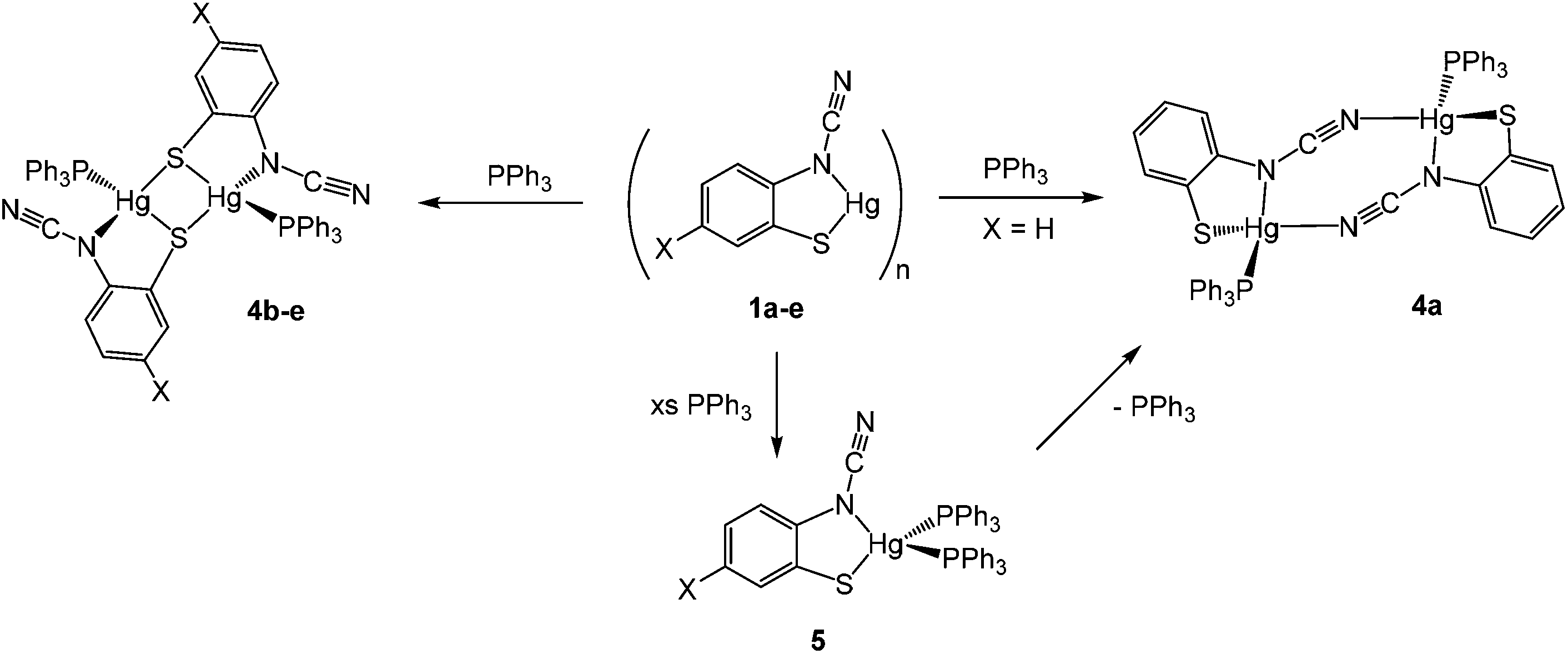
We began our exploration of the chemistry of 1a–e by trying to break down the supposed polymeric framework upon reaction with PPh3. This strategy was successful and in all cases addition of ca. 2 equivalents of PPh3 resulted in formation of adducts [Hg{SC6H3XN(CN)}(PPh3)]2 in high (60–83%) yields, being isolated as pale yellow air-stable solids (Scheme 4). Phosphine coordination was easily confirmed in the 31P{1H} NMR spectra, all showing a broad singlet resonance at ca. 25–35 ppm and in the IR spectrum the distinct CN vibration was still apparent.
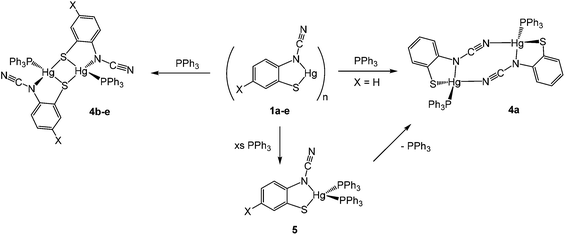 |
| | Scheme 4 Reactions of 1a–e with PPh3. | |
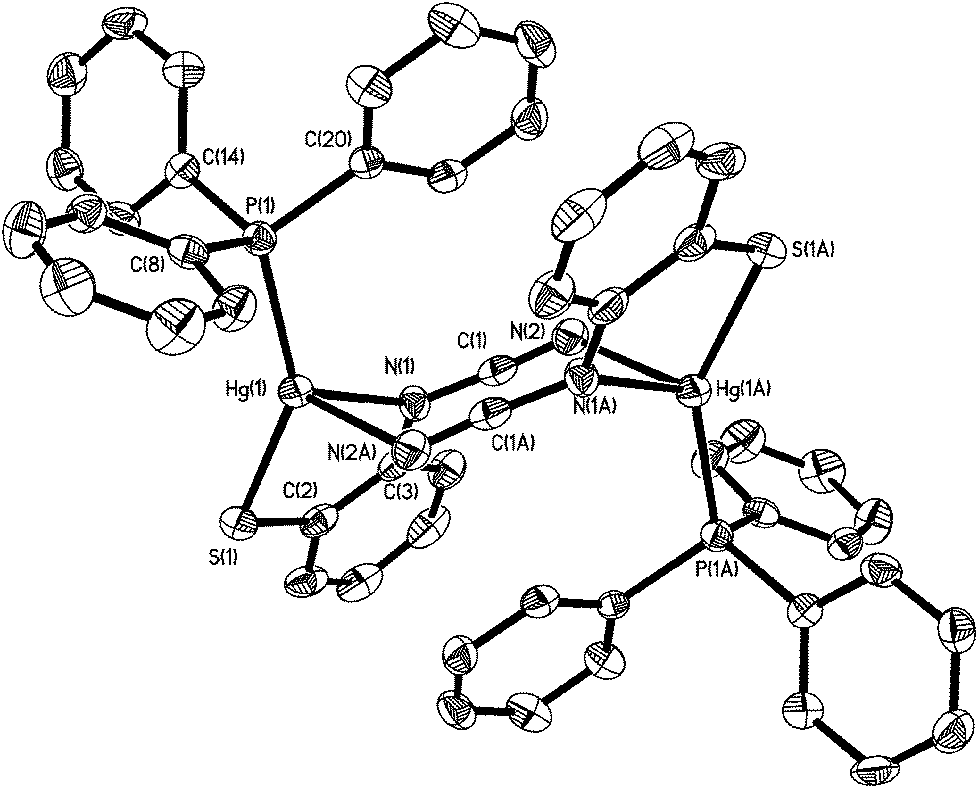
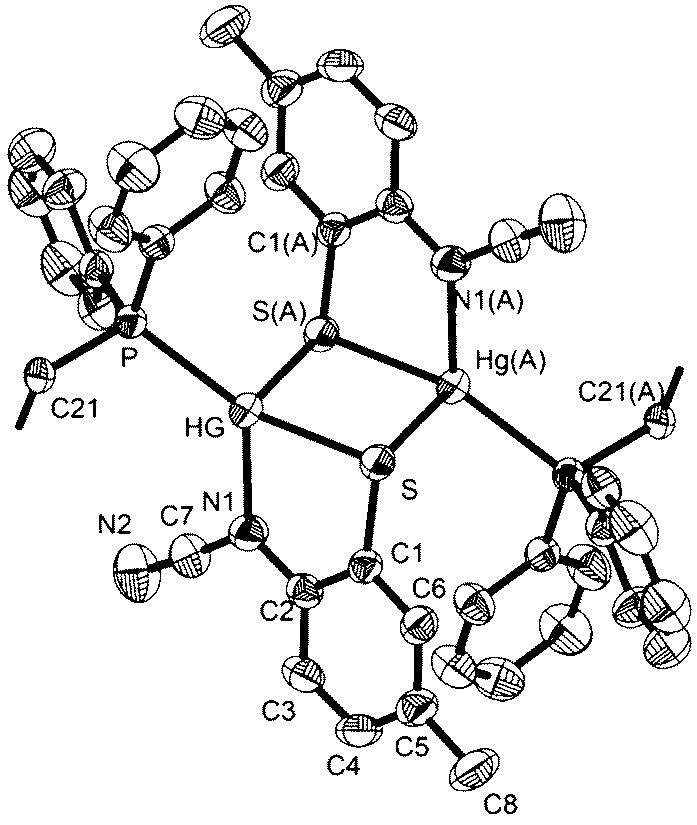
Crystal structures were carried out on 4a, 4b and 4d and these revealed that the solid-state structure, and in some cases the coordination mode of the ocap ligand was dependent upon the nature of the ocap ligand. The molecular structure of [Hg{SC6H4N(CN)}(PPh3)]2 (4a) is shown in Fig. 2.8 It is a centrosymmetric dimer in which the coordination geometry at each Hg(II) centre is a distorted N2PS tetrahedron. The ortho-cyano-aminothiophenolate ligands adopts coordination mode B (Chart 2) chelating to one mercury [N1–Hg1–S1 81.76(11)°], while bridging to a second through coordination of cyanide. This gives an eight-membered Hg2N4C2 ring which, because of the linear nature of the N–CN subunit, is akin to a chair configuration of a six-membered ring. As expected, the two Hg–N distances differ significantly, the longer Hg1–N2A of 2.517(5) Å being akin to a simple mercury–amide interaction. Perhaps most importantly, adoption of this bonding mode has little effect on the bond distances within the N–CN moiety which still displays long and short N–C interactions [N1–C1 1.287(6), N2–C1 1.153(6) Å]. This is also reflected in the observation of a strong CN stretch at 2140 cm−1 in the IR spectrum.
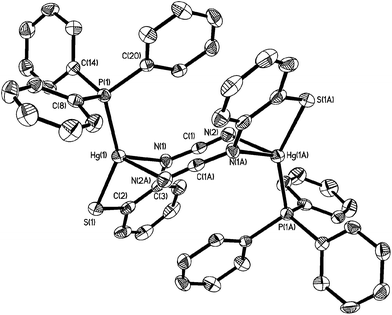 |
| | Fig. 2 Molecular structure of [Hg{SC6H4N(CN)}(PPh3)]24a.8 Thermal ellipsoids are displayed at 50% probability and hydrogen atoms omitted for clarity. Selected bond lengths (Å) and bond angles (°): Hg1–N1 2.312(4), Hg1–S1 2.352(2), Hg1–P1 2.373(2), Hg1–N2A 2.517(5), N1–C1 1.287(6), N2–C1 1.153(6), S1–Hg1–N1 81.76(11), N1–Hg1–N2A 91.86(14), S1–Hg1–P1 147.76(5), N1–Hg1–P1 122.04(11), N1–C1–N2 176.2(6). | |
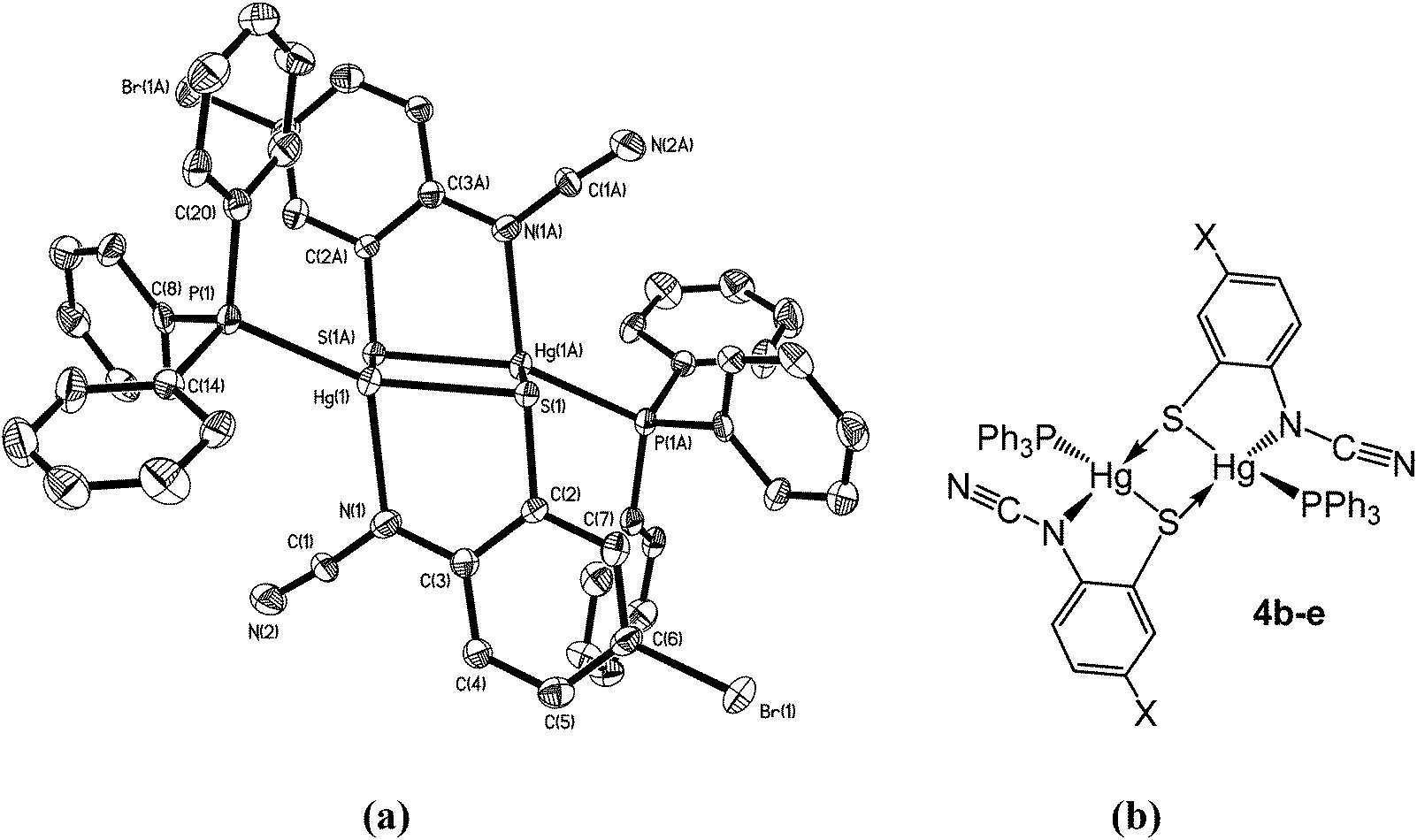
Complexes 4b–e have a different solid-state structure to that in 4a, the ocap ligands adopting coordination mode C (Chart 2). The molecular structure of [Hg{SC6H3BrN(CN)}(PPh3)]2 (4d) is shown in Fig. 3.8 It is again a centrosymmetric dimer, but unlike 4a consists of a central Hg2S2 core with each mercury centre also being coordinated to one phosphine and a nitrogen of an N–CN moiety. Both Hg–S and Hg–N bond lengths are slightly shorter than those in 4a and the Hg–P distance is longer. The N–CN subunit is linear [N1–C1–N2 174.6(7)°] and shows long [N1–C1 1.287(8) Å] and short [N2–C1 1.154(8) Å] N–C bonds. Bridging of the thiolate ligands is highly asymmetric [Hg1–S1 2.411(2), Hg1–S1A 2.871(2) Å], suggesting a better Lewis model is that shown in Fig. 3b.
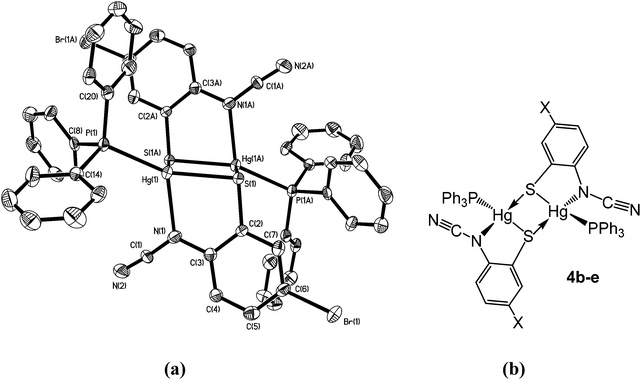 |
| | Fig. 3 (a) Molecular structure of 4d.8 Thermal ellipsoids are displayed at 50% probability and hydrogen atoms omitted for clarity. Selected bond lengths (Å) and bond angles (°): Hg1–N1 2.266(5), Hg1–S1 2.411(2), Hg1–S1A 2.871(2), Hg1–P1 2.272(2), N1–C1 1.287(8), N2–C1 1.154(8); Hg1–S1–Hg1A 83.57(5), S1–Hg1–S1A 96.43(5), S1–Hg1–N1 80.76(14), P1–Hg–S1 151.40(6), P1–Hg1–N1 122.19(13), N1–C1–N2 174.6(7); (b) line drawing highlighting the bonding within 4b–e. | |
Reasons for the adoption of different binding modes of ocap are not obvious. It is hard to attribute the difference to steric effects as only minor changes are made at a ring position, which appears not to show and significant intra- or intermolecular interactions in either form. In the solid state, the ν(CN) vibration in the IR spectrum varies little between different binding types (4a 2140, 4b–c 2142 cm−1) although for 4d it is seen at 2163 cm−1. In solution the 31P{1H} NMR chemical shifts vary a little more (4a 36.5, 4b–d 25.4–29.0 ppm) but 4e at 35.6 ppm points away from a differentiation of bonding types. Likely, binding modes B and C have similar energies and it may be secondary intermolecular interactions which serve to dictate the coordination mode adopted.
Addition of a slight excess of PPh3 to either 1a or 4a and heating in CHCl3 resulted in formation of a new complex believed to be the bis(phosphine) adduct, [Hg{SC6H4N(CN)}(PPh3)2] (5) (Scheme 4). Elemental analysis was consistent with this formulation and the 1H NMR spectrum showed a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of phosphine and ocap ligands. In the 31P{1H} NMR spectrum a broad singlet at 29.2 ppm can be compared to that at 36.5 ppm in 4a. We propose a mononuclear structure akin to that found for [HgCl2(PPh3)2]11 in which the Hg(II) centre is a distorted tetrahedron. We have been unable to confirm this by X-ray crystallography and the broad nature of the 31P{1H} NMR spectra leads us to suggest that there is an equilibrium between 5 and a three coordinate [Hg{SC6H4N(CN)}(PPh3)] which can in turn dimerize. For example, we note that with bulky phosphine ligands both 4- and 3-coordinate dihalide complexes can be isolated.12 The broad nature of the 31P{1H} NMR spectra is also the likely reason we have been unable to observe 31P–199Hg couplings.13
:1 ratio of phosphine and ocap ligands. In the 31P{1H} NMR spectrum a broad singlet at 29.2 ppm can be compared to that at 36.5 ppm in 4a. We propose a mononuclear structure akin to that found for [HgCl2(PPh3)2]11 in which the Hg(II) centre is a distorted tetrahedron. We have been unable to confirm this by X-ray crystallography and the broad nature of the 31P{1H} NMR spectra leads us to suggest that there is an equilibrium between 5 and a three coordinate [Hg{SC6H4N(CN)}(PPh3)] which can in turn dimerize. For example, we note that with bulky phosphine ligands both 4- and 3-coordinate dihalide complexes can be isolated.12 The broad nature of the 31P{1H} NMR spectra is also the likely reason we have been unable to observe 31P–199Hg couplings.13
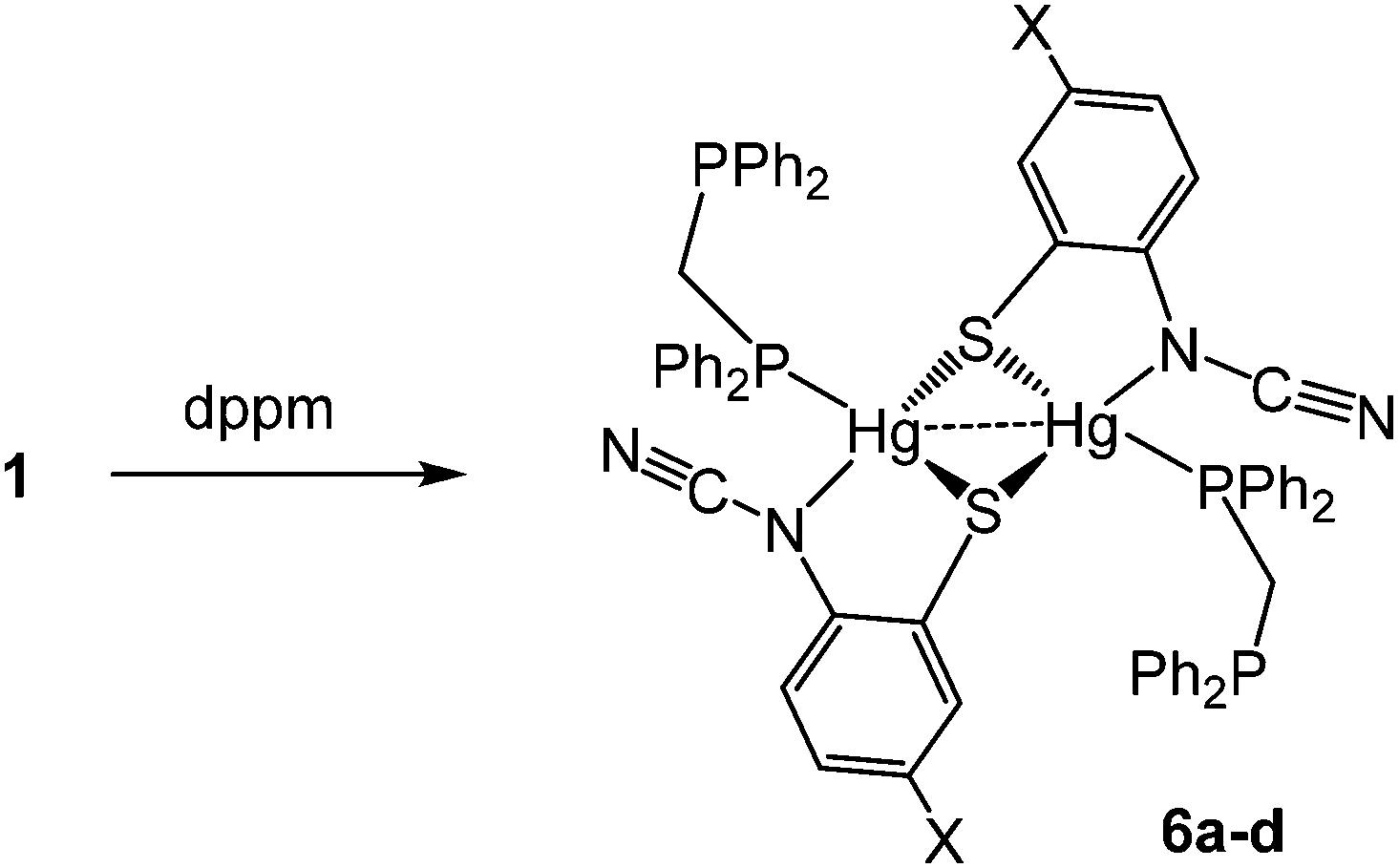
(iv) Reactions with bis(diphenylphosphino)methane (dppm): synthesis of binuclear [Hg{SC6H3XN(CN)}(κ1-dppm)]2 (6)
We next studied reactions of 1 with diphosphines, expecting to generate mononuclear complexes of the type [Hg{SC6H3XN(CN)}(κ2-diphosphine)] in which ocap adopts coordination mode A. We began with the small bite-angle bis(diphenylphosphino)methane (dppm) which normally acts as a bridging ligand but can also bind in a chelate or monodentate fashion.14 Reactions with equimolar amounts of 1a–d in EtOH at room temperature led to isolation of [Hg{SC6H3XN(CN)}(κ1-dppm)]2 (6a–d) as pale yellow solids in ca. 60–85% yields (Scheme 5).
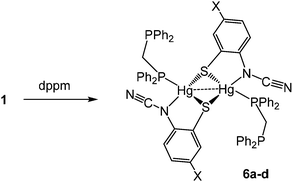 |
| | Scheme 5 Reactions of 1a–d with dppm. | |
The 31P{1H} NMR spectrum clearly shows that the diphosphine is monodentate, two doublets at 9.9 and −2.6 ppm (JPP 29 Hz) for 6a being consistent with coordinated and uncoordinated PPh2 groups respectively. We have also not been able to observe 31P–199Hg couplings for 6 despite the “sharp” nature of the 31P signals and this may be due to a slow exchange of bound and free phosphorus atoms. The proposed structure was confirmed by a preliminary crystallographic study of 6a (ESI Fig. 1†) which also confirms the adoption of binding mode C in which sulfur bridges the Hg⋯Hg vector. The monodentate nature of the dppm in 6 has literature precedent. Peringer and co-workers have studied reactions of dppm with a range of Hg(II) salts,15–18 for example, addition to [Hg(O3SCF3)2] affords [Hg(κ1-dppm)2][O3SCF3]2.16 Here the two ends of the diphosphine exchange on the NMR timescale18 presumably via a three-coordinate intermediate. No such interconversion of the two ends of the diphosphine are seen in 6. Indeed, in an attempt to disrupt the binuclear framework and force the dppm ligand to adopt a chelate or bridging mode, 6a was heated at reflux in CHCl3 for 8 h leading (after cooling) to the formation of green-yellow crystals identified by X-ray diffraction (see ESI†) as the known complex [Hg2(μ-Cl)(μ-dppm)2Cl2]Cl.19 Thus, rather than breaking the dimercury moiety, coordination of the second phosphorus centre has led to elimination of the ocap ligands.
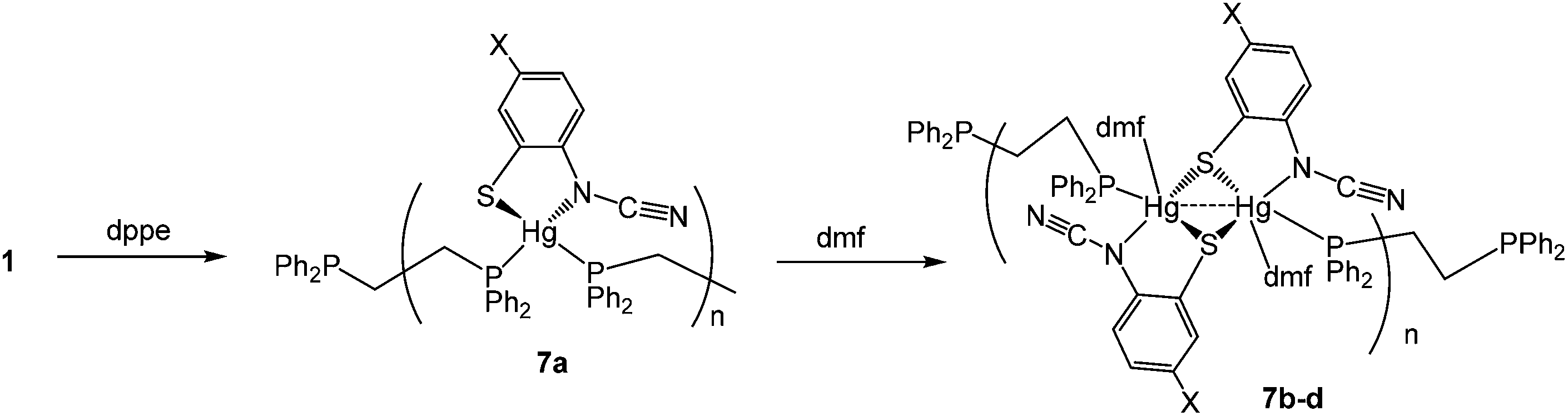
(v) Reactions with 1,2-bis(diphenylphosphino)ethane (dppe): synthesis of coordination polymers [Hg{SC6H3XN(CN)}(μ–κ1,κ1-dppe)]n (7a) and [Hg(DMF){SC6H3XN(CN)}(μ–κ1,κ1-dppe)]n (7b–d)
We next turned our attention to more flexible diphosphines, a number of which have been shown to afford chelate complexes of the type [HgX2(κ2-diphosphine)].16,20,21 Firstly, we explored the chemistry of 1,2-bis(diphenylphosphino)ethane (dppe). In contrast to reactions of Hg(II) salts with dppm, addition of dppe to [Hg(O3SCF3)2] yields the bis(chelate) [Hg(κ2-dppe)2][O3SCF3]2,16 while with HgI2 the coordination polymer [HgI2(H2O)(μ–κ1,κ1-dppe)]n is formed, in which the dppe ligand links together 5-coordinate Hg(II) centres.22
Reactions of 1 with dppe led to the isolation of coordination polymers 7a–d (Scheme 6) in which the diphosphine bridges between mercury centres. We were able to crystallise 7a from CHCl3, but in contrast 7b–d were not particularly soluble in CHCl3 and hence all were recrystallised from DMF. After recrystallisation elemental analysis consistently showed the presence of one equivalent of DMF per mercury. This was borne out by the crystallographic characterisation of 7b as shown in Fig. 4.
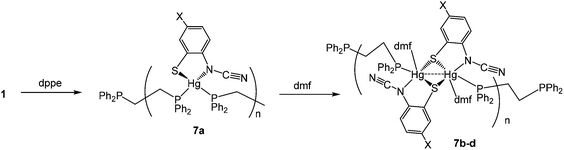 |
| | Scheme 6 Reactions of 1a–d with dppe followed by recrystallisation from DMF. | |
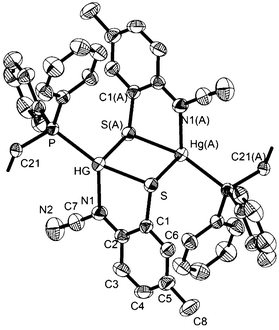 |
| | Fig. 4 Molecular structure of [Hg{SC6H3MeN(CN)}(DMF)(μ–κ1,κ1-dppe)]n (7b). Thermal ellipsoids at the 50% level and hydrogen atoms omitted for clarity and disordered DMF are omitted for clarity. Selected bond lengths (Å) and bond angles (°): Hg–N1 2.275(3), Hg–S 2.4370(9), Hg(A)–S 2.9350(9), Hg–O 2.880(5), Hg–S–Hg(A) 87.12 (3), N1–Hg–P 128.61(9), N1–Hg–S 81.13(9), S–Hg–P 146.39(3), S(A)–Hg–S 92.88(3), S(A)–Hg–P 102.32(3). Symmetry operator A: 1 − x, 1 − y, 1 − z. | |
We did obtain preliminary crystallographic data for 7a, and while of low quality, the molecular structure showing a coordination polymer consisting of 4-coordinate distorted tetrahedral Hg(II) centres linked by dppe ligands was established. In contrast in 7b, each Hg(II) centre is a 5-coordinate distorted trigonal bipyramid, pairs of which are spanned by ocap ligands which adopt the bridging thiolate mode C and it is these Hg2 centres that are linked by the diphosphine to give the polymeric structure. In many respects this mimics the structure of dppm derivatives 6 but differs in that each Hg(II) centre is also bound by DMF. The Hg–O bond distance of 2.882(5) Å might be considered at the upper end of such interactions but nevertheless coordination of DMF is clearly shown by the trigonal bipyramidal nature of the Hg(II) centre the DMF and one of the sulfur ligands occupying the axial sites [S–Hg–O 172.47°]. Coordinated DMF is retained upon redissolution, with 7b–d each showing a pair of doublets in the 31P{1H} NMR spectrum, for example at 27.5 and 36.5 (JPP 36 Hz) for 7b.
We tentatively suggest that in all cases the initially formed product has the structure adopted by 7a and upon redissolution in DMF they rearrange to form the DMF adducts, which are subsequently retained in CDCl3. The surprising feature of this transformation is not the coordination of the DMF, but the increased hapticity of the ortho-cyano-aminothiophenolate ligand; one might expect that addition of another ligand would lead to a reduction in hapticity if it changed at all. This presents evidence that, in solution, coordination polymers may breakdown to yield (at least in part) mononuclear fragments [Hg{SC6H3XN(CN)}(dppe)] which can reassemble to give the observed coordination polymers.
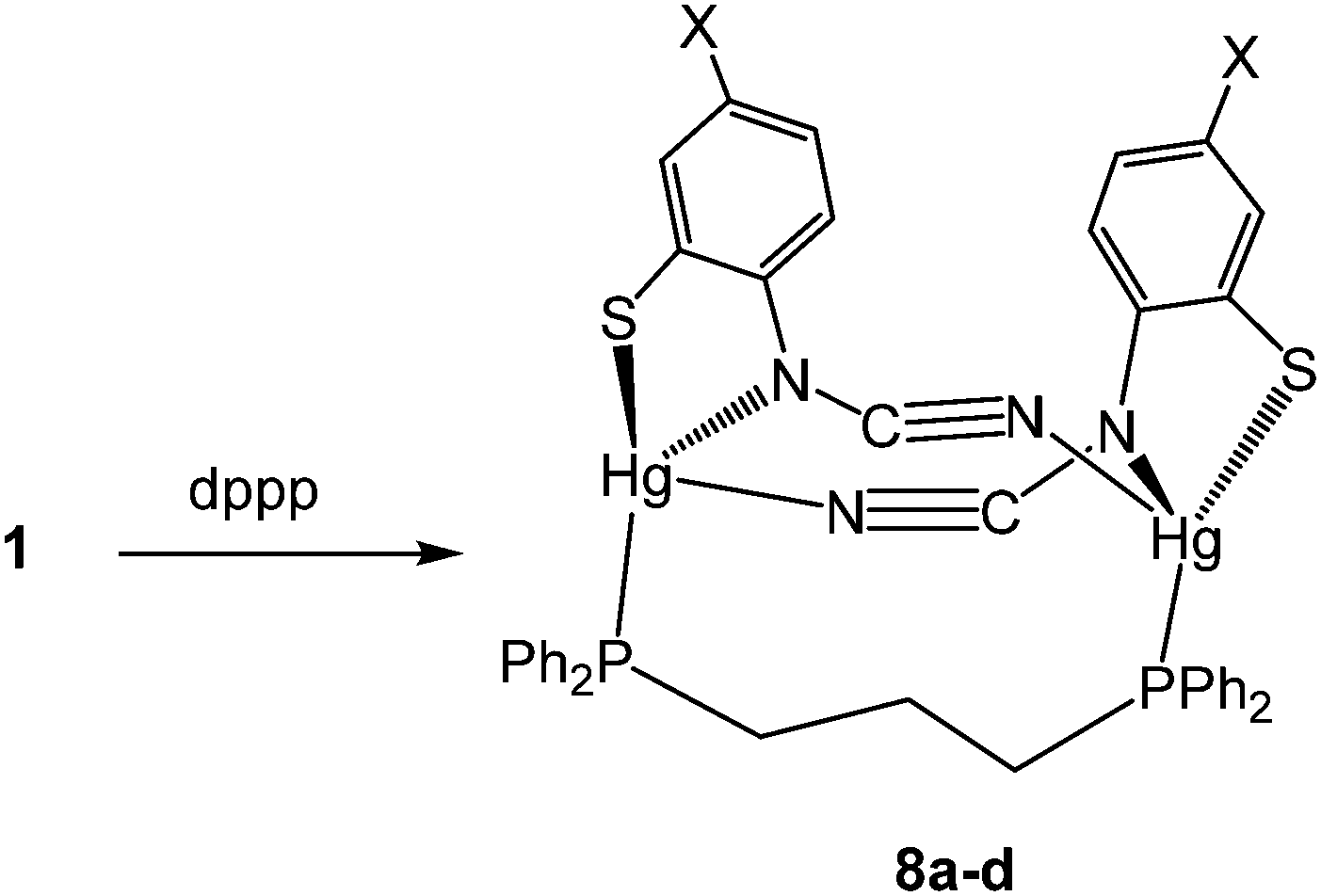
(vi) Reactions with 1,3-bis(diphenylphosphino)propane (dppp): synthesis of dimeric [Hg2{SC6H3XN(CN)}2(μ,κ1,κ1-dppp)] (8)
As far as we are aware, reactions of 1,3-bis(diphenylphosphino)propane (dppp) with Hg(II) salts have not been previously investigated. Alyea and co-workers reported details of a closely related diphosphine, Ph2PCH2SiMe2CH2PPh2, whereby the central methylene unit has been replaced by SiMe2,13b and this leads to formation of the chelate complexes [HgX2(κ2-Ph2PCH2SiMe2CH2PPh2)] which were crystallographically characterised.
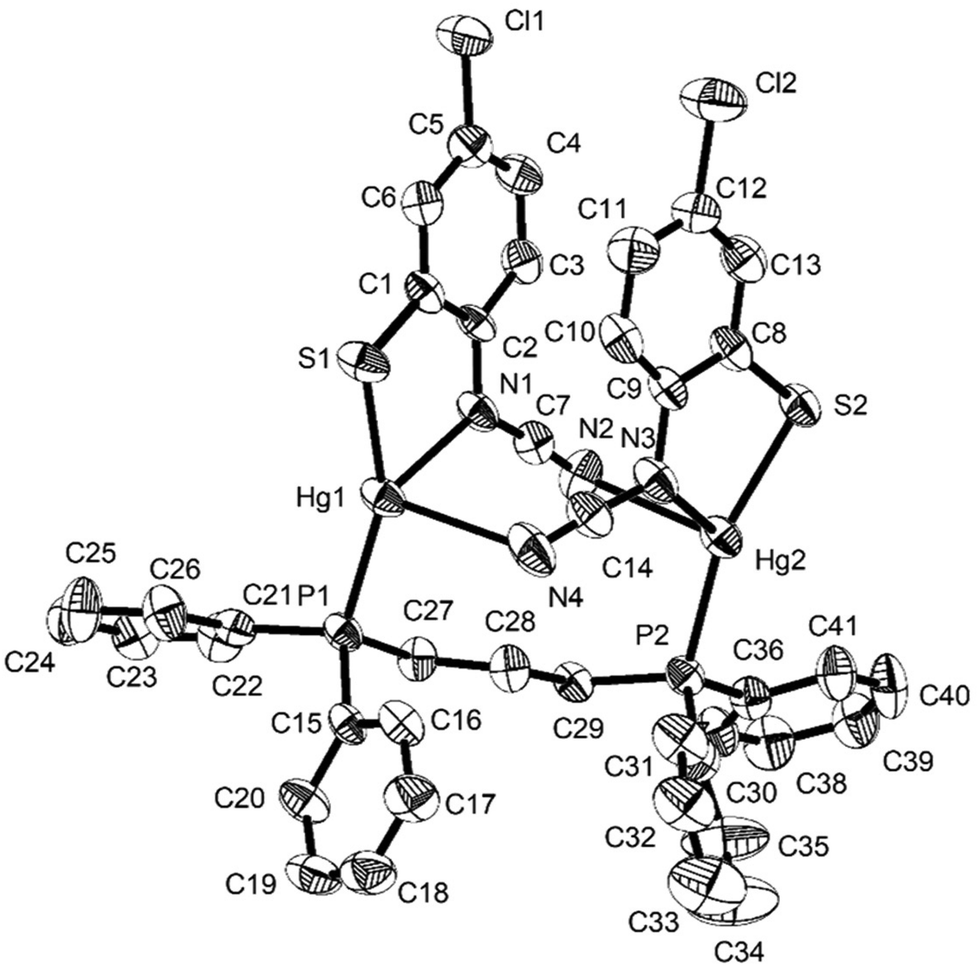
Reactions of 1a–d with equimolar amounts of dppp afforded [Hg2{SC6H3XN(CN)}2(μ,κ1,κ1-dppp)] (8a–d) in high yields (70–80%) (Scheme 7). Crystal structures were carried out on 8b–c and as they are not significantly different only that for 8c is discussed here. Unexpectedly these are binuclear complexes in which Hg⋯Hg vector is spanned by a diphosphine and two ocap ligands. The latter bind in mode B (Chart 2) acting as a chelate to one metal centre and linking to the second through coordination of the CN moiety. Binding of the dppp constricts the two ocap ligands to adopt a syn rather than the anti-conformation seen in 4a (see above). This does not affect the bite angle significantly [S1–Hg1–N1 81.05(17), S2–Hg2–N3 81.46(18)°].
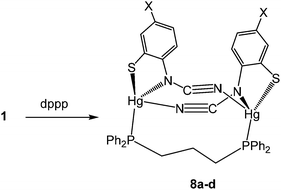 |
| | Scheme 7 Reactions of 1 with dppp. | |
In the solid-state, the two phosphorus centres in 8b–c are non-equivalent and this is retained in solution, 31P{1H} NMR spectra consisting of a pair of doublets; in 8c at 21.5 and 36.5 ppm (JPP 41 Hz). In contrast, both 8a and 8d display only a singlet resonance, although it is broad. We cannot say for certain that the same structure is adopted in these complexes. The different 31P NMR spectra may result from a different structural type of due to the flexibility of the core, leading to either solution equivalence or near equivalence of the two phosphorus nuclei. Unfortunately, in the absence of structural data for either 8a or 8d we are unable to differentiate between these possibilities.
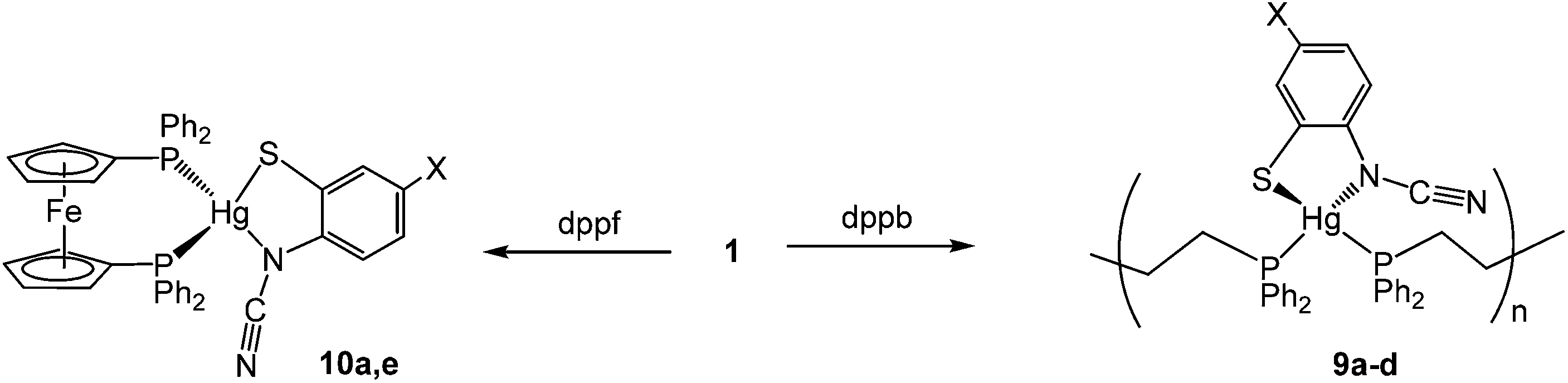
(vii) Reactions with 1,4-bis(diphenylphosphino)butane (dppb): synthesis of coordination polymers [Hg{SC6H3XN(CN)}(μ–κ1,κ1-dppb)]n (9a–d)
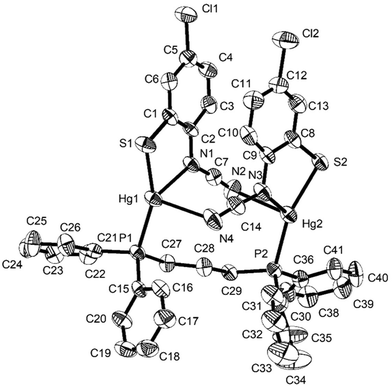
Reactions of 1a–d with bis(diphenylphosphino)butane (dppb) proceeded at room temperature over 1 h to afford coordination polymers [Hg{SC6H3XN(CN)}(μ–κ1,κ1-dppb)]n (9a–d) (Scheme 8). This is confirmed by the molecular structure of [Hg{SC6H3MeN(CN)}{Ph2P(CH2)4PPh2}]n (9b) (Fig. 6)8 which is similar to that of 7a (Fig. 5), consisting of a 1D coordination polymer in which tetrahedral Hg(II) centres are linked by diphosphines. The ocap ligand binds in a simple chelating fashion (AChart 2) subtending and angle of 81.13(9)° at mercury. Both Hg–S and Hg–N bond lengths of 2.437(1) and 2.275(3) Å respectively are within the expected ranges. This binding mode is akin to that previously found for Hg(II) 1,2-benzenedithiolate complexes such as [Hg(SC6H4S)2][NEt4]2.23 This is in accord with the linear nature of the N–CN unit [N1–C7–N2 177.45(5)°] and the significant difference in the nitrogen–carbon bonds [N1–C7 1.307(5), N2–C7 1.149(6) Å]. The structure of 9b is akin to that of [Hg(SC3S3S)(Ph2PCH2CH2PPh2)]n reported by McKenzie and co-workers20 in which the dithiolate ligand subtends an angle of 89.56(8)° at the metal centre and Hg–S bonds are 2.560(3) and 2.530(3) Å. The 31P{1H} NMR spectra of 9b–d all show a pair of doublets suggesting that the inequivalence of the phosphorus centres observed in the solid-state for 9b is maintained in solution; although notably 9a shows only a single resonance, albeit broad.
 |
| | Scheme 8 Reactions of 1 with dppb and dppf. | |
 |
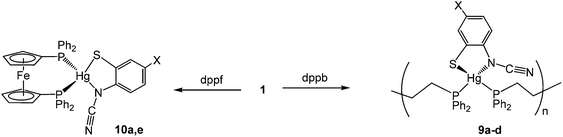
| | Fig. 5 Molecular structure of [Hg{SC6H3ClN(CN)}2(μ-dppp)] (8c). Thermal ellipsoids at the 50% level and hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and bond angles (°): Hg1–N1 2.367(6), Hg1–N4 2.835(6), Hg2–N2 2.723(6), Hg2–N3 2.249(8), Hg1–S1 2.372(2), Hg2–S2 2.372(2), Hg1–P1 2.395(2), Hg2–P2 2.385(2), Hg1⋯.Hg2 5.481(2), N1–C7 1.312(11), N2–C7 1.149(11), N3–C14 1.280(12), N4–C14 1.173(12); N1–Hg1–S1 81.06(18), N1–Hg1–P1 114.72(18), S1–Hg1–P1 157.36(8), N3–Hg2–S2 81.6(2), N3–Hg2–P2 122.5(2), S2–Hg2–P2 155.74(8). | |
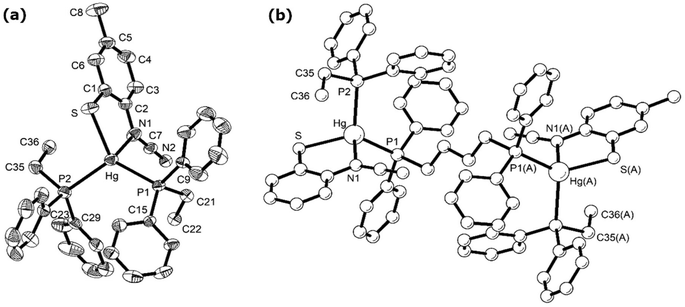 |
| | Fig. 6 Molecular structure of 9b.8 Thermal ellipsoids at the 50% level and hydrogen atoms are omitted for clarity. (a) Showing repeating unit, (b) showing section of the polymeric chain. Selected bond lengths (Å) and bond angles (°): Hg–N1 2.332(4), Hg–S 2.4616(13), Hg–P1 2.4650(12), Hg–P2 2.5522(13); S–Hg–N1 80.72(11), S–Hg–P(1) 131.83(5), S–Hg–P(2) 106.56(4), P1–Hg–P2 117.88(4). Symmetry operator A: −x, 1 − y, 1 − z. | |
(viii) Reactions with 1,1′-bis(diphenylphosphino)ferrocene (dppf)
The diphosphine 1,1′-bis(diphenylphosphino)ferrocene (dppf) is known to be extremely flexible and able to bind to metals in a range of different ways. Most relevant to our work is the observation that addition of dppf to a range of mercuric halides, HgX2 (X = Cl, Br, I, CN)24 affords crystallographically characterised mononuclear [HgX2(κ2-dppf)] (X = Cl, Br) in which the diphosphine binds in a chelate fashion.20,25 Reactions of 1a and 1e with one equivalent of dppf afforded 10a and 10e respectively as analytically pure brown-yellow solids in good yields. Each shows a singlet in the 31P{1H} NMR spectrum consistent with the equivalence of the two phosphorus centres. This does not unequivocally differentiate between mononuclear and coordination polymer structures, however, in light of the mononuclear nature of [HgCl2(κ2-dppf)] and the simple nature of the cyclopentadienyl resonances (two apparent singlets in each case) then we favour assignment as mononuclear species (Scheme 8). We have been unable to confirm this crystallographically.
3. Summary and conclusions
In this contribution we have described the unexpected ring-opening dehydrogenation of 2-aminobenzothiazoles (abt) at a Hg(II) centre, resulting in the facile preparation of otherwise inaccessible ortho-cyano-aminothiophenolate (ocap) complexes of the type, [Hg{SC6H3XN(CN)}]n. Their molecular structures have not been unequivocally established, but their insolubility strongly suggests that they are coordination polymers, although related cyclic oligomers cannot be completely excluded. Precise mechanistic details of the ring-open dehydrogenation remain unknown, but analysis of structural parameters for Hg-bound abt complexes suggest that a zwitterionic resonance hybrid which leads to enhanced acidity of the amino-proton(s) may be important. This is further supported by the isolation of an intermediate 2 in which a pair of deprotonated 2-aminobenzothiazoles bridge a Hg⋯Hg vector. The ring-opening transformation appears to be specific to acetate complexes, and this may be a result of the reversible nature of the N–H bond scission-formation. Thus, with the weak acid, HOAc, the transformation lies to the left-hand side, while for stronger acids (HX, X = Cl, Br etc.) it lies to the right-hand side.
Addition of phosphines to [Hg{SC6H3XN(CN)}]n results in scission of the polymeric (or oligomeric) structure and a series of mono- or binuclear Hg(II) complexes, which show four different coordination modes of the bound ocap ligands. We rely heavily on single crystal XRD for this insight, and it may be that some of these structures are interconverting in solution; something that requires further investigation. Perhaps the most unexpected of these structures are the small-bite angle diphosphine complexes, [Hg{SC6H3XN(CN)}(κ1-dppm)]2 in which the ocap ligands bridge the Hg2 centre at the expense of the diphosphine, which normally binds to metals through both phosphorus atoms. In related work, we have shown upon transmetalation of ocap from Hg(II) to Pt(II) and Pd(II), the ligand exclusively adopts a chelating mode.26 Binding of ocap ligands to redox-active transition metal centres also allows us to better study their ability to act in a redox-active manner akin to closely related dithiolene and ortho-aminothiophenolate ligands as discussed in the introduction.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
We thank the University of Tikrit and King's College London for partial support of this work. S. B.-M. thanks Erasmus Mundus for a Postdoctoral Fellowship and S. G. thanks the Commonwealth Scholarship Commission for the award of a Commonwealth Scholarship. We thank Professor Derek A. Tocher (UCL) and Dr Georgia Orton (University of Birmingham) for crystallographic advice.
References
-
(a) W. Kaim and B. Schwederski, Coord. Chem. Rev., 2010, 254, 1580–1588 CrossRef CAS;
(b) W. Kaim, Inorg. Chem., 2011, 50, 9752–9765 CrossRef CAS PubMed;
(c) O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC;
(d) D. L. J. Broere, R. Plessius and J. I. van der Vlugt, Chem. Soc. Rev., 2015, 44, 6886–6915 RSC.
-
(a) N. Robertson and L. Cronin, Coord. Chem. Rev., 2002, 227, 93–127 CrossRef CAS;
(b) J. A. McCleverty, Prog. Inorg. Chem., 1968, 10, 49–221 CAS;
(c) R. Eisenberg and H. B. Gray, Inorg. Chem., 2011, 50, 9741–9751 CrossRef CAS PubMed;
(d) R. Kato, Chem. Rev., 2004, 104, 5319–5346 CrossRef CAS PubMed.
-
(a) S. R. Presow, M. Ghosh, E. Bill, T. Weyhermüller and K. Wieghardt, Inorg. Chim. Acta, 2011, 374, 226–239 CrossRef CAS;
(b) P. Ghosh, E. Bill, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 2003, 125, 3967–3969 CrossRef CAS PubMed;
(c) P. Ghosh, A. Begum, E. Bill, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 2003, 42, 3208–3215 CrossRef CAS PubMed;
(d) S. Sproules and K. Wieghardt, Coord. Chem. Rev., 2010, 254, 1358–1382 CrossRef CAS;
(e) D. Herebian, E. Bothe, E. Bill, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 2001, 123, 10012–10023 CrossRef CAS PubMed.
-
(a) W.-F. Liaw, N.-H. Lee, C.-M. Chen, G.-H. Lee and S.-M. Peng, J. Am. Chem. Soc., 2000, 122, 488–494 CrossRef CAS;
(b) W.-F. Liaw, C.-M. Lee, G.-H. Lee and S.-M. Peng, Inorg. Chem., 1998, 37, 6396–6398 CrossRef CAS;
(c) W.-F. Liaw, C.-K. Hsieh, G.-Y. Lin and G.-H. Lee, Inorg. Chem., 2001, 40, 4468–4475 CrossRef PubMed.
-
(a) D. Sellman, S. Emig, F. W. Heinemann and F. Knoch, Angew. Chem., Int. Ed. Engl., 1997, 36, 1201–1203 CrossRef;
(b) D. Sellman, S. Emig and F. W. Heinemann, Angew. Chem., Int. Ed. Engl., 1997, 36, 1734–1736 CrossRef.
-
(a) P. Kumar, D. J. SantaLucia, K. Kaniewska-Laskowska, S. V. Lindeman, A. Ozarowski, J. Krzystek, M. Ozerov, J. Telser, J. F. Berry and A. T. Fiedler, Inorg. Chem., 2020, 59, 16178–16193 CrossRef CAS PubMed;
(b) M. Ebadi and A. B. P. Lever, Inorg. Chem., 1999, 38, 467–474 CrossRef CAS PubMed;
(c) J. Vincente, M. T. Chicote, M. D. Bermudez, P. G. Jones, C. Fittschen and G. M. Sheldrick, J. Chem. Soc., Dalton Trans., 1986, 2361–2366 RSC;
(d) K. Sorasaenee, J. R. Galán-Mascarós and K. R. Dunbar, Inorg. Chem., 2002, 41, 433–436 CrossRef CAS PubMed;
(e) J. C. Noveron, M. M. Olmstead and P. K. Mascharak, Inorg. Chem., 1998, 37, 1138–1139 CrossRef CAS PubMed;
(f) A. Majumber and S. Sarkar, Inorg. Chim. Acta, 2009, 362, 3493–3501 CrossRef;
(g) S. Patra, B. Sarkar, S. M. Mobin, W. Kaim and G. K. Lahiri, Inorg. Chem., 2003, 42, 6469–6473 CrossRef CAS PubMed.
-
(a) R. J. Crutchly and M. L. Naklicki, Inorg. Chem., 1989, 28, 1955–1958 CrossRef;
(b) R. J. Crutchly, Coord. Chem. Rev., 2001, 219–221, 125–155 CrossRef.
- S. A. Al-Jibori, A. A. Irzoqi, E. G. H. Al-Saraj, A. S. M. Al-Janabi, S. Basak-Modi, S. Ghosh, K. Merzweiler, C. Wagner, H. Schmidt and G. Hogarth, Dalton Trans., 2015, 44, 14217–14219 RSC.
-
(a) E. C. Wang, J. Li, Y.-L. Li, E.-C. Yang and X.-J. Zhao, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2011, 41, 791–797 CrossRef CAS;
(b) A. Giusti and G. Peyronel, Spectrochim. Acta, Part A, 1982, 38, 975–979 CrossRef.
- S. A. Al-Jibori, Z. S. Afandi, K. Merzweiler, C. Wagner, H. Schmidt, S. Basak-Modi and G. Hogarth, Polyhedron, 2014, 81, 442–449 CrossRef CAS.
-
(a) T. S. Lobana, M. K. Sandhu, M. R. Snow and E. R. T. Tiekink, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 179–181 CrossRef;
(b) N. A. Bell, T. D. Dee, M. Goldstein, P. J. McKenna and I. W. Nowell, Inorg. Chim. Acta, 1983, 71, 135–140 CrossRef CAS.
-
(a) N. A. Bell, S. J. Coles, C. P. Constable, M. B. Hursthouse, R. Mansor and N. J. Salvin, Polyhedron, 2002, 21, 1845–1855 CrossRef CAS;
(b) J. Burt, W. Levason and G. Reid, Coord. Chem. Rev., 2014, 260, 65–115 CrossRef CAS.
-
(a) P. A. W. Dean and R. S. Srivastava, Can. J. Chem., 1985, 63, 2829–2839 CrossRef CAS;
(b) E. C. Alyea, G. Ferguson, R. P. Shakya and P. R. Meehan, Inorg. Chem., 1997, 36, 4749–4752 CrossRef CAS PubMed;
(c) W. Schuh, G. Haegele, R. Olschner, A. Lindner, P. Dvortsak, H. Kopacka, K. Wurst and P. Peringer, J. Chem. Soc., Dalton Trans., 2002, 19–27 RSC.
- R. J. Puddephatt, Chem. Soc. Rev., 1983, 12, 99–127 RSC.
- M. Lusser and P. Perringer, Chem. Ber., 1985, 118, 2140–2143 CrossRef CAS.
- P. Perringer and M. Lusser, Inorg. Chem., 1985, 24, 109–110 CrossRef.
- B. Hämmerle, E. P. Müller, D. L. Wilkinson, G. Müller and P. Peringer, J. Chem. Soc., Chem. Commun., 1989, 1527–1528 RSC.
- M. Lusser and P. Perringer, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1985, 40, 1417–1418 CrossRef.
- P. D. Harvey, K. T. Aye, K. Hierso, E. Isabel, I. Lognot, Y. Mugnier and F. D. Rochon, Inorg. Chem., 1994, 33, 5981–5982 CrossRef CAS.
-
(a) J. McGinley, V. McKee and C. J. McKenzie, Acta Crystallogr., Sect. C: Cryst. Struct. Commun, 1998, 54, 345–347 CrossRef;
(b) T. R. Jenson, J. McGinley, V. McKee and C. J. McKenzie, Acta Chem. Scand., 1998, 52, 622–626 CrossRef.
- M. Camalli, F. Caruso and L. Zambonelli, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 48, 2468–2470 CrossRef.
- X. Li, Y. Liu, Y. Hao, X.-J. Yang and B. Wu, Inorg. Chem. Commun., 2010, 13, 511–513 CrossRef.
- K. Baba, T. Okamura, H. Yamamoto, T. Yamamoto and N. Ueyama, Inorg. Chem., 2008, 47, 2837–2848 CrossRef CAS PubMed.
- K. R. Mann, W. H. Morrison, Jnr. and D. N. Hendrickson, Inorg. Chem., 1974, 13, 1180–1185 CrossRef CAS.
- B. T. Jahromi, A. N. Kharat and A. Bakhoda, J. Coord. Chem., 2013, 66, 2290–2296 CrossRef CAS.
-
S. A. Al-Jibori, A. A. Irzoqi, A. S. M. Al-Janabi, A. I. A. Al-Nassiry, S. Basak-Modi, S. Ghosh, C. Wagner and G. Hogarth, manuscript in preparation.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental methods and characterising data, along with details of the X-ray crystallographic structure determinations. CCDC 2097812–2097815 and 2112641. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt00391k |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Amenah I. A.
Al-Nassiry
b,
Sucharita
Basak-Modi
d,
Shishir
Ghosh
d,
Christoph
Wagner
e and
Graeme
Hogarth
c,
Amenah I. A.
Al-Nassiry
b,
Sucharita
Basak-Modi
d,
Shishir
Ghosh
d,
Christoph
Wagner
e and
Graeme
Hogarth