
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHow change in chirality prevents β-amyloid type interaction in a protonated cyclic dipeptide dimer†
Katia
Le Barbu-Debus
 ,
Ariel
Pérez-Mellor‡
,
Valéria
Lepère
and
Anne
Zehnacker
*
,
Ariel
Pérez-Mellor‡
,
Valéria
Lepère
and
Anne
Zehnacker
*
Institut des Sciences Moléculaires d’Orsay (ISMO), CNRS, Université Paris-Saclay, F-91405 Orsay, France. E-mail: anne.zehnacker-rentien@universite-paris-saclay.fr
First published on 9th August 2022
Abstract
The protonated dimers of the diketopiperazine dipeptide cyclo (LPhe-LHis) and cyclo (LPhe-DHis) are studied by laser spectroscopy combined with mass spectrometry to shed light on the influence of stereochemistry on the clustering propensity of cyclic dipeptides. The marked spectroscopic differences experimentally observed in the hydride stretch region are well accounted for by the results of DFT calculations. Both diastereomeric protonated dimers involve a strong ionic hydrogen bond from the protonated imidazole ring of one monomer to the neutral imidazole nitrogen of the other. While this strong interaction is accompanied by a single NH⋯O hydrogen bond between the amide functions of the two moieties for the protonated dimer of cyclo (LPhe-DHis), that of cyclo (LPhe-LHis) involves two NH⋯O interactions, forming the motif of an antiparallel β sheet. Therefore, a change in chirality of the residue prevents the formation of the β sheet pattern observed in the amyloid type aggregation. These results emphasize the peculiar role of the histidine residue in peptide structure and interaction.
Introduction
Peptide aggregation is associated with amyloid-related diseases such as Alzheimer's. The aggregation propensity depends on the environment, the amino-acid sequence (primary structure) and the protein secondary structure.1,2 Although the cross-β amyloid structure is the most frequently encountered pattern in the amyloid fibres, it was shown recently that other structures, including antiparallel β sheets, could play a role in the aggregation process.1,3,4 Proteins and peptides involved in amyloidosis have been the subject of numerous in vitro studies using X-ray crystallography, electron diffraction, or NMR.1,2 An alternative approach consists of studying short model peptides isolated in the gas phase to shed light on the factors that govern the non-covalent interactions (NCI) and, more precisely, the hydrogen bonds (HB) that control the molecular shape.5,6 Neutral species have been studied under supersonic conditions by conformer-specific spectroscopy while room temperature or cryogenic ion trap studies shed light on the structure of protonated species or ion-core complexes.7–22 Controlling the HB network by chemical design has paved the way to a wealth of interactions and structures, not accessible to pure natural α peptides. Original structures have emerged in β or γ peptides, or in artificial foldamers built from 2-amino cyclohexane carboxylic acid,23–29 in which the bias introduced by the alicyclic ring results in stereochemical constraints that in turn modify the HB propensity.24 The difference in HB propensity is observed for both intermolecular HB in the solid and intramolecular HB in jet-cooled monomers.26,30 Introducing a heteroatom X in the ring stabilises intra-residues HB thanks to an additional NH⋯X interaction.31 The conformational preference of small peptides is also modified by molecular interactions such as hydration,32 or cluster formation.33 Several studies have focused on the formation of β-sheets resulting in the aggregation of peptides. For example, capped phenylalanine-based peptides form dimers linked by β-sheet type NH⋯O interactions.10,34 β-sheet formation was also observed in relatively large fragments of model amyloidogenic peptides. In addition, β-sheets were observed in oligomers of 6-residue fibril-forming peptide segments by combining ion mobility and IR spectroscopy.35 The first steps of amyloid formation have been studied also in large systems under cryogenic ion trap conditions on the example of a model amyloidogenic peptide derived from the protein transthyretin.33 However, less attention has been paid to the role of chirality in the aggregation of peptides. We focus here on the formation of protonated dimers of cyclo(Phe-His), a diketopiperazine-based cyclic dipeptide. This study aims to assess the influence of the chirality of the residues on the formation of a β-sheet type interaction. The absolute configuration of the residues indeed influences the structure of cyclic dipeptides.36–41 Cyclic dipeptides differ from linear ones in several aspects. First, the steric bias due to the ring imposes the non-usual cis conformation of the peptide bonds. It has been shown that amyloid-forming peptides contain amides with a cis conformation.42 Cyclic dipeptides offer therefore a good model for studying the influence of the cis conformation of the peptide bond on aggregation. The cis conformation favours indeed the formation of two strong NH⋯O hydrogen bonds in the gas-phase dimer of cyclo (Phe-Phe)43 or its crystal,38,44 akin to an antiparallel β sheet. Second, the molecule is deprived of the amine and carboxylic functions present in peptides, preventing protonation on the N-terminus. As a result, protonation of cyclic dipeptides usually happens on the amide CO, which results in the limited effect of chirality on their collision-induced dissociation (CID) efficiency.45–47 Histidine-containing peptides stand out by the basic nature of the His residue, which is protonated on the imidazole ring.48 This peculiar protonation site makes histidine very sensitive to solvation. The electronic spectrum recorded by UV photodissociation spectroscopy in a cryogenic ion trap shows a marked bathochromic shift relative to the aqueous solution.49 Histidine also plays a particular role in the CID of peptides.50,51 For example, doubly-charged peptides containing His at the N-terminus show enhanced backbone cleavage at the C-terminus relative to peptides with no His.52 In contrast to peptides containing aliphatic residues, peptides containing histidine show diketopiperazine or lactam in addition to oxazolone fragments.51,53,54 We recently showed that the CID of cyclo (Phe-His)H+ shows a very high sensitivity of CO loss efficiency to the chirality of the residues, due to the proton shuttling mechanism from the imidazole substituent to the amide.45 We determine here the structure of the protonated dimer of cyclo (LPhe-LHis) and cyclo (LPhe-DHis), noted in short (c-LL)2H+ and (c-LD)2H+, in a room-temperature ion trap, by Infra-Red Multiple Photons Dissociation (IRMPD) and compare them to quantum chemical calculations in the frame of the density functional theory (DFT). We aim to evidence how a change in the absolute configuration of the residue makes it possible or not to form a β-sheet type interaction between the two peptides.Experimental and theoretical methods
(a) Nomenclature
The structure of the protonated cyclo (Phe-His) monomer is shown in Chart 1. The nomenclature and numbering of atoms are those used previously.45 In short, each aromatic ring may adopt one of the three following orientations, defined by the dihedral angles τ(NCCC), namely, two gauche and one trans forms, denoted as g+, g− and t, respectively. g+, g− and t correspond to τ(NCCC) of around 60°, −60° and 180°, respectively, for the L residue, with opposite values for the D residue. In a g+ structure, the aromatic substituent is folded over the DKP ring while it is extended outwards in g−.38,55 The trans geometry also corresponds to an extended aromatic ring, but with its plane almost perpendicular to the DKP ring. In addition, the two nitrogen atoms of the imidazole ring are named according to the IUPAC Compendium of Chemical Terminology, as proposed for HisH+.48 The nitrogen atom close to the peptide ring is denoted by Nπ, while the farthest one is called Nτ, as depicted in Chart 1. The neutral monomer exists in two tautomeric forms shown in Chart 1, namely, the NπH or NτH tautomer depending on the location of the hydrogen. The two entities of the protonated dimer are differentiated by their charge state. The atoms belonging to the protonated moiety will be denoted by “P” in subscript, while those of the neutral moiety will be denoted by “N”. For example, the carbon C1 of the neutral part will be denoted CN1 and the nitrogen Nπ of the protonated part will be called NPπ. | ||
| Chart 1 (a) Structure of protonated cyclo LPhe-LHis and numbering of the atoms. (b) Structure of the NπH-tautomer (left) and NτH-tautomer (right) of neutral cyclo LPhe-LHis. | ||
(b) Experimental methods
Tandem mass spectrometry experiments were performed in the room temperature 7T FT-ICR hybrid mass spectrometer (Bruker, Apex Qe) equipped with an Apollo II electrospray ionization source of the SMAS (Spectrométrie de Masse, Analyse et Spectroscopie) facility at the Centre Laser Infrarouge d’Orsay (CLIO).56–58 The protonated dimers were generated by electrospray of slightly acidified 50 μM solutions in a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture of methanol and water. The dipeptides were obtained from Novopep (China) (98% purity). The details of the experiments can be found in previous publications.45,46,59–61 Briefly, the ions were mass-selected in a quadrupole mass filter, thermalized in the hexapole linear trap by colliding with argon, and transferred to the FT-ICR cell.
:50 mixture of methanol and water. The dipeptides were obtained from Novopep (China) (98% purity). The details of the experiments can be found in previous publications.45,46,59–61 Briefly, the ions were mass-selected in a quadrupole mass filter, thermalized in the hexapole linear trap by colliding with argon, and transferred to the FT-ICR cell.
Vibrational spectra were obtained by means of Infrared Multiple Photon Dissociation (IRMPD).62 The IRMPD spectra were obtained by irradiating the mass-selected and thermalized ions in the ICR cell and monitoring the Ln of the total fragmentation efficiency Φ = −Ln(P/(F + P)) as a function of the IR wavelength, with P and F being the abundance of the parent and the sum of the fragments, respectively. The spectra were recorded in the fingerprint region (800–1800 cm−1 region) by mildly focusing the CLIO free-electron laser (FEL) (25 Hz repetition rate) by a 2000 mm Ag-protected spherical mirror to a beam diameter of ∼1 mm at the interaction zone.57 The spectral bandwidth (full width at half-maximum FWHM) was about 7 cm−1, with a power of 1.6 to 1 W from 800 to 1800 cm−1. For the 3 μm spectral region, an IR Optical Parametric Oscillator (LaserVision OPO, 5–10 mJ pulse−1) was slightly focused by the same spherical mirror. The irradiation time was carefully adjusted to avoid the saturation of the most intense transitions. No collisional cooling could occur between two laser pulses during the irradiation of the ions in the FT-ICR cell due to the very low pressure there (<10−9 mbar).
(c) Computational methods
The potential energy surface (PES) was explored using the Monte-Carlo Multiple Minima procedure in the MacroModel suite and the Maestro interface of the Schrödinger package.63 For each protonated dimer (c-LL)2H+ and (c-LD)2H+, the two tautomers of the neutral moiety, namely, NπH and NτH, were considered. Two protonation sites were also considered, setting a formal charge +1 on NπH+ or NτH+. Five simulations, using different sampling methods of the “advanced conformational search” option and the OPLS2005 force field,64 were performed for each structure. The window in energy was fixed to 10 kcal mol−1. The number of isomers was then reduced using the “redundant option” included in Macromodel program. The used root-mean-square deviation was chosen so that between 60 and 80 conformers were obtained at the end of the process, for the different explorations.All the geometries obtained at this stage were fully optimized in the gas phase in the frame of the density functional theory (DFT) using the TURBOMOLE package.65,66 The calculations were performed with the b97-d functional including dispersion corrections67,68 and the TZVPP basis set,69 which is a good compromise between accuracy and calculation time for systems of this size with a large number of isomers. The resolution of the identity was used to reduce the computation time.70 All minima beyond 5 kcal mol−1 were then re-optimised at the ri-b97-d-D3BJ/def2-TZVPPD level of theory.68,71 The calculations were performed in both the gas phase and solution. Solvation was accounted for by the “cosmo” keyword, using a dielectric constant of water ε = 80, to reproduce the conditions of an aqueous solution prior to the electrospray. The harmonic frequencies were calculated at the same level of theory and scaled by a factor of 0.978 to account for basis set incompleteness and anharmonicity. The spectra were simulated by convoluting the scaled harmonic frequencies by a Gaussian line shape of 10 cm−1 FWMH. In what follows, we give the energies corrected by the zero-point-energy (ZPE) at 0 K in vacuum (ΔE) as well as the Gibbs free energies at room temperature in vacuum (ΔG) and in solution (ΔGsol). The complexes retained for the assignment were re-optimised using the B3LYP functional with D3 dispersion corrections and the 6-311++G(d,p) basis sets,71–74 using the Gaussian16 B01 software.75 The frequencies were calculated at the same level of theory and scaled by 0.955 as for the monomer. The results are identical to those discussed here and are shown in Fig. S1 of the ESI,† for the 3 μm region.
Results and discussion
(a) Experimental results
The vibrational spectrum of the (c-LL)2H+ and (c-LD)2H+ protonated dimers have been recorded in both the fingerprint and the OH/NH/CH stretch region by monitoring the fragment at m/z 284, i.e. the protonated monomer, which is the only fragment observed by CID. The fingerprint region is identical for the two diastereomers and will be discussed later. In contrast, the 3000–3600 cm−1 region shows striking differences between (c-LL)2H+ and (c-LD)2H+, as shown in Fig. 1.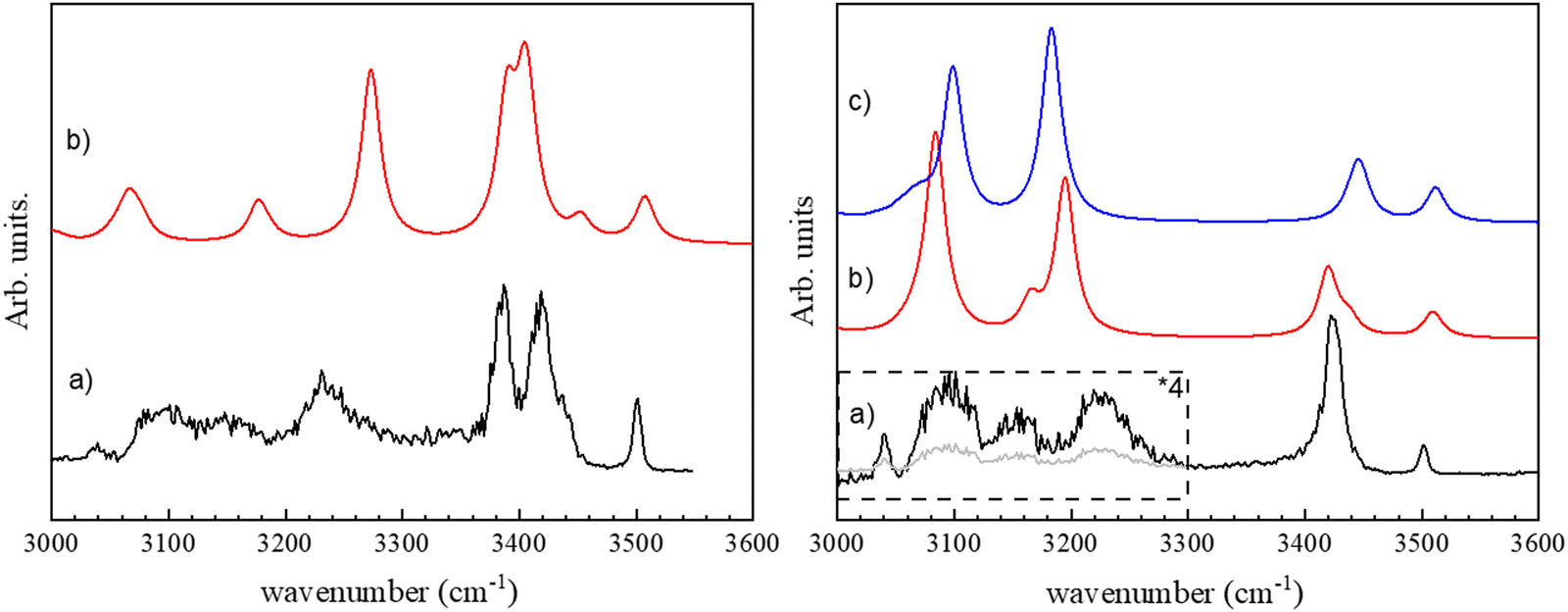 | ||
| Fig. 1 Left: (a) Experimental IRMPD spectrum of c-(LD)2H+ in the region of 3 μm. (b) Simulated spectrum for π-(c-LD)2H+. Right: (a) Experimental IRMPD spectrum of c-(LL)2H+. (b) Simulated spectrum for π-(c-LL)2H+. (c) Simulated spectrum for τ-(c-LL)2H+ (see text). | ||
The major difference between the (c-LL)2H+ and (c-LD)2H+ protonated dimers is the presence of two intense transitions located at 3385 and 3418 cm−1 for (c-LD)2H+, which contrasts with the single intense feature observed for (c-LL)2H+ at 3422 cm−1. The other transitions in this region are similar for the two diastereomers, with a weak band at 3500 cm−1 and three broad bands in the low-frequency range, at 3225, 3155 and 3090 cm−1. The broad transition observed at ∼3230 cm−1 also has larger relative intensity and a different shape in (c-LD)2H+compared to (c-LL)2H+.
(b) Calculated structures and assignment
The spectra of all calculated structures were compared to the experiment by visual examination. The simulated spectra showing the best match with the experiment are shown in Fig. 1. The corresponding calculated structures are given in Fig. 2 and the most relevant structural parameters are listed in Table 1. | ||
| Fig. 2 Protonated dimers accounting for the experimental spectrum, calculated at the ri-b97-d-D3BJ/def2-TZVPPD level of theory. (a) π-(c-LD)2H+, (b) π-(c-LD)2H+, and (c) τ-(c-LD)2H+. Hydrogen bonds are indicated by dashed lines in the 3D structures (top) and by arrows in the 2D schemes (bottom). The strong ionic NH+⋯N interaction is shown in red, the neutral NH⋯O interactions in green, the NH⋯π interactions in purple, and the intramolecular NH⋯O interaction in blue. | ||
| Structure | Charge state of monomers | Newman projection | τ 1 (°) | τ 2 (°) | Distance between the centres of the aromatic rings (Å) | Intramolecular interaction | Intermolecular interaction |
|---|---|---|---|---|---|---|---|
| π-(cLD)2H+ | Neutral (N) | gD+gL− | −54 | −73 | 6.91 | CN3αH⋯πNPhe (3.82 Å) | NP19H⋯πNPhe (2.91 Å) |
| NN19H⋯πNHis (3.42 Å) | NN18H⋯OP17C (1.96 Å; 160°) | ||||||
| Protonated (P) | gD+gL+ | −63 | 62 | 7.03 | CP3αH⋯πPPhe (2.50 Å) | NPτH+⋯NNπ (1.68 Å; 168°) | |
| NPπH⋯OP16 (2.10 Å) | |||||||
| π-(cLL)2H+ | Neutral (N) | gL+gL+ | 72 | 65 | 4.50 | πNPhe⋯πNHis (4.50 Å) | NN19H⋯OP16 (1.77 Å; 175°) |
| NP18H⋯ON17 (1.97 Å; 161°) | |||||||
| Protonated (P) | gL+gL+ | 66 | 73 | 3.87 | πPPhe⋯πPHis (3.87 Å) | NPπH+⋯NNπ (1.72 Å; 156°) | |
| τ-(cLL)2H+ | Neutral (N) | gL+gL+ | 50 | 54 | 6.75 | NNπ⋯ON16 (2.20 Å) | NN19H⋯OP16 (1.96 Å; 171°) |
| NP18H⋯ON17 (1.82 Å, 172°) | |||||||
| Protonated (P) | gL+gL+ | 61 | 51 | 3.64 | πPPhe⋯πPHis (3.87 Å) | NPπH+⋯NNτ (1.68 Å; 166°) | |
Only one structure among those calculated accounts for the spectrum of (c-LD)2H+, namely π-(c-LD)2H+ (see Fig. 2a). In contrast, the spectrum of (c-LL)2H+ can be simulated using any of the calculated (c-LL)2H+ structures shown in Fig. 2b and c, namely, π-(c-LL)2H+ and τ-(c-LL)2H+. It should be noticed that none of the calculated structures accounting for the experimental results is the most stable protonated dimer. In terms of ΔE, π-(c-LL)2H+ or τ-(c-LL)2H+ are indeed destabilised by 3.4 and 4.0 kcal mol−1, respectively, relative to the most stable calculated form, and π-(c-LD)2H+ by 1.1 kcal mol−1. A similar situation is encountered for ΔG with π-(c-LL)2H+ or τ-(c-LL)2H+ destabilised by 3.7 kcal mol−1 and π-(c-LD)2H+ by 2.0 kcal mol−1. Solvation does not substantially modify the energetics and ΔGsol amounts to 6.3 and 6.2 kcal mol−1 for π-(c-LL)2H+ or τ-(c-LL)2H+, respectively, and 2.6 kcal mol−1 for π-(c-LD)2H+. We can conclude from these values that solvation does not favour the observed complexes, but an increase in temperature does. It is therefore possible that high temperature and transient conditions during the electro-spray ionisation process are responsible for the observed structures. Still, the assignment is unambiguous due to the excellent match between simulated and observed spectra. The spectrum of the most stable calculated protonated dimer does not match the experiment and is shown in Fig. S2 of the ESI.†
The proton is located on the imidazole ring in all the calculated structures, whether (c-LL)2H+ or (c-LD)2H+. This is identical to what has been observed in the protonated histidine monomer or its complexes48,76 and in the protonated cyclo (Phe-His) monomer.45,49 Moreover, the phenyl ring of Phe is always folded over the DKP ring (g+ geometry) for (c-LL)2H+ and (c-LD)2H+ alike and whether the subunit within the dimer is neutral or protonated. The histidine aromatic ring of the protonated moiety is also folded on the aromatic ring (g+ geometry). This contrasts with the protonated monomers, shown in Fig. S3 of the ESI,† in which His is in t geometry to allow the formation of the intramolecular NH+⋯O hydrogen bond. The latter is broken in the dimer to the benefit of the strong intermolecular ionic NH+⋯N interaction. The histidine aromatic ring of the neutral moiety is also g+ in both (c-LL)2H+ calculated conformers. The folded conformation of all the aromatic ring results in very compact structures that allow maximising the hydrogen bonds and optimising the dispersion. π-(c-LD)2H+ stands out by the g− conformation of the His aromatic substituent. This results in a less compact geometry, with an extended imidazole conformation of the imidazole ring that is necessary for the concomitant formation of the ionic NPτH+⋯NNπ interaction and the NPπH⋯ON16 hydrogen bridge.
As seen in Fig. 2, the (c-LL)2H+ and (c-LD)2H+ dimers have in common a strong ionic intermolecular hydrogen bond NH+⋯N linking the two moieties. (c-LL)2H+ and (c-LD)2H+ differ by the location of the NH involved in the ionic hydrogen bond, on Nτ for (c-LD)2H+ and Nπ for (c-LL)2H+. Notwithstanding this difference, the characteristics of the ionic hydrogen bond are similar in all the structures shown in Fig. 2. Although strong, this hydrogen bond cannot be seen as a completely shared proton. The NH bond length is indeed of the order of 1.07 Å vs. ∼1.7 Å for the NH+⋯N distance. The hydrogen bond is not far from linearity (∼155–170°). In all cases, NBO calculations indicate that the positive charge is mostly localized (∼0.65) on the NH involved in the ionic hydrogen bond. The neutral moiety corresponds to a different tautomer for the different calculated dimers. π-(c-LD)2H+ or π-(c-LL)2H+ contain the most stable neutral NτH tautomer, whose structure has been determined by rotational spectroscopy in the gas phase.77 In τ-(c-LL)2H+, it is the less stable NπH tautomer, akin to the 5-imidazole tautomer.
The most striking difference between (c-LL)2H+ and (c-LD)2H+ is the pattern of the additional hydrogen bonds that stabilise the dimer. In π-(c-LD)2H+, a single additional neutral NN18H⋯OP17 intermolecular hydrogen bond is formed between the two peptide rings, bridging the amide NH of the neutral Phe residue to the amide CO of the protonated Phe residue. An additional NPπH⋯OP16 intramolecular interaction within the protonated moiety is also present, reminiscent of what is observed in the protonated monomer.45 In addition, N19PH forms a weak NH⋯π hydrogen bond with the benzene ring of the neutral moiety.
In contrast, the (c-LL)2H+ dimers accounting for the experimental spectrum involve two NH⋯OC hydrogen bonds. This pattern has been observed in the solid state of cyclo (Phe–Phe) or its dimer under supersonic expansion conditions.78 The two dimers, namely, π-(c-LL)2H+ and τ-(c-LL)2H+ have very similar structures. They both show the major ionic NPπH+⋯N interaction described above. However, they differ in the tautomer of the neutral subunit acting as an acceptor in this intermolecular H bond. For π-(c-LL)2H+, the acceptor nitrogen is NNπ, as observed in (c-LD)2H+, while for τ-(c-LL)2H+ it is NNτ. In both π-(c-LL)2H+ and τ-(c-LL)2H+, the two peptide rings are linked by two head-to-head NH⋯O hydrogen bonds, forming an antiparallel β-sheet pattern. For π-(c-LL)2H+ and τ-(c-LL)2H+ alike, one of the NH⋯OC bonds is formed from the phenylalanine NH group of the protonated moiety to the phenylalanine CO group of the neutral. The other NH⋯OC bonds is formed from the histidine NH group of the neutral moiety to the histidine CO group of the protonated part.
We can now assign the experimental spectra at the light of the structures described above. The experimental spectrum of (c-LD)2H+ (see Fig. 1a) can be safely assigned to π-(c-LD)2H+. The medium intensity transition observed at 3500 cm−1 is assigned to the neutral imidazole ν(NNτH) calculated at 3508 cm−1. The free ν(NτH) in histidine-containing peptides also appears in the same range between 3500 and 3520 cm−1.79 The ν(NH) frequency observed here is similar but slightly lower than the free ν(NH) frequency of imidazole trapped in helium droplets (3517.8 cm−1),80 in jet-cooled imidazole complexes (3515–3520 cm−1) or imidazole containing peptides.79,81,82
The intense experimental doublet at 3385 and 3418 cm−1 is well reproduced by the simulations. The higher-energy transition of the doublet is assigned to the NPπH+ vibration of the protonated imidazole that gains intensity through the intramolecular NH⋯O interaction. The lower-energy component of the doublet is assigned to the amide ν(NP19H) of the histidine residue of the protonated sub-unit, which gains intensity due to the NH⋯π intermolecular interaction with the benzene ring of the neutral sub-unit. The shoulder on the high-energy side of the doublet at 3437 cm−1 is assigned to a free amide ν(NP18H). The broad band at 3233 cm−1 is attributed to the amide ν(NN18H) of the phenylalanine residue involved in the intermolecular NN18H⋯OP17H bond, calculated at 3274 cm−1. Its intensity is weaker in the experiment than in the simulation because of the low energy of the laser in this range and the band broadening resulting from the strong HB formation. The very weak features below 3200 cm−1 are due to ν(CH) stretches. The frequency of the protonated imidazole ν(NPτH+) involved in the intermolecular NPτH+⋯NNπ hydrogen bond is calculated at 2454 cm−1 due to its ionic character and is out of the wavelength range of the IR OPO used as a light source. Last, the weak and broad absorption bands at ∼3180 and 3050 cm−1 are due to the aromatic ν(CH) of the imidazole and phenyl moieties, respectively.
The calculated spectra of π-(c-LL)2H+ and τ-(c-LL)2H+ are very similar in the hydride stretch region. We shall first compare the experimental spectrum to that simulated for π-(c-LL)2H+. The experimental band at 3501 cm−1 corresponds to ν(NNτH) of the neutral imidazole calculated at 3510 cm−1, similarly to (c-LD)2H+. The single intense band at 3424 cm−1 is attributed to ν(NPτH) of the protonated imidazole, whose frequency is downshifted and intensity is increased due to NH⋯π intramolecular interaction with the phenylalanine aromatic ring. The observed frequency is slightly lower than that observed in the imidazole/benzene complex (3445 cm−1), which also involves an NH⋯π interaction,83 probably because of a partial delocalisation of the positive charge in the imidazole ring. The two experimental broad bands at 3224 and 3092 cm−1 are assigned to the two amide ν(NH) involved in the β-sheet pattern. The vibrational modes calculated at 3084 and 3196 cm−1 are mainly localised on the NN19H of the neutral moiety and NP18H of the protonated moiety, respectively. This is reminiscent of what is observed in the solid-state of DKP dipeptides. Crystallographic results unambiguously show the presence of antiparallel β-sheets with two different NH⋯O distances, even in symmetrical systems like cyclo (Phe–Phe), resulting in two different frequencies. The shoulders at 3170 cm−1 that appear in the same range in the calculated spectrum are due to imidazole ν(CH). The frequency of the NπH+ involved in the ionic intermolecular hydrogen bond, calculated at 2590 cm−1, cannot be observed experimentally as it was the case for (c-LD)2H+.
Comparing the experimental spectrum to τ-(c-LL)2H+ leads to the following assignment: the experimental band at 3501 cm−1 is associated with the transition calculated at 3510 cm−1 corresponding to the ν(NPτH) of the protonated imidazole, a value close to that observed in protonated histidine (3482 cm−1).49 The intense band at 3424 cm−1 is attributed to ν(NNπH) of the neutral imidazole calculated at 3445 cm−1. The transition is shifted down in frequency and gains intensity relative to its counterpart in π-(c-LL)2H+ due to an intramolecular NNπH⋯ON16 interaction. It should be underlined that the NNπH⋯ON16 interaction is only possible for τ-(c-LL)2H+ but not in π-(c-LL)2H+ because NNτH is too far from the HB donor. Finally, the two broad experimental bands at 3224 and 3092 cm−1 correspond to the two intense calculated transitions at 3099 and 3183 cm−1. They stem from the amide ν(NH) involved in the intermolecular NH⋯O hydrogen bonds. As observed in π-(c-LL)2H+, the two bonded amide ν(NH) are localized in τ-(c-LL)2H+. Their frequency order is inverted relative to π-(c-LL)2H+. The ν(NP18H) located on NP18H⋯ON17 is now lower in frequency, reflecting an inversion in the NH⋯O distances (see Table 1). As described for (c-LD)2H+, the ν(CH) appear in the same range and are responsible for the experimental shoulder at 3090 cm−1. The frequency of the NPπH+ involved in the ionic intermolecular hydrogen bond is calculated at 2521 cm−1 and cannot be experimentally observed, it is slightly lower in τ-(c-LL)2H+ than π-(c-LL)2H+.
The experimental spectrum in the 3 μm region does not allow assigning unambiguously the observed protonated (c-LL)2H+ dimer to π-(c-LL)2H+ or τ-(c-LL)2H+. We cannot make the assignment based on relative energies as both protonated (c-LL)2H+ dimers have similar Gibbs energy. Therefore, we have also recorded the IRMPD spectrum in the fingerprint region. It is shown in Fig. 3 together with the calculated spectra of π-(c-LL)2H+ and τ-(c-LL)2H+. In contrast to the 3 μm region, the calculated fingerprint region show spectral differences, allowing us to preferentially assign the observed complex to π-(c-LL)2H+. Indeed, the transitions observed at 1422 and 1327 cm−1 are better accounted for by the calculated spectrum of π-(c-LL)2H+. Moreover, the calculated transition at 1080 cm−1 and the shoulder at 1620 cm−1 seem to be experimentally observed at 1090 cm−1 and 1620 cm−1, respectively. It should be noted that the triplet observed in the amide I region of the monomer, which was characteristic of the NH+⋯O intramolecular interaction, is not observed here, which confirms the assignment.45
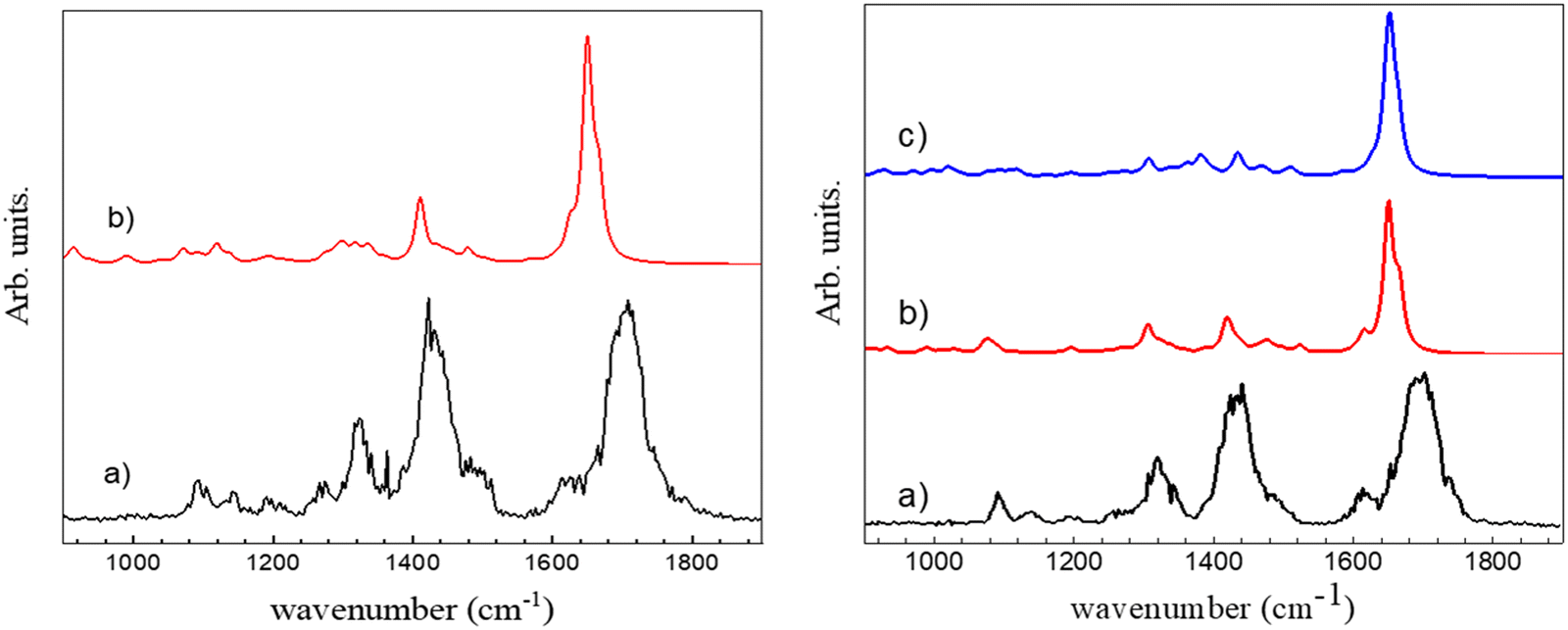 | ||
| Fig. 3 Left: (a) Experimental IRMPD spectrum of c-(LD)2H+ in the fingerprint region. (b) Simulated spectrum for π-(c-LD)2H+. Right: (a) Experimental IRMPD spectrum of c-(LL)2H+ in the fingerprint region. (b) Simulated spectrum for π-(c-LL)2H+. (c) Simulated spectrum for τ-(c-LL)2H+. | ||
Conclusion
The results presented here lead to the conclusion that the effects of stereochemistry on the protonated dimer of cyclo (Phe–His) strongly contrast with those observed in the protonated monomer. The c-(LL)H+ and c-(LD)H+ protonated monomers show identical structures, which cannot be distinguished by their vibrational spectroscopy in a room-temperature ion trap. However, they display very different CID efficiency, in particular for the CO loss channel, due to the shuttling mechanism of the proton localized on the imidazole.45 In contrast, (c-LL)2H+ and (c-LD)2H+ have very different spectroscopic signatures that prove unambiguously that an antiparallel β-sheet interaction takes place in (c-LL)2H+ and not in (c-LD)2H+. (c-LL)2H+ and (c-LD)2H+ differ much more in their spectroscopic signatures than systems showing a single strong interaction like a shared proton, as observed for example in the protonated alanine-camphor dimer, whose floppiness prevents the formation of stereoselective additional interaction points.84 They also show much more specificity than chiral amino acids85 like glutamic acid dimers or other simple systems like sodium core methyl tartrate dimers, although the latter display the multiple interaction points that are thought to be necessary for chiral recognition.86 This selectivity probably arises partially from the diastereomerism being intramolecular in nature, in contrast to the diastereomeric complexes mentioned above, which might result in stronger effects due to reduced flexibility.61 This may also be due to the peculiar properties of histidine. Indeed, the strong sensitivity to chirality observed here strongly contrasts with what is observed for the cyclo (Tyr-Pro) protonated dimer, the structure of which is identical for identical or opposite absolute configurations of the residues.46 These results support the peculiar role of histidine in the structure and fragmentation of peptides.51,87 The differences between (c-LL)2H+ and (c-LD)2H+ are very marked, and do not involve weak ancillary interactions as seen in other systems but the presence or not of an antiparallel β-sheet.61,88 While Nπ of the neutral part acts as an acceptor in the ionic NH+⋯N hydrogen bond for both protonated dimers, as it does in neutral imidazole or His-containing peptides,77,79 the two diastereomer differ in the nature of the NH+ group of the protonated moiety that acts as a donor. It is NτH+ in c-(LD)2H+ while it is NπH+ for (c-LL)2H+. Last, the complexes observed in the ion trap are not the most stable calculated structures. Trapping higher energy conformers is frequently observed in experiments coupling mass spectrometry and laser spectroscopy and results from kinetic trapping of species entropically favoured at the high temperatures achieved during the electro-spray ionisation process.89–92 It may result from a non-thermodynamic distribution of the electrospray due partly to the high but non-evaluable temperature and from the important role of the solvent in the aggregation process right before the vaporization. Continuum solvation models used in this work are not sufficient to account for the modification of relative energies by solvation. This point must be especially true in the case of histidine-containing peptides due to their large sensitivity to solvation and deserves further theoretical investigation.49Conflicts of interest
There are no conflict to declare.Acknowledgements
We thank Dr Ivan Alata for experimental assistance. We thank Dr D. Scuderi and the CLIO team for technical assistance and helpful discussions. We acknowledge the computing centre MésoLUM managed by ISMO (UMR8214) and LPGP (UMR8578), University Paris-Saclay (France). The research described here has been supported by the French National Research Agency (ANR; Grant ANR-17-CE29-0008 “TUNIFOLD-S”).References
- R. Nelson, M. R. Sawaya, M. Balbirnie, A. O. Madsen, C. Riekel, R. Grothe and D. Eisenberg, Nature, 2005, 435, 773–778 CrossRef CAS.
- O. S. Makin and L. C. Serpell, FEBS J., 2005, 272, 5950–5961 CrossRef CAS PubMed.
- R. Gallardo, N. A. Ranson and S. E. Radford, Curr. Opin. Struct. Biol., 2020, 60, 7–16 CrossRef CAS PubMed.
- M. R. Sawaya, S. Sambashivan, R. Nelson, M. I. Ivanova, S. A. Sievers, M. I. Apostol, M. J. Thompson, M. Balbirnie, J. J. W. Wiltzius, H. T. McFarlane, A. O. Madsen, C. Riekel and D. Eisenberg, Nature, 2007, 447, 453–457 CrossRef CAS.
- K. Schwing and M. Gerhards, Int. Rev. Phys. Chem., 2016, 35, 569–677 Search PubMed.
- E. Gloaguen, M. Mons, K. Schwing and M. Gerhards, Chem. Rev., 2020, 120, 12490–12562 CrossRef CAS.
- B. C. Dian, A. Longarte, S. Mercier, D. A. Evans, D. J. Wales and T. S. Zwier, J. Chem. Phys., 2002, 117, 10688–10702 CrossRef CAS.
- W. H. James, E. E. Baquero, S. H. Choi, S. H. Gellman and T. S. Zwier, J. Phys. Chem. A, 2010, 114, 1581–1591 CrossRef CAS.
- J. R. Gord, P. S. Walsh, B. F. Fisher, S. H. Gellman and T. S. Zwier, J. Phys. Chem. B, 2014, 118, 8246–8256 CrossRef CAS PubMed.
- M. Gerhards, C. Unterberg, A. Gerlach and A. Jansen, Phys. Chem. Chem. Phys., 2004, 6, 2682–2690 RSC.
- H. Fricke, G. Schafer, T. Schrader and M. Gerhards, Phys. Chem. Chem. Phys., 2007, 9, 4592–4597 RSC.
- E. Gloaguen and M. Mons, in Gas-Phase Ir Spectroscopy and Structure of Biological Molecules, ed. A. M. Rijs and J. Oomens, 2015, vol. 364, pp. 225–270 Search PubMed.
- Y. Loquais, E. Gloaguen, S. Habka, V. Vaquero-Vara, V. Brenner, B. Tardivel and M. Mons, J. Phys. Chem. A, 2015, 119, 5932–5941 CrossRef CAS PubMed.
- S. Chakraborty, K. Yamada, S. Ishiuchi and M. Fujii, Chem. Phys. Lett., 2012, 531, 41–45 CrossRef CAS.
- H. Otaki, K. Yagi, S. Ishiuchi, M. Fujii and Y. Sugita, J. Phys. Chem. B, 2016, 120, 10199–10213 CrossRef CAS PubMed.
- A. Abo-Riziq, J. E. Bushnell, B. Crews, M. Callahan, L. Grace and M. S. De Vries, Chem. Phys. Lett., 2006, 431, 227–230 CrossRef CAS.
- A. Abo-Riziq, L. Grace, B. Crews, M. P. Callahan, T. van Mourik and M. S. D. Vries, J. Phys. Chem. A, 2011, 115, 6077–6087 CrossRef CAS PubMed.
- J. A. Stearns, M. Guidi, O. V. Boyarkin and T. R. Rizzo, J. Chem. Phys., 2007, 127, 154322 CrossRef PubMed.
- T. K. Roy, V. Kopysov, N. S. Nagornova, T. R. Rizzo, O. V. Boyarkin and R. B. Gerber, ChemPhysChem, 2015, 16, 1374–1378 CrossRef CAS PubMed.
- R. C. Dunbar, G. Berden and J. Oomens, Int. J. Mass Spectrom., 2013, 354–355, 356–364 CrossRef CAS.
- R. C. Dunbar, J. Oomens, G. Berden, J. K.-C. Lau, U. H. Verkerk, A. C. Hopkinson and K. W. M. Siu, J. Phys. Chem. A, 2013, 117, 5335–5343 CrossRef CAS PubMed.
- T. D. Vaden, S. A. N. Gowers, T. S. J. A. de Boer, J. D. Steill, J. Oomens and L. C. Snoek, J. Am. Chem. Soc., 2008, 130, 14640–14650 CrossRef CAS PubMed.
- W. H. James, III, E. G. Buchanan, L. Guo, S. H. Gellman and T. S. Zwier, J. Phys. Chem. A, 2011, 115, 11960–11970 CrossRef PubMed.
- R. Kusaka, D. Zhang, P. S. Walsh, J. R. Gord, B. F. Fisher, S. H. Gellman and T. S. Zwier, J. Phys. Chem. A, 2013, 117, 10847–10862 CrossRef CAS.
- P. S. Walsh, R. Kusaka, E. G. Buchanan, W. H. James, III, B. F. Fisher, S. H. Gellman and T. S. Zwier, J. Phys. Chem. A, 2013, 117, 12350–12362 CrossRef CAS PubMed.
- M. Alauddin, E. Gloaguen, V. Brenner, B. Tardivel, M. Mons, A. Zehnacker-Rentien, V. Declerck and D. J. Aitken, Chem. – Eur. J., 2015, 21, 16479–16493 CrossRef CAS PubMed.
- Z. Imani, V. R. Mundlapati, G. Goldsztejn, V. Brenner, E. Gloaguen, R. Guillot, J. P. Baltaze, K. Le Barbu-Debus, S. Robin, A. Zehnacker, M. Mons and D. J. Aitken, Chem. Sci., 2020, 11, 9191–9197 RSC.
- V. R. Mundlapati, Z. Imani, G. Goldsztejn, E. Gloaguen, V. Brenner, K. Le Barbu-Debus, A. Zehnacker-Rentien, J. P. Baltaze, S. Robin, M. Mons and D. J. Aitken, Amino Acids, 2021, 53, 621–633 CrossRef CAS PubMed.
- K. N. Blodgett, G. Jang, S. Kim, M. K. Kim, S. H. Choi and T. S. Zwier, J. Phys. Chem. A, 2020, 124, 5856–5870 CrossRef CAS PubMed.
- V. Declerck, A. Pérez-Mellor, R. Guillot, D. J. Aitken, M. Mons and A. Zehnacker, Chirality, 2019, 31, 547–560 CrossRef CAS PubMed.
- V. R. Mundlapati, Z. Imani, V. C. D'Mello, V. Brenner, E. Gloaguen, J. P. Baltaze, S. Robin, M. Mons and D. J. Aitken, Chem. Sci., 2021, 12, 14826–14832 RSC.
- H. S. Biswal, Y. Loquais, B. Tardivel, E. Gloaguen and M. Mons, J. Am. Chem. Soc., 2011, 133, 3931–3942 CrossRef CAS.
- J. Ujma, V. Kopysov, N. S. Nagornova, L. G. Migas, M. G. Lizio, E. W. Blanch, C. MacPhee, O. V. Boyarkin and P. E. Barran, Angew. Chem., Int. Ed., 2018, 57, 213–217 CrossRef CAS PubMed.
- M. Gerhards and C. Unterberg, Phys. Chem. Chem. Phys., 2002, 4, 1760–1765 RSC.
- J. Seo, W. Hoffmann, S. Warnke, X. Huang, S. Gewinner, W. Schollkopf, M. T. Bowers, G. von Helden and K. Pagel, Nat. Chem., 2017, 9, 39–44 CrossRef CAS PubMed.
- A. Pérez-Mellor, K. Le Barbu-Debus and A. Zehnacker, Chirality, 2020, 32, 693–703 CrossRef PubMed.
- A. Pérez-Mellor, I. Alata, V. Lepère and A. Zehnacker, J. Phys. Chem. B, 2019, 123, 6023–6033 CrossRef PubMed.
- A. Pérez Mellor and A. Zehnacker, in Physical Chemistry of Cold Gas-Phase Functional Molecules and Clusters, ed. T. Ebata and M. Fujii, Springer, Singapore, 2019, pp. 63–87 Search PubMed.
- A. Pérez-Mellor, I. Alata, V. Lepere and A. Zehnacker, J. Mol. Spectrosc., 2018, 349, 71–84 CrossRef.
- F. BenNasr, A. Pérez-Mellor, I. Alata, V. Lepere, N. E. Jaidane and A. Zehnacker, Faraday Discuss., 2018, 212, 399–419 RSC.
- A. Pérez-Mellor and A. Zehnacker, Chirality, 2017, 29, 89–96 CrossRef PubMed.
- P. T. Lansbury, P. R. Costa, J. M. Griffiths, E. J. Simon, M. Auger, K. J. Halverson, D. A. Kocisko, Z. S. Hendsch, T. T. Ashburn, R. G. S. Spencer, B. Tidor and R. G. Griffin, Nat. Struct. Biol., 1995, 2, 990–998 CrossRef CAS PubMed.
- S. Bakels, M. P. Gaigeot and A. M. Rijs, Chem. Rev., 2020, 120, 3233–3260 CrossRef CAS PubMed.
- M. Gdaniec and B. Liberek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 1343–1345 CrossRef.
- A. Pérez-Mellor, K. Le Barbu-Debus, V. Lepere, I. Alata, R. Spezia and A. Zehnacker, Eur. Phys. J. D, 2021, 75, 165 CrossRef.
- A. Pérez-Mellor, I. Alata, V. Lepère, R. Spezia and A. Zehnacker-Rentien, Int. J. Mass Spectrom., 2021, 465, 116590 CrossRef.
- A. Pérez Mellor and R. Spezia, J. Chem. Phys., 2021, 155, 124103 CrossRef PubMed.
- M. Citir, C. S. Hinton, J. Oomens, J. D. Steill and P. B. Armentrout, Int. J. Mass Spectrom., 2012, 330, 6–15 CrossRef.
- A. Y. Pereverzev, V. Kopysov and O. V. Boyarkin, Angew. Chem., Int. Ed., 2017, 56, 15639–15643 CrossRef CAS.
- G. Tsaprailis, H. Nair, W. Zhong, K. Kuppannan, J. H. Futrell and V. H. Wysocki, Anal. Chem., 2004, 76, 2083–2094 CrossRef CAS PubMed.
- A. C. Gucinski, J. Chamot-Rooke, E. Nicol, A. Somogyi and V. H. Wysocki, J. Phys. Chem. A, 2012, 116, 4296–4304 CrossRef CAS PubMed.
- Y. Y. Huang, V. H. Wysocki, D. L. Tabb and J. R. Yates, Int. J. Mass Spectrom., 2002, 219, 233–244 CrossRef CAS.
- B. R. Perkins, J. Chamot-Rooke, S. H. Yoon, A. C. Gucinski, A. Somogyi and V. H. Wysocki, J. Am. Chem. Soc., 2009, 131, 17528–17529 CrossRef CAS PubMed.
- J. Cautereels, J. Giribaldi, C. Enjalbal and F. Blockhuys, Rapid Commun. Mass Spectrom., 2020, 34, e8778 CrossRef CAS PubMed.
- P. J. Milne, D. W. Oliver and H. M. Roos, J. Crystallogr. Spectrosc. Res., 1992, 22, 643–649 CrossRef CAS.
- P. Maitre, S. Le Caer, A. Simon, W. Jones, J. Lemaire, H. Mestdagh, M. Heninger, G. Mauclaire, P. Boissel, R. Prazeres, F. Glotin and J. M. Ortega, Nucl. Instrum. Methods Phys. Res., Sect. A, 2003, 507, 541–546 CrossRef CAS.
- R. Prazeres, F. Glotin, C. Insa, D. A. Jaroszynski and J. M. Ortega, Eur. Phys. J. D, 1998, 3, 87–93 CrossRef CAS.
- J. M. Bakker, T. Besson, J. Lemaire, D. Scuderi and P. Maitre, J. Phys. Chem. A, 2007, 111, 13415–13424 CrossRef CAS PubMed.
- I. Alata, A. Pérez-Mellor, F. Ben Nasr, D. Scuderi, V. Steinmetz, F. Gobert, N. E. Jaïdane and A. Zehnacker-Rentien, J. Phys. Chem. A, 2017, 121, 7130–7138 CrossRef CAS PubMed.
- P. Maitre, D. Scuderi, D. Corinti, B. Chiavarino, M. E. Crestoni and S. Fornarini, Chem. Rev., 2020, 120, 3261–3295 CrossRef CAS PubMed.
- A. Zehnacker, Int. Rev. Phys. Chem., 2014, 33, 151–207 Search PubMed.
- N. C. Polfer, Chem. Soc. Rev., 2011, 40, 2211–2221 RSC.
- MacroModel version 9.8, Schrödinger, LLC, New York, NY, 2010 Search PubMed.
- E. Harder, W. Damm, J. Maple, C. Wu, M. Reboul, J. Y. Xiang, L. Wang, D. Lupyan, M. K. Dahlgren, J. L. Knight, J. W. Kaus, D. S. Cerutti, G. Krilov, W. L. Jorgensen, R. Abel and R. A. Friesner, J. Chem. Theory Comput., 2016, 12, 281–296 CrossRef CAS PubMed.
- R. Ahlrichs, M. Bar, M. Haser, H. Horn and C. Kolmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- Turbomole V6.6 2014, A development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH Turbomole GmbH, 2014.
- A. D. Becke, J. Chem. Phys., 1997, 107, 8554–8560 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 91–100 CAS.
- L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- M. D. Halls, J. Velkovski and H. B. Schlegel, Theor. Chem. Acc., 2001, 105, 413 Search PubMed.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, 2016 Search PubMed.
- C. P. McNary, Y. W. Nei, P. Maitre, M. T. Rodgers and P. B. Armentrout, Phys. Chem. Chem. Phys., 2019, 21, 12625–12639 RSC.
- C. Bermudez, S. Mata, C. Cabezas and J. L. Alonso, Angew. Chem., Int. Ed., 2014, 53, 11015–11018 CrossRef CAS PubMed.
- S. Bakels, I. Stroganova and A. M. Rijs, Phys. Chem. Chem. Phys., 2021, 23, 20945–20956 RSC.
- W. Y. Sohn, S. Habka, E. Gloaguen and M. Mons, Phys. Chem. Chem. Phys., 2017, 19, 17128–17142 RSC.
- M. Y. Choi and R. E. Miller, J. Phys. Chem. A, 2006, 110, 9344–9351 CrossRef CAS PubMed.
- S. Kumar, A. Mukherjee and A. Das, J. Phys. Chem. A, 2012, 116, 11573–11580 CrossRef CAS PubMed.
- A. Bhattacherjee and S. Wategaonkar, Phys. Chem. Chem. Phys., 2015, 17, 20080–20092 RSC.
- M. A. Trachsel, P. Ottigeri, H. M. Frey, C. Pfaffen, A. Bihlmeier, W. Klopper and S. Leutwyler, J. Phys. Chem. B, 2015, 119, 7778–7790 CrossRef CAS PubMed.
- A. Sen, K. Le Barbu-Debus, D. Scuderi and A. Zehnacker-Rentien, Chirality, 2013, 25, 436–443 CrossRef CAS PubMed.
- M. Poline, O. Rebrov, M. Larsson and V. Zhaunerchyk, Chirality, 2020, 32, 359–369 CrossRef CAS PubMed.
- K. Le Barbu-Debus, D. Scuderi, V. Lepere and A. Zehnacker, J. Mol. Struct., 2020, 1205, 127583 CrossRef CAS.
- F. Turecek, T. W. Chung, C. L. Moss, J. A. Wyer, A. Ehlerding, A. I. S. Holm, H. Zettergren, S. B. Nielsen, P. Hvelplund, J. Chamot-Rooke, B. Bythell and B. Paizs, J. Am. Chem. Soc., 2010, 132, 10728–10740 CrossRef CAS PubMed.
- D. Scuderi, K. Le Barbu-Debus and A. Zehnacker, Phys. Chem. Chem. Phys., 2011, 13, 17916–17929 RSC.
- S. S. Lee, J. U. Lee, J. H. Oh, S. Park, Y. Hong, B. K. Min, H. H. L. Lee, H. I. Kim, X. L. Kong, S. Lee and H. B. Oh, Phys. Chem. Chem. Phys., 2018, 20, 30428–30436 RSC.
- L. Voronina and T. R. Rizzo, Phys. Chem. Chem. Phys., 2015, 17, 25828–25836 RSC.
- E. Bodo, A. Ciavardini, A. Giardini, A. Paladini, S. Piccirillo, F. Rondino and D. Scuderi, Chem. Phys., 2012, 398, 124–128 CrossRef CAS.
- K. Hirata, Y. Mori, S. I. Ishiuchi, M. Fujii and A. Zehnacker, Phys. Chem. Chem. Phys., 2020, 22, 24887–24894 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp03110h |
| ‡ Current address: Department of Physical Chemistry, University of Geneva, 1211 Geneva, Switzerland. |
| This journal is © the Owner Societies 2022 |