
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePotent strategy towards strongly emissive nitroaromatics through a weakly electron-deficient core†
Bartłomiej
Sadowski‡
 a,
Marzena
Kaliszewska
b,
Yevgen M.
Poronik
a,
Małgorzata
Czichy
c,
Patryk
Janasik
c,
Marzena
Banasiewicz
d,
Dominik
Mierzwa
a,
Wojciech
Gadomski
b,
Trevor D.
Lohrey
ef,
John A.
Clark
g,
Mieczysław
Łapkowski
*c,
Bolesław
Kozankiewicz
*d,
Valentine I.
Vullev
*ghij,
Andrzej L.
Sobolewski
*d,
Piotr
Piatkowski
*b and
Daniel T.
Gryko
*a
a,
Marzena
Kaliszewska
b,
Yevgen M.
Poronik
a,
Małgorzata
Czichy
c,
Patryk
Janasik
c,
Marzena
Banasiewicz
d,
Dominik
Mierzwa
a,
Wojciech
Gadomski
b,
Trevor D.
Lohrey
ef,
John A.
Clark
g,
Mieczysław
Łapkowski
*c,
Bolesław
Kozankiewicz
*d,
Valentine I.
Vullev
*ghij,
Andrzej L.
Sobolewski
*d,
Piotr
Piatkowski
*b and
Daniel T.
Gryko
*a
aInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: dtgryko@icho.edu.pl
bFaculty of Chemistry, University of Warsaw, Zwirki i Wigury 101, 02-089 Warsaw, Poland. E-mail: piatek@chem.uw.edu.pl
cFaculty of Chemistry, Silesian University of Technology, Strzody 9, 44-100 Gliwice, Poland. E-mail: mieczyslaw.lapkowski@polsl.pl
dInstitute of Physics, Polish Academy of Sciences, Aleja Lotnikow 32/46, 02-668 Warsaw, Poland. E-mail: sobola@ifpan.edu.pl; kozank@ifpan.edu.pl
eDepartment of Chemistry, University of California, Berkeley, 420 Latimer Hall, Berkeley, CA, USA
fChemical Sciences Division, Lawrence Berkeley National Laboratory, 1 Cyclotron Road, Berkeley, CA, USA
gDepartment of Bioengineering, University of California, Riverside, 900 University Ave., Riverside, CA 92521, USA. E-mail: vullev@ucr.edu
hDepartment of Chemistry, University of California, Riverside, 900 University Ave., Riverside, CA 92521, USA
iDepartment of Biochemistry, University of California, Riverside, 900 University Ave., Riverside, CA 92521, USA
jMaterials Science and Engineering Program, University of California, Riverside, 900 University Ave., Riverside, CA 92521, USA
First published on 29th September 2021
Abstract
Nitroaromatics seldom fluoresce. The importance of electron-deficient (n-type) conjugates, however, has inspired a number of strategies for suppressing the emission-quenching effects of the strongly electron-withdrawing nitro group. Here, we demonstrate how such strategies yield fluorescent nitroaryl derivatives of dipyrrolonaphthyridinedione (DPND). Nitro groups near the DPND core quench its fluorescence. Conversely, nitro groups placed farther from the core allow some of the highest fluorescence quantum yields ever recorded for nitroaromatics. This strategy of preventing the known processes that compete with photoemission, however, leads to the emergence of unprecedented alternative mechanisms for fluorescence quenching, involving transitions to dark nπ* singlet states and aborted photochemistry. Forming nπ* triplet states from ππ* singlets is a classical pathway for fluorescence quenching. In nitro-DPNDs, however, these ππ* and nπ* excited states are both singlets, and they are common for nitroaryl conjugates. Understanding the excited-state dynamics of such nitroaromatics is crucial for designing strongly fluorescent electron-deficient conjugates.
Introduction
The nitro group is well known for its ability to quench fluorescence when appended to a fluorophore skeleton.1–5 It effectively reduces the emission brightness from one- and two-photon excitation – a crucial consideration for the design and implementation of fluorescent probes for biomedical imaging.6,7 Nevertheless, the immense electron-withdrawing strength of the nitro group makes it a highly attractive substituent for the pursuit of electron-deficient, i.e., n-type, organic conjugates and potent photooxidants.8–11 Decoupling the nitro substituents from the states involved in radiative transitions leads to fluorescence with substantial quantum yields (Φfl) (Fig. 1A);12,13 however, such a strategy negates the benefits that may be gained from the electron-withdrawing strength of this substituent.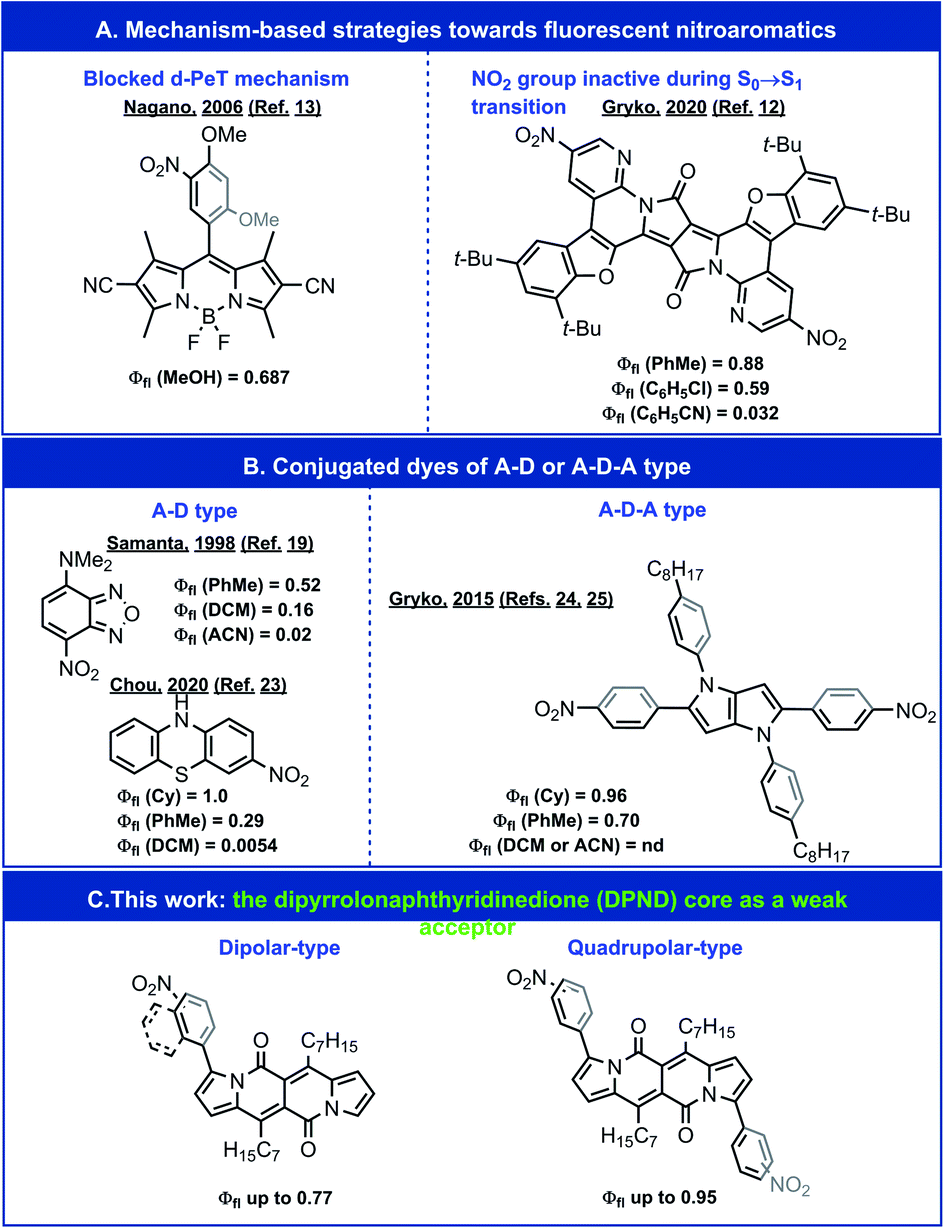 | ||
| Fig. 1 Exemplary highly emissive nitroaromatics. A – an acceptor moiety containing NO2 group. D – a donor moiety. nd – not detected. | ||
Despite their importance, many aspects of the relationship between molecular structure and fluorescence intensity of nitroaromatics still remain puzzling. For a wide range of simple nitro-containing derivatives, such as nitrobenzene,14 1-nitropyrene,15 1-nitronaphthalene,16 and 9-nitroanthracene,17 triplet manifolds, i.e., intersystem crossing (ISC), present the most common pathway for energy dissipation, leading to a drastic decrease in the lifetimes of the fluorescent excited states. Conversely, the strongly electron-withdrawing nitro group induces a charge-transfer (CT) character in the excited states, which tends to reduce the rate of ISC.18 In fact, for about half a century, amine derivatives of nitrobenzodioxazole (NBD) have been favoured as small fluorescent probes for biological samples (Fig. 1B).19,20 A recent expansion of the variety of amino-NBD analogues widens their working range to the near infrared spectral region.21 Expanding this push–pull strategy to other nitroaromatics can also result in some observable fluorescence (Fig. 1B).22,23
Inducing a CT character in the excited states, however, presents an alternative set of challenges. (1) An increase in the CT character, usually accompanied by dihedral twisting, can spatially separate the natural transition orbitals (NTOs), which decreases the oscillator strength of the radiative decays to negligibly low values and renders such CT states dark. (2) Medium polarity stabilizes polar CT states and brings them closer to the potential-energy surface (PES) of the ground state, which opens pathways for efficient deactivation via internal conversion (IC). These features warrant tuning of the CT character of the excited states in order to balance its benefits with the limitations it imposes.
Transferring this strategy for dipolar donor–acceptor (D–A) conjugates to chromophores with quadrupolar symmetry, e.g., acceptor–donor–acceptor (A–D–A) structures, presents an alternative paradigm for fluorescent nitroaromatics.24 In addition to their importance for non-linear optical applications, quadrupolar dyes tend to manifest pronounced solvatofluorochromism.24–28 That is, medium polarity has no effect on their absorption maxima because of the quadrupolar ground-state symmetry, while CT upon photoexcitation leads to symmetry breaking, leading to dipolar fluorescent states and polarity-induced bathochromic shifts of the emission.29 This property is crucially beneficial for imaging where the excitation wavelength is invariant to the media, while the fluorescence acts as a reporter for the polarity of the surrounding microenvironment.
For A–D–A dyes, tuning the coupling between electron-donating chromophore cores (D) and the nitro groups, which are components of the acceptors (A), allows for quantitative Φfl in solvents with low polarity.24 Media with even moderate polarity, such as dichloromethane (DCM), however, quenches the fluorescence of these nitroaromatic dyes.24
We sought, and now report, an effective approach to improving the emission properties of fluorophores containing nitro groups. In addition to tuning the electronic coupling of the core with the nitro groups, we focus on the utility of weakening the electron donor in D–A and A–D–A dyes. Our design of a series of nitroaromatic D–A and A–D–A conjugates comprise a weakly electron-donating dipyrrolonaphthyridinedione30 (DPND) as a core (Fig. 1C). In addition to the high Φfl values that we observe in DCM, many of these nitro-DPND conjugates fluoresce even in polar solvents, such as acetonitrile (ACN). Theoretical analysis reveals that for about half of these dyes, a low-lying nπ* singlet state, rather than a dark CT state, provides pathways for IC. Also, the dihedral twisting along the PES of the lowest singlet excited state accompanies the formation of a new bond that breaks after the IC transition to the ground state. Despite the years of discussion and hypotheses about such transient covalent interactions along excited-state reaction pathways,31,32 it was only recently that experimental proof for such processes became possible to obtain for relatively simple structures.33,34 Mechanisms involving these types of aborted photochemical reactions are unprecedented in the study of deactivation pathways in photoexcited nitroaromatics. Along with the observed fluorescence for polar media, this result presents a key consideration in the pursuit of n-type organic materials and photooxidants.
Results and discussion
Design and synthesis
DPNDs are a series of cross-conjugated heterocyclic chromophores with unique photophysical and electrochemical properties that allow them to act as week electron donors or weak electron acceptors,30,35–37 making them an excellent choice for designing the D–A and A–D–A conjugates for this study. Biaryl-like linkages enable weak coupling between the nitro group(s) and the DPND core (Fig. 2). Pd-catalyzed direct arylation35 of DPND 1 with the corresponding aryl bromides allows us to obtain the D–A and A–D–A conjugates with nitro groups at ortho, meta, para and peri positions with respect to the core (see ESI† for synthetic details). This shift in the position of the nitro groups varies their electronic coupling with the DPND moiety.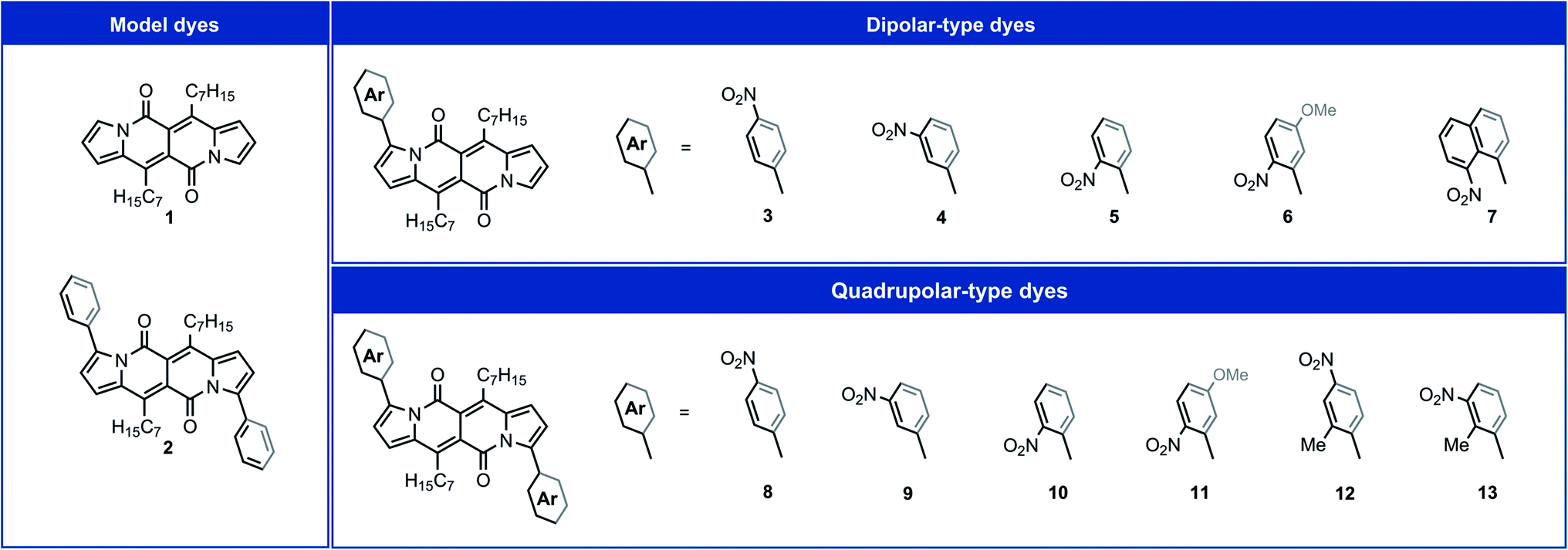 | ||
| Fig. 2 The molecular structures of DPNDs considered in this work. | ||
X-ray crystallography for DPNDs 7, 9 and 12 unambiguously shows twisted conformations of the constructed dyes (Fig. S1, S2 and Table S1 in ESI†). The average dihedral angles between adjacent aryl moieties in 9 and 12 are 51.6° and 56.1°, respectively. These values are slightly smaller than that for 2 (58.9°),38 suggesting planarity-inducing weak electronic communication between the nitro-enriched aryls and the DPND core.
Steady-state optical absorption and emission
Four aprotic solvents, n-hexane (HEX), 1,2-dichlorobenzene (DCB), dichloromethane (DCM), and acetonitrile (ACN), allowed for the exploration of the effects of medium polarity on the optical properties of the DPNDs 1–13 (Fig. 3, Table S2, and Fig. S3–S15†). In addition, we employed two ester solvents, propyl butyrate (PB) and sucrose octaacetate (SOA), for testing the effects of medium viscosity on the spectral properties of the analysed dyes.39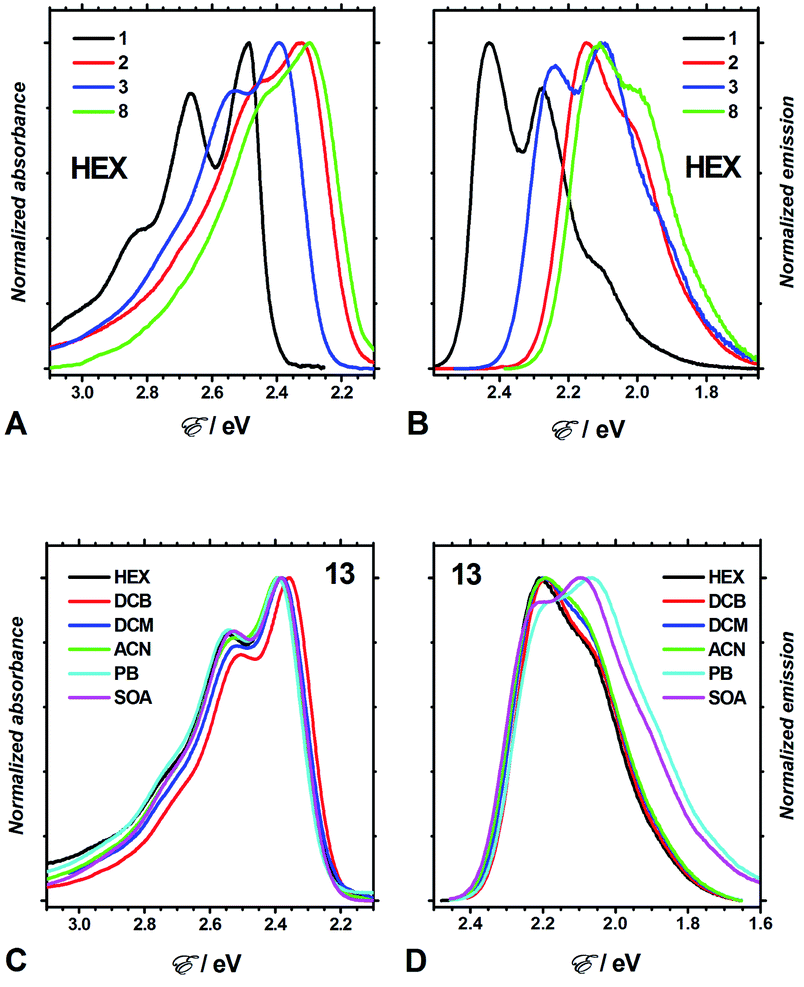 | ||
| Fig. 3 Optical absorption and emission spectra of DPND dyes for various solvents. (A and B) Absorption and fluorescence spectra of 1, 2, 3 and 8 for hexane (λex = 450 nm for 1 and 2; 480 nm for 3; and 510 nm for 8). (C and D) Solvent dependence of the absorption and fluorescence spectra of 13 (λex = 470 nm for HEX; and 505 nm for the rest of the solvents). | ||
Consistent with extension of the π-conjugated framework, substitution of the DPND core with peripheral phenyl or nitrophenyl groups bathochromically shifts the absorption and emission maxima (Fig. 3A, B, 4A and B). The absorption and emission maxima (Eabs and Efl, respectively) of the dinitrophenyl conjugates, i.e., 8–13, cluster around those of 2 for each of the solvents (Fig. 4B). The spectral maxima of the para-nitro, 8, and ortho-nitro, 10 and 11, conjugates are similar or bathochromically shifted in comparison with those of 2. Conversely, Eabs and Efl of the meta-nitro dyes, 9 and 13, are larger than those of 2 (Fig. 4B). These findings correlate with donor–acceptor electronic coupling between meta substituents that is weaker than that between para and ortho ones.40 Placing methyl groups at the ortho positions, such as in 12 and 13, causes hypsochromic shifts in comparison with the non-methylated analogues, 8 and 9, respectively (Fig. 4B). These data are consistent with a sterically-induced increase in the dihedral between nitrophenyl and the DPND core, which decreases their electronic coupling.
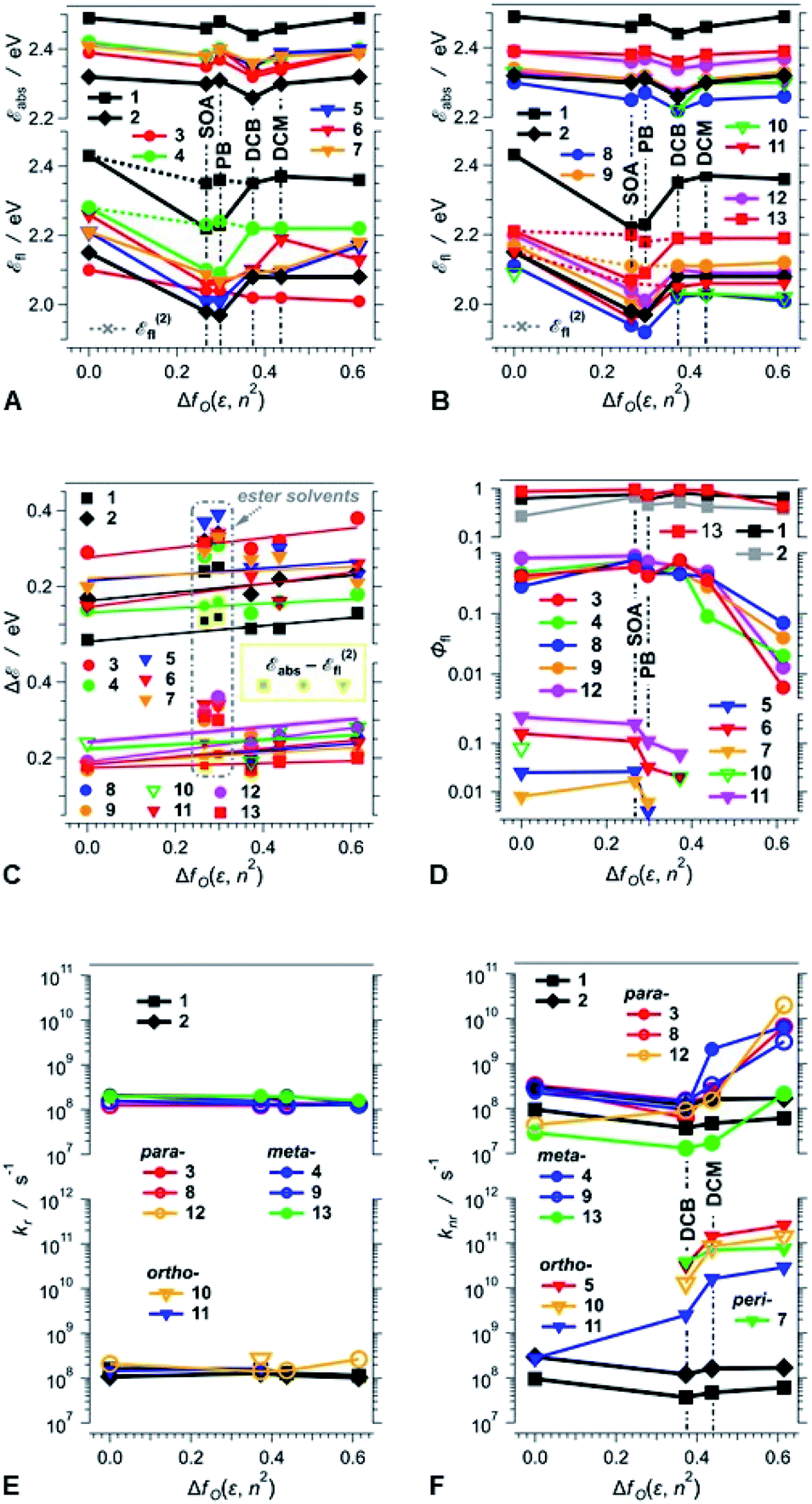 | ||
| Fig. 4 Dependence of the photophysical properties of the DPND dyes on the solvent polarity expressed in terms of the Onsager function, fO(x) = 2(x − 1)/(2x + 1), fO(ε, n2) = fO(ε) − fO(n2). (A and B) Dependence of the absorption and emission maxima, Eabs and Efl, respectively, on fO(ε, n2). For the ester solvents, PB and SOA, the conversion of the fluorescence spectra from wavelength to energy scales, i.e., F(E) = F(λ)E−5, shifts the order of the vibronic maxima, i.e., the highest (hypsochromically positioned) peaks in a wavelength scale becomes the second highest, E(2)fl, in an energy scale. The dotted lines connect with E(2)fl for PB and SOA. (C) Polarity effects on the Stokes' shifts, ΔE = Eabs − Efl. For PB and SOA, The yellow-framed markers represent the Stokes' shifts calculated using E(2)fl. The lines represent the best linear fits of ΔE vs. fO(ε, n2) for HEX, DCB, DCM and ACN. (D) Polarity effects on the fluorescence quantum yields, Φfl. (E and F) Polarity dependence of the radiative, kr, and non-radiative, knr, rate constants estimated from the fluorescence quantum yields and the excited-state lifetimes (τ) for samples manifesting Φfl >10−4, i.e., kr = Φflτ−1 and knr = (1 − Φfl)τ−1. For weakly fluorescent compounds, i.e., (1 − Φfl) ≈ 1, knr ≈ τ−1. | ||
The spectral maxima of the mono-nitrophenyl DPNDs show similar trends (Fig. 4A). In comparison to the mono- and di-ortho-nitrophenyl conjugates, 5 and 10, adding methoxy substituents at para-positions relative to the nitro groups, as in 6 and 11, causes slight hypsochromic shifts (Fig. 4A and B). This suggests a weakening of the effects of the nitro groups induced by the electron-donating ones. This weakening effect can also provide a plausible explanation for the deviated optical behaviour of 13, i.e., 13 displays the most hypsochromic spectral maxima among the di-nitrophenyl DPNDs (Fig. 4B). Namely, methyls are weakly electron-donating, which may lessen the effect of the nitro groups in 13.
These solvent effects are quite similar for all dyes 1–13 (Fig. 4A and B). Hence, attaching nitroaryl substituents to the DPND core does not enhance the polarity effects on the energy gaps between the ground and excited states involved in the radiative transitions.
An increase in medium polarity slightly increases the Stokes' shifts (ΔE) of the DPNDs (Fig. 4C), indicating that the fluorescent S1 state is more polarized than the relaxed ground state. The slopes of the linear fits of ΔE vs. fO(ε, n2), however, are relatively small. Lippert–Mataga–Ooshika analysis41–43 reveals that the difference between the electric dipole moments, Δμ, of the S0 and the fluorescent excited states is about 2 D for dyes 4, 5, 7 and 13, containing nitro groups on the ortho or meta positions, which is similar to Δμ for the DPND 1 with no aryl substituents (Table S4†). On the other hand, dyes with two phenyl or para-nitrophenyl substituents (2, 8, 11 and 12) exhibit Δμ values between 4 and 6 D. These values of Δμ are consistent with a breaking of ground-state quadrupole electronic symmetry and the formation of dipolar excited states. Considering the relatively large size of these molecules, Δμ values of 2 to 6 D, corresponding to 0.4 to 1.2e Å, do not indicate well-defined charge separation. The nitroaryl substituents, therefore, do not appear to induce a significant CT character in the emissive S1 states of the DPNDs under study.
Three distinct patterns of behaviour emerge from the polarity effects on the fluorescence quantum yields of the DPND dyes (Fig. 4D):
First, the fluorescence quantum yields of 1, 2 and 13 are relatively large, i.e., between 0.3 and 1, and show no correlation with solvent polarity (Fig. 4D). The lack of marked solvent effects on the optical properties of 13 is consistent with its structure: (1) the nitro groups are at the meta positions of the phenyl substituents, causing weakened coupling with the DPND core, (2) the ortho methyl groups enhance the dihedral angles, further decreasing the coupling, and (3) the electron-donating methyls also dampen the effect of the nitro groups. Therefore, 13 behaves similarly to the DPNDs without nitroaryl substituents, exhibiting exceptionally large Φfl even for polar media, i.e., Φfl of 13 for DCM and ACN is 0.92 and 0.43, respectively.
Second, when in non-polar media, 3, 4, 8, 9 and 12 are strongly fluorescent with Φfl ≳ 0.3 (Fig. 4D). As the solvent polarity increases beyond that of DCB, Φfl drops by an order of magnitude or more (Fig. 4D). This behaviour of the DPNDs with para and meta nitro groups is consistent with a polarity-induced transition to a dark state that provides pathways for non-radiative deactivation.
Third, the DPND derivatives 5, 6, 7, 10 and 11, with ortho or peri nitro groups on their aryl substituents, are weakly fluorescent even in non-polar media (Fig. 4D). An increase in solvent polarity makes their emission barely detectable and their fluorescence quantum yields unquantifiable. This fluorescence quenching is usual for chromophores with 2-nitrophenyl substituents.24 It is clear that placing the nitro groups at the ortho and peri positions next to the DPND core provides: (1) efficient pathways for intersystem crossing, as we previously described and (2) through-space pathways for forward and back CT that can efficiently deactivate the S1 to the S0 state. While the former can account for the weak fluorescence in the non-polar solvents, the latter is consistent with polarity-induced emission quenching.
Comparison between the results obtained in liquid PB versus solid SOA reveals that the medium viscosity has a small impact on the photophysics of the DPNDs (Fig. 4 and Table S2†). The values of Φfl for SOA are larger than those for PB (Table S2†). For the mono-substituted ortho and peri derivatives, 5, 6 and 7, Φfl for SOA exceeds Φfl for PB by a factor of approximately three to seven. For all meta and para conjugates, on the other hand, these differences between Φfl for SOA and PB are smaller than a factor of two, which is on par with the trends observed for 1 and 2, which do not contain nitro groups (Table S2†). These results show that nitro groups at ortho or peri positions enhance the susceptibility of the DPNDs toward medium viscosity. Nevertheless, these viscosity effects are considerably smaller than those observed as a function of the solvent polarity.
Nanosecond time-resolved emission spectroscopy
Time-correlated single photon counting (TCSPC) reveals mono-exponential nanosecond emission decays for all samples with relatively large Φfl (Fig. 5). This feature suggests the presence of a single fluorescent species in each of the samples.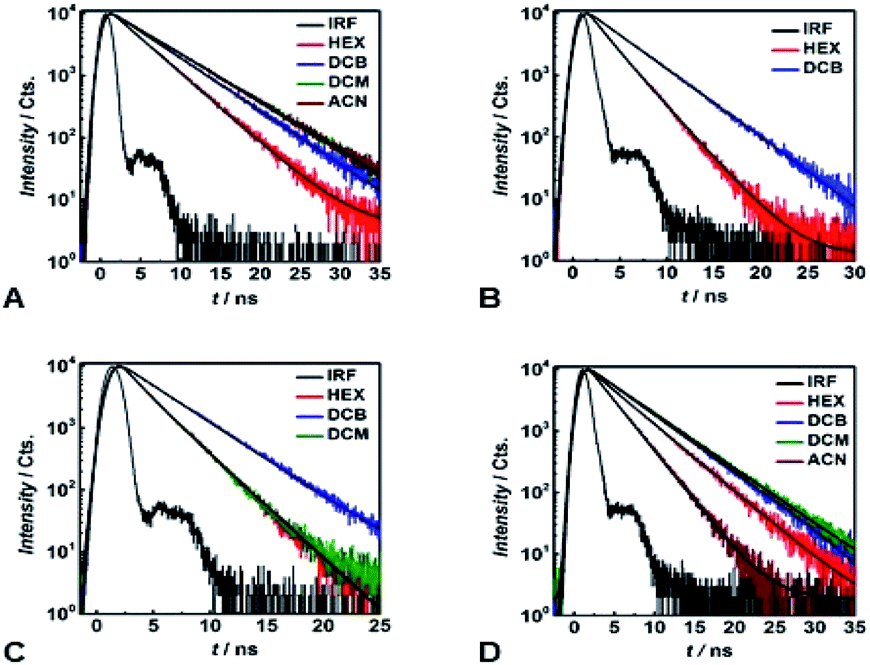 | ||
| Fig. 5 Emission decays of (A) 1, (B) 4, (C) 9 and (D) 13 in various solvents (λex = 460 nm; the decays are recorded at λem, corresponding to the fluorescence maxima; IRF = instrument response function, representing the temporal profile of the excitation pulse, which is recorded by monitoring the Rayleigh scattering of non-fluorescent media at λem = λex). Black solid lines represent monoexponential fits of the experimental data. | ||
All compounds show quite similar radiative-decay rate constants (kr) with values clustering around 108 s−1 that are invariant with medium polarity (Fig. 4E). For the non-radiative-decay rate constants (knr) of the nitroaryl DPNDs, on the other hand, medium polarity induces up to a three orders of magnitude increase, while knr of 1 and 2 exhibit negligible solvent dependence (Fig. 4F). Among them, the ortho- and peri-nitroaryl DPNDs, such as 5, 7, 10 and 11, exhibit knr of about 1011 s−1 and larger in polar media (Fig. 4F). Even for 13, where the coupling of the nitro groups with the core is weak, knr increases by an order of magnitude upon transition from a non-polar to polar solvent (Fig. 4F). These findings unequivocally show that the non-radiative decay rates govern the photophysical trends observed for these DPNDs.
The substantial enhancements of knr by solvent polarity are consistent with transitions to dark, polarized states that provide pathways for non-radiative deactivation. The susceptibility of knr for the ortho- and peri-nitro derivatives, i.e., 5, 6, 7, 10 and 11, to solvent polarity is considerably stronger than that for the para- and meta-nitro ones, i.e., 3, 4, 8, 9, 12 and 13. This solvent dependence of knr suggests that the dark states of 5, 6, 7, 10 and 11 are more polarized than those of 3, 4, 8, 9, 12 and 13. Therefore, such dark states of the ortho- and peri-nitro DPNDs most plausibly have a pronounced CT character. Conversely, we cannot unequivocally claim a well-defined CT character for the dark states of the para- and meta-nitro derivatives.
Transient-absorption spectroscopy
We conducted pump-probe TA studies on DCB, DCM and ACN solvent media, with sample concentrations required to exceed about 10 times those employed for the fluorescence measurements. Each of the TA spectra of the thirteen DPNDs shows four main features (Fig. 6): (1) a band with negative ΔA around 530 to 550 nm, ascribed to ground-state bleaching (B); (2) a broad band between about 550 and 700 nm, also with negative ΔA, ascribed to stimulated emission (SE), as suggested by its overlap with the fluorescence spectra; (3) a weak broad TA band in the red-NIR spectral region (λ >650 nm), ascribed to electronic absorption (EA) involving S1 → Sn transitions; and (4) a TA band at around 550 to 600 nm that the global-fit (GF) analysis makes especially apparent (Fig. 7).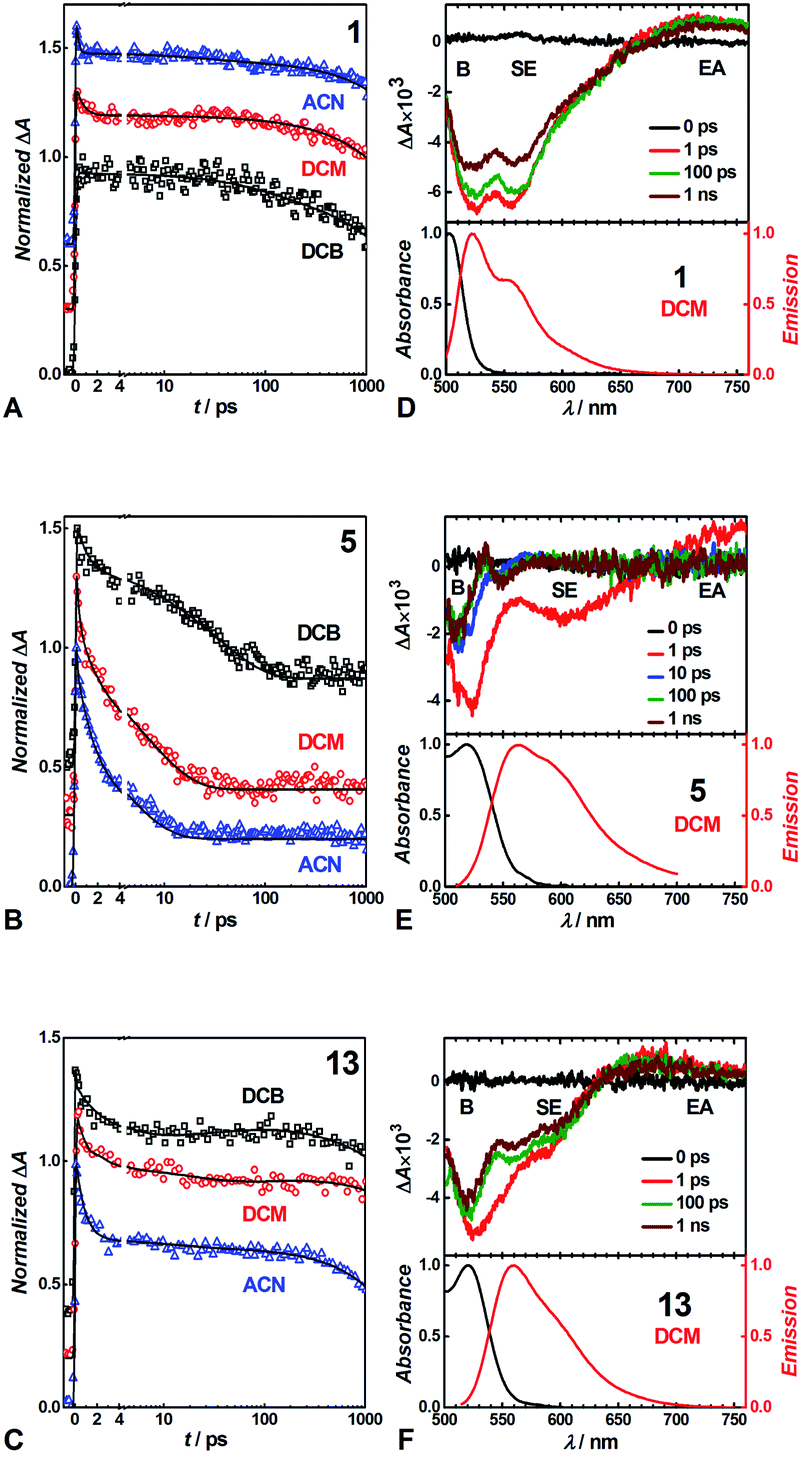 | ||
| Fig. 6 Transient absorption decays normalized to 1 of (A) 1, (B) 5 and (C) 13 in different solvents at observation wavelength of 520 nm. Transient absorption spectra at different pump-probe time delays (upper panel) and time-integrated absorption and emission spectra normalized to 1 (lower panel) of (D) 1, (E) 5 and (F) 13 in DCM. The transient absorption signals were recorded upon excitation at 480 nm. Solid lines are from global multiexponential fits to the experimental data. For clarity the transient absorption decays were offset on the y-axis. B – bleach band, SE – stimulated emission, EA – excited state absorption. | ||
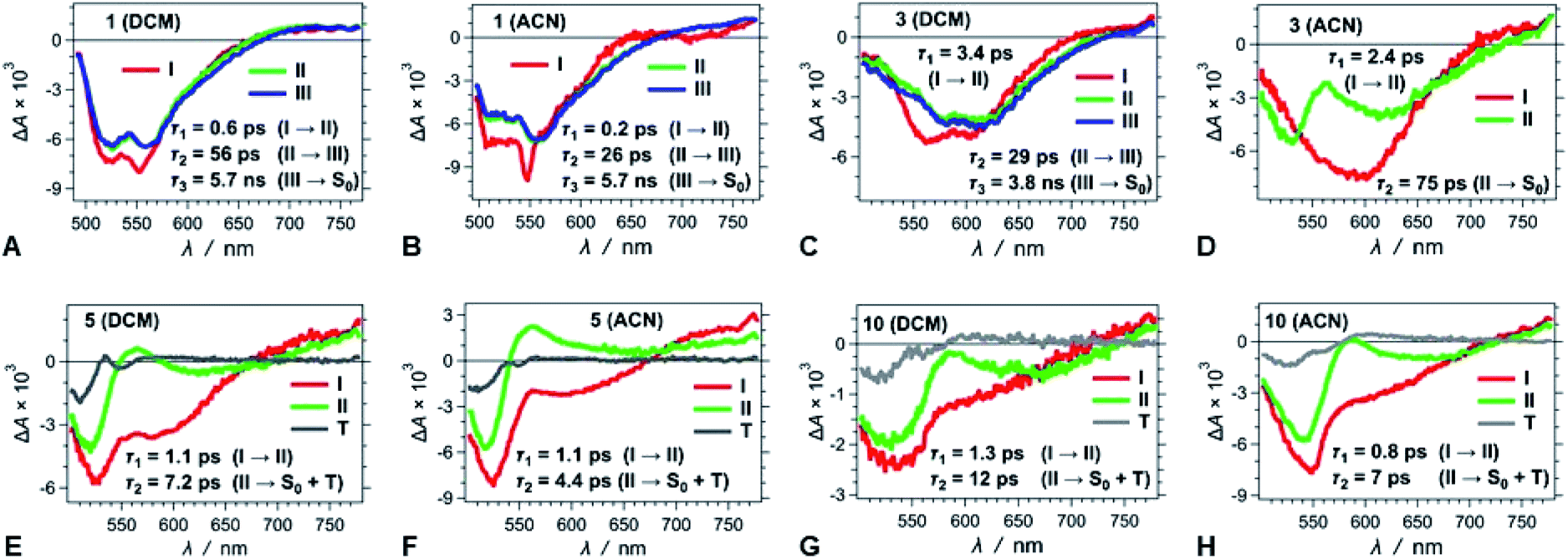 | ||
| Fig. 7 TA spectra of (A–D) strongly fluorescent and (E–H) weakly and non-fluorescent DPNDs, depicting the transitions associated with the lifetimes, τi, obtained from GF analys. | ||
Despite the similarities in the spectral shapes, the TA dynamics vary widely among the different dyes and solvents, as GF analysis reveals. The nanosecond lifetimes obtained from TCSPC, along with two additional picosecond exponential terms, describe well the TA dynamics of the dyes (Table S3†). In all cases, sub-picosecond or picosecond lifetimes, τ1, smaller than 6 ps, characterize the fastest of the components. These fast transitions reveal the emergence of a 550–600 nm TA band that overlaps with the B and the SE signals, while completely or partially maintaining the red-NIR absorption (Fig. 6 and 7). A bathochromic shift in the SE band can also induce a decrease in ΔA in the red-NIR spectral region. These (sub-)picosecond transitions are consistent with the relaxation of the Franck–Condon (FC) states to the fluorescent S1 structures. That is, the red-NIR broad TA, along with the SE characterize the FC states. Conversely, the 550–600 nm absorption band, along with bathochromically shifted emission and the same red-NIR TA, originate from the relaxed S1 state that undergoes radiative deactivation.
For the strongly fluorescent 1, 2 and 13, as well as for 3, 4, 8, 9 and 12 in low-polarity solvents, τ2 represents intermediate lifetimes, ranging between about 20 and 130 ps, which correspond to minute changes in the spectra of the relaxed emissive S1 state. Most of these changes encompass a small growth of the 550–600 nm TA band or a slight SE bathochromic shift (Fig. 7A–D). Along with the fast (sub-)picosecond components, these intermediate τ2 lifetimes suggest a degree of heterogeneity in the kinetics of relaxation to the emissive S1 states. Furthermore, solvent polarity induces a decrease in these intermediate lifetimes (Table S3†), which is consistent with the fluorescent S1 states tending to be more polar than the S0 as revealed by Lippert–Mataga–Ooshika analysis (Fig. 4C).
The longest lifetimes, i.e., τ3 obtained from triexponential fits and τ2 – from biexponential ones, characterize the decays of the emissive S1 states: these are the most important values for understanding the fluorescence properties of these dyes. The slowest decays of the para- and meta-nitro derivatives, along with those of 1 and 2, represent transitions solely to S0. For 1, 2 and 13, solvent polarity does not truly affect τ3 (Table S3†). This observation is consistent with minimal to no involvement of CT in the S1 → S0 transitions, which is valid even for 13, where the nitro groups are quite decoupled from the DPND core.
For the para and the other meta-substituted DPNDs, on the other hand, an increase in solvent polarity accelerates the S1 → S0 decays. This trend is especially pronounced in ACN, which produces S1 lifetimes of about 50–300 ps, i.e., significantly shorter than the nanosecond ones observed in HEX or DCB (Table S3†). This drastic decrease in the S1 lifetimes originates from a polarity-induced increase in knr for these compounds (Fig. 4F). These results are consistent with a S1 → S0 transition through a short-lived dark CT state. Indeed, accumulation of a dark intermediate (essential for detecting its TA features) is impossible when its formation is slower than its decay.
The GF analyses of the ortho- and peri-nitro derivatives required an introduction of infinity components, ΔA(λ, t = ∞), originating from transients with lifetimes significantly exceeding the dynamic range of the pump-probe technique. These long-lived species show broad weak TA that extends to 700 nm, and a peak at about 550 nm that overlaps with the ground-state bleach (Fig. 7E–H). The lack of nanosecond fluorescence signal from these compounds indicates that the ΔA(λ, t = ∞) transients correspond to dark states, i.e., either to long-lived CT states responsible for the polarity-induced fluorescence quenching, or to triplets. Indeed, medium polarity enhances the formation of intramolecular CT states.44 An increase in medium polarity, however, tends to decrease the ΔA amplitudes of these long-lived transients (Fig. 7E–H), precluding CT as the cause of their formation. Furthermore, placing nitro groups at ortho positions of phenyl substituents dramatically enhances the rates of ISC,24 which is consistent with a triplet configuration in the long-lived transients that 5, 6, 7, 10 and 11 produce.
While the TA analysis provided key insights about the excited-state dynamics of the DPNDs, there remained an open question as to the nature of the polarity-induced fluorescence quenching. Transitions through polar intermediates could explain the observed solvent-dependence trends. The rates of deactivation of such intermediates exceeding the rates of their formation, however, rendered them undetectable. Therefore, we resorted to computational studies to further elucidate the modes of deactivation the nitro-aryl substituted DPNDs.
Computational studies
Second-order Møller–Plesset (MP2)45 perturbation theory allowed us to obtain ab initio estimates of the ground-state equilibrium geometries. For optimization of the excited states and evaluation of vertical excitation, we employed second-order algebraic diagrammatic construction, ADC(2).46 The ADC(2) method provides a balanced description of intramolecular CT states, which makes it particularly advantageous for the analysis of organic nitro compounds.For each of the dyes, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) involved in the lowest electronic transitions have, respectively, a π and π* character, and are mainly localized on the DPND core (Fig. 8 and S69†). The radiative ππ* transitions between the S0 and S1 states are strongly allowed, with oscillator strengths for optical absorption and emission close to unity (Fig. S69†). The computed energies for the vertical absorption and fluorescence agree well with the experimental results (Fig. S69†).
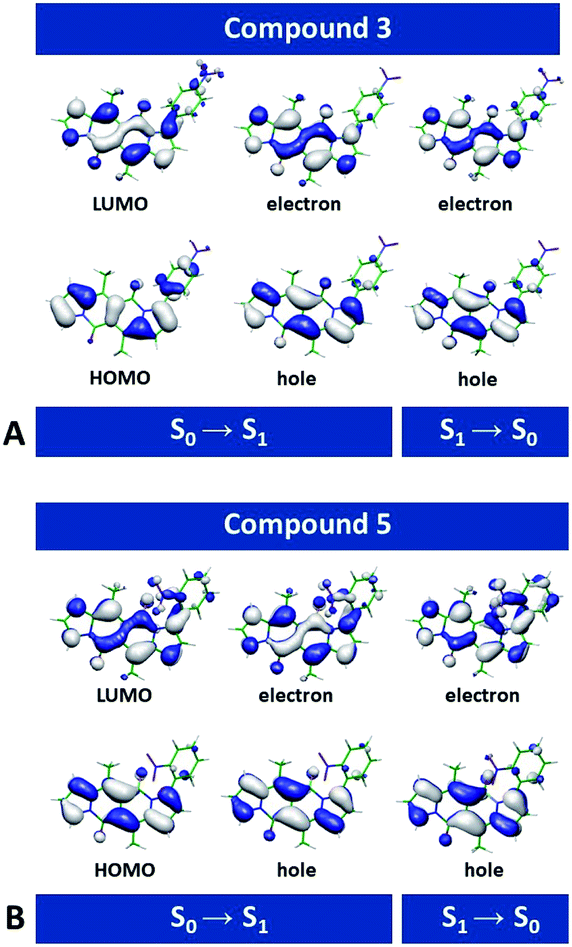 | ||
| Fig. 8 Frontier and natural transition orbitals (NTOs) involved in the S0 → S1 and S1 → S0 radiative transitions, computed at the ADC(2)/cc-pVDZ level of theory for (A) 3 and (B) 5. The long alkyl chains are truncated to methyls (isovalue = 0.03). The electron NTOs closely resemble the LUMOs, and the hole NTOs – the HOMOs. It is consistent with the HOMO → LUMO and LUMO → HOMO transitions providing the principal contributions to the absorption and the fluorescence. | ||
Removal of one nitrophenyl substituent from disubstituted DPNDs has a minor effect on their spectral properties, as a comparison between 3 and 8 illustrates (Fig. S69†). This finding agrees with the experimental results and with the localization of the radiative-transition NTOs on the DPND polycyclic ring system (Fig. 8 and S69†).
Despite this orbital localization on the DPND unit, a detailed analysis of the LUMOs shows their tendency to extend over the ortho- and peri-nitroaryl substituents. That is, optical excitation of these systems shifts some of the DPND electronic density to the nitroaryl moieties. While an increase in the dihedral angle between the aryl substituent and the DPND core slightly elevates the S1 energy, it noticeably shortens the distance between one of the oxygens of the nitro group and the closest quaternary carbon on the pyrrole ring. Therefore, we follow this carbon–oxygen distance (RCO) along the S1 PES and compute the relaxed scans along it (blue squares, Fig. 9A. At a critical RCO of about 2.4 Å, the PES of the 1ππ* state crosses to the PES of another state (red triangles, Fig. 9A). This new state has a pronounced CT character as revealed by its NTOs (Fig. 9A), and the dipole moment of 3.93 D at the 1ππ* equilibrium geometry rises to 10.95 D at RCO = 1.85 Å.
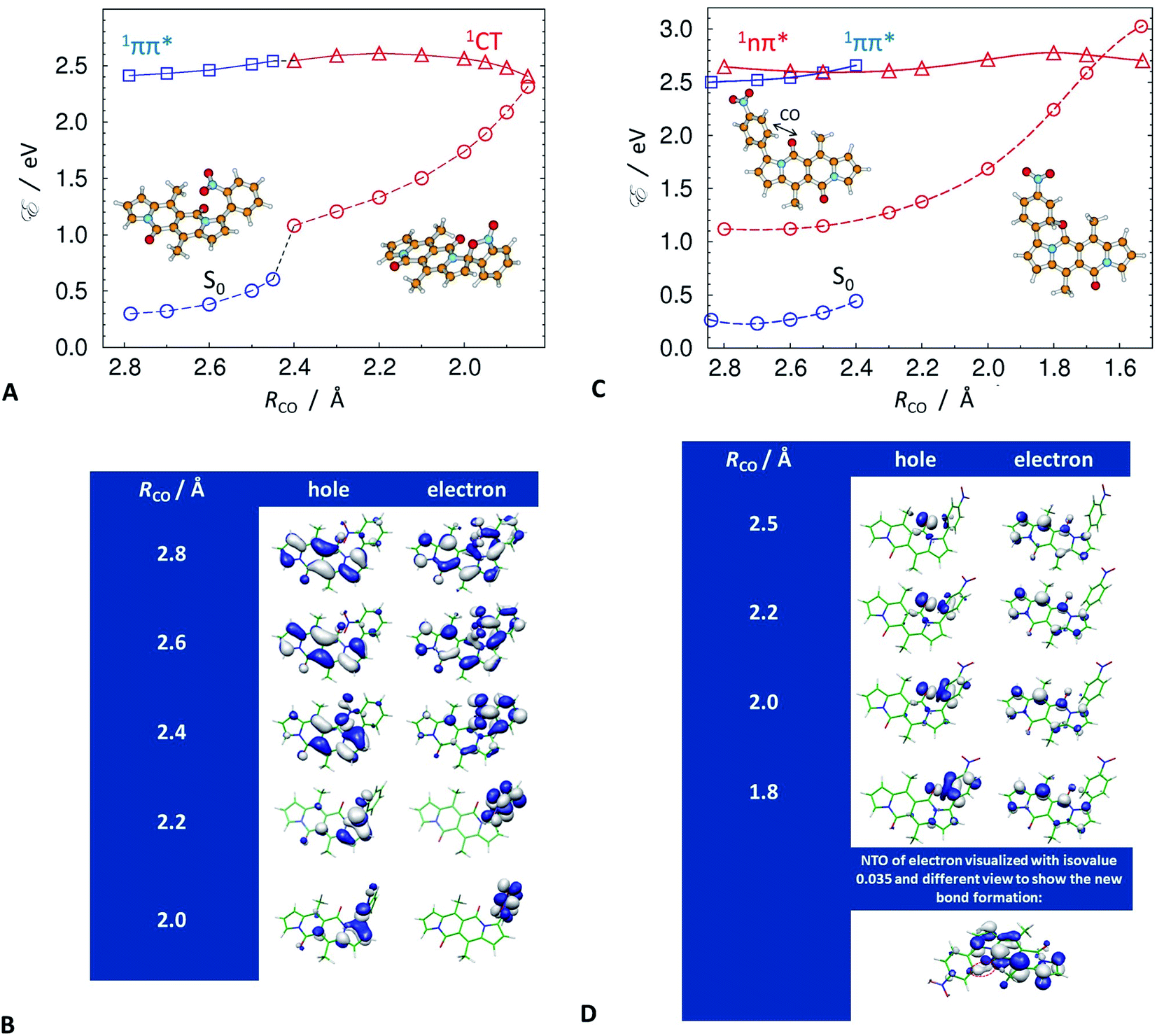 | ||
| Fig. 9 Potential-energy profiles (PEPs) along the carbon–oxygen distances, RCO, and the corresponding S1 → S0 NTOs (isovalues = 0.03, unless indicated otherwise) optimized with the ADC(2)/def-SV(P) method in the lowest excited singlet states of (A and B) 5 and (C and D) 3. (A and C) Circles connected by dashed line denote vertical energy of the ground state computed at the geometry of the respective excited state. For 5, (A) locally-excited, 1ππ*, state – blue squares, and the dark charge-transfer, 1CT, state – red triangles; and (B) NTOs along RCO. For 3, (C) locally-excited, 1ππ*, state – blue squares, and the dark 1nπ* state – red triangles; and (D) NTOs along RCO, where the last bottom structure depicts the electron NTO at isovalue of 0.035 to visualize the formation of the carbon–oxygen covalent bond, as circled with a red dashed line. | ||
As expected, the electron NTO of the 1CT state localizes around the nitro group. Upon decreasing RCO, however, there is a surprising density shift, leading to localization of the hole NTO around the pyrrole carbon and the nitro group (Fig. 9B). These changes prime the structure for covalent bonding between the pyrrole carbon and the nitro group oxygen. After passing through a small barrier at RCO ≈ 2.2 Å, a further decrease in RCO stabilizes the 1CT state (Fig. 9A and B). At RCO ≈ 1.85 Å, 1CT crosses the PES of the ground state (Fig. 9A). This 1CT/S0 intersection provides a pathway for an efficient non-adiabatic transition to the ground state. These findings unequivocally confirm the conclusions from the spectroscopy studies, which indicate the involvement of a dark CT state in the non-radiative S1 deactivation of the ortho and peri nitroaryl DPND derivatives.
At the geometry of the 1CT/S0 intersection, the formation of the carbon–oxygen covalent bond commences. Passing this intersection may lead to the formation of a photoproduct. A preferred trajectory in the ground state, involving the breaking of the temporarily-formed carbon–oxygen bond, however, restores the initial S0 structure. In other words, an aborted photochemical reaction provides a channel for non-radiative decay of the S1 state via internal conversion (IC).
The symmetric augmentation of the DPND core with two ortho-nitrophenyls in 10 raises the energy of the reactive 1CT state with respect to the 1ππ* one (Fig. S71, Tables S7 and S8†). The photochemical reaction breaks the molecular symmetry and polarizes the system. The dipole of 4.24 D in the 1ππ* state increases to 6.51 D in the 1CT state at RCO = 2.3 Å, and doubles to 12.73 D as RCO decreases to 1.85 Å.
The same non-radiative decay channel operates in the peri-nitronaphthyl derivative, 7, showing that the mechanism is not limited to just ortho-nitrophenyl conjugates. Furthermore, the naphthyl group provides additional stabilization to the reactive 1CT state (Fig. S70†).
The para and meta derivatives, wherein the nitro groups are not as close to the DPND core, are not prone to forming such 1CT states. Nonetheless, theoretical exploration of the PESs of the two lowest singlet excited states of 3, and of the other strongly fluorescent nitroaryl DPNDs, reveals unexpected photoreactivity. At the S0 geometry, S1 and S2 are 1ππ and 1nπ states, respectively, and the energy gap between them is relatively small, i.e., on the order of 0.1 eV (Fig. 9C). An S0 → 1nπ* optical transition is not dipole allowed. Nevertheless, vibronic coupling permits some intensity from the closely lying 1ππ* state. All of the investigated compounds have this second-excited 1nπ* state, regardless of the position of the nitro groups on the aryl substituents (Table S9†).
The optimized molecular structures show an elongation of the carbon–oxygen bond of the carbonyl group in the DPND core in the 1nπ* state. On average, this bond length changes from 1.23 Å in the 1ππ* state to 1.38 Å in 1nπ* state, and brings the carbonyl oxygen close to the ortho carbons of the adjacent electron-deficient nitrophenyl substituent. We, therefore, selected this interatomic distance between the nitrophenyl carbon and the carbonyl oxygen to define RCO for the analysis of the strongly fluorescent conjugates.
A decrease in RCO destabilizes the 1ππ* state and stabilizes the 1nπ* state. Along RCO, the potential-energy profiles (PEPs) of the lowest ππ* and nπ* singlet states of 3 cross at RCO = 2.5 Å (Fig. 9C). Independent geometry optimization of different electronic states along a selected/distinguished intramolecular coordinate results in different their geometric displacements along the remaining 3N-7 intramolecular coordinates. In result one-dimensional potential-energy (PE) profiles plotted vs. selected reaction coordinate are differently displaced in remaining coordinates and their visualized intersections are "apparent" (projected in 1-D). The real intersection between two PE profiles means that energies of both electronic states were computed at the same molecular geometry. The intramolecular coordinates of the 1ππ* geometries differ from those of the 1nπ* ones. The two vertical PEPs of the ground state, computed at these geometries, illustrate this difference (Fig. 9C).
The last stable point optimized in the 1ππ* structure, at RCO = 2.4 Å, represents an upper estimate of the barrier between the two excited states. Once the 1nπ* state becomes S1 and is populated, it has access to a conical intersection (CI) with S0 at RCO ≈ 1.7 Å, protected by a barrier of about 0.2 eV at RCO ≈ 1.8 Å (Fig. 9C). The 1nπ*/S0 CI provides a pathway for nonradiative deactivation of the S1 state. Extension of the reaction pathway beyond the intersection with the ground state results in optimization of a metastable open-shell diradical structure. Concurrently, decreasing RCO along the 1nπ* PEP induces an increase in the NTO densities around the DPND carbonyl and the nitrophenyl (Fig. 9D). As a result, a non-nodal segment emerges in the electron NTO between the nitrophenyl carbon and the carbonyl oxygen.
While the 1nπ* structures are not as polar as the 1CT states of the ortho derivatives, an RCO decrease still increases the S1 dipole, e.g., for 3, μS1 = 3.53 D at RCO = 2.5 Å, and μS1 = 6.93 D at RCO = 1.8 Å. Therefore, an increase in solvent polarity stabilizes the 1nπ* structures with reduced RCO more than it does the locally excited (LE) 1ππ* state, which lowers the barriers along the S1 PES and favours the non-radiative transition through the 1nπ*/S0 CI. This finding is consistent with the polarity-induced quenching of the emission of even the strongly fluorescent nitroaryl conjugates, except of 13 where the nitro group is practically completely decoupled from the DPND core (Fig. 4D and F).
Discussion
Efficient ISC, involving 1ππ* and 1nπ* singlet and triplet states, is one of the most prevalent reasons why nitro compounds do not fluoresce. The high density of triplet states and closely situated singlet excited states with 1ππ* and 1nπ* character ensure non-radiative decay via ISC. Photophysical and computational studies of nitroaryl DPNDs reveal for the first time how the two lowest 1ππ* and 1nπ* singlet excited states, on their own, can provide a pathway for non-radiative deactivation. This mechanism of decay through a dark 1nπ* state is universal for all nitroaryl DPNDs under study, and the threshold for such non-radiative deactivation channels depends on the energy gap between the 1ππ* and 1nπ* states (Table S9†).When other channels dominate the non-radiative deactivation, the 1ππ* → 1nπ* → S0 mechanism is undetectable, as the ortho and peri derivatives reveal, where ISC and a transition through a dark 1CT state are responsible for the fluorescence quenching. Orthogonality of ortho-nitrophenyl substituents with the DPND core fosters states with strong spin–orbit coupling that have negligible dependence on the solvent polarity.24 Polar environments, however, stabilize the highly polarized CT states considerably more than they do the less polar 1ππ* states, resulting in the observed enhancement of knr (Fig. 4F and Table S2†).
In both IC decay mechanisms, the reactivity of the 1nπ* and 1CT states induces a temporary formation of covalent bonds along the S1 PESs toward the CIs. Ultimately, these covalent bonds break upon relaxation to the ground state (Fig. 10). Based on ab initio calculations, in the 1990s Domcke and Sobolewski proposed the concept of a low-barrier photoreaction pathway interrupted by a CI with the ground state as a mechanism for ultrafast excited-state deactivation.31 Such photochemical reactions, aborted at CIs between the PESs of S1 and S0 states, offer efficient channels for ultrafast IC, leading to recovery of the reactant after photoexcitation.47 For example, the quenching of deleterious photochemical reactions by ultrafast IC is important for the high intrinsic photostability of DNA bases as well as sugars and peptides.48–50 Herein, interactions of the nucleophilic oxygens of the nitro group (and the DPND carbonyls) with electrophilic carbons of the pyrroles (and the nitroaryl substituents) present a key paradigm for fluorescence quenching via aborted photochemistry (Fig. 10).
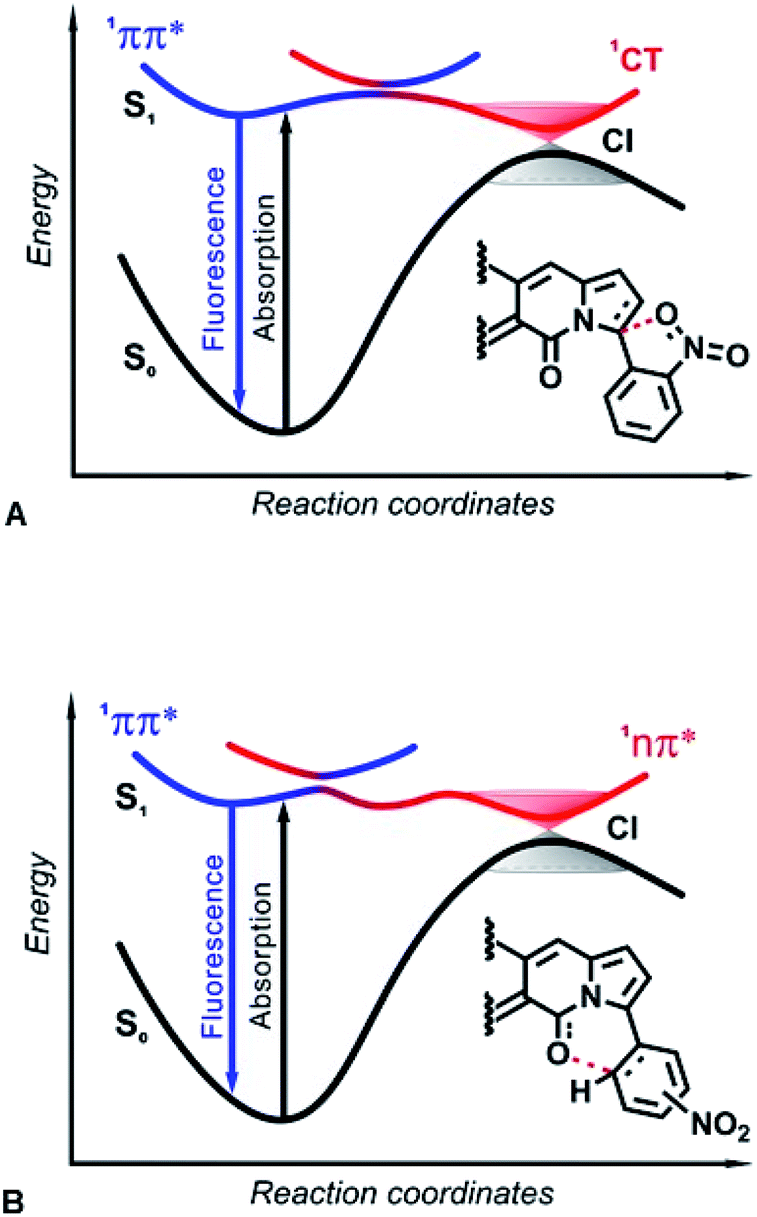 | ||
| Fig. 10 Excited-state dynamics of nitroaryl-substituted DPNDs. The formed LE 1ππ* states either radiatively decay to S0, or non-adiabatically transfer to dark states that form CIs with S0 providing pathways for non-radiative deactivation. (A) Placing the nitro groups close to the DPND core, such as in the ortho and peri derivatives, accommodates the formation of dark CT states and the non-radiative deactivation assumes a 1ππ* → 1CT → S0 pathway. (B) When the nitro groups are not spatially close to the DPND core, such as in the para and meta derivatives, the energy level of the FC second singlet excited state, with a 1nπ* character, decreases along the reaction coordinate, opening a 1ππ* → 1nπ* → S0 pathway for non-radiative deactivation. | ||
Conclusions
Weakening the electron-donating capabilities of a DPND chromophore core while balancing its coupling with nitro groups attached to its aryl substituents produces fluorescent compounds. Many of these dyes sustain their emission in polar solvents, unlike other nitroaromatics. The implemented molecular designs suppress modes of non-radiative decay that are common in nitroaromatic compounds. This suppression leads to the emergence of alternative pathways of non-radiative deactivation involving aborted photochemistry. Along with dihedral degrees of freedom, the nucleophilic oxygens and electrophilic carbons of nitroaryl derivatives render such compounds prone to intramolecular photochemical reactions. When conical intersections interrupt the trajectories of these reactions, internal conversion leads to restoration of the initial ground state and molecular structure. This sequence of processes represents a completely unexplored mechanism for the non-radiative deactivation of nitroaromatics. These results set a key photophysical paradigm, providing important guidelines for the design and engineering of new classes of fluorescent nitroaromatics.Data availability
Data associated with this article, including experimental procedures, compound characterization, steady-state absorption and emission along with the time-resolved pump-probe transient absorption details and computational analysis details are available in the ESI.†Author contributions
B. S., D. T. G. and P. P. conceived the project; B. S. designed, synthesized and characterized all nitroaromatics; P. P. carried out time-integrated and time-resolved spectroscopic studies, analyzed and interpreted spectroscopic results; M. K., Y. M. P., M. C., P. J., T. D. L. and M. B. performed physicochemical investigation; J. A. C. analyzed the transient absorption results; A. L. S. conducted the computational studies; W. G. and D. M. and helped in the conduction of the project; B. K., M. Ł. and V. I. V. co-directed the project; V. I. V. was instrumental in writing the final version of the manuscript. All authors contributed to writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was financially supported by the Polish National Science Centre, Poland (PRELUDIUM 2016/23/N/ST5/00054, HARMONIA 2016/22/M/ST5/00431, 2017/25/B/ST5/02488 to P. J. and SONATA 2017/26/D/ST3/00910 to P. P.), the Foundation for Polish Science (TEAM POIR.04.04.00-00-3CF4/16-00 and START scholarship no. 076.2020 to B. S.), the Polish National Agency for Academic Exchange (fellowship to B. S. within the Bekker programme PPN/BEK/2019/1/00256), the USA National Science Foundation (grant number CHE 1800602, and AGEP supplement supporting J. A. C.), the American Chemical Society Petroleum Research Fund (grant number 60651-ND4), the USA National Institutes of Health, National Eye Institute (grant R01 EY027440) and European Commission (CHAIR 860762). We extend our gratitude to Dr Simon J. Teat for training and guidance through our crystallography studies at the Advanced Light Source (ALS), Lawrence Berkeley National Laboratory, supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.Notes and references
- M.-C. Chen, D.-G. Chen and P.-T. Chou, ChemPlusChem, 2021, 86, 11–27 CrossRef CAS PubMed.
- D. J. Cowley, Helv. Chim. Acta, 1978, 61, 184–197 CrossRef CAS.
- B. Bursa, D. Wróbel, B. Barszcz, M. Kotkowiak, O. Vakuliuk, D. T. Gryko, Ł. Kolanowski, M. Baraniak and G. Lota, Phys. Chem. Chem. Phys., 2016, 18, 7216–7228 RSC.
- N. I. Rtishchev, D. V. Samoilov, V. P. Martynova and A. V. El’tsov, Russ. J. Gen. Chem., 2001, 71, 12 Search PubMed.
- E. F. Plaza-Medina, W. Rodríguez-Córdoba and J. Peon, J. Phys. Chem. A, 2011, 115, 9782–9789 CrossRef CAS PubMed.
- Z. Cheng, E. Kuru, A. Sachdeva and M. Vendrell, Nat. Rev. Chem., 2020, 4, 275–290 CrossRef CAS.
- A. Morozumi, M. Kamiya, S. Uno, K. Umezawa, R. Kojima, T. Yoshihara, S. Tobita and Y. Urano, J. Am. Chem. Soc., 2020, 0c00451 Search PubMed.
- T. M. Wilson, M. J. Tauber and M. R. Wasielewski, J. Am. Chem. Soc., 2009, 131, 8952–8957 CrossRef CAS PubMed.
- M. Stolar and T. Baumgartner, Phys. Chem. Chem. Phys., 2013, 15, 9007 RSC.
- Q. Yue, W. Liu and X. Zhu, J. Am. Chem. Soc., 2020, 142, 11613–11628 CrossRef CAS PubMed.
- A. Ashcraft, K. Liu, A. Mukhopadhyay, V. Paulino, C. Liu, B. Bernard, D. Husainy, T. Phan and J.-H. Olivier, Angew. Chem., Int. Ed., 2020, 59, 7487–7493 CrossRef CAS PubMed.
- K. Skonieczny, I. Papadopoulos, D. Thiel, K. Gutkowski, P. Haines, P. M. McCosker, A. D. Laurent, P. A. Keller, T. Clark, D. Jacquemin, D. M. Guldi and D. T. Gryko, Angew. Chem., Int. Ed., 2020, 59, 16104–16113 CrossRef CAS PubMed.
- T. Ueno, Y. Urano, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2006, 128, 10640–10641 CrossRef CAS PubMed.
- J.-M. Mewes, V. Jovanović, C. M. Marian and A. Dreuw, Phys. Chem. Chem. Phys., 2014, 16, 12393–12406 RSC.
- C. E. Crespo-Hernández, G. Burdzinski and R. Arce, J. Phys. Chem. A, 2008, 112, 6313–6319 CrossRef PubMed.
- J. S. Zugazagoitia, C. X. Almora-Díaz and J. Peon, J. Phys. Chem. A, 2008, 112, 358–365 CrossRef CAS PubMed.
- E. F. Plaza-Medina, W. Rodríguez-Córdoba, R. Morales-Cueto and J. Peon, J. Phys. Chem. A, 2011, 115, 577–585 CrossRef CAS PubMed.
- J. P. Zobel, J. J. Nogueira and L. González, Chem.–Eur. J., 2018, 24, 5379–5387 CrossRef CAS PubMed.
- S. Saha and A. Samanta, J. Phys. Chem. A, 1998, 102, 7903–7912 CrossRef CAS.
- P. B. Ghosh and M. W. Whitehouse, Biochem. J., 1968, 108, 155–156 CrossRef CAS PubMed.
- S. Benson, A. Fernandez, N. D. Barth, F. de Moliner, M. H. Horrocks, C. S. Herrington, J. L. Abad, A. Delgado, L. Kelly, Z. Chang, Y. Feng, M. Nishiura, Y. Hori, K. Kikuchi and M. Vendrell, Angew. Chem., Int. Ed., 2019, 58, 6911–6915 CrossRef CAS PubMed.
- R. López-Arteaga, A. B. Stephansen, C. A. Guarin, T. I. Sølling and J. Peon, J. Phys. Chem. B, 2013, 117, 9947–9955 CrossRef PubMed.
- M.-C. Chen, Y.-L. Lee, Z.-X. Huang, D.-G. Chen and P.-T. Chou, Chem.–Eur. J., 2020, 26, 7124–7130 CrossRef CAS PubMed.
- Y. M. Poronik, G. V. Baryshnikov, I. Deperasińska, E. M. Espinoza, J. A. Clark, H. Ågren, D. T. Gryko and V. I. Vullev, Commun. Chem., 2020, 3, 190 CrossRef CAS.
- D. H. Friese, A. Mikhaylov, M. Krzeszewski, Y. M. Poronik, A. Rebane, K. Ruud and D. T. Gryko, Chem.–Eur. J., 2015, 21, 18364–18374 CrossRef CAS PubMed.
- Ł. G. Łukasiewicz, H. G. Ryu, A. Mikhaylov, C. Azarias, M. Banasiewicz, B. Kozankiewicz, K. H. Ahn, D. Jacquemin, A. Rebane and D. T. Gryko, Chem.–Asian J., 2017, 12, 1736–1748 CrossRef PubMed.
- M. Tasior, G. Clermont, M. Blanchard-Desce, D. Jacquemin and D. T. Gryko, Chem.–Eur. J., 2019, 25, 598–608 CrossRef CAS PubMed.
- Z. Szakács, M. Tasior, D. T. Gryko and E. Vauthey, ChemPhysChem, 2020, 21, 1718–1730 CrossRef PubMed.
- B. Dereka, A. Rosspeintner, M. Krzeszewski, D. T. Gryko and E. Vauthey, Angew. Chem., Int. Ed., 2016, 55, 15624–15628 CrossRef CAS PubMed.
- M. Grzybowski, I. Deperasińska, M. Chotkowski, M. Banasiewicz, A. Makarewicz, B. Kozankiewicz and D. T. Gryko, Chem. Commun., 2016, 52, 5108–5111 RSC.
- A. L. Sobolewski, C. Woywod and W. Domcke, J. Chem. Phys., 1993, 98, 5627–5641 CrossRef CAS.
- I. J. Palmer, I. N. Ragazos, F. Bernardi, M. Olivucci and M. A. Robb, J. Am. Chem. Soc., 1993, 115, 673–682 CrossRef CAS.
- W. Domcke and A. L. Sobolewski, Science, 2020, 368, 820–821 CrossRef CAS PubMed.
- J. Yang, X. Zhu, J. P. F. Nunes, J. K. Yu, R. M. Parrish, T. J. A. Wolf, M. Centurion, M. Gühr, R. Li, Y. Liu, B. Moore, M. Niebuhr, S. Park, X. Shen, S. Weathersby, T. Weinacht, T. J. Martinez and X. Wang, Science, 2020, 368, 885–889 CrossRef CAS PubMed.
- B. Sadowski, M. F. Rode and D. T. Gryko, Chem.–Eur. J., 2018, 24, 855–864 CrossRef CAS PubMed.
- B. Sadowski, M. Loebnitz, D. R. Dombrowski, D. H. Friese and D. T. Gryko, J. Org. Chem., 2018, 83, 11645–11653 CrossRef CAS PubMed.
- L. Wang, L. Lin, J. Yang, Y. Wu, H. Wang, J. Zhu, J. Yao and H. Fu, J. Am. Chem. Soc., 2020, 142, 10235–10239 CrossRef CAS PubMed.
- B. Sadowski, S.-H. Su, T.-C. Lin, T. D. Lohrey, I. Deperasińska, P.-T. Chou and D. T. Gryko, J. Mater. Chem. C, 2018, 6, 12306–12313 RSC.
- G. Jones, D. Yan, J. Hu, J. Wan, B. Xia and V. I. Vullev, J. Phys. Chem. B, 2007, 111, 6921–6929 CrossRef CAS PubMed.
- A. L. Thompson, T.-S. Ahn, K. R. J. Thomas, S. Thayumanavan, T. J. Martínez and C. J. Bardeen, J. Am. Chem. Soc., 2005, 127, 16348–16349 CrossRef CAS PubMed.
- N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1956, 29, 465–470 CrossRef CAS.
- E. Lippert, Zeitschrift für Naturforschung A, 1955, 10, 541–545 CrossRef.
- Y. Ooshika, J. Phys. Soc. Jpn., 1954, 9, 594–602 CrossRef.
- J. B. Derr, J. Tamayo, J. A. Clark, M. Morales, M. F. Mayther, E. M. Espinoza, K. Rybicka-Jasińska and V. I. Vullev, Phys. Chem. Chem. Phys., 2020, 22, 21583–21629 RSC.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef.
- A. B. Trofimov and J. Schirmer, J. Phys. B: At., Mol. Opt. Phys., 1995, 28, 2299–2324 CrossRef CAS.
- I. Schapiro, F. Melaccio, E. N. Laricheva and M. Olivucci, Photochem. Photobiol. Sci., 2011, 10, 867 CrossRef CAS PubMed.
- A. Sinicropi, W. M. Nau and M. Olivucci, Photochem. Photobiol. Sci., 2002, 1, 537–546 CrossRef CAS PubMed.
- D. Tuna, A. L. Sobolewski and W. Domcke, Phys. Chem. Chem. Phys., 2014, 16, 38–47 RSC.
- D. Shemesh, A. L. Sobolewski and W. Domcke, J. Am. Chem. Soc., 2009, 131, 1374–1375 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2019018–2019020. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03670j |
| ‡ Present address: Institut für Organische und Biomolekulare Chemie, Georg-August-Universität, Tammannstrasse 2, 37077 Göttingen, Germany. |
| This journal is © The Royal Society of Chemistry 2021 |