
pH-Sensitive nanocarrier assisted delivery of adenosine to treat osteoporotic bone loss†
Hunter
Newman‡
ab,
Jiaul
Hoque‡
b,
Yu-Ru V.
Shih
b,
Gabrielle
Marushack
c,
Unghyeon
Ko
b,
Gavin
Gonzales
bc and
Shyni
Varghese
 *abc
*abc
aDepartment of Mechanical Engineering and Materials Science, Duke University, Durham, NC 27710, USA. E-mail: shyni.varghese@duke.edu
bDepartment of Orthopaedic Surgery, Duke University School of Medicine, Durham, NC 27710, USA
cDepartment of Biomedical Engineering, Duke University, Durham, NC 27710, USA
First published on 1st August 2022
Abstract
Bone tissue undergoes continuous remodeling via osteoclast-mediated bone resorption and osteoblast-mediated bone formation. An imbalance in this process with enhanced osteoclastic activity can lead to excessive bone resorption, resulting in bone thinning. Once activated, osteoclasts bind to the bone surface and acidify the local niche. This acidic environment could serve as a potential trigger for the delivery of therapeutic agents into the osteoporotic bone tissue. To this end, we developed a pH-responsive nanocarrier-based drug delivery system that binds to the bone tissue and delivers an osteoanabolic molecule, adenosine. Adenosine is incorporated into a hyaluronic acid (HA)-based nanocarrier through a pH-sensitive ketal group. The HA-nanocarrier is further functionalized with alendronate moieties to improve binding to the bone tissues. Systemic administration of the nanocarrier containing adenosine attenuated bone loss in ovariectomized mice and showed comparable bone qualities to that of healthy mice. Delivery of osteoanabolic small molecules that can contribute to bone formation and inhibit excessive osteoclast activity by leveraging the tissue-specific milieu could serve as viable therapeutics for osteoporosis.
Introduction
Osteoporosis, a metabolic disorder characterized by loss of bone mass is estimated to cause over 9 million fractures per year globally.1,2 Though historically considered to primarily affect postmenopausal women, osteoporosis also affects men. For instance, one in five men and one in three women over the age of 50 are expected to experience osteoporosis-related fractures.3 In addition to being a major cause of fractures, osteoporosis also contributes to patients becoming bedridden and even mortality.4 Clinically, both antiresorptive and anabolic agents are used to treat osteoporotic bone loss.5–8 However, the most common treatment is bisphosphonate-based antiresorptive agents, such as alendronate and risedronate, which induce osteoclast apoptosis and prevent bone loss.9–12 Anabolic agents, such as teriparatide, are typically used in patients with a high risk of fracture.13,14 Despite their ability to prevent bone loss, both antiresorptive and anabolic agents are associated with significant risks, such as osteonecrosis of the jaw and cardiovascular events.15–17 New therapeutic regimens that lower the risk of osteoporosis and prevent bone loss could have a major public health impact. In particular, interventions that could abate progressive bone loss and prevent fractures are highly needed. Bone tissue undergoes continuous remodeling via bone resorption and bone formation; an imbalance in this process with enhanced osteoclastic activity leading to bone resorption is a cellular characteristic of osteoporosis.18,19 Osteoclasts, large multinucleated cells, degrade bone tissue through the secretion of acids and proteases.20 Once activated, osteoclasts bound to the bone surface and acidify the local microenvironment through the release of hydrochloric acid.21,22 The presence of hydrogen ions could reduce the pH of the bone microenvironment to as low as ∼4.5–6.8.23–27 This allows for the mineral to degrade, creating resorption crypts for proteases such as cathepsin K to further digest the extracellular matrix.21 An acidic microenvironment has been shown to increase osteoclast activity while reducing osteoblast activity.28,29 A recent study by Lin et al. has shown that mere neutralization of the acidic bone tissue microenvironment with sodium bicarbonate can reduce osteoporotic bone loss.30Previously, we have shown a pathological association between adenosine signaling and estrogen deficient-induced osteoporotic bone loss in mice.31 Adenosine is a naturally occurring osteoanabolic molecule that promotes osteoblastogenesis and decreases osteoclastogenesis.32,33 Systemic administration of adenosine or A2B agonists has shown to be effective in attenuating osteoporotic bone loss in an ovariectomy-induced mouse model of osteoporosis.31,34 Although delivery of adenosine is a promising therapeutic strategy to address bone loss, systemic administration of adenosine is challenging due to the ubiquitous nature of adenosine receptors and its short half-life in circulation.35 Encapsulation of drugs within nanocarriers and tissue targeting are some of the approaches widely used to improve the delivery of biomolecules and drugs.36,37 Advances, such as cell or tissue-specific delivery strategies, are used to further improve the outcome of drug delivery; one such approach is stimuli-responsive delivery.38–40 Herein, we leverage the acidic microenvironment of the osteoporotic bone tissue as a trigger to promote site-specific delivery of adenosine. To achieve pH-responsive release, adenosine was conjugated into a hyaluronic acid-based nanocarrier via a ketal functional moiety. The nanocarrier was also functionalized with alendronate, second-generation bisphosphate, to promote binding to the bone tissue.41,42 We examined the efficacy of the pH-sensitive nanocarrier-mediated release of adenosine to mitigate osteoporotic bone loss in vivo, in ovariectomized mice (Fig. 1a).
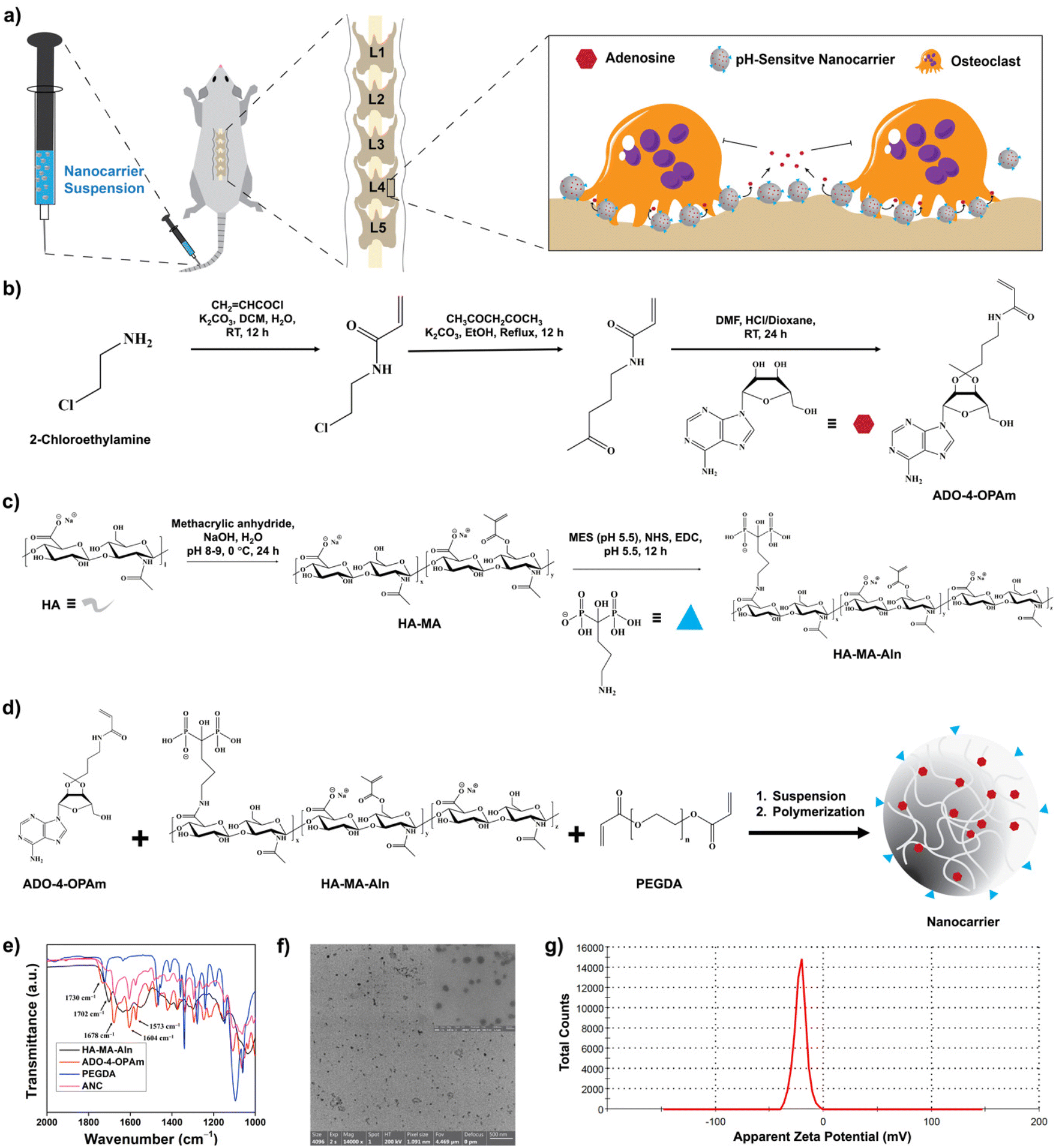 | ||
| Fig. 1 Synthesis and characterization of pH-sensitive bone targeting nanocarriers. (a) Schematic representation of the pH-sensitive bone targeting nanocarrier to deliver adenosine to osteoporotic bone. (b) Reaction scheme of the synthesis of 4-OPAm conjugated with adenosine. (c) Reaction scheme for the methacrylation of hyaluronic acid and alendronate conjugation (HA-MA-Aln). (d) Synthetic scheme of bone targeting nanocarriers containing adenosine (ANC). (e) FTIR spectra of various reaction products and the nanocarrier, ANC. (f) Transmission electron microscopy (TEM) images of the nanocarrier (ANC) recorded at 200 kV. High resolution image of the ANC (inset). (g) Zeta potential of the ANC. | ||
Results
Synthesis and characterization of nanocarriers
Adenosine was conjugated to N-(4-oxopentyl)acrylamide (4-OPAm) containing a ketone functional group and reacted with hyaluronic acid (HA) to generate adenosine-loaded HA nanocarriers as shown in Fig. 1. Specifically, 2-chloroethylamine (2-CEA) was first reacted with acryl chloride to obtain N-(2-chloroethyl)acrylamide (2-CEAm). Proton nuclear magnetic resonance (1H NMR) spectrum of 2-CEAm revealed the presence of olefinic protons at 5.67, 6.12, and 6.33 ppm confirming the presence of acrylamide group in the product, 2-CEAm (Fig. S1, ESI†). Next, 2-CEAm was reacted with 2,4-pentanedione to obtain 4-OPAm, thus introducing a ketone group into the molecule. Fourier-transform infrared (FTIR) spectrum of 4-OPAm showed an absorption peak at 1705 cm−1, which is characteristic of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency of the ketone group (Fig. S2, ESI†). Finally, adenosine was conjugated to 4-OPAm where the ketone group of 4-OPAm reacted with the vicinal diol groups of adenosine (Fig. 1b). NMR spectrum of the reaction product confirmed conjugation of adenosine to 4-OPAm (Fig. S3, ESI†); the spectra showed peaks at 6.05, 6.11, and 6.23 ppm corresponding to olefinic protons of the acrylamide group of 4-OPAm, peaks at 7.84 and 8.30 ppm corresponding to the aromatic protons of the adenine ring, and peaks at 3.59–5.22 ppm corresponding to protons of the sugar ring of adenosine. The synthesis details and characterization of the adenosine conjugated 4-OPAm are provided in the Experimental section and ESI (Fig. S1–S3†).
O stretching frequency of the ketone group (Fig. S2, ESI†). Finally, adenosine was conjugated to 4-OPAm where the ketone group of 4-OPAm reacted with the vicinal diol groups of adenosine (Fig. 1b). NMR spectrum of the reaction product confirmed conjugation of adenosine to 4-OPAm (Fig. S3, ESI†); the spectra showed peaks at 6.05, 6.11, and 6.23 ppm corresponding to olefinic protons of the acrylamide group of 4-OPAm, peaks at 7.84 and 8.30 ppm corresponding to the aromatic protons of the adenine ring, and peaks at 3.59–5.22 ppm corresponding to protons of the sugar ring of adenosine. The synthesis details and characterization of the adenosine conjugated 4-OPAm are provided in the Experimental section and ESI (Fig. S1–S3†).
Hyaluronic acid (HA) was reacted with methacrylic anhydride and sodium alendronate to introduce polymerizable methacrylate (MA) and bone-targeting alendronate (Aln) groups, respectively, as described in the Experimental section and ESI (Fig. 1c and Fig. S4–S6†).34 As estimated from the 1H NMR spectroscopy, ∼30 ± 2% of methacrylation and ∼18 ± 1% of alendronate conjugation were achieved per dimeric repeating unit of HA (Fig. S4, ESI†). The methacrylated-HA with bone binding alendronate (HA-MA-Aln) was then copolymerized with the adenosine conjugated 4-OPAm via emulsion suspension polymerization to generate nanocarriers loaded with adenosine (ANC) (Fig. 1d). To prevent aggregation, the nanocarriers were modified with poly(ethylene glycol) (i.e., PEGylation) as detailed in experimental methods. Nanocarriers without adenosine (NC) were generated and used as controls.
The nanocarriers were characterized via a combination of ultraviolet-visible (UV-vis) absorption spectroscopy, FTIR spectroscopy, dynamic light scattering (DLS), and zeta potential measurements. FTIR spectra of the nanocarriers showed absorptions at 1730 cm−1, 1702 cm−1, and 1678 cm−1 which are characteristics of ester CO stretching frequency of PEGDA, ester CO stretching frequency of HA-MA-Aln, and amide CO stretching frequency of 4-OPAm, respectively. Absorption at 1604 cm−1, corresponding to the CC stretching frequency of the benzene ring, indicates the presence of adenosine (Fig. 1e). The size of the nanocarrier containing adenosine was determined via dynamic light scattering (DLS). The average hydrodynamic diameter of the nanocarriers was found to be ∼98.7 ± 19.3 nm in phosphate buffer (Fig. S7, ESI†). The nanocarriers were furthered characterized by transmission electron microscopy (TEM) imaging (Fig. 1f), and the average diameter of the nanocarrier was found to be 60.1 ± 19.7 nm. The adenosine loaded nanocarriers were also characterized by UV-visible spectra. The UV-Vis spectra of the nanocarrier showed an absorption peak at around 260 nm corresponding to the adenine nucleobase, which further confirms successful conjugation of adenosine (Fig. S8, ESI†). The surface charge of the nanocarrier was determined using zetasizer, which showed a net negative charge of −21.8 ± 1.9 mV (Fig. 1g). The net negative charge is due to the anionic hyaluronic acid and alendronate moieties.43 Nanocarriers labelled with fluorescent dye cyanine 7 (Cy7) were also synthesized and used to examine both bone binding efficiency and in vivo distribution (Fig. S9, ESI†).
Adenosine loading and pH-dependent release
To quantify the adenosine content in the ANC, the nanocarrier was suspended in an acetate buffer of pH 3.5 for 24 hours. The amount of adenosine, determined by the UV-visible absorption spectroscopy, was found to be 412 μg mg−1 of the nanocarrier. In order to test pH-triggered adenosine release from the ANC, the release kinetics were determined at different pHs (pH 5.0, 5.5, 6.0, 6.8 and 7.4) over a period of 21 days, which showed a pH-dependent release of adenosine (Fig. 2a). The ANCs at all pH conditions showed first order release kinetics, albeit varying rate of release (Fig. 2b). Within 12 hours, the ANCs at pH 5.0 and 5.5 showed significantly greater adenosine release compared to those at higher pHs. Although the ANCs incubated at higher pHs (pH 6.0–7.4) showed no significant difference in the rate of adenosine release prior to day 1, significant differences were observed at later time points (Fig. 2c). After 3 days, ∼96.5% of adenosine was found to be released at pH 5.0, ∼85.7% at pH 5.5, ∼62.5% at pH 6.0, ∼50.7% at pH 6.8, and ∼40.5% at pH 7.4 (Fig. 2a). Incubation of the ANCs at lower pHs resulted in complete release of adenosine within 10 days (Fig. 2a), while those at pH 6.0 showed almost complete release of adenosine by 21 days. In contrast, only partial release of adenosine was observed for those incubated at higher pHs; around, 68.9% of release at pH 7.4, and 83.0% of release at pH 6.8 (Fig. 2a).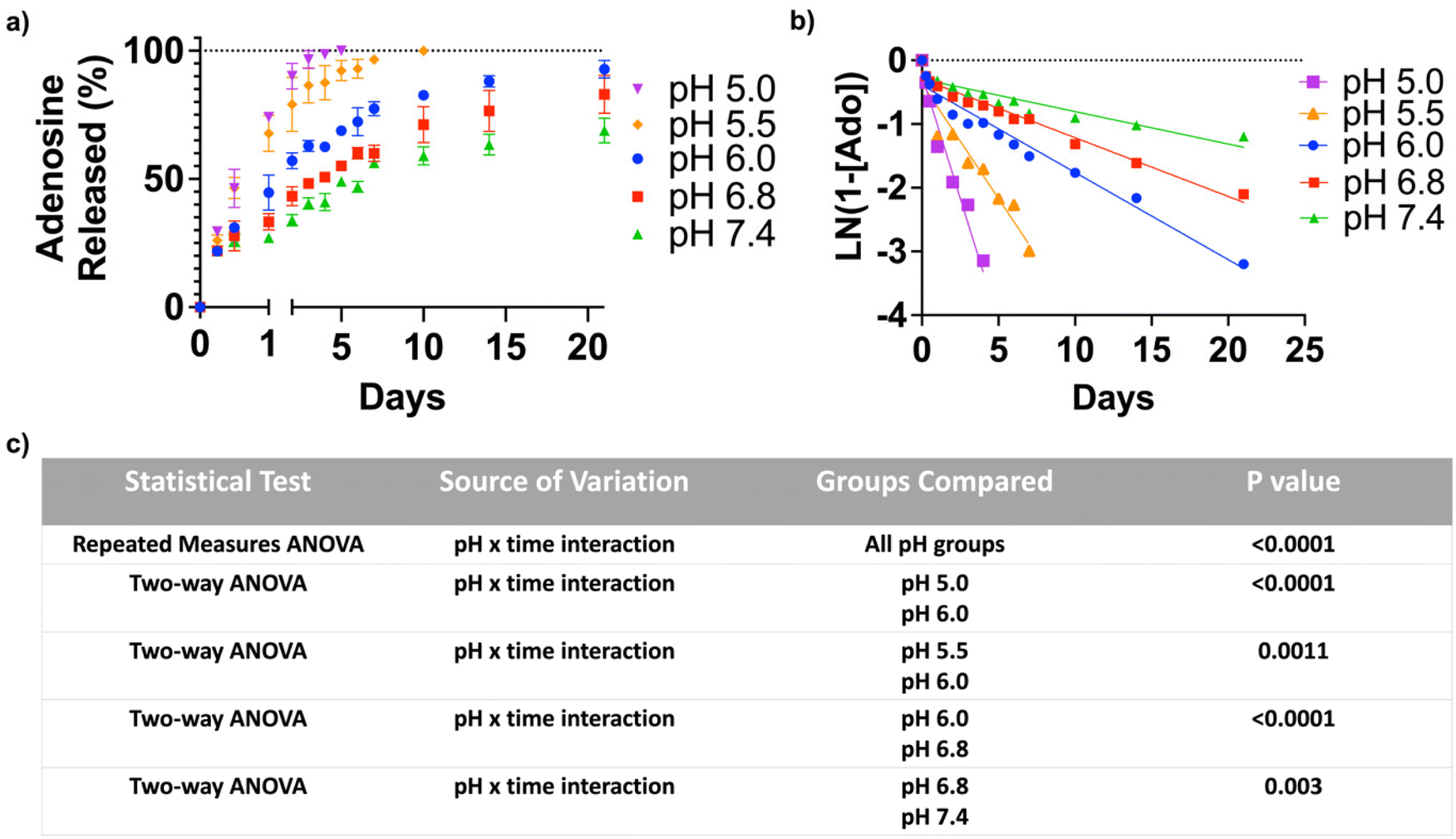 | ||
| Fig. 2 pH-mediated release of adenosine from the nanocarriers, ANC. (a) Cumulative release of adenosine from the ANC in 0.5 M PBS at various pHs (pH 5.0, 5.5, 6.0, 6.8, and 7.4). (b) First-order release kinetics for the adenosine release from the ANC at different pHs. (c) Statistical analysis of the adenosine release at different pHs. (n = 3). | ||
Adenosine release from the nanocarrier inhibits osteoclastogenesis and promotes osteoblastogenesis
The effect of adenosine released from the nanocarrier on osteoclast and osteoblast function was examined in vitro. To study osteoclastogenesis and osteoblastogenesis, we used bone marrow-derived primary monocytes and osteoprogenitor cells, respectively. To minimize the confounding effect from continuous dosing of the bone marrow derived cells with the nanocarriers, we used conditioned medium generated from the nanocarriers to examine the effect adenosine release on cell functions.44 Specifically, the cells were cultured in a conditioned medium collected from the nanocarriers with (ANC) and without (NC) adenosine and incubated either at pH 6.5 or 7.4. Presence of adenosine in the collected medium at day 1 was examined by UV-visible spectroscopy, which estimated ∼45 μg mL−1 in the ANC group at pH 6.5 while no adenosine was detected in the NC group. For osteoclastogenesis, the monocyte-derived osteoclasts were cultured in an osteoclast induction medium containing the conditioned medium from the nanocarriers (NC and ANC at different pHs) for 10 days. Macrophages in the growth medium, osteoclast induction medium devoid of conditioned medium, and osteoclast induction medium supplemented with 45 μg mL−1 of adenosine were used as controls. An upregulation of acid phosphatase 5, tartrate-resistant (Acp5) gene expression was observed in cells cultured in osteoclast induction medium, but Acp5 expression was downregulated in medium supplemented with adenosine (Fig. 3a). Similar to the medium containing adenosine, the cells in medium supplemented with ANC conditioned medium showed downregulation of Acp5 with the conditioned medium from pH 6.5 exhibiting higher downregulation compared to that from pH 7.4 (Fig. 3a). A similar pattern was also observed with cathepsin K (Ctsk) gene expression, where the conditioned medium from ANC at pH 6.5 showed lower levels of Ctsk expression but had no effect when the conditioned medium corresponding to pH 7.4 was used (Fig. 3b). Results from the tartrate-resistant acid phosphatase (TRAP) staining correlated with the gene expression (Fig. 3c). We further investigated the bone resorptive activity of osteoclasts through a pit assay (Fig. 3d). Bovine bone slices were similarly cultured in the conditioned medium collected from the nanocarriers with (ANC) and without (NC) adenosine and incubated either at pH 6.5 or 7.4 for 10 days with a daily change of medium. Following 10-day culture, the cells were removed by sonication. Bone slices were then stained with toluidine blue to detect osteoclast pits. Cells undergoing osteoclast differentiation demonstrated higher resorption that was attenuated when the medium was supplemented with adenosine (Fig. 3d). The slices cultured with the ANC conditioned medium from pH 6.5 showed a decrease in resorption area compared to the slices cultured in conditioned medium from the corresponding NC cultures (Fig. 3d).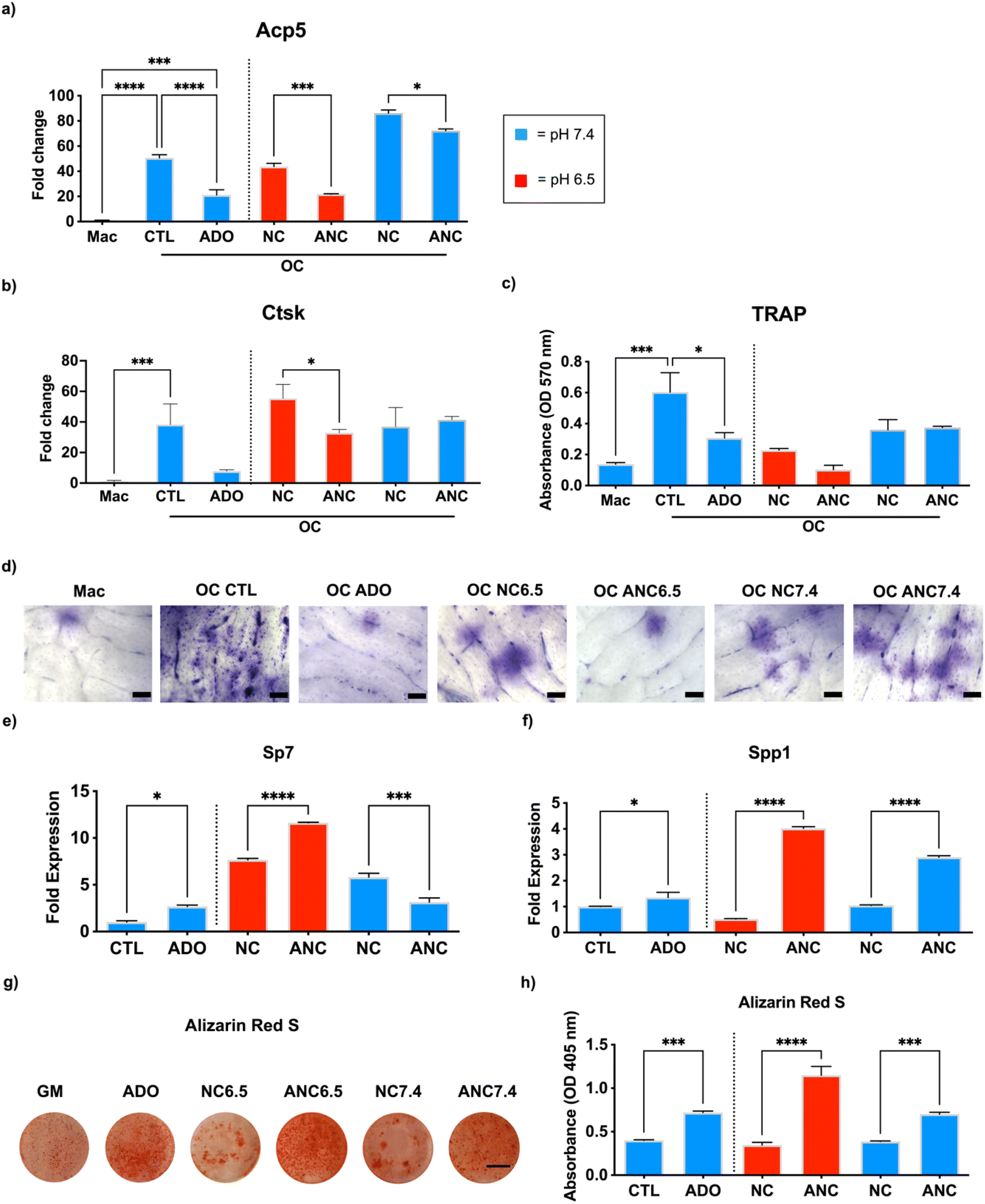 | ||
| Fig. 3 Adenosine release from the nanocarrier (ANC) inhibits osteoclastogenesis and promotes osteoblastogenesis. Macrophages were cultured in a growth medium (Mac) and osteoclasts in the osteoclast induction medium (CTL), osteoclast induction medium supplemented with 45 μg mL−1 adenosine (ADO), and osteoclast induction medium involving a conditioned medium generated from the nanocarriers (NC and ANC) maintained at different pHs (6.5 and 7.4). Expression levels of osteoclastic genes (a) Acp5 and (b) Ctsk were quantified via RT-qPCR. (c) TRAP staining quantification of the cultures at different conditions. (d) Bone resorptive activity of osteoclasts at different conditions; scale bar: 100 μm. Bone marrow mesenchymal stem cells were cultured in growth medium (CTL), growth medium supplemented with 45 μg mL−1 adenosine (ADO), and growth medium involving conditioned medium generated from the nanocarriers (NC and ANC) maintained at different pHs (6.5 and 7.4). Culture condition-dependent osteogenic differentiation was examined for Sp7 (e) and Spp1 (f) via RT-qPCR, as well as alizarin red S staining (g and h); scale bar: 5 mm. Fold expression was normalized to the housekeeping gene and compared to control. *p < 0.05, **p < 0.005, ***p < 0.0005, ****p < 0.0001. | ||
No such decrease was observed with the conditioned medium generated from both ANC and NC at pH 7.4. We examined the short-term cytotoxicity of the nanocarriers on osteoclasts by incubating the cells in medium supplemented with NC or ANC of varying concentrations (100 μg mL−1, 200 μg mL−1, and 500 μg mL−1) for 24 hours. Live/dead analysis of osteoclasts following their exposure showed minimal cell death (Fig. S10, ESI†).
Similar to osteoclastogenesis, we examined the effect of nanocarrier-assisted adenosine delivery on osteoblastogenesis. We cultured the bone marrow mesenchymal stem cells (BMMSCs) for 14 days with a daily change of medium. Osteogenic differentiation was examined through osteoblast gene expression and extent of mineralization. The osteoblast Sp7 transcription factor (Sp7/Osterix) and ECM secreted phosphoprotein (Spp1/Osteopontin) increased for cells cultured in growth medium supplemented with adenosine (Fig. 3e and f). Similarly, cells cultured in ANC conditioned medium from pH 6.5 showed significantly higher Sp7 and Spp1 expression compared to those cultured in NC conditioned medium (Fig. 3e and f). Among the two pH conditions, cells exposed to conditioned medium from pH 6.5 expressed higher levels of osteogenic markers. A similar trend was seen in the alizarin red S staining and quantification for mineralization, in which medium supplemented with adenosine increased mineralization compared to control. Cells cultured in medium supplemented with conditioned medium from ANC at pH 6.5 had significantly higher mineralization compared to conditioned medium from NC at pH 6.5 and ANC at pH 7.4 (Fig. 3g and h). Cytotoxicity analyses by live/dead assay, following short-term exposure of BMMSCS to medium containing either NC or ANC for 24 hours, showed minimal cell death at all nanocarrier concentrations (100 μg mL−1, 200 μg mL−1, and 500 μg mL−1) (Fig. S11, ESI†). To verify that the inhibition of osteoclastogenesis was not attributed to alendronate, we tested the release of alendronate from the nanocarriers, and NMR analysis of the incubation medium showed no presence of alendronate (Fig. S12, ESI†).
Bone targeting efficacy and biodistribution of the nanocarrier
The bone binding efficiency of the nanocarriers was investigated both in vitro and in vivo using Cy7-conjugated ANCs. Femur bone chips incubated with PBS supplemented with ANC (0 mg mL−1, 0.5 mg mL−1, and 5 mg mL−1) were imaged with an in vivo imaging system (IVIS) and quantified for fluorescence intensity (Fig. 4a and b). As expected, only the bone chips incubated with ANCs showed fluorescence signal, demonstrating successful binding of the ANCs to bone tissue (Fig. 4b).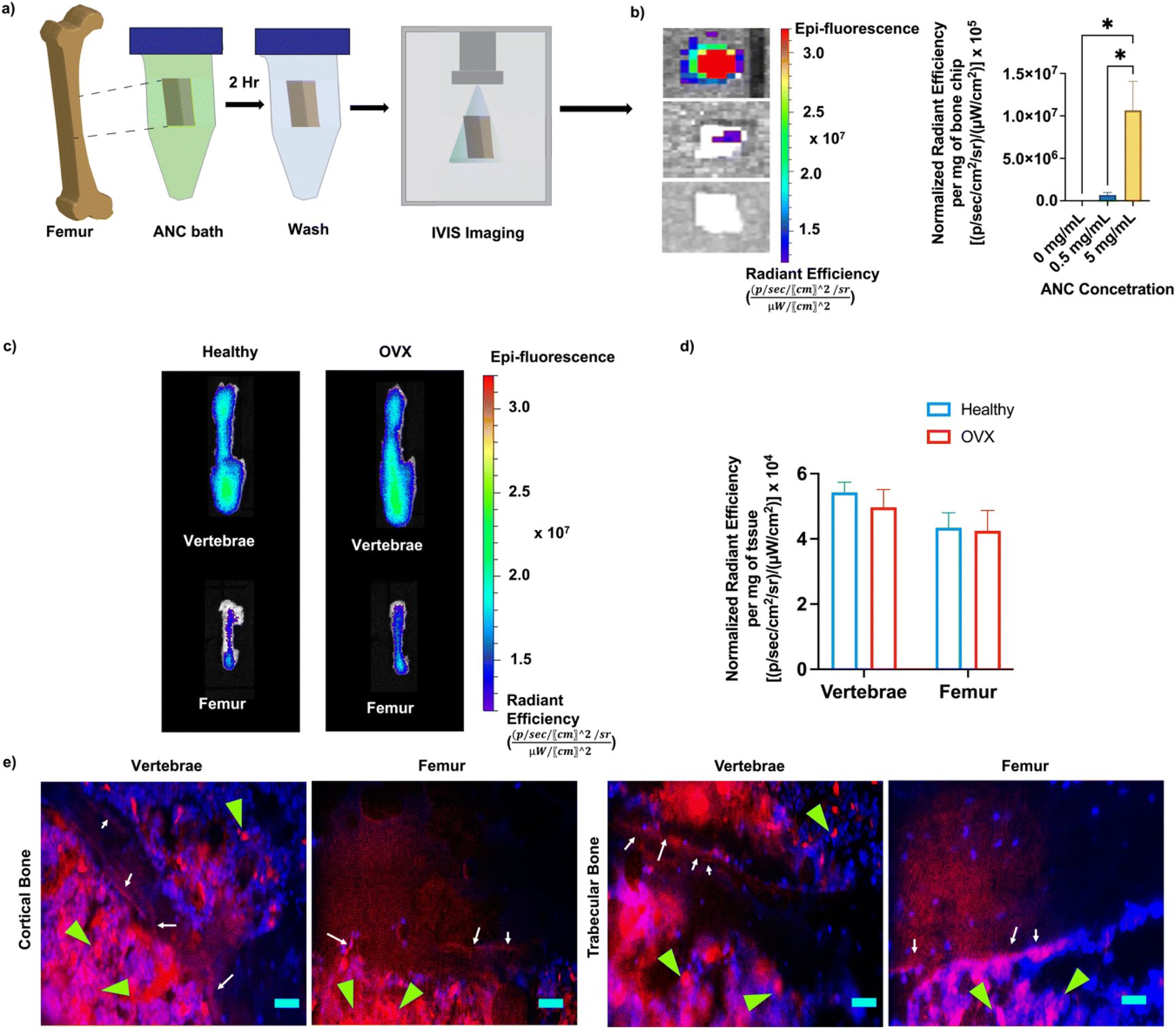 | ||
| Fig. 4 Bone binding efficacy of adenosine containing nanocarrier (ANC). (a) Schematic of the experimental design assessing binding affinity of the ANC to femur bone chips. Femur bone chips were incubated in PBS containing different concentrations of ANC (0, 0.5 or 5 mg mL−1 (n = 3). (b) Representative IVIS images of the bone chips and corresponding radiant efficiency expressed per milligram of the bone chip. (c) IVIS images of the vertebrae and femur for the ANC in healthy and OVX mice (n = 4–5, representative image). (d) Radiant efficiency of fluorescence in the vertebrae and femur represented per milligram of the tissue in healthy and OVX mice (n = 4–5). (e) Immunofluorescence images of L4 vertebrae and femur with cortical and trabecular bone tissue sections. White arrows indicate bone lining with ANC. Green arrows indicate ANCs distributed in the bone marrow, (n = 4–5 per group; scale bar: 50 μm). *p < 0.05. | ||
The role of alendronate functionalization in promoting bone binding of the nanocarriers has been demonstrated in earlier studies.34,45 To evaluate bone binding capability and in vivo biodistribution of the nanocarrier containing alendronate in healthy and osteoporotic bone, fluorescently labeled ANCs were injected systemically into healthy and osteoporotic mice. The distribution of the nanocarrier was determined within the femur and vertebrae 72 hours post-injection, an experimental time chosen based on our prior study.34 The nanocarriers were found to accumulate in both the femur and vertebrae (Fig. 4c). No significant difference in the nanocarrier signal was observed between the healthy and osteoporotic bone (Fig. 4d). Localization of the nanocarriers to the bone tissue was further examined through non-decalcified bone tissue sections. The vertebral and femur sections showed the presence of the nanocarriers at the bone-to-marrow interface as well as in the bone marrow (Fig. 4e). Consistent with prior studies, presence of nanocarriers was also found in other organs – liver, kidney, spleen, heart, lungs, and muscle (Fig. S13, ESI†).34,46 Hematoxylin and eosin staining of the liver tissue sections, the organ with the highest accumulation, showed no obvious atypical-cell morphology (Fig. S14, ESI†).
Adenosine-loaded nanocarrier attenuates bone loss in ovariectomized mice
In order to examine the effectiveness of the pH-responsive adenosine delivery from the nanocarriers to attenuate osteoporotic bone loss, a mouse model of ovariectomy (OVX)-induced osteoporosis was utilized. Four different groups were studied: healthy control without OVX (H), OVX without treatment (O), OVX treated with the nanocarrier (NC), and OVX treated with adenosine containing nanocarrier (ANC). After 6 weeks of OVX surgery, the nanocarriers were administered once a week for 8 weeks, and the bone tissues were characterized 2 weeks later as described in Fig. 5a. Micro-computed tomography (micro-CT) was used to quantify the vertebral trabecular bone parameters such as bone volume over total volume (BV/TV), bone mineral density (BMD), trabecular number (Tb. N), trabecular spacing (Tb. Sp.), trabecular thickness (Tb. Th.), and connectivity density (Conn. D.). Micro-CT reconstructed images showed higher bone loss in the OVX group and those treated with NC, compared to the OVX group treated with ANC and the healthy group (Fig. 5b). Vertebral tissue sections were stained with hematoxylin and eosin (H & E) and displayed an increased magnitude and interconnected trabecular bone in the healthy and ANC when compared to OVX and NC groups (Fig. S15, ESI†). Micro-CT analysis of overall bone parameters of BV/TV and BMD was significantly lower for the OVX group compared to the healthy mice (Fig. 5c). The ANC treated group showed an increase in BV/TV and BMD values compared to the OVX group and did not significantly differ from the healthy group. The other trabecular bone parameters such as trabecular number, trabecular thickness, connectivity density, and trabecular spacing further demonstrated the beneficial effect of adenosine in attenuating osteoporotic bone loss. The healthy group and ANC group exhibited higher values of Tb. N, Conn. D., and Tb. Th. and lower values of Tb. Sp. when compared to the OVX and NC groups (Fig. 5c). We also varied the frequency of ANC treatment and assessed its effect on attenuating bone loss. Ovariectomized mice subjected to monthly (1/month), biweekly (2/month), or weekly (4/month) ANC dosing for 8 weeks were compared. Analyses for various bone parameters (BV/TV, BMD, Tb. N., Tb. Th., Conn. D., Tb. Sp.) showed an increasing trend in bone loss attenuation with an increase in treatment frequency (Fig. S16, ESI†).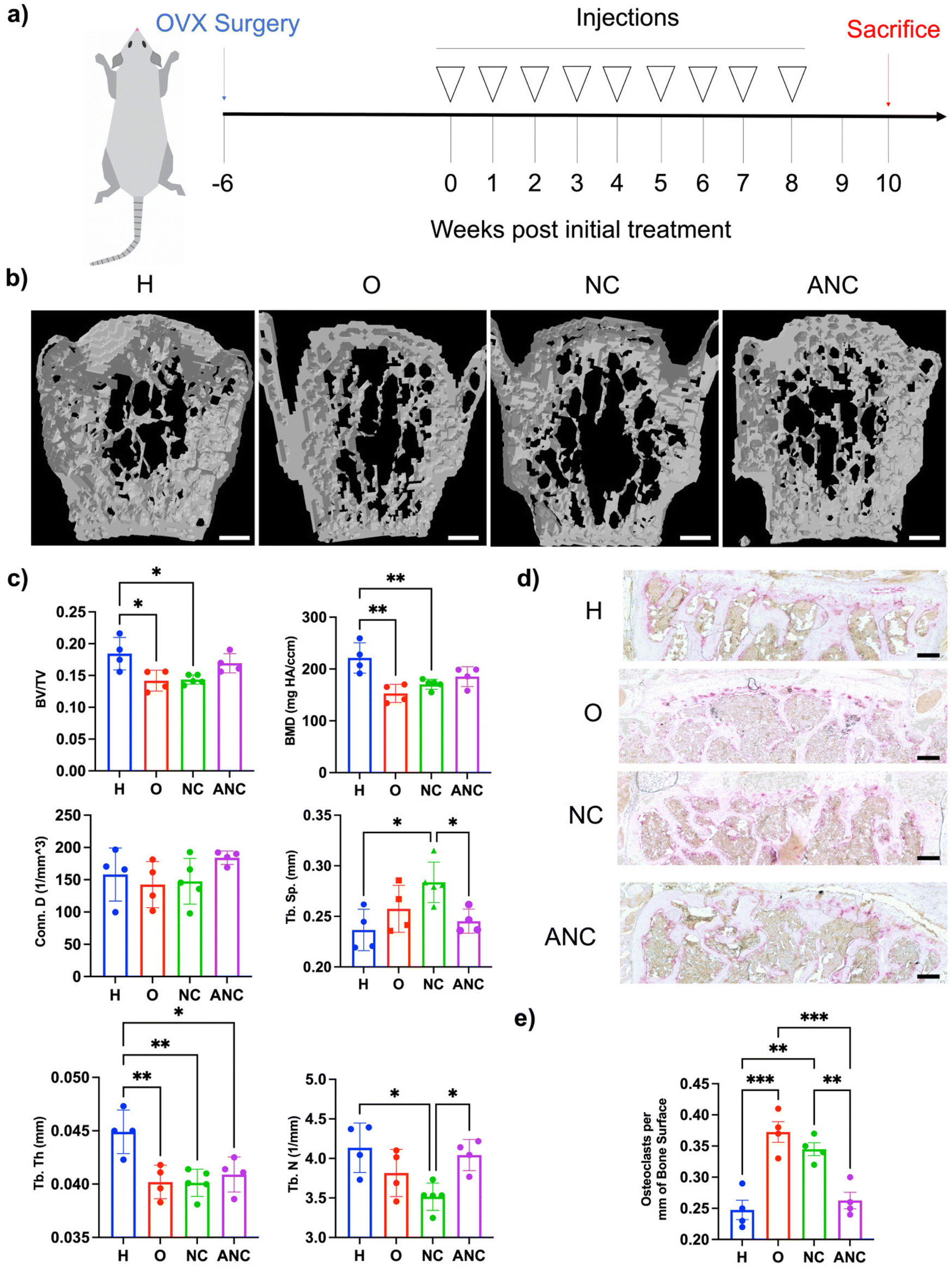 | ||
| Fig. 5 pH-assisted delivery of adenosine from the nanocarriers to mitigate vertebral bone loss in ovariectomized mice. (a) Schematic showing the experimental timeline and frequency of nanocarrier administration. (b) Reconstructed micro-CT images of vertebrae (scale bar: 500 μm). (c) Quantification of the micro-CT images: ratio of bone volume (BV/TV); bone mineral density (BMD); connectivity density (Conn. D.); trabecular spacing (Tb. Sp.); trabecular thickness (Tb. Th.); trabecular number (Tb. N). (d) Tartrate-resistant acid phosphatase (TRAP; red) staining of the vertebrae. (e) Quantification of the TRAP staining per bone surface (scale bar: 100 μm). n = 4–5 per group. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. H: healthy control group without OVX; O: OVX without treatment; NC: OVX treated with nanocarrier without adenosine (NC); ANC: OVX treated with adenosine containing nanocarrier (ANC). | ||
To examine adenosine mediated changes in osteoclasts, vertebral tissue sections were stained for tartrate-resistant acid phosphatase (TRAP) (Fig. 5d). Significantly fewer osteoclasts were observed in the healthy group with a mean value of 0.248 ± 0.015 osteoclasts per bone surface (mm) than the OVX group which had a mean value of 0.373 + 0.017 osteoclasts per bone surface (mm) (Fig. 5e). The ANC treated group had significantly fewer osteoclasts with a mean value of 0.263 ± 0.013 osteoclasts per bone surface (mm) and was found to be comparable to the healthy group. The NC treated group did not show any significant reduction in the number of osteoclasts compared to the OVX group.
Adenosine-loaded nanocarriers promote new bone formation and increase bone mechanical strength in ovariectomized mice
In order to assess adenosine-mediated new bone formation, two fluorescently labeled dyes (a green fluorescent dye, calcein, and a red fluorescent dye, alizarin, both of which are known to bind to the calcium ions and thus label newly mineralizing bone) were systemically administered 14 days apart. Undecalcified femur sections were collected and used to quantify bone formation rate (BFR, μm day−1) and mineral apposition rate (MAR, μm day−1). The tissue sections showed a clear difference in the size of the gap between the green and red fluorescent dyes for both the healthy and those treated with adenosine (ANC group), whereas no such clear separation of fluorescence labelling was observed for the OVX and NC groups (Fig. 6a). Quantification of the tissue sections showed BFR and MAR values of 0.755 ± 0.087 and 0.791 ± 0.085 for the ANC group, which was significantly higher than the OVX (0.179 ± 0.081, 0.249 ± 0.104) and NC (0.142 ± 0.071, 0.152 ± 0.078) groups. The values for the ANC group were similar to the healthy group (Fig. 6b and c). The changes in bone mechanical properties following the treatment were determined by analyzing the tibia's ability to bear maximum load and the tissue stiffness. The maximum load of the tibia belonging to the healthy group was significantly higher than the OVX group (Fig. 6d). The load-withstanding ability of the OVX group increased with the adenosine treatment and was found to be similar to the healthy group (Fig. 6d). None of the groups demonstrated any significant differences in bone tissue stiffness (Fig. 6e). We also examined varying the ANC treatment frequency and its effect on bone formation. We compared monthly (1/month), biweekly (2/month), and weekly (4/month) ANC dosing in OVX mice over 8 weeks. As expected, there was an increasing trend in bone formation associated with an increase in treatment frequency (Fig. S17, ESI†).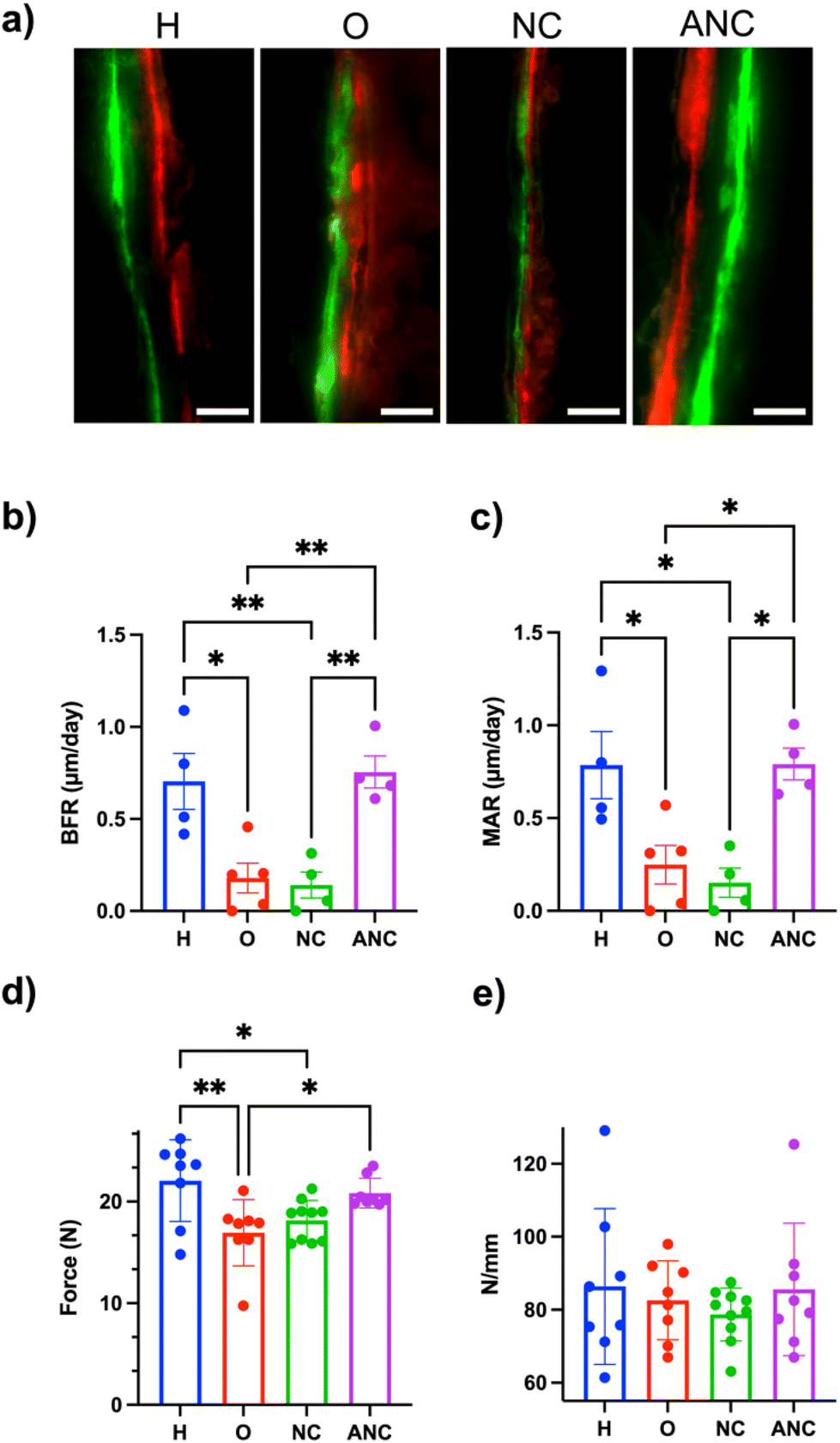 | ||
| Fig. 6 pH-assisted release of adenosine from the nanocarrier promoted new bone formation. (a) Double fluorescence bone labeling by calcein (green) and alizarin complexone (red) of the femur (n = 4–5, scale bars: 20 μm). Quantification of bone formation rate (BFR) (b) and mineral apposition rate (MAR) (c) from the images. (d) Maximum load and (e) stiffness of tibiae (n = 8–9) *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. H: healthy control group without OVX; O: OVX without treatment; NC: OVX treated with nanocarrier without adenosine; ANC: OVX treated with adenosine containing nanocarrier. | ||
Discussion
pH-Responsive drug delivery systems have been used extensively to achieve tissue-specific delivery of therapeutics, especially utilizing the local acidic microenvironment.47–49 Several pH-responsive chemical functional groups such as esters, carbonates, ketals, hydrazones, and imines have been utilized to deliver therapeutics.50–53 In this study, we leveraged the acidic microenvironment of the osteoporotic bone tissue as a trigger to release adenosine from a nanocarrier. Adenosine is an osteoanabolic molecule that promotes osteoblastogenesis and decreases osteoclastogenesis.31–34,54,55 Though the nanocarriers showed a pH-dependent release in vitro, some level of adenosine release was also observed at physiological pH. This is possibly due to the intrinsic acidic nature of hyaluronic acid and alendronate moieties present in the nanocarriers. In vitro cell culture studies showed that adenosine-loaded nanocarriers exposed to pH 6.5 promoted osteoblastogenesis and inhibited osteoclastogenesis but were not as effective when exposed to pH 7.4. The pit assay further demonstrated that nanocarriers exposed to pH 6.5 reduced osteoclastic resorption area, however not at physiological pH. The pH-responsive effect could be attributed to the higher amount of adenosine released at pH 6.5 compared to 7.4.Previously, we have shown that while alendronate functionalization can improve accumulation of nanocarriers into the bone, the nanocarriers were also found in other organs.34 Albeit the liver displaying the highest accumulation of nanocarrier, histological analysis showed no obvious toxicity. In the case of bone tissue, the presence of nanocarriers was observed both in the marrow and bone-marrow interface. The localization of the nanocarrier at the bone-to-marrow interface could be due to the affinity of alendronate to bind to the hydroxyapatite minerals. Despite the bone loss in osteoporotic tissue, the nanocarriers showed no difference in bone binding.
The bone loss in ovariectomized mice was attenuated by using the pH-sensitive nanocarrier loaded with adenosine. The analyses showed comparable vertebral bone parameters and increased mechanical strength of the tibia for the adenosine treated animals with that of healthy animals. The mitigated bone loss is partly due to the inhibition of osteoclast activity. In addition, extracellular adenosine also contributes to bone formation by increasing osteoblastogenesis, which is supported by the results from the bone double-labeling studies. The sequential administration of fluorochromes that are preferentially taken up by the calcification front showed a clear separation in healthy cohorts and those treated with adenosine loaded nanocarriers, suggesting new bone formation.56 Lack of such separation in OVX and those treated with nanocarriers devoid of adenosine suggest minimal new bone formation. Despite the presence of alendronate molecules, the cohorts that received NC had bone loss similar to OVX mice which could be due to the low amount of alendronate present in the nanocarriers. The animals received ∼1.8–2.0 mg per kg per week of alendronate, which is roughly 25-fold less than the therapeutic regimen of freely administered alendronate used to treat OVX mice.57,58 The alendronate is conjugated to the HA via an amide bond and did not undergo cleavage and release from the nanocarrier. Results from the NC group clearly suggest that the beneficial effect observed from the ANC treatment is solely due to adenosine.
The ovariectomized mouse model was investigated due to its significance in the clinic. However, there are other forms of osteoporosis, such as secondary or age-related osteoporosis, that need to be studied as the release profile of adenosine from the nanocarrier could be affected by age or health conditions.59 The nanocarrier was distributed in multiple organs and improvements should be made to further reduce off-target accumulation.
Conclusion
In summary, a pH-sensitive nanocarrier system was developed to deliver osteoanabolic adenosine molecules to treat osteoporotic bone loss. Specifically, we leverage the acidic microenvironment of the osteoporotic bone tissues as a trigger to release the adenosine molecule. Adenosine was incorporated into the nanocarrier via a ketal moiety, which hydrolyzes preferentially in acidic conditions. Systemic administration of the adenosine-loaded nanocarriers containing alendronate moieties enabled their accumulation in the bone tissue and reduced bone loss in ovariectomized mice while promoting new bone formation.Experimental
Materials
Hyaluronic acid (HA, molecular weight 40 kDa, HA40K-5) was purchased from Lifecore, USA. Methacrylic anhydride (276685), N-hydroxysuccinimide (NHS, 130672), sodium hydroxide (795429), adenosine (A4036), and mineral oil (M5904) were obtained from Millipore Sigma, USA. 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC·HCl, D1601), sodium alendronate trihydrate (J61397), and 2-chloroethylamine hydrochloride (2-CEA, A14455) were purchased from Alfa Aesar, USA. Cyanine 7.0 amine (550C0) was purchased from Lumiprobe. Dialysis bags (Molecular weight cut off, MWCO of 2.0 and 3.5 kDa) were obtained from Spectrum, USA. ABIL EM90 surfactant (420095-L-151) was obtained from Universal PreservA Chem Inc., Germany. Hexane, acetone, ethanol, and dimethyl sulfoxide (DMSO) were purchased from Millipore-Sigma, USA; the solvents were of ACS or spectroscopic grade. Genesys 10S UV-vis spectrometer was used to record the absorbance spectra. FTIR and NMR spectra were recorded using Thermo Electron Nicolet 8700 FTIR spectrometer and FFSC 500 MHz Agilent/Varian Inova spectrometer, respectively. The Thermo Fisher Scientific Talos F200X instrument was used to obtain the transmission electron microscopy (TEM) images.Synthesis of hyaluronic acid methacrylate (HA-MA)
Photopolymerizable methacrylate groups were introduced into HA via esterification of the hydroxyl groups by reacting HA with methacrylic anhydride.34 Briefly, HA (∼600 mg) was dissolved in deionized water. The solution was placed over an ice bath and allowed to cool for 30 minutes. Methacrylic anhydride (∼4.4 mL) was added to the HA solution and pH of the reaction mixture was maintained at 8–8.5 by adding 5 M NaOH. The reaction was continued for 24 hours at 4 °C. Ethanol-acetone mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was added to precipitate the product, HA-MA. The product was filtered and washed multiple times with an ethanol-acetone mixture. The product was dissolved in deionized water and dialyzed against deionized water for 4 days using a cellulose acetate dialysis membrane with a MWCO of 3.5 kDa. The polymer solution was freeze-dried and stored at −20 °C. The product, HA-MA, was characterized by using a combination of FTIR and 1H NMR spectroscopy. FTIR spectra of the freeze-dried HA-MA was recorded using ZnSe crystal via attenuated total reflection (ATR) mode. 1H NMR spectrum was recorded by dissolving 3–4 mg of HA-MA in deuterium oxide (D2O) at 25 °C.
:1) was added to precipitate the product, HA-MA. The product was filtered and washed multiple times with an ethanol-acetone mixture. The product was dissolved in deionized water and dialyzed against deionized water for 4 days using a cellulose acetate dialysis membrane with a MWCO of 3.5 kDa. The polymer solution was freeze-dried and stored at −20 °C. The product, HA-MA, was characterized by using a combination of FTIR and 1H NMR spectroscopy. FTIR spectra of the freeze-dried HA-MA was recorded using ZnSe crystal via attenuated total reflection (ATR) mode. 1H NMR spectrum was recorded by dissolving 3–4 mg of HA-MA in deuterium oxide (D2O) at 25 °C.
Synthesis of alendronate-conjugated HA-MA (HA-MA-Aln)
Bone targeting alendronate (Aln) moieties were conjugated to the HA-MA via amide coupling reaction between the carboxylic acid groups of HA-MA and the primary amine group of alendronate (Aln).34 Briefly, HA-MA (∼400 mg) was dissolved in 40 mL of 2-(N-morpholino)ethanesulfonic acid (MES) buffer of pH 5.5. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl, ∼175 mg) and N-hydroxysuccinimide (NHS, ∼105 mg) were added to the HA-MA solution at 15 minutes intervals. Sodium alendronate trihydrate (∼74.2 mg) was added to the reaction mixture and was continued for 12 hours at 25 °C. The mixture was dialyzed using dialysis membrane (MWCO 3.5 kDa) against deionized water for 4 days, and the resulting solution was lyophilized to obtain alendronate conjugated HA-MA (HA-MA-Aln). HA-MA-Aln was characterized by using FTIR and 1H NMR spectroscopy. FTIR spectrum was recorded via attenuated total reflection (ATR) mode using ZnSe crystal. 1H NMR spectrum was recorded by dissolving 3–4 mg of the polymer samples in deuterium oxide (D2O) at 25 °C.Synthesis of adenosine conjugated N-(4-oxopentyl)acrylamide (4-OPAm)
2-Chloroethylamine hydrochloride (2-CEA, ∼5 g) was dissolved in dichloromethane (120 mL). Potassium carbonate (K2CO3, ∼15 g) dissolved in water was added to the above organic solution. Acryloyl chloride (4.3 mL) was then added to the reaction mixture dropwise at 4 °C and the reaction was continued for 24 hours at room temperature. The reaction mixture was washed with 1 M HCl saturated sodium bicarbonate solution and saturated sodium chloride solution. The final organic layer was passed through anhydrous sodium sulfate and dried using a rotary-evaporator. The resulted N-(2-chloroethyl)acrylamide (2-CEAm) (4.5 g) was dissolved in 50 mL of ethanol containing 2,4-pentadione (5 g), and to this anhydrous K2CO3 (∼7.5 g) was added. The reaction mixture was refluxed for 24 hours and cooled to room temperature. It was then filtered through Whatman filter paper, and the resulting filtrate was dried using a rotary-evaporator. The dried residue was dissolved in 20 mL DI water and extracted using chloroform (CHCl3, 5 × 50 mL). The resulting product in CHCl3 was passed through anhydrous sodium sulfate and dried using a rotary-evaporator yielding the product of N-(4-oxopentyl)acrylamide (4-OPAm). Next, adenosine was conjugated to 4-OPAm by reacting the ketone group of 4-OPAm with the vicinal diol groups of adenosine.60 Briefly, 4-OPAm (5 g), adenosine (6.7 g), and triethyl orthoformate (4.2 mL) were dissolved in 20 mL of N,N-dimethylformamide (DMF). The reaction mixture was purged with argon gas and then 6 mL of 4 M HCl in 1,4-dioxane was added, continuing the reaction at room temperature for 24 hours. The reaction mixture was then diluted in 100 mL DCM and washed with saturated sodium bicarbonate solution. The organic phase was then concentrated using a rotary-evaporator and the product was precipitated in 1:1 hexane:diethylether mixture. The precipitate was centrifuged at 7000 rpm for about 10 minutes and followed by diethylether washing. The precipitate was placed in a vacuum-oven at 40 °C overnight and allowed to dry in order to obtain adenosine conjugated 4-OPAm. The intermediate compounds (2-CEA, 2-CEAm, and 4-OPAm) and adenosine conjugated 4-OPAm were characterized via a combination of 1H NMR and FTIR spectroscopy. FTIR was performed using ZnSe crystal via attenuated total reflection (ATR) mode. For 2-CEA, 2-CEAm, and 4-OPAm, 1H NMR spectra were recorded by dissolving 3–4 mg of the compounds in deuterated chloroform (CDCl3) at 25 °C. For adenosine conjugated 4-OPAm, the spectrum was recorded by dissolving 3–4 mg of the compound in CDCl3 at 25 °C.
Nanocarrier synthesis and purification
The nanocarrier was synthesized via inverse emulsion photopolymerization. Adenosine conjugated 4-OPAm (337.5 mg) was dissolved in DMSO at 150 mg mL−1. HA-MA-Aln (37.5 mg, 150 mg mL−1) and PEGDA (37.5 mg, 150 mg mL−1) were dissolved in 500 μL of 0.1 M phosphate buffer at pH 7.4. The adenosine conjugated 4-OPAm solution was then added to the solution of HA-MA-Aln and PEGDA. Lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) dissolved in 0.1 M phosphate buffer of pH 7.4 at 50 mg mL−1 was added (0.125 mL) to the reaction mixture. The resulting solution was purged with argon gas and added to a continuous phase consisting of mineral oil with 10% ABIL EM90 surfactant and sonicated using a probe sonicator for 180 seconds at 100 W output voltage. Following sonication, the resulting nano-emulsion was UV-irradiated for 15 minutes under constant stirring at a speed of 300 rpm, and the resulting nanoparticles were precipitated using a 1:1 mixture of hexane and isopropanol. The precipitate was centrifuged at 7000 rpm for 10 minutes and washed extensively with 1:1 hexane:isopropanol (4 × 30 mL). The precipitate was further washed with pure isopropanol and suspended in 5 mL DI water (pH ∼ 8.0). The aqueous suspension was immediately flash frozen and freeze-dried to obtain adenosine loaded nanocarriers (ANC). The same method was used to synthesize nanocarrier devoid of adenosine (NC) by using 4-OPAm instead of adenosine conjugated 4-OPAm.
Characterization of the nanocarriers
The nanocarriers were characterized through a combination of FTIR, UV-visible spectroscopy, dynamic light scattering (DLS), transmission electron microscopy (TEM), and zeta potential measurements. FTIR spectroscopy of the freeze dried nanocarriers was recorded using an attenuated total reflectance (ATR) with ZnSe crystal. To record UV-visible absorption spectra, freeze-dried nanocarrier was suspended in 1:1 water:ethanol mixture, and absorbance was measured at 200–800 nm. The average hydrodynamic diameter of the nanocarriers was determined by dynamic light scattering (DLS, Wyatt Technology DynaPro PlateReader). Nanocarriers were suspended in phosphate buffer of pH of 7.4, at 50 μg mL−1. The nanocarrier suspension (40 μL) was transferred to a clear bottom black 96-well plate. The measurements were carried out at 25 °C, with a laser of 833 nm, and each individual acquisition consisted of an average intensity of ten different measurements. For transmission electron microscopy (TEM) images, 2 mg of the freeze-dried nanocarriers (ANC) were suspended in 1 mL of water and subsequently diluted to ∼300 μg mL−1 in 1:6 water:ethanol mixture. The nanocarrier suspension (3 μL) was drop-casted on formvar/carbon 300 mesh carbon grid (FCF300-CU, Electron Microscopy Sciences) and dried in a vacuum-oven at 50 °C. Images were acquired by using a Thermo Fisher Scientific Talos F200X instrument operated at 200 kV. TEM images were captured on a 4K Ceta CMOS camera. All signal processing were performed using standard routines within the Thermo Fisher Velox software package. The surface charge of the nanocarriers was determined by measuring the zeta potential using a Zetasizer Nano Zs (Malvern Instrument, UK). The nanocarriers were dispersed in ultrapure DI water at 25 μg mL−1 and filtered using a 0.45 μm syringe filter. The filtered solution (1 mL) was transferred into a cuvette, and the zeta potential was measured at room temperature with an average of 100 runs. Each measurement was repeated eight times using a folded capillary zeta cell 1070. The Smoluchowski equation was used to calculate the zeta potential of the nanocarriers. To determine the release of alendronate, if any, the nanocarriers (NC, ∼5 mg) were suspended in 0.5 mL of DI water of pH 6.8 and immediately placed in a dialysis bag (MWCO of 2 kDa). The dialysis bags were sealed and placed in a 15 mL Falcon tube containing 9.5 mL of DI water of pH 6.8. Following incubation at 37 °C for 72 hours, the dialysis bags were removed, and the incubating medium was freeze-dried. The 1H NMR spectrums of the freeze-dried product were recorded and analyzed for alendronate content.
pH dependent adenosine release
Adenosine containing nanocarriers (ANC, ∼5 mg) were suspended in 0.5 mL of 0.5 M phosphate buffer with either pH of 5.0, 5.5, 6.0, 6.8, or 7.4 (n = 3 per group) and immediately placed in dialysis membrane (MWCO of 2 kDa). The dialysis bags were sealed and placed in a 15 mL Falcon tube containing 9.5 mL of phosphate buffer of the same pH. At predetermined time intervals (0 hours, 6 hours, 12 hours, 24 hours, 48 hours, 72 hours, 96 hours, 120 hours, 144 hours, 168 hours, 240 hours, 336 hours, 504 hours), 2 mL of the buffer solution surrounding the bag was collected and replenished with 2 mL of fresh buffer solution of the same pH. The amount of adenosine released in the buffer solutions of different pHs at different time intervals was determined by UV-visible spectroscopy at a wavelength of 260 nm. Standard calibration curves of free adenosine in various buffers were generated at 260 nm for a concentration range of 2–125 μg mL−1 and were used to determine the amount of released adenosine in the buffer solutions. The cumulative amount of adenosine was plotted at different pH conditions.Conditioned medium from nanocarriers
The nanocarriers (NC and ANC, ∼5 mg) were suspended in 1.0 mL of alpha-minimal essential medium (α-MEM) at either pH 6.5 or 7.4 (n = 3 per group) and immediately placed in a dialysis bag (2 kDa MWCO). The bags were sealed and placed in 50 mL Falcon tubes containing α-MEM (19 mL) of the appropriate pH. At 24-hour intervals, 4 mL of the medium surrounding the bag was collected and replenished with 4 mL of fresh medium. The conditioned medium collected was sterilized using a 0.2 μm filter and used for cell culture studies.Animal and cell isolation studies
All animal studies were performed with the approval of Institutional Animal Care and Use Committee (IACUC) (A151-20-07) at Duke University and in accordance with the guidelines of the National Institutes of Health (NIH). 12-week-old female C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME) were used for cell isolation and in vivo studies.Isolation and culture of mouse mononuclear cells (MNCs)
MNCs were isolated as previously described.31 Briefly, vertebrae were harvested and crushed in a harvest buffer [FBS (1% v/v) in PBS] to release the bone marrow (BM). BM was passed through a 40 μm cell strainer and centrifuged at 200g for 5 minutes to collect cell pellet. Cells resuspended in harvest buffer, were gently layered onto Ficoll-Paque PLUS (GE Healthcare, Marlborough, MA) at 1:1 ratio. Then the solutions were centrifuged without rotor acceleration and deceleration at 200g for 15 minutes. The opaque layer was collected and centrifuged at 200g for 5 minutes in harvest buffer. MNCs were seeded in a 24-well plate at a density of 100000 cells per cm2 and cultured in macrophage induction medium (MIM) [α-MEM supplemented with 10% FBS, 10000 U mL−1 penicillin/streptomycin, prostaglandin E2 (PGE2; 10−7 M; Santa Cruz Biotechnology, Dallas, TX), and Macrophage-Colony Stimulating Factor (M-CSF; 10 ng mL−1; PeproTech, Rocky Hill, NJ)]. To examine the effect of adenosine released from the nanocarrier on osteoclastogenesis, MNCs cultured for 6 days in macrophage induction medium were further differentiated to osteoclasts for 10 days using either osteoclast induction medium [macrophage induction medium supplemented with RANKL (10 ng mL−1; PeproTech)], osteoclast induction medium containing adenosine (45 μg ml−1), or osteoclast induction medium generated using the conditioned medium collected from NC (pH 6.5 and 7.4) or ANC (pH 6.5 and 7.4). For the osteoclast induction medium containing conditioned medium, α-MEM collected from the NC and ANC at different pHs were supplemented with 10% FBS, 10000 U mL−1 penicillin/streptomycin, prostaglandin E2 (PGE2; 10−7 M), M-CSF Macrophage-Colony Stimulating Factor (M-CSF; 10 ng mL−1), and RANKL (10 ng mL−1; PeproTech).
TRAP staining
Tartrate-resistant acid phosphatase (TRAP) staining and quantification were performed as previously described with a slight modification.34 TRAP staining was accomplished by fixing cells in 4% paraformaldehyde (PFA) for 10 minutes at room temperature. Cells rinsed with distilled water were incubated in 0.2 M acetate buffer containing 50 mM sodium l-tartrate dibasic dihydrate at pH 5.0 for 20 minutes at 25 °C followed by incubation with naphthol AS-MX phosphate disodium salt (Millipore-Sigma, N5000-1G; 0.5 mg mL−1) and Fast Red TR Salt 1,5-naphthalenedisulfonate (Millipore-Sigma, F6760-5G; 1.1 mg mL−1) for 90 minutes at 37 °C devoid of light. To quantify the TRAP staining, cells were incubated in 500 μL in Triton X-100 in PBS (2% v/v) for 1 week to release the stains. The absorption spectrum was recorded at 200–800 nm using a UV-visible spectrophotometer (Genesys 10S). Subsequently, the OD value of the solution was quantified using the absorption wavelength of TRAP at 570 nm with a spectrophotometer (Tecan Infinite 200 PRO).In vitro osteoclast pit assay
The osteoclast pit assay was conducted as previously described, and adapted to culture with the conditioned medium from NC and ANC.61 In brief, MNCs were seeded on bovine bone slices (Biovendor, DT-1BON1000-96) at 100000 cells per cm2 and cultured in macrophage induction medium [α-MEM, containing 10% FBS, 10000 U mL−1 penicillin/streptomycin, 10−7 M prostaglandin E2 (PGE2), and 10 ng mL−1 Macrophage-Colony Stimulating Factor (M-CSF)] for 6 days. Subsequently, the macrophage cultures were induced to undergo osteoclast differentiation by using either osteoclast induction medium [macrophage induction medium supplemented with RANKL (10 ng mL−1)], osteoclast induction medium containing adenosine (45 μg mL−1), or osteoclast medium generated from the conditioned medium from NC or ANC at pH 6.5 or 7.4. For the osteoclast induction medium containing conditioned medium, α-MEM collected from the NC and ANC at different pHs were supplemented with 10% FBS, 10000 U mL−1 penicillin/streptomycin, PGE2 (10−7 M), M-CSF (10 ng mL−1), and RANKL (10 ng mL−1). Bone slices were fixed with glutaraldehyde (2.5% in 1× PBS) for 30 minutes at 25 °C, placed in distilled water, and sonicated for 30 minutes at 40 Hz to dislodge the cells. The bone slices were then stained with 1% toluidine blue O (Millipore Sigma, T3260-5G) in 1% sodium borate for 4 minutes, rinsed in distilled water, and air dried. Images were taken with Keyence BZ-X700 microscope.
Isolation and culture of mouse bone marrow mesenchymal stromal cells (BMMSCs)
BMMSCs were isolated as previously described.31,62 Briefly, the mouse vertebrae were harvested, crushed and harvested in buffer [fetal bovine serum (FBS, 1% v/v) in PBS] to release the bone marrow (BM). Then, this solution was filtered through a 40 μm cell strainer and centrifuged at ∼923 rpm. Cells were seeded in a twenty-four well plate at 1000000 cells per cm2, and ultimately cultured in growth medium (GM) containing α-MEM, FBS (10%, v/v), penicillin/streptomycin (10000 U mL−1; 1%, v/v) in humidified incubator at 37 °C and 5% CO2. After 3 days, medium was replaced and cultured for six days prior to passage. For passaging, cells were detached using a scraper and sub-cultured at a density of 8000 cells per cm2. The following experiments were performed after 1 passage. BMMSCs cultured for 14 days in either growth medium (GM), GM supplemented with adenosine (45 μg mL−1), or GM generated with conditioned medium collected from NC (pH 6.5 and 7.4) or ANC (pH 6.5 and 7.4). For the growth medium containing conditioned medium, α-MEM collected from the NC and ANC at different pHs were supplemented with 10% FBS and 10000 U mL−1 penicillin/streptomycin. For mineralization, cells were further cultured in phosphate medium (GM supplemented with 4 mM monobasic sodium phosphate and 4 mM dibasic sodium phosphate at 1:1 ratio) for 4 days.
Alizarin red S staining
Alizarin red S staining was performed as described previously with slight modifications, and quantified.63,64 Cells were fixed in 4% paraformaldehyde (PFA) for 10 minutes at 25 °C, rinsed with DI water, and stained with 2% alizarin red S solution at pH 4.2 for 20 minutes at 25 °C. The stained wells were rinsed with DI water and imaged. To quantify mineralization using alizarin red S staining, 500 μL of 0.5 M HCl was added to the wells for 10 minutes at 25 °C with shaking. The solutions were placed into tubes, heated for 10 minutes at 85 °C, and centrifuged at 12631 rpm for 15 minutes. Subsequently, the supernatant (∼100 μL) was transferred to a 96-well clear bottom plate, and UV-vis spectrophotometer absorbance was measured at 405 nm.
Quantitative reverse transcription PCR (RT-qPCR)
After culture completion, cells were analyzed for osteogenic differentiation through quantitative real-time polymerase chain reaction (RT-qPCR).31 Nucleic acids were extracted with TRIzol (Thermo Fisher Scientific, 15596026), phase-separated in chloroform, and precipitated using isopropanol. 1 μg of RNA was reverse transcribed using iScript cDNA Synthesis Kit (Bio-Rad, 1708890).34 Quantitative PCR was performed using iTaq Universal SYBR green reagent (Bio-Rad, 1725120) with denaturation for 30 seconds at 95 °C for one cycle, and amplification (denaturation + annealing/extension) for 5 seconds at 95 °C, and for 40 cycles at 60 °C for 30 seconds on a polymerase chain reaction (PCR) cycler (Bio-Rad, 1851148). The mouse primer sequences used are: Sp7 (forward, TGCCT GACTC CTTGG GACC; reverse, TAGTG AGCTT CTTCC TCAAG CA), Spp1 (forward, AAACC AGCCA AGGTA AGCCT; reverse, TCAGT CACTT TCACC GGGAG), Acp5 (forward, CAGCA GCCCA AAATG CCT; reverse, TTTTG AGCCA GGACA GCTGA), Ctsk (forward, CACCC TTAGT CTTCC GCTCA; reverse, CTTGA ACACC CACAT CCTGC T), and 18S ribosomal RNA (forward, ACCAG AGCGA AAGCA TTTGC CA; reverse, ATCGC CAGTC GGCAT CGTTT AT).34 Each gene target expression level was normalized to the housekeeping gene (expressed as 2−ΔΔCt values). The expression levels were normalized to the controls (GM for osteogenesis and MIM for osteoclastogenesis) and presented as fold change.Live dead assay
Primary BMMSCs seeded at 6000 cells per cm2 were differentiated to osteoblasts for 14 days. Primary MNCs seeded at 100000 cells per cm2 were differentiated into osteoclasts in chamber slides. Nanocarrier (NC and ANC) suspensions were made in growth medium (MEM with 10% v/v FBS) at different concentrations (100 μg mL−1, 200 μg mL−1, and 500 μg mL−1). ∼500 μL of the nanocarrier suspension was added to each chamber containing cells and incubated at 37 °C in a 5% CO2 atmosphere humidified incubator. Following 24 hours of incubation, cells were washed with PBS, and exposed to a mixture of calcein AM (1 μM; ThermoFisher C3100MP) and propidium iodide (1.5 μM; ThermoFisher P3566) dissolved in GM. Slides were incubated in a 5% CO2 atmosphere humidified incubator for ∼15 minutes. The medium containing the dyes were then removed, and GM without dye was added. Images were obtained using a Keyence BZ-X700 microscope.
In vitro bone binding affinity
The nanocarrier bone tissue binding capability was assessed in vitro using femur bone chips from 8-week-old female C57BL/6J mice (Jackson Laboratory) (n = 3). The femur bone marrow was flushed, and the bone was cut to obtain ∼2–3 mm length slices. Various concentrations of the Cy7-conjugated nanocarriers (0 mg mL−1, 0.5 mg mL−1, and 5 mg mL−1) were generated in α-MEM medium containing 10% FBS. The chips were incubated with 200 μL of the suspension for 2 hours under constant shaking at 37 °C. The bone slices were removed and washed with PBS. An in vivo imaging system [IVIS Kinetics system (excitation filter, 745 nm; emission filter, ICG; excitation time, 100 milliseconds)] was used to record fluorescence intensity. The normalized radiant efficiency [(p per second per cm2 per sr)/(μW cm−2)] was normalized by the surface area of the bone chip and expressed as radiant efficiency/mm2.In vivo administration of nanocarrier
C57BL/6J ovariectomized female mice (12 weeks old; Jackson Laboratory, Bar Harbor, ME) were used for the study. Animal grouping and treatment included: mice with no OVX surgery, (healthy, H), mice with OVX surgery (O), OVX mice treated with NC without adenosine (NC), OVX mice treated with adenosine containing NC (ANC) by tail vein injection (n = 4–5 per group). Administration of the nanocarriers started 6 weeks after OVX surgery, at which point osteoporotic bone loss is observed.31 The treatment was continued once a week for 10 weeks. Mice were either treated with 90 mg kg−1 body weight of NC, or 120 mg kg−1 body weight of ANC. The higher weight in the ANC group was used to ensure that the animals received the same amount of nanocarriers.In vivo biodistribution
The Cy7-conjugated ANC was suspended in sterile saline (0.9% NaCl; Hospira). A single dose of ∼2.1 nM dye concentration was injected intravenously through the tail vein. After 72 hours post intravenous injection, the mice were anesthetized with isoflurane. An IVIS imaging system was used to acquire whole-body images. Following IVIS imaging, the animals were sacked and major tissues were harvested (vertebra, femur, tibia, liver, muscle, spleen, lungs, kidneys). The organ weight was recorded, and then the samples were imaged using IVIS. Data analysis was carried out using Living Image software (PerkinElmer). Finally, the results were expressed as radiant efficiency/gram of the organ. In order to analyze the distribution of the nanocarrier within the bone tissue, the L4 lumbar vertebrae and femur were removed, and placed in 4% PFA at 4 °C overnight. Following fixation, samples were incubated in 30% sucrose, embedded in cryomatrix (ThermoFisher; Epredia 6769006) and cryosections (10 μm) were obtained using CryoJane tape transfer system (with a Leica cryotome). The tissue section nuclei were stained with ProLong diamond antifade mountant containing DAPI (Invitrogen, P36971). The Zeiss Axio Observer Z1 microscope was used to obtain fluorescence images. The images of sections were imaged for both healthy and OVX groups (n = 4 per group). Sections were imaged using a 710/75 band pass excitation filter and 810/90 nm band pass emission filter and shown in pseudo red color. DAPI was imaged at 365 nm excitation wavelength and 445/50 nm band pass emission filter.Microcomputed tomography
The L3–5 vertebrae and femur were harvested, fixed in 4% paraformaldehyde (PFA) for 1 day at 4 °C, and washed with PBS. The fixed samples were placed in centrifuge tubes containing styrofoam spacers, placed in a μ-CT scanner (vivaCT 80, Scanco Medical, Wayne, PA). Samples were scanned at 55 keV with a pixel resolution of 10.4 μm. Reconstruction was made of the images using μ-CT Evaluation Program V6.6 (Scanco Medical). Bone mineral density was quantified and expressed as percentage of bone volume (BV) per total volume (TV) (BV/TV) using the phantom as a reference from 100 contiguous slices. Trabecular number (Tb. N), trabecular spacing (Tb. Sp), connectivity density (Conn. D), and trabecular thickness (Tb. Th) were quantified through μ-CT Evaluation Program V6.6 (Scanco Medical) software. Vertebral-3D model images were acquired using CTAn software (n = 4–5 per group).Histological staining
Vertebral samples were fixed using 4% paraformaldehyde (PFA) at 4 °C for 24 hours and decalcified using 14% ethylenediaminetetracetic acid (EDTA, pH 7.4) for 2 weeks at 4 °C. Liver samples were fixed using 4% PFA at 4 °C for 24 hours. The samples were gradually dehydrated using various concentrations of ethanol, then incubated in Citrisolv (Decon Laboratories, 125160) until equilibrium. After dehydration, samples immersed in a mixture of 50% (v/v) Citrisolv and 50% (w/w) paraffin (General Data Healthcare, H-PF) were placed at 70 °C for 30 minutes. These samples were embedded in paraffin and sectioned to obtain 5 μm sections with a rotary microtome (Leica, RM2255). Prior to staining, sections were deparaffinized. Hematoxylin and eosin (H&E) staining was performed via incubating the samples in hematoxylin solution (Ricca Chemical) for 1 minute followed by incubation in Eosin-Y solution (Richard-Allan Scientific) for 1 minute. Sections were gradually dehydrated using various concentrations of ethanol until equilibrium was reached. Tartrate-resistant acid phosphatase (TRAP) staining was performed by incubating rehydrated sections in acetate buffer (0.2 M) containing sodium L-tartrate dibasic dihydrate (50 mM) at pH 5.0 for 20 minutes at room temperature followed by incubating with naphthol AS-MX phosphate disodium salt (Sigma, N5000-1G; 0.5 mg mL−1) and Fast Red TR Salt 1,5-naphthalenedisulfonate (Sigma, F6760-5G; 1.1 mg mL−1) dissolved in the same buffer at 37 °C for 1.5 hours. Sections were mounted using Cytoseal Mountant 60 (Epredia, 23-244257) and imaged using a Keyence BZ-X700 microscope. For quantification, the number of TRAP positive cells were counted per L4 vertebrae and divided by the length of total bone surface.Bone labeling
Animals were administered with 10 mg kg−1 body weight calcein (Sigma-Aldrich, 154071-48-4) 31 days prior to sacrifice and 30 mg kg−1 body weight of alizarin-3-methyliminodiacetic acid (Sigma-Aldrich, 3952-78-1) 14 days post-administration of calcein (n = 4–5 per group). Harvested femurs were fixed in 4% paraformaldehyde (PFA) at 4 °C for 24 hours and stored in 70% ethanol. Samples were incubated in 30% sucrose for 1 day and embedded in cryomatrix. Then the samples were cryo-sectioned with CryoJane Tape transfer system, and cortical bone was imaged for flourescent bone labeling. Bone formation rate (BFR) and mineral apposition rate (MAR) were calculated from parameters measured from images using ImageJ software. BFR = MAR × (Mineralizing surface, MS/bone surface, BS). MAR = (interlabel width, irL. Wi)/time interval. Where interlabel width is the distance between the double fluorescent labels. (MS/BS) = (dL. Pm + (0.5 × sL. Pm))/B. Pm, where perimeter of double labeled bone (dL. Pm) plus perimeter of one half of the singly labeled bone (0.5 × sL. Pm) is expressed as a fraction of the total bone perimeter (B. Pm).Mechanical measurement
Tibiae were used to measure the mechanical properties. After removing the soft tissues, tibia samples were covered in PBS containing tissue and frozen at −20 °C (n = 8–10 per group). Samples were placed at room temperature an hour before the measurement. Four-point bending measurements using an Electroforce 3220 (TA Instruments, New Castle, DE) instrument with 225 N load cell was performed. Samples were aligned on the fixtures and the load was applied perpendicular to the principal axis of the tibia. Span length of the bottom support was about 9.2 mm with the top span length at 2.8 mm. The bending test was performed using a displacement control mode with a loading rate of 0.025 mm s−1. Load-displacement data was acquired at a data acquisition rate of 10 Hz. The highest load (maximum load) experienced by the samples before was determined from using load-displacement graphs. Bending stiffness was calculated as the slope of the linear region in the load vs. displacement graph.Statistical analyses
All numerical data are expressed as means plus or minus standard deviation. Data were subjected to one-way analysis of variance (ANOVA), two-way repeated measures ANOVA, or two-tailed Student's t-test with post hoc Tukey-Kramer test for multiple comparisons. P-Values of less than 0.05 were considered statistically significant and indicated with an asterisk. All statistical analyses were performed with either GraphPad Prism 9.1.0 or JMP.Author contributions
Hunter Newman: conceptualization, methodology, investigation, data curation, validation, formal analysis, writing-original draft, writing-reviewing & editing. Jiaul Hoque: conceptualization, methodology, investigation, data curation, validation, formal analysis, writing-original draft, writing-reviewing & editing. Yu-Ru V. Shih: investigation, formal analysis, writing-reviewing & editing. Gabrielle Marushack: investigation, formal analysis. Unghyeon Ko: investigation, formal analysis. Gavin Gonzales: investigation, formal analysis. Shyni Varghese: conceptualization, supervision, visualization, writing-review & editing, and funding acquisition.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the financial support from National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (NIH) under Award Numbers NIH RO1 AR071552 and NIH RO1 AR079189. The TEM images were acquired at the Analytical Instrumentation Facility (AIF) at North Carolina State University, which is supported by the State of North Carolina and the National Science Foundation (award number ECCS-1542015) and the National Science Foundation (DMR-1726294) which supported the instrument acquisition.References
- J. M. Lane, L. Russell and S. N. Khan, Clin. Orthop. Relat. Res., 2000, 139–150, DOI:10.1097/00003086-200003000-00016.
- O. Johnell and J. A. Kanis, Osteoporosis Int., 2006, 17, 1726–1733 CrossRef CAS PubMed.
- C. B. Johnston and M. Dagar, Med. Clin. North Am., 2020, 104, 873–884 CrossRef PubMed.
- K. N. Tu, J. D. Lie, C. K. V. Wan, M. Cameron, A. G. Austel, J. K. Nguyen, K. Van and D. Hyun, P T, 2018, 43, 92–104 Search PubMed.
- Y. Yu, H. Newman, L. Shen, D. Sharma, G. Hu, A. J. Mirando, H. Zhang, E. Knudsen, G.-F. Zhang, M. J. Hilton and C. M. Karner, Cell Metab., 2019, 29, 966–978 CrossRef CAS PubMed.
- E. W. Yu, R. Kumbhani, E. Siwila-Sackman, M. DeLelys, F. I. Preffer, B. Z. Leder and J. Y. Wu, J. Bone Miner. Res., 2014, 29, 1380–1386 CrossRef CAS PubMed.
- M. Girotra, M. R. Rubin and J. P. Bilezikian, Treat. Endocrinol., 2006, 5, 347–358 CrossRef CAS PubMed.
- A. V. Haas and M. S. LeBoff, J. Endocr. Soc., 2018, 2, 922–932 CrossRef CAS PubMed.
- M. F. Moreau, C. Guillet, P. Massin, S. Chevalier, H. Gascan, M. F. Basle and D. Chappard, Biochem. Pharmacol., 2007, 73, 718–723 CrossRef CAS PubMed.
- D. E. Hughes, K. R. Wright, H. L. Uy, A. Sasaki, T. Yoneda, G. D. Roodman, G. R. Mundy and B. F. Boyce, J. Bone Miner. Res., 1995, 10, 1478–1487 CrossRef CAS PubMed.
- A. A. Reszka, J. M. Halasy-Nagy, P. J. Masarachia and G. A. Rodan, J. Biol. Chem., 1999, 274, 34967–34973 CrossRef CAS PubMed.
- M. T. Drake, B. L. Clarke and S. Khosla, Mayo Clin. Proc., 2008, 83, 1032–1045 CrossRef CAS PubMed.
- R. Phipps, B. H. Mitlak, D. B. Burr and M. R. Allen, in Basic and Applied Bone Biology, ed. D. B. Burr and M. R. Allen, Academic Press, 2nd edn, 2019, pp. 389–410. DOI:10.1016/B978-0-12-813259-3.00021-X.
- N. Nair, M. Liu and A. Cohen, in Marcus and Feldman's Osteoporosis, ed. D. W. Dempster, J. A. Cauley, M. L. Bouxsein and F. Cosman, Academic Press, 5th edn, 2021, pp. 951–968. DOI:10.1016/B978-0-12-813073-5.00038-1.
- G. Russow, D. Jahn, J. Appelt, S. Mardian, S. Tsitsilonis and J. Keller, Int. J. Mol. Sci., 2018, 20, 83 CrossRef PubMed.
- K. A. Kennel and M. T. Drake, Mayo Clin. Proc., 2009, 84, 632–637 CrossRef CAS PubMed ; quiz 638.
- J. M. O'Neal, T. Diab, M. R. Allen, B. Vidakovic, D. B. Burr and R. E. Guldberg, Bone, 2010, 47, 241–247 CrossRef PubMed.
- X. Chen, Z. Wang, N. Duan, G. Zhu, E. M. Schwarz and C. Xie, Connect. Tissue Res., 2018, 59, 99–107 CrossRef CAS PubMed.
- K. Matsuoka, K. A. Park, M. Ito, K. Ikeda and S. Takeshita, J. Bone Miner. Res., 2014, 29, 1522–1530 CrossRef CAS PubMed.
- T. Ono and T. Nakashima, Histochem. Cell Biol., 2018, 149, 325–341 CrossRef CAS PubMed.
- B. F. Boyce, Z. Yao and L. Xing, Crit. Rev. Eukaryotic Gene Expression, 2009, 19, 171–180 CrossRef CAS PubMed.
- S. L. Teitelbaum and F. P. Ross, Nat. Rev. Genet., 2003, 4, 638–649 CrossRef CAS PubMed.
- T. R. Arnett, J. Nutr., 2008, 138, 415S–418S CrossRef CAS PubMed.
- I. A. Silver, R. J. Murrills and D. J. Etherington, Exp. Cell Res., 1988, 175, 266–276 CrossRef CAS PubMed.
- A. Nishiguchi and T. Taguchi, Biomacromolecules, 2019, 20, 1385–1393 CrossRef CAS PubMed.
- S. Avnet, G. Di Pompo, S. Lemma and N. Baldini, Cancer Metastasis Rev., 2019, 38, 133–147 CrossRef PubMed.
- Y. Han, X. You, W. Xing, Z. Zhang and W. Zou, Bone Res., 2018, 6, 16 CrossRef PubMed.
- F. L. Yuan, M. H. Xu, X. Li, H. Xinlong, W. Fang and J. Dong, Front. Physiol., 2016, 7, 222 Search PubMed.
- A. Brandao-Burch, J. C. Utting, I. R. Orriss and T. R. Arnett, Calcif. Tissue Int., 2005, 77, 167–174 CrossRef CAS PubMed.
- X. Lin, Q. Wang, C. Gu, M. Li, K. Chen, P. Chen, Z. Tang, X. Liu, H. Pan, Z. Liu, R. Tang and S. Fan, J. Am. Chem. Soc., 2020, 142, 17543–17556 CrossRef CAS PubMed.
- Y. V. Shih, M. Liu, S. K. Kwon, M. Iida, Y. Gong, N. Sangaj and S. Varghese, Sci. Adv., 2019, 5, eaax1387 CrossRef CAS PubMed.
- A. Mediero, F. M. Kara, T. Wilder and B. N. Cronstein, Am. J. Pathol., 2012, 180, 775–786 CrossRef CAS PubMed.
- A. Mediero and B. N. Cronstein, Trends Endocrinol. Metab., 2013, 24, 290–300 CrossRef CAS PubMed.
- J. Hoque, Y. V. Shih, Y. Zeng, H. Newman, N. Sangaj, N. Arjunji and S. Varghese, Biomaterials, 2021, 273, 120819 CrossRef CAS PubMed.
- M. Kazemzadeh-Narbat, N. Annabi, A. Tamayol, R. Oklu, A. Ghanem and A. Khademhosseini, J. Drug Targeting, 2015, 23, 580–596 CrossRef CAS PubMed.
- F. U. Din, W. Aman, I. Ullah, O. S. Qureshi, O. Mustapha, S. Shafique and A. Zeb, Int. J. Nanomed., 2017, 12, 7291–7309 CrossRef PubMed.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. D. P. Rodriguez-Torres, L. S. Acosta-Torres, L. A. Diaz-Torres, R. Grillo, M. K. Swamy, S. Sharma, S. Habtemariam and H. S. Shin, J. Nanobiotechnol., 2018, 16, 71 CrossRef PubMed.
- R. Jia, L. Teng, L. Gao, T. Su, L. Fu, Z. Qiu and Y. Bi, Int. J. Nanomed., 2021, 16, 1525–1551 CrossRef PubMed.
- S. H. Pham, Y. Choi and J. Choi, Pharmaceutics, 2020, 12, 630 CrossRef CAS PubMed.
- T. T. Lee, J. R. García, J. I. Paez, A. Singh, E. A. Phelps, S. Weis, Z. Shafiq, A. Shekaran, A. Del Campo and A. J. García, Nat. Mater., 2015, 14, 352–360 CrossRef CAS PubMed.
- A. J. Roelofs, K. Thompson, F. H. Ebetino, M. J. Rogers and F. P. Coxon, Curr. Pharm. Des., 2010, 16, 2950–2960 CrossRef CAS PubMed.
- I. Lambrinoudaki, G. Christodoulakos and D. Botsis, Ann. N. Y. Acad. Sci., 2006, 1092, 397–402 CrossRef CAS PubMed.
- M. Karimi, A. Ghasemi, P. Sahandi Zangabad, R. Rahighi, S. M. Moosavi Basri, H. Mirshekari, M. Amiri, Z. Shafaei Pishabad, A. Aslani, M. Bozorgomid, D. Ghosh, A. Beyzavi, A. Vaseghi, A. R. Aref, L. Haghani, S. Bahrami and M. R. Hamblin, Chem. Soc. Rev., 2016, 45, 1457–1501 RSC.
- B. M. Jarai and C. A. Fromen, Adv. NanoBiomed. Res., 2022, 2, 2100127 CrossRef.
- D. A. Heller, Y. Levi, J. M. Pelet, J. C. Doloff, J. Wallas, G. W. Pratt, S. Jiang, G. Sahay, A. Schroeder, J. E. Schroeder, Y. Chyan, C. Zurenko, W. Querbes, M. Manzano, D. S. Kohane, R. Langer and D. G. Anderson, Adv. Mater., 2013, 25, 1449–1454 CrossRef CAS PubMed.
- D. A. Heller, Y. Levi, J. M. Pelet, J. C. Doloff, J. Wallas, G. W. Pratt, S. Jiang, G. Sahay, A. Schroeder, J. E. Schroeder, Y. Chyan, C. Zurenko, W. Querbes, M. Manzano, D. S. Kohane, R. Langer and D. G. Anderson, Adv. Mater., 2013, 25, 1449–1454 CrossRef CAS PubMed.
- D. Liu, F. Yang, F. Xiong and N. Gu, Theranostics, 2016, 6, 1306–1323 CrossRef CAS PubMed.
- W. Wu, L. Luo, Y. Wang, Q. Wu, H. B. Dai, J. S. Li, C. Durkan, N. Wang and G. X. Wang, Theranostics, 2018, 8, 3038–3058 CrossRef CAS PubMed.
- W. Gao, J. M. Chan and O. C. Farokhzad, Mol. Pharm., 2010, 7, 1913–1920 CrossRef CAS PubMed.
- A. B. Cook and P. Decuzzi, ACS Nano, 2021, 15, 2068–2098 CrossRef CAS PubMed.
- N. Yu, Y. Xu, T. Liu, H. Zhong, Z. Xu, T. Ji, H. Zou, J. Mu, Z. Chen, X. J. Liang, L. Shi, D. S. Kohane and S. Guo, Nat. Commun., 2021, 12, 5532 CrossRef CAS PubMed.
- Y. Shin, P. Husni, K. Kang, D. Lee, S. Lee, E. Lee, Y. Youn and K. Oh, Pharmaceutics, 2021, 13, 725 CrossRef CAS PubMed.
- Z. Cao, W. Li, R. Liu, X. Li, H. Li, L. Liu, Y. Chen, C. Lv and Y. Liu, Biomed. Pharmacother., 2019, 118, 109340 CrossRef CAS PubMed.
- M. Takedachi, H. Oohara, B. J. Smith, M. Iyama, M. Kobashi, K. Maeda, C. L. Long, M. B. Humphrey, B. J. Stoecker, S. Toyosawa, L. F. Thompson and S. Murakami, J. Cell Physiol., 2012, 227, 2622–2631 CrossRef CAS PubMed.
- A. Mediero, M. Perez-Aso and B. N. Cronstein, Br. J. Pharmacol., 2013, 169, 1372–1388 CrossRef CAS PubMed.
- R. G. Erben, in Handbook of Histology Methods for Bone and Cartilage, ed. Y. H. An and K. L. Martin, Humana Press, Totowa, NJ, 2003, pp. 99–117. DOI:10.1007/978-1-59259-417-7_5.
- H. Sasaki, N. Miyakoshi, Y. Kasukawa, S. Maekawa, H. Noguchi, K. Kamo and Y. Shimada, J. Bone Miner. Metab., 2010, 28, 403–409 CrossRef CAS PubMed.
- S. Y. Chen, H. T. Yu, J. P. Kao, C. C. Yang, S. S. Chiang, D. O. Mishchuk, J. L. Mau and C. M. Slupsky, PLoS One, 2014, 9, e106559 CrossRef PubMed.
- T. Sozen, L. Ozisik and N. C. Basaran, Eur. J. Rheumatol., 2017, 4, 46–56 CrossRef PubMed.
- X. Liu, C. Corciulo, S. Arabagian, A. Ulman and B. N. Cronstein, Sci. Rep., 2019, 9, 7430–7430 CrossRef PubMed.
- A. Vesprey and W. Yang, Bio-Protoc., 2016, 6(12), e1836 CrossRef PubMed.
- Y.-R. Shih, H. Kang, V. Rao, Y.-J. Chiu, S. K. Kwon and S. Varghese, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5419–5424 CrossRef CAS PubMed.
- S. Varghese, N. S. Hwang, A. Ferran, A. Hillel, P. Theprungsirikul, A. C. Canver, Z. Zhang, J. Gearhart and J. Elisseeff, Stem Cells, 2010, 28, 765–774 CrossRef CAS PubMed.
- H. Kang, Y.-R. V. Shih, M. Nakasaki, H. Kabra and S. Varghese, Sci. Adv., 2016, 2, e1600691 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm00843b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |