
Paving the way towards continuous biosensing by implementing affinity-based nanoswitches on state-dependent readout platforms
Annelies
Dillen
 and
Jeroen
Lammertyn
*
and
Jeroen
Lammertyn
*
KU Leuven, Department of Biosystems – Biosensors Group, Willem de Croylaan 42, Box 2428, 3001, Leuven, Belgium. E-mail: jeroen.lammertyn@kuleuven.be
First published on 15th February 2022
Abstract
Continuous biosensors provide real-time information about biochemical processes occurring in the environment of interest and are therefore highly desirable in research, diagnostics and industrial settings. Although remarkable progress has been made in the field of biosensing, most biosensors still rely on batch processes and, thus, are not suited to perform continuous measurements. Recently, however, it has been shown that by combining affinity-based nanoswitches with state-dependent readout platforms, the necessity for batch processes can be overcome and affinity-based continuous biosensing can be achieved. In this review, we first provide an overview of affinity-based continuous biosensing and discuss the required components to achieve this goal. More specifically, we summarize the strategies that have been applied to develop and tune both protein and nucleic acid-based switches, as well as readout strategies that can be applied in combination with the former. Afterwards, biosensors in which both elements were already integrated and hence enabled continuous measurements are reviewed. We also discuss the challenges and opportunities associated with each approach and therefore believe this review can help to encourage and guide future research towards continuous biosensing.
 Annelies Dillen | Annelies Dillen obtained a Master in Bioscience engineering (KU Leuven) in 2018 for which she submitted a thesis on the development of digital assays for single exosome detection in cancer diagnostics. In the same year she started her PhD in the Biosensors group in the division Mechatronics, Biostatistics and Sensors (MeBioS) of the Biosystems department at KU Leuven, where she is currently working on fiber optic SPR sensors combined with DNA nanotechnology, to develop a new concept for continuous biosensing. |
 Jeroen Lammertyn | Jeroen Lammertyn is full professor in bionanotechnology at the KU Leuven. He founded the Biosensors group in 2005. The group closely follows the emerging field of food and medical diagnostics and is active in bio-assay development, optical biosensors, and microfluidics. He is (co-)author of >300 peer reviewed research papers, conference papers and book chapters. He acts as reviewer for many international peer reviewed journals, is co-inventor of 10 patent applications and founder of the spin-off company FOx Biosystems. He is also chairman of the DIATECH 2014 and 2020 International Conferences on Novel technologies for in vitro Diagnostics in Leuven. |
1. Introduction
Real-time, continuous biosensing is gaining more and more attention in healthcare,1,2 biotechnology,3,4 and food and beverage processing,5,6 as it provides instant and continuous information about biochemical processes occurring in the environment of interest, thereby allowing direct and even feedback-controlled intervention.7 Furthermore, by continuously monitoring target molecules in their respective environment, the necessity for batch measurements can be overcome, which is often a tedious and time-consuming process, as samples frequently have to be withdrawn, processed, transported to the sensing setup, and analyzed before information about the target concentration can be obtained.7 Although remarkable progress has been made in the field of biosensing since the development of the first biosensor in 1956,8 most biosensors do not satisfy the requirements for continuous biosensing, being that they (i) must not require externally added reagents, (ii) cannot generate products, (iii) cannot rely on multistep processes such as washing, incubation or regeneration steps, (iv) must exhibit a dynamic response and (v) must be stable for a long time period in the environment of interest.7,9 Glucose and other enzymatic sensors are the best-known examples of biosensors that are capable of continuously monitoring specific target concentrations,10 but as their detection strategy relies on the reactivity of the target itself, it cannot be applied for non-reactive targets.7,9,11 Affinity-based biosensors on the other hand, represent a more general approach for biosensing. However, they generally rely on multistep processes including washing, incubation or regeneration steps to translate the binding of an analyte to the bioreceptors into a detectable output, which limits their use for continuous biosensing.7,9 Recently, it was shown that by combining affinity-based nanoswitches with a state-dependent readout platform, the aforementioned limitations could be overcome and affinity-based continuous biosensing could be achieved.11–17 Therefore, in this review, we will discuss both crucial components separately and will explain how these can be applied to develop affinity-based continuous biosensors.First, we explain the concept of affinity-based nanoswitches in terms of continuous biosensing and provide an overview of the approaches to transform affinity-binders into the former. Second, we review readout strategies that are suitable to be applied in combination with the affinity-based nanoswitches, to develop an integrated biosensor for continuous monitoring. Finally, we discuss the platforms that have already integrated both elements, thereby overcoming the abovementioned challenges and enabling continuous biosensing.
2. Elements to enable affinity-based continuous biosensing
2.1. Affinity-based nanoswitches
Affinity-based nanoswitches are entities, composed of affinity-binders, that undergo a reversible conformational change upon target binding. Due to the conformational change, target binding can directly be translated into a detectable output (by using a state-dependent readout platform as will be explained in the next section), without multistep processes or exogenous reagents. Furthermore, as this conformational change is reversible, the switch can react to changing target concentrations and a dynamic response can thus be achieved.7,9 In this manner, the abovementioned limitations can be overcome and therefore, affinity-based nanoswitches are key elements for continuous biosensors. The development and use of these switches is inspired by nature, where switching molecules sense and react to biological cues in a variety of ways. The amount of targets for which naturally occurring switches exist is however limited, and therefore, strategies have been described to develop synthetic switches from affinity binders, which we subdivide here in the following categories.18–20The first approach is the development of a clamp, as shown in Fig. 1A and 2A, for which two affinity-binders are tethered together, so the switch will shift from a loose to a more rigid state when both receptors are bound to the target. In order to develop a switch using this approach for multivalent targets, two copies of the same binder can be used, as they can bind to different subunits of the target molecule.21 For monovalent targets however, either two affinity binders that can bind to distinct epitopes of the target molecule, or one affinity binder recognizing the target molecule and a second one binding to the target-receptor-complex, are required.18,20,22 This strategy is highly modular and thus allows for straightforward design of affinity-based nanoswitches.21 However, as small ligands cannot easily be detected using clamping due to their limited recognition domains, affinity-based nanoswitches have also been engineered based on competition, which relies on the attachment of a competitor to the bioreceptor (Fig. 1B and 2B). The competitor will bind to the target recognition zone, so when the target is present, it will compete with and lead to the displacement of the competitor and thus conformational change of the switch. Next to the modularity and generalizability of this approach, the overall affinity of the switch can be rationally tuned without requiring changes to the native bioreceptor itself, by adjusting the binding strength of the competitor. However, by using a competitor, the overall affinity of the target towards the switch will be lower than its affinity for the native bioreceptor.18,20,23–26 Lastly, synthetic switches can be developed by introducing an alternative folding, as depicted in Fig. 1C and 2C. In this strategy, the affinity-binder needs to be adapted so it can fold into two well-defined structures, of which one is stabilized by target binding. As for this approach, the bioreceptor itself needs to be tailored to undergo a conformational change upon target binding, the design process is often challenging and requires empirical optimization, which is why this strategy is less generally applicable.18,20,24,25,27
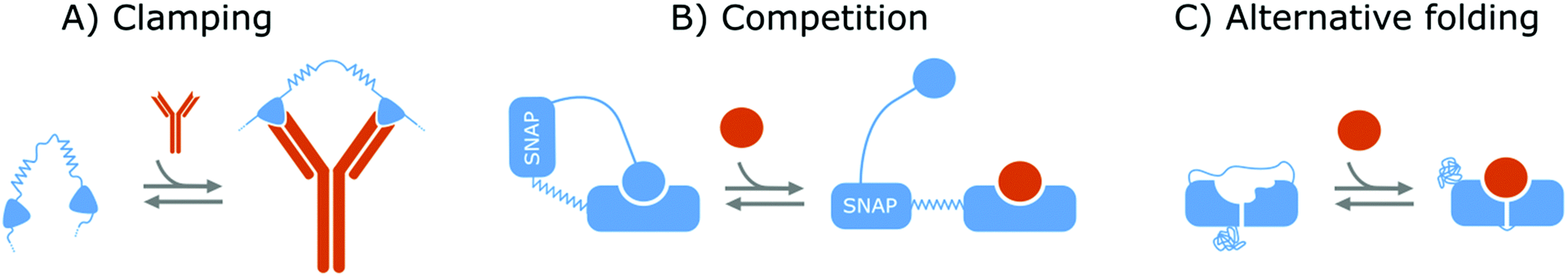 | ||
| Fig. 1 Strategies to create protein switches. (A) Clamping: Two protein binders are tethered together via a flexible linker. When the target is present, both will bind the target and a rigid structure will be formed. (B) Competition: The protein binder is linked to an intramolecular ligand via a SNAP-tag and stiff linker. In this manner, the target will compete with the ligand, leading to its displacement and thus an extended conformation of the protein switch. (C) Alternative folding: Part of the protein binder is duplicated and linked to its opposite end so the protein can fold into two distinct conformations. Furthermore, a mutation is inserted into the duplicated part and as a result, the target can only bind one of both conformations. In this way, the protein binder can switch between its two conformations of which one is stabilized by target binding. A protein switch can also be developed via alternative folding by destabilizing the protein structure, so folding is only achieved in presence of the target molecule. For all strategies, the switches are depicted in blue while the target molecules are portrayed in orange. | ||
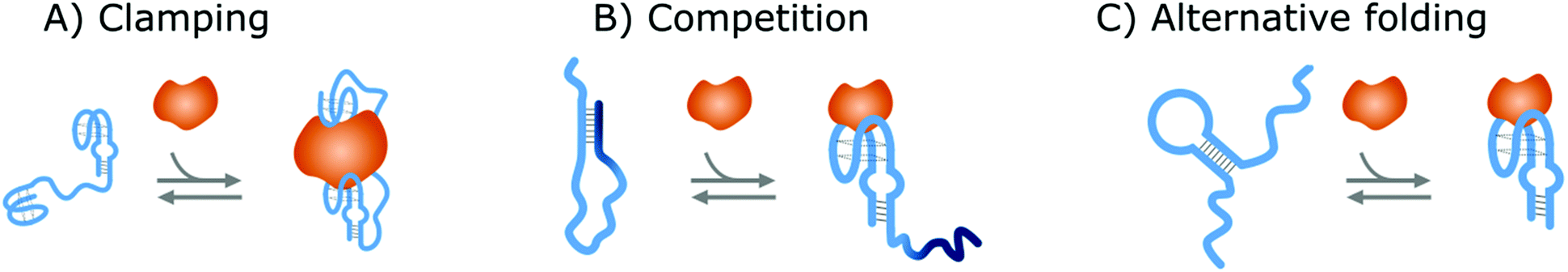 | ||
| Fig. 2 Strategies to create NA switches. (A) Clamping: Two NA receptors are tethered together via a flexible linker. When the target is present, both will bind the target and a more rigid structure will be formed. (B) Competition: The NA receptor (light blue) is linked to a NA competitor (dark blue). In this way, the target will compete with the competitor, leading to its displacement and thus a conformational change of the NA switch. (C) Alternative folding: The secondary structure of the NA receptor to which the target binds is destabilized or an alternative folding of the receptor is introduced. In this manner, the NA receptor can switch between its two conformations of which one is stabilized by target binding. For all strategies, the switches are depicted in blue while the target molecules are portrayed in orange. | ||
Using these strategies, both protein and nucleic acid (NA) switches have been developed. As both molecules are structurally and chemically different, the engineering of protein or NA switches requires a different expertise and is even conducted in different research fields (i.e. protein engineering and DNA nanotechnology, respectively). Therefore, the abovementioned strategies to create either protein or NA switches will be discussed in more detail separately. Afterwards, the development of NA–protein hybrid switches will be discussed shortly.
Importantly, for affinity-based switches suitable for continuous monitoring, all components of the switch need to be linked together, as no components can be added as external reagents. Therefore, we will only discuss the switches that fulfill this requirement.
To engineer such protein switches, the abovementioned strategies have been widely applied. For instance, protein-based clamps have been constructed for antibody detection by the group of Maarten Merkx by connecting two copies of the respective antibody binding epitope, as presented in Fig. 1A. This construct was developed for antibodies against HIV (anti-HIV1-p17),29–32 EGFR (cetuximab),30,33 hemagglutinin31,32 and dengue virus (type I),31,32 thereby demonstrating the suitability of this approach for antibody detection. Notably, the binding epitopes were connected by a glycine-serine linker, which are commonly applied flexible spacers, and are therefore desirable to develop a protein clamp that switches between a flexible and rigid state upon target binding.34 The abovementioned strategy was also applied by Adamson et al. by connecting two Affimer proteins35 that bind to human C-reactive protein (a protein biomarker), Herceptin (a therapeutic antibody) or the cow pea mosaic virus (a plant virus) respectively, thereby further demonstrating the generalizability of this approach to develop protein switches for multivalent targets.21 Moreover, protein clamps have also been developed for various peptides, by sandwiching them between two distinct peptide binding interfaces that were selected through directed interface evolution.36–38 Additionally, a Zn(II)-binding clamp was developed by connecting two metal-binding domains (Atox1 and WD4).39–41 Altogether, clamping proved to be a straightforward and general strategy to develop protein switches for multivalent targets, but remains less widespread for small and monovalent targets, due to the need for two distinct epitopes and affinity binders.
Therefore, protein switches have also been engineered based on competition as depicted in Fig. 1B. To achieve this, the group of Kai Johnsson covalently attached protein binders to their ligands via an intramolecular tether and SNAP-tag (a commercially available self-labeling protein tag), so when the target molecule is present, the intramolecular ligand is displaced by the target and the protein switch adopted an extended conformation.42 Importantly, to ensure an extended conformation upon target binding, the linker between the binder and SNAP tag should not be too flexible. Therefore, although glycine/serine linkers proved useful for the development of clamps, polyproline linkers were applied in this format, as they are more rigid and thus better suited for the development of competitive protein switches. Furthermore, the intramolecular tethers should be long enough to allow binding of the intramolecular ligand and thus closing of the switch. Another important consideration is the affinity of the protein binder for its intramolecular ligand, as this should be high enough to ensure the closed state is predominant in the absence of target molecules, whereas it should be low enough to allow competition with the respective target, for the desired target concentration range. As a result, the overall affinity of the protein switch for its target depends on the affinity between the applied protein binder and the intramolecular ligand as well as the affinity between the protein binder and its target, of which the first can be tuned to adjust the concentration range to which the switch will respond. Using this strategy, the research group of Kai Johnsson was able to develop protein switches for the detection of small molecules such as glutamate,43 acetylcholine,44 sulfonamides,42,45 γ-aminobutyric acid46 as well as for clinically relevant drugs including anti-epileptics (topiramate),47 immunosuppressants (sirolimus, tacrolimus and cyclosporine A),47 anti-arrhythmics (digoxin),47 anticancer agents (methotrexate),47,48 bronchodilators (theophylline)48 and drugs against malaria (quinine).48
Furthermore, as many naturally occurring protein switches are based on alternative folding, this strategy has also been applied to develop their synthetic counterparts.18–20 In particular, to engineer protein switches using alternative folding, either the structure of the protein can be destabilized, so the protein only folds upon target binding,49 or the topology of the polypeptide chain of the respective protein binder itself can be altered, so the protein can fold into two distinct conformations, of which one is stabilized by target binding. For instance, Kevin Plaxco and coworkers inserted switching properties in the FynSH3 protein using the first alternative folding strategy. More specifically, they truncated the respective protein (four-residue carboxy-terminal truncation), which led to the destabilization of FynSH3 in absence of the target molecule. As a result, the population FynSH3 successfully shifted from an unfolded to a folded conformation upon binding of the VSL12 peptide target.49,50 To induce an alternative folding using the latter strategy, the research group of Stewart N. Loh duplicated part of the protein that recognizes the target molecule and attached it to the opposite side of the protein allowing its folding either into its native or alternative conformation. Furthermore, they inserted a mutation in the duplicated segment, so target molecules could only bind to the native state and as a result, an equilibrium was formed between the two conformational states of the protein binder, of which one (native) was stabilized by binding of the target molecule,28,51–54 as presented in Fig. 1C. Using this approach, protein binders were successfully converted into protein switches for the detection of Ca(II)51,53 and ribose.54
Notably, when engineering protein switches, most often the reporter molecules and switching properties are engineered into the protein binder simultaneously. However, as the respective labels depend on the applied readout strategy, they will be discussed separately in section 2.2.
Although the broad chemical diversity of amino acids provides a wide functionality to proteins, which enabled the development of protein switches with various in- and outputs, due to this diversity, the behavior of proteins cannot be easily predicted. As a result, the development of protein switches can be challenging and commonly relies on extensive empirical optimization and is often limited to binders for which structural information is available.18,20
In fact, NA switches have been engineered by tethering two aptamers or single-stranded NA (ssNA) probes together, via a flexible linker. This was achieved either through covalent attachment, using DNA,22,56–59 RNA,60 PEG,59,61 phosphoramidite linkers62 or larger scale structures,11 or through hybridization of ssNA linkers.63 In this manner NA clamps were successfully formed that switched from a flexible to a more rigid state upon target binding,64–66 as shown in Fig. 2A. Using this clamping strategy, NA switches have been developed for protein targets including the hepatitis C virus non-structural protein 3,60 vitronectin,63 thrombin11,22,56–58,61,62 and vascular endothelial growth factor22,59 as well as DNA targets.11 Although the design and development of NA clamps proved to be straightforward, their application remains limited and is mostly applied to thrombin, as this is the best-known example for which two well established aptamers targeting distinct epitopes are available.67
In contrast to clamping, competition is a widespread strategy for the development of NA switches from aptamers, as the predictability of Watson–Crick basepairing provides a straightforward approach to design and tune a competitor, consisting of ssNA that is partially complementary to the target binding region of the aptamer,23,26 as presented in Fig. 2B. Importantly, in contrast to many protein switches developed via competition, the competitors of these NA switches thus do not consist of the respective (modified) target itself. These switches were termed duplexed aptamers in a previous review paper by Munzar et al. (2019)23 and have been developed by linking the competitor NA strands and aptamers again either covalently using ssNA spacers68–96 or PEG,97–100 or via hybridization to NA linkers101,102 or even DNA origami structures.103 Using this strategy, NA switches have been developed for the detection of various targets including thrombin,68,71–74,102,104 protein tyrosine kinase-7,74–78,100 immunoglobin heavy mu chain,89 nucleolin,86,103 theophylline,88 platelet-derived growth factor (BB),84,85 lysozyme,81,82 IFN-Y,69,80,90 TNF-α,69 IgE101 ATP,70,87,91–99 AMP,79,81 ADP83 and GTP.83 This broad availability proves the generalizability of this approach for a wide range of targets. Note that these only represent the duplexed aptamers in which the competitor is linked to the aptamer itself, but that many more duplexed aptamers have been described in which the competitor is added as an external reagent which further demonstrates their widespread application.23,26,105 However, the latter do not fulfill the requirements for continuous biosensing and thus will not be discussed further. Importantly, as was also the case for protein switches developed by competition, the binding strength between the competitor and NA receptor needs to be carefully tuned. Therefore, in our previous work, we developed a model that predicts how the competitor affects the final switching properties of duplexed aptamers.26 This model allows to rationally tune the performance of duplexed aptamers and thereby reduce the amount of empirical optimization required to develop new NA switches from NA bioreceptors based on competitive displacement. Furthermore, although most competitive NA switches were developed starting from a previously described aptamer, Nutiu et al. adapted the standard SELEX procedure (systemic evolution of ligands by exponentially enrichment), which is commonly used for the in vitro selection of aptamers for specific targets, to directly select NA switches based on competition from random sequences. Using this strategy, they successfully selected NA switches for the detection of ATP and GTP.106 Altogether, the development of NA switches based on competitive displacement proved to be a straightforward and general strategy suitable for a variety of target molecules.
Finally, NA switches have been constructed based on the introduction of an alternative fold from both ssNA probes107 and aptamers,24,27 commonly described as molecular beacons or structure-switching aptamers (SSAs) respectively, of which the latter is depicted in Fig. 2C. To develop SSAs, the group of Kevin Plaxco either destabilized the tertiary structure of the respective aptamer or introduced an alternative fold, similar as was described for protein switches.27 Notably, to do this, the tertiary structure of the aptamer needs to be known. Using this strategy, they were able to develop NA switches for the detection of proteins,108,109 drugs12,15–17,110–117 and other small molecules,118 including thrombin,108 the TATA binding protein (transcription factor),109 doxorubicin,12,17,112,113 kanamycin,12,17,114 tobramycin,16,17,115,117 gentamycin,17 irinotecan,110 vancomycin,111 cocaine15,112,114 and melamine.118 Although SSAs have successfully been developed using this strategy, the engineering process remains unpredictable and is mostly based on trial and error, thereby undermining the widespread adaptation of this type of NA switch.23 Molecular beacons are commonly applied to detect NAs, due to their ease in design and development, as all interactions are based on hybridization. They were first described by Tyagi and Kramer in 1996![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 119 and are composed of a ssNA probe equipped with hairpin formation sequences on each end. In this manner, a hairpin loop is formed in absence of the NA target, which opens when the respective target sequence binds. This format has been widely applied throughout the years and proven its applicability for the detection of NA.120–128
119 and are composed of a ssNA probe equipped with hairpin formation sequences on each end. In this manner, a hairpin loop is formed in absence of the NA target, which opens when the respective target sequence binds. This format has been widely applied throughout the years and proven its applicability for the detection of NA.120–128
Altogether, NAs are suitable components for the development of affinity-based nanoswitches due to their ease of synthesis and use, and their highly predictable thermodynamic behavior.23,129,130 Furthermore, due to the progress in aptamer selection processes (SELEX), the amount of NA receptors for various targets keeps increasing.131–133 Consequently, NA switches have already been widely applied for bioassay development55,134,135 and play an important role in the development of continuous biosensors.
In a similar format as the protein clamps developed by the group of Maarten Merkx (shown in Fig. 1A), the group of Francesco Ricci developed an NA–protein hybrid switch for the detection of the anti-HIV and anti-flag antibodies as presented in Fig. 3A. Therefore, they designed a DNA hairpin with ssDNA tails, that are hybridized with ssDNA equipped with the ligands of the respective antibodies. In this manner, when the target antibodies are present, they will bind to their respective ligands and thereby open the DNA hairpin.138 With this format, they could achieve the same advantages as discussed in section 2.1.1. for the protein clamps developed by the group of Maarten Merkx. On top of that, their format allows (i) straightforward control over the conformational change and (ii) tuning of the switch thermodynamics, by altering the basepairing of the hairpin stem, which further demonstrates the potential of NA–protein hybrid switches.137,138 Furthermore, they showed that this format can be applied for the detection of monovalent targets (anti-Dig Fab fragment and DNA-binding transcription factor TBP), as well, on the condition that the simultaneous binding of two target molecules causes enough steric hindrance to open the DNA hairpin.138
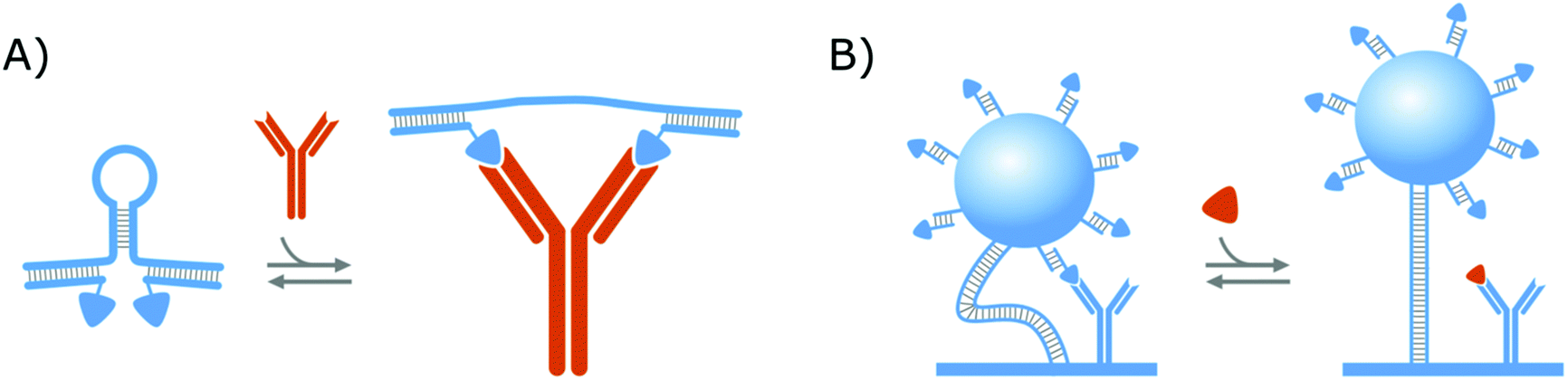 | ||
| Fig. 3 Strategies to create NA–protein hybrid switches. (A) Clamping: Antibody ligands were attached via hybridization to a DNA hairpin. In this manner, when target antibodies were present, they would bind to their respective ligands and the DNA hairpin would be opened. (B) Competition: Switches are composed of microparticles, which were immobilized on the surface via dsDNA tethers. The microparticles were equipped with a (creatinine) competitor via hybridization, whereas the surface was equipped with protein bioreceptors (antibodies). In this manner, when the target molecule (creatinine) was present, they would bind to the protein bioreceptors and the switches became less restricted in their movement. For all strategies, the switches are depicted in blue while the target molecules are portrayed in orange. | ||
Aside from the abovementioned NA–protein hybrid clamps, a competitive hybrid switch has also been developed by the research group of Menno Prins. Herein, DNA was applied both as a tether to link a microparticle to the sensor surface, as well as a linking group, to attach the competitor of the protein bioreceptors,14,139 as shown in Fig. 3B. This switch was developed for creatinine and will be elaborated further in section 3.2, where we will discuss biosensing strategies that applied affinity-based nanoswitches to enable continuous measurements.
The abovementioned examples clearly show how the advantages of proteins and NAs can be combined for the development of affinity-based nanoswitches. Notably, to do this, knowledge about both protein engineering and DNA nanotechnology is required, which might be a reason why the amount of NA–protein hybrid switches remains limited. In summary, although the potential of NA–protein hybrid switches is in our opinion currently still underexplored, we believe that this strategy can further help in broadening the library of available affinity-based nanoswitches and provides a highly versatile and modular technique for the development of affinity-based switches, suitable for continuous biosensing.
with KD[M] the dissociation constant and
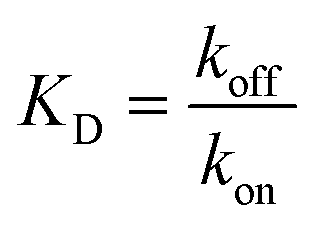
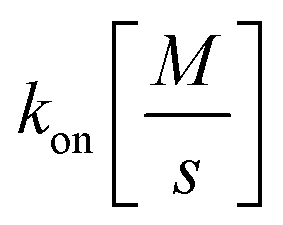 and
and 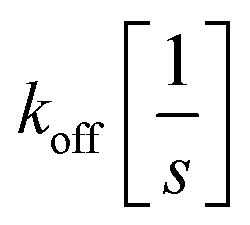 the association and dissociation rate, respectively, a lower dissociation constant is associated with a lower dissociation or higher association rate and thus less dissociation of the switch-target complex.140 Consequently, for conditions in which the reaction rates are the limiting step of the (un)binding, the higher the affinity, the longer it will take for a new equilibrium to be formed and thus the slower the dynamic response. Notably, here, KD represents the overall dissociation constant of the switch for its target, which depends on but is not the same as the dissociation constant of the applied bioreceptor for its target.
the association and dissociation rate, respectively, a lower dissociation constant is associated with a lower dissociation or higher association rate and thus less dissociation of the switch-target complex.140 Consequently, for conditions in which the reaction rates are the limiting step of the (un)binding, the higher the affinity, the longer it will take for a new equilibrium to be formed and thus the slower the dynamic response. Notably, here, KD represents the overall dissociation constant of the switch for its target, which depends on but is not the same as the dissociation constant of the applied bioreceptor for its target.
As both the sensitivity and dynamic response are important parameters for the performance of a continuous biosensor, attention should be paid to this trade-off during the design process of the affinity-based nanoswitches, for which the time-resolution and sensitivity that is required for the respective application should be taken into account. Notably, the choice of the strategy to achieve switching will affect the performance of the continuous biosensor and will even more determine how the abovementioned trade-off can be tuned. For example, when creating an affinity clamp, the overall binding strength between the switch and its target is generally increased compared to the binding strength of the stand-alone bioreceptors due to their increased avidity, which is aside from switching properties a main reason for the development of many clamps.22,64 Furthermore, for both protein and NA clamps, it was shown that by altering the length or stiffness of the linkers that attach both binders, the affinity of the switch can be tuned.34,39,142,143 More specifically, the length and stiffness of the linkers will affect both the magnitude of the conformational change as well as the local effective concentration of the respective binders, which both affect the final signal that is generated for a certain target concentration.34,39 This strategy was applied to enhance the affinity of protein and NA clamps for the detection of Ca(II),144 K(I),145 Zn(II)142 and immunoglobin heavy mu chain,143 by screening different lengths and compositions of glycine-serine and PEG-linkers respectively.142–145 In this manner, the affinity could be enhanced from the pM to fM,142 low to mid nM,144 low to mid mM and low to mid μM143 range for the Zn(II),142 Ca(II),144 K(I)145 and immunoglobin heavy mu chain143 binding switches respectively.
In contrast, by using a competitor, the overall affinity of the resulting competitive switches will be lower than the affinity of the applied native bioreceptor. However, this strategy allows rational and straightforward tuning of the final switch performance, as it was previously described how the final performance of competitive switches can be tuned by altering the binding strength between the bioreceptor and competitor. As a result, the overall affinity of competitive switches can be tuned by adapting the competitor's affinity for the bioreceptor.25,26 For example, Xue et al.48 modified the structure of the intramolecular ligands in competitive protein switches for antibodies against methotrexate, theophylline and quinine, to lower their LOD. They did this by taking advantage of structural information available on the interaction between the antibodies and drugs, and in this manner could specifically remove or shift the position of the groups taking place in the binding.48 As a result they were able to enhance the affinity from low to mid μM range for all three antibodies. Notably, the binding strength between the bioreceptor and competitor is not only dependent on the affinity between both, but also on their local effective concentration, meaning that the length or stiffness of the linker attaching both can also be changed to tune the overall affinity,23 similar as for clamping. In fact, Wilson et al.141 successfully developed a range of competitive NA switches with distinct affinities for the detection of ATP (ranging from 10 μM to 40 mM) by screening both (i) different linker lengths between the aptamer and competitors (ranging from poly T(23) to poly T(43)) and (ii) different lengths of competing NA strands (ranging from 6 to 9 complementary nucleotides).141
Lastly, when an alternative fold is introduced to develop affinity-based nanoswitches, the overall affinity of the switch for the target can be tuned by adjusting the equilibrium between the native and the alternative conformational state of the switch, as both states have a distinct affinity for the target, by stabilizing or destabilizing either state (e.g. by inducing mutations in the oligo or peptide sequence).27,52 This was done by Porchetta et al.,146 who destabilized the folded state of a SSA for cocaine, so the equilibrium would shift more to the unfolded state. Therefore, based on the secondary structure of the SSA, which is predicted to be a three-way junction, they designed three SSA variants in which either (i) mutations were inserted, (ii) the terminal stem was shortened or (iii) the sequence was circularly permutated. Notably, the target binding region was kept conserved throughout the variants to ensure target binding was not hindered. In this manner, they obtained three SSA variants with a distinct, lower affinity (KD of 23, 82 and 1390 μM) compared to the original SSA (KD = 0.5 μM).146 Similarly, as the equilibrium of their FynSH3 protein switch shifted almost completely to the folded state at high ionic strength conditions (present in whole blood), Kurnik et al.49 destabilized the folded state of the respective switch by performing an I50L substitution in the hydrophobic core of the protein. In this manner, they successfully altered the overall affinity and obtained target-responsiveness at high ionic strength conditions.49 Importantly, by introducing an alternative fold, the overall affinity of switches developed through this strategy will be lower compared to the affinity of the applied bioreceptor,147 similar as for competitive switches.
In summary, depending on the final application, particular care should be given to the trade-off between the sensitivity and dynamic response of the resulting biosensor, which will be partly determined by the applied strategy to transform affinity-binders into affinity-based nanoswitches. Importantly, although the affinity of the obtained switches is often characterized, this is not the case for the kinetics and dynamic response. Therefore, in order to apply these switches for continuous measurements, more research is required in the future to characterize and tune the kinetics as well.
Importantly, although we discussed the dynamic response and sensitivity of affinity-based switches, other performance parameters, such as the selectivity, are also important to take into account when developing a continuous biosensor. More specifically, as the selectivity of the affinity-based switches is mainly determined by the selectivity of the applied bioreceptors, care should be taken regarding this parameter when choosing which bioreceptor to apply.148 Notably, as a thorough discussion about the selectivity of affinity-based bioreceptors is out of the scope of this review, it will not be discussed further.
Besides, to increase the stability of NA bioreceptors, both pre- and post-SELEX modification strategies have been described. Notably, although an advantage of NAs compared to proteins is their high stability in various conditions, they are susceptible to nuclease degradation, and therefore, most of the reported modifications are meant to protect the NAs from the latter. More specifically, chemical modifications can be made post-SELEX to the sugar backbone, phosphate linkages or 3′ and/or 5′ ends, which all lead to an improved stability.155,157–162 Furthermore, SELEX can be performed starting from a modified NA library (pre-SELEX), such as xenobiotic nucleic acid (XNA), threose nucleic acid (TNA), locked nucleic acids (LNA), fluoroarabino nucleic acid (FANA) and hexitol nucleic acid (HNA) amongst others, which are more stable against nuclease degradation.163–166 Importantly, as NAs are chemically synthesized, they can be modified easily.157 Notably, not only the inherent stability of NAs, but also the stability of aptamer folding is important, as their tertiary structure is responsible for target recognition. The tertiary structure of NAs is affected by the surrounding conditions such as pH, temperature or ionic strength and therefore, SELEX can be performed in those respective conditions to ensure target recognition in the environment of interest.131,160
We will not provide a detailed description of the abovementioned strategies to improve both protein and NA stability here, as it is out of the scope of this review and already discussed elsewhere.156–161
2.2. State-dependent readout platforms
In the previous section, affinity-based nanoswitches were discussed as an essential tool for continuous biosensing, as they respond to target binding by undergoing a conformational change.7,9 However, in order to translate this conformational change into a measurable output, a readout platform that is able to distinguish the different states of the switch (state-dependent), is required. Furthermore, the switches need to be labeled with reporter groups, depending on the respective readout platform, as will be discussed further. In this manner, a distinct signal will be generated for the bound and unbound state, so the obtained output can be linked to the target concentration.9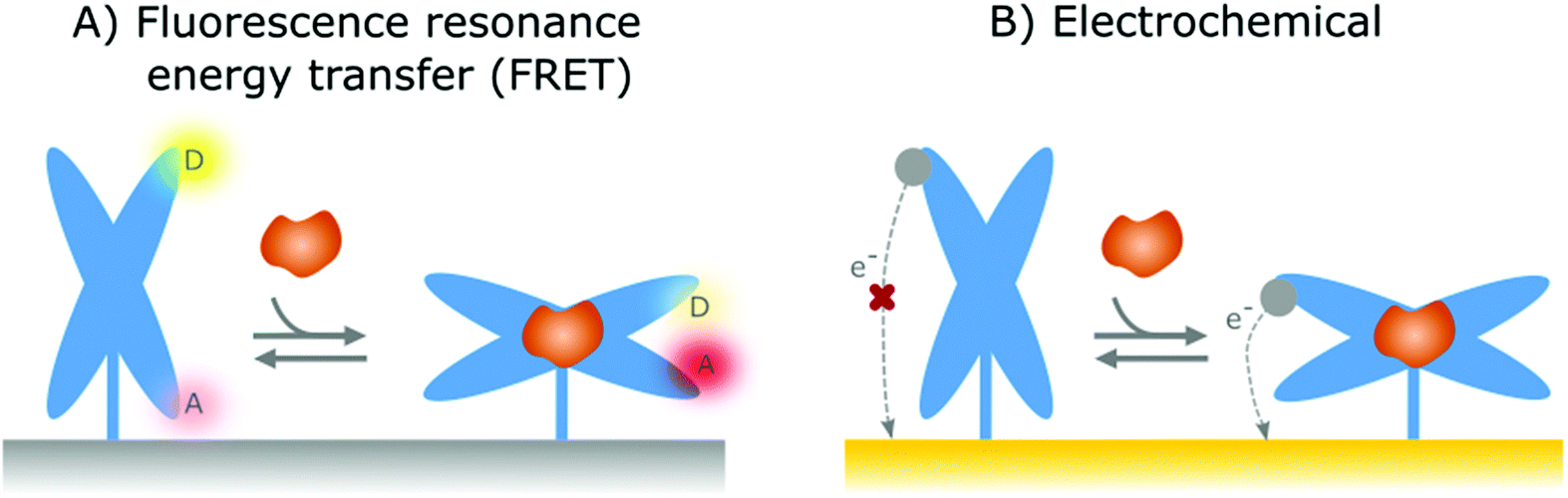 | ||
| Fig. 4 Representation of the two most widely applied state-dependent readout strategies in combination with affinity-based nanoswitches. (A) Fluorescence resonance energy transfer (FRET): by labelling the switches with both a donor and acceptor fluorophore, a distinct fluorescent signal will be generated for the bound and unbound state of the switch, as the distance between both fluorophores gets altered due to the conformational change. Indeed, when the donor and acceptor fluorophores are in close proximity, energy will be transferred from the donor to the acceptor molecule, leading to a fluorescent signal originating from the acceptor molecule. As the energy transfer efficiency is highly dependent on the distance between both fluorophores, the energy transfer will decrease upon increasing distance between both and as a result, the fluorescent signal originating from the donor fluorophore will be released at higher distances. (B) Electrochemical: The affinity-based nanoswitches are labelled with a redox reporter. In this manner, when the switches undergo a conformational change after target binding, the redox reporter will be brought closer to the surface, thereby enhancing the electron transfer efficiency between the reporter and sensor surface and thus generating a detectable signal. | ||
In contrast to FRET, electrochemical readout platforms have already been frequently applied in combination with surface-immobilized, affinity-based nanoswitches (NA and hybrid switches).15–17,68,69,80,90,91,101,102,108–118,125,137,171,172 Using this platform, a state-dependent readout can be obtained by immobilizing the switches on the sensor surface and labelling them with a redox reporter, as depicted in Fig. 4B. Since the electron transfer efficiency between the reporter and sensor surface is highly dependent on the distance between both, a distinguishable signal can be obtained for the bound and unbound state of the respective switches, provided that the conformational change caused by target binding alters the distance between the reporter and sensor surface.27 The respective signal can be obtained using various electrochemical interrogation methods, including square-wave voltammetry,12,16,17,49,69,80,90,101,109–116,118,137,150,173,174 differential pulse voltammetry,68,125,171 alternating current voltammetry,15,102,108 impedance spectroscopy91 and chronoamperometry117 among others. Interestingly, as electrochemical sensors are easy and cheap to manufacture, can be miniaturized, and can easily be combined with further electronic readout and processing, they already have found widespread applications in the field of continuous biosensing.175
In Table 1, the readout strategies that have currently been combined with affinity-based nanoswitches are summarized, together with the applied switching strategy, target molecule, obtained KD and LOD values, as well as the matrices in which measurements were performed. Importantly, only readout strategies for which the switches were attached to the sensor surface are provided, as these are in accordance with the requirements for continuous biosensing applications. As can be seen in this table, this strategy proved to be suitable for the detection of a variety of target molecules including proteins, NA and small molecules in a wide range of complex matrices. Notably, until now, mostly NA switches have been applied, whereas the amount of protein switches that have been attached to a sensor surface remains limited. However, as protein switches already proved to be suitable for real-time intracellular imaging,168 we believe that they are promising for surface-based continuous biosensing as well. Moreover, although many more state-dependent readout platforms have been combined with affinity-based nanoswitches, from this summary, it becomes clear that electrochemical sensors are most widespread in this field. Importantly, not all biosensors listed in Table 1 have been applied for continuous measurements. Indeed, although in theory they consist of the required components to develop a continuous biosensor, this often simply was not the final objective. In particular, reagentless biosensors, intended for batch measurements, are a major achievement in itself, and already entail significant improvements compared to conventional affinity-based assays.20,27 Furthermore, even when all components required for continuous biosensing are combined, important challenges still need to be tackled to turn a reagentless biosensor for batch measurements into a continuous biosensor, being the long-term stability of the sensor and its dynamic response, as discussed in the following and previous sections, respectively. The biosensors indicated in Table 1 that have been applied for continuous measurements will be discussed in more detail in section 3. Notably, the readout strategies summarized in Table 1 are not the only possible choices when developing a continuous biosensor, but only represent an overview of what has been reported in literature. In fact, more already existing strategies or newly developed approaches could be applied for this purpose, on the condition that they provide a distinguishable signal when the switches are in their bound or unbound states respectively.
| Switch strategy | Readout | Target | Batch/continuous | K D | LOD | Matrix | Ref. | |
|---|---|---|---|---|---|---|---|---|
| a The respective matrix in which the KD and LOD values were obtained, in case measurements were performed in multiple matrices. | ||||||||
| Protein | Alternative fold | Square-wave voltammetry | VSL12 | Continuous | 76 ± 4 μM | 2.5 μM | Buffera, whole blood | 49 |
| p85a-2 | Batch | 470 ± 50 μM | — | Buffer | ||||
| NA | Clamping | Surface plasmon resonance | Thrombin | Batch | 0.06 nM | — | Buffer | 176 |
| Particle mobility sensing | Thrombin (bovine) | Continuous | 170 ± 56 nM | — | Buffer | 11 | ||
| Thrombin (human) | 250 ± 60 pM | — | Buffer | |||||
| ssDNA | Continuous | 49 ± 10 pM | — | Buffera, filtered blood plasma | 11 | |||
| 680 ± 268 pM | — | PBS buffer | 139 | |||||
| 524 ± 257 pM | — | Filtered blood plasma | ||||||
| Thickness shear mode acoustic method | Thrombin | Batch | 2.3 ± 0.7 nM | 0.4 nM | Buffer | 177 | ||
| Competition | Differential pulse voltammetry | Thrombin | Batch | — | 11 nM | Buffer | 68 | |
| Alternating current voltammetry | — | Buffer | 102 | |||||
| Square wave voltammetry | IFN-Y | Batch | — | — | Red blood cell-depleted whole blood, cell suspension | 69 | ||
| <60 pM | Red blood cell-depleted whole blood | 80 | ||||||
| 5 ng ml−1 | Cell suspension | 90 | ||||||
| TNF-α | Batch | — | 0.58 nM | Buffera, red blood cell-depleted whole blood, cell suspension | 69 | |||
| IgE | Batch | — | 60 pM | Buffera, 10× diluted serum | 101 | |||
| Impedance spectroscopy | ATP | Batch | — | — | Buffer | 91 | ||
| Particle mobility sensing | ssDNA | Continuous | 150 ± 50 nM | — | Buffer | 14 | ||
| Continuous | 35 ± 9 nM | — | Buffer | 139 | ||||
| Alternative folding | FRET | ssDNA | Batch | — | — | Buffer | 121 | |
| — | 0.1 pmol | Buffer | 122 | |||||
| — | 10 nM | Buffer | 124 | |||||
| ssRNA | — | 100 pM | Buffer | 123 | ||||
| — | — | Intracellular (HeLa cell) | 120 | |||||
| Differential pulse voltammetry | ssDNA | Batch | — | — | Buffer | 125 | ||
| dsDNA | — | 275 pM | Buffer | 171 | ||||
| Alternating current voltammetry | Thrombin | Batch | — | — | Buffer, 2× diluted fetal calf serum | 108 | ||
| Cocaine | Continuous | 100 ± 9 μM | — | Fetal bovine serum | 15 | |||
| Chrono-amperometry | Tobramycin | Continuous | — | — | Rat whole blood (in vivo) | 117 | ||
| Square-wave voltammetry | Doxorubycin | Continuous | 824 ± 18 nM | 10 nM | Buffera, human whole blood, rat whole blood (ex and in vivo) | 12, 17 and 113 | ||
| 69.2 μM | — | Whole blood | 112 | |||||
| Batch | 3.98 μM | — | Serum | |||||
| Kanamycin | Continuous | 920 μM | — | Buffera, rat whole blood (ex and in vivo) | 12, 17, 112 and 114 | |||
| Cocaine | Continuous | 395 μM | — | Whole blood | 112 | |||
| Batch | 182 μM | — | Serum | |||||
| Continuous | — | — | Whole blood | 114 | ||||
| Tobramycin | Continuous | 250 μM | — | Rat whole blood (ex and in vivo) | 16, 17, 115 and 116 | |||
| Irenotican | Continuous | 58 ± 17 μM | — | Buffer | 110 | |||
| 331 ± 57 | Rat whole blood (ex and in vivo) | |||||||
| Gentamycin | Continuous | — | — | Rat whole blood (in vivo) | 17 | |||
| Melamine | Batch | 490 ± 10 μM | — | Buffer | 118 | |||
| Continuous | 463 ± 10 μM | — | Milk | |||||
| Vancomycin | Continuous | 45.5 ± 2.2 μM | — | Rat whole blood (ex and in vivo) | 111 | |||
| Transcription factor (TATA-binding protein) | Batch | 121 ± 20 nM | Buffera, HeLa nuclear extracta | 109 | ||||
| NGAL | Continuous | — | 2/3.5 nM | Urine (artificial/human) | 173 | |||
| Ochratoxin A | Batch | 6.5 ± 0.6 μM | — | Buffer | 150 | |||
| 19 ± 2 μM | — | Unprocessed iced coffee | ||||||
| Phenylalanine | Continuous | — | — | Buffer, whole blood | 174 | |||
| Surface plasmon fluorescence spectroscopy | ssDNA | Batch | — | 500 pM | Buffer | 126 | ||
| Surface-enhanced Raman scattering | ssDNA | Batch | — | — | Buffer | 127 | ||
| NA–protein hybdrid | Clamping | Square wave voltammetry | Anti-DNP antibody | Batch | 3.6 nM | 0.6 nM | Buffer | 137 |
| Anti-Dig antibody | 8 nM | 2 nM | Whole blood | |||||
| Anti-HIV antibody | 33 nM | 9 nM | Whole blood | |||||
| Competition | Particle mobility sensing | Creatinine | Continuous | 25 ± 7 μM | 13.2 μM | Buffer | 14 | |
| 66 ± 8 μM | — | Buffer | 139 | |||||
3. Innovative concepts for continuous biosensing
In the following section, we will discuss biosensors in which affinity-based nanoswitches have been successfully combined with a state-dependent readout platform to achieve continuous measurements of varying target concentrations.3.1. Electrochemical aptamer-based sensors
The electrochemical aptamer-based sensor (EA-B), developed in the research group of Kevin Plaxco, was the first platform to be applied for affinity-based continuous biosensing. Herein, SSAs, labelled with a redox reporter, are applied on an electrochemical sensor (using square wave, alternating current or cyclic voltammetry). These probes undergo a reversible conformational change after target binding, thereby altering the distance between the redox reporter and the biosensor surface, as depicted in Fig. 5A, which provides a dynamic signal, dependent on the target concentration. Using this strategy, continuous monitoring of small molecules, including drugs such as Doxorubicin,12,17,112,113 Kanamycin,12,17 Cocaine,15,112,114 Tobramycin,16,17,115–117 Irenotican,110 Gentamycin,17 and Vancomycin,111 could be achieved, of which an example can be seen in Fig. 5B.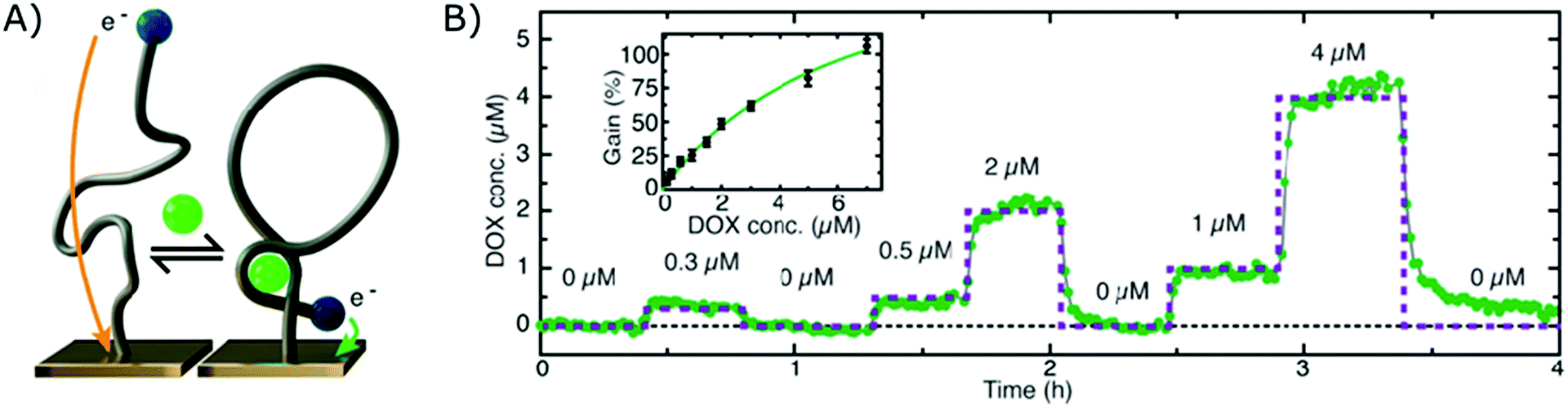 | ||
| Fig. 5 (A) Working principle of the electrochemical aptamer-based sensor (EA-B): SSAs, equipped with a redox reporter, are immobilized on an electrochemical sensor. Upon target binding, the SSAs will undergo a conformational change, thereby bringing the redox reporter closer to the sensor surface. This conformational change enables electron transfer, which provides a detectable signal.12 (B) Dynamic signal obtained by the EA-B platform for Doxorubicin detection. Adapted with permission from ref. 12. Copyright (2013) The American Association for the Advancement of Science. | ||
In their first report on the use of this platform for continuous biosensing in 2009, cocaine concentrations were monitored in undiluted blood serum.15 Since then, much progress has been made to further enhance the performance and application of these sensors, such as the inclusion of a second reporter114 and the application of a biomimetic monolayer to increase the stability of the respective sensor.113 In this manner, they could successfully reduce drift due to fouling of the sensor surface when performing multi-hour measurements in whole blood.113,114 Furthermore, they tuned the affinity of SSAs as elaborated in section 2.1.4. and in this manner could tune, extend and narrow the dynamic range of the final EA-B sensor.146,183,184 Moreover, by using chronoamperometrical interrogation, they achieved measurements with a 300 ms time resolution.119 The group also increased the surface area of the working electrode by roughening the gold surface, which resulted in a higher signal to noise ratio.117 Later, the surface area was further increased by employing nanoporous gold, allowing miniaturization of the sensor.185 Due to these improvements, the EA-B sensor could be applied in situ (in vivo) for multi-hour measurements of therapeutic drug concentrations with (sub)second resolution.110,117 On top of that, to close the gap between drug administration and therapeutic drug monitoring, the EA-B sensor was successfully applied for feedback-controlled drug administration.111 Furthermore, aside from the application of the EA-B sensor for therapeutic drug monitoring, the sensor was also applied to continuously monitor a protein biomarker for acute renal injury (NGAL) in urine173 and toxic substances (melamine, ochratoxin A) for molecular quality control in food and beverage streams.118,150
In 2020, the Plaxco group supplemented their arsenal of SSAs applied on the electrochemical platform for continuous biosensing with a protein switch as well, to further expand the range of targets to be detected. The protein switch was engineered through the inclusion of an alternative fold from the SH3 domain from human Fyn kinase (FynSH3), as discussed in section 2.1.1, so a binding-induced conformational change could be achieved.49
Altogether, the EA-B sensor enables continuous biosensing directly in the environment of interest (in situ) with a fast time-resolution (subsecond). However, as mentioned before, a trade-off between dynamic response and detection limit can generally be expected. Indeed, although the dynamic response of the EA-B sensor is fast, the detection limit of the EA-B sensors is relatively high (μM–mM). This is however not necessarily a limitation, as the detection limit suffices for reported applications of the EA-B sensor.
Currently, there are 2 companies working on the commercialization of the EA-B sensors, being Nanogenecs and Eccrine Systems, Inc.,186 of which the latter is commercializing these sensors for continuous biosensing, and aims to ‘individualize the prescription of medicine with a breakthrough platform for remote, non-invasive, and quantitative medication monitoring through sweat’.186
3.2. Biomarker monitoring by particle mobility sensing
A second biosensor in which affinity-based nanoswitches have been combined with a state-dependent readout system to achieve continuous biosensing, was developed by the group of Menno Prins. The switches applied on their sensor are NA-based affinity clamps, composed of a microparticle tethered to a surface via dsDNA. Both the particle and surface are equipped with NA bioreceptors (ssDNA probes or aptamers), so when the target is present, a sandwich will be formed and the switch will undergo a conformational change, as presented in Fig. 6A. Digital state-change events of the particles are observed by tracking the mobility of each particle through microscopy. More specifically, when the switches are unbound, the particles will move according to Brownian motion and are only restricted in their movement by the dsDNA tether, with which they are linked to the surface. When they are however bound to the target, a sandwich is formed, and the particles are more restricted in their movement as they are linked to the surface both via the tether and the formed sandwich. In this manner, the mobility patterns, which are shown in Fig. 6A for both the bound and unbound state, can be clearly discriminated. Furthermore, by following the activity of the sensor (which is defined by the average amount of state-changes occurring per switch during a certain time-interval), the analyte concentration can be determined and continuously followed, as shown in Fig. 6B.11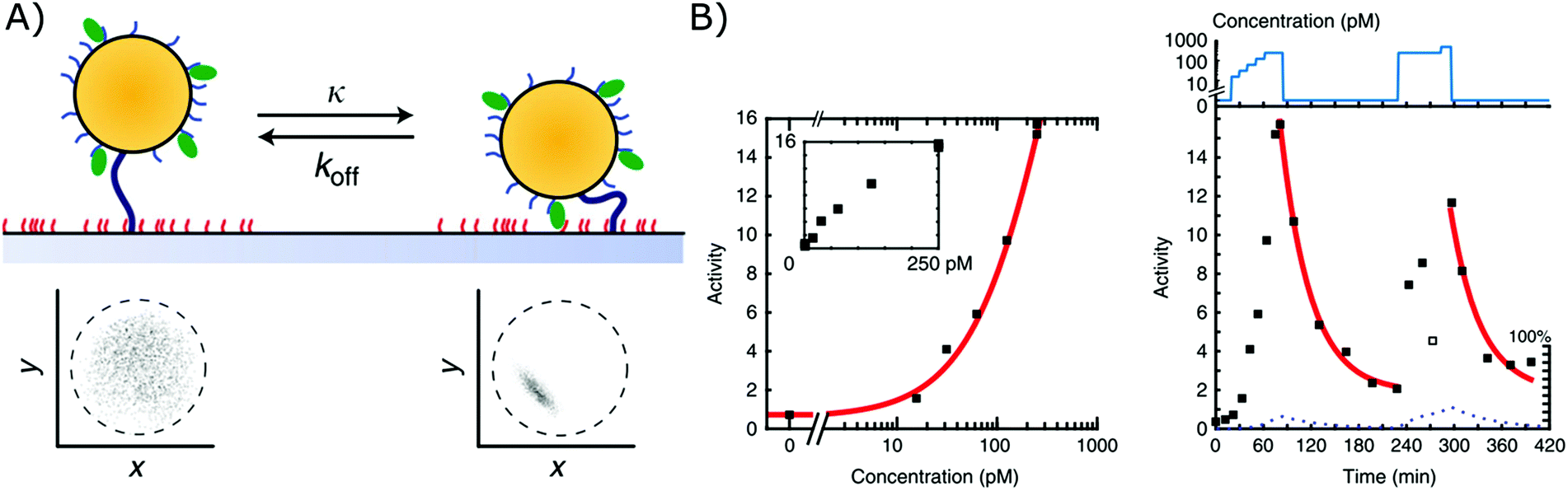 | ||
| Fig. 6 (A) Affinity clamps composed of a microparticle, tethered to a sensor surface, both equipped with bioreceptors (top). The bound and unbound states of the switch can be discriminated through their mobility patterns (below), which are obtained by following the position of a single particle over time.11 (B) The target concentration is linked to the activity of the sensor as shown in the left graph. Herein, the activity is determined as the average amount of state-changes occurring per switch, during a certain time-interval. By following the activity of the sensor over time, a dynamic signal, dependent on the target concentration, can be obtained (right graph). Adapted with permission from ref. 11 (CC BY 4.0).187 | ||
Using this approach, continuous monitoring of both ssDNA and protein (thrombin) targets could be achieved in filtered blood plasma, obtained by applying a porous membrane to the sensor setup (as discussed in section 2.2.2), of which the results can be seen in Fig. 6B for ssDNA detection.11 Furthermore, by utilizing the dissociation rate of each single particle as a signature to identify a kinetic subpopulation, multiplexing of target molecules with a significantly different dissociation rate (ssDNA with different length and sequence) could be achieved.13
Although the proposed combination of the particle mobility sensing platform with affinity clamps proved to be suitable for continuous biosensing of both ssDNA and protein targets, it does not allow detection of small molecules, as the size of such analytes restricts the use of a sandwich conformation. Therefore, similar NA and NA–protein hybrid switches were developed via competition,14 for the detection of both creatinine and a short ssDNA oligo. The switches were again composed of a microparticle linked to the sensor surface. However, only one was equipped with a bioreceptor (antibody or ssDNA probes), whereas the other was linked to the competitor, being (i) a creatinine molecule or (ii) a DNA strand containing the same sequence as the short oligonucleotide target, for the detection of creatinine and DNA respectively. The resulting NA–protein hybrid switch for creatinine was depicted in Fig. 3B. Using these competitive switches, they were able to successfully detect creatinine and DNA with μM and nM detection limits respectively.14 Furthermore, corresponding to the importance of long-term stability of a continuous biosensor as discussed above, they improved the control and durability of their platform by functionalizing the respective switches via click-chemistry to a bottlebrush polymer (PLL-g-PEG).139
Overall, by tracking the mobility of affinity-based switches, continuous biosensing of DNA, proteins and small molecules could be achieved with a low detection limit (pM–μM), due to the single molecule resolution of the platform.11 Importantly, although it was stated before that a lower target affinity generally provides a lower sensitivity, this is not the case for this readout strategy. Indeed, low affinity capture elements can be applied to achieve a low detection limit, as the obtained signal is not determined by the average amount of bound switches for a certain target concentration, but by the amount of state-changes the switches will undergo during a certain time interval. Moreover, information about the binding kinetics could be obtained, which allowed multiplex detection.13 A limitation of the proposed strategy however arises from the fact that the readout strategy is based on microscopy. As a result, measurements cannot be performed in opaque samples (such as whole blood) or directly in the environment of interest (in situ). Rather part of the sample needs to be transported over the sensor and can potentially be transported back to the environment of interest (in-line). Furthermore, in order to determine the activity of the sensor (and thus the target concentration), the state-changes of the switches need to be followed over a certain timeframe and therefore, the time-resolution is limited by the duration this timeframe.11
The biosensor for biomarker monitoring by particle mobility sensing is currently in the process of being commercialized by Helia Biomonitoring, a spin-off company of Eindhoven University of Technology, with a vision of ‘real-time biomolecular sensing for closed-loop control in healthcare and industry’.188
4. Conclusions and future perspectives
Continuous biosensors can provide real-time information about biochemical processes occurring in the environment of interest, which is highly desirable in both research, diagnostic, as well as industry settings. However, the amount of commercially available biosensors for this purpose remain limited and are all based on the enzymatic conversion of the target molecules. Affinity-based biosensors are not dependent on the reactivity of the target itself and can therefore be applied for a wider range of targets, such as DNA, proteins, small molecules etc.7 Recently, it was shown that affinity-based continuous biosensors can be developed by combining affinity-based nanoswitches, with a state-dependent readout platform.11,15Three main strategies have been applied to develop affinity-based nanoswitches from affinity binders, being (i) the development of a clamp,18,22 (ii) linking the affinity binder to a competitor18,23–25 and (iii) the introduction of an alternative fold.18,24,25,27 Using these approaches, both DNA and protein switches have successfully been developed. Importantly, as both components are structurally and chemically different, they require a different expertise and provide distinct opportunities for switch development. Generally, due to its chemical simplicity, the thermodynamic behavior of NA is predictable, which eases the design and development of NA switches.55 On the other hand, as amino acids have a broader chemical diversity than nucleic acids, a broader functionality can be obtained using protein switches.18 Furthermore, by combining both components, their strengths can be combined, and NA–protein hybrid switches can thus be engineered with a broad functionality and high predictability.136,137 However, in our opinion, the potential of hybrid switches currently remains underexplored. Moreover, further improvements of current predictive software for both DNA and protein engineering are required to allow rational tuning of affinity-based nanoswitches and thereby ease their design and development of affinity-based nanoswitches for a specific application in the future.
To convert the conformational change of affinity-based nanoswitches into a detectable signal, they should be combined with a state-dependent readout strategy, which provides a distinct signal for both conformational states of the affinity-based nanoswitches. In this manner, the conformational changes of the switches can be linked to the target concentration.9 Furthermore, as the switches itself cannot be applied as external reagents, due to the requirements for continuous biosensing, all components should be attached to the sensor surface.7,9 Although many affinity-based nanoswitches have already been combined with state-dependent readout platforms, resulting in reagentless biosensors,27 up to now, only 2 research groups have integrated both components for continuous biosensing.11,15 Indeed, although in theory it might seem easy to combine the required components to develop a continuous biosensor, in practice, it takes more than that. More specifically, depending on the final application, the affinity-based nanoswitches must be carefully tuned to obtain the required sensitivity and dynamic response.7,9 Furthermore, the switches and sensor surface must be stable in the environment of interest for the duration of the measurement, so the provided signal does not suffer from drift over time.7,9,148 Although much progress has already been made to overcome each of the aforementioned difficulties, it remains challenging to integrate all solutions into one continuous biosensor. We believe however, that due to the ever-improving fields of protein engineering and DNA nanotechnology and by bringing together expertise from those and other fields, the field of continuous biosensing holds much more potential in the future.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work has received funding from the Research Foundation Flanders (FWO SB/1SC8519N) and European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 955623 (H2020-MSCA-CONSENSE).References
- M. C. Frost and M. E. Meyerhoff, Annu. Rev. Anal. Chem., 2015, 8, 171–192 CrossRef CAS PubMed.
- O. S. Alwan and K. P. Rao, Healthc. Technol. Lett., 2017, 4, 142–144 CrossRef PubMed.
- N. D. Lourenço, J. A. Lopes, C. F. Almeida, M. C. Sarraguça and H. M. Pinheiro, Anal. Bioanal. Chem., 2012, 404, 1211–1237 CrossRef PubMed.
- A. Guerra, M. Von Stosch and J. Glassey, Crit. Rev. Biotechnol., 2019, 39, 289–305 CrossRef CAS PubMed.
- R. Desmarais, D. Anderson, J. Downey, O. Wesstrom, M. Rice, A. Worley, S. Wise, F. Tappen, J. L. Cawley and N. Andersen, Inline monitoring aids in food safety and quality Available online: https://www.foodengineeringmag.com/articles/90659-inline-monitoring-aids-in-food-safety-and-quality (accessed on May 27, 2020).
- J. Meseguer and J. Quevedo, Real-time monitoring and control in water systems in: Real-time monitoring and operational control of drinking-water systems, Springer, 2017 Search PubMed.
- J. Qu, A. Dillen, W. Saeys, J. Lammertyn and D. Spasic, Anal. Chim. Acta, 2019, 1104, 10–27 CrossRef PubMed.
- R. Renneberg, D. Pfeiffer, F. Lisdat and G. Wilson, Frieder Scheller and the Short History of Biosensors in: Biosensing for the 21st Century, Springer, Berlin, 2008 Search PubMed.
- K. W. Plaxco and H. T. Soh, Trends Biotechnol., 2011, 29, 1–5 CrossRef CAS PubMed.
- K. Pandit, Indian J. Endocrinol. Metab., 2012, 16, 263–266 CrossRef PubMed.
- E. W. A. Visser, J. Yan, L. J. Van Ijzendoorn and M. W. J. Prins, Nat. Commun., 2018, 9, 1–10 CrossRef CAS PubMed.
- B. S. Ferguson, D. A. Hoggarth, D. Maliniak, K. Ploense, R. J. White, N. Woodward, K. Hsieh, A. J. Bonham, M. Eisenstein, T. E. Kippin, K. W. Plaxco and H. T. Soh, Sci. Transl. Med., 2013, 5, 213–165 Search PubMed.
- R. M. Lubken, A. M. De Jong and M. W. J. Prins, Nano Lett., 2020, 20, 2296–2302 CrossRef CAS PubMed.
- J. Yan, L. Van Smeden, M. Merkx, P. Zijlstra and M. W. J. Prins, ACS Sens., 2020, 5, 1168–1176 CrossRef CAS PubMed.
- J. S. Swensen, Y. Xiao, B. S. Ferguson, A. A. Lubin, R. Y. Lai, A. J. Heeger, K. W. Plaxco and H. Tom Soh, J. Am. Chem. Soc., 2009, 131, 4262–4266 CrossRef CAS PubMed.
- N. Arroyo-Currás, G. Ortega, D. A. Copp, K. L. Ploense, Z. A. Plaxco, T. E. Kippin, P. Hespanha and K. W. Plaxco, ACS Pharmacol. Transl. Sci., 2018, 1, 110–118 CrossRef PubMed.
- N. Arroyo-Currás, J. Somerson, P. A. Vieira, K. L. Ploense, T. E. Kippin and K. W. Plaxco, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 645–650 CrossRef PubMed.
- V. Stein and K. Alexandrov, Manifesting Synth. Biol. Synth., 2015, 33, 101–110 CAS.
- A. Vallée-Bélisle and K. W. Plaxco, Curr. Opin. Struct. Biol., 2010, 20, 518–526 CrossRef PubMed.
- H. Adamson and L. J. C. Jeuken, ACS Sens., 2020, 5, 3001–3012 CrossRef CAS PubMed.
- H. Adamson, M. O. Ajayi, E. Campbell, E. Brachi, C. Tiede, A. A. Tang, T. L. Adams, R. Ford, A. Davidson, M. Johnson, M. J. Mcpherson, D. C. Tomlinson and L. J. C. Jeuken, ACS Sens., 2019, 4, 3014–3022 CrossRef CAS PubMed.
- H. Hasegawa, K.-I. Taira, K. Sode and K. Ikebukuro, Sensors, 2008, 8, 1090–1098 CrossRef CAS PubMed.
- J. D. Munzar, A. Ng and D. Juncker, Chem. Soc. Rev., 2019, 48, 1390–1419 RSC.
- T. A. Feagin, N. Maganzini and H. T. Soh, ACS Sens., 2018, 3, 1611–1615 CrossRef CAS PubMed.
- H. Farrants, J. Hiblot, R. Griss and K. Johnsson, Rational Design and Applications of Semisynthetic Modular Biosensors: SNIFITs and LUCIDs in: Synthetic Protein Switches, 2017 Search PubMed.
- A. Dillen, W. Vandezande, D. Daems and J. Lammertyn, Anal. Bioanal. Chem., 2021, 413, 4739–4750 CrossRef CAS PubMed.
- L. R. Schoukroun-Barnes, F. C. Macazo, B. Gutierrez, J. Lottermoser, J. Liu and R. J. White, Annu. Rev. Anal. Chem., 2016, 9, 163–181 CrossRef CAS PubMed.
- M. M. Stratton and S. N. Loh, Protein Sci., 2011, 20, 19–29 CrossRef CAS PubMed.
- M. V. Golynskiy, W. F. Rurup and M. Merkx, ChemBioChem, 2010, 11, 2264–2267 CrossRef CAS PubMed.
- Y. Ni, R. Arts and M. Merkx, ACS Sens., 2018, 4, 20–25 CrossRef PubMed.
- R. Arts, I. Den Hartog, S. E. Zijlema, V. Thijssen, S. H. E. Van Der Beelen and M. Merkx, Anal. Chem., 2016, 88, 4525–4532 CrossRef CAS PubMed.
- K. Tomimuro, K. Tenda, Y. Ni, Y. Hiruta, M. Merkx and D. Citterio, ACS Sens., 2020, 5, 1786–1794 CrossRef CAS PubMed.
- M. Van Rosmalen, Y. Ni, D. F. M. Vervoort, R. Arts, S. K. J. Ludwig and M. Merkx, Anal. Chem., 2018, 90, 3592–3599 CrossRef CAS PubMed.
- E. Ceballos-Alcantarilla and M. Merkx, Understanding and applications of Ser/Gly linkers in protein engineering in: Linkers in Biomacromolecules, Academic Press Inc., 2021, vol. 647 Search PubMed.
- C. Tiede, R. Bedford, S. J. Heseltine, G. Smith, I. Wijetunga, R. Ross, D. Alqallaf, A. P. E. Roberts, A. Balls, A. Curd, R. E. Hughes, H. Martin, S. R. Needham, L. C. Zanetti-Domingues, Y. Sadigh, T. P. Peacock, A. A. Tang, N. Gibson, H. Kyle, G. W. Platt, N. Ingram, T. Taylor, L. P. Coletta, I. Manfield, M. Knowles, S. Bell, F. Esteves, A. Maqbool, R. K. Prasad, M. Drinkhill, R. S. Bon, V. Patel, S. A. Goodchild, M. Martin-Fernandez, R. J. Owens, J. E. Nettleship, M. E. Webb, M. Harrison, J. D. Lippiat, S. Ponnambalam, M. Peckham, A. Smith, P. K. Ferrigno, M. Johnson, M. J. McPherson and D. C. Tomlinson, eLife, 2017, 6, 1–35 CrossRef PubMed.
- J. Huang, K. Makabe, M. Biancalana, A. Koide and S. Koide, J. Mol. Biol., 2009, 392, 1221–1231 CrossRef CAS PubMed.
- J. Huang, A. Koide, K. Makabe and S. Koide, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6578–6583 CrossRef CAS PubMed.
- J. Huang and S. Koide, ACS Chem. Biol., 2010, 5, 273–277 CrossRef CAS PubMed.
- S. J. Aper and M. Merkx, ACS Synth. Biol., 2016, 5, 698–709 CrossRef CAS PubMed.
- E. M. W. M. Van Dongen, L. M. Dekkers, K. Spijker, E. W. Meijer, L. W. J. Klomp and M. Merkx, J. Am. Chem. Soc., 2006, 128, 10754–10762 CrossRef CAS PubMed.
- J. L. Vinkenborg, T. J. Nicolson, E. A. Bellomo, M. S. Koay, G. A. Rutter and M. Merkx, Nat. Methods, 2009, 6, 737 CrossRef CAS PubMed.
- M. A. Brun, K.-T. Tan, E. Nakata, M. J. Hinner and K. Johnsson, J. Am. Chem. Soc., 2009, 131, 5873–5884 CrossRef CAS PubMed.
- M. A. Brun, K.-T. Tan, R. Griss, A. Kielkowska, L. Reymond and K. Johnsson, J. Am. Chem. Soc., 2012, 134, 16 CrossRef PubMed.
- A. Schena, K. Johnsson, A. Schena and K. Johnsson, Angew. Chem., Int. Ed., 2014, 53, 1302–1305 CrossRef CAS PubMed.
- M. A. Brun, R. Griss, L. Reymond, K.-T. Tan, J. Piguet, R. J. R. W. Peters, H. Vogel and K. Johnsson, J. Am. Chem. Soc., 2011, 133, 16235–16242 CrossRef CAS PubMed.
- A. Masharina, L. Reymond, D. Maurel, K. Umezawa and K. Johnsson, J. Am. Chem. Soc., 2012, 134, 56 CrossRef PubMed.
- R. Griss, A. Schena, L. Reymond, L. Patiny, D. Werner, C. Tinberg, D. Baker and K. Johnsson, Nat. Chem. Biol., 2014, 10, 598–604 CrossRef CAS PubMed.
- L. Xue, Q. Yu, R. Griss, A. Schena and K. Johnsson, Angew. Chem., Int. Ed., 2017, 56, 7112–7116 CrossRef CAS PubMed.
- M. Kurnik, E. Z. Pang and K. W. Plaxco, Angew. Chem., Int. Ed, 2020, 59, 1–5 CrossRef PubMed.
- J. E. Kohn and K. W. Plaxco, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10841–10845 CrossRef CAS PubMed.
- M. M. Stratton, D. M. Mitrea and S. N. Loh, ACS Chem. Biol., 2008, 3, 723–732 CrossRef CAS PubMed.
- J.-H. Ha and S. N. Loh, Construction of Allosteric Protein Switches by Alternate Frame Folding and Intermolecular Fragment Exchange in: Synthetic Protein Switches Methods and Protocols, Humana Press, 2017 Search PubMed.
- A. J. Degrave, J.-H. Ha, S. N. Loh and L. T. Chong, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed.
- J.-H. Ha, S. A. Shinsky and S. N. Loh, Biochemistry, 2013, 52, 600–612 CrossRef CAS PubMed.
- S. G. Harroun, C. Prévost-Tremblay, D. Lauzon, A. Desrosiers, X. Wang, L. Pedro and A. Vallée-Bélisle, Nanoscale, 2018, 10, 4607–4641 RSC.
- K. M. Ahmad, Y. Xiao and H. Tom Soh, Nucleic Acids Res., 2012, 40, 11777–11783 CrossRef CAS PubMed.
- J. Müller, D. Freitag, G. Mayer and B. Pötzsch, J. Thromb. Haemostasis, 2008, 6, 2105–2112 CrossRef PubMed.
- J. Müller, B. Wulffen, B. Pötzsch and G. Mayer, ChemBioChem, 2007, 8, 2223–2226 CrossRef PubMed.
- C. Riccardi, D. Musumeci, C. Platella, R. Gaglione, A. Arciello and D. Montesarchio, Int. J. Mol. Sci., 2020, 21, 2–24 CrossRef.
- T. Umehara, K. Fukuda, F. Nishikawa, M. Kohara, T. Hasegawa and S. Nishikawa, J. Biochem., 2005, 137, 339–347 CrossRef CAS PubMed.
- Q. W. Hughes, B. T. Le, G. Gilmore, R. I. Baker and R. N. Veedu, Molecules, 2017, 22, 1–8 CrossRef PubMed.
- Y. Kim, Z. Cao and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5664–5669 CrossRef CAS PubMed.
- C. H. Stuart, K. R. Riley, O. Boyacioglu, D. M. Herpai, W. Debinski, S. Qasem, F. C. Marini, C. L. Colyer and W. H. Gmeiner, Mol. Ther. – Nucleic Acids, 2016, 5, 1–9 Search PubMed.
- C. Riccardi, E. Napolitano, D. Musumeci and D. Montesarchio, Molecules, 2020, 25, 7–15 CrossRef PubMed.
- D. Musumeci and D. Montesarchio, Pharmacol. Ther., 2012, 136, 202–215 CrossRef CAS PubMed.
- M. Vorobyeva, P. Vorobjev and A. Venyaminova, Molecules, 2016, 21, 14–16 CrossRef PubMed.
- B. Deng, Y. Lin, C. Wang, F. Li, Z. Wang, H. Zhang, X.-F. Li and X. C. Le, Anal. Chim. Acta, 2014, 837, 1–15 CrossRef CAS PubMed.
- G. S. Bang, S. Cho and B.-G. Kim, Biosens. Bioelectron., 2005, 21, 863–870 CrossRef CAS PubMed.
- Y. Liu, T. Kwa and A. Revzin, Biomaterials, 2012, 33, 1–18 CrossRef PubMed.
- V. C. Özalp and T. Schäfer, Chem. – Eur. J., 2011, 17, 9893–9896 CrossRef PubMed.
- N. Hamaguchi, A. Ellington and M. Stanton, Anal. Biochem., 2001, 294, 126–131 CrossRef CAS PubMed.
- L. Xue, X. Zhou and D. Xing, Anal. Chem., 2012, 84, 3507–3513 CrossRef CAS PubMed.
- X. Shi, J. Wen, Y. Li, Y. Zheng, J. Zhou, X. Li and H.-Z. Yu, ACS Appl. Mater. Interfaces, 2014, 6, 21788–21797 CrossRef CAS PubMed.
- H. Shi, X. He, K. Wang, X. Wu, X. Ye, Q. Guo, W. Tan, Z. Qing, X. Yang and B. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3900–3905 CrossRef CAS PubMed.
- Y.-M. Wang, Z. Wu, S.-J. Liu and X. Chu, Anal. Chem., 2015, 87, 6470–6474 CrossRef CAS PubMed.
- S. Lin, W. Gao, Z. Tian, C. Yang, L. Lu, J.-L. Mergny, C.-H. Leung and D.-L. Ma, Chem. Sci., 2015, 6, 4284–4290 RSC.
- L. Li, Q. Wang, J. Feng, L. Tong and B. Tang, Anal. Chem., 2014, 86, 5101–5107 CrossRef CAS PubMed.
- X. Zhang, K. Xiao, L. Cheng, H. Chen, B. Liu, S. Zhang and J. Kong, Anal. Chem., 2014, 86, 5567–5572 CrossRef CAS PubMed.
- G. Pelossof, R. Tel-Vered, J. Elbaz and I. Willner, Anal. Chem., 2010, 82, 4396–4402 CrossRef CAS PubMed.
- Y. Liu, J. Yan, M. C. Howland, T. Kwa and A. Revzin, Anal. Chem., 2011, 83, 8286–8292 CrossRef CAS PubMed.
- C. Teller, S. Shimron and I. Willner, Anal. Chem., 2009, 81, 9114–9119 CrossRef CAS PubMed.
- C. Chen, J. Zhao, J. Jiang and R. Yu, Talanta, 2012, 101, 357–361 CrossRef CAS PubMed.
- C. Furutani, K. Shinomiya, Y. Aoyama, K. Yamada and S. Sando, Mol. BioSyst., 2010, 6, 1569–1571 RSC.
- X. Tan, Y. Wang, B. A. Armitage and M. P. Bruchez, Anal. Chem., 2014, 86, 10864–10869 CrossRef CAS PubMed.
- L. Yang, C. W. Fung, E. J. Cho and A. D. Ellington, Clin. Microbiol. Rev., 2006, 79, 3329 Search PubMed.
- F. J. Hernandez, L. I. Hernandez, A. Pinto, T. Schäfer and V. C. Özalp, Chem. Commun., 2013, 49, 1285–1287 RSC.
- R. M. Dirks and N. A. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15275–15278 CrossRef CAS PubMed.
- S. Xie and S. P. Walton, Anal. Chim. Acta, 2009, 638, 213–219 CrossRef CAS PubMed.
- J. F. Georges, X. Liu, J. Eschbacher, J. Nichols, M. A. Mooney and A. Joy, PLoS One, 2015, 10, 1–12 CrossRef PubMed.
- Q. Zhou, T. Kwa, Y. Gao, Y. Liu, A. Rahimian and A. Revzin, Lab Chip, 2014, 14, 276 RSC.
- T. Goda and Y. Miyahara, Biosens. Bioelectron., 2011, 26, 3949–3952 CrossRef CAS PubMed.
- R. E. Armstrong and G. F. Strouse, Bioconjugate Chem., 2014, 25, 1769–1776 CrossRef CAS PubMed.
- S. Liu, Y. Wang, C. Zhang, Y. Lin and F. Li, Chem. Commun., 2013, 49, 2337 Search PubMed.
- J. Zhu, L. Zhang, Z. Zhou, S. Dong and E. Wang, Chem. Commun., 2014, 50, 3323 Search PubMed.
- X. Cong and M. Nilsen-Hamilton, Biochemistry, 2005, 44, 7945–7954 CrossRef CAS PubMed.
- Y. Shen, W. Chiuman, J. D. Brennan and Y. Li, ChemBioChem, 2006, 7, 1343–1348 CrossRef CAS PubMed.
- Z. Tang, P. Mallikaratchy, R. Yang, Y. Kim, Z. Zhu, H. Wang and W. Tan, J. Am. Chem. Soc., 2008, 130, 11268–11269 CrossRef CAS PubMed.
- C. Wu, T. Chen, D. Han, M. You, L. Peng, S. Cansiz, G. Zhu, C. Li, X. Xiong, E. Jimenez, C. James Yang and W. Tan, ACS Nano, 2013, 7, 5724–5731 CrossRef CAS PubMed.
- L. J. Nielsen, L. F. Olsen and V. C. Ozalp, ACS Nano, 2010, 4, 4361–4370 CrossRef CAS PubMed.
- H. Shi, D. Li, F. Xu, X. He, K. Wang, X. Ye, J. Tang and C. He, Analyst, 2014, 139, 4181–4184 RSC.
- B. Jiang, F. Li, C. Yang, J. Xie, Y. Xiang and R. Yuan, Anal. Chem., 2015, 87, 3094–3098 CrossRef CAS PubMed.
- Y. Xiao, B. D. Piorek, K. W. Plaxco and A. J. Heeger, J. Am. Chem. Soc., 2005, 127, 17990–17991 CrossRef CAS PubMed.
- S. Li, Q. Jiang, S. Liu, Y. Zhang, Y. Tian, C. Song, J. Wang, Y. Zou, G. J. Anderson, J.-Y. Han, Y. Chang, Y. Liu, C. Zhang, L. Chen, G. Zhou, G. Nie, H. Yan, B. Ding and Y. Zhao, Nat. Biotechnol., 2018, 36, 258–268 CrossRef CAS PubMed.
- J. Zhuang, Y. He, G. Chen and D. Tang, Electrochem. Commun., 2014, 47, 25–28 CrossRef CAS.
- A. Dillen, A. Mohrbacher and J. Lammertyn, ACS Sens., 2021, 6, 3684 Search PubMed.
- R. Nutiu and Y. Li, Angew. Chem., Int. Ed., 2005, 44, 1061–1065 CrossRef CAS PubMed.
- G. Goel, A. Kumar, A. K. Puniya, W. Chen and K. Singh, J. Appl. Microbiol., 2005, 99, 435–442 CrossRef CAS PubMed.
- Y. Xiao, A. A. Lubin, A. J. Heeger and K. W. Plaxco, Angew. Chem., Int. Ed., 2005, 44, 5456–5459 CrossRef CAS PubMed.
- A. J. Bonham, K. Hsieh, B. S. Ferguson, A. Valleé-Bélisle, F. Ricci, H. T. Soh and K. W. Plaxco, J. Am. Chem. Soc., 2012, 134, 3346–3348 CrossRef CAS PubMed.
- A. Idili, N. Arroyo-Currás, K. L. Ploense, A. T. Csordas, M. Kuwahara, T. E. Kippin and K. W. Plaxco, Chem. Sci., 2019, 10, 8164–8170 RSC.
- P. Dauphin-Ducharme, K. Yang, N. Netzahualcóyotl, A.-C. Currás, K. L. Ploense, Y. Zhang, J. Gerson, M. Kurnik, T. E. Kippin, M. N. Stojanovic and K. W. Plaxco, ACS Sens., 2019, 4, 2832–2837 CrossRef CAS PubMed.
- H. Li, P. Dauphin-Ducharme, G. Ortega and K. W. Plaxco, J. Am. Chem. Soc., 2017, 139, 11207–11213 CrossRef CAS PubMed.
- H. Li, P. Dauphin-Ducharme, N. Arroyo-Currás, C. H. Tran, P. A. Vieira, S. Li, C. Shin, J. Somerson, T. E. Kippin and K. W. Plaxco, Angew. Chem., Int. Ed., 2017, 56, 7492–7495 CrossRef CAS PubMed.
- H. Li, N. Arroyo-Currás, D. Kang, F. Ricci and K. W. Plaxco, J. Am. Chem. Soc., 2016, 138, 15809–15812 CrossRef CAS PubMed.
- N. Arroyo-Currás, K. Scida, K. L. Ploense, T. E. Kippin and K. W. Plaxco, Anal. Chem., 2017, 89, 12185–12191 CrossRef PubMed.
- P. A. Vieira, C. B. Shin, N. Arroyo-Currás, G. Ortega, W. Li, A. A. Keller, K. W. Plaxco and T. E. Kippin, Front. Mol. Biosci., 2019, 6, 1–10 CrossRef PubMed.
- N. Arroyo-Currás, P. Dauphin-Ducharme, G. Ortega, K. L. Ploense, T. E. Kippin and K. W. Plaxco, ACS Sens., 2018, 3, 360–366 CrossRef PubMed.
- H. Li, J. Somerson, F. Xia and K. W. Plaxco, Anal. Chem., 2018, 90, 10641–10645 CrossRef CAS PubMed.
- S. Tyagi and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303–306 CrossRef CAS PubMed.
- T. Kihara, N. Yoshida, T. Kitagawa, C. Nakamura, N. Nakamura and J. Miyake, Biosens. Bioelectron., 2010, 26, 1449–1454 CrossRef CAS PubMed.
- G. Yao and W. Tan, Anal. Biochem., 2004, 331, 216–223 CrossRef CAS PubMed.
- U. Uddayasankar and U. J. Krull, Anal. Chim. Acta, 2013, 803, 113–122 CrossRef CAS PubMed.
- X. Dai, W. Yang, E. Firlar, S. A. E. Marras and M. Libera, Soft Matter, 2012, 8, 3036 RSC.
- H. Du, M. D. Disney, B. L. Miller and T. D. Krauss, J. Am. Chem. Soc., 2003, 125, 4012–4013 CrossRef CAS PubMed.
- H. Nasef, V. Beni and C. K. O′sullivan, J. Electroanal. Chem., 2011, 662, 322–327 CrossRef CAS.
- Q. Su, D. Wesner, H. Schö and G. Nö, Langmuir, 2014, 30, 14360–14367 CrossRef CAS PubMed.
- H. T. Ngo, H.-N. Wang, A. M. Fales and T. Vo-Dinh, Anal. Chem., 2013, 85, 6378–6383 CrossRef CAS PubMed.
- Molecular Beacons, ed. C. J. Yang and W. Tan, Springer, 2013 Search PubMed.
- N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2017, 3, 1–23 Search PubMed.
- S. Tombelli, M. Minunni and M. Mascini, Biosens. Bioelectron., 2005, 20, 2424–2434 CrossRef CAS PubMed.
- Z. Zhuo, Y. Yu, M. Wang, J. Li, Z. Zhang, J. Liu, X. Wu, A. Lu, G. Zhang and B. Zhang, Int. J. Mol. Sci., 2017, 18, 2–19 Search PubMed.
- J. Tan, M. Zhao, J. Wang, Z. Li, L. Liang, L. Zhang, Q. Yuan and W. Tan, Angew. Chem., Int. Ed., 2019, 58, 1621–1625 CrossRef CAS PubMed.
- D. Shangguan, Y. Li, Z. Tang, Z. C. Cao, H. W. Chen, P. Mallikaratchy, K. Sefah, J. Yang and W. Tan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11838–11843 CrossRef CAS PubMed.
- R. Nutiu and Y. Li, J. Am. Chem. Soc., 2003, 125, 4771–4778 CrossRef CAS PubMed.
- F. Wang, X. Liu and I. Willner, Angew. Chem., Int. Ed., 2015, 54, 1098–1129 CrossRef CAS PubMed.
- Y. Zhang, C. Ge, C. Zhu and K. Salaita, Nat. Commun., 2014, 5, 1–10 CAS.
- A. Valleé-Beíisle, F. Ricci, T. Uzawa, F. Xia and K. W. Plaxco, J. Am. Chem. Soc., 2012, 134, 15197–15200 CrossRef PubMed.
- S. Ranallo, M. Rossetti, K. W. Plaxco, A. Vallée-Bélisle and F. Ricci, Angew. Chem., Int. Ed., 2015, 54, 13214–13218 CrossRef CAS PubMed.
- Y.-T. Lin, R. Vermaas, J. Yan, A. M. De Jong and M. W. J. Prins, ACS Sens., 2021, 6, 1980–1986 CrossRef CAS PubMed.
- J. Corzo, Biochem. Mol. Biol. Educ., 2006, 34, 413–416 CrossRef CAS PubMed.
- B. D. Wilson, A. A. Hariri, I. A. P. Thompson, M. Eisenstein and H. T. Soh, Nat. Commun., 2019, 10, 1–9 CrossRef CAS PubMed.
- E. M. W. M. Van Dongen, T. H. Evers, L. M. Dekkers, E. W. Meijer, L. W. J. Klomp and M. Merkx, J. Am. Chem. Soc., 2007, 129, 3494–3495 CrossRef CAS PubMed.
- P. R. Mallikaratchy, A. Ruggiero, J. R. Gardner, V. Kuryavyi, W. F. Maguire, M. L. Heaney, M. R. McDevitt, D. J. Patel and D. A. Scheinberg, Nucleic Acids Res., 2011, 39, 2458–2469 CrossRef CAS PubMed.
- K. Horikawa, Y. Yamada, T. Matsuda, K. Kobayashi, M. Hashimoto, T. Matsu-ura, A. Miyawaki, T. Michikawa, K. Mikoshiba and T. Nagai, Nat. Methods, 2010, 7, 729–732 CrossRef CAS PubMed.
- H. Bischof, M. Rehberg, S. Stryeck, K. Artinger, E. Eroglu, M. Waldeck-Weiermair, B. Gottschalk, R. Rost, A. T. Deak, T. Niedrist, N. Vujic, H. Lindermuth, R. Prassl, B. Pelzmann, K. Groschner, D. Kratky, K. Eller, A. R. Rosenkranz, T. Madl, N. Plesnila, W. F. Graier and R. Malli, Nat. Commun., 2017, 8, 1–12 CrossRef CAS PubMed.
- A. Porchetta, A. Vallée-Bélisle, K. W. Plaxco and F. Ricci, J. Am. Chem. Soc., 2012, 134, 20601–20604 CrossRef CAS PubMed.
- S. Bissonnette, E. Del Grosso, A. J. Simon, K. W. Plaxco, F. Ricci and A. Valleé-Bélisle, ACS Sens., 2020, 5, 1937–1942 CrossRef CAS PubMed.
- N. Bhalla, P. Jolly, N. Formisano and P. Estrela, Essays Biochem., 2016, 60, 1–8 CrossRef PubMed.
- H.-B. Huang, J.-M. Peng, L. Weng, C.-Y. Wang, W. Jiang and B. Du, Ann. Intensive Care, 2017, 7, 1–10 CrossRef PubMed.
- J. Somerson and K. W. Plaxco, Molecules, 2018, 23, 1–7 CrossRef PubMed.
- H. Ma, C. Ó'Fágáin and R. O′kennedy, Biochimie, 2020, 117, 213–225 CrossRef PubMed.
- S. Warszawski, A. B. Katz, R. Lipsh, L. Khmelnitsky, G. Ben Nissanid, G. Javittid, O. Dym, T. Unger, O. Knop, S. Albeck, R. Diskinid, D. Fass, M. Sharon and S. J. Fleishmanid, PLOS Comput. Biol., 2019, 15, 1–24 CrossRef PubMed.
- S. Wang, M. Liu, D. Zeng, W. Qiu, P. Ma, Y. Yu, H. Chang and Z. Sun, Proteins, 2014, 82, 2620–2630 CrossRef CAS PubMed.
- S. Ni, H. Yao, L. Wang, J. Lu, F. Jiang, A. Lu and G. Zhang, Int. J. Mol. Sci., 2017, 18, 1–21 Search PubMed.
- C. Kratschmer and M. Levy, Nucleic Acid Ther., 2017, 27, 335–344 CrossRef CAS PubMed.
- P. G. Chandler, S. S. Broendum, B. T. Riley, M. A. Spence, C. J. Jackson and A. M. Buckle, Strategies for Increasing Protein Stability in: Protein Nanotechnology, Humana, New York, 2020, vol. 2073 Search PubMed.
- J. P. Elskens, J. M. Elskens and A. Madder, Int. J. Mol. Sci., 2020, 21, 1–31 Search PubMed.
- S. Ni, H. Yao, L. Wang, J. Lu, F. Jiang, A. Lu and G. Zhang, Int. J. Mol. Sci., 2017, 18, 1–21 Search PubMed.
- K. E. Maier and M. Levy, Mol. Ther. – Methods Clin. Dev., 2016, 3, 16014 CrossRef PubMed.
- S. Y. Tan, C. Acquah, A. Sidhu, C. M. Ongkudon, L. S. Yon and M. K. Danquah, Crit. Rev. Anal. Chem., 2016, 46, 521–537 CrossRef CAS PubMed.
- S. Gao, X. Zheng, B. Jiao and L. Wang, Anal. Bioanal. Chem., 2016, 408, 4567–4573 CrossRef CAS PubMed.
- S. Ni, Z. Zhuo, Y. Pan, Y. Yu, F. Li, J. Liu, L. Wang, X. Wu, D. Li, Y. Wan, L. Zhang, Z. Yang, B.-T. Zhang, A. Lu and G. Zhang, Appl. Mater. Interfaces, 2021, 13, 9500–9519 CrossRef CAS PubMed.
- M. A. Vorobyeva, A. S. Davydova, P. E. Vorobjev, D. V. Pyshnyi and A. G. Venyaminova, Int. J. Mol. Sci., 2018, 19, 1–21 Search PubMed.
- E. Eremeeva, A. Fikatas, L. Margamuljana, M. Abramov, D. Schols, E. Groaz and P. Herdewijn, Nucleic Acids Res., 2019, 47, 4927–4939 CrossRef CAS PubMed.
- M. R. Dunn, C. M. Mccloskey, P. Buckley, K. Rhea and J. C. Chaput, J. Am. Chem. Soc., 2020, 142, 7721–7724 CrossRef CAS PubMed.
- M. Varada, M. Aher, N. Erande, V. A. Kumar and M. Fernandes, ACS Omega, 2020, 5, 498–506 CrossRef CAS PubMed.
- M. Merkx, M. V. Golynskiy, L. H. Lindenburg and J. L. Vinkenborg, Biochem. Soc. Trans., 2013, 41, 1201–1205 CrossRef CAS PubMed.
- Fluorescent Protein-Based Biosensors Methods and Protocols, ed. J. Zhang, Q. Ni and R. H. Newman, Humana Press, 2014 Search PubMed.
- W. M. Keck, J. Cell Biol., 2003, 160, 629–633 CrossRef PubMed.
- Principles of Fluorescence Spectroscopy, ed. J. R. Lakowicz, Springer, 3rd edn, 2006 Search PubMed.
- X. Miao, X. Guo, Z. Xiao and L. Ling, Biosens. Bioelectron., 2014, 59, 54–57 CrossRef CAS PubMed.
- B. S. Ferguson, D. A. Hoggarth, D. Maliniak, K. Ploense, R. J. White, N. Woodward, K. Hsieh, A. J. Bonham, M. Eisenstein, T. Kippin, K. W. Plaxco and H. T. Soh, Sci. Transl. Med., 2013, 5, 213–165 Search PubMed.
- C. Parolo, A. Idili, G. Ortega, A. Csordas, A. Hsu, N. Netzahualcóyotl, A.-C. Currás, Q. Yang, B. S. Ferguson, J. Wang and K. W. Plaxco, ACS Sens., 2020, 5, 1877–1881 CrossRef CAS PubMed.
- A. Idili, J. Gerson, T. Kippin and K. W. Plaxco, Anal. Chem., 2021, 93, 4023–4032 CrossRef CAS PubMed.
- D. Grieshaber, R. MacKenzie, J. Vörös and E. Reimhult, Sensors, 2008, 8, 1400–1458 CrossRef CAS PubMed.
- H. Hasegawa, K.-I. Taira, K. Sode and K. Ikebukuro, Sensors, 2008, 8, 1090–1098 CrossRef CAS PubMed.
- T. Hianik, I. Grman and I. Karpisova, Chem. Commun., 2009, 6303–6305 RSC.
- S. Campuzano, M. Pedrero, P. Yáñez-Sedeño and J. M. Pingarrón, Int. J. Mol. Sci., 2019, 20, 1–19 Search PubMed.
- P.-H. Lin and B.-R. Li, Analyst, 2020, 145, 1110–1120 RSC.
- J. Xu and H. Lee, Chemosensors, 2020, 8, 66 CrossRef CAS.
- J. Breault-Turcot and J.-F. Masson, Chem. Sci., 2015, 6, 4247–4254 RSC.
- G. Rong, S. R. Corrie and H. A. Clark, ACS Sens., 2018, 2, 327–338 CrossRef PubMed.
- A. Idili, K. W. Plaxco, A. Vallée-Bélisle and F. Ricci, ACS Nano, 2013, 7, 10863–10869 CrossRef CAS PubMed.
- F. Ricci, A. Valleé-Beíisle, A. J. Simon, A. Porchetta, K. W. Plaxco, B. Science and E. Program, Acc. Chem. Res., 2016, 49, 21 CrossRef PubMed.
- A. M. Downs, J. Gerson, M. Nur Hossain, K. Ploense, M. Pham, H.-B. Kraatz, T. Kippin and K. W. Plaxco, ACS Sens., 2021, 6, 2299–2306 CrossRef CAS PubMed.
- Eccrine Systems, Inc., https://www.eccrinesystems.com/.
- Creative Commons Attribution 4.0, http://creativecommons.org/licenses/by/4.0/.
- Helia Biomonitoring, https://www.heliabiomonitoring.com/.
| This journal is © The Royal Society of Chemistry 2022 |