
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect decarboxylation of ten-eleven translocation-produced 5-carboxylcytosine in mammalian genomes forms a new mechanism for active DNA demethylation†
Yang
Feng
ab,
Juan-Juan
Chen
a,
Neng-Bin
Xie
a,
Jiang-Hui
Ding
a,
Xue-Jiao
You
a,
Wan-Bing
Tao
a,
Xiaoxue
Zhang
c,
Chengqi
Yi
 c,
Xiang
Zhou
a,
Bi-Feng
Yuan
*ab and
Yu-Qi
Feng
ab
c,
Xiang
Zhou
a,
Bi-Feng
Yuan
*ab and
Yu-Qi
Feng
ab
aSauvage Center for Molecular Sciences, Department of Chemistry, Wuhan University, Wuhan 430072, China. E-mail: bfyuan@whu.edu.cn
bSchool of Health Sciences, Wuhan University, Wuhan 430071, China
cState Key Laboratory of Protein and Plant Gene Research, School of Life Sciences, Peking University, Beijing 100871, China
First published on 21st July 2021
Abstract
DNA cytosine methylation (5-methylcytosine, 5mC) is the most important epigenetic mark in higher eukaryotes. 5mC in genomes is dynamically controlled by writers and erasers. DNA (cytosine-5)-methyltransferases (DNMTs) are responsible for the generation and maintenance of 5mC in genomes. Active demethylation of 5-methylcytosine (5mC) is achieved by ten-eleven translocation (TET) dioxygenase-mediated oxidation of 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC). 5fC and 5caC are further processed by thymine DNA glycosylase (TDG)-initiated base excision repair (BER) to restore unmodified cytosines. The TET-TDG-BER pathway could cause the production of DNA strand breaks and therefore jeopardize the integrity of genomes. Here, we investigated the direct decarboxylation of 5caC in mammalian genomes by using metabolic labeling with 2′-fluorinated 5caC (F-5caC) and mass spectrometry analysis. Our results clearly demonstrated the decarboxylation of 5caC occurring in mammalian genomes, which unveiled that, in addition to the TET-TDG-BER pathway, the direct decarboxylation of TET-produced 5caC constituted a new pathway for active demethylation of 5mC in mammalian genomes.
Introduction
DNA cytosine methylation (5-methylcytosine, 5mC) is the most extensively studied epigenetic modification that can regulate gene expressions in higher eukaryotic genomes.1,2 5mC is involved in various biological processes, including cell differentiation, X-chromosome inactivation, and genomic imprinting. DNA (cytosine-5)-methyltransferases (DNMTs) are responsible for the generation and maintenance of 5mC in genomes of higher eukaryotes.3,4 The active demethylation of 5mC in the genomes of mammalian cells has been established through the consecutive oxidation of 5mC by ten-eleven translocation (TET) dioxygenases with the production of 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC).5–11 Then the N-glycosidic bonds of 5fC and 5caC can be cleaved by thymine DNA glycosylase (TDG) to form abasic sites (AP-sites), which are removed via a base excision repair (BER) pathway to restore the unmodified cytosines.12–15The active demethylation of 5mC via the TET-TDG-BER pathway is complicated and requires multiple steps of reaction and more than 10 different kinds of enzymes, such as TETs, DNA glycosylases, AP lyase, DNA polymerases, and ligases.16,17 In addition, the TET-TDG-BER pathway involves DNA strand breaks and could potentially jeopardize the integrity of genomes.18 In this respect, some other potential 5mC demethylation mechanisms have been investigated in recent years. A previous report demonstrated that deficiency in TDG would not affect the active demethylation of 5mC in mouse zygotes, suggesting the potential existence of the 5mC demethylation pathway that is independent of TDG.19
A possible pathway for reversal of 5mC could be achieved through the decarboxylation of TET-generated 5caC to restore cytosine. In this pathway, DNA strand breaks that occur in the TET-TDG-BER pathway will be avoided since TDG and BER are not involved. Indeed, it was previously reported that 5fC and 5caC could undergo the direct deformylation (for 5fC) and decarboxylation (for 5caC) mediated by thiols to generate cytosine in vitro.20 In addition, it was reported that bacterial and mammalian DNA (cytosine-5)-methyltransferases were capable of decarboxylating 5caC in synthesized 5caC-containing DNA in vitro,21 and treatment of synthesized 5caC-containing DNA with nuclear extracts of mouse embryonic stem cells could decarboxylate 5caC to cytosine.22 Furthermore, the isoorotate decarboxylase that was able to convert 5-carboxyluracil to uracil in fungi also showed certain activity in the decarboxylation of 5caC to cytosine.23 These studies clearly demonstrated the direct deformylation of 5fC and decarboxylation of 5caC would happen in vitro.
These in vitro studies stimulated the investigation of the direct C–C bond cleavage of 5fC and 5caC in vivo. Actually, the direct deformylation of 5fC to cytosine via C–C bond cleavage in genomes of mammalian cells has been reported.24,25 We recently also investigated the potential decarboxylation of 5caC in cell extracts and in live cells using a synthesized 5caC-containing DNA.26 We observed the direct decarboxylation of 5caC to cytosine in the synthesized 5caC-containing DNA that was transfected into human cells.26 However, whether this phenomenon of the direct decarboxylation of 5caC in synthesized 5caC-containing DNA also occurs in mammalian genomes in vivo is still unknown.
Here we investigated the decarboxylation of 5caC in mammalian genomes by using metabolic labeling and mass spectrometry analysis. Our results demonstrated that the decarboxylation of 5caC also occurred in mammalian genomes, which revealed that the TET-produced 5caC decarboxylation pathway also contributes to active 5mC demethylation in mammalian cells.
Experimental section
Chemicals and reagents
2′-Deoxyadenosine (dA), thymidine (T), 2′-deoxycytidine (dC), 2′-deoxyguanosine (dG), adenosine (rA), uridine (U), cytidine (rC), and guanosine (rG) were purchased from Sigma-Aldrich (Beijing, China). 5-Methyl-2′-deoxycytidine (5mC), 5-hydroxymethyl-2′-deoxycytidine (5hmC), 5-formyl-2′-deoxycytidine (5fC), and 5-carboxyl-2′-deoxycytidine (5caC) were purchased from Berry & Associates (Dexter, MI, USA). 2′-Fluorinated deoxycytidine (F-dC) was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). 2′-Fluorinated 5-carboxyl-deoxycytidine (F-5caC) was purchased from RC ChemTec Co. Ltd. (Wuhan, China) and further purified in the current study (detailed procedure can be found in the ESI†). 2′-Fluorinated 5-methyl-deoxycytidine (F-5mC) was prepared in the current study (detailed preparation procedure can be found in the ESI†).Venom phosphodiesterase I was purchased from Sigma-Aldrich (Beijing, China). S1 nuclease, DNase I, RNase A, and calf intestinal alkaline phosphatase (CIAP) were obtained from Takara Biotechnology Co. Ltd. (Dalian, China). Dulbecco's Modified Eagle medium (DMEM), RPMI-1640 medium, fetal bovine serum and RNase T were purchased from Thermo-Fisher Scientific (Waltham, MA, USA). LCMS-grade methanol (MeOH) and acetonitrile (ACN) were obtained from FTSCI Co., Ltd. (Wuhan, China).
Cell culture and isolation of genomic DNA
The human embryonic kidney epithelial cell line (HEK293T), human leukemic cell line (Jurkat-T), human breast adenocarcinoma cell line (MCF-7), and mouse neuroblastoma N2a cell line (Neuro-2a) were obtained from the China Center for Type Culture Collection (Wuhan, China). The TDG knockout HEK293T cell line was generated according to our previous study using a CRISPR-Cas9 system.27 HEK293T, TDG−/− HEK293T, MCF-7, and Neuro-2a cells were cultured in DMEM medium; Jurkat-T cells were maintained in RPMI-1640 medium at 37 °C in a 5% CO2 atmosphere. The medium included 10% fetal bovine serum, 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin. Isolation of genomic DNA was conducted using a Tissue DNA Kit (Omega Bio-Tek Inc., Norcross, GA, USA) according to the manufacturer's instruction.In vitro decarboxylation assay
The nuclear extracts of TDG−/− HEK293T cells were obtained using a Nuclear Extraction kit (Abcam, Cambridge, MA). Briefly, the TDG−/− HEK293T cells were collected by centrifugation at 1500 rpm below 4 °C for 10 min and then washed twice with PBS to remove the medium and fetal bovine serum. The cell pellet was resuspended in 1 mL of 1× Pre-Extraction buffer containing DTT and protease inhibitor cocktail. The mixture was incubated on ice for 10 min, then vortexed vigorously for 10 s and centrifuged for 1 min at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. After removing the cytoplasmic extract from the nuclear pellet, 100 μL of extraction buffer supplied DTT and protease inhibitor cocktail was added. The extract was incubated on ice for 15 min with vortex (5 s) every 3 min and then centrifuged for 10 min at 14000 rpm at 4 °C. The nuclear extracts were obtained by collecting the supernatant.
000 rpm. After removing the cytoplasmic extract from the nuclear pellet, 100 μL of extraction buffer supplied DTT and protease inhibitor cocktail was added. The extract was incubated on ice for 15 min with vortex (5 s) every 3 min and then centrifuged for 10 min at 14000 rpm at 4 °C. The nuclear extracts were obtained by collecting the supernatant.
For in vitro decarboxylation assay, 20 μg of genomic DNA and 100 μL of nuclear extracts from the TDG−/− HEK293T cells were incubated at 37 °C for 3 h. Then 0.5 μL proteinase K (20 mg mL−1) was added into the mixture and incubated at 55 °C for 2 h. The solution was incubated at 95 °C for 10 min to inactivate proteinase K. The resulting solution was extracted with chloroform twice, and then the aqueous layer was collected and lyophilized to dryness. The resulting DNA was enzymatically digested to nucleosides followed by LC-ESI-MS/MS analysis.
Metabolic labeling experiments
For metabolic labeling experiments, cells (HEK293T, TDG−/− HEK293T, Jurkat-T, MCF-7, and Neuro-2a cells) were cultured in DMEM medium (or RPMI-1640 medium) supplemented with diverse concentrations of F-5caC (100 μM to 400 μM) or F-dC (2 nM to 500 nM) to metabolically label genomic DNA for 3 d. The time-course experiment was conducted by culturing HEK293T cells in DMEM medium containing 300 μM of F-5caC and the cells were harvested at different time points ranging from 0.25 h to 72 h.Enzymatic digestion of DNA
Enzymatic digestion of genomic DNA was carried out according to previous reports.28–31 The detailed enzymatic digestion procedure can be found in the ESI.† The resulting nucleosides were analyzed by LC-ESI-MS/MS.Extraction of nucleoside/nucleotide pool
HEK293T cells were cultured in DMEM medium supplemented with 300 μM of F-5caC for different times. The cells were harvested and washed twice with PBS to remove the medium and fetal bovine serum. 500 μL of ice-cold 50% acetonitrile (v/v) was added dropwise to the cell pellet and vortexed. The mixture was kept on ice for 10 min and then centrifuged for 10 min at 14000 rpm at 4 °C to remove the insoluble fraction. The supernatant was collected and lyophilized to dryness, reconstituted in 1.5 mL H2O and purified using Supel-Select SPE HLB tubes (Sigma-Aldrich, Beijing, China). Prior to use, the tubes were equilibrated with 3 mL methanol and 3 mL acidified H2O (with HCl to pH 4.0). The pH of the sample was adjusted to 4.0 by adding HCl and then 1.5 mL acidic solution was loaded into the tubes. After washing with 10 mL of H2O, the nucleosides and nucleotides were eluted with 3 mL methanol/acetonitrile (1:1), lyophilized to dryness and reconstituted in 26 μL H2O. 3 μL of 10× buffer (500 mM Tris–HCl, 100 mM NaCl, 10 mM MgCl2, 10 mM ZnSO4, pH 7.0), and1 μL of calf intestinal alkaline phosphatase (30 U μL−1) were added into the mixture, and incubated at 37 °C for 1 h. The resulting solution was extracted with chloroform twice and subjected to LC-ESI-MS/MS analysis.
LC-ESI-MS/MS analysis
Quantitative LC-ESI-MS/MS analysis of digested nucleosides was conducted on a system consisting of a Shimadzu 8045 mass spectrometer (Kyoto, Japan) with an electrospray ionization (ESI) source coupled with a Shimadzu LC-30AD UPLC system (Tokyo, Japan). The chromatographic separation of nucleosides was performed on a Thermo Accucore C18 column (150 mm × 2.1 mm i.d., 2.6 μm) at 35 °C. Nucleosides were separated by a 12 min gradient with the use of 0.05% (v/v) formic acid in H2O (A) and acetonitrile (B) as the mobile phases at a flow rate of 0.3 mL min−1.Gradients of 0–1.5 min 5% B, 1.5–3 min 5 to 40% B, 3–5 min 40% B, 5–5.5 min 40 to 5% B, and 5.5–12 min 5% B were employed. As for the in vivo decarboxylation assay, a Waters Acquity UPLC® HSS T3 column (100 mm × 2.1 mm i.d., 1.8 μm) was used for the chromatographic separation and 0.05% (v/v) formic acid in H2O and methanol were employed as the mobile phases. The nucleosides were detected with multiple-reaction monitoring (MRM) in the positive-ion mode. The mass spectrometric parameters for MRM analysis were optimized by direct injection and the optimized conditions are listed in ESI Table S1.†Expression and purification of recombinant TDG
The N-terminal 6× His-tagged human TDG expression construct of pET21a ampicillin-resistant vector was obtained from Tsingke Biotechnology Co. Ltd. (Beijing, China). The plasmid was transformed into E. coli Rostta-gami (DE3) competent cells. The cells were grown to an optical density (OD600) of 0.5 at 37 °C and the protein expression was induced with the addition of 0.25 mM IPTG for 16 h at 25 °C. The E. coli cells were lysed by pulsed ultrasonication and centrifuged at 13000 rpm for 1 h at 4 °C. The His-tagged recombinant TDG protein was purified using HisSep Ni-NTA agarose resin (Yeasen Biotechnology Co. Ltd) according to manufacturer's protocol. The purified protein was further concentrated by ultrafiltration (Merck Millipore Ltd) and stored at −80 °C.
TDG activity assay
For in vitro TDG activity assay, 0.5 μM of double-stranded fluorescein labeled DNA containing T/G mismatch was incubated with TDG protein for 3 h at 37 °C in a 10 μL solution containing 10 mM Tris–HCl (pH 8.0), 1 mM EDTA, and 0.05% BSA. DNA was purified by precipitation with ethanol and then suspended in 10 μL H2O. 2 μL of 1 M NaOH was added to the solution and heated to 95 °C for 10 min. Then 36 μL of formamide was added and heated to 95 °C for 10 min and kept in ice-water immediately. The resulting DNA was analyzed by 20% denaturing polyacrylamide gel electrophoresis.To evaluate the potential cleavage of the N-glycosidic bond of F-5caC, 10 μg of F-5caC labeled genomic DNA from HEK293T cells was incubated with 3 μM or 6 μM recombinant TDG protein. After incubation at 37 °C for 3 h, proteinase K (0.1 mg mL−1) was added into the reaction solution and incubated at 55 °C for additional 2 h. To inactivate proteinase K, the solution was incubated at 95 °C for 10 min. The resulting solution was extracted with chloroform twice, and then the aqueous layer was collected and lyophilized to dryness for subsequent enzymatic digestion and LC-ESI-MS/MS analysis.
Statistical analysis
The experimental data were processed with SPSS 19.0 software (SPSS Inc., USA). We used a paired t test to evaluate the difference between samples. All p values were two-sided and p values < 0.05 were considered to have statistical significance.Results
Direct decarboxylation of 5caC in genomic DNA using nuclear extracts
In this study, we aimed to explore the direct decarboxylation of 5caC within mammalian genomes. To this end, we started the study with in vitro decarboxylation assay. We examined the decarboxylation of 5caC in genomes of HEK293T cells by treating the isolated genomic DNA with nuclear extracts (Fig. 1a). The levels of 5fC and 5caC were higher in the genomic DNA of TDG−/− HEK293T cells compared to wild-type (wt) HEK293T cells (ESI Fig. S1 and S2†). The quantification results showed that the level of 5caC was 7.5 × 10−7 per dC in the TDG−/− HEK293T cells, while no detectable 5caC was observed in the wt HEK293T cells (ESI Fig. S2†). Thus, we used the genomic DNA from the TDG−/− HEK293T cells to carry out the in vitro decarboxylation assay.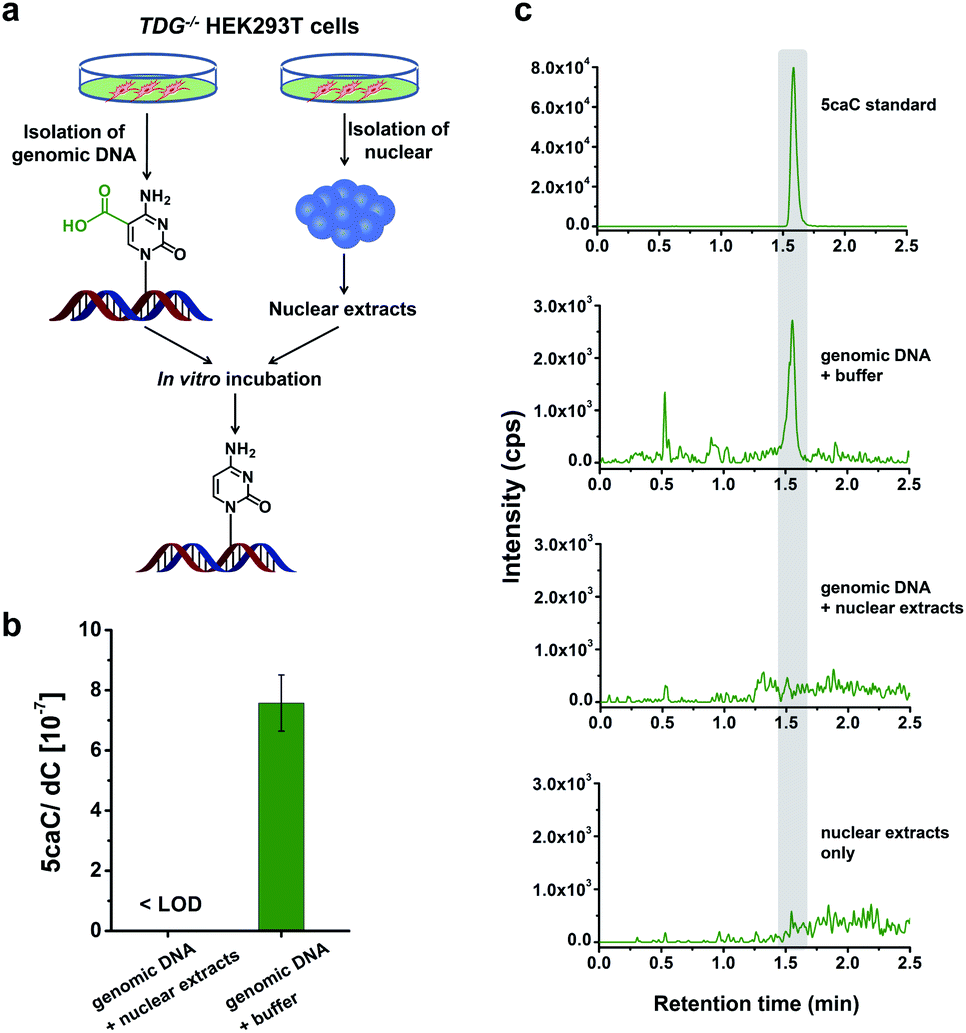 | ||
| Fig. 1 In vitro decarboxylation assay. (a) Schematic overview of the in vitro decarboxylation assay. Genomic DNA from TDG−/− HEK293T cells was incubated with the nuclear extracts generated from TDG−/− HEK293T cells. (b) Quantification of the level of 5caC in genomic DNA upon incubation with nuclear extracts from TDG−/− HEK293T cells or buffer. (c) Extracted-ion chromatograms for monitoring 5caC from genomic DNA with different treatments with LC-ESI-MS/MS analysis. | ||
The isolated genomic DNA from the TDG−/− HEK293T cells was incubated with nuclear extracts that were generated from the TDG−/− HEK293T cells (Fig. 1a). After incubation at 37 °C for 3 h, the genomic DNA was enzymatically digested for LC-ESI-MS/MS analysis. The results showed that 5caC was dramatically decreased to an undetectable level in genomic DNA upon incubation with nuclear extracts; however, 5caC was still clearly observed in the control sample of genomic DNA that was treated with the buffer (Fig. 1b and c, ESI Fig. S3†). No detectable 5caC was observed in the nuclear extracts (Fig. 1c and ESI Fig. S3†), excluding the interference from the background. We also monitored the content change of 5hmC and 5fC beside 5caC in genomic DNA. The results showed that there was no obvious decrease of the level of 5hmC or 5fC (ESI Fig. S4†), indicating the high specificity of the decarboxylation of 5caC in genomic DNA. Collectively, the results demonstrated the direct decarboxylation of 5caC within mammalian genomes using nuclear extracts.
In vivo decarboxylation assay by F-5caC metabolic labeling
The direct decarboxylation of 5caC within genomic DNA using nuclear extracts inspired us to further explore the decarboxylation of 5caC in vivo. To distinctly trace the endogenous decarboxylation of 5caC in genomes, we first utilized 2′-fluorinated 5-carboxyl-deoxycytidine (F-5caC) to metabolically label genomic DNA. Upon feeding of F-5caC to mammalian cells, the nucleoside of F-5caC would be converted into triphosphate and then incorporated into genomic DNA. Then we can evaluate the direct decarboxylation of F-5caC within genomes by examining the decarboxylated product of 2′-fluorinated dC (F-dC) and the potentially remethylated product of 2′-fluorinated 5mC (F-5mC) (Fig. 2a).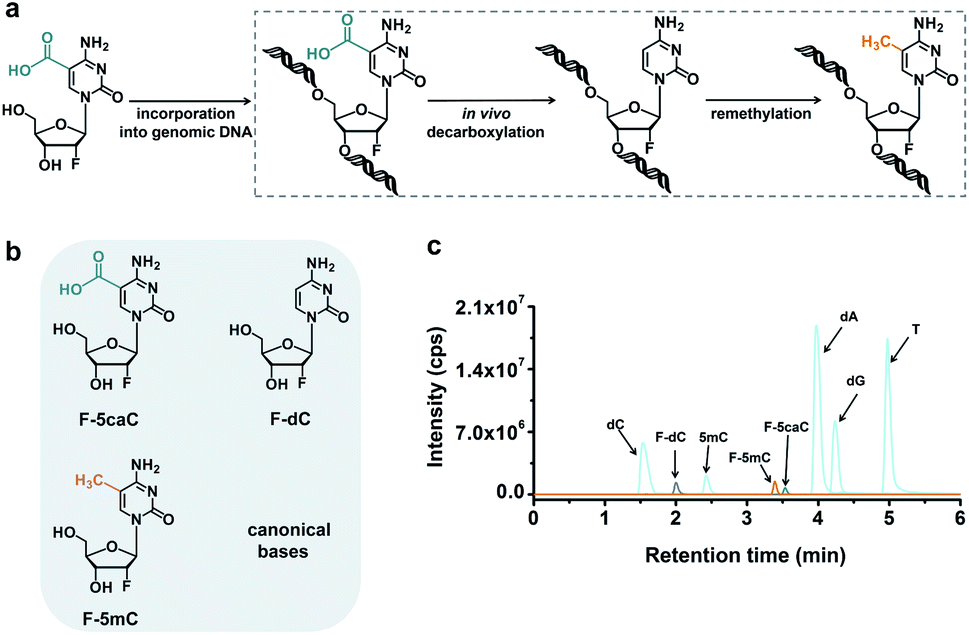 | ||
| Fig. 2 F-5caC metabolic labeling monitored by mass spectrometry. (a) Schematic overview of the feeding experiment using F-5caC. Upon feeding of F-5caC to mammalian cells, the nucleoside of F-5caC would be converted into triphosphate and then incorporated into genomic DNA. F-5caC within genomic DNA could undergo decarboxylation to produce F-dC and the remethylated product of F-5mC. (b) Chemical structures of F-5caC, F-dC, and F-5mC. (c) Extracted-ion chromatograms of F-5caC, F-dC, F-5mC and canonical nucleosides with LC-ESI-MS/MS analysis. | ||
The metabolic labeling of F-5caC requires the F-5caC to contain no even trace level of F-dC. If there is F-dC contamination in F-5caC, F-dC will also be incorporated into genomic DNA in a similar manner to F-5caC. This contaminated F-dC would lead to false results since the F-dC incorporated into genomes through DNA replication cannot be distinguished from the F-dC originating from the direct decarboxylation of F-5caC. Therefore, we carried out additional purification for F-5caC by HPLC to ensure its high purity. The retention time of F-dC (13.7 min) is far away from that of F-5caC (21.2 min) (ESI Fig. S5†). Purification of F-5caC was performed by collecting the F-5caC peak. The LC/HRMS and NMR analysis showed no detectable F-dC in the HPLC-purified F-5caC (ESI Fig. S6 and S7†). In addition, no detectable F-5caC was observed in the F-dC standard (ESI Fig. S8 and S9†). These highly pure compounds of F-dC and F-5caC guaranteed the subsequent accurate and reliable analysis. Moreover, we prepared a F-5mC standard (ESI Fig. S10†). These 2′-fluorinated dC derivatives (F-dC, F-5caC, and F-5mC) could be well separated from each other and distinguished from other canonical nucleosides (Fig. 2b and c), which offered the basis to evaluate the decarboxylation of 5caC in vivo by metabolic labeling and LC-ESI-MS/MS analysis.
We cultured HEK293T cells in the presence of diverse concentrations of F-5caC for 3 d, and then isolated the genomic DNA and analyzed the nucleosides by LC-ESI-MS/MS. The results showed that, in addition to F-5caC, F-dC was clearly detected (Fig. 3a, b, d, and e). Moreover, we also detected F-5mC upon feeding of F-5caC (Fig. 3c and f), suggesting that the decarboxylated product of F-dC was remethylated to form F-5mC in the genomes. Calibration curves were constructed to quantify these 2′-fluorinated dC derivatives (ESI Fig. S11†). The quantification results showed that the contents of F-5caC and F-dC were increased along with the increased concentrations of F-5caC (Fig. 4a, b and ESI Fig. S12†). The level of F-dC was approximately 20-fold higher than that of F-5caC in genomic DNA (Fig. 4a and b), indicating that the conversion rate of F-5caC to F-dC was relatively high. It should be noted that neither F-5caC nor F-dC was detected from the control cells without F-5caC feeding or being treated with DMSO (ESI Fig. S12†). Moreover, neither F-dA nor F-dG was detected, indicating that there is no potential decarboxylation-independent pathway for the conversion of F-5caC to F-dC. Collectively, these experiments demonstrated that F-5caC could be decarboxylated to form F-dC within mammalian genomes in vivo.
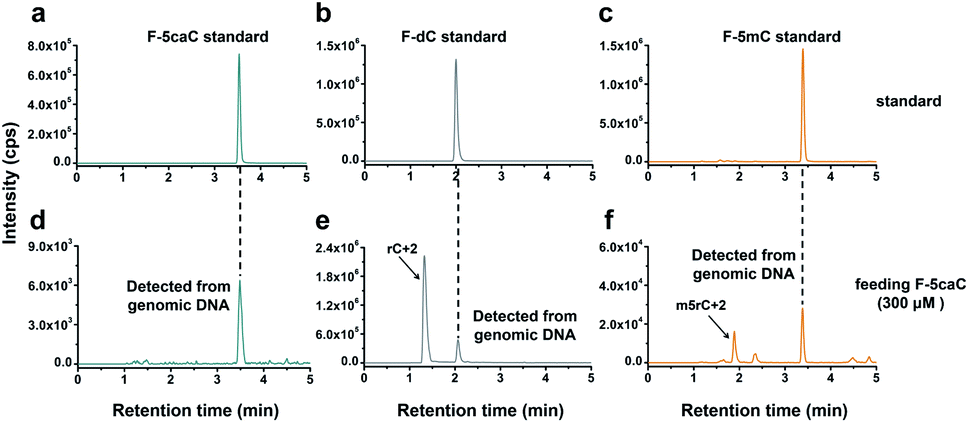 | ||
| Fig. 3 Determination of F-5caC, F-dC, and F-5mC within mammalian genomes upon feeding of F-5caC by LC-ESI-MS/MS analysis. (a–c) Extracted-ion chromatograms of F-5caC, F-dC, and F-5mC standards. (d–f) Extracted-ion chromatograms of F-5caC, F-dC, and F-5mC detected from genomic DNA of HEK293T cells upon feeding of 300 μM of F-5caC. The peaks of rC + 2 and m5rC + 2 represented the natural isotope peaks of RNA cytidine and 5-methylcytidine, respectively. | ||
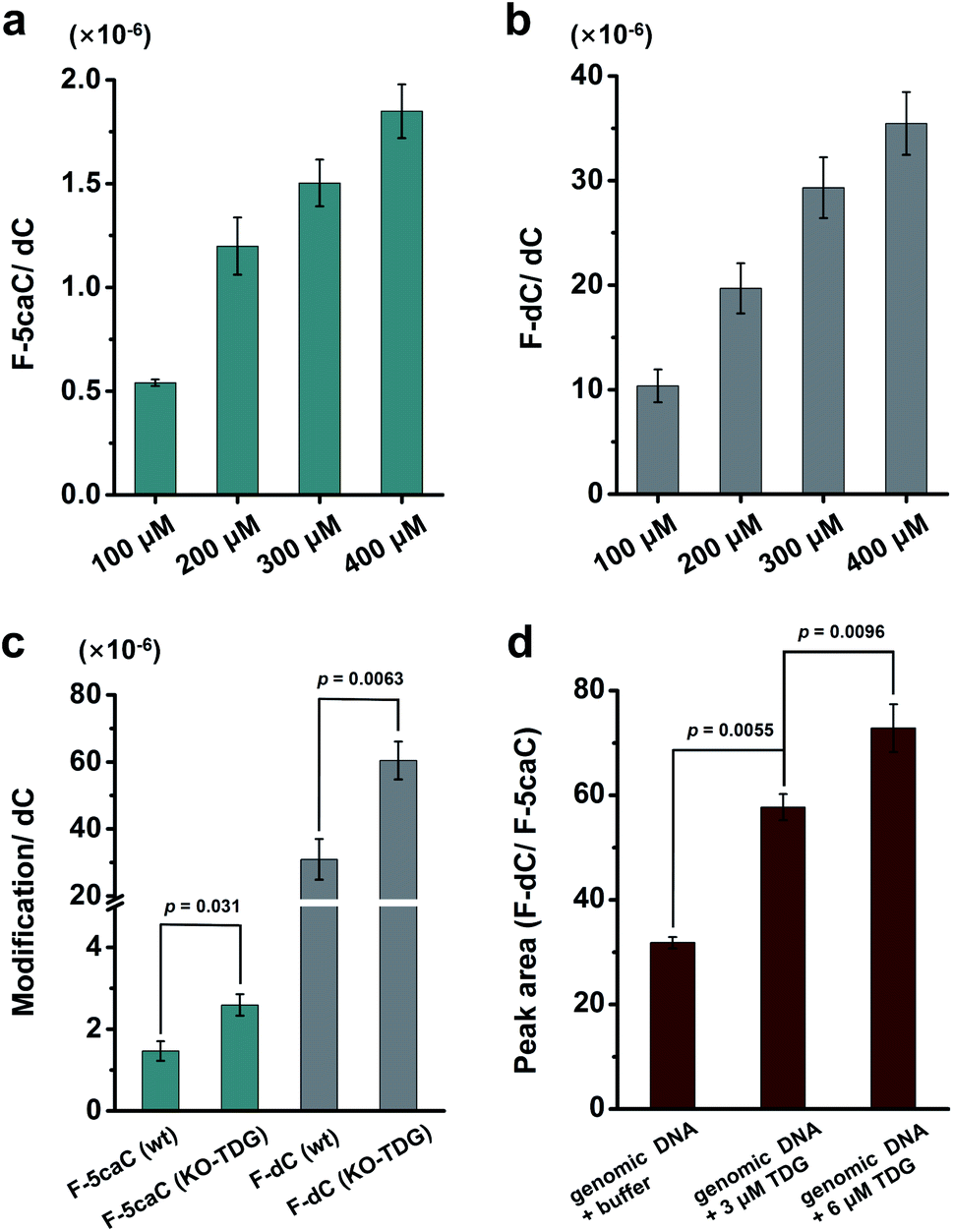 | ||
| Fig. 4 In vivo decarboxylation. (a, b) Quantification of levels of F-5caC and F-dC in genomic DNA upon feeding of F-5caC with diverse concentrations to HEK293T cells. (c) Quantification of the levels of F-5caC and F-dC in genomic DNA upon feeding of 300 μM of F-5caC to wt or TDG−/− HEK293T cells. (d) Peak area ratio of F-dC/F-5caC in F-5caC (300 μM) metabolically labeled genomic DNA upon incubation with the recombinant human TDG protein or buffer. | ||
We further evaluated the decarboxylation of F-5caC in TDG−/− HEK293T cells (ESI Fig. S13†). The result showed that the level of F-5caC was increased in the TDG−/− HEK293T cells (2.6 × 10−6 per dC) compared to that in the wt HEK293T cells (1.5 × 10−6 per dC) (Fig. 4c). Similarly, the level of F-dC in the TDG−/− HEK293T cells (60.4 × 10−6 per dC) was also significantly higher than that in the wt HEK293T cells (30.9 × 10−6 per dC) (Fig. 4c). The increased level of F-5caC in the TDG−/− HEK293T cells could be attributed to the compromised pathway of TET-TDG-BER because the deficiency of TDG led to the accumulation of F-5caC. Along this line, a larger amount of F-5caC in the TDG−/− HEK293T cells could be converted to F-dC, which therefore led to a higher level of F-dC in TDG−/− than in the wt HEK293T cells.
The above interpretation is built on the basis that TDG could, at least to some extent, recognize and cleave the N-glycosidic bond of 5caC. However, the substitution of the 2′-hydrogen by the F group might affect the cleavage of the N-glycosidic bond of F-5caC by the TDG protein. We then expressed and purified the human TDG protein (ESI Fig. S14†) and carried out the in vitro experiment to investigate whether the TDG protein could cleave the N-glycosidic bond of F-5caC. The activity of the recombinant TDG protein was confirmed by using a FAM-labeled duplexed DNA containing T/G mismatch (ESI Fig. S15†). Upon incubation of the TDG protein with F-5caC metabolically labeled genomic DNA, the level of F-5caC was significantly decreased, but there was no significant level change of F-dC (ESI Fig. S16†). The incubation of F-5caC metabolically labeled genomic DNA with the recombinant TDG protein led to the increased ratio of F-dC/F-5caC from 31.8 (incubation with buffer) to 57.8 (incubation with 3 μM TDG) and 72.8 (incubation with 6 μM TDG) (Fig. 4d). In addition, we also determined the kinetics of TDG in catalyzing F-5caC-DNA and 5caC-DNA. The results showed that the measured Vmax/Km (min−1 nM−1) was 4.1 × 10−3 and 66.3 × 10−3 for F-5caC-DNA and 5caC-DNA, respectively (ESI Fig. S17†), suggesting the activity of TDG toward F-5caC-DNA was weaker than toward the 5caC-DNA substrate. Although the crystal structure showed no obvious interaction between TDG and F-5caC,32 our in vitro experiment suggested that the recombinant TDG protein could recognize and cleave the N-glycosidic bond of F-5caC under the tested conditions. It's clear that a substantial amount of F-5caC was still left after incubation with the TDG protein (ESI Fig. S16†), indicating that the substitution of the 2′-hydrogen by the F group would attenuate the cleavage, but not abolish the activity of TDG.
The cleavage of F-5caC in genomic DNA by TDG will theoretically generate a 2′-fluorinated AP (F-AP) site. We further detected F-AP from the genomic DNA of HEK293T cells upon feeding of F-5caC. Since F-AP carries an aldehyde group, we used the Girard's reagent P to derivatize the F-AP to increase the LC-ESI-MS/MS detection performance according to our previous study.33,34 2-Deoxy-D-ribose was employed to evaluate the derivatization reaction conditions (ESI Fig. S18a†). The high-resolution mass spectrometry analysis showed that the desired derivative of Girard's reagent P labeled 2-deoxy-D-ribose was obtained (ESI Fig. S18b and S18c†). To achieve the best derivatization, we optimized the reaction conditions (ESI Fig. S19†). Under the optimized derivatization conditions, F-AP could be clearly observed in wide-type HEK293T cells, but not in TDG−/− HEK293T cells (ESI Fig. S20†), further indicating that the generated F-AP should be from the cleavage of F-5caC by TDG.
F-5caC is decarboxylated to F-dC within mammalian genomes
The metabolic labeling of F-5caC is based on the fact that F-5caC is converted into triphosphate and then incorporated into genomic DNA. However, if F-5caC is first decarboxylated to F-dC in soluble nucleoside/nucleotide pool, the free nucleoside of F-dC could also be converted into triphosphate and then incorporated into genomic DNA (Fig. 5a). Thus, the detected F-dC from genomes may not reflect the direct decarboxylation of F-5caC within genomes. To evaluate this possibility of the potential decarboxylation of F-5caC in soluble nucleoside/nucleotide pool, we extracted the soluble nucleoside/nucleotide from cells upon feeding of F-5caC for different times (ESI Fig. S21†). LC-ESI-MS/MS analysis showed that F-5caC was clearly detected; however, F-dC was undetectable at any time points (Fig. 5b and c; ESI Fig. S22†), excluding the possibility of the decarboxylation of F-5caC in the soluble nucleoside/nucleotide pool.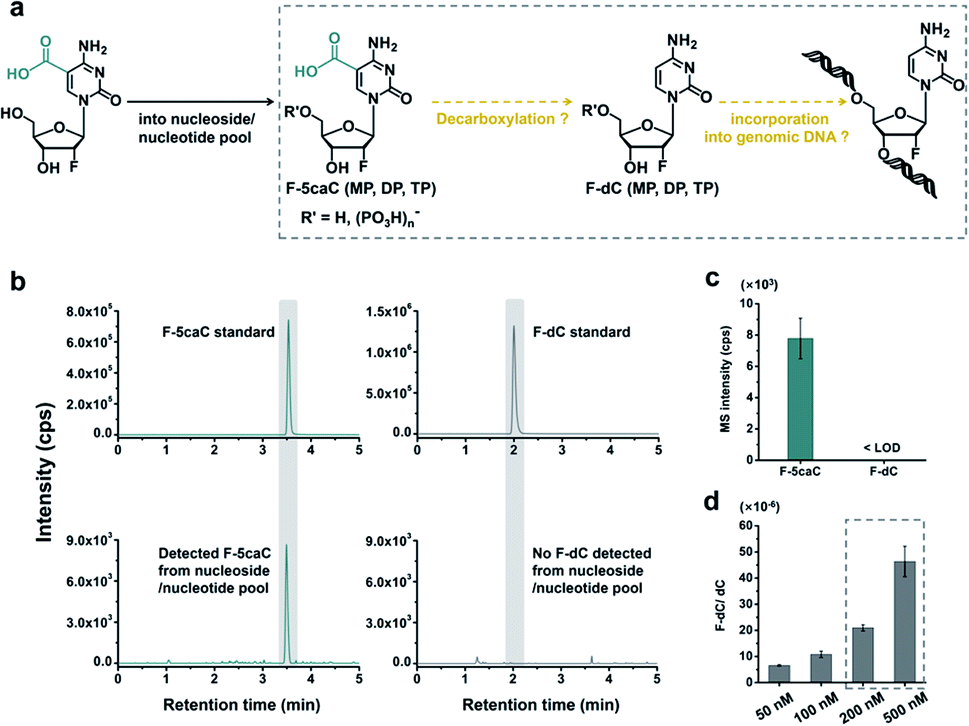 | ||
| Fig. 5 F-5caC was not decarboxylated to F-dC in soluble nucleoside/nucleotide pool. (a) Schematic overview of the assumed pathway of decarboxylation of F-5caC in nucleoside/nucleotide soluble pool. F-5caC (MP, DP, and TP) and F-dC (MP, DP, and TP) refer to the form of nucleoside, monophosphate, diphosphate, and triphosphate, respectively. (b) Extracted-ion chromatograms for monitoring F-5caC and F-dC in the soluble pool. (c) Quantitative data of F-5caC and F-dC in the soluble pool after feeding 300 μM of F-5caC for 3 d. (d) Quantification of F-dC level in genomic DNA after feeding of F-dC with diverse concentrations to HEK293T cells for 3 d. | ||
We further investigated whether the inability to detect F-dC in the soluble pool was due to the capability for the detection of F-dC being beyond the instrumental detection limit. We found that to achieve the similar level of detected F-dC in genomic DNA with feeding of 300 μM of F-5caC (Fig. 4b) we required a concentration of 200–500 nM F-dC in the soluble pool (Fig. 5d and ESI Fig. S23†). However, as low as 50 nM of F-dC in the soluble pool can be distinctly detected (ESI Fig. S23†), indicating that the inability to detect F-dC in the soluble pool was not because of the low instrumental detection sensitivity, but indeed the absence of conversion of F-5caC to F-dC in the soluble pool. We also examined the potentially spontaneous decarboxylation of F-5caC in medium and during genomic DNA processing. The results showed no detectable F-dC in medium or during the processing of genomic DNA (ESI Fig. S24†). Collectively, the results supported that the direct decarboxylation of F-5caC occurred within mammalian genomes, but not in soluble nucleoside/nucleotide pool or during the processing of genomic DNA.
Direct decarboxylation is a rapid process and occurs in diverse mammalian cells
To study the time dependence of the direct decarboxylation process, we fed HEK293T cells with F-5caC and measured the genomic levels of F-5caC, F-dC, and F-5mC. The results showed that F-5caC could be detected at 0.25 h, along with a time-dependent increase from 0.25 h to 1 h (Fig. 6a, ESI Fig. S25a†). F-dC could be detected at 1 h (Fig. 6b, ESI Fig. S25b†), suggesting that the direct decarboxylation is a rapid process. The remethylated product of F-5mC could be detected at 2 h (Fig. 6c and ESI Fig. S25c†). The level of F-5caC kept stable while the levels of F-dC and F-5mC increased with extended feeding time. It can be observed that the level of F-dC reached a plateau at ∼8 h, while F-5mC reached a plateau at ∼24 h. The quantification results showed that the level of F-5mC is ∼3% relative to F-dC, which is comparable to the proportion of natural 5mC in genomic DNA of HEK293T cells.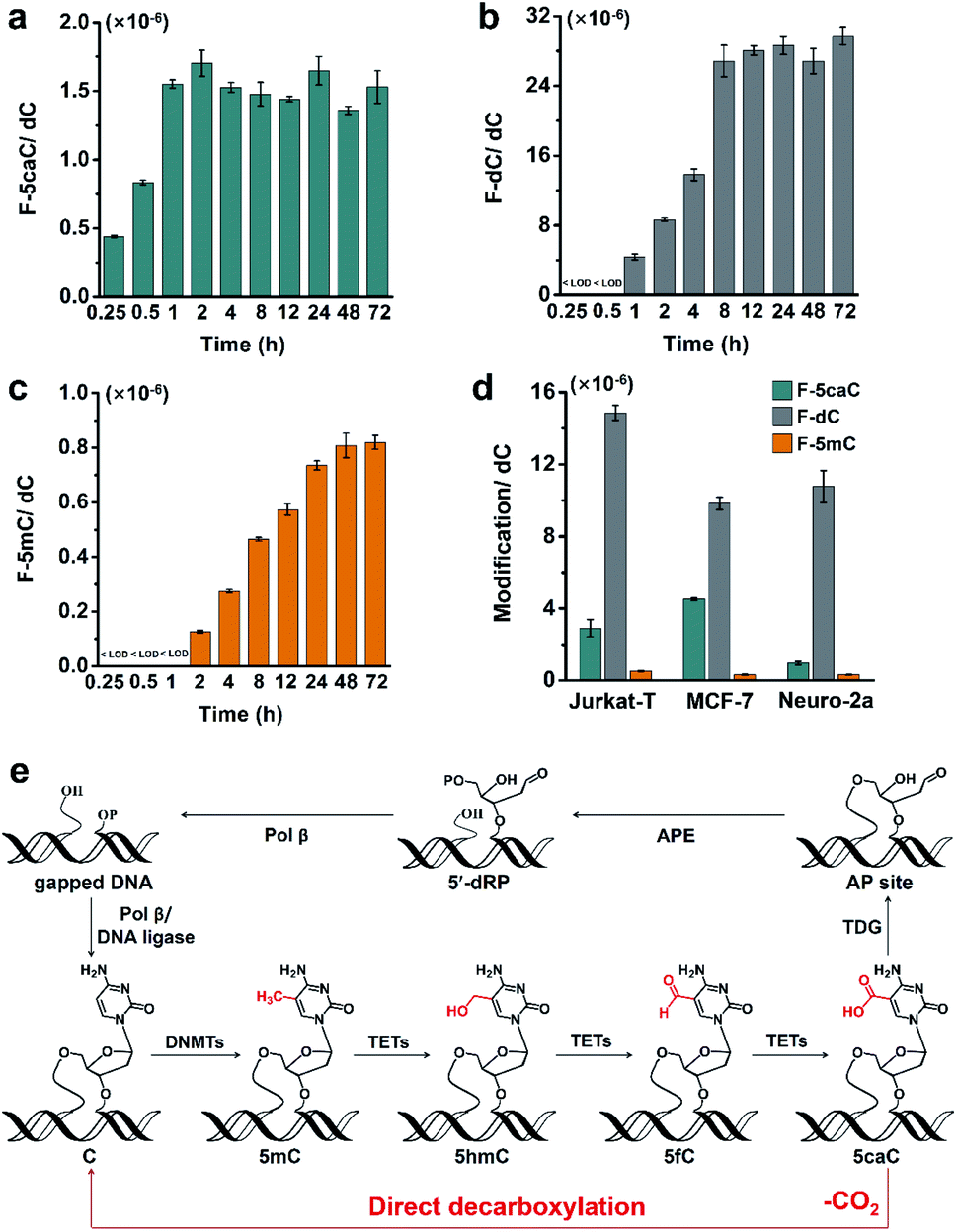 | ||
| Fig. 6 Decarboxylation of F-5caC is a rapid process and occurs in diverse mammalian cells. (a–c) Quantification of F-5caC, F-dC, and F-5mC levels in genomic DNA of HEK293T cells upon feeding of 300 μM of F-5caC for different times. (d) Quantification of F-5caC, F-dC, and F-5mC levels in genomic DNA of Jurkat-T, MCF-7, and Neuro-2a cells upon feeding of 300 μM of F-5caC for 3 d. (e) Schematic illustration of the active 5mC demethylation through two pathways: the direct decarboxylation of TET-produced 5caC to cytosine in mammalian genomes established in the current study and the previous TET-TDG-BER pathway. | ||
In addition to HEK293T cells, we also evaluated the direct decarboxylation of 5caC in different cell lines by F-5caC metabolic labeling. The results showed that both F-dC and F-5mC were detected in all these cell lines (Fig. 6d and ESI Fig. S26†), suggesting that direct decarboxylation of F-5caC with genomes is widespread in mammalian cells. Taken together, we reveal that the direct decarboxylation of TET-produced 5caC to cytosine in genomes constitutes a new mechanism for active 5mC demethylation, which provides an additional regulation layer for the gene expression besides the TET-TDG-BER pathway (Fig. 6e).
Discussion
To date, several DNA demethylation mechanisms have been proposed. The most prevalent mechanism is the TET-TDG-BER pathway. Our previous study demonstrated the direct decarboxylation of 5caC to cytosine in the synthesized 5caC-containing DNA that was transfected into human cells. It is reasonable to speculate that an active 5mC demethylation mechanism without the involvement of the TET-TDG-BER pathway may also exist in genomes. In the current study, we demonstrated the decarboxylation of 5caC occurring in mammalian genomes by using metabolic labeling and mass spectrometry analysis, which established an intragenomic demethylation process independent of the TDG-BER system.Although we confirmed the occurrence of direct decarboxylation of 5caC to dC within mammalian genomes in vivo, the enzymes which mediated the decarboxylation reaction remain to be identified. The well-studied DNA methyltransferases, which can convert 5caC to dC in vitro, show reduced activity in the physiological levels of SAM. Some other known decarboxylases including orotate and isoorotate decarboxylase exhibited weak decarboxylation activity to 5caC in vitro, suggesting that the mammalian homologues of these decarboxylase could be potential decarboxylase toward 5caC. The identification of specific binding proteins for 5caC by a proteomics-based strategy in the future study may provide clues for the candidate decarboxylases of 5caC.
The decarboxylation of 5caC in genomes provides a novel mechanism for the active DNA demethylation. The active DNA demethylation through the TET-TDG-BER pathway entails many enzymes and reactions. In contrast, the direct decarboxylation of TET-produced 5caC only needs one step reaction and DNA strand breaks will not be produced in this pathway, and is more cost-effective in terms of the consumption of energy and biomass. In addition, the pathway of the direct decarboxylation of TET-produced 5caC offers a useful backup system in scenarios where the TDG-BER system is damaged.
Conclusions
In summary, we investigated the direct decarboxylation of 5caC in mammalian genomes by treating 5caC-containing genomic DNA with nuclear extracts or utilizing F-5caC metabolic labeling coupled with mass spectrometry analysis. The quantitative data demonstrated that 5caC was efficiently decarboxylated into dC with mammalian genomes. The direct decarboxylation of 5caC is a rapid process and occurs in various mammalian cell lines. Taken together, we suggested that the TET-mediated oxidation of 5mC followed by direct decarboxylation of 5caC constitutes a novel pathway for active DNA demethylation in mammalian genomes.Author contributions
Y. F., and B. F. Y. designed the experiments and interpreted the data. Y. F., J. J. C., and N. B. X. performed the synthesis and purification of F-5caC, F-dC, and F-5mC compounds, and carried out the metabolic labeling, mass spectrometry analysis, and TDG protein expression and purification. J. H. D., X. J. Y., and W. B. T. performed the cell culturing and DNA isolation. X. Z. and C. Y. generated the TDG−/− HEK293T cells. X. Z. and Y. Q. F. performed the mass spectrometry analysis and interpreted the data. Y. F. and B. F. Y. wrote the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22074110, 21635006, and 21721005).References
- C. Luo, P. Hajkova and J. R. Ecker, Science, 2018, 361, 1336–1340 CrossRef CAS PubMed.
- T. Liu, C. J. Ma, B. F. Yuan and Y. Q. Feng, Sci. China: Chem., 2018, 61, 381–392 CrossRef CAS.
- K. Chen, B. S. Zhao and C. He, Cell Chem. Biol., 2016, 23, 74–85 CrossRef CAS PubMed.
- B. F. Yuan, Chem. Res. Toxicol., 2020, 33, 695–708 Search PubMed.
- S. Kriaucionis and N. Heintz, Science, 2009, 324, 929–930 CrossRef CAS.
- M. Tahiliani, K. P. Koh, Y. Shen, W. A. Pastor, H. Bandukwala, Y. Brudno, S. Agarwal, L. M. Iyer, D. R. Liu, L. Aravind and A. Rao, Science, 2009, 324, 930–935 CrossRef CAS PubMed.
- Y. F. He, B. Z. Li, Z. Li, P. Liu, Y. Wang, Q. Tang, J. Ding, Y. Jia, Z. Chen, L. Li, Y. Sun, X. Li, Q. Dai, C. X. Song, K. Zhang, C. He and G. L. Xu, Science, 2011, 333, 1303–1307 CrossRef CAS PubMed.
- S. Ito, L. Shen, Q. Dai, S. C. Wu, L. B. Collins, J. A. Swenberg, C. He and Y. Zhang, Science, 2011, 333, 1300–1303 CrossRef CAS.
- R. Yin, S. Q. Mao, B. Zhao, Z. Chong, Y. Yang, C. Zhao, D. Zhang, H. Huang, J. Gao, Z. Li, Y. Jiao, C. Li, S. Liu, D. Wu, W. Gu, Y. G. Yang, G. L. Xu and H. Wang, J. Am. Chem. Soc., 2013, 135, 10396–10403 CrossRef CAS.
- S. Liu, J. Wang, Y. Su, C. Guerrero, Y. Zeng, D. Mitra, P. J. Brooks, D. E. Fisher, H. Song and Y. Wang, Nucleic Acids Res., 2013, 41, 6421–6429 CrossRef CAS PubMed.
- C. B. Qi, J. H. Ding, B. F. Yuan and Y. Q. Feng, Chin. Chem. Lett., 2019, 30, 1618–1626 CrossRef CAS.
- X. Lu, B. S. Zhao and C. He, Chem. Rev., 2015, 115, 2225–2239 CrossRef CAS.
- X. Wu and Y. Zhang, Nat. Rev. Genet., 2017, 18, 517–534 CrossRef CAS PubMed.
- F. Spada, S. Schiffers, A. Kirchner, Y. Zhang, G. Arista, O. Kosmatchev, E. Korytiakova, R. Rahimoff, C. Ebert and T. Carell, Nat. Chem. Biol., 2020, 16, 1411–1419 CrossRef CAS PubMed.
- Q. Wang, J. H. Ding, J. Xiong, Y. Feng, B. F. Yuan and Y. Q. Feng, Chin. Chem. Lett., 2021 DOI:10.1016/j.cclet.2021.05.020.
- A. R. Weber, C. Krawczyk, A. B. Robertson, A. Kusnierczyk, C. B. Vagbo, D. Schuermann, A. Klungland and P. Schar, Nat. Commun., 2016, 7, 10806 CrossRef CAS.
- L. Shen, H. Wu, D. Diep, S. Yamaguchi, A. C. D'Alessio, H. L. Fung, K. Zhang and Y. Zhang, Cell, 2013, 153, 692–706 CrossRef CAS.
- Z. J. Liu, S. Martinez Cuesta, P. van Delft and S. Balasubramanian, Nat. Chem., 2019, 11, 629–637 CrossRef CAS PubMed.
- F. Guo, X. Li, D. Liang, T. Li, P. Zhu, H. Guo, X. Wu, L. Wen, T. P. Gu, B. Hu, C. P. Walsh, J. Li, F. Tang and G. L. Xu, Cell Stem Cell, 2014, 15, 447–458 CrossRef CAS PubMed.
- S. Schiesser, T. Pfaffeneder, K. Sadeghian, B. Hackner, B. Steigenberger, A. S. Schroder, J. Steinbacher, G. Kashiwazaki, G. Hofner, K. T. Wanner, C. Ochsenfeld and T. Carell, J. Am. Chem. Soc., 2013, 135, 14593–14599 CrossRef CAS.
- Z. Liutkeviciute, E. Kriukiene, J. Licyte, M. Rudyte, G. Urbanaviciute and S. Klimasauskas, J. Am. Chem. Soc., 2014, 136, 5884–5887 CrossRef CAS.
- S. Schiesser, B. Hackner, T. Pfaffeneder, M. Muller, C. Hagemeier, M. Truss and T. Carell, Angew. Chem., Int. Ed., 2012, 51, 6516–6520 CrossRef CAS.
- S. Xu, W. Li, J. Zhu, R. Wang, Z. Li, G. L. Xu and J. Ding, Cell Res., 2013, 23, 1296–1309 CrossRef CAS.
- K. Iwan, R. Rahimoff, A. Kirchner, F. Spada, A. S. Schroder, O. Kosmatchev, S. Ferizaj, J. Steinbacher, E. Parsa, M. Muller and T. Carell, Nat. Chem. Biol., 2018, 14, 72–78 CrossRef CAS.
- A. Schon, E. Kaminska, F. Schelter, E. Ponkkonen, E. Korytiakova, S. Schiffers and T. Carell, Angew. Chem., Int. Ed., 2020, 59, 5591–5594 CrossRef PubMed.
- Y. Feng, N. B. Xie, W. B. Tao, J. H. Ding, X. J. You, C. J. Ma, X. Zhang, C. Yi, X. Zhou, B. F. Yuan and Y. Q. Feng, CCS Chem., 2020, 2, 994–1008 Search PubMed.
- X. Shu, M. Liu, Z. Lu, C. Zhu, H. Meng, S. Huang, X. Zhang and C. Yi, Nat. Chem. Biol., 2018, 14, 680–687 CrossRef CAS PubMed.
- J. Xiong, T. T. Ye, C. J. Ma, Q. Y. Cheng, B. F. Yuan and Y. Q. Feng, Nucleic Acids Res., 2019, 47, 1268–1277 CrossRef CAS PubMed.
- C. B. Qi, H. P. Jiang, J. Xiong, B. F. Yuan and Y. Q. Feng, Chin. Chem. Lett., 2019, 30, 553–557 CrossRef CAS.
- M. Y. Cheng, X. J. You, J. H. Ding, Y. Dai, M. Y. Chen, B. F. Yuan and Y. Q. Feng, Chem. Sci., 2021, 12, 8149–8156 RSC.
- Y. Dai, C. B. Qi, Y. Feng, Q. Y. Cheng, F. L. Liu, M. Y. Cheng, B. F. Yuan and Y. Q. Feng, Anal. Chem., 2021, 93, 6938–6946 CrossRef CAS PubMed.
- L. Zhang, X. Lu, J. Lu, H. Liang, Q. Dai, G. L. Xu, C. Luo, H. Jiang and C. He, Nat. Chem. Biol., 2012, 8, 328–330 CrossRef CAS.
- Y. Tang, J. Xiong, H. P. Jiang, S. J. Zheng, Y. Q. Feng and B. F. Yuan, Anal. Chem., 2014, 86, 7764–7772 CrossRef CAS PubMed.
- M. D. Lan, B. F. Yuan and Y. Q. Feng, Chin. Chem. Lett., 2019, 30, 1–6 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: HPLC purification of F-5caC; preparation of F-5mC; characterization of F-5caC, F-dC, and F-5mC by high-resolution LC/MS; characterization of F-5caC and F-dC by NMR; nucleoside stability assay; enzymatic digestion of DNA; kinetics study; derivatization of AP; detection of F-AP in genomic DNA; Table S1; Fig. S1–S26. See DOI: 10.1039/d1sc02161c |
| This journal is © The Royal Society of Chemistry 2021 |