
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evaluation of laser direct infrared imaging for rapid analysis of pharmaceutical tablets†
Hannah
Carruthers
 ab,
Don
Clark
b,
Fiona C.
Clarke
b,
Karen
Faulds
a and
Duncan
Graham
*a
ab,
Don
Clark
b,
Fiona C.
Clarke
b,
Karen
Faulds
a and
Duncan
Graham
*a
aUniversity of Strathclyde, Department of Pure and Applied Chemistry, George Street, Glasgow, G1 1RD, UK. E-mail: duncan.graham@strath.ac.uk
bPfizer Ltd., Ramsgate Road, Sandwich, CT19 9NJ, UK
First published on 25th April 2022
Abstract
Vibrational spectroscopic chemical imaging is an important tool in the pharmaceutical industry for characterising the spatial distribution of components within final drug products. The applicability of these techniques is currently limited by the long data acquisition times required to obtain high-definition chemical images of a sample surface. Advancements in quantum cascade laser (QCL) technology have provided an exciting new opportunity for infrared (IR) imaging. Instead of collecting a full IR spectrum at each point, it is possible to focus on distinct spectral bands to reduce imaging data collection time. This study explores a laser direct infrared (LDIR) chemical imaging approach that couples QCL technology with rapid scanning optics to provide high-definition chemical images at an order of magnitude faster than traditional imaging techniques. The capabilities of LDIR chemical imaging were evaluated for pharmaceutical formulations and compared with other established spectroscopic chemical imaging techniques including Raman, near-infrared (NIR) and scanning electron microscopy-energy dispersive X-ray (SEM-EDX) spectroscopy with regards to data acquisition time and image quality. The study showed that LDIR imaging provided high-definition component distribution maps comparable to Raman and SEM-EDX at orders of magnitude faster in terms of time. The ability to obtain high-definition chemical images of the whole tablet surface in relatively fast time frames indicates LDIR imaging could be a promising tool in the pharmaceutical industry to rapidly characterise the size and distribution of components within tablets and could help enhance drug product manufacturing understanding.
Introduction
Vibrational spectroscopic chemical imaging is an important tool in the pharmaceutical industry for characterising the size and spatial arrangement of components within final drug products.1–7 Raman and near-infrared (NIR) chemical imaging are generally the most commonly used techniques with the choice of vibrational spectroscopy depending on the chemical nature of the formulation, time available for analysis and the spatial resolution required to obtain the desired information.8,9 Raman spectroscopy offers high-definition chemical images with detailed domain morphological information at the expense of long data collection times, while the fast data acquisition time achievable by NIR provides a less precise method to rapidly characterise the distribution of components within tablets.9Recently, the combination of Raman chemical imaging with serial sectioning has enabled full visualisation of the three-dimensional (3D) microstructure of a tablet system.10 The applicability of this method is limited by the data collection time required to obtain high-definition two-dimensional (2D) chemical images of the exposed surface. To overcome the limitations of current vibrational spectroscopic chemical imaging techniques, this study assesses the capabilities of novel laser direct infrared (LDIR) chemical imaging that offers the opportunity of obtaining high-definition chemical images of the sample surface at an order of magnitude faster.
Traditionally, chemical mapping experiments involve the collection of spectral information at a number of spatial points by collecting a full spectrum at every point. To reduce data collection times, research has focused on techniques that probe specific vibrational transitions to obtain distribution maps of each component, providing all components have unique spectral bands. These methods require the identity of all components in the tablet to be known and thus are not suitable for all pharmaceutical applications, such as reverse engineering and foreign matter identification.
Global imaging techniques combining infrared (IR) spectroscopy and focal plane array (FPA) detectors have been previously explored for obtaining rapid chemical images of a sample.11 Unlike point-mapping systems, discussed in this paper, chemical images of the sample surface at each specified wavenumber are obtained by irradiating the whole sample simultaneously. There are numerous challenges associated with global imaging approaches, including but not limited to, poorer spectral and spatial resolution.2,12,13 There is, therefore, great interest in developing point-mapping techniques that can probe specific vibrational transitions.
Stimulated Raman scattering (SRS)14 and coherent anti-Stokes Raman scattering (CARS)15 have the ability to channel specific molecular vibrations and thus have been explored as a point-mapping method to rapidly obtain component distribution maps of solid dosage forms. SRS has shown to provide spectral information comparable to confocal Raman mapping 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold faster and could be a potential tool for high-speed screening of pharmaceutical tablets.14 CARS, however, suffers from a non-resonant background and instead offers inferior chemical selectivity relative to confocal Raman mapping. Fluorescence and autofluorescence are some of the main challenges associated with Raman spectroscopy and should be considered during experimental design. Although Raman mapping is considered a non-destructive technique, sample heating from the finely tuned laser can cause sample burning and thus particular care should be taken when imaging single unique samples for troubleshooting investigations.
000-fold faster and could be a potential tool for high-speed screening of pharmaceutical tablets.14 CARS, however, suffers from a non-resonant background and instead offers inferior chemical selectivity relative to confocal Raman mapping. Fluorescence and autofluorescence are some of the main challenges associated with Raman spectroscopy and should be considered during experimental design. Although Raman mapping is considered a non-destructive technique, sample heating from the finely tuned laser can cause sample burning and thus particular care should be taken when imaging single unique samples for troubleshooting investigations.
Recent advancements in quantum cascade laser (QCL) technology have driven the QCL laser to becoming the leading laser source in the mid-infrared (mid-IR) region.16 Coupling a QCL with an IR microscope provides an exciting new opportunity to obtain rapid spatially resolved spectral information of a sample (LDIR imaging).17 Instead of obtaining a full spectrum at every point, it is possible to focus on specific spectral bands to decrease the imaging time by an order of magnitude. Full formulation details must be known prior to analysis to construct a reference library for identifying unique spectral bands for each material.
Sacré et al.18 recently evaluated the LDIR imaging approach for assessing the homogeneity of components within a model pharmaceutical formulation and compared the data with confocal Raman mapping. The study demonstrated that both techniques achieved comparable results, however the LDIR produced data with the same spatial resolution 32-fold faster indicating LDIR is a promising technique for pharmaceutical analysis. The model system analysed in this study, however, contained two active pharmaceutical ingredients (APIs) at relatively high compositions (20% w/w each) resulting in relatively large component domains distributed across the tablet matrix. The capabilities of LDIR for analysing more complex systems containing a low concentration of API more typical to many real pharmaceutical formulations have not yet been explored.
The aim of this study is to assess the capabilities of LDIR imaging for analysing more complex systems and provide a comparison with other established spectroscopic imaging techniques such as Raman, NIR and SEM-EDX with regards to data acquisition time and image quality. A simplified model system containing an API (5% w/w) and two excipients (15% w/w and 80% w/w) which all differed in their elemental composition was initially explored by all four techniques. SEM-EDX analysis was used as an alternative surface imaging technique to confirm the spatial arrangement of components and compare with the vibrational spectroscopic chemical images. A real pharmaceutical tablet containing the same API was then examined by all methods to determine the capabilities of LDIR imaging for examination of real formulations.
Experimental
Sample formulation
:0.15:0.05 w/w ratio, respectively. The raw materials were weighed using a METTLER TOLEDO® XP205 analytical balance and the combined mixture was blended using a TURBULA® shaker-mixer (Glen Mills Inc, New Jersey, USA) at a rate of 46 rotations per minute for 5 minutes. A Specac Atlas Auto T8 wafer press (Specac Ltd, Orpington, UK) was used to compact the blend into a wafer. An A2 scoop of the formulation was inserted into a 10 mm die and compressed to 1 tonne, held for one minute and medium release to 0 tonne.
Sample preparation
To prevent movement during analysis, all samples were adhered to a microscope slide using a cyanoacrylate adhesive. Each sample was milled using a Leica EM Rapid Tablet Mill (Leica, Wetzlar, Germany) to produce an optically flat surface.Chemical imaging
A summary of the data acquisition parameters used for all vibrational spectroscopic chemical imaging methods are displayed in Table 1.| LDIR | Raman | NIR | |
|---|---|---|---|
| Instrument name | Agilent 8700 LDIR | WITec Alpha 500+ CRM | PerkinElmer Spotlight 400N FT-NIR |
| Light source | QCL | 785 nm laser | — |
| Spectral range/cm−1 | 975–1800 | 132.5–1910 | 3900–7600 |
| Spectral resolution/cm−1 | 1 | 1 | 16 |
| Detector | MCT | CCD | InGaAs |
| Objective | 0.72 NA‡ | 20 × 0.46 NA | 15 × 0.60 NA Cassegrainian |
| Step size/μm | 10 | 10 | 25 |
| Acquisition time/s | — | 0.1 | — |
| Number of scans per spectrum | — | 1 | 4 |
| Total mapping time | ∼17 s | ∼3.5 hours | ∼13 min |
The imaging method was developed using Agilent Clarity Software (version 1.4.10). A reference library of components was built by obtaining raw component spectra from compacts of the pure components. A total of 10 spectra were collected for each component which was averaged to provide a reference spectrum for each material. Based on the reference spectra, a single peak ratio analysis was developed for each component in the formulation. A unique peak and baseline position was automatically identified for each component in the library using the Clarity software and verified by visual inspection. The unique peak and baseline wavenumbers chosen for each component in both formulations are provided in Table 2. All data were collected with a spatial pixel size of 10 μm. The data output was a component distribution image for each library component. The intensity of each pixel in the component distribution image is a ratio of the peak and baseline intensity. The distribution component images for all components were combined together to provide a false-colour image of the sample.
| Formulation | Component | Peak wavenumber/cm−1 | Baseline wavenumber/cm−1 |
|---|---|---|---|
| 3-Component model system | MCC | 1058 | 1132 |
| Saccharin | 1718 | 1752 | |
| HBr salt API | 1198 | 1160 | |
| Pharmaceutical tablet | HBr salt API | 1134 | 1034 |
| MCC | 1158 | 1132 | |
| Lactose monohydrate | 1034 | 1052 | |
| Croscarmellose sodium | 1586 | 1684 |
Chemical images were prepared using WITec Project 5.3+ Raman mapping software. All datasets were treated with True Component Analysis which uses a basis analysis algorithm to fit the measured spectrum at each pixel as a linear combination of the reference library spectra using a least squares method.19–21 A reference library of raw materials was built containing 1000 μm × 1000 μm Raman chemical images of compacts of the pure components. Each chemical image contained 1600 spectra which was averaged to constitute a reference spectrum for each material. The average raw component spectra were used to build a True Component reference library. Application of the True Component linear combination model to the hyperspectral dataset resulted in a component distribution image for each library component. The intensity of each pixel in the distribution image is determined by degree of membership to a particular component by fitting the spectral response at the specific pixel with the reference library spectra. This was given an arbitrary value between 0 and 1, where a score value of 0 represents the absence of a component in a pixel and a score value of 1 demonstrates 100% presence a component. False-colour chemical images were obtained by combining the classification images for each component.
Chemical images were prepared using ISys® 5.0 chemical imaging software. All datasets were treated with Partial Least Squares Discriminant Analysis (PLS-DA) II. A reference library of raw materials was built containing 1000 μm × 1000 μm NIR chemical images of compacts of the pure components. Each chemical image contained 1600 spectra which was used to build a PLS classification model. Application of the respective PLS model to the chemical images of the sample resulted in a classification score image for each library component. The intensity of each pixel in the classification score image is determined by degree of membership to a particular class (component) by comparing the spectral response at the specific pixel with the reference library spectra. False-colour chemical images were obtained by the combining the classification images for each component.
Quantitative domain size statistics
Quantitative domain size statistics were calculated using FIJI22 image processing software. Binary images of the individual components were obtained by applying a suitable threshold to the single component distribution maps. The ‘Analyze Particles’ command was used to count and measure the domains present in the binary images. The number of domains and the percentage area covered was measured by counting the number of domains present and the proportion of the image covered by domains, respectively. The size of each domain was calculated by measuring the longest distance between any two points within the domain boundary (Feret diameter).Results and discussion
3-Component model system
A three-component model system composed of one API (5% w/w) and two excipients (80% w/w and 15% w/w) was used to simulate a real tablet formulation. The model system was designed to contain low concentrations of API and one excipient to evaluate the ability of each vibrational spectroscopic technique for imaging the spatial distribution of more complex systems that are typical of real tablet formulations. A hydrobromide salt API (C22H27BrN202S), MCC (C14H26O11) and saccharin (C7H5NO3S) were chosen for the individual components due to each material differing in their chemical nature and elemental composition and thus could be uniquely identifiable by both spectroscopy and SEM-EDX analysis. The LDIR, Raman and NIR reference spectra of each component is shown in Fig. 1.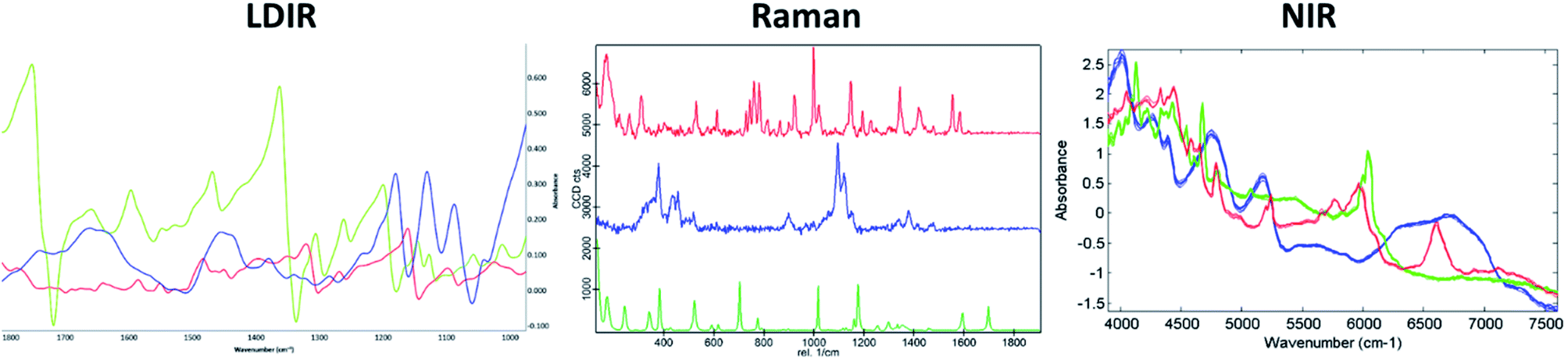 | ||
| Fig. 1 The LDIR (left), Raman (centre) and NIR (right) spectra of hydrobromide salt API (red), microcrystalline cellulose (blue) and saccharin (green). | ||
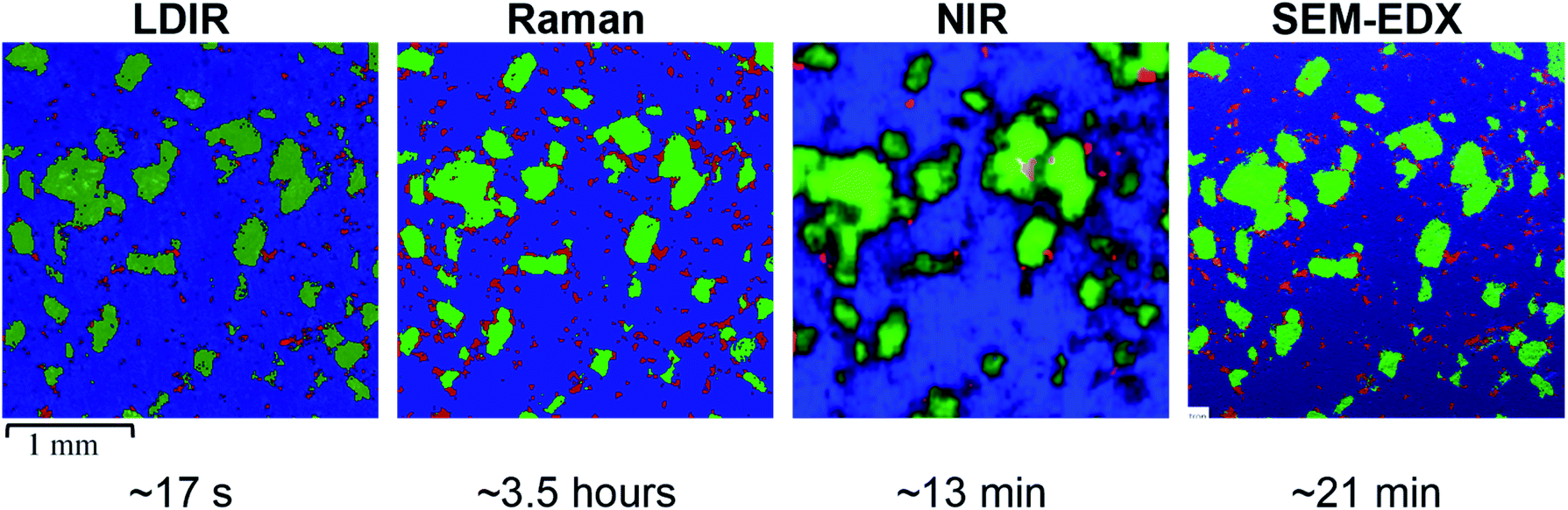 | ||
| Fig. 2 (Left) LDIR, (left centre) Raman, (right centre) NIR and (right) SEM-EDX chemical images of the 3-component model system, where blue = microcrystalline cellulose, green = saccharin and red = hydrobromide salt API. Each chemical image represents a 3000 μm × 3000 μm area of sample and the total data collection time for each technique is displayed below the respective chemical image. | ||
Initial inspection of the images reveals a comparable spatial distribution of components for the LDIR, Raman and SEM-EDX images. There is a notable difference in the quality of the image obtained by NIR compared with the other techniques. In our previous work, we found the combination of the lower spatial resolution offered by commercial NIR instruments with the lack of confocality results in pixelated images with ill-defined domain boundaries.9 Up until now the relatively rapid data collection times compared with Raman mapping has provided NIR as a popular tool to rapidly characterise the spatial distribution of components within tablets.
Closer inspection of the LDIR, Raman and SEM-EDX chemical images reveals very similar domain size and shape of the excipient components however there appears to be some variation in the number and size of API domains. A higher number of larger API domains are present in the Raman image compared with the LDIR data. The distribution of API within the SEM-EDX map is more comparable with the Raman data however there still remains a subtle variation such that despite the position and shape of the domains remain the same, the domains appear slightly smaller in the SEM-EDX data.
As suggested previously, the size, shape and distribution of the MCC and saccharin domains appear comparable across the LDIR, Raman and SEM-EDX images. The biggest variation is seen in the size and distribution of the API domains. Overall, the Raman and SEM-EDX data appear most comparable to one another, however there are some subtle differences in the size of the API domains. The domains present in the Raman data appear to be slightly overestimated. Due to the selection rules for Raman spectroscopy, highly conjugated molecules are characteristically strongly Raman active.23 Many APIs contain a high level of conjugation and thus Raman spectroscopy is usually the preferred technique for API detection.6,24 The API used in the model formulation is a highly conjugated salt and thus exhibits a relatively stronger Raman response compared with the major excipient (MCC) which is an aliphatic organic material. The difference in the Raman signal intensity across the two components may result in mixed pixels on the API-MCC domain boundary being incorrectly assigned. If a spectrum contains spectral bands from both the hydrobromide salt API and MCC, the higher intensity peaks from the API molecule may overpower the relatively weaker MCC bands resulting in the pixel being classified as API even if in reality the sample spot contains a higher proportion of MCC. As a result, the concentration of API in the Raman data is slightly overestimated as the domains appear somewhat larger than their absolute true size. The difference in the position of the domain boundary is limited by the spatial step size of the experiment and therefore for this study the maximum increase in domain radius will be 10 μm. The other excipient in the formulation, saccharin, is also a highly conjugated molecule and exhibits a strong Raman response however, there doesn't appear to be an overestimation in the Raman data. Saccharin exists as relatively large domains in the tablet matrix compared with the small particle size of the API and thus there will be a smaller number of pixels which are likely to be mixed components. To improve the discrimination of the API domains, the spatial step size could be decreased, however this will increase the data collection time by an order of magnitude which will reduce the applicability of Raman mapping. The subtle differences in the size of the API domains with the relatively faster acquisition time is a good compromise to still obtain high-definition chemical images of the spatial distribution of components within tablets.
For the LDIR data, there appears to be more variation in the distribution of API compared with the Raman map. Comparison of the chemical images provided by LDIR and SEM-EDX reveals that there are a number of the small API domains absent in the LDIR data. The size of the domains present is also underestimated in the LDIR image and thus this consequently causes some variation in the domain shape. Although both Raman and IR spectroscopy involve the interaction of radiation with molecular vibrations, the selection rules of transitions differ and as a result molecular bonds that are highly Raman active tend to have a poor IR response and vice versa.25 Generally, X–H σ-bonds are good examples of strong IR-active vibrations. Many excipients are σ-bonded organic systems and therefore usually exhibit a strong IR response. As a result, for this formulation, the organic excipients exhibit a stronger IR signal relative to the highly aromatic API. Each pixel in the LDIR component distribution maps represent the ratio of the peak and baseline position chosen for each component in the formulation. For this particular formulation, there was no obvious unique peak for the hydrobromide salt API component and instead an alternative position on the spectral band was chosen as the unique peak wavenumber (refer to Table 2). Consequently, the difference in the intensity of the peak and baseline position is relatively small and thus in combination with the relatively poorer IR response from the API it may be more challenging to detect using this method. The requirement for this method to have unique spectral bands for each component does pose challenges for this approach, however despite this the resultant LDIR image does provide a comparable distribution of components and produces data at an order of magnitude faster than all other techniques.
As previously stated, there are notable differences in the size and distribution of domains for all components in the NIR data relative to all the other techniques. The NIR data provides poor discrimination between domains of each component and thus does not provide an accurate representation of the size and shape of each domain. The distribution of the organic excipients (MCC and saccharin) is generally comparable to the other techniques, however the poor discrimination between components results in an overestimation of the size of these domains. However, for the smallest and lowest concentration component, hydrobromide salt API, the NIR data has not been able to spectrally identify all the domains that are present in the images obtained by the other techniques. The NIR data is collected using a poorer spatial resolution of 25 μm relative to the 10 μm used for the Raman and LDIR experiments and thus it is likely that the majority of the API domains are below the limit of detection for this method. The API used in the model formulation is a hydrobromide salt and has a characteristically relatively weaker NIR spectrum relative to the other components which are organic excipients. As previously discussed in our earlier work,9 NIR has a deeper depth of penetration and thus it is possible that the weak NIR response of the API from the sample surface is being overpowered by the stronger response from the excipients in the tablet core resulting in the large ill-defined MCC and saccharin domains.
| LDIR | Raman | NIR | SEM-EDX | |
|---|---|---|---|---|
| Number of domains | 195 | 437 | 23 | 386 |
| % area covered | 1.8 | 6.9 | 0.9 | 5.5 |
| Average Feret diameter/μm | 41.2 | 55.8 | 87.1 | 52.8 |
As suggested previously, the size and distribution of the API domains in the Raman image are most comparable to the SEM-EDX data. The average Raman Feret diameter is slightly overestimated compared with the SEM-EDX value, however, only varies by 3 μm and thus is relatively insignificant. The biggest variation is in the number of domains present. The larger number of domains in the Raman data is most likely due to the API exhibiting a relatively stronger Raman scattering compared with the major excipient (MCC) and therefore will dominate the spectrum of a mixed component pixel even if the API is not the highest concentration component. Despite this, this, the Raman data still exhibits a similar % area covered (+1.4%) to the SEM-EDX data which is also consistent with the formulation.
A bigger variation is demonstrated in the LDIR data. There are fewer API domains which are of a smaller size compared with the SEM-EDX data resulting in an underestimation of the percentage area covered (−3.7%). The average Feret diameter provided by the LDIR image is underestimated by 11.6 μm relative to the SEM-EDX data. There is a more notable variation in the number of domains such that the LDIR image contains only ∼50% of the domains present in the SEM-EDX image. As discussed previously, the combination of the relatively weaker IR absorbance of the API with the small intensity difference of the API peak and baseline intensity makes API detection more challenging for this technique. Comparison of the LDIR and NIR quantitative statistics demonstrates the LDIR data does provide a more accurate representation of the sample and offers an improvement of both data collection time and image quality compared with NIR chemical imaging.
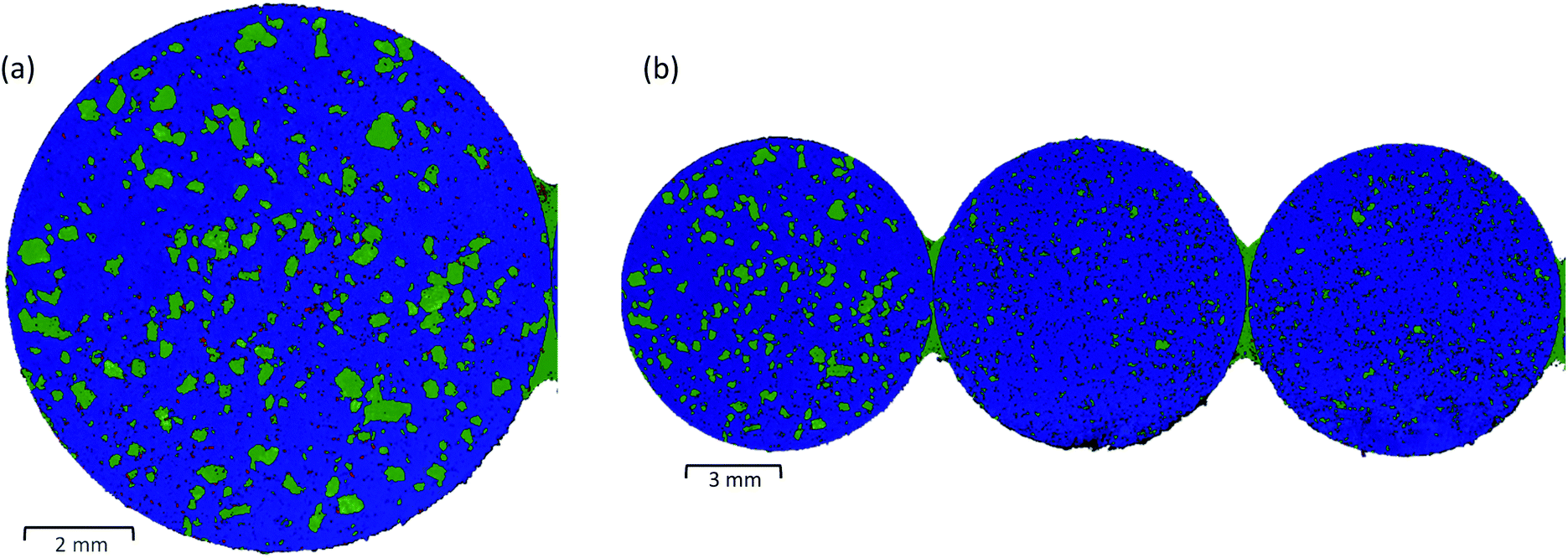 | ||
| Fig. 3 LDIR chemical images of (a) a full single tablet and (b) three full tablets of the 3-component model system, where blue = microcrystalline cellulose, green = saccharin and red = hydrobromide salt API. | ||
The speed of LDIR imaging enables a single microscope slide to be examined at a time. Depending on the tablet size, it is possible to mount multiple tablets and examine all samples in a single experiment, eliminating the need to repeat the same experimental setup for each specimen. Fig. 3(b) displays a single chemical image for three tablets of the three-component model system collected using the same experimental parameters (10 μm XY spatial step size, total data collection time = ∼9 minutes). For this experiment, model tablets were produced using two different grades of saccharin. The LDIR image revealed a distinct difference in the saccharin domain size for the first tablet (grade 1) compared with the second and third tablets (grade 2) and demonstrates that the system could be a powerful tool for manufacturing process understanding to rapidly characterise differences in the size and spatial distribution of components for formulations that have been processed under different manufacturing routes.
Pharmaceutical tablet
To further evaluate the relative capabilities of each vibrational spectroscopic mapping technique for pharmaceutical analysis, a real pharmaceutical tablet was examined. A commercially available tablet used to treat migraines was chosen for this study due to the formulation containing common excipients at concentrations typically used in tablet formulations. Each tablet contained 80 mg of a hydrobromide salt API. Excipients in the core included MCC, lactose monohydrate, croscarmellose sodium and magnesium stearate. The LDIR, Raman and NIR reference spectra of each component are shown in Fig. 4.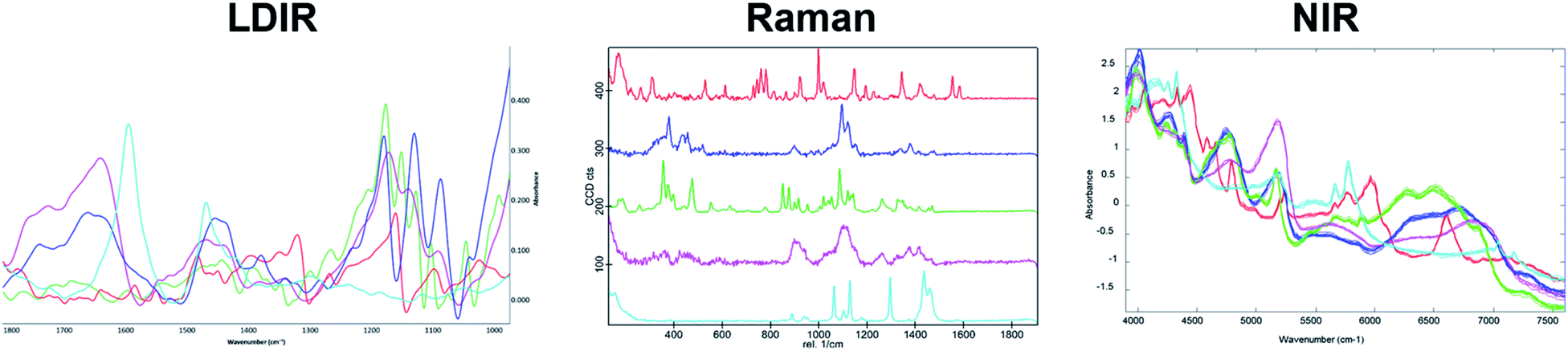 | ||
| Fig. 4 The LDIR (left), Raman (centre) and NIR (right) spectra of the raw materials in the pharmaceutical tablet formulation where red = hydrobromide salt API, blue = microcrystalline cellulose, green = lactose monohydrate, magenta = croscarmellose sodium and cyan = magnesium stearate. | ||
Magnesium stearate is a commonly used lubricating agent in many formulations and typically displays a film-forming propensity on the surfaces of the API and excipient domains within the drug matrix.26 The combination of a low concentration (1.25% w/w) with, under normal circumstances, a lack of large domains makes it very difficult to identify spectral bands corresponding to magnesium stearate within imaging datasets.27 Advanced multivariate analysis approaches are usually required to attempt to identify spectral bands corresponding to magnesium stearate within imaging spectra, with success dependent on the particular sample matrix analysed.27,28 For this formulation, no spectral bands corresponding to magnesium stearate could be identified by any of the four imaging techniques, including SEM-EDX analysis, suggesting the magnesium stearate is so well dispersed it is below the limit of detection for all techniques. For the LDIR imaging experiment, magnesium stearate was removed from the reference library to avoid obtaining ambiguous or misleading component distribution maps where noise could be mistaken for the signal of magnesium stearate.
SEM-EDX was used as a comparison tool however due to the sample being a real formulation, each material did not exhibit unique atoms and thus MCC and lactose monohydrate could not be distinguished from one another. As these are the major components, SEM-EDX was used as a tool to map the size and distribution of the minor components (hydrobromide salt API and croscarmellose sodium).
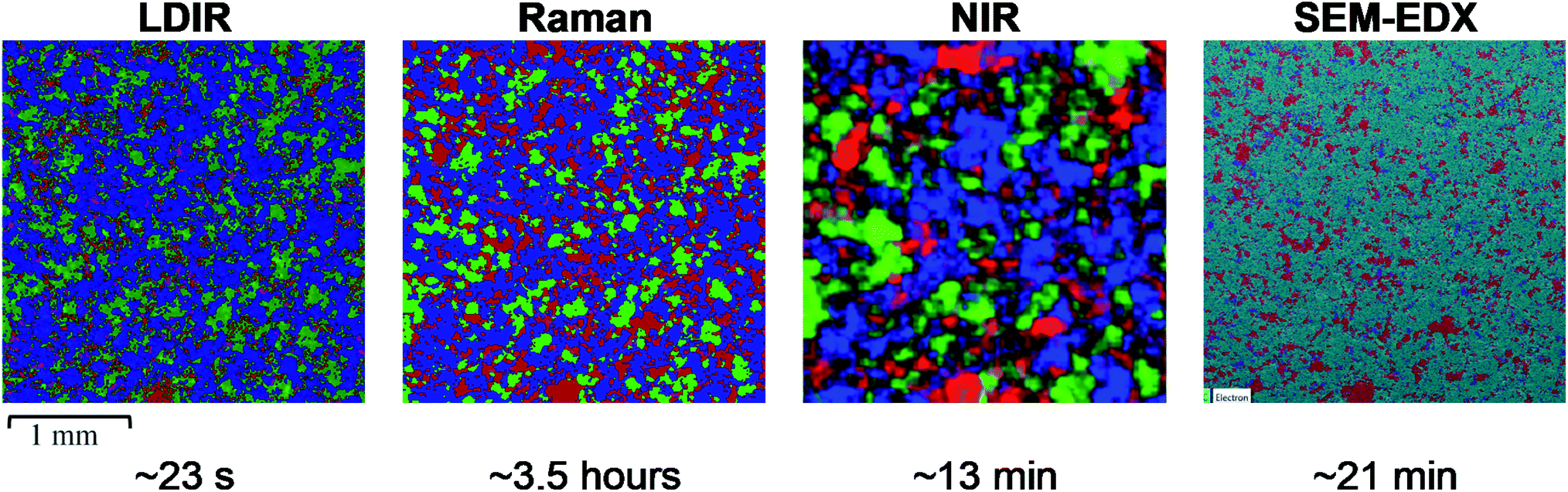 | ||
| Fig. 5 (Left) LDIR, (left centre) Raman, (right centre) NIR and (right) SEM-EDX chemical images of the pharmaceutical tablet, where blue = microcrystalline cellulose, green = lactose monohydrate, red = hydrobromide salt API and magenta = croscarmellose sodium. For the SEM-EDX image cyan = MCC and lactose monohydrate as due to their chemical nature, they could not be individually identified. Each chemical image represents a 3000 μm × 3000 μm area of sample and the total data collection time for each technique is displayed below the respective chemical image. | ||
Initial inspection of the chemical images reveals a similar distribution of components for the LDIR, Raman and SEM-EDX data. Similarly, to the three-component model system data, the size and distribution of API domains appear to be slightly overestimated in the Raman data and underestimated in the LDIR data compared with the SEM-EDX map. Some variation is also seen in the size and shape of the lactose monohydrate domains. In the Raman data, the lactose monohydrate domains appear to exist as discrete, well-resolved domains, whereas there is less discrimination between the domains in the LDIR image. Unfortunately, due to the chemical nature of the components in the formulation, SEM-EDX was unable to discriminate between the MCC and lactose monohydrate components and thus was unable to provide a gold standard reference for these components. Despite this, the variation shown across the Raman and LDIR images is relatively small and both images still demonstrate a similar distribution of components. As expected from previous studies, there are notable differences in the size and distribution of components provided by the NIR image and further demonstrates the superiority of the LDIR, Raman and SEM-EDX imaging techniques with regards to obtaining high-definition chemical distribution maps of tablet systems.
Both the Raman and LDIR chemical images provide a comparable spatial distribution of the MCC component. A similar size and shape of domains can be seen across both images. A larger variation is demonstrated for the lactose monohydrate component. As discussed previously, the domains present in the Raman data exist as well resolved, discrete domains, however the LDIR image reveals less discrimination between domains. Although, the exact distribution of lactose monohydrate could not be confirmed using an alternative imaging technique, the poorer discrimination between domains may be due to poorer chemical selectivity provided by the LDIR instrument relative to Raman due to only probing one unique wavenumber for each component. Similarly, to the three-component model system, for the hydrobromide salt API component, the Raman image overestimates its concentration. For this formulation, however, the distribution of API provided by the LDIR imaging appears more comparable with the SEM-EDX map indicating that the reduced chemical selectivity provided by the LDIR for highly conjugated APIs may be more suitable for analysing complex real pharmaceutical formulations containing a higher concentration of API and a greater number of components. Both the Raman and LDIR chemical images display a similar size and distribution of croscarmellose sodium. Comparison with the SEM-EDX map shows the Raman and LDIR images contain a greater number of low intensity pixels which could be noise incorrectly assigned as croscarmellose sodium. Both techniques, however, provide a similar distribution map and thus neither technique provided a superior chemical selectivity for croscarmellose sodium.
Despite the variations discussed above, the distribution of components provided by LDIR is comparable to the Raman data and superior to the NIR chemical image. The ability to achieve the LDIR data at orders of magnitude faster than both techniques indicates LDIR could be a promising imaging technique for pharmaceutical analysis of tablets.
 | ||
| Fig. 6 A LDIR chemical image of a full single tablet of pharmaceutical formulation, where blue = microcrystalline cellulose, green = lactose monohydrate, red = hydrobromide salt API and magenta = croscarmellose sodium. | ||
Conclusions
LDIR imaging has been demonstrated to be a promising technique for rapidly characterising the spatial distribution of components within pharmaceutical tablets. Comparison with other established spectroscopic chemical imaging techniques showed LDIR imaging provides high-definition component distribution maps comparable to Raman and SEM-EDX at orders of magnitude faster. Fluorescence is a major challenge associated with obtaining chemical images using Raman spectroscopy and usually requires spectral pre-processing to remove this component. Non-white samples suffer from an elevated fluorescence contribution and usually results in low-quality spectra. LDIR, NIR and SEM-EDX spectroscopy offer advantages for such samples. LDIR imaging also provides a better discrimination between components compared to NIR chemical imaging, which is routinely used to rapidly characterise the spatial distribution of components within solid oral dosage forms. The LDIR chemical imaging approach provides the opportunity to obtain detailed domain morphology information in a fraction of the time and would be ideal for routine analysis or as a rapid screening tool to characterise differences in the size and spatial arrangement of components for troubleshooting investigations.The requirement that formulation details must be known prior to analysis and each component must exhibit unique spectral bands limits the capabilities of this technique for several applications, such as reverse engineering. LDIR imaging may need to be used in combination with Raman or other traditional spectroscopic mapping techniques that can collect a full spectrum at each position to provide a baseline of the spatial distribution of components within new formulations or out-of-specification tablets.
LDIR imaging enables the full exposed surface of multiple tablet samples to be examined simultaneously within a greatly reduced time frame and provides an exciting opportunity to reduce the long experimental times currently required for 3D vibrational spectroscopic chemical imaging of pharmaceutical tablets. By providing a means to rapidly characterise the spatial distribution of components within a whole tablet system, the effect of each processing condition on the final drug product can be understood and could be used to enhance drug product development.
Data availability statement
All data underpinning this publication are openly available from the University of Strathclyde KnowledgeBase at: https://doi.org/10.15129/1bab3334-79be-4f88-bf3c-2efa6e4ac45f.Author contributions
Hannah Carruthers: conceptualization, formal analysis, investigation, methodology, writing – original draft. Don Clark: conceptualization, supervision, writing – review & editing. Fiona Clark: supervision. Karen Faulds: conceptualization, supervision. Duncan Graham: conceptualization, supervision, writing – review & editing.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
This work was supported by Global Technology and Engineering, Pfizer Global Supply. Agilent is acknowledged for the loan of their LDIR imaging system. David Troiani is acknowledged for providing technical instrument support for the LDIR imaging system.Notes and references
- A. A. Gowen, C. P. O'Donnell, P. J. Cullen and S. E. J. Bell, Eur. J. Pharm. Biopharm., 2008, 69, 10–22 CrossRef CAS PubMed.
- D. Clark, M. Henson, F. LaPlant, S. Šašić and L. Zhang, in Applications of Vibrational Spectroscopy in Pharmaceutical Research and Development, ed. D. E. Pivonka, J. M. Chalmers and P. R. Griffiths, John Wiley & Sons Ltd., Chichester, UK, 2007, pp. 309–335, DOI:10.1002/9780470027325.s8913.
- K. C. Gordon and C. M. McGoverin, Int. J. Pharm., 2011, 417, 151–162 CrossRef CAS PubMed.
- L. R. Hilden, C. J. Pommier, S. I. F. Badawy and E. M. Friedman, Int. J. Pharm., 2008, 353, 283–290 CAS.
- R. C. Lyon, D. S. Lester, E. N. Lewis, E. Lee, L. X. Yu, E. H. Jefferson and A. S. Hussain, AAPS PharmSciTech, 2002, 3, E17 CrossRef PubMed.
- C. Gendrin, Y. Roggo and C. Collet, J. Pharm. Biomed. Anal., 2008, 48, 533–553 CrossRef CAS PubMed.
- D. Clark, M. Henson, F. LaPlant, S. Šašić and L. Zhang, in Handbook of Vibrational Spectroscopy, John Wiley & Sons, London, 2007, pp. 309–335, DOI:10.1002/9780470027325.s8913.
- A. V. Ewing and S. G. Kazarian, Spectrochim. Acta, Part A, 2018, 197, 10–29 CrossRef CAS PubMed.
- H. Carruthers, D. Clark, F. Clarke, K. Faulds and D. Graham, Appl. Spectrosc., 2021, 75, 178–188 CrossRef CAS PubMed.
- H. Carruthers, D. Clark, F. Clarke, K. Faulds and D. Graham, J. Raman Spectrosc., 2022 DOI:10.1002/jrs.6337 , Early View.
- O. Kolomiets, U. Hoffmann, P. Geladi and H. W. Siesler, Appl. Spectrosc., 2008, 62, 1200–1208 CrossRef CAS PubMed.
- C. Ravn, E. Skibsted and R. Bro, J. Pharm. Biomed. Anal., 2008, 48, 554–561 CrossRef CAS PubMed.
- C. Ravn, Near Infrared Chemical Imaging in Formulation Development of Solid Dosage Forms, University of Copenhagen, 2009 Search PubMed.
- M. N. Slipchenko, H. Chen, D. R. Ely, Y. Jung, M. T. Carvajal and J. X. Cheng, Analyst, 2010, 135, 2613–2619 RSC.
- C. M. Hartshorn, Y. J. Lee, C. H. Camp, Z. Liu, J. Heddleston, N. Canfield, T. A. Rhodes, A. R. Hight Walker, P. J. Marsac and M. T. Cicerone, Anal. Chem., 2013, 85, 8102–8111 CrossRef CAS PubMed.
- M. Razeghi, W. Zhou, S. Slivken, Q.-Y. Lu, D. Wu and R. McClintock, Appl. Opt., 2017, 56, H30–H44 CrossRef CAS PubMed.
- K. Yeh, S. Kenkel, J.-N. Liu and R. Bhargava, Anal. Chem., 2015, 87, 485–493 CrossRef CAS PubMed.
- P. Y. Sacré, M. Alaoui Mansouri, C. De Bleye, L. Coïc, P. Hubert and E. Ziemons, Int. J. Pharm., 2022, 612, 121373 CrossRef PubMed.
- I. Benito-González, M. Martínez-Sanz, A. López-Rubio and L. G. Gómez-Mascaraque, J. Raman Spectrosc., 2020, 51, 2022–2035 CrossRef.
- U. Schmidt, S. Hild, W. Ibach and O. Hollricher, Macromol. Symp., 2005, 230, 133–143 CrossRef CAS.
- T. Dieing and W. Ibach, in Confocal Raman Microscopy, ed. J. Toporski, T. Dieing and O. Hollricher, Springer International Publishing, Cham, 2018, vol. 66, pp. 89–120 Search PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld and B. J. N. m. Schmid, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- J. R. Ferraro, K. Nakamoto and C. W. Brown, in Introductory Raman Spectroscopy, ed. J. R. Ferraro, K. Nakamoto and C. W. Brown, Academic Press, San Diego, 2nd edn, 2003, pp. 1–94, DOI:10.1016/B978-012254105-6/50004-4.
- S. Mazurek and R. Szostak, J. Pharm. Biomed. Anal., 2006, 40, 1225–1230 CrossRef CAS PubMed.
- P. J. Larkin, in Infrared and Raman Spectroscopy, ed. P. J. Larkin, Elsevier, 2nd edn, 2018, pp. 1–5, DOI:10.1016/B978-0-12-804162-8.00001-X.
- G. Morin and L. Briens, AAPS PharmSciTech, 2013, 14, 1158–1168 CrossRef CAS PubMed.
- S. Šašiċ, P. Ojakovo, M. Warman and T. Sanghvi, Appl. Spectrosc., 2013, 67, 1073–1079 CrossRef PubMed.
- J. Li and Y. Wu, Lubricants, 2014, 2, 21–43 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ay00471b |
| ‡ The exact objective used in the LDIR imaging system could not be disclosed by Agilent Technologies. |
| This journal is © The Royal Society of Chemistry 2022 |