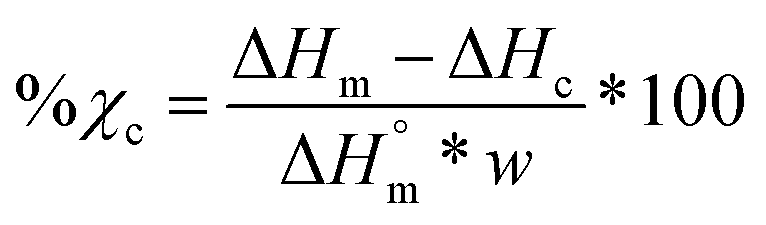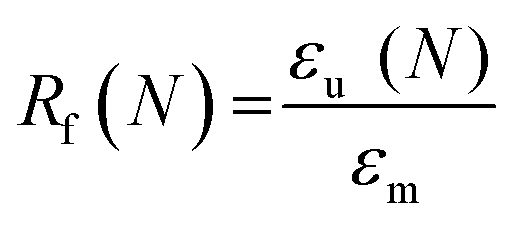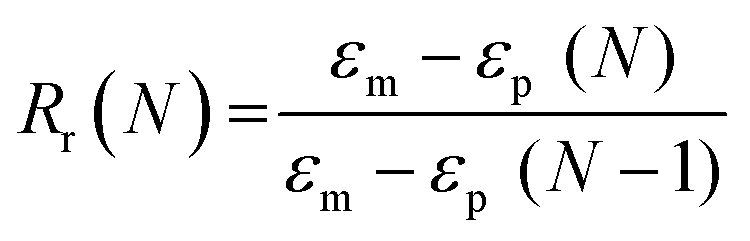Shape memory polymer (SMP) scaffolds with improved self-fitting properties†
Received
25th December 2020
, Accepted 8th April 2021
First published on 15th April 2021
Abstract
“Self-fitting” shape memory polymer (SMP) scaffolds prepared as semi-interpenetrating networks (semi-IPNs) with crosslinked linear-poly(ε-caprolactone)-diacrylate (PCL-DA, Mn ∼10 kg mol−1) and linear-poly(L-lactic acid) (PLLA, Mn ∼15 kg mol−1) [75/25 wt%] exhibited robust mechanical properties and accelerated degradation rates versus a PCL-DA scaffold control. However, their potential to treat irregular craniomaxillofacial (CMF) bone defects is limited by their relatively high fitting temperature (Tfit ∼55 °C; related to the Tm of PCL) required for shape recovery (i.e. expansion) and subsequent shape fixation during press fitting of the scaffold, which can be harmful to surrounding tissue. Additionally, the viscosity of the solvent-based precursor solutions, cast over a fused salt template during fabrication, can limit scaffold size. Thus, in this work, analogous semi-IPN SMP scaffolds were formed with a 4-arm star-PCL-tetracryalate (star-PCL-TA) (Mn ∼10 kg mol−1) and star-PLLA (Mn ∼15 kg mol−1). To assess the impact of a star-polymer architecture, four semi-IPN compositions were prepared: linear-PCL-DA/linear-PLLA (L/L), linear-PCL-DA/star-PLLA (L/S), star-PCL-TA/linear-PLLA (S/L) and star-PCL-TA/star-PLLA (S/S). Two PCL controls were also prepared: LPCL (i.e. 100% linear-PCL-DA) and SPCL (i.e. 100% star-PCL-TA). The S/S semi-IPN scaffold exhibited particularly desirable properties. In addition to achieving a lower, tissue-safe Tfit (∼45 °C), it exhibited the fastest rate of degradation which is anticipated to more favourably permit neotissue infiltration. The radial expansion pressure exerted by the S/S semi-IPN scaffold at Tfit was greater than that of LPCL, which is expected to enhance osseointegration and mechanical stability. The intrinsic viscosity of the S/S semi-IPN macromer solution was also reduced such that larger scaffold specimens could be prepared.
Introduction
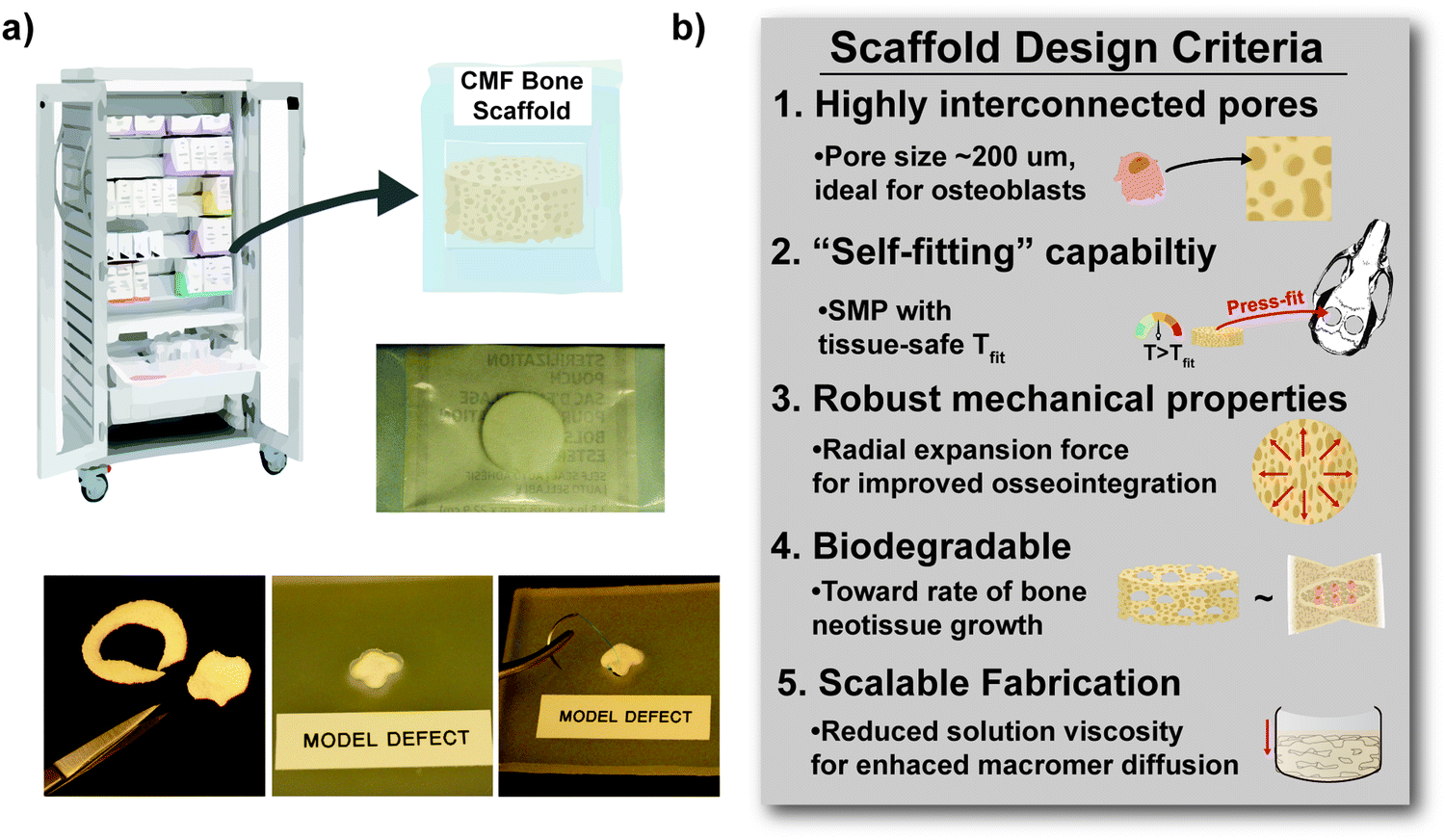
A major limitation of biologic and alloplastic grafts used to treat irregularly shaped cranio-maxillofacial (CMF) bone defects is the difficulty in achieving sufficient bone-to-graft contact, essential for osseointegration and healing.1,2 Autografting remains the clinical “gold standard”, but in addition to the demands of surgical harvesting, bone graft rigidity contributes to poor shaping and tissue contact, ultimately leading to graft resorption.3–5 Synthetic CMF bone graft substitutes, including ceramic injectables6 and bone cements,7 utilize in situ curing to achieve a defect-specific fit. However, they are limited by risks associated with brittle mechanical properties (leading to post-surgical fracture), exothermic curing (leading to tissue damage), and shrinkage post-cure (leading to poor bone-to-graft contact).8–10 PEEK implants can be formed with patient- specific geometry via 3D printing, but are non-regenerative.7,11 Thus, an off-the-shelf regenerative scaffold material that can readily achieve conformal fit into irregular CMF bone defects is expected to improve healing outcomes.
We have previously reported “self-fitting” scaffolds based on thermoresponsive shape memory polymers (SMPs) as a regenerative approach to treat CMF bone defects.12–16 Porous SMP scaffolds were prepared from linear-poly(ε-caprolactone)-diacrylate (linear-PCL-DA, Mn ∼10 kg mol−1) by photocuring a solvent-based macromer solution over a fused salt template followed by aqueous extraction of the template (i.e. solvent-casting/particulate leaching, SCPL). For such PCL SMP scaffolds, covalent cross-links act as netpoints and PCL lamellae act as switching segments. In a surgical setting, the PCL scaffolds could be warmed in saline to their “fitting temperature” (Tfit ∼55 °C, related to Tm,PCL), causing the PCL lamellae to begin to melt and the scaffold to subsequently soften. It could thus be readily press-fitted into the defect site as shape recovery would drive expansion of the scaffold to the perimeter. Then, as the scaffold would cool to body temperature (T < Tfit), the PCL lamellae would re-crystallize and return the scaffold to its relatively rigid state with the scaffold fixed into the shape of the defect. Importantly, the PCL SMP scaffolds displayed high shape fixity and recovery, non-brittle mechanical properties, and high pore interconnectivity.12–15 Increasing the rigidity of the PCL scaffolds would improve structural support in the early stages of healing and increasing the rate of degradation would promote osseointegration and regeneration.17–21 Thus, thermoplastic linear-poly(L-lactic acid) (PLLA, ∼15 kg mol−1) was incorporated into thermoset linear-PCL-DA networks to yield linear-PCL-DA/linear-PLLA semi-interpenetrating network (semi-IPN) scaffolds.16,22 A semi-IPN scaffold prepared with 75/25 wt% PCL/PLLA maintained SMP behavior (Tfit ∼55 °C), but demonstrated an increased modulus and accelerated degradation rate compared to the linear-PCL-DA control. The faster degradation of the linear-PCL-DA/linear-PLLA semi-IPNs was linked to polymer phase separation.16,23,24 Likewise, phase separation has been shown to impact mechanical and degradation properties of polyester blends.25–28
Further improvements to mechanical and degradation properties of PCL-based SMP scaffolds, as well as reducing the Tfit (to avoid possible tissue damage) and reducing macromer solution viscosity (to aid in scaffold fabrication), would be a significant enhancement in their utility. Because of their unique thermal, degradative, mechanical, and rheological properties,29–31star-polymer analogues may offer distinct advantages to the PCL/PLLA semi-IPN scaffolds. Tm values of multi-arm polymers are typically reduced due in part to their more sterically hindered architectures.32–34 Biodegradable star polyesters have also been employed to refine degradation behavior.32,35,36 Additionally, star-polymers are associated with reduced hydrodynamic volumes which affects dispersion and interfacial macromolecular interactions.37,38 Thus, star-polymers have been used to improve miscibility and resulting toughness of blends and polymer nanocomposites.39–42 Lastly, star-polymers are well known for having reduced dilute solution viscosities due to less chain entanglements relative to their linear counterparts.43,44 In the fabrication of SMP scaffolds, during solvent casting of the macromer solution over a fused salt template, this could aide in diffusion such that larger scaffold specimens could be readily prepared.
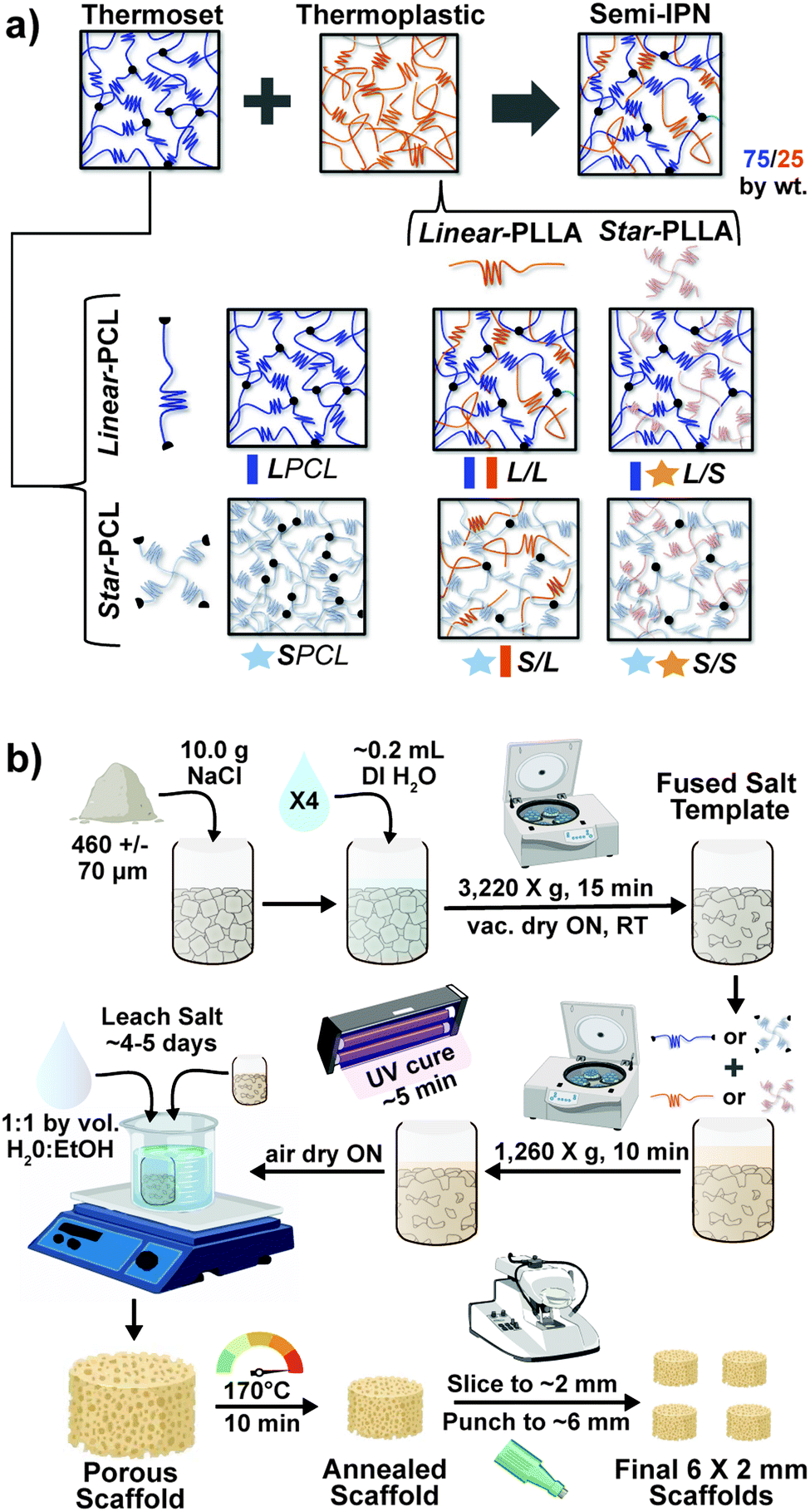
Herein, towards favorable tuning of semi-IPN scaffold properties, the impact of a crosslinkable 4-arm star-PCL analogue and a thermoplastic 4-arm star-PLLA was assessed. Specifically, scaffold compositions were systematically made with combinations of linear-PCL-DA or star-PCL-tetracryalate (star-PCL-TA) and linear-PLLA or star-PLLA: linear-PCL-DA/linear-PLLA (L/L), linear-PCL-DA/star-PLLA (L/S), star-PCL-TA/linear-PLLA (S/L) and star-PCL-TA/star-PLLA (Fig. 1a). The ratio of PCL/PLLA was maintained at 75/25 wt%, that of the L/L semi-IPN previously shown to best enhance compressive modulus and degradation rate versus the linear-PCL-DA control (LPCL) (i.e. 100% PCL-DA).16,22 In addition to the LPCL control, a star-PCL-TA control (SPCL) (i.e. 100% PCL-TA) was also prepared. All scafffolds were prepared with the same SCPL protocol to generate scaffolds with similar pore size and interconnectivity (Fig. 1b). The resulting SMP scaffolds were assessed for their thermal, degradative, mechanical, and shape memory properties. The solution viscosity of macromer solutions used in the SCPL fabrication process was also examined and select compositions were used to fabricate scaffolds with larger dimensions.
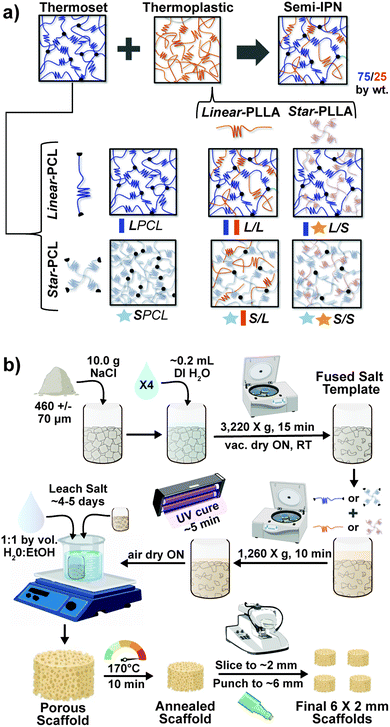 |
| | Fig. 1 (a) Four semi-IPN SMP scaffolds were prepared with combinations of linear-PCL-DA or star-PCL-TA and linear-PLLA or star-PLLA (75/25 PCL/PLLA). Two 100% PCL controls were also prepared from linear-PCL-DA or star-PCL-TA. (b) All SMP scaffolds were prepared via solvent-casting/particulate leaching (SCPL) whereby a designated solvent-based macromer solution was sequentially cast over a fused salt template, UV-cured, and the template extracted to yield highly interconnected pores. | |
Experimental
Materials
Linear-PCL-diol (Mn = 10 kg mol−1 per manufacturer specifications), 4-(dimethylamino)pyridine (DMAP), triethylamine (Et3N), acryloyl chloride, potassium carbonate (K2CO3), anhydrous magnesium sulfate (MgSO4), sodium chloride (NaCl, salt), (3S)-cis-3,6-dimethyl-1,4-dioxane-2,5-dione (L-lactide), ε-caprolactone, pentaerythritol, tin(II) 2-ethylhexanoate (Sn(Oct)2), ethylene glycol, 2,2-dimethoxy-2-phenyl acetophenone (DMP), 1-vinyl-2-pyrrolidinone (NVP), sodium hydroxide (NaOH), deuterated chloroform (CDCl3), and solvents were purchased from Sigma-Aldrich. All solvents and ethylene glycol were dried over 4 Å molecular sieves, all reagents were vacuum dried overnight (ON), and all glassware and stir bars were dried at 120 °C ON prior to use. Salt was sieved using an ASTM E-11 no. 40 and no. 35 sieves with 425 μm and 500 μm openings respectively; scanning electron microscopy (SEM) and ImageJ showed an average salt size of 460 ± 70 μm.
Methods
Syntheses.
All reactions were run under a nitrogen (N2) atmosphere with a Teflon-covered stir bar. Following purification, polymer structures (including % acrylation, architecture, and Mn) were confirmed with 1H NMR spectroscopy (Inova 500 MHz spectrometer in FT-mode with CDCl3 as the standard). Polymer thermal properties were determined using differential scanning calorimetry (DSC, TA Instruments Q100) as described below.
Thermoplastic linear- and star-PLLA (Mn ∼15 kg mol−1) were synthesized via ring opening polymerizations (ROPs) according to an established protocol.45L-Lactide (6.0 g), alcohol initiator, and Sn(Oct)2 catalyst were allowed to react ON at 120 °C. The alcohol initiator was varied from difunctional ethylene glycol to tetrafunctional pentaerythritol to achieve a linear- and star-PLLA architecture, respectively. Mn was controlled via molar equivalence of monomer to initiator (104![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, [M]:[I]). The crude products were dissolved in a minimal amount of chloroform and were precipitated into methanol. Final products were filtered and vacuum dried (RT, ON, 30 in. Hg) to obtain purified linear- and star-PLLA. Target Mn and architecture were verified using 1H NMR end group analysis (CHδ = 5.2 ppm in repeat unit compared to terminal CHδ = 3.7 ppm). The following thermal transitions were observed for linear-PLLA [Tg = 45 °C, Tm = 155 °C, 50% crystallinity] and star-PLLA [Tg = 49 °C, Tm = 152 °C, 15% crystallinity] (Fig. S1, ESI†).
:1, [M]:[I]). The crude products were dissolved in a minimal amount of chloroform and were precipitated into methanol. Final products were filtered and vacuum dried (RT, ON, 30 in. Hg) to obtain purified linear- and star-PLLA. Target Mn and architecture were verified using 1H NMR end group analysis (CHδ = 5.2 ppm in repeat unit compared to terminal CHδ = 3.7 ppm). The following thermal transitions were observed for linear-PLLA [Tg = 45 °C, Tm = 155 °C, 50% crystallinity] and star-PLLA [Tg = 49 °C, Tm = 152 °C, 15% crystallinity] (Fig. S1, ESI†).
Star-PCL-tetrol was synthesized via ROP (analogous to that described above) with a target Mn of ∼10 kg mol−1 to match that of linear-PCL-diol (Mn = 10 kg mol−1; Sigma-Aldrich). The ε-caprolactone (25.0 g), pentaerythritol (88:1, [M]:[I]) and Sn(Oct)2 were combined and were allowed to react ON at 120 °C. The crude product was re-dissolved and precipitated as described above to yield purified star-PCL-tetrol. The target Mn and architecture were verified via1H NMR end group analysis (CH2δ = 4.1 ppm in repeat unit compared to terminal CH2δ = 3.7 ppm). Thermal transitions were determined for both the linear-PCL-diol [Tg = −65 °C, Tm = 53 °C, 48% crystallinity] and the star-PCL-tetrol [Tg = −63 °C, Tm = 50 °C, 45% crystallinity] (Fig. S2, ESI†).
Linear-PCL-diol and star-PCL-tetrol were acrylated to form photo-crosslinkable linear-PCL-DA and star-PCL-TA macromers, respectively, using established acrylation protocols.15 Briefly, linear-PCL-diol (20.0 g, 2.0 mmol) was combined with DMAP (6.6 mg) serving as the catalyst and they were dissolved in dichloromethane (DCM, 0.17 g mL−1). After purging with N2, triethylamine (4.0 mmol) and acryloyl chloride (8.0 mmol) were added to the flask and the reaction was left to stir at RT for 30 min. An analogous procedure was followed for the star-PCL-tetrol but molar ratios were doubled to account for the 4 end groups [DMAP = 13.2 mg, triethylamine = 8.0 mmol, acryloyl chloride = 16.0 mmol]. Established work-up procedures were followed to obtain linear-PCL-DA and star-PCL-TA.15 Percent acrylation was confirmed via1H NMR end group analysis (CH2δ = 4.1 ppm in repeat unit, compared to acrylate protons CH = CH2δ = 5.6, 6.1 and 6.4 ppm) to be >85% for both linear-PCL-DA and star-PCL-TA (Fig. S3, ESI†).
Fabrication
Scaffolds.
Porous scaffolds were prepared via SCPL, based on a previous report,15 employing a fused salt template for pore interconnectivity. Sieved NaCl (10.0 g, 460 ± 70 μm) was placed in a 20 mL scintillation vial (I.D. = 25 mm) and DI water (7.5 wt%) was added in four portions followed by manual stirring with a spatula after each addition. The wet salt was pressed with a glass rod and the vials were centrifuged (15 min, 3220 × g). The opened vials were air dried for ∼1 h and were subsequently vacuum dried (RT, ON, 30 in. Hg).
Next, macromer solutions were prepared by dissolving a designated macromer or combination of two macromers (75/25 wt% ratio) in DCM (0.15 g total per mL DCM). Photoinitiator solution (10 wt% DMP in NVP) was then added at 15 vol%. To each salt template ∼5 mL of macromer solution was added and the vials were centrifuged (10 min, 1260 × g) to promote macromer solution diffusion throughout the template. To crosslink acrylated macromers, opened vials were then exposed to UV light for 5 min (UV-Transilluminator, 6 mW cm−2, 365 nm) followed by air drying in a fume hood ON. To remove the salt template, vials were then placed in a solution of water and ethanol (1:1 by vol.) for ∼5 days with daily solution changes. Resulting porous scaffolds were air dried ON, and finally heat treated (170 °C, 10 min, 30 in. Hg). The dried scaffolds (d ∼12 mm) were sliced into three specimens (t ∼2 mm) (Vibratome, Leica VT 1000 S) and were biopsy punched (Integra Miltex, 6 mm). Final specimen dimensions were d ∼6 mm × t ∼ 2 mm.
Solid films.
Analogous solid films of each scaffold composition were prepared for % porosity calculations and to evaluate polymer miscibility in film cross-sections. A macromer solution (25 wt% total polymer in DCM), combined with the aforementioned photoinitiator solution (15 vol%), was added to a circular silicone mold (d ∼45 mm × t ∼2 mm; McMaster-Carr) secured between 2 glass slides. The mold was then exposed to UV-light (UV Transilluminator, 6 mW cm−2, 365 nm) for 3 min on each side. The swollen films were air dried ON followed by vacuum drying (RT, 4 h, 30 in. Hg), soaking in ethanol while placed atop a shaker table (150 rpm, 3 h), air drying ON, and finally, were heat treated (170 °C, 30 min, 30 in. Hg). Films were punched to form disc specimens (d ∼5 mm × t ∼1.1 mm) used for testing.
Scaffold sol content
Scaffolds (d ∼6 mm × t ∼2 mm; N = 3) were each submerged in 10 mL of DCM in a scintillation vial. Sealed vials were placed atop a shaker table (150 rpm, 48 h) and scaffolds were subsequently rinsed with DCM, air dried, and dried under vacuum (RT, ON, 30 in. Hg). Initial and final mass values were used to calculate % sol content.
Thermal gravimetric analysis (TGA)
TGA (TA Instruments Q50) of scaffolds (∼10 mg; N = 1) was performed under N2 from RT to 500 °C (heating rate = 10 °C min−1) using platinum pans.
% Porosity
The percent porosity of scaffolds (N = 3) was determined gravimetrically using eqn (1):| | 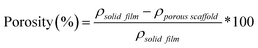 | (1) |
where ρporous scaffold is the density of the final scaffold specimens and ρsolid films is the density of analogous solid film samples.
Pore size
Scaffold pore interconnectivity and pore size were evaluated with SEM (JEOL JCM-5000 Neoscope, accelerating voltage ∼10 kV) following coating with Au-Pt (∼4 nm). Scaffold images (n = 4) were analyzed using image analysis software (Image J); measurements (N = 30) were taken from pores along the diaganol midline to determine average pore size.
Thermal transitions and % crystallinity
Differential scanning calorimetry (DSC; TA Instruments Q100) was used to determine Tg, Tm, and % crystallinity of PCL and PLLA polymers prior to scaffold fabrication. Specimens (∼10 mg; N = 3) were sealed in hermetic pans and heated at a rate of 10 °C min−1, and values were taken from the second cycle to erase thermal history. The onset and midpoint of Tm,PCL and Tm,PLLA was determined using TA Universal Analysis software from the onset and the maximum of the endothermic melt peak, respectively. Percent crystallinity was determined with eqn (2):| | 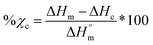 | (2) |
where is ΔHm is the enthalpy of fusion taken from the integral of the endothermic melt peak, ΔHc is the enthalpy of crystallization from the exothermic cold crystallization peak and  is the theoretical value for 100% crystalline PCL (139.5 J g−1)46 or PLLA (93.0 J g−1).47
is the theoretical value for 100% crystalline PCL (139.5 J g−1)46 or PLLA (93.0 J g−1).47
Scaffolds (N = 3) were likewise examined but using a heating rate of 5 °C min−1 and using the first cycle to examine the impact of fabrication. For semi-IPNs (PCL/PLLA 75/25 wt%), a correction factor to account for polymer wt% was included in % crystallinity calculations according to eqn (3):
| | 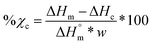 | (3) |
where
w is the mass fraction of the designated polymer species (
i.e. w = 0.75 for PCL and
w = 0.25 for PLLA in semi-IPN compositions).
Degradation
Degradation tests were performed under base-catalyzed conditions (0.2 M NaOH) according to ASTM F1635. Scaffold specimens (d ∼6 mm × t ∼2 mm; N = 3 per time point) were each submerged in 10 mL of the basic solution in a sealed glass vial and maintained in an incubator (VWR Benchtop Shaking Incubator Model 1570) at 37 °C and 60 rpm. At each of the five designated time points (24, 48, 72, 120, and 168 h), samples were removed, thoroughly rinsed with DI water, blotted, and finally dried under vacuum (RT, ON, 30 in. Hg). Specimen mass was measured to examine gravimetric mass loss.
Compressive mechanical properties
Scaffold specimens (d ∼6 mm × t ∼2 mm; N = 3) underwent static compression testing (Instron 5944) at RT. Specimens were subjected to a constant strain (1.5 mm min−1) up to 85% strain. Due to their non-brittle nature, no specimen fractured. The average compressive modulus (E), strength (CS), and toughness were reported: E was determined from the initial linear region (≤10% ε). CS was determined from the stress at 85% strain. Toughness values were calculated from the area of the stress–strain curves up to 85% strain.
Shape memory properties
Self-fitting behavior in model defect.
Scaffold specimens (d ∼6 mm × t ∼2 mm; N = 3) were evaluated for their “self-fitting” ability using a model defect representative of a rat calvarial defect.48,49 From an ultra-high-molecular-weight polyethyle (UHMWPE) sheet (McMaster-Carr, t ∼2 mm), a circular defect (d ∼5 mm) was created with a drill press (Grizzly G7948). A “fitting temperature” (Tfit) was determined as the saline temperature that, after 1 min of submersion, consistently produced a scaffold that was malleable to the touch. A hot plate equipped with a digital temperature probe (Heidolph, MR HEI-TEC) was used to the warm saline in 1 °C intervals within a given scaffold's onset to midpoint Tm,PCL range (i.e. 50–56 °C for linear-PCL-based and 42–50 °C for star-PCL-based scaffolds). Tfit was determined to be ∼55 °C (for linear-PCL-based scaffolds) and ∼45 °C (for star-PCL-based scaffolds). Next, each scaffold specimen was subjected to the following protocol: (1) submerged into saline previously heated to the designated Tfit and maintained for 1 min; (2) removed and immediately press-fitted into a model defect (at RT); (3) maintained in the model defect for 2 min to fix the new temporary shape; (4) removed from the defect (pushing out by hand), allowed to sit for 2 min; (5) re-submerged into the saline bath at Tfit for 1 min to elicit shape recovery, removed, allowed to cool at RT for 2 min. At key points during this sequence, the scaffold diameter was measured using electronic calipers to quantify scaffold strain (ε). Steps 1–5 were repeated to determine shape fixity (Rf) and shape recovery (Rr) over a second cycle. From this process, the Rf and shape recovery Rr for the first (N = 1) and second (N = 2) cycles were calculated, according to eqn (4) and (5), respectively:| | 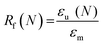 | (4) |
| | 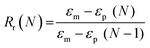 | (5) |
where εm is the maximum strain following step 2, εu (N) is the strain in the stress-free state following step 3, and εp is the final recovered strain following step 4. Strain values were determined via electronic caliper measurements.
Radial pressure during shape recovery.
Scaffold discs (d ∼6 mm × t ∼2 mm; N = 5) were subjected to radial mechanical testing (Instron 5965 equipped with a Blockwise RJA62 J-Crimp Radial Compression Station), to determine the radial pressure exerted during shape recovery at a scaffold's Tfit (LPCL, L/L, L/S at 55 °C and SPCL, S/L, S/S at 45 °C). This was intended to mimic shape recovery during self-fitting of the scaffold specimen into a d ∼5 mm defect. Specimens were loaded into the bore set to an initial d ∼6.5 mm at RT. The temperature was then increased to the designated Tfit and maintained for 3 min. Next, the bore diameter was reduced from 6.5 mm to 5 mm at a rate of 1 mm min−1. Force was monitored throughout the procedure, and total radial force (TRF) was calculated and converted to radial pressure based on exact scaffold dimensions.50
Solution viscosity and scaffold scale-up
Solution viscosity.
The complex viscosity [η*] of each scaffold macromer precursor solutions (N = 3) was measured as a function of frequency (100 Hz to 0.1 Hz, Anton Parr MCR 301). Macromer solutions (0.15 g per mL of DCM) were comprised of linear-PCL-diol or star-PCL-tetrol (i.e. non-acrylated) and no photoinitiator solution to avoid cross-linking during the test. To determine the intrinsic viscosity, the η* data was extrapolated to a theoretical zero shear rate (0 Hz).
Solution diffusion through salt template.
Select macromer solutions (L/L and S/S), containing dye, were used to assess differences in the rate of diffusion through a salt template. To aide inspection of diffusion, salt templates with a somewhat higher heights were prepared as above but with 15.0 g of sieved salt. Macromer solutions (∼7.5 mL) were prepared with designated macromers (i.e. linear-PCL-DA and linear-PLLA or star-PCL-TA and star-PLLA), 15 vol% photoinitiator solution, and a few drops of food coloring. With two salt templates placed side-by-side, each macromer solution was gently poured over the template simultaneously and diffusion captured via video. The process was repeated in triplicate.
Scaled-up scaffold fabrication.
The L/L and S/S compositions were again selected to fabricate larger scaffolds due to their lowered solution viscosities. A 5 mm hole (diamond core drill bit, Marshalltown) was drilled into the bottom of a 100 mL beaker (I.D. = 43.6 mm) to aid in macromer solution diffusion. Each 100 mL beaker was filled with 50.0 g of salt and 7.5 wt% water was incorporated over 4 additions with mechanical mixing following each addition. A smaller beaker was used to manually push the wet salt down and the salt molds were vacuum dried (RT, 30 in. Hg., ON). Macromer solutions were prepared (∼15 mL) according to that described above for fabrication of smaller scaffolds. Once mixed, macromer solution was poured on top of the fused salt mold and was allowed to sit for ∼3 min to permit diffusion; aluminum foil covered the beaker to prevent premature UV curing and solvent evaporation. Following UV-cure (InetlliRay 400, 50% intensity) for 10 min, specimens were allowed to dry in a fume hood (48 h) and were then soaked in a 1:1 DI water:ethanol solution with daily solution changes. Dried scaffolds were then annealed and sliced into 2 mm specimens, as described above for the smaller scaffolds. Note, both types of scaffolds were maintained at their full diameter for size comparisons (i.e. no biopsy punch was used). Photos were taken throughout the procedure and low magnification optical microscopy (Leica DM 6B; 5X) was performed on scaffold specimens to broadly examine pore morphology. The procedure was performed in triplicate and scaffolds were measured with electronic calipers to quantify dimensional changes. SEM (JEOL JCM-5000 Neoscope, accelerating voltage ∼10 kV, Au-Pt coating ∼4 nm) with energy dispersive X-ray spectroscopy (EDS, Oxford Instruments) elemental mapping was also performed to confirm complete porogen leaching from larger constructs.
Statistical analyses
All data was reported as the average ± standard deviation. ANOVA tests were performed and if there was a statistical difference, t-tests were performed against the LPCL control. T-tests were also used to make direct comparisons between compositions of interest, which will be specified for each result discussed. For mechanical testing, interquartile range tests were performed and values that were determined as being outliers were removed from the data (final N ≥5). For rheology data, linear regression was used to extrapolate complex viscosity to zero shear. Regression analyses were only performed up to 1 Hz to achieve R2 >0.5 and zero shear viscosities were reported as averages ± the standard error.
Results and discussion
Macromer synthesis
Linear- and star-PLLA (Mn ∼ 15 kg mol−1) (Fig. S1, ESI†) as well as linear-PCL-diol and star-PCL-tetrol (Mn ∼10 kg mol−1) (Fig. S2, ESI†) were characterized. Star macromer Mn was selected to match previously studied linear macromers in order to rule out Mn as a variable. As described above, 1H NMR end group analysis was used to determine Mn and confirm architecture (i.e. terminal group protons were approximately doubled for star precursors). DSC was used to determine thermal transitions and % crystallinity, with differences in thermal properties used to further validate precursor architecture. The Tg and Tm values as well as % crystallinity varied for the linear-PLLA (Tg ∼45 °C, Tm ∼155 °C, ∼50%) versus the star-PLLA (Tg ∼49 °C, Tm ∼152 °C, ∼15%). Likewise, differences were observed for the Tg, Tm, and % crystallinity values of the linear-PCL diol (Tg ∼ −65 °C, Tm ∼53 °C, ∼48%) and the star-PCL tetrol (Tg ∼ −63 °C, Tm ∼50 °C, ∼45%). Subsequently, the linear-PCL diol and star-PCL tetrol were successfully acrylated (>85%) to yield linear-PCL-DA and star-PCL-TA, respectively (Fig. S3, ESI†).
Scaffold fabrication
Fabricated scaffolds were characterized in various ways to ensure effective cross-linking (sol content), to confirm the targeted PCL/PLLA wt% ratio of 75/:25 (TGA), and to quantify pore size and % porosity (SEM and density calculations, respectively). Sol content values for 100% PCL controls [LPCL and SPCL] was just 2–4%, further indicating successful cross-linking (i.e. >95%) (Fig. S4, ESI†). All semi-IPN scaffolds displayed sol content values <29%, similar to the controls when the thermoplastic PLLA (incorporated at 25 wt%) was considered. Additionally, the TGA thermograms of semi-IPNs all showed ∼25 wt% mass loss from 250–350 °C that corresponded to the 25 wt% PLLA contained (Fig. S5, ESI†). Thus, the PLLA did not diminish linear-PCL-DA or star-PCL-TA cross-linking and the targeted 75/25 wt% ratio of PCL/PLLA was maintained. Finally, SEM imaging and analysis confirmed the targeted pore interconnectivity and ∼220 μm average pore size (Fig. S6a, ESI†), within the range associated with osteogenesis.51 Porosity calculations revealed that all scaffolds were similarly ∼60% (Fig. S6b, ESI†).
Scaffold thermal properties
PCL Tm.
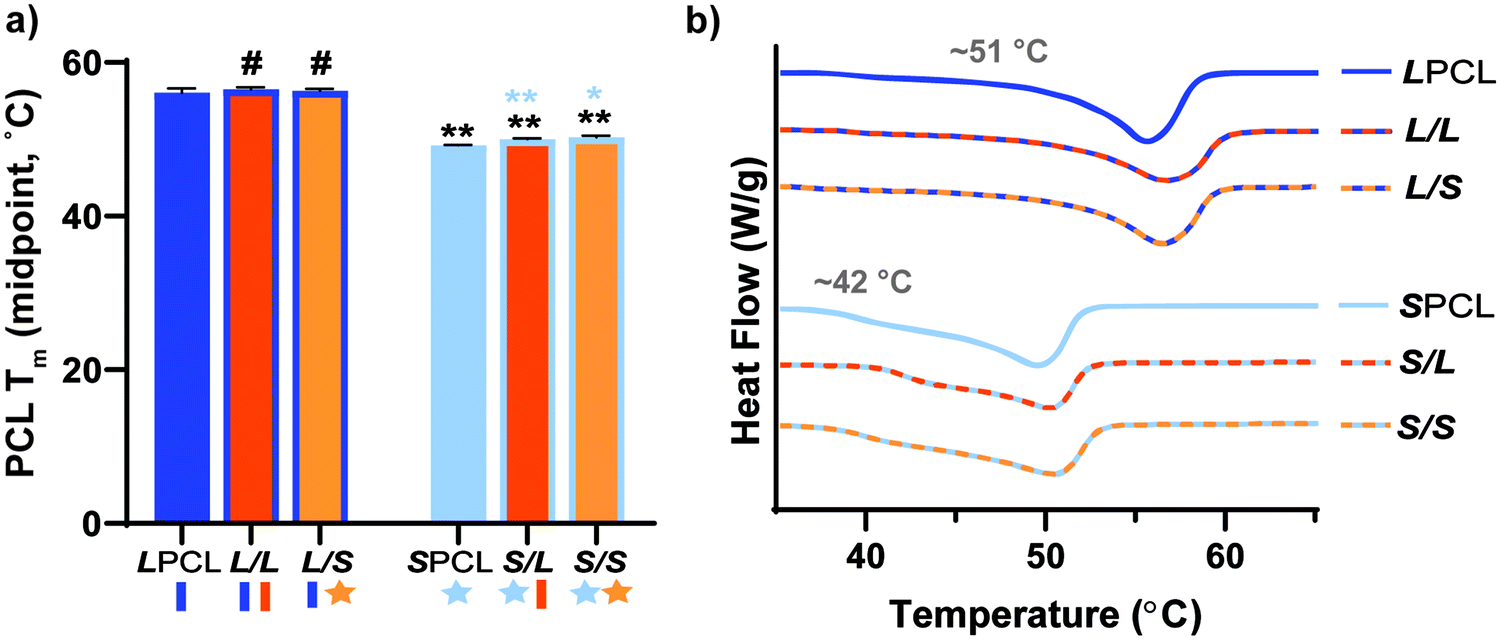
The midpoint melting temperature of PCL (Tm,PCL) represents the temperature to which the scaffold must be heated to confer maximum shape recovery, key to self-fitting into the bone defect. The Tm values were quantified for all scaffold compositions (Fig. 2a and Table S1, ESI†). Notably, the midpoint Tm,PCL values were significantly reduced (∼6 °C) for star-PCL-based versus linear-PCL-based scaffolds. The LPCL scaffold had a Tm,PCL ∼56 °C (midpoint) that was maintained following incorporation of linear- or star-PLLA to form L/L and L/S semi-IPN scaffolds, respectively. In contrast, for the SPCL scaffold, the Tm,PCL (midpoint) was significantly reduced to ∼50 °C. These values were maintained with the incorporation of linear- or star-PLLA to form S/L and S/S semi-IPNS, respectively. As is discussed later, star-PCL-based scaffolds begin to soften and undergo self-fitting in model defects at temperatures below Tm,PCL ∼50 °C (midpoint), due to the fact that the onset melting temperature of PCL is just ∼42 °C (Fig. 2b and Table S1, ESI†). This presented a unique way to afford a tunable Tm,PCL in a chemically cross-linked PCL scaffold. In contrast, we previously tuned linear PCL Mn (∼10 kg mol−1 and ∼5 kg mol−1), but this did not yield appreciable differences in scaffold Tm,PCL (56.2 ± 0.4 and 54.4 ± 0.6 °C, respectively).16 In this way, star-PCL-based compositions are expected to improve tissue safety during self-fitting into bone defects.
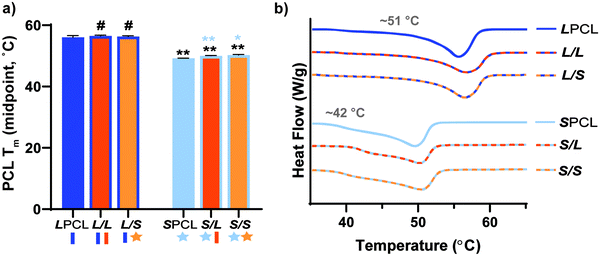 |
| | Fig. 2 (a) Midpoint tm of PCL of scaffolds; *p < 0.05, **p < 0.01, #p > 0.05. Note: the black color-coded statistics are comparisons to LPCL and blue color-coded statistics are comparisons to SPCL. (b) Representative thermogram for each scaffold composition. | |
PCL crystallinity.
PCL crystalline lamellae are the origin of shape memory behavior, and self-fitting behavior, and further have a significant impact on degradation and mechanical properties. Thus, scaffold PCL % crystallinity was quantified from DSC (Fig. S7a and Table S1, ESI†). For LPCL, PCL % crystallinity was ∼42%. When corrected for weight % in semi-IPN compositions (PCL/PLLA, 75/25 wt%), PCL % crystallinity was maintained for linear-PCL-based semi-IPNs (i.e.L/L and L/S). In the case of SPCL, PCL crystallinity was significantly reduced to ∼30%. As described later, the PCL % crystallinity of all scaffolds was sufficient to retain similarly shape recovery and shape fixity. However, the addition of linear- or star-PLLA to form S/L and S/S semi-IPNs resulted in increased PCL crystallinity of ∼34% and ∼39% (with S/S similar to the LPCL control), respectively.
PLLA crystallinity.
PLLA crystallinity can also impact scaffold degradation and mechanical properties. The previously reported L/L semi-IPN scaffold exhibited PLLA crystallinity (∼38%) and Tm,PLLA (midpoint) (164 °C) (Table S1 and Fig. S7b, ESI†). When star-PLLA was incorporated into the linear-PCL-DA network, the resulting S/L semi-IPN scaffold exhibited significantly decreased PLLA crystallinity (∼20%, ∼158 °C). For star-PCL-based semi-IPNs, the PLLA crystallinity was somewhat intermediate: S/L (∼23%, ∼160 °C) and S/S (∼25%, ∼157 °C), but was not statistically significant compared to the L/L. Thus, versus the L/L semi-IPN scaffolds, the S/L, S/L, and S/S had somewhat diminished PLLA crystallinity and is considered in analysis of degradation and mechanical properties.
Degradation behavior
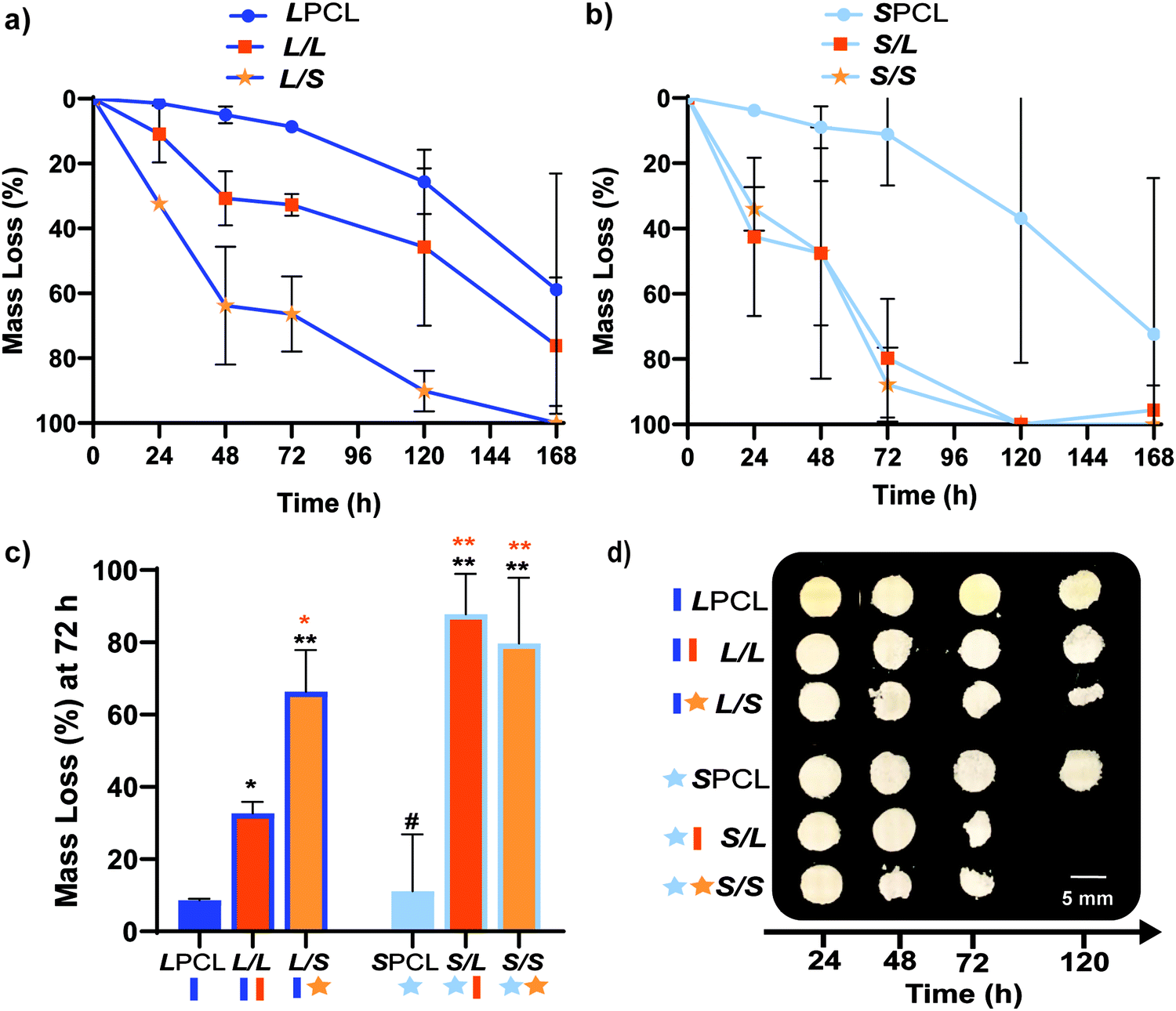
Previously, we reported that the L/L semi-IPN scaffold degraded significantly faster than the LPCL control.16,22,23 Further acceleration of degradation is anticipated to favorably allow neotissue formation as well as osteogenesis.17,19–21 This present study revealed that the L/S semi-IPN degraded faster than the L/L semi-IPN (Fig. 3a). In the case of star-PCL-based compositions, the SPCL scaffold degraded slowly, similar to LPCL (Fig. 3b). However, the S/L and S/S semi-IPNs degraded faster and generally similar to each other. By examining mass loss at the 72 h timepoint (Fig. 3c) as well as images of specimens at increasing time points (Fig. 3d), it is clear that S/L and S/S exhibited the most rapid rate of mass loss, even faster than L/S. Notably, mass loss at earlier timepoints (48 h) was greater for S/SversusS/L. While reduced levels of PCL and/or PLLA % crystallinity of semi-IPNs (Table S1 and Figure S7, ESI†) would be predicted to increase their rate of degradation, these properties were not always correlative. For instance, the L/S and S/S showed similar PCL % crystallinity (∼40%), and the L/S showed a lower PLLA % crystallinity (∼20% compared to 25%), but the S/S degraded significantly faster than the L/S. Thus, PCL/PLLA phase separation was considered, as this has been known to contribute to accelerated degradation of blends25,26 and semi-IPNs.16,23,24 SEM of analogous solid films demonstrated distinct morphologies for each composition (Fig. S8, ESI†). Both 100% PCL controls [LPCL and SPCL] showed a uniform morphology as expected based on their chemical homogeneity. The L/L (i.e. slowest degrading semi-IPN) also showed minimal signs of phase separation. However, all other semi-IPNs [L/S, S/L and S/S] showed greater evidence of coalescence, indicative of greater phase separation or immiscibility.24,52,53 Further, these new semi-IPN scaffolds demonstrate the potential to both accelerate and tune scaffold degradation rates based on phase separation. The current results were limited to base-catalyzed conditions, known to impact polyester degradation kinetics.54 Thus, future studies wherein scaffold degradation is assessed in vitro under physiological conditions as well as in vivo would be informative. PCL has been known to degrade in vivo over the course of ∼2 years,55,56 but these faster degrading scaffolds are expected to more closely mimic the timescale of CMF bone regeneration (3 to 6 months).21 As rates of regeneration can vary due to patient age and other factors,57 the tunability of these scaffolds’ degradation rates may be advantageous.
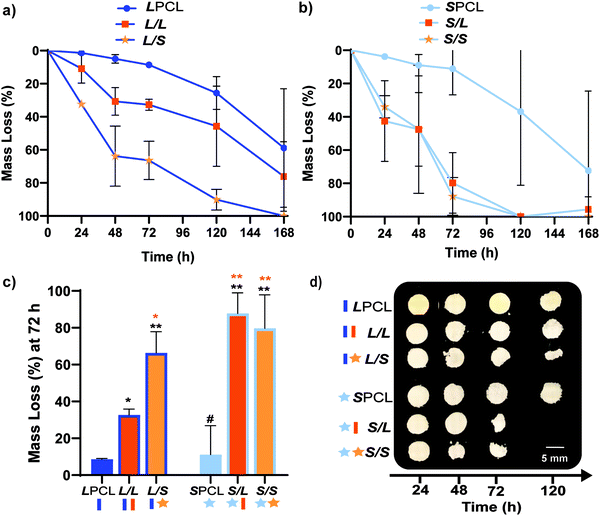 |
| | Fig. 3 Gravimetric mass loss over time for base-catalyzed degradation studies (0.2 M NaOH, 37 °C, 60 rpm) for (a) linear-PCL-based and (b) star-PCL-based scaffolds. (c) Mass loss at 72 h was compared for all scaffold compositions; *p < 0.05, **p < 0.01, #p > 0.05. Note: the black color-coded statistics were compared to LPCL control while orange color-coded statistics were compared to the L/L composition. (d) Representative photos of specimens at different timepoints during degradation study. | |
Mechanical, shape memory, and radial expansion pressure properties
Mechanical properties.
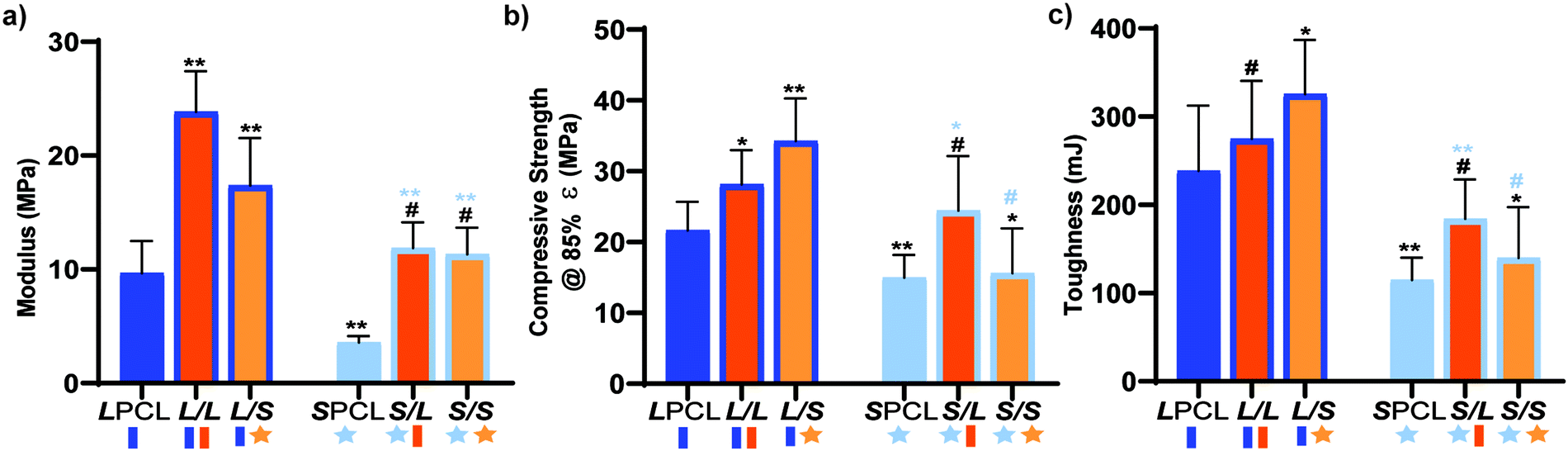
Mechanically robust SMP scaffolds are expected to afford superior outcomes in the treatment of bone defects. Static compressive testing was performed to assess the mechanical properties of the SMP scaffolds. For linear-PCL-based compositions, versus the LPCL control (∼9.65 MPa), the modulus (E) was significantly increased for both the L/L (∼23.8 MPa) and L/S (∼17.4 MPa) semi-IPNs (Fig. 4a and Table S2, ESI†). In terms of star-PCL-based compositions, for the SPCL control (∼3.57 MPa), E was significantly lower than the LPCL. This was attributed to the former's reduced PCL % crystallinity, in spite of having a higher relative cross-link density. However, versus the SPCL, E was increased for the S/L (∼11.9 MPa) and S/S (∼11.3 MPa) semi-IPNs, similar to the LPCL control. All semi-IPNS exhibited higher E values versus the 100% PCL controls, but the E values of L/L and L/S were higher than that of S/L and S/S. Similar trends generally emerged for compressive strength (CS) (Fig. 4b) as well as for toughness (Fig. 4c). No scaffold fractured during the test (i.e. withstood 85% strain), indicative of their non-brittle behavior that is desirable in the intended application of CMF bone defect treatment. Moreover, all scaffold compositions demonstrated robust mechanical properties for handling and press-fitting. Of all compositions, the L/S semi-IPN exhibited the greatest CS and toughness, while the S/L semi-IPN exhibited enhanced CS and toughness versus the SPCL control. Thus, a star-architecture affords certain semi-IPNs (L/S and S/L) with particularly notable mechanical properties.
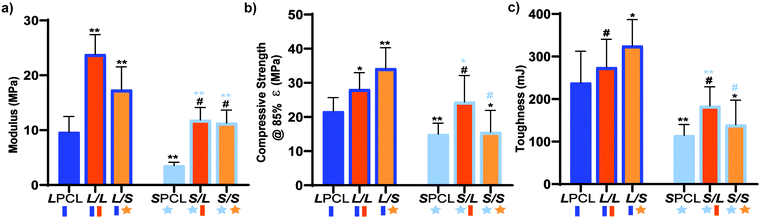 |
| | Fig. 4 Compressive mechanical properties were compared including (a) E, (b) CS, and (c) toughness; *p < 0.05, **p < 0.01, #p > 0.05. Note: the black color-coded statistics are compared to the LPCL and light blue color-coded statistics are compared to SPCL. | |
Self-fitting properties.
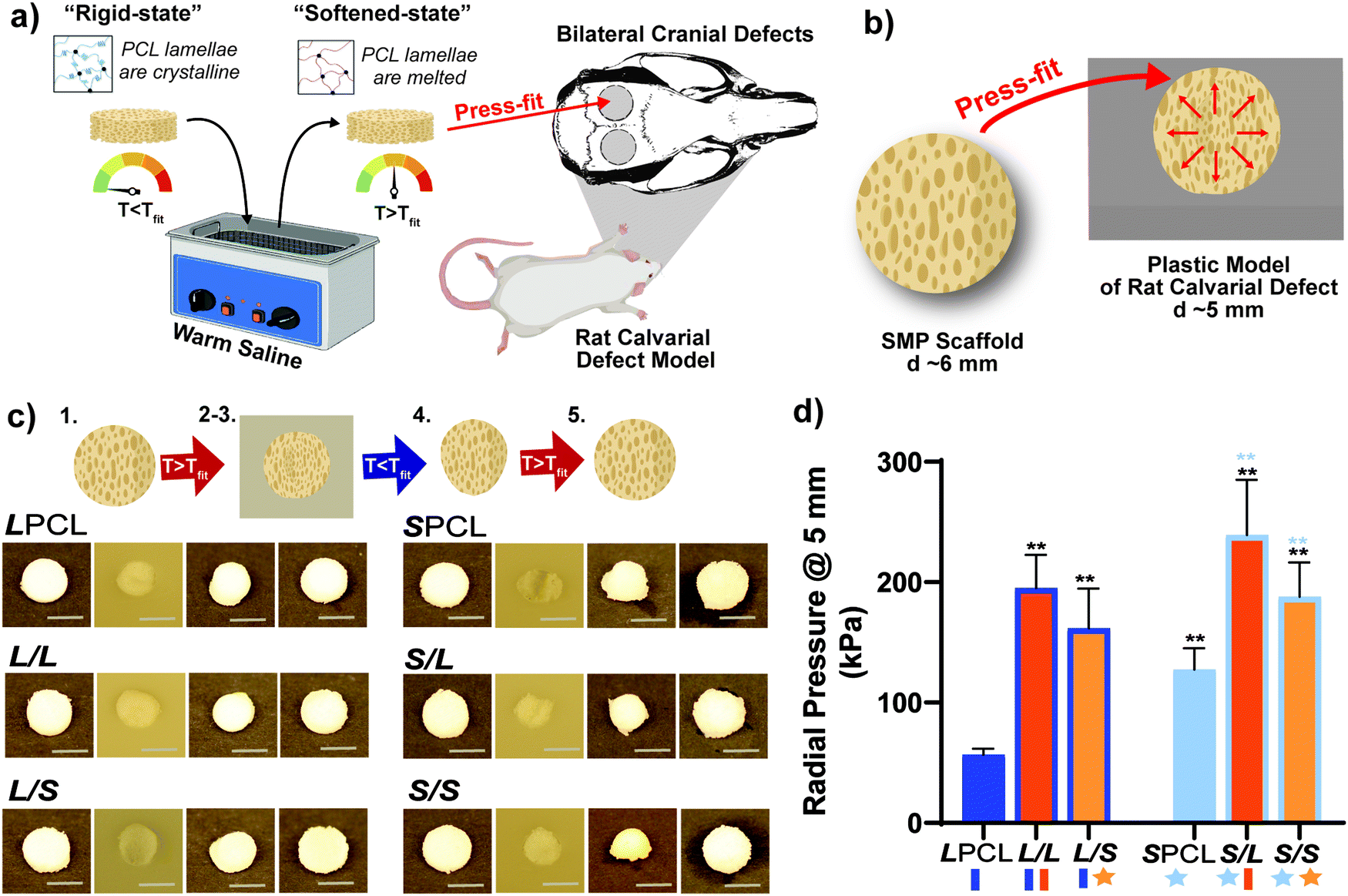
Scaffold specimens (d ∼6 mm × t ∼2 mm) were press-fitted into a plastic model defect (d ∼5 mm × t ∼2 mm). This defect represented a rat bilateral calvarial defect model of the same dimensions, typically used as an entry-level model for bone defect healing studies.49,58 A slighter larger scaffold diameter was selected to promote contact along the defect perimeter. Herein, scaffolds were fitted in the same fashion envisioned a clinical setting (Fig. 5a and b). A Tfit was the minimum saline bath temperature that in just 1 minute produced a softened, malleable scaffold: ∼55 °C for linear-PCL-based and ∼45 °C for star-PCL-based scaffolds. A sequence of steps was used to assess self-fitting and ultimately quantify Rf and Rr (Fig. 5c and Fig. S9, ESI†). Following submersion in saline at Tfit for 1 minute [step 1], all scaffolds were successfully press-fitted into defects (i.e. expanded via shape recovery to fill the defect) [step 2]. After just 2 minutes within the defect, scaffolds returned to their relatively rigid state (i.e. underwent shape fixation in new shape within the defect) [step 3]. Next, scaffolds were removed from the defect and allowed to sit for 2 min (to determine shape fixity) [step 4] and reheated at Tfit in saline for 1 minute (to determine shape recovery) [step 5]. For both cycles, these values were consistently at or near 100% for all scaffolds. These results further validate that the semi-IPN design, based on any combination of linear-PCL-DA or star-PCL-TA and both linear-PLLA or star-PLLA, does not compromise shape memory behavior. However, as osteonecrosis begins to occur with exposure to temperatures ≥50 °C,59 the lower Tfit of star-PCL-based scaffolds (i.e.SPCL, S/L, and S/S) is more “tissue-safe”. Furthermore, the observed Tfit of 45 °C is considered ideal for self-fitting CMF bone scaffolds, as it is sufficiently above Tbody and so exists in a rigid state within the defect to support healing.
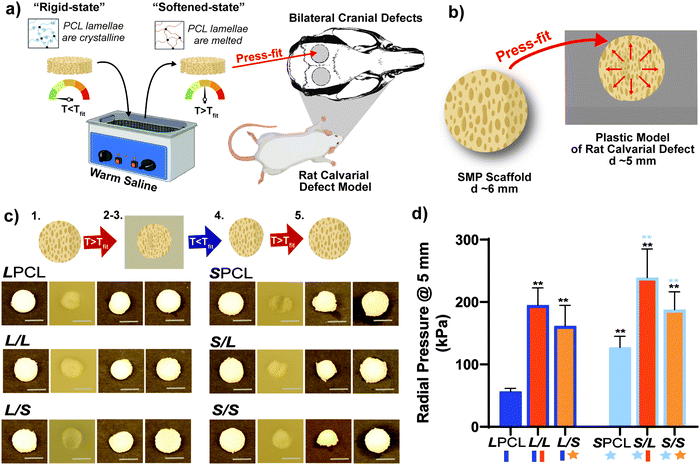 |
| | Fig. 5 (a) Shape memory testing was performed to mimic a bilateral rat calvarial defect model in vivo study. (b) Scaffolds were designed to be slightly larger than the model defect, so the warm scaffold will exert a force on the defect edges, as shown in the schematic. (c) All compositions were able to be press-fitted into a plastic model defect and demonstrated excellent shape fixity/recovery. Protocol: following submersion in saline at Tfit for 1 minute [step 1], all scaffolds were successfully press-fitted into defects (i.e. expanded via shape recovery to fill the defect) [step 2]. After just 2 minutes within the defect, scaffolds returned to their relatively rigid state (i.e. underwent shape fixation in new shape within the defect) [step 3]. Next, scaffolds were removed from the defect and allowed to sit for 2 min (to determine shape fixity) [step 4] and reheated at Tfit in saline for 1 minute (to determine shape recovery) [step 5]. (d) Radial expansion pressure tested at Tfit; *p < 0.05, **p < 0.01. Note: black color-coded statistics are compared to LPCL and blue color-coded statistics are compared to SPCL. | |
Radial pressure.
For the first time, we report the radial pressures exerted by the SMP scaffolds during self-fitting at their Tfit to quantify the force exerted by the scaffold against the defect edges, driven by shape recovery (Fig. 5d). The pressure was monitored while a scaffold (d ∼6mm × t ∼2 mm), initially loaded into a bore (d ∼6.5 mm) at RT, was heated to its Tfit and the bore diameter then reduced to that of a calvarial defect (d ∼5 mm). Versus the LPCL control (∼57 kPa), radial pressure significantly increased for the L/L (∼195 kPa) and L/S (∼162 kPa) semi-IPNs, attributed to the rigid PLLA. The radial pressure of the SPCL (∼127 kPa) was also much higher than the LPCL, which may be attributed to its higher crosslink density. A further substantial increase in radial pressure was noted for the S/L (∼239 kPa) and S/S (∼188 kPa) versus the SPCL, again stemming from the rigid PLLA. Thus, the substantial gains in radial pressure (versus the LPCL control) observed for the SPCL and all semi-IPNs affords improved scaffold expansion toward defect edges during self-fitting, which is anticipated to promote osseointegration and overall implant stability in vivo.
Solution viscosity and scaffold scale-up
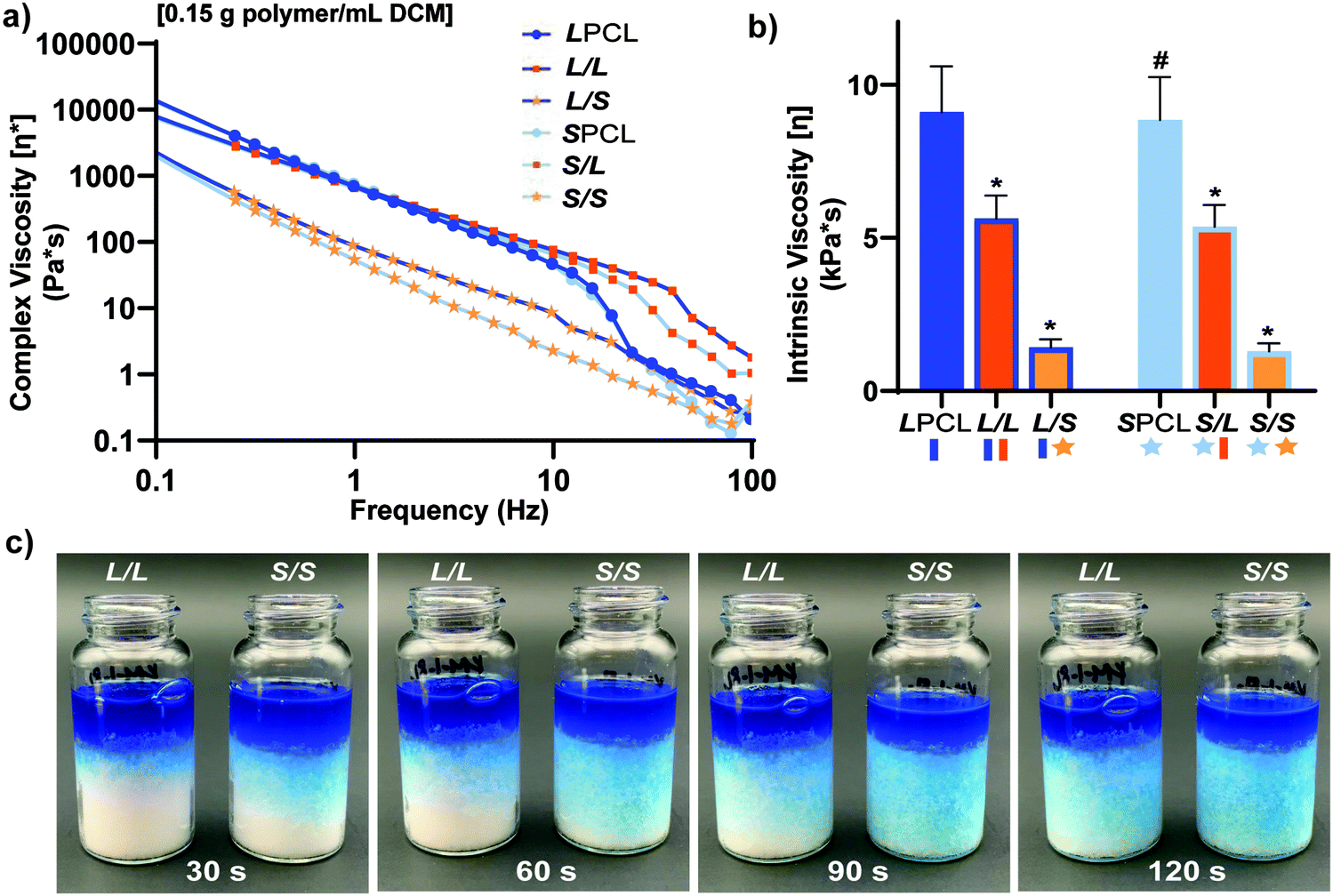
In the aforementioned analyses, SMP scaffolds were prepared with a diameter of ∼6 mm (biopsy punch of a scaffold with d ∼12 mm); this size is appropriate for bilateral rat calvarial defect studies. However, larger scaffolds are necessary for critically-sized defects in animal models (up to d ∼22 mm)60 and eventually for human patients. While centrifugation to drive diffusion is permissible for small scaffolds that are prepared in scintillation vials, this is not the case for larger scaffolds. Because star-polymers are known to have a lowered solution viscosity,43,44 we expected that SMP scaffolds prepared with such would more readily permit the preparation of larger specimens. First, the complex viscosity [η*] of scaffold precursor solutions were determined over a frequency sweep (Fig. 6a) and the intrinsic viscosity calculated by extrapolation to a zero-shear rate (Fig. 6b). Both 100% PCL controls (LPCL and SPCL), exhibited a relatively high intrinsic viscosity (∼9 kPa*s). For semi-IPN macromer solutions containing linear-PCL, intrinsic viscosity was reduced with star-PLLA (L/S; ∼1 kPa*s) versus with linear-PLLA (L/L; ∼6 kPa*s). Semi-IPN macromer solutions based on star-PCL were likewise reduced, particularly with star-PLLA (S/S; ∼1 kPa*s) versus with linear-PLLA (S/L; ∼6 kPa*s). Because of their relatively high and low intrinsic viscosities, respectively, L/L and S/S semi-IPN macromer solutions were selected to prepare larger scaffold specimens. First, using fused salt templates prepared in scintillation vials, diffusion of the precursor solutions containing food coloring was monitored (Fig. 6c and Video S1, ESI†). Owing to its lower intrinsic viscosity, the S/S solution diffused more quickly to the bottom of the template (∼90 s) versus the L/L solution (>120 s). Next, L/L and S/S were prepared as actual scaled-up, “larger” scaffolds, using 100 mL beakers (50.0 g salt). Analogous “regular” scaffolds were prepared in the 20 mL vials (10.0 g salt), but the diameter was not reduced from ∼12 mm using a biopsy punch. Thus, the “larger” scaffolds had a diameter and volume that was 2X and 5X, respectively, that of the “regular” scaffolds (Fig. S10a and b, ESI†). For the “large” S/S scaffolds, a total of four 2 mm thick specimens (i.e. slices) could be harvested versus just three 2 mm thick slices for the “larger” L/L scaffolds (Fig. 7). This stemmed from a lack of diffusion, wherein the L/L macromer solution did not reach the bottom of the mold, rendering the bottom portion deficient. While density did not change according to gravimetric analysis (Fig. S10c, ESI†), low magnification optical microscopy revealed that S/S demonstrated superior uniformity of pores throughout versus the L/L. Full porogen leaching has been previously noted as a limitation in SCPL fabrication;61 however, herein the NaCl porogen used in fabrication was shown to be fully removed even from “larger” scaffolds, likely owing to the use of a fused salt template resulting in interconnected pores. This was validated via SEM and EDS mapping to show that the scaffolds did not contain any appreciable amount of Na or Cl (Fig. S11, ESI†). Lastly, as a further indicator of their utility as a surgical product to treat bone defects, the S/S scaffold was able to be trimmed with a scissor and also sutured (Fig. 8a).
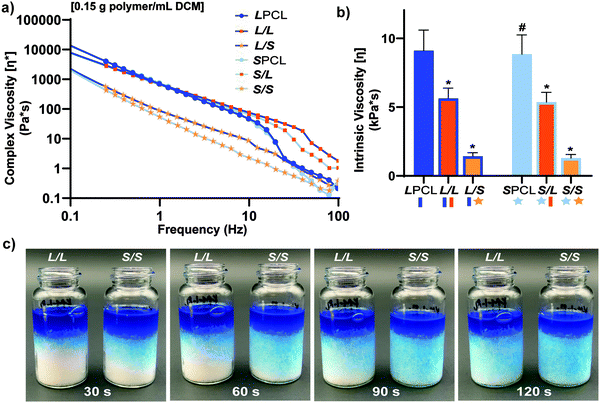 |
| | Fig. 6 For scaffold precursor solutions: (a) Complex viscosity [η*] versus frequency, (b) intrinsic viscosity (*p < 0.05, **p < 0.01 compared to LPCL). (c) L/L and S/S semi-IPN macromer solution diffusion through a template using 15.0 g salt in a scintillation vial. | |
 |
| | Fig. 7 Photos and optical microscopy (5x) of scaled-up, “large” L/L and S/S scaffolds (d ∼24 mm) demonstrating superior macromer diffusion, and more uniform pores, for the S/S composition. | |
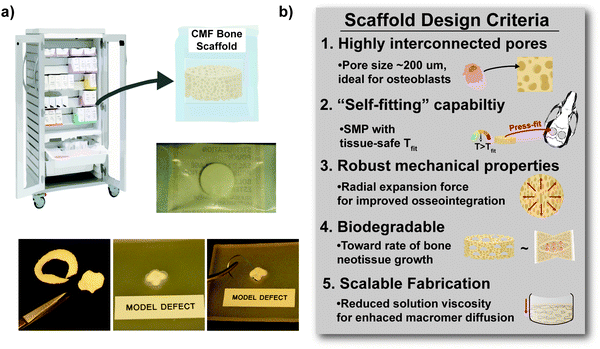 |
| | Fig. 8 (a) The scaled-up, larger scaffold specimens (d ∼24 mm × t ∼2 mm) were able to be easily cut to custom defect geometries with scissors and could hold a suture. (b) The S/S semi-IPN (i.e. comprised of star-PCL-TA and star-PLLA) achieved five scaffold design criteria intended for an off-the-shelf surgical product to heal bone defects. | |
Conclusions
Towards improving the utility of “self-fitting” SMP scaffolds, semi-IPN compositions were prepared with star-polymer architectures. Originally prepared from linear-PCL-DA and linear-PLLA (75/25 wt%), the L/L semi-IPN exhibited improved rigidity and accelerated degradation versus linear-PCL-DA (LPCL). In this work, the semi-IPN based on star-PCL-TA and star-PLLA (S/S) (75/25 wt%) exhibited distinct advantages and fulfilled key criteria as a surgical product to treat CMF bone defects (Fig. 8b). The pore size (∼220 μm) and pore interconnectivity, to promote osteogenesis and to favorably allow neotissue infiltration, was maintained using the SCPL fabrication protocol. While this study was limited to in vitro material characterization, the LPCL control scaffold had been previously shown to support osteogenesis, which was improved with the addition of cell adhesion motifs and bioactive coatings.12,13 The new scaffold compositions are expected to yield favorable and potentially improved results in such cell culture studies. Importantly, self-fitting of the S/S semi-IPN scaffold could be performed at a more tissue-safe, lower Tfit (∼45 °C) versus for the L/L semi-IPN scaffold (∼55 °C). The S/S semi-IPN exhibited similar rigidity versus the original LPCL, although it was somewhat less rigid and strong versus the L/L semi-IPN. Despite this, radial pressure during shape recovery at Tfit for the S/S semi-IPN was shown to be significantly improved versus for the LPCL and was similar to that of the L/L semi-IPN. This ability to expand with greater force toward the defect edges during self-fitting is expected to improve scaffold osseointegration and implant stability prior to healing. Additionally, the S/S semi-IPN exhibited even faster degradation versus the L/L semi-IPN, and so is expected to better promote neotissue infiltration. Finally, the reduced intrinsic viscosity of S/S semi-IPN precursor solution improved its diffusion through the salt template (in the absence of centrifiguation), permitting larger scafffolds to be prepared. Thus, star-polymer architectures were successfully leveraged to create “self-fitting” SMP scaffolds with properties better suited for treatment of CMF bone defects.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by NIH NIDCR 1R01DE025886-01A1. The use of the Texas A&M Microscopy and Imaging Center is acknowledged. SEM acquisition was supported in part by the National Science Foundation under Grant No. DBI-0116835.
References
- S. Parithimarkalaignan and T. V. Padmanabhan, J. Indian Prosthodont. Soc., 2013, 13, 2–6 CrossRef CAS PubMed.
- K. Doi, T. Kubo, Y. Makihara, H. Oue, K. Morita, Y. Oki, S. Kajihara and K. Tsuga, J. Appl. Oral Sci., 2016, 24, 325–331 CrossRef CAS PubMed.
- E. Neovius and T. Engstrand, J. Plast. Reconstr. Aesthet. Surg., 2010, 63, 1615–1623 CrossRef PubMed.
- J. H. Phillips and B. A. Rahn, Plast. Reconstr. Surg., 1990, 85, 891–897 CrossRef CAS PubMed.
- H. G. Moghadam, Implant Dent., 2009, 18 Search PubMed.
- K. L. Low, S. H. Tan, S. H. S. Zein, J. A. Roether, V. Mouriño and A. R. Boccaccini, J. Biomed. Mater. Res., Part B, 2010, 94B, 273–286 CAS.
- A. M. Shah, H. Jung and S. Skirboll, Neurosurg. Focus, 2014, 36, E19 Search PubMed.
- A. B. Lennon and P. J. Prendergast, J. Biomech., 2002, 35, 311–321 CrossRef CAS PubMed.
- D. C. Lobb, B. R. DeGeorge and A. B. Chhabra, J. Hand Surg., 2019, 44, 497–505.e492 CrossRef PubMed.
- J. F. Orr, N. J. Dunne and J. C. Quinn, Biomaterials, 2003, 24, 2933–2940 CrossRef CAS PubMed.
- A. Haleem and M. Javaid, Clin. Epidemiol. Glob. Health, 2019, 7, 571–577 CrossRef.
- D. Zhang, O. J. George, K. M. Petersen, A. C. Jimenez-Vergara, M. S. Hahn and M. A. Grunlan, Acta Biomater., 2014, 10, 4597–4605 CrossRef CAS PubMed.
- J. D. Erndt-Marino, D. J. Munoz-Pinto, S. Samavedi, A. C. Jimenez-Vergara, P. Diaz-Rodriguez, L. Woodard, D. Zhang, M. A. Grunlan and M. S. Hahn, ACS Biomater. Sci. Eng., 2015, 1, 1220–1230 CrossRef CAS PubMed.
- D. Zhang, K. M. Petersen and M. A. Grunlan, ACS Appl. Mater. Interfaces, 2013, 5, 186–191 CrossRef CAS PubMed.
- L. N. Nail, D. Zhang, J. L. Reinhard and M. A. Grunlan, JoVE, 2015, e52981, DOI:10.3791/52981.
- L. N. Woodard, K. T. Kmetz, A. A. Roth, V. M. Page and M. A. Grunlan, Biomacromolecules, 2017, 18, 4075–4083 CrossRef CAS PubMed.
- S.-H. Park, E. S. Gil, H. J. Kim, K. Lee and D. L. Kaplan, Biomaterials, 2010, 31, 6162–6172 CrossRef CAS PubMed.
- S. J. Hollister, Nat. Mater., 2005, 4, 518–524 CrossRef CAS PubMed.
- R. Langer and D. A. Tirrell, Nature, 2004, 428, 487–492 CrossRef CAS PubMed.
- B. Dhandayuthapani, Y. Yoshida, T. Maekawa and D. S. Kumar, Int. J. Polym. Sci., 2011, 2011, 290602 Search PubMed.
- S. Bose, M. Roy and A. Bandyopadhyay, Trends Biotechnol., 2012, 30, 546–554 CrossRef CAS PubMed.
- L. N. Woodard, V. M. Page, K. T. Kmetz and M. A. Grunlan, Macromol. Rapid Commun., 2016, 37, 1972–1977 CrossRef CAS PubMed.
- L. N. Woodard and M. A. Grunlan, ACS Biomater. Sci. Eng., 2019, 5, 498–508 CrossRef CAS PubMed.
- M. R. Pfau, K. G. McKinzey, A. A. Roth and M. A. Grunlan, Biomacromolecules, 2020, 21, 2493–2501 CrossRef CAS PubMed.
- H. Tsuji and Y. Ikada, J. Appl. Polym. Sci., 1998, 67, 405–415 CrossRef CAS.
- V. Arias, A. Höglund, K. Odelius and A.-C. Albertsson, Biomacromolecules, 2014, 15, 391–402 CrossRef CAS PubMed.
- A. Ostafinska, I. Fortelny, M. Nevoralova, J. Hodan, J. Kredatusova and M. Slouf, RSC Adv., 2015, 5, 98971–98982 RSC.
- J. Urquijo, G. Guerrica-Echevarría and J. I. Eguiazábal, J. Appl. Polym. Sci., 2015, 132, 42641 CrossRef.
- W. Wu, W. Wang and J. Li, Prog. Polym. Sci., 2015, 46, 55–85 CrossRef CAS.
- A. Jahandideh and K. Muthukumarappan, Eur. Polym. J., 2017, 87, 360–379 CrossRef CAS.
- A. Michalski, M. Brzezinski, G. Lapienis and T. Biela, Prog. Polym. Sci., 2019, 89, 159–212 CrossRef CAS.
- Y. Li and T. Kissel, Polymer, 1998, 39, 4421–4427 CrossRef CAS.
- E. S. Kim, B. C. Kim and S. H. Kim, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 939–946 CrossRef CAS.
- A. Ghoorchian and N. B. Holland, Biomacromolecules, 2011, 12, 4022–4029 CrossRef CAS PubMed.
- A. Breitenbach, Y. X. Li and T. Kissel, J. Controlled Release, 2000, 64, 167–178 CrossRef CAS PubMed.
- J. Burke, R. Donno, R. d’Arcy, S. Cartmell and N. Tirelli, Biomacromolecules, 2017, 18, 728–739 CrossRef CAS PubMed.
- B. Atthoff, M. Trollsås, H. Claesson and J. L. Hedrick, Macromol. Chem. Phys., 1999, 200, 1333–1339 CrossRef CAS.
- C.-F. Huang, S.-W. Kuo, H.-C. Lin, J.-K. Chen, Y.-K. Chen, H. Xu and F.-C. Chang, Polymer, 2004, 45, 5913–5921 CrossRef CAS.
- P. E. Theodorakis, A. Avgeropoulos, J. J. Freire, M. Kosmas and C. Vlahos, Macromolecules, 2006, 39, 4235–4239 CrossRef CAS.
- C. Singh and A. C. Balazs, Polym. Int., 2000, 49, 469–471 CrossRef CAS.
- X. Bian, B. Zhang, B. Sun, Z. Sun, S. Xiang, G. Li and X. Chen, Polym. Eng. Sci., 2016, 56, 1125–1137 CrossRef CAS.
- S. C. Mauck, S. Wang, W. Ding, B. J. Rohde, C. K. Fortune, G. Yang, S.-K. Ahn and M. L. Robertson, Macromolecules, 2016, 49, 1605–1615 CrossRef CAS.
- Y. Lu, L. An and Z.-G. Wang, Macromolecules, 2013, 46, 5731–5740 CrossRef CAS.
- J. F. Douglas, J. Roovers and K. F. Freed, Macromolecules, 1990, 23, 4168–4180 CrossRef CAS.
- S. Kaihara, S. Matsumura, A. G. Mikos and J. P. Fisher, Nat. Protoc., 2007, 2, 2767–2771 CrossRef CAS PubMed.
- C. G. Pitt, F. I. Chasalow, Y. M. Hibionada, D. M. Klimas and A. Schindler, J. Appl. Polym. Sci., 1981, 26, 3779–3787 CrossRef CAS.
- K. Fukushima, Polym. Degrad. Stab., 2011, v. 96, pp. 2120 CrossRef -2129-2011 v.2196 no. 2112.
- C. Zong, D. Xue, W. Yuan, W. Wang, D. Shen, X. Tong, D. Shi, L. Liu, Q. Zheng, C. Gao and J. Wang, Eur. Cells Mater., 2010, 20, 109–120 CrossRef CAS PubMed.
- Y. Shirakata, T. Nakamura, Y. Shinohara, K. Taniyama, K. Sakoda, T. Yoshimoto and K. Noguchi, J. Mater. Sci.: Mater. Med., 2014, 25, 899–908 CrossRef CAS PubMed.
- M. A. Wierzbicki, J. Bryant, M. W. Miller, B. Keller and D. J. Maitland, J. Mech. Behav. Biomed. Mater., 2016, 59, 156–167 CrossRef PubMed.
- N. Abbasi, S. Hamlet, R. M. Love and N.-T. Nguyen, J. Sci. Adv. Mater. Med., 2020, 5, 1–9 Search PubMed.
- S.-P. Lyu, F. S. Bates and C. W. Macosko, AIChE J., 2000, 46, 229–238 CrossRef CAS.
- E. Van Hemelrijck, P. Van Puyvelde, S. Velankar, C. W. Macosko and P. Moldenaers, J. Rheol., 2003, 48, 143–158 CrossRef.
- F. v. Burkersroda, L. Schedl and A. Göpferich, Biomaterials, 2002, 23, 4221–4231 CrossRef.
- H. Sun, L. Mei, C. Song, X. Cui and P. Wang, Biomaterials, 2006, 27, 1735–1740 CrossRef CAS PubMed.
- S. Doppalapudi, A. Jain, W. Khan and A. J. Domb, Polym. Adv. Technol., 2014, 25, 427–435 CrossRef CAS.
- M. A. Velasco, C. A. Narváez-Tovar and D. A. Garzón-Alvarado, BioMed Res. Int., 2015, 2015, 729076 Search PubMed.
- C. Bosch, B. Melsen and K. Vargervik, J. Craniofac. Surg., 1998, 9, 310–316 CrossRef CAS PubMed.
- C. Timon and C. Keady, Cureus, 2019, 11, e5226–e5226 Search PubMed.
- C. Szpalski, J. Barr, M. Wetterau, P. B. Saadeh and S. M. Warren, Neurosurg. Focus, 2010, 29, E8 Search PubMed.
- H. Janik and M. Marzec, Mater. Sci. Eng., C, 2015, 48, 586–591 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tb02987d |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a and
Melissa A.
Grunlan
a and
Melissa A.
Grunlan
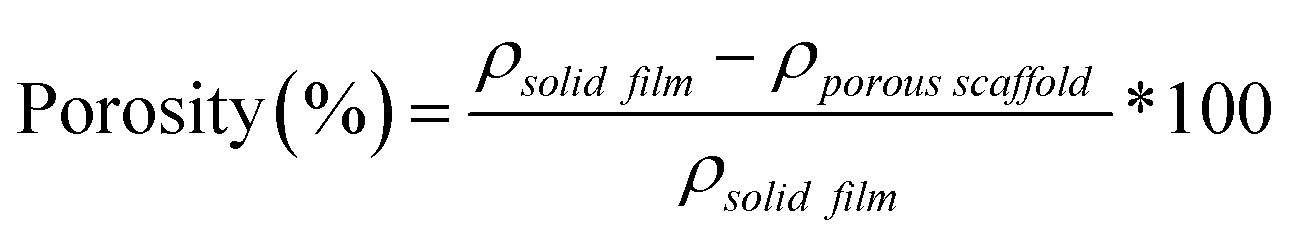
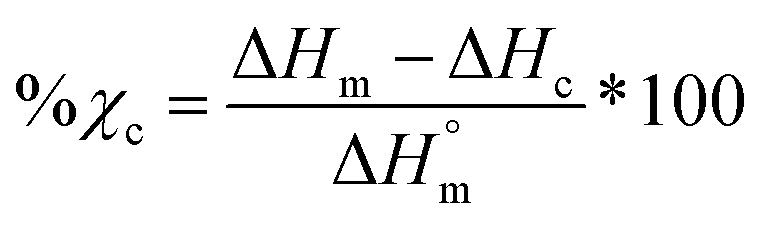
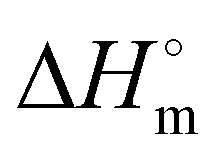 is the theoretical value for 100% crystalline PCL (139.5 J g−1)46 or PLLA (93.0 J g−1).47
is the theoretical value for 100% crystalline PCL (139.5 J g−1)46 or PLLA (93.0 J g−1).47