
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral polymer hosts for circularly polarized electroluminescence devices†
Jayeon
Hong‡
a,
Sangsub
Kim‡
b,
Gyurim
Park
a,
Yongmoon
Lee
b,
Hyungchae
Kim
b,
Sungjin
Kim
c,
Tae-Woo
Lee
 cd,
Changsoon
Kim
*b and
Youngmin
You
*a
cd,
Changsoon
Kim
*b and
Youngmin
You
*a
aDivision of Chemical Engineering and Materials Science, Graduate Program in System Health Science and Engineering, Ewha Womans University, Seoul 03760, Republic of Korea. E-mail: odds2@ewha.ac.kr
bGraduate School of Convergence Science and Technology, Inter-University Semiconductor Research Center, Seoul National University, Seoul 08826, Republic of Korea. E-mail: changsoon@snu.ac.kr
cDepartment of Materials Science and Engineering, Seoul National University, Seoul 08826, Republic of Korea
dSchool of Chemical and Biological Engineering, Research Institute of Advanced Materials, Institute of Engineering Research, Nano Systems Institute (NSI), Seoul National University, Seoul 08826, Republic of Korea
First published on 21st May 2021
Abstract
Polymer electroluminescence devices producing circularly polarized luminescence (CP PLEDs) have valuable photonic applications. The fabrication of a CP PLED requires a polymer host that provides the appropriate chiral environment around the emitting dopant. However, chemical strategies for the design of chiral polymer hosts remain underdeveloped. We have developed new polymer hosts for CP PLED applications. These polymers were prepared through a free-radical polymerization of 3-vinylcarbazole with a chiral N-alkyl unit. This chiral unit forces the carbazole repeat units to form mutually helical half-sandwich conformers with preferred (P)-helical sense along the polymer main chain. Electronic circular dichroism measurements demonstrate the occurrence of chirality transfer from chiral monomers to achiral monomers during chain growth. The (P)-helical-sense-enriched polymer interacts diastereoselectively with an enantiomeric pair of new phosphorescent (R)- and (S)-dopants. The magnitude of the Kuhn dissymmetry factor (gabs) for the (P)-helically-enriched polymer film doped with the (R)-dopant was found to be one order of magnitude higher than that of the film doped with the (S)-dopant. Photoluminescence dissymmetry factors (gPL) of the order of 10−3 were recorded for the doped films, but the magnitude of diastereomeric enhancement decreased to that of gabs. The chiral polymer host permits faster energy transfer to the phosphorescent dopants than the achiral polymer host. Our photophysical and morphological investigations indicate that the acceleration in the chiral polymer host is due to its longer Förster radius and improved compatibility with the dopants. Finally, multilayer CP PLEDs were fabricated and evaluated. Devices based on the chiral polymer host with the (R)- and (S)-dopants exhibit electroluminescence dissymmetry factors (gEL) of 1.09 × 10−4 and −1.02 × 10−4 at a wavelength of 540 nm, respectively. Although challenges remain in the development of polymer hosts for CP PLEDs, our research demonstrates that chiroptical performances can be amplified by using chiral polymer hosts.
Introduction
Circularly polarized luminescence (CPL) refers to the differential emission between left- (LCPL) and right-handed circularly polarized light (RCPL). The two handedness of CPL has unique photonic utility: it makes possible the fabrication of 3D optical displays,1,2 bioprobes,3–5 biolabels,6 spintronic devices,7,8 security tags,9 lasers,10 enantioselective sensors,11,12 quantum encryption,13–15 and photocatalysts for asymmetric syntheses.16 This utility is expected to be maximized by the development of CPL sources with high brightness and large dissymmetry factor values. Of the various classes of CPL sources,17–33 polymer light-emitting devices producing inherent CPL (CP PLEDs) are particularly valuable. The relevant polymers are compatible with solution processes, which enables the fabrication of devices with large active areas at low cost. The majority of CP PLEDs developed to date contain CPL-active small molecular dopants that are doped into CPL-inactive polymers.20,21,25,29,34–37 Intrinsically CPL-active polymers based on poly(fluorene),22,38–46 poly(p-phenylenevinylene),30,47 poly(p-phenyleneethylene),48 poly(silane),49,50 poly(thiophene),51,52 and poly(dibenzofulvene)53 have been developed.Recent studies have found that the performances of CP PLEDs can be improved by providing an external asymmetric milieu around the emitting center. Di Nuzzo and co-workers obtained CP electroluminescence with a high electroluminescence dissymmetry factor (|gEL|, gEL = 2(ILCPL − IRCPL)/(ILCPL + IRCPL) where ILCPL and IRCPL are electroluminescence intensities of LCPL and RCPL, respectively) of 0.8 from PLEDs employing poly(fluorene-alt-benzothiadiazole) with tethered chiral alkyl branches.54 The high |gEL| value is attributed to the extrinsic polarization provided by chiral cholesteric phases formed upon thermal annealing of the polymer. Kim and co-workers obtained a very high |gEL| value of 1.13 from multilayer PLEDs.34 In these devices, a rubbed poly(imide) layer and a non-emissive chiral dopant are present in order to convert linearly polarized luminescence into CPL. Despite these high |gEL| values, these strategies have several intrinsic drawbacks. The presence of insulating additives and multidomains can adversely influence charge carrier behavior. In addition, extrinsic modulation relies on the macroscopic formation of chiral phases and is likely to be sensitive on the film-forming conditions.
Inspired by the above studies, we decided to explore the idea that a chiral-at-main chain polymer host could be used to create an asymmetric milieu at the molecular level. In particular, we were intrigued by the idea that a chiral polymer could exert diastereoselective effects on the chiroptical activities of homochiral dopants (Fig. 1). The molecular structures of our polymer hosts are based on poly(3-vinylcarbazole). The carbazole unit has been widely employed in polymer hosts because of its favorable characteristics, including a large charge carrier mobility, and a wide bandgap and a high triplet-state energies.55,56 Previous studies have investigated chiral polymers bearing carbazoles, but with a focus on their non-chiroptical properties.57–67 CPL-emitting polymers with a carbazole emissive unit have recently been developed.68,69 However, carbazole-based polymer hosts have not previously been utilized in CP PLEDs. Our polymer structure is intended to generate an intrinsic helical sense between adjacent carbazoles. Carbazole possesses a chiral (R)-α-phenylisopropyl moiety at its nitrogen atom. We expected that this chiral substituent would exert an asymmetric bias during polymerization, favoring one helical sense between the two adjacent carbazoles. A copolymer was also synthesized using achiral monomers to examine the chirality transfer during the course of its chain growth.
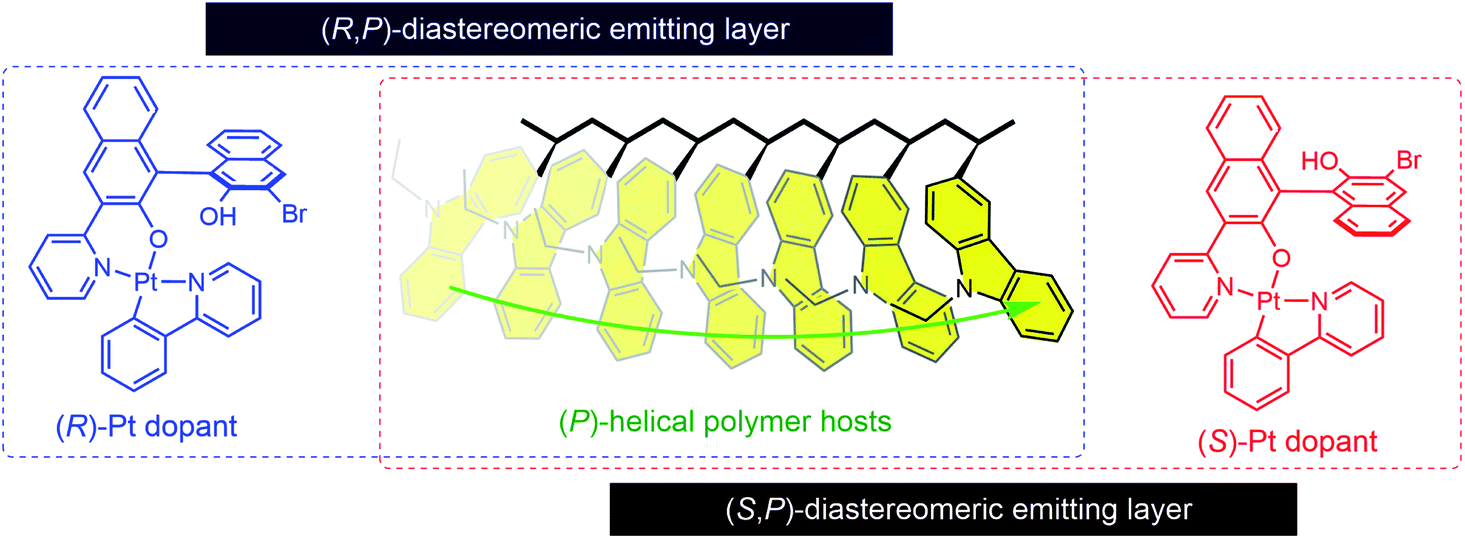 | ||
| Fig. 1 Diastereomeric interactions between dopants and polymer hosts with enriched helical sense. | ||
This strategy has particular potential because it requires neither chiral additives nor post-thermal treatments. Herein, we report our research into the development of chiral polymer hosts for use in CP PLEDs. The polymers were characterized by employing a variety of techniques, including structural, thermal, and spectroscopic methods. An enantiomeric pair of phosphorescent dopants based on cyclometalated Pt(II) complexes were newly created for our study. The diastereomeric interactions between the polymer hosts and the phosphorescent dopants were examined, with particular focus on chiral amplification and chirality-controlled energy transfer. Finally, CP PLEDs employing the chiral or achiral polymer hosts were fabricated, and their CP electroluminescence behaviors were compared. Our results demonstrate that chiroptical performances can be amplified by using chiral polymer hosts, while such an amplification is obscured within devices. This study undertook the first systematic research on chiral-at-main chain polymer hosts for CP PLEDs, which can provide insights that will be useful for the future development of CP PLEDs.
Results and discussion
Syntheses and structural characterizations of the polymer hosts
Our syntheses commenced with the tosylation of (R)-1-phenyl-2-propanol. The tosylate (1 in Scheme 1) was reacted with carbazole in the presence of potassium hydroxide and 18-crown-6. A Vilsmeier–Haack formylation reaction was then used to install a formyl unit on the carbazole adduct. Finally, Wittig-type olefination was performed on the aldehyde by using methyltriphenylphosphonium bromide and potassium carbonate to furnish the chiral monomer (4). The achiral monomer (5) was also prepared through olefination of the commercially available N-ethyl-3-carbazolecarboxaldehyde. The details of the syntheses are presented in the ESI section.† The chemical structures of the monomers and their precursors were characterized with standard spectroscopic techniques, including 1H and 13C{1H} NMR spectroscopy and high-resolution mass spectrometry. The spectroscopic identification data were found to be fully consistent with the proposed structures (ESI).†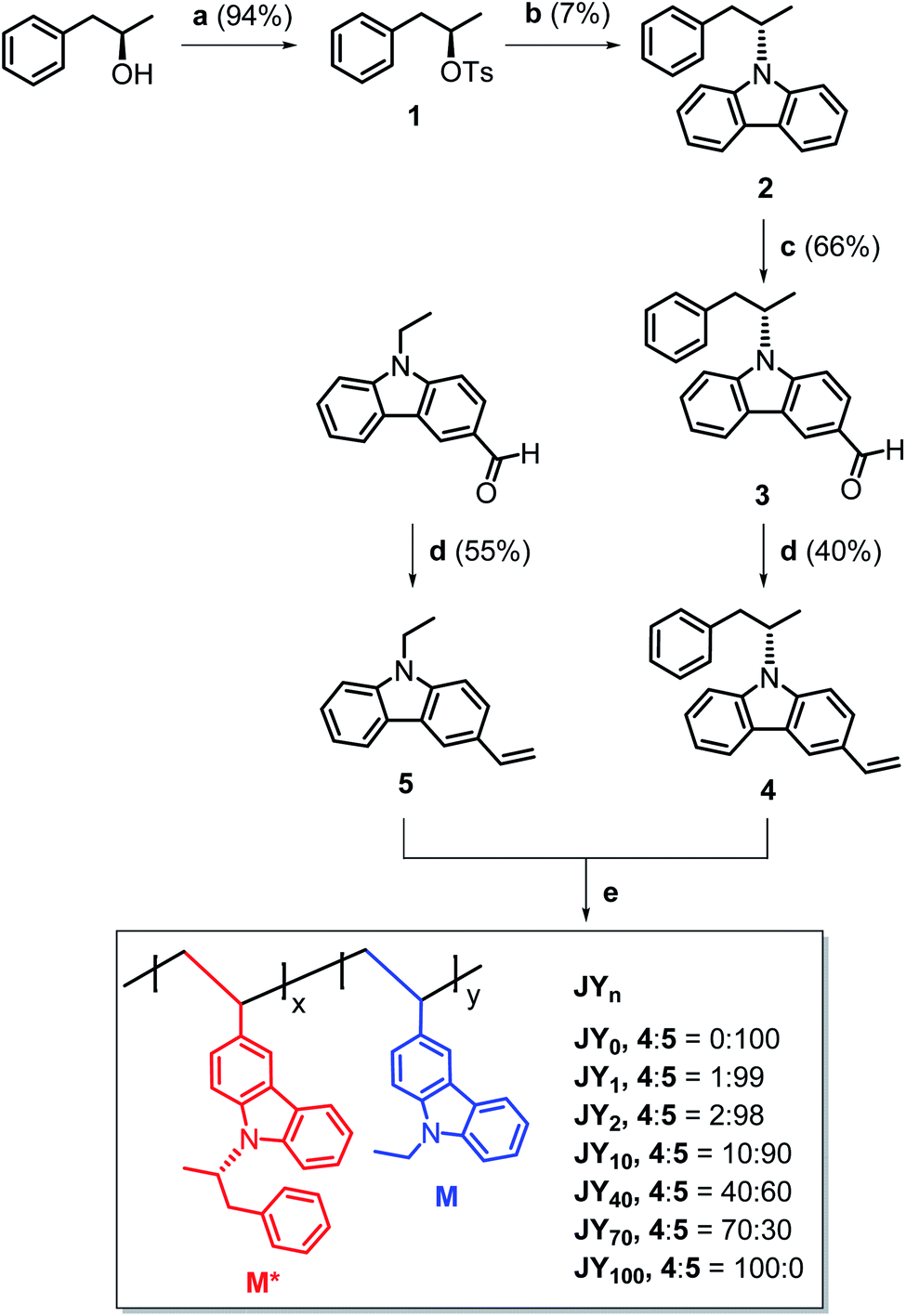 | ||
Scheme 1 Syntheses of the polymer hosts (JYn). n is the mole percentage of the chiral monomer (M*) in the feed. Conditions: (a), TsCl, pyridine, Ar, rt, 1 day; (b), (i) carbazole, KOH, 18-crown-6, DMSO, 80 °C, 1 h, (ii) 1, 80 °C, 2 days; (c), (i) POCl3, DMF, 0 °C, Ar, 1 h, (ii) 2, 110 °C, Ar, 1 day; (d), Ph3PCH3Br, K2CO3, 1,4-dioxane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 12:1 (v/v), reflux, Ar, 4 days; (e), 4:5 = 1–0:1 (mol/mol), 2 mol% AIBN, degassed NMP, 65 °C, 2 days. :H2O = 12:1 (v/v), reflux, Ar, 4 days; (e), 4:5 = 1–0:1 (mol/mol), 2 mol% AIBN, degassed NMP, 65 °C, 2 days. | ||
Conventional free-radical polymerization was performed with 4 and 5. A NMP solution containing the monomers and 2 mol% azobisisobutyronitrile (AIBN) initiator was thoroughly degassed with repeated vacuum–freeze–thaw cycles, and then heated at 65 °C for two days. The comonomer feed ratio of 4:5 was varied from 0:100 to 100:0. The resulting homopolymers (JY0 and JY100) and statistical copolymers (JY1, JY2, JY10, JY40, and JY70) were purified through repeated precipitation in methanol. Here, the subscript n in JYn refers to the mole percentage of 4 in its monomer feed. As summarized in Table 1, the conversion yields are in the range 68–80%. The conversion yield decreases with n, which is probably due to the steric hindrance exerted by the (R)-α-phenylisopropyl unit in 4. The number-averaged molecular weight (Mn) values of the copolymers were determined with GPC by using a Shodex® standard to be in the range 8700–13000 Da. Their polydispersity index (PDI) values are in the range 1.8–2.5, which is typical of free-radical polymerizations. Although it is tempting to correlate n with Mn, the two parameters do not exhibit a clear proportionality. This absence of a proportionality and the inverse relationship between n and the conversion yield, suggest that chain growth between chiral monomers is disfavored.
| Comonomer feed ratio ([4]:[5]) |
Yielda (%) | Repeat unit ratiob ([M*]:[M]) |
T g (°C) | T d,90 (°C) | M n | M w | PDIg | |
|---|---|---|---|---|---|---|---|---|
| a Conversion yields. b The chiral (M*) and achiral (M) repeat units molar ratio determined with 1H NMR spectroscopy. c Glass transition temperature. d Temperature at which 10% decomposition occurs (heating rate = 10 °C min−1). e Number average molecular weight. f Weight average molecular weight. The Mn and Mw values were determined with gel permeation chromatography (THF, Shodex® standard) techniques. g Polydispersity index, Mw/Mn. | ||||||||
| JY0 | 0:100 |
80 | 0:100 |
194 | 408 | 13000 |
30000 |
2.3 |
| JY1 | 1:99 |
79 | 2:98 |
190 | 411 | 10000 |
18000 |
1.8 |
| JY2 | 2:98 |
82 | 4:96 |
190 | 411 | 10000 |
18000 |
1.8 |
| JY10 | 10:90 |
71 | 7:93 |
170 | 401 | 7400 | 14000 |
2.0 |
| JY40 | 40:60 |
72 | 30:70 |
164 | 398 | 8700 | 19000 |
2.2 |
| JY70 | 70:30 |
73 | 53:47 |
150 | 401 | 8800 | 21000 |
2.4 |
| JY100 | 100:0 |
68 | 100:0 |
140 | 399 | 12000 |
30000 |
2.5 |
1H NMR spectroscopy (300 MHz, CD2Cl2) was employed to determine the ratio of the numbers of chiral (M*) and achiral (M) repeat units. As shown in Fig. 2a, the proton peaks belonging to M* and M are distinguishable. As expected, increasing the feed ratio of 4 yields a greater fraction of M* in the polymer chains. However, there is a concave deviation from linearity in the relationship between these two parameters (Fig. 2b); the fraction of M* in the polymer chain (i.e., [M*]/([M*] + [M])) is lower than the fraction of 4 in the monomer feed (i.e., [4]/([4] + [5])). This nonlinearity indicates that the reactivity with respect to polymerization of 4 is inferior to that of 5. The glass transition temperature (Tg, 10 °C min−1) of JY0 was determined to be 194 °C, and found to decrease in proportion with n (Fig. 2c and Table 1). This inverse relationship is probably due to the increase in free volume provided by the bulky (R)-α-phenylisopropyl unit in M*. In contrast, the degradation temperature, defined here as the temperature when the residual weight reaches 90% (Td,90), varies only very slightly with n. This result indicates that the presence of the chiral group in the copolymers has insignificant effects on their thermal stability (Fig. 2d and Table 1).
 | ||
| Fig. 2 (a) 1H NMR (300 MHz, CD2Cl2) spectra of the JYn polymers (6 mg mL−1). The peak marked with an asterisk is due to residual solvent. The structure on the right shows the assignments for Ha, Hb and Hc. The ratio of the integrated values of Hb and Hc corresponds to the ratio of the numbers of chiral (M*) and achiral (M) repeat units. (b) Fraction of chiral repeat units in the polymer (i.e., [M*]/([M*] + [M])) as a function of the mole fraction of the chiral monomer in the feed (i.e., [4]/([4] + [5])). (c) Differential scanning calorimetry results recorded during the first scan of the JYn polymers (heating rate = 10 °C min−1). (d) Thermogravimetric analyses for the JYn polymers (heating rate = 10 °C min−1). The structural and thermal parameters are summarized in Table 1. | ||
Chiroptical properties of polymer hosts
The host actions of the JYn polymers are likely to be sensitive to the helical sense. This claim is based on the fact that the mutual disposition of the carbazoles dictates the chiroptical properties of the polymers. An axial chirality (i.e., helical sense) of carbazole pendants emerges when the main chain is isotactic with the carbazole pendants must be helically disposed due to steric interactions. This carbazolyl conformer can have in either a right-handed or a left-handed helical sense, which we define as (P)- and (M)-axial chirality, respectively. In contrast, axial chirality is not present when the main chain is syndiotactic. Fig. 3a displays the UV-vis absorption spectra of neat films of the JYn polymers spin-coated onto quartz substrates. The spectra contain three major bands with peak wavelengths at 352, 308, and 265 nm. Based on quantum chemical calculations for carbazole, Belletête et al. assigned the longest (i.e., 352 nm) and medium (i.e., 308 nm) wavelength absorption bands to the short- and long-axis electronic transitions, respectively (see the structures in the inset in Fig. 3a).70 These bands also appear in the spectrum of a 10 μM model compound of M* (i.e., 2 in Scheme 1), which indicates that they are among the intrinsic transitions of monomeric carbazole. The absence of any new absorption bands excludes ground-state interactions in the JYn polymers.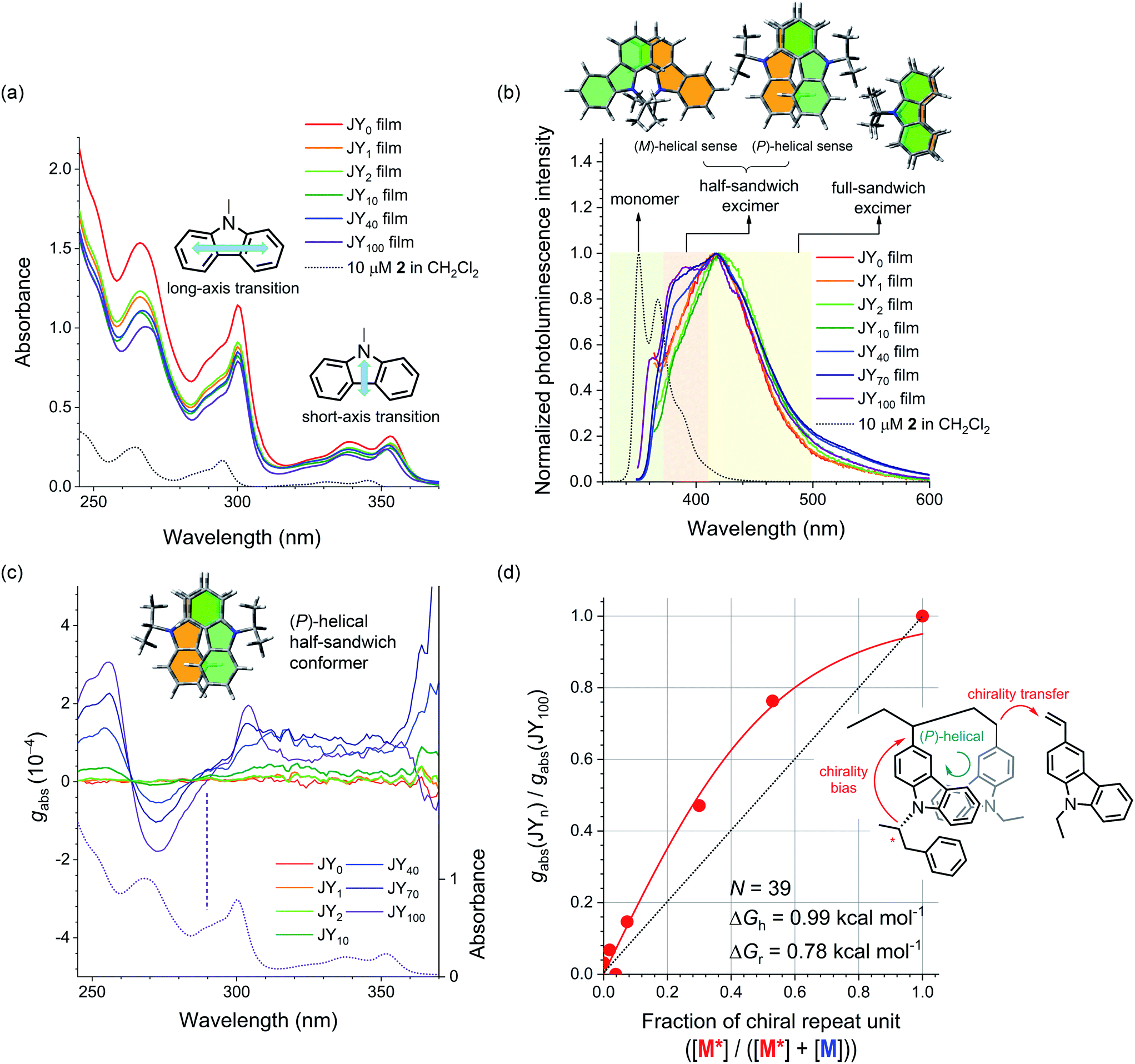 | ||
| Fig. 3 (a) UV-vis absorption spectra of neat films of the JYn polymers spin-coated onto quartz substrates. The black dotted curve is the UV-vis absorption spectrum of a 10 μM solution of 2 (CH2Cl2). The insets show the short- and long-axis transitions of carbazole. (b) Fluorescence spectra of neat films of the JYn polymers (λex = 339 nm). The black dotted curve is the fluorescence spectrum of a 10 μM solution of 2 (CH2Cl2). The colored regions highlight the monomeric emission of carbazole (green) and the carbazole excimer emissions of a half-sandwich conformer (beige) and a full-sandwich conformer (yellow). The structures at the top depict the two half-sandwich conformers and the full-sandwich conformer. (c) Plots of the Kuhn dissymmetry factor (gabs) as a function of wavelength for neat films of the JYn polymers cast onto quartz substrates. See ESI, Fig. S5† for the corresponding ECD spectra. The dotted curve is the UV-vis absorption spectrum of a neat film of the JY100 polymer. The positive Cotton effect in the long-axis transition reflects the (P)-helical sense of the half-sandwich conformer of carbazoles (see the inset). (d) A plot of the normalized gabs values (λ = 304 nm) for the JYn polymers (i.e., gabs(JYn)/gabs(JY100)) as a function of the chiral repeat unit fraction in the polymers (i.e., [M*]/([M*] + [M])). The red curve is the nonlinear least-squares fit of the data to eqn (1). The inset shows the chirality transfer from the chiral monomer to the achiral monomer during the polymerization (see the inset). | ||
In contrast to the UV-vis absorption spectra, the fluorescence spectra of 2 and the JYn polymers differ significantly (Fig. 3b). The fluorescence spectrum of a 10 μM solution of 2 (CH2Cl2) retains the characteristic vibronic progressions of carbazole with a peak wavelength of 350 nm. This spectral signature disappears in the fluorescence spectra of JYn, and a broad emission band emerges with a bathochromically shifted peak wavelength at ca. 420 nm. The fluorescence spectra also contain shoulder bands in the region 380–384 nm. The peak and shoulder emissions can be assigned to originate from the excimers of a full-sandwich carbazole pair and a half-sandwich carbazole pair, respectively (see the structures in the inset in Fig. 3b), on the basis of the earlier study by Johnson.71 The excimer assignments are supported by long rise profiles of the transient photoluminescence signals at 420 nm (ESI, Fig. S1†).72–77 The full-sandwich excimers are observed for fully eclipsed conformers of carbazoles along the polymer chain, whereas the half-sandwich excimers are observed for staggered or half-eclipsed conformers. Note that the intensity of the shoulder band increases with n; this trend indicates that the bulky substituent in the chiral repeat unit preferentially disfavors the formation of the fully eclipsed conformers of full-sandwich excimer fluorescence.
The presence of half-sandwich excimers points to an axially chiral alignment of carbazoles along the polymer main chain. To identify their helical sense, electronic circular dichroism (ECD) spectra of the JYn polymer films were obtained. Fig. 3c displays the normalized ECD curves, which were constructed with the Kuhn dissymmetry factor (gabs). gabs is defined as gabs = 2(εLCPL − εRCPL)/(εLCPL + εRCPL), where εLCPL and εRCPL are the molar absorbance for the left- and right-handed circularly polarized light, respectively.78,79 Bisignate signals emerge for JYn for n > 2. Negative and positive couplets are evident in the crossing points of the abscissa at 263 and 290 nm, respectively. The silence of the bisignate couplets for JY1–2 indicates insufficient induction of helical sense. Comparison of these spectra with the UV-vis absorption spectra reveals that the latter couplet corresponds to the long-axis transition of carbazole. This observation indicates that there is asymmetric coupling between the long-axis-transition excitons of the vicinal carbazoles along the polymer main chain. This conclusion is supported by the absence of ECD signaling at 290 nm for a highly concentrated solution of the model compound of M* (i.e., 2; ESI, Fig. S2a†). Interchain asymmetric exciton coupling is also excluded because gabs is virtually constant for JY100 solutions with various concentrations (0.017–0.17 mg mL−1 in 1,2-dichloroethane; ESI, Fig. S2b†).
The conformation of the carbazoles can be assigned to the (P)-helical sense, according to Nakanishi and Harada's exciton chirality rule.80,81 The gabs values of the couplets increase with n, with the largest |gabs| value of 3.1 × 10−4 found for JY100 at 255 nm. Note that this proportionality also parallels that of the relative fluorescence intensity of the half-sandwich excimer. This parallel suggests that both the ECD and the excimer fluorescence originate from the helical carbazole conformers. This notion is supported by a decrease in the ECD signals of JY100 (0.028 mg mL−1 in 1,2-dichloroethane) at elevated temperatures (ESI, Fig. S3†). The temperature dependence is fully reversible, which points to its conformational nature.
Finally, we determined whether chirality transfer occurs along the polymer main chain. To test this hypothesis, we plotted gabs (λ = 304 nm) normalized to that of JY100 as a function of the fraction of M* (i.e., [M*]/([M*] + [M])). As shown in Fig. 3d, a convex deviation from linearity is found: the normalized gabs is located above the linear relationship to the fraction of M*. This nonlinearity indicates the presence of chirality interactions between M* and the achiral monomer 5 during the polymerization. This chirality interaction is of an intrachain origin,82,83 because N-ethylcarbazole films doped with 2 (0.5–15 wt%) do not show any ECD signaling (ESI, Fig. S4†). Such chirality interaction is expected to involve transfer of the point chirality in the carbazole pendant to the polymer main chain under the chiral random field exerted by (R)-α-phenylisopropyl, followed by propagation of the chirality along the main chain during the course of chain growth (see Fig. 3d). The first chirality transfer is governed by the chiral bias energy (ΔGh), and the second chirality transfer must overcome the chirality reversal energy (ΔGr). Adopting Selinger's solutions to the Ising model,84–86 we fitted the normalized gabs curve to the following equation:
 | (1) |
In eqn (1), R is the gas constant, T is 300 K, and N is the degree of polymerization. Note that N ranges from 39 for JY100 to 59 for JY0, but its variation does not alter the fit results. Our nonlinear least-squares fit returned ΔGh = 0.99 kcal mol−1 and ΔGr = 0.78 kcal mol−1. Although other combinations of ΔGh and ΔGr are possible, this ΔGh value is similar to that of copolymers based on poly(isocyanate) with achiral and chiral alkyl branches, whereas ΔGr is one order of magnitude smaller than that of poly(isocyanate).87 The lower driving force for forming a chiral domain (i.e., small ΔGr) in JYn is expected considering that free-radical polymerization usually exhibits low stereoselectivity. Table 2 lists the photophysical and chiroptical parameters of JYn.
| λ abs (nm) | λ em (nm) | g abs (10−4, λobsd = 304 nm) | τ obs (ns) | PLQYf | k r (107 s−1) | k nr (107 s−1) | |
|---|---|---|---|---|---|---|---|
| a Neat films of JYn spin-cast onto quartz substrates. b Absorption peak wavelengths. c Emission peak wavelengths. λex = 339 nm. sh = shoulder. d Kuhn dissymmetry factor determined at a wavelength of 304 nm. e Photoluminescence lifetimes determined at λem by using the time-correlated single-photon-counting techniques after picosecond pulsed laser excitation at 377 nm (temporal resolution = 0.025 ns). f Absolute PLQY values determined with an integrating sphere. These determinations were performed in triplicate with fresh samples. g The radiative decay rate (kr) was calculated with the equation kr = PLQY/τobs. h The non-radiative decay rate (knr) was calculated with the expression (1 − PLQY)/τobs. | |||||||
| JY0 | 308, 353 | 418 | 0.063 | 15 | 0.18 (±0.046) | 1.2 | 5.5 |
| JY1 | 308, 353 | 418 | 0.13 | 15 | 0.12 (±0.004) | 0.73 | 5.9 |
| JY2 | 308, 353 | 420 | 0.056 | 15 | 0.16 (±0.027) | 1.1 | 5.6 |
| JY10 | 308, 353 | 420 | 0.29 | 9.1 | 0.23 (±0.029) | 2.5 | 8.5 |
| JY40 | 308, 353 | 384 (sh), 417 | 0.92 | 12 | 0.20 (±0.025) | 1.7 | 6.7 |
| JY70 | 308, 352 | 382 (sh), 417 | 1.5 | 10 | 0.26 (±0.015) | 2.6 | 7.4 |
| JY100 | 308, 352 | 380 (sh), 417 | 2.0 | 15 | 0.26 (±0.008) | 1.7 | 4.9 |
Diastereomeric interactions between the polymer hosts and the phosphorescent dopant
To assess the utility of our polymer hosts, we synthesized an enantiomeric pair of phosphorescent Pt(II) complex dopants, (R)- and (S)-Pt (Scheme 2). The molecular structure of the dopants is based on a square planar complex of Pt(II), where the ligands with C^N and N^O motifs are coordinated with Pt. The N^O ligand contains axially chiral 1,1′-bi-2-naphthol, which endows the complexes with chiroptical properties. (R)- and (S)-Pt were prepared with a two-step synthesis. A Pd(0)-catalyzed Stille reaction between homochiral 3,3′-dibromo-1,1′-bi-2-naphthol and 2-(tributylstannyl)pyridine furnished a monopyridinylated product. The reaction did not proceed further to a bipyridinylated product, even in the presence of excess amounts of the catalyst and 2-(tributylstannyl)pyridine. Finally, the monopyridinylated ligand was reacted with a μ-dichloro dinuclear Pt(II) precursor in the presence of potassium carbonate to form (R)- or (S)-Pt. The presence of residual bromo and hydroxy groups in the Pt(II) complexes is deleterious to the performance of devices. The synthetic details and spectroscopic identification data are summarized in ESI.† The photophysical parameters of the Pt dopants are listed in Table 3. | ||
| Scheme 2 Syntheses of the phosphorescent chiral dopants, (R)- and (S)-Pt. | ||
| λ abs (nm) | λ em (nm) | g abs (10−3, λobs = 372 nm) | τ obs (μs) | PLQYf | k r (104 s−1) | k nr (105 s−1) | |
|---|---|---|---|---|---|---|---|
| a 10 μM in Ar-saturated 1,2-dichloroethane. b Absorption peak wavelengths. c Emission peak wavelengths. λex = 400 nm. d Kuhn dissymmetry factor determined at a wavelength of 372 nm. See ESI, Fig. S7 for the spectra. e Photoluminescence lifetimes of 100 μM Pt in Ar-saturated 1,2-dichloroethane determined with nonlinear least-squares fits of photoluminescence traces recorded at a wavelength of 590 nm employing time-correlated single-photon-counting techniques after picosecond pulsed laser excitation at 377 nm (temporal resolution = 10 ns). f Photoluminescence quantum yield determined relative to the 9,10-diphenylanthracene standard (PLQY = 1.00, toluene).88 g The radiative decay rate (kr) was calculated with the equation kr = PLQY/τobs. h The non-radiative decay rate (knr) was calculated with the expression (1 − PLQY)/τobs. | |||||||
| (R)-Pt | 262, 320, 400 | 590 | −2.1 | 21 | 0.25 | 1.2 | 3.6 |
| (S)-Pt | 262, 320, 400 | 590 | 2.3 | 21 | 0.24 | 1.1 | 3.6 |
We now investigated the intermolecular chirality interactions in the binary mixtures of these enantiomeric dopants and chiral polymer hosts. The maximum chiroptical properties are found for JY100. We thus chose JY100 and its achiral control polymer, JY0, for the rest of our studies. We aimed to determine whether the chiroptical behaviors of the Pt(II) dopants are influenced by the local chiral environments provided by the JY100 host. As shown in Fig. 4, the UV-vis absorption spectra of the neat films of JY0 and JY100 are barely affected by the presence of 10 wt% (R)- or (S)-Pt, indicating there is little or no formation of ground-state species between the host and dopant. As expected, the ECD signals of the JY0 films doped with (R)- and (S)-Pt (i.e., (R)-Pt:JY0 and (S)-Pt:JY0, respectively) are mirror images (Fig. 4a). Two major bands with peaks at 316 nm (|gabs| = 1.37 × 10−4) and 372 nm (|gabs| = 2.2 × 10−4) are evident. The former and the latter overlap with the ligand-centered (LC) transition bands and the metal-to-ligand charge-transfer (MLCT) transition bands of the Pt(II) dopants, respectively. The magnitudes of gabs of the MLCT transition bands are not sensitive to the configuration of the Pt dopants, which implies that the observed chiroptical properties are inherent to the dopants.
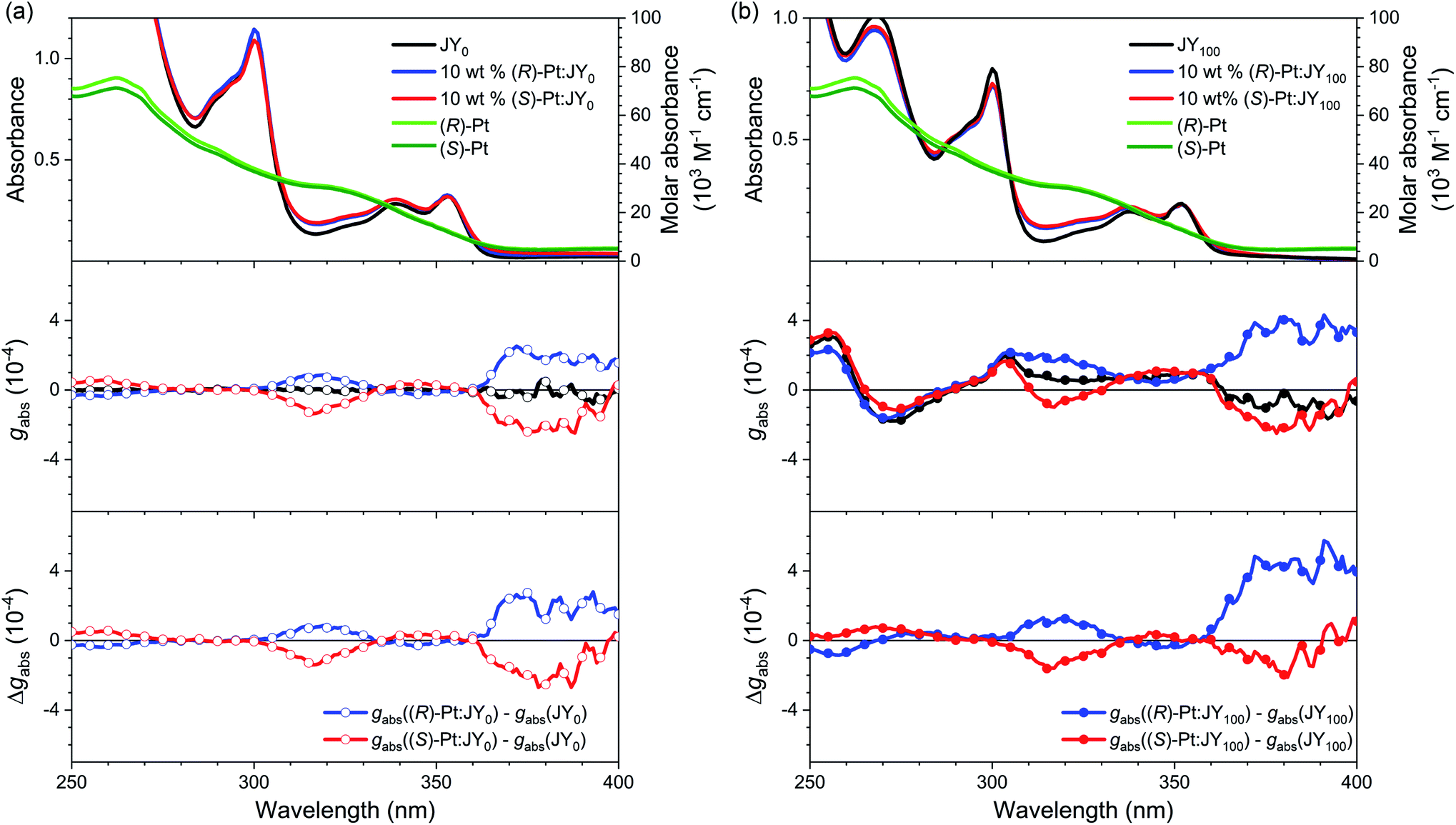 | ||
| Fig. 4 (a) UV-vis absorption (top), gabs (middle), and the gabs difference spectra (bottom; Δgabs, gabs(10 wt% Pt:JY0) − gabs(JY0)) for the JY0 films (quartz substrates) in the absence and presence of 10 wt% (R)-Pt or 10 wt% (S)-Pt. (b) UV-vis absorption (top), gabs (middle), and the gabs difference spectra (bottom; Δgabs, gabs(10 wt% Pt:JY100) − gabs(JY100)) for the JY100 films (quartz substrates) in the absence and presence of 10 wt% (R)-Pt or 10 wt% (S)-Pt. | ||
The ECD signals of the Pt:JY100 films are dominated by those of JY100, but the contributions from LC and MLCT regions of the Pt dopants are evident. As shown in Fig. 4b, the gabs profiles of (R)- and (S)-Pt in JY100 deviate from the mirror-image relationship found for JY100. Subtracting the gabs curve of the JY100 neat film from those of the doped films (i.e., Δgabs) provides more decisive evidence for dissymmetric ECD signals (see the bottom panel in Fig. 4b). The Δgabs value of (R)-Pt:JY100 is 5.7 × 10−4 at 391 nm which is greater than that of (R)-Pt:JY0 (2.5 × 10−4). On the contrary, an opposite behavior is found for (S)-Pt: the Δgabs value of (S)-Pt:JY100 is 0.50 × 10−4 at 391 nm which is smaller than that for (S)-Pt:JY0 (1.7 × 10−4). This stereoselectivity points to the presence of diastereoselective interactions between the chiral polymer host and the Pt dopants. Chiral repeat units are probably responsible for the diastereomeric effects, because gabs of the MLCT transition band increases with the fraction of chiral repeat units (i.e., M*) in JYn (ESI, Fig. S6†).
The CPL behaviors of the Pt-doped JY films were characterized by using our home-made instrument based on a photoelastic modulator and a lock-in amplifier (see ESI†). Fig. 5 compares the photoluminescence and CPL spectra of JY0 and JY100 doped with 30 wt% (R)- or (S)-Pt. The films produce broad emissions due to the MLCT transition of the Pt dopants; the spectra arising from the solutions are identical (ESI, Fig. S7†). The presence of CPL in the same wavelength range of the photoluminescence spectra indicates that the Pt dopants are responsible for its emergence. The signs of the photoluminescence dissymmetry factor (gPL, gPL = 2(PILCPL − PIRCPL)/(PILCPL + PIRCPL) where PILCPL and PIRCPL are photoluminescence intensities of LCPL and RCPL, respectively) for the (R)-Pt:JY0 and (S)-Pt:JY0 films are opposite, and are identical to those of the MLCT transition bands in the ECD spectra (Fig. 4). These results indicate that common electronic states with different spin configurations (i.e., singlet and triplet MLCT transition state) are involved in the absorption and photoluminescence transitions. A CPL spectrum obtained for a 100 mM solution of (R)-Pt is virtually identical to that of the (R)-Pt:JY0 film (ESI, Fig. S8†). The maximum gPL values of the (R)-Pt:JY100 and (S)-Pt:JY100 films are 1.0 × 10−3 (λem = 540 nm) and −0.94 × 10−3 (λem = 545 nm), respectively, which are slightly larger than the values of the corresponding films with the JY0 host ((R)-Pt:JY0, 0.65 × 10−3; (S)-Pt:JY0, −0.73 × 10−3). There is possibly a diastereoselective effect in the gPL spectra (see the bottom panel of Fig. 5). The differences between the |gPL| values of the (R)-Pt:JY films (i.e., the blue solid and empty circles) are greater than the differences between those of (S)-Pt:JY films (i.e., the red solid and empty circles) around 550 nm, but the magnitudes of the differences are low.
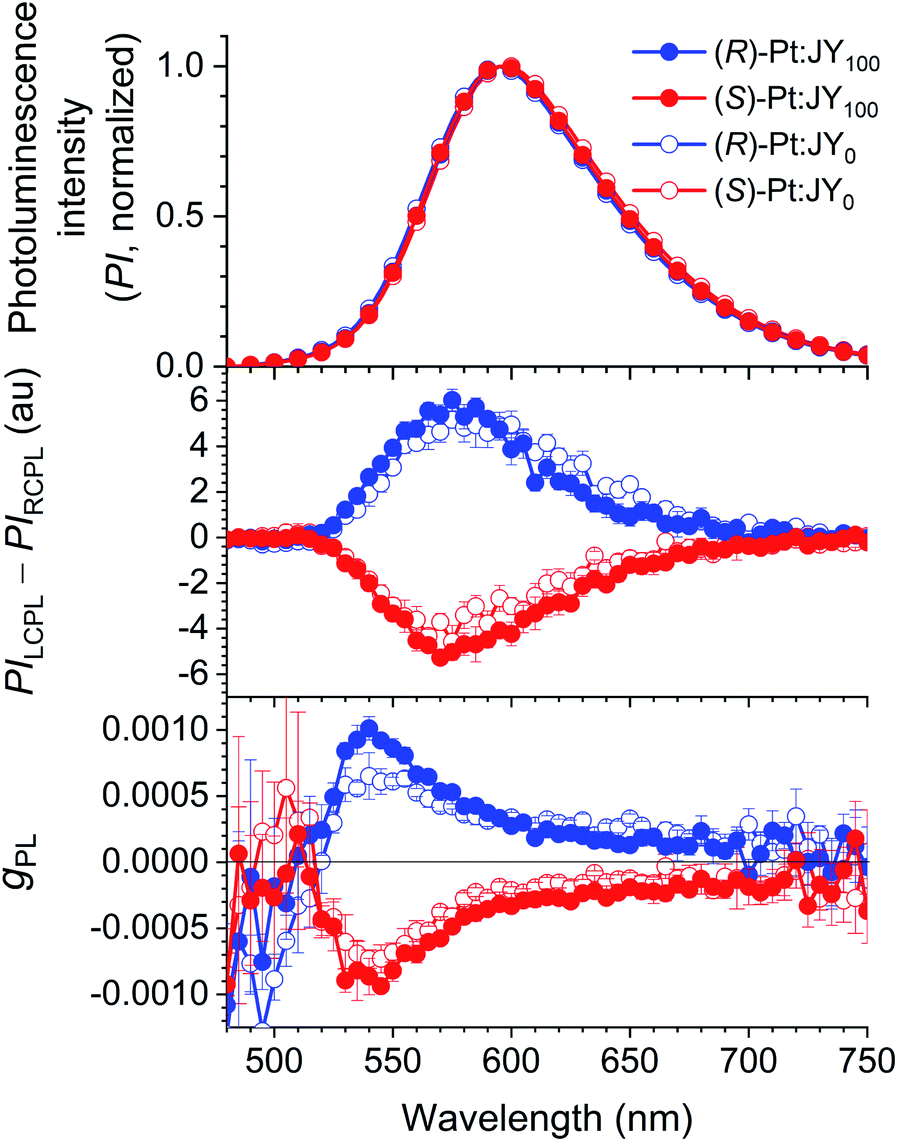 | ||
| Fig. 5 Circularly polarized luminescence of JY films doped with 30 wt% Pt dopants. Top, Photoluminescence spectra; middle, circularly polarized luminescence spectra; bottom, the variations in gPL with wavelength. | ||
One of key requirements for the host is the ability to trap excitons within a dopant. The formation of dopant excitons occurs through two pathways: one is Langevin recombination on a host, followed by exciton (energy) transfer to a dopant; the other is Shockley–Read–Hall recombination on a dopant. The exergonic energy transfer from the JY hosts to Pt dopants was directly monitored by using steady-state and transient photoluminescence techniques. A neat film of JY0 was found to exhibit excimer emission of carbazoles at 428 nm upon photoexcitation at 339 nm. The host emission decreases in intensity with increases in the concentration of (R)-Pt (0–10 wt%), with the emergence of sensitized dopant emission at a peak wavelength of 586 nm (Fig. 6a). This decrease-and-increase profile indicates the presence of an active energy donor/energy acceptor pair. The titration isotherms of I0/I, where I0 and I are the host emission intensities (λem = 420 nm) in the absence and presence of the dopant, respectively, follow rise profiles due to energy transfer (Fig. 6c). Qualitatively similar, but faster rises are observed for JY100 films doped with various concentrations of (R)-Pt (0–10 wt%). This result indicates that the energy transfer from JY100 to the Pt dopants occurs more rapidly than that from JY0. The results for JYn films with (S)-Pt are shown in ESI, Fig. S9.†
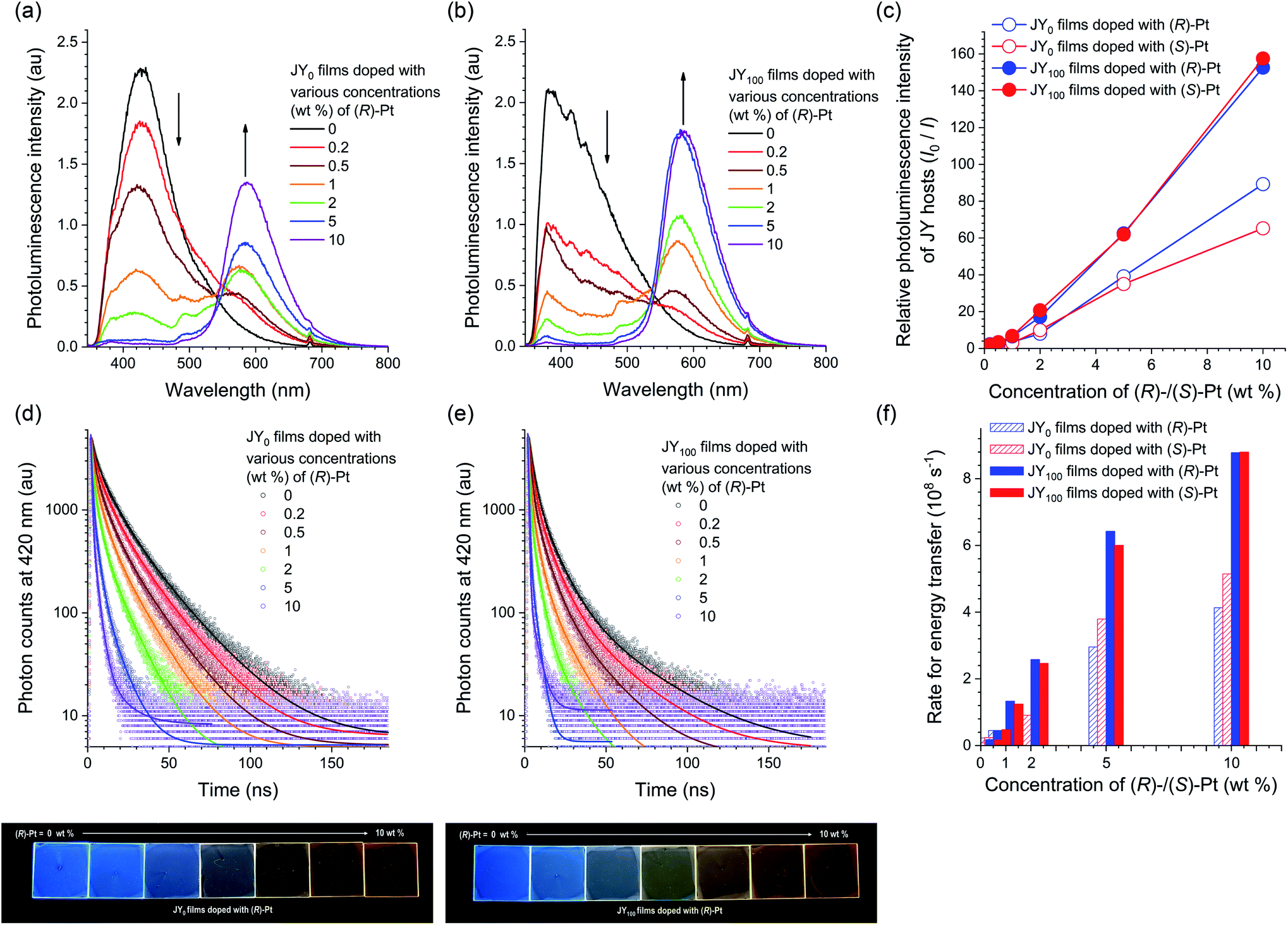 | ||
| Fig. 6 (a and b) Photoluminescence spectra (λex = 339 nm) for JY0 (a) and JY100 (b) films doped with various concentrations (0–10 wt%) of (R)-Pt. The spectra of the films doped with (S)-Pt are shown in ESI, Fig. S9.† (c) Ratios of the photoluminescence intensities of the JYn hosts in the absence over the presence of increased concentrations of the Pt dopants (I0/I). (d and e) Photoluminescence decay traces (λobs = 420 nm) of JY0 (d) and JY100 (e) films doped with various concentrations (0–10 wt%) of (R)-Pt obtained after picosecond pulsed laser photoexcitation at 377 nm (temporal resolution = 25 ps). The solid curves are nonlinear least-squares fits to a biexponential decay model. The decay traces of the films doped with (S)-Pt are shown in ESI, Fig. S9.† The decay traces of dopant emissions recorded at a wavelength of 590 nm are shown in ESI, Fig. S10.† The bottom photographs show the photoluminescence emissions of JY0 (left) and JY100 (right) films doped with various concentrations of (R)-Pt visualized under photoirradiation at a wavelength of 365 nm. (f) Rates of energy transfer (kET) from the JY hosts to the Pt dopants. | ||
The faster energy transfer in the case of JY100 was also demonstrated by using transient photoluminescence experiments. The decay traces of the host emission were monitored at a wavelength of 420 nm after picosecond pulsed laser excitation at 377 nm. As shown in Fig. 6d and e, the transient emission signals decay following a biexponential decay model, which is probably due to film inhomogeneity (vide infra). The average photoluminescence lifetime (τ) of a neat film of JY0 is 10 ns, which is typical of excimer fluorescence. τ decreases with increasing the concentration of the Pt dopant due to energy transfer. The apparent energy transfer rate (kET), which was calculated with the relationship kET = 1/τ − 1/τ0 where τ and τ0 are photoluminescence lifetimes in the presence and absence of the dopant, respectively, increases with the dopant concentration. It was found that kET is greater for JY100 in the tested concentration range (i.e., 0.2–10 wt%). A possible explanation for these different energy transfer behaviors could invoke the photophysical and structural properties of the JY hosts. Actually, the Förster radii of the JY0 and JY100 films calculated based on a refractive index of 1.047 and the spectral overlap integrals with the Pt dopants are 25 and 27 Å, respectively. The longer Förster radius of JY100 is probably due to its greater PLQY (Table 2), and is consistent with its faster energy transfer behavior.
In addition, our microscopic experiments suggest that the phase homogeneity of JY100 might also contribute to its superior energy transfer behavior. Fig. 7 compares confocal laser-scanning fluorescence micrographs of JY0 and JY100 films doped with identical concentrations (10, 30, and 50 wt%) of (S)-Pt. The bright puncta correspond to aggregates of (S)-Pt. It can be seen that the JY0 films contain more dopant aggregates than the JY100 films. The poor phase homogeneity in JY0 is also evident in the transmission electron micrographs (note the black dots in the inset images in Fig. 7c and d). The energy dispersive X-ray spectra confirm that there is Pt enrichment in the aggregates. In contrast, phase separation is effectively suppressed in the JY100 films. The different phase homogeneity is likely due to the differed free volumes of polymers. The bulky (R)-α-phenylisopropyl moiety retards the packing of polymer chains in JY100, providing more rooms for the dissolution of the Pt dopants. The better phase homogeneity of JY100 permits the molecular level dispersion of the dopants within the Förster radius of the host. These results indicate that the faster energy transfer of JY100 originates from both a longer Förster radius and improved compatibility with the Pt dopants. Finally, the energy transfer rates to (R)- and (S)-Pt are the same in JY100. The lack of chirality-discriminated energy transfer is expected, as |gPL| values of the hosts (<10−3) and of the |gabs| values of the dopants (∼10−4) are very small.
 | ||
| Fig. 7 (a and b) Confocal laser-scanning fluorescence micrographs (λex = 405 nm, λobs = 500–700 nm) for JY0 (a) and JY100 (b) films (quartz substrates) doped with various concentrations (10, 30 and 50 wt%) of (S)-Pt: top panels, fluorescence images; bottom panels, bright-field images. Scale bar = 100 μm. (c and d) Energy dispersive X-ray spectra and the average atomic percentage compositions of JY0 (c) and JY100 (d) films doped with 50 wt% (S)-Pt (200 mesh carbon films on copper). The atomic percentage compositions of Pt were quantified for Cliff Lorimer thin ratio sections: 1.4% Pt for (S)-Pt:JY0 films; 1.5% Pt for (S)-Pt:JY100 films. The insets show TEM images of the JY films doped with 50 wt% (S)-Pt. Scale bar = 1 μm. | ||
The Shockley–Read–Hall recombination has the following electrochemical requirements: (i) the oxidation potential (Eox) of the host > Eox of the dopant, and (ii) the reduction potential (Ered) of the host < Ered of the dopant. The redox potentials for the JYn hosts and the Pt dopants were determined with cyclic voltammetry (CV) and differential pulse voltammetry (DPV). Fig. 8 displays the voltammograms of Ar-saturated THF containing JY0, JY100, and (R)- and (S)-Pt. JY0 and JY100 exhibit irreversible oxidation at 1.25 and 1.22 V vs. standard calomel electrode (SCE), respectively. These peaks are close to Eox of poly(N-vinylcarbazole) (1.39 V vs. SCE; ESI, Fig. S11†), which indicates that carbazole is responsible for the oxidation. The Eox value of (R)- and (S)-Pt is 0.88 V vs. SCE, which is more cathodic than the Eox values of the JY hosts. Therefore, hole trapping within the Pt dopants is thermodynamically favored with positive driving forces of 0.37 and 0.34 eV for JY0 and JY100, respectively. The Ered values calculated by subtracting the optical bandgap energies (3.43 eV for the JY hosts and 2.48 eV for the Pt dopants) from the Eox values are −2.18 V (JY0), −2.21 V (JY100), and −1.60 V ((R)- and (S)-Pt) vs. SCE. The Ered value of the Pt complexes is more anodic than those of the JY hosts, which suggests that electron trapping by the dopants will be effective with thermodynamic driving forces of 0.58 eV (JY0) and 0.61 eV (JY100). These electrochemical results indicate that the JYn polymers will effectively confine charge carriers within the Pt dopants.
 | ||
| Fig. 8 Cyclic (solid curves) and differential pulse (dotted lines) voltammograms of Ar-saturated THF solutions of JY100 (red, 0.63 mg mL−1), JY0 (blue, 0.43 mg mL−1), and (R)- (brown) and (S)-Pt (green, 2 mM). 0.10 M Bu4NPF6 was used as the supporting electrolyte. A Pt disk and a Pt wire served as the working and counter electrodes, respectively. A Ag/AgNO3 pseudo reference electrode was used. Scan rates: 0.1 V s−1 (CV) and 0.004 V s−1 (DPV). The grey dotted line corresponds to blank signals (i.e., Ar-saturated THF containing 0.10 M Bu4NPF6). | ||
Circularly polarized electroluminescence
In the final stage of our study, we fabricated PLEDs with the following emitting layers (EMLs): (R)-Pt:JY0, (S)-Pt:JY0, (R)-Pt:JY100, and (S)-Pt:JY100. Fig. 9a depicts the energy-level diagram of our PLEDs with the configuration glass/ITO/50 nm GraHIL/50 nm EML/50 nm TPBi/1 nm LiF/100 nm Al (TPBi, 1,3,5-tris(N-phenylbenzimidazole-2-yl)benzene). To facilitate hole injection into the polymer hosts with deep levels of HOMO (the highest occupied molecular orbital, 6.0 eV), a conducting polymer hole injection layer denoted GraHIL, which is composed of poly(3,4-ethylenedioxythiophene):poly (styrenesulfonate) (PEDOT:PSS) and tetrafluoroethylene-perfluoro-3,6-dioxa-4-methyl-7-octenesulphonic acid, was used.89 The dopant concentration in the EMLs was chosen to be 30 wt% to maximize the dopant emission (ESI, Fig. S12†). Fig. 9c and d show the current density–voltage–luminance (J–V–L) and external quantum efficiency (EQE)–J characteristics of the PLEDs, respectively. The values of V at J = 5 mA cm−2 range from 11.4 to 12.0 V, which suggests that, even with the use of GraHIL, hole injection into JY0 and JY100 is somewhat inefficient. Nevertheless, all devices exhibit normal electroluminescence operation reliable for device characterization: the electroluminescence spectra recorded at J = 1.6 mA cm−2 consist of dopant emission only (Fig. 9b), and are almost identical to one another and the photoluminescence spectra of the corresponding films (Fig. 5). The EQE–J curves are typical of phosphorescent PLEDs, although the peak EQE values are much smaller than those of state-of-the-art devices (Fig. 9d).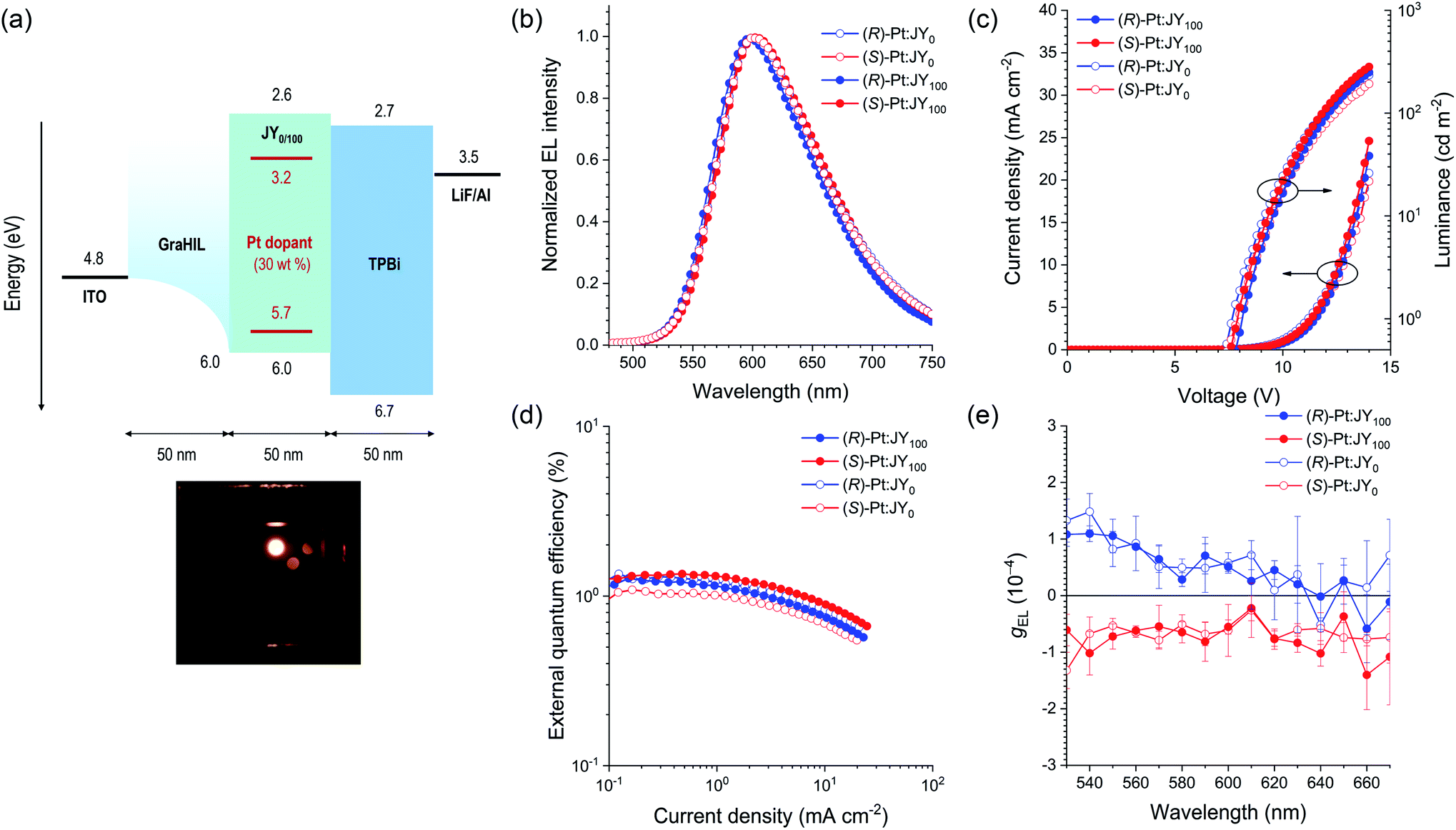 | ||
| Fig. 9 (a) Energy-level diagram and device structure for the PLEDs with (R)- or (S)-Pt:JYn emitting layers. The positions of the energy levels of the polymer hosts and the Pt dopants were determined with cyclic and differential pulse voltammetry, and the positions of the other levels have previously been reported.89–91 The bottom photo shows the electroluminescence of the device. (b) Electroluminescence spectra recorded at a current density of 1.6 mA cm−2. (c) Current density–voltage–luminance and (d) external quantum efficiency–current density characteristics of the PLEDs. (e) Spectra of gEL recorded at a current density of 2.2 mA cm−2 averaged over 20 measurements. The error bars represent standard deviations. | ||
g EL was measured as functions of the emission wavelength (Fig. 9e). For each device, the sign of gEL is the same as that of gPL over the entire range of emission wavelengths. The magnitude of gEL, however, is smaller than that of gPL by a factor of ca. 10. This difference is probably due to the presence of the metal electrodes in the PLEDs; CPL emitted toward the metal electrode reverses its handedness upon reflection at the metal surface,21 and thus interferes with the CPL emitted toward the glass substrate with the opposite handedness. This hypothesis is consistent with our results for the EMLs deposited on quartz substrates, which shows that the signs of gPL measured on the film and substrate sides are the same (ESI, Fig. S13†). Although the (R)-Pt:JY100 film has lager gPL values than the (R)-Pt:JY0 film in the spectral region λem < 590 nm, this enhancement is not observed for gEL; the gEL values of the PLEDs with (R)-Pt:JY100 and (R)-Pt:JY0 EMLs are similar. The lack of enhancement in gEL is possibly due to the multilayer structure of the PLEDs; the presence of the metal electrode, in particular, provides an optical environment that is different from that of the samples used in the photoluminescence studies (i.e., the EMLs on quartz substrates). It is anticipated that careful optical modeling of the PLEDs will enable diastereoselective enhancement in the CP electroluminescence.
Conclusions
We have developed and evaluated chiral polymer hosts for use in CP PLEDs. The polymers were synthesized through free radical polymerization of 3-vinylcarbazole. The vinyl monomer was modified to contain either the chiral (R)-α-phenylisopropyl moiety or the achiral ethyl group. The homopolymers and copolymers of the chiral and achiral monomers were structurally, thermally, and spectroscopically analyzed. Our analyses demonstrate that transfer of chirality from the chiral moiety in the monomer to the polymer main chain occurs during chain growth. In particular, the carbazole pendants form half-sandwich conformers with a mutually (P)-helical sense. This helical sense is not affected by variations in temperature or the concentrations of the polymer solutions, which indicates that the helical half-sandwich conformers are of intrachain origin. To evaluate the performances of the hosts, a novel enantiomeric pair of phosphorescent dopants was synthesized. We found gabs increased for the diastereomeric mixture of the (R)-dopant and the (P)-helical polymer, whereas gabs decreased for the diastereomeric mixture of the (S)-dopant and the (P)-helical polymer. Similar enhancements in gPL are also observed. The chiral polymer host exhibits faster energy transfer to the dopants than the achiral polymer host, due to its longer Förster radius and better compatibility with the dopants. However, the energy transfer from the polymer host does not depend on the chirality of materials. This result indicates that the contribution of the chirality-controlled excited-state interactions between the host and dopant is negligible. The PLED based on the (P)-helical polymer and the (R)-dopant was found to exhibit a gEL value of 1.09 × 10−4 (λem = 540 nm) and an EQE of 1.2%. This study has demonstrated that selection of the chiral host enables the tuning of the chiroptical properties of the emitting layers in electroluminescence devices, although its effects remain marginal. Our synthetic strategy provides particular benefits over previous approaches that rely on phase separation and insulating chiral additives.Author contributions
J. H. designed and performed most of the experiments, analyzed the data, and wrote the manuscript. S. K. obtained the CPL spectra, and fabricated and tested the devices. G. P. synthesized the Pt dopants. Y. L. and H. K. assisted the CPL measurements and the device experiments. S. K. synthesized GraHIL. T.-W. L. supervised the work at Seoul National University. C. K. and Y. Y. coordinated all of the experiments and analyses, and co-wrote the manuscript. All authors contributed to discussion on the study, and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a grant from the Samsung Research Funding Center for Future Technology (SRFC-MA1602-03).Notes and references
- M. Schadt, Annu. Rev. Mater. Sci., 1997, 27, 305–379 CrossRef CAS.
- T. Kim, S. Kim, M.-J. Gim and S.-W. Choi, J. Korean Phys. Soc., 2013, 62, 713–717 CrossRef CAS.
- G. Muller, Dalton Trans., 2009, 9692–9707 RSC.
- J. Yuasa, T. Ohno, H. Tsumatori, R. Shiba, H. Kamikubo, M. Kataoka, Y. Hasegawa and T. Kawai, Chem. Commun., 2013, 49, 4604–4606 RSC.
- H. Koike, K. Nozaki and M. Iwamura, Chem.–Asian J., 2020, 15, 85–90 CrossRef CAS PubMed.
- C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925–937 CrossRef CAS PubMed.
- R. Naaman and D. H. Waldeck, J. Phys. Chem. Lett., 2012, 3, 2178–2187 CrossRef CAS PubMed.
- R. Naaman, Y. Paltiel and D. H. Waldeck, Nat. Rev. Chem., 2019, 3, 250–260 CrossRef CAS.
- R. Carr, N. H. Evans and D. Parker, Chem. Soc. Rev., 2012, 41, 7673–7686 RSC.
- S. Furumi, Chem. Rec., 2010, 10, 394–408 CAS.
- F. Song, G. Wei, X. Jiang, F. Li, C. Zhu and Y. Cheng, Chem. Commun., 2013, 49, 5772–5774 RSC.
- M. Seitz, E. G. Moore, A. J. Ingram, G. Muller and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 15468–15470 CrossRef CAS PubMed.
- C. Wagenknecht, C.-M. Li, A. Reingruber, X.-H. Bao, A. Goebel, Y.-A. Chen, Q. Zhang, K. Chen and J.-W. Pan, Nat. Photonics, 2010, 4, 549–552 CrossRef CAS.
- J. F. Sherson, H. Krauter, R. K. Olsson, B. Julsgaard, K. Hammerer, I. Cirac and E. S. Polzik, Nature, 2006, 443, 557–560 CrossRef CAS PubMed.
- H. Li, H. Li, W. Wang, Y. Tao, S. Wang, Q. Yang, Y. Jiang, C. Zheng, W. Huang and R. Chen, Angew. Chem., Int. Ed., 2020, 59, 4756–4762 CrossRef CAS PubMed.
- R. J. Cave, Science, 2009, 323, 1435–1436 CrossRef CAS PubMed.
- X.-F. Luo, H.-B. Han, Z.-P. Yan, Z.-G. Wu, J. Su, J.-W. Zou, Z.-Q. Zhu, Y.-X. Zheng and J.-L. Zuo, ACS Appl. Mater. Interfaces, 2020, 12, 23172–23180 CrossRef CAS PubMed.
- Y. Wang, Y. Zhang, W. Hu, Y. Quan, Y. Li and Y. Cheng, ACS Appl. Mater. Interfaces, 2019, 11, 26165–26173 CrossRef CAS PubMed.
- F. Song, Z. Xu, Q. Zhang, Z. Zhao, H. Zhang, W. Zhao, Z. Qiu, C. Qi, H. Zhang, H. H. Y. Sung, I. D. Williams, J. W. Y. Lam, Z. Zhao, A. Qin, D. Ma and B. Z. Tang, Adv. Funct. Mater., 2018, 28, 1800051 CrossRef.
- F. Zinna, M. Pasini, F. Galeotti, C. Botta, L. Di Bari and U. Giovanella, Adv. Funct. Mater., 2017, 27, 1603719 CrossRef.
- F. Zinna, U. Giovanella and L. Di Bari, Adv. Mater., 2015, 27, 1791–1795 CrossRef CAS PubMed.
- M. Oda, H.-G. Nothofer, G. Lieser, U. Scherf, S. C. J. Meskers and D. Neher, Adv. Mater., 2000, 12, 362–365 CrossRef CAS.
- Z. -G. Wu, H. -B. Han, Z. -P. Yan, X. -F. Luo, Y. Wang, Y. -X. Zheng, J. -L. Zuo and Y. Pan, Adv. Mater., 2019, 31, 1900524 CrossRef PubMed.
- Y. Yang, R. C. da Costa, D.-M. Smilgies, A. J. Campbell and M. J. Fuchter, Adv. Mater., 2013, 25, 2624–2628 CrossRef CAS PubMed.
- J. Han, S. Guo, J. Wang, L. Wei, Y. Zhuang, S. Liu, Q. Zhao, X. Zhang and W. Huang, Adv. Opt. Mater., 2017, 5, 1700359 CrossRef.
- M. Li, S.-H. Li, D. Zhang, M. Cai, L. Duan, M.-K. Fung and C.-F. Chen, Angew. Chem., Int. Ed., 2018, 57, 2889–2893 CrossRef CAS PubMed.
- M. Li, Y.-F. Wang, D. Zhang, L. Duan and C.-F. Chen, Angew. Chem., Int. Ed., 2020, 59, 3500–3504 CrossRef CAS PubMed.
- Z.-P. Yan, X.-F. Luo, W.-Q. Liu, Z.-G. Wu, X. Liang, K. Liao, Y. Wang, Y.-X. Zheng, L. Zhou, J.-L. Zuo, Y. Pan and H. Zhang, Chem.–Eur. J., 2019, 25, 5672–5676 CrossRef CAS PubMed.
- J. R. Brandt, X. Wang, Y. Yang, A. J. Campbell and M. J. Fuchter, J. Am. Chem. Soc., 2016, 138, 9743–9746 CrossRef CAS PubMed.
- E. Peeters, M. P. T. Christiaans, R. A. J. Janssen, H. F. M. Schoo, H. P. J. M. Dekkers and E. W. Meijer, J. Am. Chem. Soc., 1997, 119, 9909–9910 CrossRef CAS.
- Y. Zhang, X. Zhang, H. Zhang, Y. Xiao, Y. Quan, S. Ye and Y. Cheng, J. Phys. Chem. C, 2019, 123, 24746–24753 CrossRef CAS.
- T.-Y. Li, Y.-M. Jing, X. Liu, Y. Zhao, L. Shi, Z. Tang, Y.-X. Zheng and J.-L. Zuo, Sci. Rep., 2015, 5, 14912 CrossRef CAS PubMed.
- D.-W. Zhang, M. Li and C.-F. Chen, Chem. Soc. Rev., 2020, 49, 1331–1343 RSC.
- D.-M. Lee, J.-W. Song, Y.-J. Lee, C.-J. Yu and J.-H. Kim, Adv. Mater., 2017, 29, 1700907 CrossRef PubMed.
- H. Sakai, S. Shinto, J. Kumar, Y. Araki, T. Sakanoue, T. Takenobu, T. Wada, T. Kawai and T. Hasobe, J. Phys. Chem. C, 2015, 119, 13937–13947 CrossRef CAS.
- L. Wan, J. Wade, X. Shi, S. Xu, M. J. Fuchter and A. J. Campbell, ACS Appl. Mater. Interfaces, 2020, 12, 39471–39478 CrossRef CAS PubMed.
- G. Fu, Y. He, W. Li, B. Wang, X. Lu, H. He and W.-Y. Wong, J. Mater. Chem. C, 2019, 7, 13743–13747 RSC.
- M. Oda, H.-G. Nothofer, U. Scherf, V. Sunjic, D. Richter, W. Regenstein and D. Neher, Macromolecules, 2002, 35, 6792–6798 CrossRef CAS.
- X. H. Yang, D. Neher, C. Spitz, E. Zojer, J. L. Bredas, R. Guntner and U. Scherf, J. Chem. Phys., 2003, 119, 6832–6839 CrossRef CAS.
- M. R. Craig, P. Jonkheijm, S. C. J. Meskers, A. P. H. J. Schenning and E. W. Meijer, Adv. Mater., 2003, 15, 1435–1438 CrossRef CAS.
- J. Gilot, R. Abbel, G. Lakhwani, E. W. Meijer, A. P. H. J. Schenning and S. C. J. Meskers, Adv. Mater., 2010, 22, E131–E134 CrossRef CAS PubMed.
- G. Lakhwani and S. C. J. Meskers, J. Phys. Chem. Lett., 2011, 2, 1497–1501 CrossRef CAS.
- G. Lakhwani and S. C. J. Meskers, J. Phys. Chem. A, 2012, 116, 1121–1128 CrossRef CAS PubMed.
- K. Watanabe, Y. Koyama, N. Suzuki, M. Fujiki and T. Nakano, Polym. Chem., 2014, 5, 712–717 RSC.
- S. T. Duong and M. Fujiki, Polym. Chem., 2017, 8, 4673–4679 RSC.
- J. Li, X. Peng, C. Huang, Q. Qi, W.-Y. Lai and W. Huang, Polym. Chem., 2018, 9, 5278–5285 RSC.
- A. Satrijo, S. C. J. Meskers and T. M. Swager, J. Am. Chem. Soc., 2006, 128, 9030–9031 CrossRef CAS PubMed.
- J. N. Wilson, W. Steffen, T. G. McKenzie, G. Lieser, M. Oda, D. Neher and U. H. F. Bunz, J. Am. Chem. Soc., 2002, 124, 6830–6831 CrossRef CAS PubMed.
- S. Fukao and M. Fujiki, Macromolecules, 2009, 42, 8062–8067 CrossRef CAS.
- Y. Nakano, F. Ichiyanagi, M. Naito, Y. Yang and M. Fujiki, Chem. Commun., 2012, 48, 6636–6638 RSC.
- K. Watanabe, I. Osaka, S. Yorozuya and K. Akagi, Chem. Mater., 2012, 24, 1011–1024 CrossRef CAS.
- B. M. W. Langeveld-Voss, R. A. J. Janssen, M. P. T. Christiaans, S. C. J. Meskers, H. P. J. M. Dekkers and E. W. Meijer, J. Am. Chem. Soc., 1996, 118, 4908–4909 CrossRef CAS.
- K. Watanabe, T. Sakamoto, M. Taguchi, M. Fujiki and T. Nakano, Chem. Commun., 2011, 47, 10996–10998 RSC.
- D. Di Nuzzo, C. Kulkarni, B. Zhao, E. Smolinsky, F. Tassinari, S. C. J. Meskers, R. Naaman, E. W. Meijer and R. H. Friend, ACS Nano, 2017, 11, 12713–12722 CrossRef CAS PubMed.
- K. Meerholz, B. L. Volodin, Sandalphon, B. Kippelen and N. Peyghambarian, Nature, 1994, 371, 497–500 CrossRef CAS.
- Y. Kawamura, S. Yanagida and S. R. Forrest, J. Appl. Phys., 2002, 92, 87–93 CrossRef CAS.
- E. Chiellini, R. Solaro, O. Colella and A. Ledwith, Eur. Polym. J., 1978, 14, 489–496 CrossRef CAS.
- V. Percec, M. Obata, J. G. Rudick, B. B. De, M. Glodde, T. K. Bera, S. N. Magonov, V. S. K. Balagurusamy and P. A. Heiney, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3509–3533 CrossRef CAS.
- Z.-B. Zhang, M. Motonaga, M. Fujiki and C. E. McKenna, Macromolecules, 2003, 36, 6956–6958 CrossRef CAS.
- L. Angiolini, T. Benelli, L. Giorgini, F. Mauriello and E. Salatelli, Macromol. Chem. Phys., 2006, 207, 1805–1813 CrossRef CAS.
- H. Zhao, F. Sanda and T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 253–261 CrossRef CAS.
- J. Qu, R. Kawasaki, M. Shiotsuki, F. Sanda and T. Masuda, Polymer, 2007, 48, 467–476 CrossRef CAS.
- J. Qu, M. Shiotsuki, F. Sanda and T. Masuda, Macromol. Chem. Phys., 2007, 208, 823–832 CrossRef CAS.
- J. Qu, Y. Suzuki, M. Shiotsuki, F. Sanda and T. Masuda, Polymer, 2007, 48, 4628–4636 CrossRef CAS.
- F. Sanda, R. Kawasaki, M. Shiotsuki and T. Masuda, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4450–4458 CrossRef CAS.
- F. Sanda, R. Kawasaki, M. Shiotsuki, T. Takashima, A. Fujii, M. Ozaki and T. Masuda, Macromol. Chem. Phys., 2007, 208, 765–771 CrossRef CAS.
- L. Angiolini, T. Benelli, L. Giorgini, A. Golemme, F. Mauriello, E. Salatelli and R. Termine, Macromol. Chem. Phys., 2008, 209, 944–956 CrossRef CAS.
- Y. Hu, F. Song, Z. Xu, Y. Tu, H. Zhang, Q. Cheng, J. W. Y. Lam, D. Ma and B. Z. Tang, ACS Appl. Polym. Mater., 2019, 1, 221–229 CrossRef CAS.
- X. Ma, E. Abdel Azeem, X. Liu, Y. Cheng and C. Zhu, J. Mater. Chem. C, 2014, 2, 1076–1084 RSC.
- M. Belletête, M. Bedard, M. Leclerc and G. Durocher, J. Mol. Struct., 2004, 679, 9–15 CrossRef.
- G. E. Johnson, J. Chem. Phys., 1975, 62, 4697–4709 CrossRef CAS.
- Y. K. Moon, J.-S. Huh, S. Kim, S. Kim, S. Y. Yi, J.-J. Kim and Y. You, ACS Appl. Polym. Mater., 2020, 2, 604–617 CrossRef CAS.
- Y.-C. Wei, Z. Zhang, Y.-A. Chen, C.-H. Wu, Z.-Y. Liu, S.-Y. Ho, J.-C. Liu, J.-A. Lin and P.-T. Chou, Commun. Chem., 2019, 2, 10 CrossRef.
- A. J. Musser, S. K. Rajendran, K. Georgiou, L. Gai, R. T. Grant, Z. Shen, M. Cavazzini, A. Ruseckas, G. A. Turnbull, I. D. W. Samuel, J. Clark and D. G. Lidzey, J. Mater. Chem. C, 2017, 5, 8380–8389 RSC.
- A. Wrona-Piotrowicz, J. Zakrzewski, R. Métivier, A. Brosseau, A. Makal and K. Woźniak, RSC Adv., 2014, 4, 56003–56012 RSC.
- T. Kayano, S. Takayasu, K. Sato and K. Shinozaki, Chem.–Eur. J., 2014, 20, 16583–16589 CrossRef CAS.
- B. Ma, P. I. Djurovich and M. E. Thompson, Coord. Chem. Rev., 2005, 249, 1501–1510 CrossRef CAS.
- G. F. Johnson and D. J. Graves, Biochemistry, 1966, 5, 2906–2911 CrossRef CAS PubMed.
- M. Wakabayashi, S. Yokojima, T. Fukaminato, K.-i. Shiino, M. Irie and S. Nakamura, J. Phys. Chem. A, 2014, 118, 5046–5057 CrossRef CAS PubMed.
- N. Harada, S.-M. L. Chen and K. Nakanishi, J. Am. Chem. Soc., 1975, 97, 5345–5352 CrossRef CAS.
- N. Harada and K. Nakanishi, J. Am. Chem. Soc., 1968, 90, 7351–7352 CrossRef CAS.
- M. M. Green, M. P. Reidy, R. D. Johnson, G. Darling, D. J. O'Leary and G. Willson, J. Am. Chem. Soc., 1989, 111, 6452–6454 CrossRef.
- T. Ikai, S. Shimizu, S. Awata and K.-i. Shinohara, Macromolecules, 2018, 51, 2328–2334 CrossRef CAS.
- J. V. Selinger and R. L. B. Selinger, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 1997, 55, 1728–1731 CrossRef CAS.
- M. M. Green, J. -W. Park, T. Sato, A. Teramoto, S. Lifson, R. L. B. Selinger and J. V. Selinger, Angew. Chem., Int. Ed., 1999, 38, 3138–3154 CrossRef.
- V. Jain, K.-S. Cheon, K. Tang, S. Jha and M. M. Green, Isr. J. Chem., 2011, 51, 1067–1074 CrossRef CAS.
- S. K. Jha, K.-S. Cheon, M. M. Green and J. V. Selinger, J. Am. Chem. Soc., 1999, 121, 1665–1673 CrossRef CAS.
- G. Heinrich, S. Schoof and H. Gusten, J. Photochem., 1974, 3, 315–320 CrossRef CAS.
- T. -H. Han, M. R. Choi, S. -H. Woo, S. -Y. Min, C. -L. Lee and T. -W. Lee, Adv. Mater., 2012, 24, 1487–1493 CrossRef CAS PubMed.
- T. D. Anthopoulos, J. P. J. Markham, E. B. Namdas, I. D. W. Samuel, S.-C. Lo and P. L. Burn, Appl. Phys. Lett., 2003, 82, 4824–4826 CrossRef CAS.
- R. Arunchai, T. Sudyoadsuk, N. Prachumrak, S. Namuangruk, V. Promarak, M. Sukwattanasinitt and P. Rashatasakhon, New J. Chem., 2015, 39, 2807–2814 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details; Fig. S1–S34, displaying the photoluminescence decay traces of JYn films, the UV-vis absorption and ECD spectra of 2 and JY100, variable-temperature ECD spectra of JY100, the ECD spectra of JYn films, concentration-dependent ECD spectra of JY100, gabs spectra of the JYn polymers, the gabs spectra of dropcast films of N-ethylcarbazole and 2, steady-state and chiroptical spectra of the Pt dopants, the CPL spectra of a solution and films of (R)-Pt, energy transfer behaviors of JY0 with the Pt dopants, cyclic and differential pulse voltammograms of PVK, electroluminescence spectra of PLEDs, gPL spectra of JYn:Pt films, schematic representation of the CPL measurement system, gEL spectra, and 1H and 13C{1H} NMR spectra. See DOI: 10.1039/d1sc02095a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |